

Abstract
Purkinje cells (PCs) are spontaneously active neurons of the cerebellar cortex that inhibit glutamatergic projection neurons within the deep cerebellar nuclei (DCN) that provide the primary cerebellar output. Brief reductions of PC firing rapidly increase DCN neuron firing. However, prolonged reductions of PC inhibition, as seen in some disease states, certain types of transgenic mice, during optogenetic suppression of PC firing, and in acute slices of the cerebellum, do not lead to large, sustained increases in DCN firing. Here we test whether DCN neurons undergo spike frequency adaptation that could account for these properties. We perform current-clamp recordings at near physiological temperature in acute brain slices from mice of both sexes to examine how DCN neurons respond to prolonged depolarizations. DCN neuron adaptation is exceptionally slow and bidirectional. A depolarizing current step evokes large initial increases in firing that decay to ∼20% of the initial increase within ∼10 s. We find that spike frequency adaptation in DCN neurons is mediated by a novel mechanism that is independent of the most promising candidates, including calcium entry and Na+-activated potassium channels mediated by Slo2.1 and Slo2.2. Slow adaptation allows DCN neurons to gradually and bidirectionally adapt to prolonged currents but to respond linearly to current injection on rapid timescales. This suggests that an important consequence of slow adaptation is that DCN neurons respond linearly to the rate of PC firing on rapid timescales but adapt to slow firing rate changes of PCs on long timescales.
SIGNIFICANCE STATEMENT Excitatory neurons in the cerebellar nuclei provide the primary output from the cerebellum. This study finds that these neurons exhibit very slow bidirectional spike frequency adaptation that has important implications for cerebellar function. This mechanism allows neurons in the cerebellar nuclei to adapt to long-lasting changes in synaptic drive while also remaining responsive to short-term changes in excitatory or inhibitory drive.
Keywords: adaptation, cerebellar nuclei, cerebellum
Introduction
The cerebellum participates in motor tasks, motor learning, cognitive processing, social behaviors, and many other nonmotor behaviors (Buckner, 2013; Reeber et al., 2013; Van Overwalle et al., 2014; Wang et al., 2014). To understand how the cerebellum contributes to these diverse functions, it is important to understand what controls cerebellar output. Purkinje cells (PCs) of the cerebellar cortex make strong GABAergic synapses onto glutamatergic projection neurons in the deep cerebellar nuclei (DCN), which in turn provide the output of the cerebellum. PCs provide an extraordinary amount of ongoing inhibition to the DCN. PCs fire spontaneously at high frequencies in vivo (Thach, 1968; Zhou et al., 2014), and each DCN cell receives an estimated 400 Hz to 4 kHz of inhibition (40 PCs converging onto each DCN neuron firing at 10–100 Hz; Chan-Palay, 1973a, b; Person and Raman, 2011). Yet, DCN cells are not silenced by this ongoing inhibition and fire at basal rates of ∼10–50 Hz in vivo and at even higher rates during sensory or motor events (Thach, 1968, 1970, 1975; Eccles et al., 1974; McDevitt et al., 1987; LeDoux et al., 1998; Rowland and Jaeger, 2005, 2008). Synchronous firing among PCs can entrain the firing of DCN neurons and promote DCN firing (de Solages et al., 2008; Person and Raman, 2011; Han et al., 2020). Brief pauses in PC firing can also rapidly and transiently decrease inhibition to promote firing (de Solages et al., 2008; Person and Raman, 2011; Han et al., 2020).
It is not known how DCN firing is regulated on long timescales because the firing of DCN neurons has only been studied on the hundreds of milliseconds timescale (Jahnsen, 1986; Aizenman and Linden, 1999; Czubayko et al., 2001; Uusisaari et al., 2007). There are reasons to suspect that DCN neurons slowly adapt to prolonged changes in PC firing. In PC degeneration (PCD) mutant mice where most PCs degenerate, it is expected that the loss of PC synapses would lead to disinhibition and elevated spiking in DCN neurons and in neurons of the vestibular nucleus (VN), another major output nucleus of the cerebellum (Bäurle et al., 1997). However, in PCD mice, the firing rates of VN neurons are not elevated, and rotation-evoked changes in VN neurons are much smaller than expected (Bäurle et al., 1997). This suggests that VN neurons may adapt their responses to the long-term absence of PC inputs. In addition, prolonged optogenetic suppression of PC firing leads to a large transient increase in DCN neuron firing that eventually adapts to a much smaller sustained elevation in firing rate (Khan et al., 2021). Similarly, in acute brain slices, DCN neurons fire spontaneously at moderate frequencies, although it is expected that the elimination of PC inhibition would elevate the firing of DCN neurons, because most PC axons are severed in acute brain slices (Jahnsen, 1986; Llinas and Muhlethaler, 1988; Aizenman and Linden, 1999; Czubayko et al., 2001).
Here we find that the firing rate of DCN neurons strongly, bidirectionally, and very slowly adapts (within ∼10 s) to either increases or decreases in sustained current injection. The most promising mechanisms for such adaptation involve action potential-mediated calcium influx and the activation or modulation of either potassium channels or chloride channels (Ha and Cheong, 2017; Huang and Chacron, 2017), or buildup of intracellular sodium and activation of Na+-activated potassium channels (Bischoff et al., 1998; Sanchez-Vives et al., 2000a). We found that slow adaptation was not mediated by these candidate mechanisms. This indicates that a novel mechanism mediates the very slow adaptation of DCN neurons. Regardless of the mechanism, slow adaptation could allow DCN neurons to adapt to prolonged changes in PC firing, while allowing DCN neurons to have a linear relationship between firing frequency and input current on subsecond timescales.
Materials and Methods
Ethics.
All animal procedures were conducted in accordance with the National Institutes of Health and Animal Care and Use Committee guidelines and protocols approved by the Harvard Medical Area Standing Committee on Animals (animal protocol #1493).
Animals.
Postnatal day 24 (P24) to P40 C57BL/6 wild-type mice (Charles River Laboratories) of both sexes were used for most acute slice experiments. P24 to P34 animals, in which the genes for Na+-activated potassium channels Kcnt1 (corresponding to Slo2.2 or Slack protein) and Kcnt2 (corresponding to Slo2.1 or Slick protein) in double-knockout (DKO) mice of both sexes were used, (see Figs. 3, 4). These animals are referred to as “Slo 2.1/2.2 DKO” in this article.
Figure 3.

The pharmacological sensitivity and calcium dependence of spike frequency adaptation and the subsequent afterhyperpolarization. A, Average instantaneous firing frequencies evoked by 200 pA depolarizing currents for 30 s in the indicated conditions. The effects of apamin (300 nm; a blocker of SK Ca2+-activated K channels), XE-991 [10 μm; an antagonist of Kv7 channels (KCNQ, M-current)], and E4031 [a blocker of KCNH1 (ERG K channels)] were assessed. Low Ca solution consisted of 0.1 mm external Ca2+ and 4 mm external Mg2+, 0 Ca2+ solution consisted of 0 Ca2+ and 1 mm EGTA. Control, n = 18 cells (9 animals); apamin, n = 7 cells (four animals); XE991, n = 5 cells (three animals); low Ca, n = 10 cells (7 animals); 0 Ca + EGTA, n = 7 cells (6 animals). Shading indicates the SE. B, The average AHPs following the current steps in A are shown for the indicated conditions. Shading indicates the SE.
Figure 4.

Assessing the involvement of Na+-activated potassium channels in spike frequency adaptation and afterhyperpolarization. A, The effect of substituting extracellular LiCl for NaCl on responses evoked by prolonged depolarization for a representative example cell. n = 5 cells (5 animals). B, Responses evoked by prolonged depolarization in the presence of 1 mm quinidine. n = 4 cells (2 animals). Ca, Average instantaneous firing frequencies evoked by 200 pA depolarizing currents for 30 s in Slo2.1/2.2 double-knockout animals. n = 9 cells (5 animals). Cb, The average AHPs following the current steps in Slo2.1/2.2 double-knockout animals. Shading in C is the SE.
Slice preparation.
Mice were anesthetized with ketamine/xylazine/acepromazine and transcardially perfused with warm (34°C) choline-ACSF solution containing the following: 110 mm choline Cl, 2.5 mm KCl, 1.25 mm NaH2PO4, 25 mm NaHCO3, 25 mm glucose, 0.5 mm CaCl2, 7 mm MgCl2, 3.1 mm Na-pyruvate, 11.6 mm Na-ascorbate, 5 µm NBQX, and 2.5 μm 3-[(6)-2-carboxypiperazin-4-yl]-propyl-1-phosphonic acid (CPP), oxygenated with 95% O2/5% CO2. To prepare sagittal cerebellar slices, the hindbrain was removed, a cut was made down the midline of the cerebellum, and the halves of the cerebellum were glued down to the slicing chamber. The 170-µm-thick sagittal slices were cut with a vibratome (model 1200S, Leica) in warm choline-ACSF. Slices were transferred to ACSF solution containing the following (in mm): 127 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 25 glucose, 1.5 CaCl2, and 1 MgCl2, and were maintained at 34°C for 10–12 min before being moved to room temperature for 20–30 min before beginning recordings.
Electrophysiology.
Whole-cell voltage current-clamp recordings were performed on spontaneously firing, large-diameter (20–25 µm) neurons in the lateral and interposed deep cerebellar nuclei. Borosilicate glass electrodes were filled with an internal solution containing the following (in mm): 150 K-gluconate, 3 KCl, 10 HEPES, 0.5 EGTA, 3 Mg-ATP, 0.5 Na-GTP, 5 phosphocreatine-tris2, and 5 phosphocreatine-Na2, and adjusted to pH 7.2 with KOH. The osmolarity was adjusted to 300 mOsm. High-resistance (3–4 MΩ) electrodes were used to minimize the dilution of cytosolic components, and only recordings where series resistance was <15 MΩ were accepted. Series resistance was compensated up to 80% for the estimated capacitance of the cell body (5 pF). In most cases, DCN neurons spontaneously fired on breaking in at voltages between −50 to −60 mV. Hyperpolarizing current was injected to keep cells from spontaneously spiking between experimental trials. As a result, cells typically “rested” between −65 and −75 mV. Reported voltages were corrected for a liquid junction potential of 10 mV. All experiments were performed at 36°C. Synaptic transmission was blocked with 5 μm NBQX to block AMPARs, 2.5 μm (R)-CPP to block NMDARs, 5 μm SR94431 to block GABAA receptors, and 1 μm strychnine to block glycine receptors.
Additional experiments were performed to assess the possibility of the involvement of pH homeostasis using a high HEPES internal consisting of the following (in mm): 125 K-gluconate, 3 KCl, 20 HEPES, 20 HEPES-K, 0.5 EGTA, 3 Mg-ATP, 0.5 Na-GTP, 5 phosphocreatine-tris2, and 5 phosphocreatine-Na2, adjusted to pH 7.2 with KOH. We found that adaptation had similar properties when using the standard internal [decay time (tdecay) = 3.70 ± 0.90 s; adaptation ratio = 0.23 ± 0.04; n = 5 cells, N = 4 animals] and high HEPES internal (tdecay = 3.44 ± 0.42; adaptation ratio = 0.23 ± 0.04; n = 5 cells, N = 4 animals) suggesting that pH changes were unlikely to contribute to adaptation.
For each experiment seen in Figure 3, stable responses to a 200 pA current injection were first obtained in control conditions (1.5 mm Ca2+, 1.0 mm Mg2+). After 5 min, external solution was switched to begin washin. Control and washin solutions always included blockers of synaptic transmission. The following external solutions were used: “low Ca” (0.1 mm Ca2+, 4.0 mm Mg2+), “0 Ca” (0 mm Ca2+, 4.0 mm Mg2+, 1 mm EGTA), apamin (300 nm apamin added to control solution), and XE991 (10 μm XE991 added to control solution). For Slo2.1/2.2 DKO animals that lack the Na+-activated potassium channels Slo 2.1 and Slo2.2, a control external solution was used. Washin solutions were allowed to equilibrate for at least 15 min before recording responses to current injections. Responses to depolarizing current steps were collected with an intertrial interval of 60 s during baseline and washin conditions.
Dynamic-clamp experiments.
We performed dynamic-clamp experiments to determine the response of DCN neurons to changes in excitatory inputs and inhibitory inputs (see Fig. 8). Dynamic-clamp recordings were performed at 20 kHz with an ITC-18 computer interface controlled by mafPC in Igor Pro (WaveMetrics). The capacitance of DCN neurons was 70–170 pF, and the leak currents were −300 to −600 pA. We based the number of PCs that converge on each DCN neuron (40 PCs) and the properties of inhibitory conductances on previous studies (Person and Raman, 2011), with a rise time of 0.1 ms, a decay time of 2.5 ms, and an amplitude of 5 nS (reflecting depression; Khan and Regehr, 2020). We based the timing of PC firing on 10 PCs recorded in awake behaving animals, with the interspike intervals for each simulated PC determined from randomly selected interspike intervals from one of the recorded cells (four simulated PCs were based on each recorded PC). The average firing frequency was 80 Hz, and the average frequency of all PC inputs converging on a DCN neuron was ∼3 kHz. This corresponded to an average PC conductance of ∼50 nS. The reversal potential for inhibitory conductances was set at −85 mV after correcting for junction potential.
Figure 8.

Dynamic-clamp studies of the adaptation of firing during prolonged changes in excitatory and inhibitory synaptic conductances. Aa, Excitatory synaptic conductance (GE) delivered to DCN cells. See Materials and Methods for details. Ab, Inhibitory synaptic conductance (GI) delivered to DCN cells. Ac, Example of dynamic-clamp recordings of the firing response to the excitatory and inhibitory synaptic conductances in Aa and Ab. Ad, The instantaneous firing frequency (gray) and the average firing frequency (0.5 s bins, black) for the example cell in Ac are shown. B, Expanded regions of the membrane potential corresponding to the trace in Ac. C, Average firing frequency evoked by increases in excitatory conductances for different cells (gray) and the average response (black) ± SEM (gray). D, Summary data for the initial (0–30 s), peak, and steady-state firing rates; decay times; and adaptation ratios (n = 6 cells). E–H, As in A–D but for a decrease in inhibition.
Excitatory conductances were based on the AMPA component characterized by Wu and Raman (2017). They recorded mossy fiber (MF) EPSCs with a rise time of 0.28 ms, a decay time of 1.06 ms, and an amplitude of 0.4 nS (reflecting depression). They estimated that 20–600 MFs converged on each DCN neuron, with an unknown firing frequency, so the excitatory conductances were relatively unconstrained in our experiments. We adjusted the frequency of MF EPSCs so that the firing rate of DCN neurons was maintained at 20–40 Hz in the presence of the inhibitory conductance. The average baseline excitatory conductance was 20–30 nS. The reversal potential for excitatory conductances was set at 0 mV after correcting for junction potential.
We examined spike frequency adaptation by either increasing the excitatory conductance or decreasing the inhibitory conductance. For increased excitation, we maintained average inhibition at 50 nS, and after a 30 s baseline excitation (20–30 nS), we increased the excitatory conductance to 40–45 nS for 30 s before returning the excitatory conductance to the initial level. We assessed the effects of suppressing inhibition on adaptation by maintaining the excitatory conductance for 90 s and decreasing the frequency of inhibitory inputs by a factor of 2 for times 30–60 s (reducing the average inhibitory conductance from ∼50 to 25 nS).
Analysis.
Recordings were obtained using Multiclamp 700B (Molecular Devices), sampled at 50 kHz and filtered at 4 kHz, and collected in Igor Pro (WaveMetrics). Data were analyzed using custom-written scripts in MATLAB (MathWorks). Instantaneous firing rates were calculated as the reciprocal of the interspike interval. In Igor Pro, decay times for firing rate were determined from an exponential fit to the instantaneous firing frequency during depolarizations. All summary data are shown as the mean ± SEM unless otherwise indicated. Before comparing the adaptation ratio, half-decay time, peak firing, steady-state (ss) firing, afterhyperpolarization (AHP) amplitude, and AHP half-decay time between control and experimental conditions seen in Figure 5, a Shapiro–Wilk test with significance level of 0.05 was used to test whether data were normally distributed. Most data were found to be normally distributed and subsequently were compared with their baseline values obtained before the washin by a two-tailed unpaired Student's t test. In all cases, the threshold for statistical significance was set at p < 0.05. Control (baseline) steady-state firing rates seen in Figure 5 were found to be asymmetrically distributed and were subsequently compared with steady-state firing rates of the experimental conditions using a nonparametric Wilcoxon signed-rank test.
Figure 5.

The pharmacological sensitivity, calcium dependence, and dependence on Na+-activated potassium channels of spike frequency adaptation and the subsequent afterhyperpolarization for single cells. A, The steady-state/peak firing rates (ss/peak) in DCN cells after delivering 200 pA depolarizing currents for 30 s in the indicated conditions. The effects of apamin (300 nm; a blocker of SK Ca2+-activated K channels), XE991 [10 µm; an antagonist of Kv7 channels (KCNQ, M-current)], and E4031 [a blocker of KCNH2 (ERG K channels)] were assessed. Low Ca solution consisted of 0.1 mm external Ca2+ and 4 mm external Mg2+; 0 Ca2+ solution consisted of 0 Ca2+ and 1 mm EGTA. Control, n = 18 cells (9 animals); apamin, n = 7 cells (4 animals); XE991, n = 5 cells (3 animals); E4031, n = 3 cells (2 animals); low Ca, n = 10 cells (7 animals); 0 Ca + EGTA, n = 7 cells (6 animals); Slo2.1/2.2 DKO, n = 9 cells (5 animals). The ss/peak for each experimental condition was not statistically significant from that of the control, as assessed by two-tailed unpaired Student's t tests. The threshold for statistical significance was set at p < 0.05. B, Peak firing rates observed during the depolarizing current step. Conditions with statistically significant differences from control are indicated with an asterisk (low Ca, p = 0.002; 0 Ca + EGTA, p = 0.004; two-tailed unpaired Student's t tests). All other comparisons with control were not statistically significant. C, Steady-state firing rates observed during the depolarizing current step. Conditions with statistically significant differences from control are indicated with an asterisk (low Ca, p = 0.014; 0 Ca + EGTA, p = 0.00079; nonparametric Wilcoxon signed-rank test). D, Decay times for the instantaneous firing frequencies occurring during the current step. Decay time for each experimental condition was not statistically significant compare with control, as assessed by two-tailed unpaired Student's t tests. E, Amplitude of the AHP observed in DCN neurons after the cessation of the depolarizing current step. Conditions with statistically significant differences from control are indicated with an asterisk (low Ca, p = 0.049; 0 Ca + EGTA, p = 0.015; two-tailed unpaired Student's t tests). All other comparisons with control were not statistically significant. F, The half-decay times for AHPs observed after each current step. Conditions with statistically significant differences from control are indicated with an asterisk (Slo2.1/2.2 DKO, p = 0.017; two-tailed unpaired Student's t tests test). All other comparisons with control were not statistically significant.
Results
To determine whether DCN neurons adapt on long timescales, we determined the effects of prolonged depolarizing current steps on the firing of DCN neurons. We recorded in whole-cell current clamp from DCN neurons at near physiological temperatures (36°C) in acute slices from juvenile (P25 to P40) mice and injected constant depolarizing currents for 50 s. We recorded from large DCN neurons that correspond to glutamatergic projection neurons (Uusisaari et al., 2007; Turecek et al., 2016). DCN neurons initially fired at high frequencies, and their rate of firing adapted very slowly (Fig. 1). This is shown for an example cell that initially fired at 180 Hz and gradually slowed to ∼23 spikes/s (Fig. 1A). We defined the ratio of steady-state to peak firing as the adaptation ratio (steady-state firing rate/peak firing rate, ss/peak) and the tdecay as the time constant of an exponential function best fit to the firing rate response. In Figure 1A, the adaptation ratio is 0.13 and the decay time is 7.4 s. There was variability in the time course and extent of adaptation across cells (Fig. 1B,D), with an average adaptation ratio of 0.20 ± 0.01 and an average decay time of 4.9 ± 0.2 s (Fig. 1C). For each cell, an intertrial interval of 60 s was applied between the 50 s depolarizing current steps. There was substantial variability in the adaptation properties (Fig. 1D). The decay time was weakly correlated with the peak firing rate (Fig. 1E, left: tdecay = 3.57 s + 0.0075 s × peak firing, r2 = 0.08), but the adaptation ratio was not strongly influenced by the initial firing frequency (Fig. 1E, right) The decay times and adaptation ratios were not significantly different for males and females (decay time, p = 0.59; adaptation ratio, p = 0.47; two-tailed unpaired Student's t test; Fig. 1F) or for different age animals over the age range studied (rs = −0.10 for animal age vs decay time and rs = −0.25 for animal age vs adaptation ratio; Spearman's ρ; Fig. 1G). Indeed, we observed considerable variability of the properties of adaptation for different cells from the same animal. Thus, all large glutamatergic neurons in the DCN exhibit robust spike frequency adaptation, but the time course and extent of the adaptation is highly variable for different cells.
Figure 1.

DCN neurons exhibit robust, slow spike frequency adaptation. Aa, Representative cell response to a 50 s depolarizing current step (200 pA) delivered to a DCN cell in whole-cell current clamp (top), the resulting voltage trace (middle), and the instantaneous firing frequency of spiking response (bottom). Ab, Expanded views of different periods of the voltage trace in Aa are shown (top), as well as the corresponding instantaneous firing frequencies (bottom). Expanded views of the same time points as immediately above for instantaneous firing rate. B, Example instantaneous firing frequencies for three different DCN cells that were depolarized for 50 s. Decay times and adaptation were determined as above. Decay times were determined from an exponential fit to the instantaneous firing frequency during the depolarization. Adaptation ratio (ss/peak) was calculated as the ss firing rate divided by the peak firing rate (peak). C, Average response to a 50 s current step for n = 22 DCN neurons (7 animals), with the SE overlaid and shaded in gray. D, Summary data for the peak and steady-state firing rates, decay times, and adaptation ratios (n = 48 cells). Data from responses to current steps of 50 and 30 s. E, Decay times and adaptation ratios as a function of initial firing frequency. F, Decay times and adaptation ratios for male (n = 28) and female (n = 20) mice. G, Decay times and adaptation ratios as a function of age.
In many neurons, spike frequency adaptation is thought to reflect the buildup of a hyperpolarizing conductance that suppresses spiking and also leads to an AHP after spiking is terminated (Ha and Cheong, 2017; Huang and Chacron, 2017). We found that a prominent AHP was observed in DCN neurons after the cessation of spiking (Fig. 2A). To examine the relationship between the magnitude of the AHP and the extent of adaptation, we delivered depolarizing current steps with a range of durations (0.3–30 s). During 0.3 s steps, the extent of adaptation and the amplitude of the AHP were both small (Fig. 2A,B, top). As the durations of current steps were increased, adaptation became more prominent (Fig. 2B) and the amplitude of the AHP increased (Fig. 2C,D). An intertrial interval of 60 s was used between each trial for each current step duration. Longer current steps were also followed by longer-lasting AHPs, as measured by the time taken for the AHP to return halfway to the initial resting potential, which is the AHP half-recovery time (t1/2; Fig. 2C,D,F). We observed a strong negative correlation between the amplitude of the AHP and the firing rate at the end of the depolarizing step (Fig. 2E; ss/peak = 1.36 – 0.12 × AHP amplitude; r2 = 0.93), and for AHP t1/2 versus firing rate at the end of the depolarizing step (Fig. 2F; ss/peak = 0.99–0.23 × AHP t1/2, r2 = 0.89).
Figure 2.
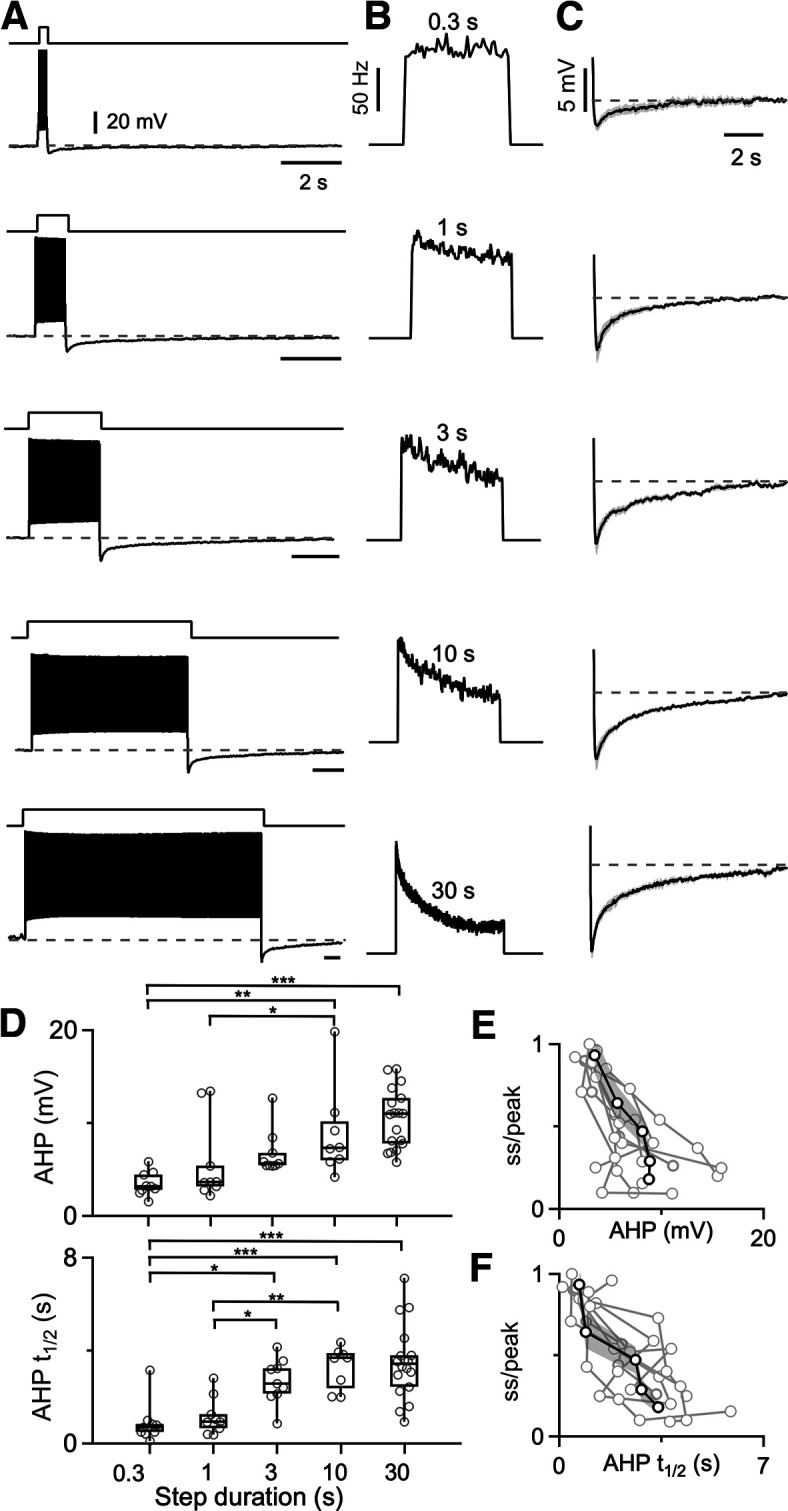
Prolonged spiking leads to a slow afterhyperpolarization that is dependent on spiking duration. A, Firing and AHP for a DCN neuron are shown in response to delivering depolarizing current steps of varying duration. B, Instantaneous firing frequencies are shown for the spiking shown in A. C, Voltage traces showing the average AHP following 0.3, 1, 3, 10, and 30 s current steps (N = 7 cells from 5 animals; SEs are in gray). D, Summaries of the amplitudes and half-recovery times of the AHPs for individual cells (circles) after current steps of the indicated durations. Half-recovery time was calculated as half the time for the AHP to return to the initial membrane potential before the step. N = 7 cells (5 animals). One-way ANOVA was performed to compare the AHP amplitude and the AHP half-recovery time of different current step durations. Statistically significant differences are indicated with asterisks. E, Summary of firing rate at the end of a depolarizing current step lasting 0.3–30 s divided by the peak firing of the step (ss/peak), as a function of the magnitude of the subsequent AHP. Responses from individual neurons are shown (light gray), with the average response overlaid in black and the SE shaded in dark gray. F, Same as in E, but ss/peak is plotted as a function of the AHP half-recovery time.
Spike frequency adaptation and afterhyperpolarizations have been shown to be mediated by a variety of mechanisms in other types of neurons. Some of these mechanisms involve channels for which selective antagonists are available. One possibility is that high-frequency firing leads to a buildup of calcium and the activation of small conductance Ca2+-activated potassium (SK) channels that suppresses firing and leads to an afterhyperpolarization (Huang and Chacron, 2017; Cui and Strowbridge, 2019). We tested the involvement of SK channels by assessing the effects of apamin, a blocker of SK channels (Stocker, 2004). Apamin had no effect on either the spike frequency adaptation or the afterhyperpolarization, indicating that SK channels are not involved (adaptation ratio in apamin vs control, p = 0.49; tdecay, p = 0.19; AHP amplitude, p = 0.44; AHP recovery time, p = 0.68; two-tailed unpaired Student's t tests; Fig. 3, purple; also see Fig. 5, purple). In other cells, M-currents that are mediated by Kv7 (KCNQ) channels, contribute to spike frequency adaptation and afterhyperpolarization (Madison and Nicoll, 1984; Gu et al., 2005; Otto et al., 2006). We used the selective blocker XE991 (Wang et al., 1998) to test the involvement of M-currents in spike frequency adaptation and afterhyperpolarization in DCN cells. XE991 did not affect either spike frequency adaptation or the AHP, indicating that M-currents are not involved (adaptation ratio in XE991 vs control, p = 0.71; tdecay, p = 0.19; AHP size, p = 0.74; AHP recovery time, p = 0.99; two-tailed unpaired Student's t tests; Fig. 3, blue; also see Fig. 5, blue). In some cell types, the ether-a-go-go-related gene (ERG) current (Kv7) contributes to spike frequency adaptation (Sacco et al., 2003; Pessia et al., 2008; Cui and Strowbridge, 2019). We tested whether ERG channels might also be involved in the slow adaptation of DCN cells. Application of the selective ERG channel blocker E-4031 had no effect on either spike frequency adaptation or afterhyperpolarization, indicating that ERG channels are not involved (adaptation ratio in E-4031 vs control, p = 0.56; tdecay, p = 0.24; AHP size, p = 0.92; AHP recovery time, p = 0.99; two-tailed unpaired Student's t tests; Fig. 3, red; also see Fig. 5, red).
Calcium entry during high-frequency firing could also activate other types of calcium-dependent conductances to produce spike frequency adaptation and afterhyperpolarization. These include Ca2+-activated chloride channels (Huang et al., 2012; Zhang et al., 2017; Wang et al., 2019). The most straightforward means of testing their involvement is to reduce calcium entry. We initially did this using an external solution (low Ca2+) that contained 0.1 mm Ca2+ and 4 mm Mg2+ rather than the standard 1.5 mm Ca2+ and 1 mm Mg2+ (Fig. 3, orange; also see Fig. 5, orange). We also explored the effects of an external solution with no added external Ca2 (0 mm Ca2+, 4 mm Mg2) supplemented with the calcium chelator EGTA (1 mm) to buffer low levels of contaminating Ca2+ (0 Ca2+; Fig. 3, yellow; also see Fig. 5, yellow). This external solution severely reduces calcium entry and is expected to disrupt any mechanism that relies on external calcium entry. The results were similar for both external solutions. Spike frequency adaptation and the duration of adaptation were still intact, although the peak firing frequency and steady-state firing frequency were both elevated (adaptation ratio in low Ca vs control, p = 0.28; tdecay, p = 0.25; peak firing frequency, p = 0.002; and steady-state firing, p = 0.014; adaptation ratio in 0 Ca + EGTA vs control, p = 0.41; tdecay, p = 0.25; peak firing frequency, p = 0.004; and steady-state firing, p = 0.00079; two-tailed unpaired Student's t tests were used for all comparisons except nonparametric use of the Wilcoxon signed-rank test for comparing steady-state firing in control vs low Ca and control vs 0 Ca + EGTA). The increases in peak and steady-state firing suggest that low Ca2+ and 0 Ca2+ external solutions increase the excitability of DCN neurons independently of spike frequency adaptation. It is possible that this arises from activation of the sodium leak channel NALCN that occurs when external Ca2+ is reduced (Chua et al., 2020). Another curious effect is that the amplitude of the AHP is reduced in low Ca2+ and to a much larger extent in 0 Ca2+. This suggests that in some instances the extent of spike frequency adaptation and the amplitude of the AHP can be decoupled.
Thus far we have eliminated the leading candidate mechanisms for spike frequency adaptation that rely on calcium entry, such as Ca2+-activated chloride channels. This leads us to the second major candidate mechanism of spike frequency adaptation, Na+-activated potassium channels. In numerous cell types, elevated firing leads to a buildup of intracellular sodium and the opening of Na+-activated potassium channels that in turn suppress firing and lead to a long-lasting afterhyperpolarization (Gustafsson and Wigström, 1983; Schwindt et al., 1989; Dryer, 1991; Kubota and Saito, 1991; Bischoff et al., 1998; Kim and McCormick, 1998; Sanchez-Vives et al., 2000a,b; Wallén et al., 2007; Barraza et al., 2009). Na+-activated potassium channels were the favored mediators of slow spike frequency adaptation and long-lasting afterhyperpolarization in the ferret primary visual cortex (Sanchez-Vives et al., 2000a,b). This hypothesis was not tested because a selective antagonist of Na-activated potassium channels was not available. Subsequent studies have established that the channels Slo2.1 (Slick) and Slo2.2 (Slack) mediate Na+-activated potassium currents (Martinez-Espinosa et al., 2015). Moreover, in situ hybridization indicates that Slo2.1 and Slo2.2 are present in neurons within the DCN (Bhattacharjee et al., 2002, 2003). For these reasons, we decided to test the hypothesis that Na+-activated potassium currents are responsible for slow adaptation and hyperpolarization in DCN neurons.
We initially explored this question by determining whether the substitution of lithium for sodium eliminated spike frequency adaptation (Fig. 4A). LiCl substitution for NaCl could allow action potential generation without engaging Na+-activated conductances (Wallén et al., 2007). If firing were preserved and adaptation eliminated, it would implicate a Na+-dependent mechanism and be consistent with the involvement of Na+-activated potassium channels. We found, however, that under these conditions, prolonged depolarizing current steps typically evoked abnormal burst-like spiking that lasted for hundreds of milliseconds to slightly more than a second, followed by a plateau depolarization. This suppression of sustained firing by lithium substitution indicates that that this approach lacks sufficient specificity to provide insight into the role of sodium influx in adaptation.
We also used pharmacological approaches to study the involvement of Na+-activated potassium channels in adaptation. When expressed in heterologous systems, Slo2.1 and Slo2.2 currents are both completely abolished by 1 mm quinidine (Bhattacharjee et al., 2003). We found that 1 mm quinidine depolarized and eliminated spiking in DCN neurons (Fig. 4B). Similar results were seen for a range of concentrations of quinidine in other cells (1 mm, n = 4 cells; 100 μm, n = 4 cells; 50 μm, n = 2 cells). This indicates that the lack of specificity of quinidine compromises it as a tool to study spike frequency adaptation in DCN neurons. The limitations of quinidine as a tool to study Na+-activated potassium channels in intact cells is consistent with the observation that it also blocks potassium rest channels, TASK and TWIK-1, and some voltage-gated sodium channels (Campbell and Williams, 1983; Kehl, 1991; Leonoudakis et al., 1998).
In the absence of suitable pharmacological tools to study Na+-activated potassium channels, we needed a different approach. Fortunately, global Slo2.1 and Slo2.2 DKO mice are viable (Martinez-Espinosa et al., 2015), and we used these mice to assess the role of Na+-activated potassium channels in adaptation. We found that in Slo 2.1/2.2 double-knockout mice, spike frequency adaptation was intact and afterhyperpolarization remained prominent (Figs. 4C, green, 5, green), establishing that they are not mediated by Na+-activated potassium channels in DCN neurons.
The slow AHP we observe has the potential to reduce excitability following high-frequency spiking and could also contribute to spike frequency adaptation. We examined this possibility by using a protocol consisting of a 200 pA current injection, followed by a 400 pA current step, and then returning to a 200 pA current (Fig. 6). An intertrial interval of 60 s was used. As shown for an example cell, the initial depolarization transiently elevated spiking that then decayed to a steady-state level, and a further increase in depolarization elevated spiking that again adapted to a new steady-state level (Fig. 6A). In response to the additional depolarization, steady-state spiking was elevated (11.5 spikes/s for 400 pA vs 5.4 spikes/s for 200 pA), and the time constant of spike adaptation was slower (6.3 s for tdecay2 vs 3.8 s for tdecay1). Although the reason for the differences in the time courses of adaptation are unclear, this observation indicates that in addition to differences in adaptation between cells, an individual cell can exhibit different time courses of adaptation. After stepping down from 400 to 200 pA, spiking stopped for 13.8 s [time taken for firing to resume (tfiring); Fig. 6A]. Seven cells and their average responses showed similar trends (Fig. 6B–D). This result indicates that adaptation in DCN neurons is bidirectional. DCN neuron firing slowly adapts during periods of elevated firing and recovers from reduced firing in a manner that is consistent with the time course of the long-lasting conductance that mediates the AHP.
Figure 6.

DCN firing bidirectionally adapts to increases and decreases in depolarization over seconds. A, Top, A three-step depolarizing current injection protocol delivered to a DCN cell, starting with an initial 200 pA depolarization for 25 s, followed by a step to 400 pA for 30 s, and then a 200 pA depolarization for 60 s. Middle, Voltage response during a current-clamp recording from a DCN neuron. Bottom, Instantaneous firing response. B, Average instantaneous firing frequency evoked by the three-step current injection protocol. N = 7 cells (5 animals). C, Summary data for the steady-state firing at the ends of the initial depolarization, for the 400 pA step, and for the 60 s recovery. Statistical significance among steady-state firing during initial, step, and recovery periods was assessed by one-way ANOVA and found to be statistically insignificant at a significance level of 0.05. D, Summary data for the decay times to reach steady state for the initial current injection (tdecay1), and for the 400 pA step (tdecay2). Decay times were determined from an exponential fit to the instantaneous firing frequency during the indicated period. In all cases, the spiking stopped when the depolarization was decreased from 400 to 200 pA, and tfiring values are summarized for each cell. Statistical significance between steady-state firing during initial, step, and recovery periods was assessed by one-way ANOVA. The only statistically significant comparison was the time taken to resume firing (tfiring) and the time to reach steady state for the initial current injection (tdecay1; p = 0.025).
The slow adaptation of DCN neurons and their lack of adaptation on rapid timescales suggests that DCN neurons respond differentially to brief and long-lasting depolarizations. We examined this possibility by delivering a series of depolarizing current steps of varying durations to DCN neurons, with amplitudes of 50, 100, 150, 200, 150, 100, and 50 pA. In all trials, the magnitude of the series of current injections was similar, but the duration of each step varied across trials. The instantaneous firing frequencies are shown for a cell for steps of 1 and 30 s (Fig. 7A), and spiking is shown for the initial 100 ms (Fig. 7B, black) and the final 100 ms (Fig. 7B, blue) of each depolarization (Fig. 7B). For 1 s steps, firing rates were similar for the first and last 100 ms of each step, consistent with minimal adaptation. Responses were very different when the duration of each step was 30 s. For step increases in the depolarizing current, the initial firing was much higher at the start of the depolarization than at the end, but when depolarizing currents were decreased, the initial firing was much lower at the start than at the end. Similar experiments were performed for 0.3, 1, 3, 10, and 30 s steps for a number of DCN neurons, with average responses as a function of time (Fig. 7C, left) and the average firing rate in the initial 100 ms (black) and the final 100 ms (blue) plotted for each current step (Fig. 7C, right). The order in which trials were delivered was randomized, and a 60 s intertrial interval was used. For 0.3–1 s steps, the firing of DCN neurons tracked the magnitude of the depolarization (Fig. 7C, top). Responses to steps that lasted 1 s most faithfully tracked the change in depolarization over time. For 3 s steps, a small amount of adaptation is apparent, and there is a small decrease in the firing rate by the end of the current step. Longer steps resulted in more adaptation and increasing divergence between initial and steady-state firing (Fig. 7C). There were no significant differences in the steady-state firing frequencies for 30 s current steps that had the same amplitude [for steps 1 and 7 (p = 0.87), for steps 2 and 6 (p = 0.87), or for steps 3 and 5 (p = 0.93)], suggesting that there is a preferred setpoint for a particular level of activation. Overall, these results indicate that for short depolarizing current steps, DCN neurons faithfully convert the magnitude of the depolarizing current into a spike rate. For long changes in the magnitude of depolarizing inputs, DCN neurons slowly adapt and remain responsive to additional changes in the magnitude of depolarizing currents.
Figure 7.
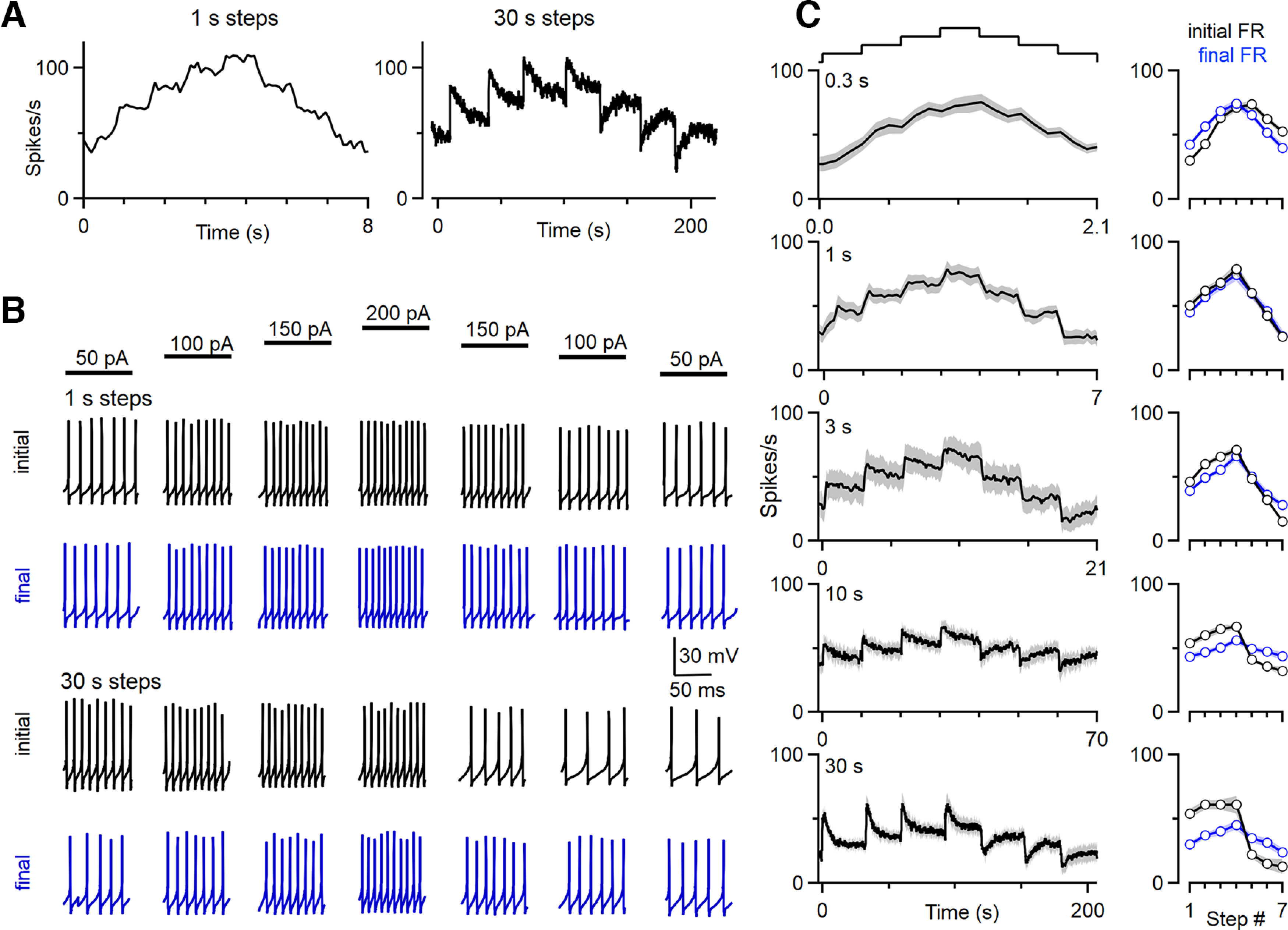
The firing of DCN neurons evoked by a series of current steps. A series of current steps of increasing and then decreasing amplitude were used to stimulate DCN neurons (C, top). For different trials, the durations of the current steps were 0.3, 1, 3, 10, and 30 s. These steps are delivered in addition to a 100 pA current used to maintain baseline firing at rates similar to those observed in vivo. A, Instantaneous firing frequencies evoked by 1 s (left) and 30 s (right) current steps are shown for a DCN neuron. B, Responses of the DCN cell in A are shown to illustrate the differences between 1 s steps and 30 s steps. The initial 100 ms (black) and the final 100 ms (blue) are shown for each current step. For 1 s steps, the firing frequencies at the start of current steps are proportional to the magnitude of the current step, and firing frequencies remained similar during current steps. For 30 s steps, firing frequencies slowly adapt, resulting in very different firing frequencies at the start and the end of each step. C, Summary of the firing responses to current steps of varying duration. Plots on the left show the average instantaneous firing frequency of DCN cells in response to the depolarizing current ramp where each step is of the indicated duration. The SE is shaded in gray. Plots on the right show the average firing rates for the initial (black) and final (blue) 100 ms (n = 7 cells, n = 6 animals).
Thus far, we have controlled firing rates by steady current injections. However, DCN neuron firing in vivo is controlled by the balance of strong synaptic excitation and inhibition. We used dynamic-clamp studies to examine the properties of spike frequency adaptation under conditions that approximate a balance of strong excitation and strong inhibition (Fig. 8). We based the properties of excitatory and inhibitory inputs on previous studies (Person and Raman, 2011; Wu and Raman, 2017) and based the timing and frequency of inputs on PC firing recorded in awake behaving mice (see Materials and Methods). We initially examined spike frequency adaptation when spiking was elevated by increasing the excitation (Fig. 8A–D). The excitatory (Fig. 8Aa) and inhibitory (Fig. 8Ab) conductances were both very noisy, and the resulting baseline firing was highly irregular (Fig. 8Ad). Increasing the average excitatory conductance from 20 to 40 nS elevated the firing, and the firing slowly adapted. The adaptation of instantaneous firing frequency is not obvious because it is so variable (Fig. 8Ad, gray), but the average firing frequency (500 ms bins) clearly shows prominent and slow adaptation (Fig. 8Ad, black). The differences in firing are also apparent in expanded views before the increase in excitation as well as at the start and end of the elevation in excitation (Fig. 8B). Following the return of the excitatory conductance to baseline levels, firing transiently stopped and slowly recovered to baseline levels. This suggests that the bidirectional spike frequency adaptation is present for conditions of strong inhibition and excitation. The properties of spike frequency adaptation for these dynamic-clamp experiments were consistent for different cells (Fig. 8C,D). We also examined the effects of decreasing inhibition and found that the effects on the firing of DCN neurons were remarkably similar for decreases in inhibition (Fig. 8E–H) and increases in excitation (Fig. 8A–D). These studies suggest that slow bidirectional adaptation is prominent for dynamic-clamp experiments with strong balanced synaptic excitation and inhibition regardless of whether spiking is elevated by an increase in excitation or a decrease in inhibition.
Discussion
Our main finding is that DCN neurons exhibit a robust, remarkably slow form of spike frequency adaptation. This adaptation is so slow that it does not impact responses on timescales of hundreds of milliseconds or several seconds. Consequently, the firing of DCN neurons can linearly respond to changes in injected current on these short timescales. However, on longer timescales, DCN neurons adapt their firing in a manner that allows them to remain responsive to subsequent changes in excitatory or inhibitory drive.
Previous studies of DCN neurons
There have been extensive studies of DCN neurons, but there have been no previous descriptions of the slow spike frequency adaptation we report here. This is likely because of differences in experimental conditions and the experimental approach. Many previous studies used short depolarizations of hundreds of milliseconds (Jahnsen, 1986; Llinas and Muhlethaler, 1988; Aizenman and Linden, 1999; Czubayko et al., 2001; Uusisaari et al., 2007; Han et al., 2014; Canto et al., 2016), and the slow spike frequency adaption we see is not apparent during such brief steps (Fig. 1Ab, left). Numerous studies also focused on much younger animals than those studied here (Aizenman and Linden, 1999; Raman et al., 2000; Czubayko et al., 2001; Uusisaari et al., 2007; Zheng and Raman, 2009), and adaptation may have different properties in young animals. Careful examination of DCN spiking evoked by somewhat more prolonged depolarizations in other studies is consistent with the slow adaptation we observe. In young rats (P16), spike frequency adaption of ∼20% occurred for a 5 s depolarization (Czubayko et al., 2001). In more mature guinea pigs, a 2 s depolarization resulted in highly variable adaptation that ranged from a 50% reduction in firing rate to no reduction at all (Jahnsen, 1986). Although the time course of adaptation was not characterized in that study, it is likely that a 2 s step was insufficiently long to reveal the full extent of spike frequency adaptation. Thus, previous studies did not use protocols that were suited to studying the slow spike frequency adaptation present in DCN neurons.
Implications for the cerebellar processing
The properties of spike frequency adaptation of DCN neurons have several implications for cerebellar processing. We found that on average DCN firing adapts to 20 ± 1% of initial firing with a decay time of 4.9 ± 0.20 s (Fig. 1), that DCN adaptation is also bidirectional (Figs. 6, 7), and that adapted cells remain remarkably sensitive to further changes in depolarization (Fig. 6, 7). On short timescales that are <1 s, DCN firing reliably encodes changes in PC firing. This would ensure that cerebellar output could preserve signals generated from short-term changes in PC activity, such as those occurring during fine movements or error signals generated on a subsecond timescale. However, adaptation makes the DCN less sensitive to slower, more prolonged changes in input activity. PCs are known to undergo long periods of modulation in their excitability and activity, such as during motor learning (Gilbert and Thach, 1977; Jirenhed et al., 2007; Rasmussen et al., 2008; Belmeguenai et al., 2010; Heiney et al., 2014; Yang and Lisberger, 2014; Grasselli et al., 2016). In these situations, DCN cells could initially generate large responses but gradually adapt to maintain a stable firing range. Without such adaptation, DCN spiking could saturate, and these neurons might be unable to respond to further changes in PC activity. When PC firing rates are increased, the adaptation of firing rates described here will act in concert with slow depression of the PC–DCN synapse (Pedroarena, 2020). Most previous behavioral studies of the cerebellum have been performed on the seconds timescale, and the adaptation described here will have little effect on the DCN responses. Future studies performed on longer timescales could provide insight into the behavioral consequences of slow spike frequency adaptation.
Spike frequency adaptation of DCN neurons is well suited to protecting against the loss of PC inhibition. Many types of ataxia involve a long-term decrease in PC inhibition of the DCN, often as a result of chronic decreases in PC firing, decreases in the strength of PC synapses, or PC death. PCs are also susceptible to hypoxia (Welsh et al., 2002; Au et al., 2015). PCs fire at such high frequencies (10–100 Hz), their inputs are sufficiently large (30 nS on average; Khan and Regehr, 2020), and so many (∼40) converge onto each DCN neuron that if PCs fire asynchronously, they provide a noisy inhibitory tone that strongly suppresses the firing of DCN neurons. If this inhibition is removed, there is expected to be a large (∼10-fold) and immediate increase in the firing of DCN neurons (Person and Raman, 2011). It is possible that if spike frequency adaptation did not occur, and if such firing-rate increases were sustained, excitotoxicity might develop and harm DCN neurons, compromising the ability of the cerebellum to influence the rest of the brain. However, DCN neurons can survive in rodent models with strong reductions in PC inhibition (Bäurle et al., 1997; Au et al., 2015), and spike frequency adaptation likely helps to prevent such large, sustained increases in DCN firing. It is clear that spike frequency adaptation cannot overcome profound deficits in PC inhibition and spare all cerebellar-dependent behaviors, as is apparent in motor deficits that accompany PC loss (Kordasiewicz and Gomez, 2007; Hurlock et al., 2008; Shakkottai et al., 2009, 2011; Hourez et al., 2011; Dougherty et al., 2012, 2013; Bosch et al., 2015; Jayabal et al., 2016; Larivière et al., 2019; Tsai et al., 2018). It is also likely that in brain slices, spike frequency adaptation prevents DCN neurons from firing at high rates in the absence of PC inputs. Finally, spike frequency adaptation likely contributes to the observation that acutely increasing PC activity or decreasing DCN activity reduces aggression (Reis and Ross, 1973; Cooper et al., 1976; Heath, 1977; Jackman et al., 2020), whereas chronically removing PC inhibition by lesioning the cerebellar cortex, which would be predicted to increase DCN firing, similarly reduces aggression (Sprague and Chambers, 1959; Berman, 1997).
Comparison to spike frequency adaptation AHP in other cell types
Spike frequency adaptation and AHPs have been described in many types of neurons. In most cases the timescales are much faster than we observed in DCN neurons, lasting tens or hundreds of milliseconds (Madison and Nicoll, 1984; Schwindt et al., 1988; Foehring et al., 1989; Nishimura et al., 1989; Pedarzani and Storm, 1993; Sawczuk et al., 1995; Fleidervish et al., 1996; Safronov and Vogel, 1996; Bischoff et al., 1998; Pineda et al., 1999.; Faber and Sah, 2002; Sacco et al., 2003; Higley and Contreras, 2006; Khawaja et al., 2007; Tzingounis et al., 2007; Hardman and Forsythe, 2009; Ha et al., 2016). There are several types of cells that exhibit slow spike frequency adaptation: visual cortical neurons, subthalamic nucleus neurons, and fast-spiking GABAergic cortical interneurons (Sanchez-Vives et al., 2000a,b; Descalzo et al., 2005; La Camera et al., 2006; Barraza et al., 2009). In V1 neurons, slow spike frequency adaptation allows neurons to adapt to constant stimuli, while preserving their ability to respond to rapidly changing stimuli (Sanchez-Vives et al., 2000a,b). In subthalamic nuclei, it has been proposed that slow adaptation removes slow trends in the average rates of inputs (Barraza et al., 2009). Major questions remain regarding the mechanisms of slow spike frequency adaptation in all types of neurons where this phenomenon has been observed. The mechanism responsible for slow adaptation of fast-spiking cortical neurons is not known (La Camera et al., 2006). In V1 neurons, an AHP produced by a potassium current is implicated, and it has been proposed that a buildup of intracellular calcium and/or sodium may activate this current (Sanchez-Vives et al., 2000a,b), and Na+-activated potassium channels were the favored mediators. In subthalamic nuclei neurons, a potassium current is also implicated, but it is thought to be independent of calcium and sodium (Barraza et al., 2009). For DCN neurons, we also think that a hyperpolarizing current is involved, but we were unable to eliminate adaptation by eliminating calcium entry, by eliminating Na+-activated potassium channels in Slo2.1/2.2 DKO mice, by blocking M-current, or by blocking SK Ca2+-activated potassium channels. It remains possible that these different types of neurons share a common mechanism of spike frequency adaptation that has yet to be determined. It is also likely that slow spike adaptation is more common than is currently appreciated. Just as we revealed slow adaptation here in DCN neurons by using prolonged depolarizations, it is likely that extending the duration of depolarization in other cell types will reveal slow adaptation in some other types of cells.
Although we have not determined the mechanisms underlying slow spike frequency adaptation, we have eliminated the most promising candidate mechanisms. Based on the correlation between the buildup of the hyperpolarizing current and the reduction in firing (Fig. 2), it seems likely that the primary mechanism is the use-dependent activation of a slow outward current. It is possible that slow sodium channel inactivation could also contribute to spike frequency adaptation (Powers et al., 1999; Barraza et al., 2009; Milescu et al., 2010). Unfortunately, we were unable to directly determine the contributions of this mechanism because we could not voltage clamp sodium currents in DCN neurons at near physiological temperatures in brain slice.
Implications for the optogenetic manipulation of activity
The bidirectional spike frequency adaptation we observe that opposes long-term changes in the firing of DCN neurons has important implications for optogenetic and chemogenetic manipulations of cerebellar outputs and deep brain stimulation of the cerebellar nuclei, which is a promising approach for treating dystonia, ataxia, essential tremor, and a variety of other neurologic and neuropsychiatric disorders (Kim et al., 2021; Miterko et al., 2021; Ponce et al., 2022). Numerous studies have used these approaches to investigate the involvement of the cerebellum and specific regions of the cerebellar cortex in behaviors (Stoodley et al., 2017; Darmohray et al., 2019; Prestori et al., 2020). We have shown previously that prolonged optogenetic suppression of PC firing results in large transient increases in the firing of DCN neurons, and then a slow adaptation of firing to slightly elevated levels (Khan et al., 2021). The properties of the spike frequency adaptation are suited to contribute to such in vivo adaptation and could also occur during sustained optogenetic increases in PC firing. Slow spike frequency adaptation has not been examined in most cell types, and it is possible that it is much more prevalent than has been appreciated and may be an important factor in optogenetic and chemogenetic studies that examine the roles of specific brain regions or cell types in behavior.
Footnotes
This work was supported by National Institutes of Health Grants R01-NS-032405 and R35-NS-097284 (to W.G.R.) and K99-NS-110978 (to C.H.C.).
The authors declare no competing financial interests.
References
- Aizenman CD, Linden DJ (1999) Regulation of the rebound depolarization and spontaneous firing patterns of deep nuclear neurons in slices of rat cerebellum. J Neurophysiol 82:1697–1709. 10.1152/jn.1999.82.4.1697 [DOI] [PubMed] [Google Scholar]
- Au AK, Chen Y, Du L, Smith CM, Manole MD, Baltagi SA, Chu CT, Aneja RK, Bayir H, Kochanek PM, Clark RSB (2015) Ischemia-induced autophagy contributes to neurodegeneration in cerebellar Purkinje cells in the developing rat brain and in primary cortical neurons in vitro. Biochim Biophys Acta 1852:1902–1911. 10.1016/j.bbadis.2015.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barraza D, Kita H, Wilson CJ (2009) Slow spike frequency adaptation in neurons of the rat subthalamic nucleus. J Neurophysiol 102:3689–3697. 10.1152/jn.00759.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bäurle J, Helmchen C, Grüsser-Cornehls U (1997) Diverse effects of Purkinje cell loss on deep cerebellar and vestibular nuclei neurons in Purkinje cell degeneration mutant mice: a possible compensatory mechanism. J Comp Neurol 384:580–596. [DOI] [PubMed] [Google Scholar]
- Belmeguenai A, Hosy E, Bengtsson F, Pedroarena CM, Piochon C, Teuling E, He Q, Ohtsuki G, De Jeu MTG, Elgersma Y, De Zeeuw CI, Jörntell H, Hansel C (2010) Intrinsic plasticity complements long-term potentiation in parallel fiber input gain control in cerebellar Purkinje cells. J Neurosci 30:13630–13643. 10.1523/JNEUROSCI.3226-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman AJ (1997) Amelioration of aggression: response to selective cerebellar lesions in the rhesus monkey. Int Rev Neurobiol 41:111–119. [PubMed] [Google Scholar]
- Bhattacharjee A, Gan L, Kaczmarek LK (2002) Localization of the Slack potassium channel in the rat central nervous system. J Comp Neurol 454:241–254. 10.1002/cne.10439 [DOI] [PubMed] [Google Scholar]
- Bhattacharjee A, Joiner WJ, Wu M, Yang Y, Sigworth FJ, Kaczmarek LK (2003) Slick (Slo2.1), a rapidly-gating sodium-activated potassium channel inhibited by ATP. J Neurosci 23:11681–11691. 10.1523/JNEUROSCI.23-37-11681.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischoff U, Vogel W, Safronov B (1998) Na+-activated K+ channels in small dorsal root ganglion neurones of rat. J Physiol 510:743–754. 10.1111/j.1469-7793.1998.743bj.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch MK, Carrasquillo Y, Ransdell JL, Kanakamedala A, Ornitz DM, Nerbonne JM (2015) Intracellular FGF14 (iFGF14) is required for spontaneous and evoked firing in cerebellar Purkinje neurons and for motor coordination and balance. J Neurosci 35:6752–6769. 10.1523/JNEUROSCI.2663-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckner RL (2013) The cerebellum and cognitive function: 25 years of insight from anatomy and neuroimaging. Neuron 80:807–815. 10.1016/j.neuron.2013.10.044 [DOI] [PubMed] [Google Scholar]
- Campbell TJ, Williams EMV (1983) Voltage- and time-dependent depression of maximum rate of depolarisation of guinea-pig ventricular action potentials by two new antiarrhythmic drugs, flecainide and lorcainide. Cardiovasc Res 17:251–258. 10.1093/cvr/17.5.251 [DOI] [PubMed] [Google Scholar]
- Canto CB, Witter L, de Zeeuw CI (2016) Whole-cell properties of cerebellar nuclei neurons in vivo. PLoS One 11:e0165887. 10.1371/journal.pone.0165887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan-Palay V (1973a) On the identification of the afferent axon terminals in the nucleus lateralis of the cerebellum an electron microscope study. Z Anat Entwicklungsgesch 142:149–186. 10.1007/BF00519720 [DOI] [PubMed] [Google Scholar]
- Chan-Palay V (1973b) Afferent axons and their relations with neurons in the nucleus lateralis of the cerebellum: a light microscopic study. Z Anat Entwicklungsgesch 142:1–21. 10.1007/BF00519873 [DOI] [PubMed] [Google Scholar]
- Chua HC, Wulf M, Weidling C, Rasmussen LP, Pless SA (2020) The NALCN channel complex is voltage sensitive and directly modulated by extracellular calcium. Sci Adv 6:eaaz3154. 10.1126/sciadv.aaz3154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper IS, Amin I, Riklan M, Waltz JM, Tung Poon P (1976) Chronic cerebellar stimulation in epilepsy. Clinical and anatomical studies. Arch Neurol 33:559–570. 10.1001/archneur.1976.00500080037006 [DOI] [PubMed] [Google Scholar]
- Cui ED, Strowbridge BW (2019) Selective attenuation of ether-a-go-go related K+ currents by endogenous acetylcholine reduces spike-frequency adaptation and network correlation. Elife 8:e44954. 10.7554/eLife.44954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czubayko U, Sultan F, Thier P, Schwarz C (2001) Two types of neurons in the rat cerebellar nuclei as distinguished by membrane potentials and intracellular fillings. J Neurophysiol 85:2017–2029. 10.1152/jn.2001.85.5.2017 [DOI] [PubMed] [Google Scholar]
- Darmohray DM, Jacobs JR, Marques HG, Carey MR (2019) Spatial and temporal locomotor learning in mouse cerebellum. Neuron 102:217–231.e4. 10.1016/j.neuron.2019.01.038 [DOI] [PubMed] [Google Scholar]
- Descalzo VF, Nowak LG, Brumberg JC, McCormick DA, Sanchez-Vives MV (2005) Slow adaptation in fast-spiking neurons of visual cortex. J Neurophysiol 93:1111–1118. 10.1152/jn.00658.2004 [DOI] [PubMed] [Google Scholar]
- de Solages C, Szapiro G, Brunel N, Hakim V, Isope P, Buisseret P, Rousseau C, Barbour B, Léna C (2008) High-frequency organization and synchrony of activity in the Purkinje cell layer of the cerebellum. Neuron 58:775–788. 10.1016/j.neuron.2008.05.008 [DOI] [PubMed] [Google Scholar]
- Dougherty SE, Reeves JL, Lucas EK, Gamble KL, Lesort M, Cowell RM (2012) Disruption of Purkinje cell function prior to huntingtin accumulation and cell loss in an animal model of Huntington disease. Exp Neurol 236:171–178. 10.1016/j.expneurol.2012.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty SE, Reeves JL, Lesort M, Detloff PJ, Cowell RM (2013) Purkinje cell dysfunction and loss in a knock-in mouse model of Huntington disease. Exp Neurol 240:96–102. 10.1016/j.expneurol.2012.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dryer SE (1991) Na(+)-activated K+ channels and voltage-evoked ionic currents in brain stem and parasympathetic neurones of the chick. J Physiol 435:513–532. 10.1113/jphysiol.1991.sp018522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eccles JC, Sabah NH, Táboříková H (1974) The pathways responsible for excitation and inhibition of fastigial neurones. Exp Brain Res 19:78–99. 10.1007/BF00233396 [DOI] [PubMed] [Google Scholar]
- Faber ESL, Sah P (2002) Physiological role of calcium-activated potassium currents in the rat lateral amygdala. J Neurosci 22:1618–1628. 10.1523/JNEUROSCI.22-05-01618.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleidervish IA, Friedman A, Gutnick MJ (1996) Slow inactivation of Na+ current and slow cumulative spike adaptation in mouse and guinea-pig neocortical neurones in slices. J Physiol 493:83–97. 10.1113/jphysiol.1996.sp021366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foehring RC, Schwindt PC, Crill WE (1989) Norepinephrine selectively reduces slow Ca2+- and Na+-mediated K+ currents in cat neocortical neurons. J Neurophysiol 61:245–256. 10.1152/jn.1989.61.2.245 [DOI] [PubMed] [Google Scholar]
- Gilbert PFC, Thach W (1977) Purkinje cell activity during motor learning. Brain Res 128:309–328. 10.1016/0006-8993(77)90997-0 [DOI] [PubMed] [Google Scholar]
- Grasselli G, He Q, Wan V, Adelman JP, Ohtsuki G, Hansel C (2016) Activity-dependent plasticity of spike pauses in cerebellar Purkinje cells. Cell Rep 14:2546–2553. 10.1016/j.celrep.2016.02.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu N, Vervaeke K, Hu H, Storm JF (2005) Kv7/KCNQ/M and HCN/h, but not KCa2/SK channels, contribute to the somatic medium after-hyperpolarization and excitability control in CA1 hippocampal pyramidal cells. J Physiol 566:689–715. 10.1113/jphysiol.2005.086835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson B, Wigström H (1983) Hyperpolarization following long-lasting tetanic activation of hippocampal pyramidal cells. Brain Res 275:159–163. 10.1016/0006-8993(83)90429-8 [DOI] [PubMed] [Google Scholar]
- Ha GE, Cheong E (2017) Spike frequency adaptation in neurons of the central nervous system. Exp Neurobiol 26:179–185. 10.5607/en.2017.26.4.179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha GE, Lee J, Kwak H, Song K, Kwon J, Jung SY, Hong J, Chang GE, Hwang EM, Shin HS, Lee CJ, Cheong E (2016) The Ca2+-activated chloride channel anoctamin-2 mediates spike-frequency adaptation and regulates sensory transmission in thalamocortical neurons. Nat Commun 7:13791. 10.1038/ncomms13791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han KS, Chen CH, Khan MM, Guo C, Regehr WG (2020) Climbing fiber synapses rapidly and transiently inhibit neighboring Purkinje cells via ephaptic coupling. Nat Neurosci 23:1399–1409. 10.1038/s41593-020-0701-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han VZ, Magnus G, Zhang Y, Wei AD, Turner EE (2014) Bidirectional modulation of deep cerebellar nuclear cells revealed by optogenetic manipulation of inhibitory inputs from Purkinje cells. Neuroscience 277:250–266. 10.1016/j.neuroscience.2014.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardman RM, Forsythe ID (2009) Ether-à-go-go-related gene K+ channels contribute to threshold excitability of mouse auditory brainstem neurons. J Physiol 587:2487–2497. 10.1113/jphysiol.2009.170548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath R (1977) Modulation of emotion with a brain pacemaker. J Nerv Ment Dis 165:300–317. [PubMed] [Google Scholar]
- Heiney SA, Wohl MP, Chettih SN, Ruffolo LI, Medina JF (2014) Cerebellar-dependent expression of motor learning during eyeblink conditioning in head-fixed mice. J Neurosci 34:14845–14853. 10.1523/JNEUROSCI.2820-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higley MJ, Contreras D (2006) Balanced excitation and inhibition determine spike timing during frequency adaptation. J Neurosci 26:448–457. 10.1523/JNEUROSCI.3506-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hourez R, Servais L, Orduz D, Gall D, Millard I, de Kerchove d'Exaerde A, Cheron G, Orr HT, Pandolfo M, Schiffmann SN (2011) Aminopyridines correct early dysfunction and delay neurodegeneration in a mouse model of spinocerebellar ataxia type. J Neurosci 31:11795–11807. 10.1523/JNEUROSCI.0905-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CG, Chacron MJ (2017) SK channel subtypes enable parallel optimized coding of behaviorally relevant stimulus attributes: a review. Channels (Austin) 11:281–304. 10.1080/19336950.2017.1299835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang WC, Xiao S, Huang F, Harfe BD, Jan YN, Jan LY (2012) Calcium-activated chloride channels (CaCCs) regulate action potential and synaptic response in hippocampal neurons. Neuron 74:179–192. 10.1016/j.neuron.2012.01.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurlock EC, McMahon A, Joho RH (2008) Purkinje-cell-restricted restoration of Kv3.3 function restores complex spikes and rescues motor coordination in Kcnc3 mutants. J Neurosci 28:4640–4648. 10.1523/JNEUROSCI.5486-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman SL, Chen CH, Offermann HL, Drew IR, Harrison BM, Bowman AM, Flick KM, Flaquer I, Regehr WG (2020) Cerebellar Purkinje cell activity modulates aggressive behavior. Elife 9:e53229. 10.7554/eLife.53229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahnsen H (1986) Electrophysiological characteristics of neurones in the guinea-pig deep cerebellar nuclei in vitro. J Physiol 372:129–147. 10.1113/jphysiol.1986.sp016001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayabal S, Chang HHV, Cullen KE, Watt AJ (2016) 4-Aminopyridine reverses ataxia and cerebellar firing deficiency in a mouse model of spinocerebellar ataxia type 6. Sci Rep 6:29489. 10.1038/srep29489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jirenhed DA, Bengtsson F, Hesslow G (2007) Acquisition, extinction, and reacquisition of a cerebellar cortical memory trace. J Neurosci 27:2493–2502. 10.1523/JNEUROSCI.4202-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehl SJ (1991) Quinidine-induced inhibition of the fast transient outward K+ current in rat melanotrophs. Br J Pharmacol 103:1807–1813. 10.1111/j.1476-5381.1991.tb09867.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan MM, Regehr WG (2020) Loss of doc2b does not influence transmission at Purkinje cell to deep nuclei synapses under physiological conditions. Elife 9:e55165. 10.7554/eLife.55165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan MM, Chen CH, Regehr WG (2021) Unusually slow spike frequency adaptation in deep cerebellar nuclei neurons preserves linear transformations on the sub-second timescale. BioRxiv. Advance online publication. Retrieved August 22, 2022. 10.1101/2021.08.10.455865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khawaja FA, Alonso AA, Bourque CW (2007) Ca2+-dependent K+ currents and spike-frequency adaptation in medial entorhinal cortex layer II stellate cells. Hippocampus 17:1143–1148. 10.1002/hipo.20365 [DOI] [PubMed] [Google Scholar]
- Kim JE, Chae S, Kim S, Jung Y-J, Kang M-G, Do Heo W, Kim D (2021) Cerebellar 5HT-2A receptor mediates stress-induced onset of dystonia. Sci Adv 7:abb5735. 10.1126/sciadv.abb5735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim U, Mccormick DA (1998) Functional and ionic properties of a slow afterhyperpolarization in ferret perigeniculate neurons in vitro. J Neurophysiol 80:1222–1235. 10.1152/jn.1998.80.3.1222 [DOI] [PubMed] [Google Scholar]
- Kordasiewicz HB, Gomez CM (2007) Molecular pathogenesis of spinocerebellar ataxia type 6. Neurotherapeutics 4:285–294. 10.1016/j.nurt.2007.01.003 [DOI] [PubMed] [Google Scholar]
- Kubota M, Saito N (1991) Sodium- and calcium-dependent conductances of neurones in the zebra finch hyperstriatum ventrale pars caudale in vitro. J Physiol 440:131–142. 10.1113/jphysiol.1991.sp018700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Camera G, Rauch A, Thurbon D, Lüscher HR, Senn W, Fusi S (2006) Multiple timescales of temporal response in pyramidal and fast spiking cortical neurons. J Neurophysiol 96:3448–3464. 10.1152/jn.00453.2006 [DOI] [PubMed] [Google Scholar]
- Larivière R, Sgarioto N, Márquez BT, Gaudet R, Choquet K, McKinney RA, Watt AJ, Brais B (2019) Sacs R272C missense homozygous mice develop an ataxia phenotype. Mol Brain 12:19. 10.1186/s13041-019-0438-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeDoux MS, Hurst DC, Lorden JF (1998) Single-unit activity of cerebellar nuclear cells in the awake genetically dystonic rat. Neuroscience 86:533–545. 10.1016/s0306-4522(98)00007-4 [DOI] [PubMed] [Google Scholar]
- Leonoudakis D, Gray AT, Winegar BD, Kindler CH, Harada M, Taylor DM, Chavez RA, Forsayeth JR, Yost CS (1998) An open rectifier potassium channel with two pore domains in tandem cloned from rat cerebellum. J Neurosci 18:868–877. 10.1523/JNEUROSCI.18-03-00868.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinas R, Muhlethaler M (1988) Electrophysiology of guinea-pig cerebellar nuclear cells in the in vitro brain stem-cerebellar preparation. J Physiol 404:241–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madison DV, Nicoll RA (1984) Control of the repetitive discharge of rat CA 1 pyramidal neurones in vitro. J Physiol 354:319–331. 10.1113/jphysiol.1984.sp015378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Espinosa PL, Wu J, Yang C, Gonzalez-Perez V, Zhou H, Liang H, Xia X-M, Lingle CJ (2015) Knockout of Slo2.2 enhances itch, abolishes K Na current, and increases action potential firing frequency in DRG neurons. Elife 4:e10013. 10.7554/eLife.10013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDevitt CJ, Ebner TJ, Bloedel JR (1987) Relationships between simultaneously recorded Purkinje cells and nuclear neurons. Brain Res 425:1–13. 10.1016/0006-8993(87)90477-x [DOI] [PubMed] [Google Scholar]
- Milescu LS, Bean BP, Smith JC (2010) Isolation of somatic Na+ currents by selective inactivation of axonal channels with a voltage prepulse. J Neurosci 30:7740–7748. 10.1523/JNEUROSCI.6136-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miterko LN, Lin T, Zhou J, van der Heijden ME, Beckinghausen J, White JJ, Sillitoe RV (2021) Neuromodulation of the cerebellum rescues movement in a mouse model of ataxia. Nat Commun 12:1295. 10.1038/s41467-021-21417-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura Y, Schwindt PC, Crill WE (1989) Electrical properties of facial motoneurons in brainstem slices from guinea pig. Brain Res 502:127–142. 10.1016/0006-8993(89)90468-X [DOI] [PubMed] [Google Scholar]
- Otto JF, Yang Y, Frankel WN, White HS, Wilcox KS (2006) A spontaneous mutation involving Kcnq2 (Kv7.2) reduces M-current density and spike frequency adaptation in mouse CA1 neurons. J Neurosci 26:2053–2059. 10.1523/JNEUROSCI.1575-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedarzani P, Storm JF (1993) PKA mediates the effects of monoamine transmitters on the K+ current underlying the slow spike frequency adaptation in hippocampal neurons. Neuron 11:1023–1035. 10.1016/0896-6273(93)90216-E [DOI] [PubMed] [Google Scholar]
- Pedroarena CM (2020) A slow short-term depression at Purkinje to deep cerebellar nuclear neuron synapses supports gain-control and linear encoding over second-long time windows. J Neurosci 40:5937–5953. 10.1523/JNEUROSCI.2078-19.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Person AL, Raman IM (2011) Purkinje neuron synchrony elicits time-locked spiking in the cerebellar nuclei. Nature 481:502–505. 10.1038/nature10732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pessia M, Servettini I, Panichi R, Guasti L, Grassi S, Arcangeli A, Wanke E, Pettorossi VE (2008) ERG voltage-gated K+ channels regulate excitability and discharge dynamics of the medial vestibular nucleus neurones. J Physiol 586:4877–4890. 10.1113/jphysiol.2008.155762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pineda JC, Galarraga E, Foehring RC (1999) Different Ca2+ source for slow AHP in completely adapting and repetitive firing pyramidal neurons. Neuroreport 10:1951–1956. 23 10.1097/00001756-199906230-00029 [DOI] [PubMed] [Google Scholar]
- Ponce GV, Klaus J, Schutter DJLG (2022) A brief history of cerebellar neurostimulation. Cerebellum 21:715–730. 10.1007/s12311-021-01310-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers RK, Sawczuk A, Musick JR, Binder MD (1999) Multiple mechanisms of spike-frequency adaptation in motoneurones. J Physiol Paris 93:101–114. 10.1016/s0928-4257(99)80141-7 [DOI] [PubMed] [Google Scholar]
- Prestori F, Montagna I, D'angelo E, Mapelli L (2020) The optogenetic revolution in cerebellar investigations. Int J Mol Sci 21:2494. 10.3390/ijms21072494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, Gustafson AE, Padgett D (2000) Ionic currents and spontaneous firing in neurons isolated from the cerebellar nuclei. J Neurosci 20:9004–9016. 10.1523/JNEUROSCI.20-24-09004.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen A, Jirenhed DA, Hesslow G (2008) Simple and complex spike firing patterns in Purkinje cells during classical conditioning. Cerebellum 7:563–566. 10.1007/s12311-008-0068-2 [DOI] [PubMed] [Google Scholar]
- Reeber SL, Otis TS, Sillitoe RV (2013) New roles for the cerebellum in health and disease. Front Syst Neurosci 7:83. 10.3389/fnsys.2013.00083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reis DJ, Ross RA (1973) Dynamic changes in brain dopamine-beta-hydroxylase activity during anterograde and retrograde reactions to injury of central noradrenergic axons. Brain Res 57:307–326. 27 10.1016/0006-8993(73)90139-X [DOI] [PubMed] [Google Scholar]
- Rowland NC, Jaeger D (2005) Coding of tactile response properties in the rat deep cerebellar nuclei. J Neurophysiol 94:1236–1251. 10.1152/jn.00285.2005 [DOI] [PubMed] [Google Scholar]
- Rowland NC, Jaeger D (2008) Responses to tactile stimulation in deep cerebellar nucleus neurons result from recurrent activation in multiple pathways. J Neurophysiol 99:704–717. 10.1152/jn.01100.2007 [DOI] [PubMed] [Google Scholar]
- Sacco T, Bruno A, Wanke E, Tempia F (2003) Functional roles of an ERG current isolated in cerebellar Purkinje neurons. J Neurophysiol 90:1817–1828. 10.1152/jn.00104.2003 [DOI] [PubMed] [Google Scholar]
- Safronov BV, Vogel W (1996) Properties and functions of Na(+)-activated K+ channels in the soma of rat motoneurones. J Physiol 497:727–734. 10.1113/jphysiol.1996.sp021803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Vives MV, Nowak LG, McCormick DA (2000a) Membrane mechanisms underlying contrast adaptation in cat area 17 in vivo. J Neurosci 20:4267–4285. 10.1523/JNEUROSCI.20-11-04267.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Vives MV, Nowak LG, McCormick DA (2000b) Cellular mechanisms of long-lasting adaptation in visual cortical neurons in vitro. J Neurosci 20:4286–4299. 10.1523/JNEUROSCI.20-11-04286.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawczuk A, Powers RK, Binder MD (1995) Spike frequency adaptation studied in hypoglossal motoneurons of the rat. J Neurophysiol 73:1799–1810. 10.1152/jn.1995.73.5.1799 [DOI] [PubMed] [Google Scholar]
- Schwindt PC, Spain WJ, Foehring RC, Chubb MC, Crill WE (1988) Slow conductances in neurons from cat sensorimotor cortex in vitro and their role in slow excitability changes. J Neurophysiol 59:450–467. 10.1152/jn.1988.59.2.450 [DOI] [PubMed] [Google Scholar]
- Schwindt PC, Spain WJ, Crill WE (1989) Long-lasting reduction of excitability by a sodium-dependent potassium current in cat neocortical neurons. J Neurophysiol 61:233–244. 10.1152/jn.1989.61.2.233 [DOI] [PubMed] [Google Scholar]
- Shakkottai VG, Xiao M, Xu L, Wong M, Nerbonne JM, Ornitz DM, Yamada KA (2009) FGF14 regulates the intrinsic excitability of cerebellar Purkinje neurons. Neurobiol Dis 33:81–88. 10.1016/j.nbd.2008.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakkottai VG, Costa M do C, Dell'Orco JM, Sankaranarayanan A, Wulff H, Paulson HL (2011) Early changes in cerebellar physiology accompany motor dysfunction in the polyglutamine disease spinocerebellar ataxia type 3. J Neurosci 31:13002–13014. 10.1523/JNEUROSCI.2789-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprague JM, Chambers W (1959) An analysis of cerebellar function in the cat, as revealed by its partial and complete destruction, and its interaction with the cerebral cortex. Arch Ital Biol 97:68–88. [Google Scholar]
- Stocker M (2004) Ca2+-activated K+ channels: molecular determinants and function of the SK family. Nat Rev Neurosci 5:758–770. 10.1038/nrn1516 [DOI] [PubMed] [Google Scholar]
- Stoodley CJ, D'Mello AM, Ellegood J, Jakkamsetti V, Liu P, Nebel MB, Gibson JM, Kelly E, Meng F, Cano CA, Pascual JM, Mostofsky SH, Lerch JP, Tsai PT (2017) Altered cerebellar connectivity in autism and cerebellar-mediated rescue of autism-related behaviors in mice. Nat Neurosci 20:1744–1751. 10.1038/s41593-017-0004-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thach WT (1968) Discharge of Purkinje and cerebellar nuclear neurons during rapidly alternating arm movements in the monkey. J Neurophysiol 31:785–797. 10.1152/jn.1968.31.5.785 [DOI] [PubMed] [Google Scholar]
- Thach WT (1970) Discharge of cerebellar neurons related to two maintained postures and two prompt movements. I. Nuclear cell output. J Neurophysiol 33:527–536. 10.1152/jn.1970.33.4.527 [DOI] [PubMed] [Google Scholar]
- Thach WT (1975) Timing of activity in cerebellar dentate nucleus and cerebral motor cortex during prompt volitional movement. Brain Res 88:233–241. 10.1016/0006-8993(75)90387-x [DOI] [PubMed] [Google Scholar]
- Tsai PT, Rudolph S, Guo C, Ellegood J, Gibson JM, Schaeffer SM, Mogavero J, Lerch JP, Regehr W, Sahin M (2018) Sensitive periods for cerebellar-mediated autistic-like behaviors. Cell Rep 25:357–367.e4. 10.1016/j.celrep.2018.09.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turecek J, Jackman SL, Regehr WG (2016) Synaptic specializations support frequency-independent Purkinje cell output from the cerebellar cortex. Cell Rep 17:3256–3268. 10.1016/j.celrep.2016.11.081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzingounis AV, Kobayashi M, Takamatsu K, Nicoll RA (2007) Hippocalcin gates the calcium activation of the slow afterhyperpolarization in hippocampal pyramidal cells. Neuron 53:487–493. 10.1016/j.neuron.2007.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uusisaari M, Obata K, Knöpfel T (2007) Morphological and electrophysiological properties of GABAergic and non-GABAergic cells in the deep cerebellar nuclei. J Neurophysiol 97:901–911. 10.1152/jn.00974.2006 [DOI] [PubMed] [Google Scholar]
- Van Overwalle F, Baetens K, Mariën P, Vandekerckhove M (2014) Social cognition and the cerebellum: a meta-analysis of over 350 fMRI studies. Neuroimage 86:554–572. 10.1016/j.neuroimage.2013.09.033 [DOI] [PubMed] [Google Scholar]
- Wallén P, Robertson B, Cangiano L, Löw P, Bhattacharjee A, Kaczmarek LK, Grillner S (2007) Sodium-dependent potassium channels of a Slack-like subtype contribute to the slow afterhyperpolarization in lamprey spinal neurons. J Physiol 585:75–90. 10.1113/jphysiol.2007.138156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HS, Pan Z, Shi W, Brown BS, Wymore RS, Cohen IS, Dixon JE, McKinnon D (1998) KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science 282:1890–1893. 10.1126/science.282.5395.1890 [DOI] [PubMed] [Google Scholar]
- Wang L, Simms J, Peters CJ, Fontaine MT, la Li K, Gill TM, Jan YN, Jan LY (2019) TMEM16B calcium-activated chloride channels regulate action potential firing in lateral septum and aggression in male mice. J Neurosci 39:7102–7117. 10.1523/JNEUROSCI.3137-18.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SSH, Kloth AD, Badura A (2014) The cerebellum, sensitive periods, and autism. Neuron 83:518–532. 10.1016/j.neuron.2014.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh JP, Yuen G, Placantonakis DG, Vu TQ, Haiss F, O'Hearn E, Molliver ME, Aicher SA (2002) Why do Purkinje cells die so easily after global brain ischemia? Aldolase C, EAAT4, and the cerebellar contribution to posthypoxic myoclonus. Adv Neurol 89:331–359. [PubMed] [Google Scholar]
- Wu Y, Raman IM (2017) Facilitation of mossy fibre-driven spiking in the cerebellar nuclei by the synchrony of inhibition. J Physiol 595:5245–5264. 10.1113/JP274321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Lisberger SG (2014) Purkinje-cell plasticity and cerebellar motor learning are graded by complex-spike duration. Nature 510:529–532. 10.1038/nature13282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Zhang Z, Xiao S, Tien J, Le S, Le T, Jan LY, Yang H (2017) Inferior olivary TMEM16B mediates cerebellar motor learning. Neuron 95:1103–1111.e4. 10.1016/j.neuron.2017.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng N, Raman IM (2009) Ca currents activated by spontaneous firing and synaptic disinhibition in neurons of the cerebellar nuclei. J Neurosci 29:9826–9838. 10.1523/JNEUROSCI.2069-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Lin Z, Voges K, Ju C, Gao Z, Bosman LWJ, Ruigrok TJ, Hoebeek FE, De Zeeuw CI, Schonewille M (2014) Cerebellar modules operate at different frequencies. Elife 3:e02536. 10.7554/eLife.02536 [DOI] [PMC free article] [PubMed] [Google Scholar]