

Abstract
The current increase in number and diversity of targeted anticancer agents poses challenges to the logistics and timeliness of molecular diagnostics (MolDx), resulting in underdiagnosis and treatment. Whole‐genome sequencing (WGS) may provide a sustainable solution for addressing current as well as future diagnostic challenges. The present study therefore aimed to prospectively assess feasibility, validity, and value of WGS in routine clinical practice. WGS was conducted independently of, and in parallel with, standard of care (SOC) diagnostics on routinely obtained tumor samples from 1,200 consecutive patients with metastatic cancer. Results from both tests were compared and discussed in a dedicated tumor board. From 1,200 patients, 1,302 samples were obtained, of which 1,216 contained tumor cells. WGS was successful in 70% (854/1,216) of samples with a median turnaround time of 11 days. Low tumor purity (<20%) was the main reason for not completing WGS. WGS identified 99.2% and SOC MolDx 99.7% of the total of 896 biomarkers found in genomic regions covered by both tests. Actionable biomarkers were found in 603/848 patients (71%). Of the 936 associated therapy options identified by WGS, 343 were identified with SOC MolDx (36.6%). Biomarker‐based therapy was started in 147 patients. WGS revealed 49 not previously identified pathogenic germline variants. Fresh‐frozen, instead of formalin‐fixed and paraffin‐embedded, sample logistics were easily adopted as experienced by the professionals involved. WGS for patients with metastatic cancer is well feasible in routine clinical practice, successfully yielding comprehensive genomic profiling for the vast majority of patients. © 2022 The Authors. The Journal of Pathology published by John Wiley & Sons Ltd on behalf of The Pathological Society of Great Britain and Ireland.
Keywords: cancer, diagnostics, DNA sequencing, whole genome sequencing Biomarker, precision oncology
Introduction
With the rapidly expanding tableau of (increasingly tumor‐agnostic) targeted therapies [1, 2, 3, 4, 5], genome‐driven cancer care has become the cornerstone of modern precision oncology [6]. However, pathology laboratories are facing increasing challenges in keeping up with the speed at which targeted drugs and their associated biomarkers are entering clinical oncology practice. These challenges exist at multiple levels. First, the rapidly expanding druggable genome requires pathology laboratories to continuously update and validate their molecular diagnostic (MolDx) arsenal to cover the latest actionable genomic alterations. In practice, this inevitably causes substantial delays in clinical implementation of newly approved biomarkers [7], and contributes to inequality of clinical care [8]. Second, indications for MolDx are still largely tumor type‐dependent [9]. This leads to a multitude of complex and often sequential tumor type‐specific diagnostic routings that are error prone and easily outdated [10]. This impedes effective MolDx for identifying rare targets in common cancers, as well as identifying therapeutic targets in less common cancers [11]. Third, the interplay between somatic mutations and (possible) germline DNA alterations is becoming increasingly important in targeted therapies, e.g. carriers of germline BReast CAncer genes (BRCA) mutations without biallelic loss of function do not respond to Poly(ADP‐Ribose) Polymerase (PARP) inhibitors [12, 13] and whole‐genome sequencing (WGS) potentially offers insight into both tumor and germline. Fourth, there is an increasing demand for more complex biomarkers like signatures for homologous repair deficiency (HRD), microsatellite instability (MSI), and tumor mutational load, which increasingly guide therapeutic decisions and can act as valid proxies of epigenetic inactivation of druggable pathways [1, 14, 15, 16]. Fifth, combined large datasets on comprehensive genomic characterization, therapeutic interventions, and patient outcome improves decision support in precision oncology.
Hence, there is a growing need for a future proof, tumor‐type‐independent, comprehensive MolDx approach for all (metastatic) cancer patients, and the current fragmented and reactive MolDx approach does not meet these standards. Although most of these challenges could partly be covered by implementing large next‐generation sequencing (NGS) panels, WGS has considerable advantages over panel sequencing. First, large NGS panels still have to be updated every couple of years. Second, since WGS is a stable test, the generated data will always be comparable in time and place and allows algorithms like Cancer of Unknown Primary Prediction Algorithm (CUPPA) and Cancer of Unknown Primary Location Resolver (CUPLR) to be implemented and improved over time [17, 18]. Third, because of its completeness, it allows for better retrospective analysis in self‐learning healthcare systems and research endeavors. Furthermore, no biomarker will be forgotten to be tested, a challenge especially relevant when the diagnosis is uncertain. WGS can successfully address the above‐mentioned challenges and would therefore offer an attractive solution [19, 20, 21], but its feasibility in routine pathology practice remains to be proven [22]. WGS has proven its feasibility in multiple pediatric centers; however, the setting of pediatric oncology centers differs substantially in volume and scale compared to adult oncology centers [21, 23, 24]. Historically, tissue handling in diagnostic pathology is based on formalin fixation and paraffin embedding (FFPE). While targeted gene panel‐based diagnostics work well with FFPE material, WGS ideally requires fresh‐frozen material to avoid FFPE‐induced sequencing artifacts and obtain genome‐wide accurate variant calls, including comprehensive copy number and structural variant calling. Implementation of WGS in routine clinical practice therefore requires collecting and working with fresh‐frozen samples in routine pathology workflows. The whole genome sequencing implementation in standard diagnostics for every cancer patient (WIDE) study (WGS Implementation in standard Diagnostics for Each cancer patient) therefore aimed to prospectively generate evidence on the feasibility and clinical validity (primary endpoints), as well as clinical value of WGS (secondary endpoint) in routine clinical practice for patients with metastatic cancer [25].
Materials and methods
Study design, setting and population
WIDE is a single‐center prospective, observational, diagnostic study in patients with (suspected) stage IV solid tumors of all occurring tumor types, approved by the Medical Ethical Committee of the Netherlands Cancer Institute (NKI) (NL68609.031.18) and conducted in concordance with the Declaration of Helsinki, Dutch law, and Good Clinical Practice. All patients provided written informed consent. Patients were eligible when biopsy, resection, or suitable cytology (e.g. pleural effusion or ascites) samples could be obtained safely as part of routine diagnostic procedures. Patients, from whom archival fresh‐frozen tumor samples were available, were also eligible if not treated in‐between with tyrosine kinase inhibitors, since this could have shifted the genomic profile by clonal selection [26]. WGS was performed at the Hartwig Medical Foundation (Amsterdam, The Netherlands; hereafter referred to as Hartwig) in parallel with and independently of standard of care (SOC) diagnostics. Depending on patient preference, declared in the informed consent, pathogenic (class 4 and 5 [27]) germline variants in genes with targeted tumor therapy implications were either reported as inherited variants, along with an offer for routine clinical genetics counseling, or as variants present in the tumor sample without reporting germline status (supplementary material, Table S1).
Germline variants without cancer‐related actionability were not investigated nor reported.
Sample collection and processing
SOC procedures aimed to collect 2–4 biopsies. A 10‐ml whole‐blood sample was drawn for sequencing germline DNA as a reference, allowing us to discriminate somatic mutations from germline DNA background variations in bioinformatic analyses. The macroscopically best biopsy was prioritized for SOC diagnostics, which, depending on the clinical question at hand, either did or did not include MolDx. This biopsy then was FFPE processed. For the SOC MolDx portfolio at the NKI, see Supplementary materials and methods and supplementary material, Tables S2–S4. Next, WGS was performed in parallel with and independently of SOC diagnostics. To this end, specimens were cryoembedded using a PrestoCHILL (Milestone Medical, Kalamazoo, MI, USA) device for 60 s at −40 °C. Subsequently, the specimens were cut into frozen sections of 5 μm thickness on coated glass then stained with hematoxylin and eosin.
Next, a pathologist microscopically assessed on frozen sections of every sample a tumor cell percentage (pTCP), and when needed demarcated a tumor area for manual microdissection. For tissue specimens, a pTCP of ≥20% and for cytology specimens ≥30% was required. Preferably within 24 h, both tumor and blood samples were shipped by courier to Hartwig for WGS. Any remaining (frozen) tissue was processed to FFPE blocks. The NKI Department of Pathology operates under ISO15189:2012 accreditation.
WGS and bioinformatics
WGS was performed at Hartwig on Illumina NovaSeq6000 platforms (Illumina, San Diego, CA, USA) with a sequencing depth of >90x for tumor DNA according to standard procedures as described previously [20, 25]. DNA isolated from blood was sequenced at an average depth of >30x. Sequencing data were analyzed with an optimized fully open source in‐house bioinformatic pipeline [28] (code available through github.com/hartwigmedical, see further details in Supplementary materials and methods). Hartwig operates under ISO17025:2015 and ISO/NEN27001 accreditation.
Reporting
The WGS report (OncoAct, Amsterdam, The Netherlands) contained all variants with a high driver likelihood [20] relevant for diagnostic purposes and cancer treatment decision‐making, further referred to as biomarkers. These encompassed mutations and amplifications/losses in 460 genes, 63 promiscuous fusion partners, and 402 known oncogenic fusions, mutational signatures (tumor mutational load, HRD, and MSI), and viral insertions (Human Papilloma virus, Epstein–Barr virus, and Merkel cell polyomavirus). Although each tumor sample was analyzed for its whole‐genome characteristics, including all genes (exons and introns) and intergenic regions, the WGS report is limited to variants with high driver likelihood in order to provide clinically manageable reports for the treating physicians. WGS, as well as SOC MolDx results, when applicable, were discussed in a weekly dedicated molecular tumor board. In case of any discrepancies, additional verification tests were performed on the original input samples used for WGS and SOC according to a predefined workflow (supplementary material, Figure S1). Results of both WGS and SOC MolDx were communicated via routine pathology reporting to the treating physician.
Continuous evaluation and improvement
The design of the study allowed for continuous evaluation and improvement of procedures, in line with ISO15189:2012. Study progress was evaluated biweekly in a multidisciplinary team involving study coordinators, pathologists, radiologists, medical oncologists, clinical geneticists, and support staff. As a result, multiple stages of the process underwent optimizations such as biopsy procedures, sample logistics (tissue and DNA handling and processing), and bioinformatics (supplementary material, Figure S2).
Sample size calculation
The objective sample size of 1,200 patients was based on the primary endpoint “clinical validation.” The aim was to detect the same variants by WGS as SOC MolDx in at least 95% of the cases with one‐sided 95% confidence. Under an assumed concordance rate of 97.5%, 624 individual genomic biomarkers were needed to achieve a lower limit of the confidence interval to be at least 95% with a power of 96%. Based on the retrospective WGS data analysis of ~3,000 patients [20], the required 624 SOC biomarkers were expected to be identifiable in 1,200 patients.
Statistical analyses
Patient and tumor characteristics, feasibility, clinical validity, and clinical value were analyzed with descriptive statistics. Categorical variables are shown as percentages or frequencies and continuous variables as medians with ranges. Analyses were performed using the Matplotlib and NumPy packages in Python, 3.7.5. [29, 30]
Results
Feasibility of WGS in routine clinical care
One thousand and two hundred patients with 32 different tumor types were included over a 22‐month period (Table 1), and 95 patients underwent >1 sampling procedure, resulting in 1,302 samples in total. Of these, 86 (7%) did not contain tumor cells. The remaining 1,216 samples, consisting of biopsies (n = 931), resections (n = 247), and cytological specimens (n = 38), entered the WGS procedure. Overall, WGS was successfully completed in 70% (854/1,216) of tumor samples (Figure 1A). In 9% (113/1,216) of samples the pTCP was <20% and consequently WGS was not started, and in 15% (181/1,216) the molecular tumor cell percentage (TCP) (mTCP, as determined by WGS data) detected by WGS was <20%, despite a pTCP ≥20%. Other reasons for dropout included poor DNA quality (technical failure) in 3% (31/1,216), low DNA yield in 2% (29/1,216), and patient‐specific circumstances (e.g. allogenic stem cell transplantation) in 0.7% (8/1,216). Over time, the feasibility of WGS improved from 50% in 111 WGS attempts during the first 3 months to 72% in 177 attempts during the last 3 months of the study, due to continuous improvements in sample retrieval, handling, and processing (Figure 1B). Direct feedback between pathologists and radiologists performing image‐guided biopsies contributed to this improvement. WGS was successfully performed with a median turnaround time (TAT) of 12 working days (range 4–52) (Figure 1C). TAT improved throughout the study down to 11 working days in the final 3 months, and 95% of the WGS results were available within 17 working days. Importantly, when WGS could not be completed, yet a clinical indication for MolDx existed, both panel sequencing and Archer fusion analysis was successful in 87% (186/214) of these cases, indicating that targeted sequencing approaches can still be performed in the majority of cases if WGS is not feasible.
Table 1.
Baseline table for all 1,200 included patients with metastatic cancer.
| Patient characteristics | N = 1,200 |
|---|---|
| Age at WGS, years | |
| Mean | 59.3 |
| Range | 18–98 |
| Sex, male:female | 43:57 |
| Primary tumor location, n (%) | |
| Lung cancer | 344 (29%) |
| Colorectal cancer | 210 (18%) |
| Breast cancer | 143 (12%) |
| Sarcoma | 80 (6.7%) |
| Other | 67 (5.6%) |
| Prostate cancer | 47 (3.9%) |
| CUP | 34 (2.8%) |
| Ovarian cancer | 32 (2.7%) |
| Melanoma | 29 (2.4%) |
| Bladder cancer | 25 (2.1%) |
| Lung NETs | 23 (1.9%) |
| Esophageal cancer | 21 (1.8%) |
| Renal cell cancer | 19 (1.6%) |
| Head and neck cancer | 14 (1.2%) |
| Stomach cancer | 13 (1.1%) |
| GEP‐NETs | 13 (1.1%) |
| Cervical cancer | 11 (0.92%) |
| GIST | 10 (0.83%) |
| Malignant mesothelioma | 9 (0.75%) |
| Urothelial cell cancer of the bladder and renal pelvis | 8 (0.67%) |
| Anal cancer | 7 (0.58%) |
| Thymoma and thymic cancer | 7 (0.58%) |
| Vulva cancer | 6 (0.50%) |
| Penile cancer | 6 (0.50%) |
| Pancreatic cancer | 5 (0.42%) |
| Endometrial cancer | 4 (0.33%) |
| Lymphoma | 4 (0.33%) |
| Cholangiocarcinoma | 3 (0.25%) |
| Thyroid cancer | 3 (0.25%) |
| Basal cell carcinoma | 2 (0.17%) |
| Hepatocellular carcinoma | 1 (0.083%) |
CUP, cancer of unknown primary; GEP‐NETs, gastroenteropancreatic neuroendocrine tumors; GIST, gastrointestinal stromal tumor; NET, neuroendocrine tumor; WGS, whole‐genome sequencing.
Figure 1.
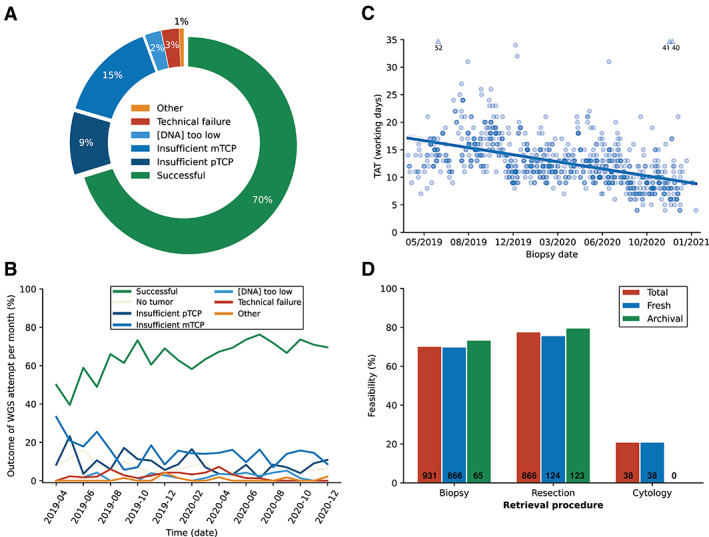
Feasibility of WGS in routine pathology practice. WGS was successfully completed in 854/1,216 (70%) samples containing tumor cells. The main reason for ineligibility for WGS was a low percentage of tumor cells in 24% of samples (9% insufficient pTCP and 15% insufficient mTCP) (A). Due to continuous optimizations during the course of the study, feasibility of WGS improved as samples with no tumor cells or insufficient TCP declined (B). WGS could be performed in a clinically acceptable time frame of a median of 12 working days. During the course of the study, the TAT decreased from 16 workings days in the first 3 months to 11 workings days in the last 3 months (C). Success rates of WGS procedures were high when using biopsy or resection specimens (70% and 77%, respectively), and could be performed both freshly obtained and fresh‐frozen archival tissue. Cytology specimens were less suitable for WGS, with a success rate of 21% (D). mTCP, molecular tumor cell percentage; pTCP, tumor cell percentage assessed by a pathologist.
Determinants of WGS feasibility
The success rates of WGS on tissues from biopsies and resections (including 187 fresh‐frozen archival tissues) were 70 and 77%, respectively, and could be performed on both freshly obtained samples and fresh‐frozen archival tissue (Figure 1D). Cytology specimens proved a less suitable source for WGS, with a success rate of 21%, mainly due to a low pTCP (68% of cases). WGS success rates differed by biopsy sites (supplementary material, Figure S3), with the highest success rates from liver (78%, n = 298), soft tissue (70%, n = 138), and lymph nodes (62%, n = 177), and the lowest success rates from lung (transthoracic biopsies 49%, n = 168), peritoneum (52%, n = 58), and bone (54%, n = 47). Details on all WGS attempts are listed in supplementary material, Table S5.
Biomarker detection
Concordance between SOC MolDx and WGS was examined by looking at all biomarkers that could in principle be detected by both methods. Any discordant result was analyzed and classified using a predefined scheme (supplementary material, Figure S1). In total, 932 biomarkers, including 766 driver mutations (SNVs/MNVs/indels), 100 copy number alterations (amplifications/deletions), 46 fusion events, 13 viral insertions, and seven genome‐wide signatures were identified for comparison between WGS and SOC MolDx (Table 2). On top of these 932 biomarkers potentially detectable by SOC MolDx, WGS detected an additional 3,860 biomarkers, including 249 genome‐wide signatures (high mutational load [n = 195], MSI [n = 8], and HRD [n = 46]) (supplementary material, Table S6). This included 2,018 biomarkers in patients who, by that time, did not have an indication for SOC MolDx and 1,842 biomarkers not covered by targeted sequencing panels in patients who did receive SOC MolDx.
Table 2.
Concordance of WGS and SOC MolDx diagnostics.
| Total (n = 914) | SNVs/MNs/indels (n = 760) | Copy number variants | Fusions (n = 46) | Viral insertions (n = 13) | ||
|---|---|---|---|---|---|---|
| Amplifications (n = 66) | Deletions (n = 31) | |||||
| True positives | 903 | 760 | 64 | 25 | 41 | 13 |
|
887 | 749 | 64 | 25 | 36 | 13 |
|
889 | 755 | 64 | 15 | 41 | 13 |
| False negatives | ||||||
|
16 | 11 | 0 | 0 | 5 | 0 |
|
14 | 4 | 0 | 10 | 0 | 0 |
| False positives | ||||||
|
3 | 0 | 0 | 0 | 3 | 0 |
|
8 | 0 | 2 | 6 | 0 | 0 |
| Post hoc analysis | ||||||
|---|---|---|---|---|---|---|
| Total (n = 880) | SNVs/MNs/indels (n = 760) | Copy number variants | Fusions (n = 46) | Viral insertions (n = 13) | ||
| Amplifications (n = 66) | Deletions (NA) | |||||
| True positives | 878 | 760 | 64 | NA | 41 | 13 |
|
871 | 754 | 64 | 40 | 13 | |
|
876 | 758 | 64 | 41 | 13 | |
| False negatives | ||||||
|
7 | 6 | 0 | NA | 1 | 0 |
|
2 | 2 | 0 | 0 | 0 | |
| False positives | NA | |||||
|
0 | 0 | 0 | 0 | 0 | |
|
2 | 0 | 2 | 0 | 0 | |
Bold is used for total numbers, e.g. in the post hoc analysis first row there are 878 true positives with 871 biomarkers detected by WGS + 7 (false negatives) − 0 (false positives).
Indels, insertion/deletions; MNVs, multi‐nucleotide variants; MolDx, molecular diagnostics; NA, not available; SNVs, single nucleotide variants; SOC, standard of care; WGS, whole‐genome sequencing.
Of the 924 true‐positive biomarkers that were diagnostically reported during the course of the study, WGS detected 904 (97.8%, two‐sided 95% CI 96.7–98.7%), along with three false‐positive calls (Figure 2A), while SOC MolDx detected 910 (98.5%, two‐sided 95% CI 97.5–99.2%), along with five false‐positive calls. As part of continuous optimization, changes in the WGS pipeline included optimized calling of splice variants and detection of fusion events. In the SOC MolDx workflow, NGS‐based calling of gene deletions was abandoned because of large numbers of false‐positive and false‐negative results observed. In a post hoc analysis with the latest versions of these pipelines, biomarker detection rates were 889/896 (99.2%, two‐sided 95% CI 98.4–99.6%) for WGS and 894/896 (99.7%, two‐sided 95% CI 99.2–99.9%) for SOC MolDx, along with zero and two false‐positives, respectively (Table 2, Figure 2B). These remaining false‐negative and –positive results were attributable to various factors (supplementary material, Table S7).
Figure 2.
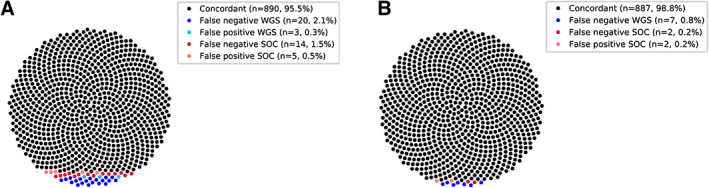
Concordance of WGS and SOC MolDx was determined in two ways. (A) by comparing WGS and SOC MolDx results as they were reported in real time during the course of the study, while in accordance with ISO 15189:2012 a continuous optimization process of bioinformatic procedures took place and (B) in a post hoc analysis of all samples using the latest optimized pipelines as these emerged by the end of the study. SOC, standard of care; WGS, whole‐genome sequencing.
Clinical value of WGS
Of 848 patients, 603 (71%) had ≥1 actionable event(s), i.e. biomarker‐based eligibility for either regular therapy or a clinical trial in the Netherlands (supplementary material, Figure S4). In 250 patients, multiple biomarker‐based therapy options were detected (supplementary material, Figure S5), resulting in a total of 936 different regular (n = 145) or experimental (n = 791) therapy options (supplementary material, Figure S6). Of these, 343 were identified with SOC MolDx (36.6%). Conversely, 593 therapy options in 431 patients remained undetected without WGS, either because SOC MolDx was not (yet) performed as part of the regular diagnostic work‐up (345 options in 241 patients), or genomic biomarker regions were not covered by SOC MolDx (248 options in 190 patients).
At a median follow‐up of 14 months, 147 out of 603 patients with actionable events (24%) had started with a biomarker‐based therapy in a regular (n = 54, 11% based on WGS‐only findings) or clinical trial setting (n = 93; 80% based on WGS‐only findings). These numbers are likely to increase, as not all patients have exhausted their regular treatment options (Figure 3 and Table 3).
Figure 3.
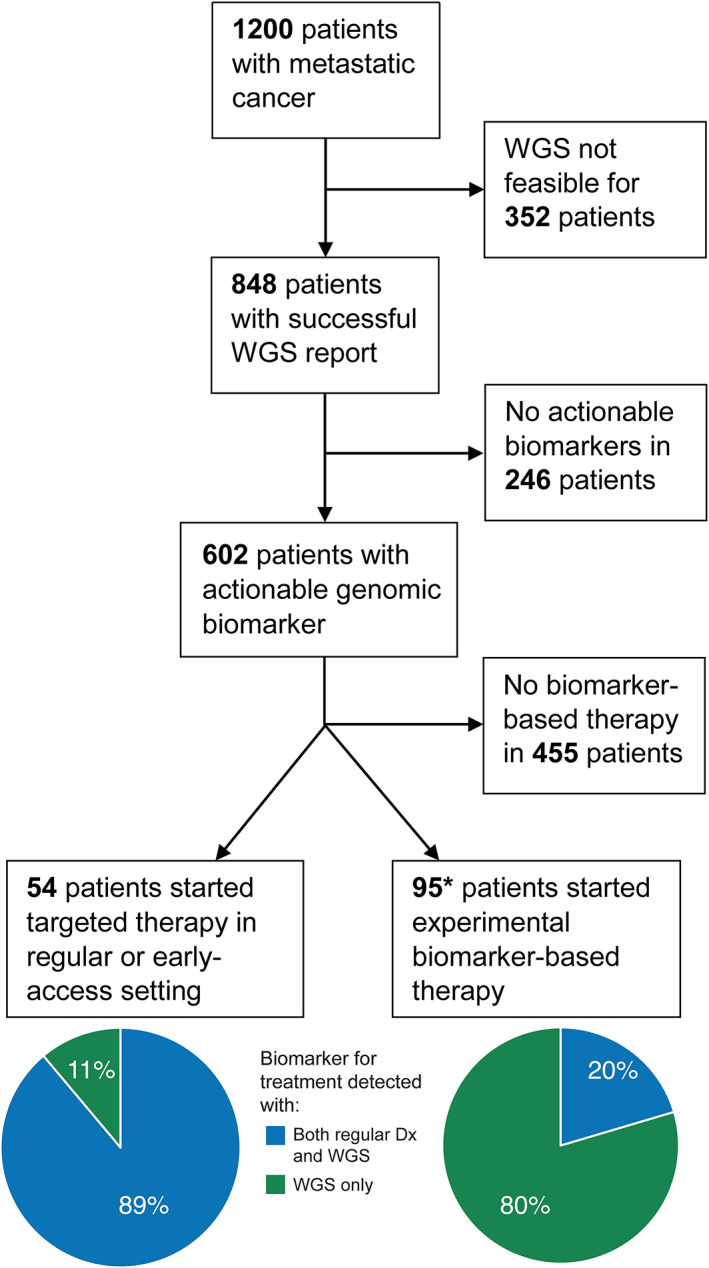
Clinical value of prospective WGS. Ultimately, 147 patients started biomarker‐based therapy at a median follow‐up of 14 months, of which two patients (*) received both biomarker‐based therapy in a regular setting and an experimental setting after progression. Dx, diagnostics, WGS, whole‐genome sequencing.
Table 3.
Treatment options in patients (who initiated therapy) based on SOC + WGS or WGS‐only results.
| Regular therapy | Number of patients |
|---|---|
| Detected with SOC + WGS | 40 |
| Detected with WGS only | 3 |
| Early access program | |
| Detected with SOC + WGS | 8 |
| Detected with WGS only | 3 |
| Clinical trials | |
| Detected with SOC + WGS | 19 |
| Detected with WGS only | 76 |
| Regular + early access program | |
| Detected with SOC + WGS | 48 |
| Detected with WGS only | 6 |
SOC, standard of care; WGS, whole‐genome sequencing.
In 70 patients, 72 pathogenic germline variants (PGVs) were detected, 23 of which had been identified before with SOC germline diagnostics (Figure 4 and supplementary material, Table S8). Interestingly, somatic second hits (biallelic mutation or loss of heterozygosity (LOH)) were present in only 41/72 (57%) patients, predominantly in cancer predisposition genes associated with the tumor type at hand (supplementary material, Figures S7, S8). Biallelic loss in the background of PGVs provided a rationale for tumor‐directed therapy in 20/39 patients with PGVs in HRD genes, and 4/7 patients with PGVs in mismatch repair (MMR) genes (supplementary material, Figure S9). In all patients with biallelic loss of function of HRD or MMR genes, genome‐wide signatures of HRD [15], and MSI, respectively, were present (supplementary material, Table S9).
Figure 4.
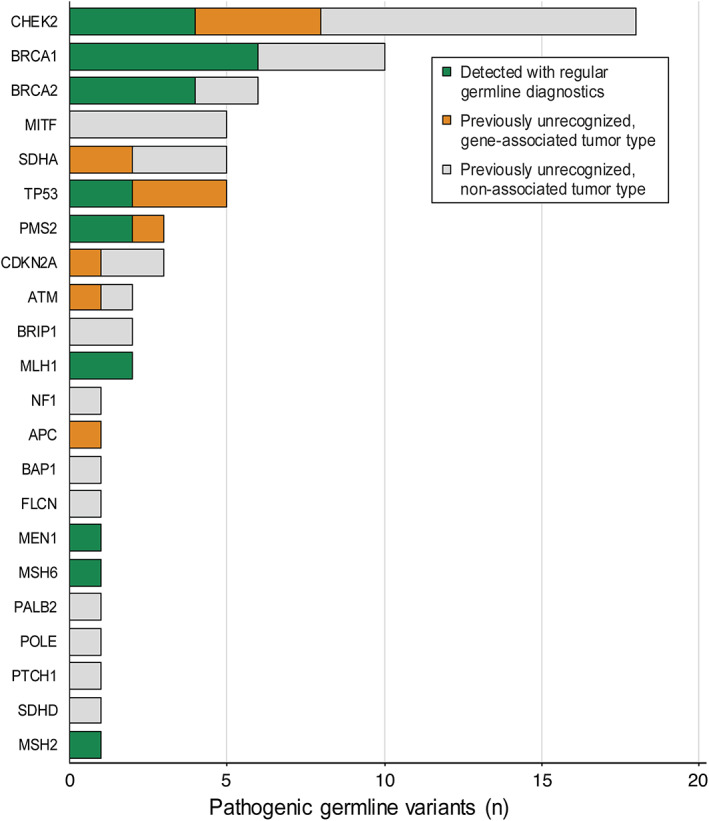
Germline variants detected by WGS. In total, 72 pathogenic germline variants (PGVs) were identified by WGS in 848 patients, of which 23 previously had been detected with SOC diagnostics. The figure shows the type and number of PGVs identified in these 848 patients and whether they were detected with SOC diagnostics or previously unrecognized.
Discussion
In modern precision oncology, we are facing a diagnostic challenge to identify all relevant genomic alterations for every individual cancer patient, the number of which increases with the rapidly expanding druggable genome. While this requires diagnostic pathology laboratories to continuously update their MolDx arsenal, in reality infrastructural delays occur in implementing assays for new biomarkers, which translates into delayed access for patients to new treatments [31]. As WGS allows for complete genomic characterization, any new DNA‐based biomarker is by definition already covered by WGS; it merely requires a small adaptation of the bioinformatics or reporting pipeline, thus providing a versatile solution to this challenge [32].
In the WIDE study we demonstrated that implementation of WGS, including adapting to fresh‐frozen instead of FFPE sample logistics, is well feasible in routine pathology and clinical practice [25]. While the current study was conducted in the setting of a dedicated comprehensive cancer center, laboratory procedures basically do not differ from other pathology laboratories operating under ISO15189 and according to professional guidelines. WGS succeeded in 71% of metastatic cancer patients within clinically acceptable timelines, even when only the second‐best sample was used [33]. Furthermore, with steadily decreasing sequencing costs, sequencing with a deeper coverage may further increase WGS feasibility in low TCP samples [28]. Importantly, in the majority of cases in which a low TCP was limiting for WGS, SOC MolDx was still feasible using the isolated fresh‐frozen DNA or remaining biopsy material from the same procedure. A sensible strategy would therefore be to use WGS when possible and panels when needed, thus providing the most comprehensive MolDx possible for every patient (supplementary material, Figure S10). The concordance between WGS and SOC MolDx of 98.8% demonstrated the clinical validity of WGS (Figure 2).
During the study, pathologists, clinical molecular biologists in pathology, medical oncologists, and clinical geneticists became more familiar with the interpretation and additional diagnostic value of WGS, especially in the context of complex differential diagnoses [18, 34]. In fact, WGS also appeared to have additional diagnostic value in germline diagnostics, with previously 49 unrecognized pathogenic germline variants in cancer susceptibility genes being identified. WGS thus encompasses valuable somatic (including genome‐wide signatures) and germline information, allowing further optimization of therapeutic strategies.
In line with other reports [20, 35], here WGS identified actionable biomarkers for regular therapy options or clinical trial allocation in 71% of patients with a WGS result. The majority of these biomarkers were not detected with current SOC diagnostic approaches, including targeted sequencing panels in selected patient populations. Moreover, comprehensive genomic characterization of tumors by WGS in combination with detailed clinical phenotyping provides a solid basis for a learning healthcare system, which is a crucial condition for deploying precision medicine to its full extent.
As an alternative to WGS, whole‐exome sequencing (WES) is occasionally proposed. Often this is a cost‐based consideration, since the laboratory logistics and bioinformatics pipeline are similar for both methods but less (costly) sequencing reagents are required. However, WGS allows the analysis of more complex tumor characteristics, including mutational signatures and MSI analysis. Reliable quantification of these characteristics also relies on the intronic and intergenic analysis, and these will not be analyzed by WES. Furthermore, detection of fusion genes, typically formed by fusions of intergenic breakpoint regions, is not possible with WES.
Evidently, cost is a crucial consideration for implementing WGS into routine pathology practice, and trade‐offs may vary between different institutions/locations. While at an individual test level, direct costs of WGS are higher than that of WES or NGS panels, a comprehensive cost versus benefit analysis is much more complex. This is being studied outside the scope of the present study and will be presented elsewhere.
In summary, the present prospective study has demonstrated that WGS‐based diagnostics is feasible in routine pathology practice and adds value for clinical decision‐making. The required adjustments in laboratory logistics were well manageable and acceptable to the healthcare professionals involved, which shows implementation hurdles in adopting WGS in routine pathology practice can be overcome. In fact, in immediate follow up to the present study, the Department of Pathology at the Netherlands Cancer Institute, in collaboration with Hartwig, has implemented WGS in routine clinical practice. This is further facilitated by the fact that recently a first provision for reimbursement of WGS in the Netherlands has been established [36].
Author contributions statement
PR, LB, AL, TB, VN, JH, HS, LK, EC, EV, GM and KM were responsible for the conceptualization of the study design. HS, EC, EV, GM and KM were involved in funding acquisition. EK and FL were responsible for sample collection and KS, LS, PR, LB, AL, TB, IR, LS, DS, JB, EB, LK, KM and GM for data collection. KS, LS, KM and GM were responsible for drafting of the article. KS and LS were responsible for the formal data analysis with aid of VN. The underlying data reported in the article has been accessed and verified by multiple authors (KS, LS and KM). All authors have read, revised, and approved the article.
Supporting information
Supplementary materials and methods
Figure S1. Predefined workflow for resolving discordant results between WGS and SOC molecular diagnostics
Figure S2. Optimizations of the workflow during the course of the study
Figure S3. Feasibility of WGS on biopsies per localization
Figure S4. Clinical value of prospective WGS: actionable events
Figure S5. Clinical value of prospective WGS: number of therapy options per patient
Figure S6 Clinical value of prospective WGS: therapy options
Figure S7. Pathogenic germline variants with somatic loss
Figure S8. Pathogenic germline variants per tumor type
Figure S9. Pathogenic germline variants with therapy options
Figure S10. Cascade strategy for molecular diagnostics
Table S1. Genes for which pathogenic germlines were analyzed
Table S2. NGS modified Ampliseq panel
Table S3. Archer Fusionplex – Lung v1.0
Table S4. Archer Fusionplex – (Expanded) Sarcoma v2.0
Table S5. Information per WGS attempt
Table S6. Additional variants detected by WGS
Table S7. Clinical validity
Table S8. Pathogenic germline variants detected by WGS
Table S9. Correlation signatures in BRCA and Lynch patients
Acknowledgements
The authors thank the following members of the Pathology Department of the Netherlands Cancer Institute: Petur Snaebjornsson, Liudmila Kodach, Maurits van Montfoort, Claudie Flohil, Elise Bekers, Laura Smit, Hester van Boven, Hugo Horlings, Bart van de Wiel, Joyce Sanders, Emilie Groen, Mijke Bol, Jacqueline van der Wal, Mathilde Almekinders, Efraim Rosenberg, Mirjam Boelens, Frans Hogervorst, Jelle Wesseling, Lisette Forrer, Annegien Broeks, Petra Nederlof, and Tom van Wezel for implementing the WGS workflow and the interpretation of sequencing data. Furthermore, we thank the laboratory technicians: Jan‐Nico Ridderbos, Kelly van Deventer, Saphira van Diest, Angelique van Dam, Sandra van der Slikke‐Meijvogel, Nicoline Woudsma, Tim Niessen, Candy van Riel, Jaschenka Houtgraaf, Irene Hegger, Selena Jevtic, Assia Bourdoudi, Unga Unmehopa, Mickey Dukic, Isis van Hallem, Maarten Breet, and Verena Koning for adopting and executing the fresh‐frozen workflow and organizing all the involved logistics. Likewise, we thank the following clinicians and research nurses: Willemijn Theelen, Marieke Vollebergh, Egbert Smit, Wanda de Kanter, Winette van der Graaf, Marloes van Dongen, Martin Rijlaarsdam, Wieneke Buikhuisen, Sandra Visser, and Judith Westra for their active role in the recruitment of patients. We thank Tarik Baetens for his role in tissue retrieval and Simone Koole, Geert Frederix, and Valesca Retèl for Health Technology Assessment advice. We thank ZonMw (the Netherlands Organization for Health Research and Development) and the Hartwig Medical Foundation for funding the study. The WIDE is funded by ZonMw, the Netherlands Organization for Health Research and Development (Project No. 446002004), including an in‐kind contribution of the Hartwig Medical Foundation. The study protocol has been independently peer‐reviewed by ZonMw. ZonMw had no role in the design nor the collection, analysis, and interpretation of the data, nor the writing of the article. Health‐RI is acknowledged for providing research infrastructure support.
Conflict of interest statement: AL reports grants from BMS, MSD, AstraZeneca, and Boehringer, nonfinancial support from Merck Serono and Roche. HS and EC report consultancy fees and support for attending meetings and traveling from Illumina. EV is member of the supervisory board of Hartwig. GM is cofounder and a board member (CSO) of CRCbioscreen BV, he has a research collaboration with CZ Health Insurances (cash matching to ZonMW grant) and he has research collaborations with Exact Sciences, Sysmex, Sentinel Ch. SpA, Personal Genome Diagnostics (PGDX), DELFi; these companies provide materials, equipment, and/or sample/genomic analyses. GM is an Advisory Board member of ‘Missie Tumor Onbekend.’ KM reports research grants from AstraZeneca and speakers' fees from MSD, Roche, AstraZeneca. and Benecke. KM received consultancy fees from Pfizer, BMS, Roche, MSD, Abbvie, AstraZeneca, Diaceutics, Lilly, Bayer, Boehringer Ingelheim and nonfinancial support from Roche, Takeda, Pfizer, PGDx and DELFi. KS, LS, PR, LB, FL, EK, TB, IR, LS, DS, VN, JB, EB, JH, and LK report no conflicts of interest.
Data availability statement
The researchers are willing to share with external qualified researchers access to patient‐level data and supporting clinical documents. These requests are reviewed and approved by an independent review committee on the basis of scientific merit. All data provided are anonymized to respect the privacy of patients who have participated in the study, in line with applicable laws and regulations. Data can be requested via https://www.hartwigmedicalfoundation.nl/applying‐for‐data/ or g.meijer@nki.nl.
References
- 1. Marcus L, Fashoyin‐Aje LA, Donoghue M, et al. FDA Approval Summary: pembrolizumab for the treatment of tumor mutational burden‐high solid tumors. Clin Cancer Res 2021; 27: 4685–4689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Marcus L, Lemery SJ, Keegan P, et al. FDA Approval Summary: pembrolizumab for the treatment of microsatellite instability‐high solid tumors. Clin Cancer Res 2019; 25: 3753–3758. [DOI] [PubMed] [Google Scholar]
- 3. Looney AM, Nawaz K, Webster RM. Tumour‐agnostic therapies. Nat Rev Drug Discov 2020; 19: 383–384. [DOI] [PubMed] [Google Scholar]
- 4. Drilon A, Laetsch TW, Kummar S, et al. Efficacy of Larotrectinib in TRK fusion‐positive cancers in adults and children. N Engl J Med 2018; 378: 731–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Doebele RC, Drilon A, Paz‐Ares L, et al. Entrectinib in patients with advanced or metastatic NTRK fusion‐positive solid tumours: integrated analysis of three phase 1‐2 trials. Lancet Oncol 2020; 21: 271–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yap TA, Johnson A, Meric‐Bernstam F. Precision medicine in oncology‐toward the integrated targeting of somatic and germline genomic aberrations. JAMA Oncol 2021; 7: 507–509. [DOI] [PubMed] [Google Scholar]
- 7. Aitken MVP, Bennet K, Tewary V, et al. Optimizing Oncology Care Through Biomarker Adoption. Parisippany, NJ: IQVIA Institute, 2020. [Google Scholar]
- 8. Lynch JA, Berse B, Rabb M, et al. Underutilization and disparities in access to EGFR testing among Medicare patients with lung cancer from 2010–2013. BMC Cancer 2018; 18: 306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mosele F, Remon J, Mateo J, et al. Recommendations for the use of next‐generation sequencing (NGS) for patients with metastatic cancers: a report from the ESMO Precision Medicine Working Group. Ann Oncol 2020; 31: 1491–1505. [DOI] [PubMed] [Google Scholar]
- 10. Steeghs EMP, Groen HJM, Schuuring E, et al. Mutation‐tailored treatment selection in non‐small cell lung cancer patients in daily clinical practice. Lung Cancer 2022; 167: 87–97. [DOI] [PubMed] [Google Scholar]
- 11. Horak P, Heining C, Kreutzfeldt S, et al. Comprehensive genomic and transcriptomic analysis for guiding therapeutic decisions in patients with rare cancers. Cancer Discov 2021; 11: 2780–2795. [DOI] [PubMed] [Google Scholar]
- 12. van der Wijngaart H, Hoes LR, van Berge Henegouwen JM, et al. Patients with biallelic BRCA1/2 inactivation respond to olaparib treatment across histologic tumor types. Clin Cancer Res 2021; 27: 6106–6114. [DOI] [PubMed] [Google Scholar]
- 13. Sokol ES, Pavlick D, Khiabanian H, et al. Pan‐cancer analysis of BRCA1 and BRCA2 genomic alterations and their association with genomic instability as measured by genome‐wide loss of heterozygosity. JCO Precis Oncol 2020; 4: 442–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Van Hoeck A, Tjoonk NH, van Boxtel R, et al. Portrait of a cancer: mutational signature analyses for cancer diagnostics. BMC Cancer 2019; 19: 457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nguyen L, W M Martens J, Van Hoeck A, et al. Pan‐cancer landscape of homologous recombination deficiency. Nat Commun 2020; 11: 5584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Davies H, Glodzik D, Morganella S, et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat Med 2017; 23: 517–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nguyen L, van Hoeck A, Cuppen E. Machine learning‐based tissue of origin classification for cancer of unknown primary diagnostics using genome‐wide mutation features. bioRxiv 2021; 2021.10.05.463244. [DOI] [PMC free article] [PubMed]
- 18. Schipper LJSP, Samsom KG, Bosch LJW, et al. 1133P Whole genome sequencing can classify diagnostically challenging tumors ESMO congress 2021 abstract book. Ann Oncol 2021; 32: S924–S925. [Google Scholar]
- 19. ICGC/TCGA Pan‐Cancer Analysis of Whole Genomes Consortium . Pan‐cancer analysis of whole genomes. Nature 2020; 578: 82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Priestley P, Baber J, Lolkema MP, et al. Pan‐cancer whole‐genome analyses of metastatic solid tumours. Nature 2019; 575: 210–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wong M, Mayoh C, Lau LMS, et al. Whole genome, transcriptome and methylome profiling enhances actionable target discovery in high‐risk pediatric cancer. Nat Med 2020; 26: 1742–1753. [DOI] [PubMed] [Google Scholar]
- 22. Nangalia J, Campbell PJ. Genome sequencing during a patient's journey through cancer. N Engl J Med 2019; 381: 2145–2156. [DOI] [PubMed] [Google Scholar]
- 23. Byrjalsen A, Hansen TVO, Stoltze UK, et al. Nationwide germline whole genome sequencing of 198 consecutive pediatric cancer patients reveals a high incidence of cancer prone syndromes. PLoS Genet 2020; 16: e1009231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Trotman J, Armstrong R, Firth H, et al. The NHS England 100,000 Genomes Project: feasibility and utility of centralised genome sequencing for children with cancer. Br J Cancer 2022; 127: 137–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Samsom KG, Bosch LJW, Schipper LJ, et al. Study protocol: Whole genome sequencing Implementation in standard Diagnostics for Every cancer patient (WIDE). BMC Med Genomics 2020; 13: 169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. van de Haar J, Hoes LR, Roepman P, et al. Limited evolution of the actionable metastatic cancer genome under therapeutic pressure. Nat Med 2021; 27: 1553–1563. [DOI] [PubMed] [Google Scholar]
- 27. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015; 17: 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Roepman P, de Bruijn E, van Lieshout S, et al. Clinical validation of whole genome sequencing for cancer diagnostics. J Mol Diagn 2021; 23: 816–833. [DOI] [PubMed] [Google Scholar]
- 29. Harris CR, Millman KJ, van der Walt SJ, et al. Array programming with NumPy. Nature 2020; 585: 357–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hunter JD. Matplotlib: a 2D graphics environment. Comput Sci Eng 2007; 9: 90–95. [Google Scholar]
- 31. van den Broek D, Hiltermann TJN, Biesma B, et al. Implementation of novel molecular biomarkers for non‐small cell lung cancer in The Netherlands: how to Deal with increasing complexity. Front Oncol 2020; 9: 1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cameron DL, Di Stefano L, Papenfuss AT. Comprehensive evaluation and characterisation of short read general‐purpose structural variant calling software. Nat Commun 2019; 10: 3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lindeman NI, Cagle PT, Aisner DL, et al. Updated molecular testing guideline for the selection of lung cancer patients for treatment with targeted tyrosine kinase inhibitors: guideline from the College of American Pathologists, the International Association for the Study of Lung Cancer, and the Association for Molecular Pathology. Arch Pathol Lab Med 2018; 142: 321–346. [DOI] [PubMed] [Google Scholar]
- 34. Schipper LJ, Monkhorst K, Samsom K, et al. Whole‐genome sequencing to improve sarcoma diagnosis and patient care. J Clin Oncol 2021; 39: 11540. [Google Scholar]
- 35. Cobain EF, Wu YM, Vats P, et al. Assessment of clinical benefit of integrative genomic profiling in advanced solid tumors. JAMA Oncol 2021; 7: 525–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zorginstituut . Moleculaire diagnostiek in de Oncologie. Diemen, The Netherlands, 2021. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary materials and methods
Figure S1. Predefined workflow for resolving discordant results between WGS and SOC molecular diagnostics
Figure S2. Optimizations of the workflow during the course of the study
Figure S3. Feasibility of WGS on biopsies per localization
Figure S4. Clinical value of prospective WGS: actionable events
Figure S5. Clinical value of prospective WGS: number of therapy options per patient
Figure S6 Clinical value of prospective WGS: therapy options
Figure S7. Pathogenic germline variants with somatic loss
Figure S8. Pathogenic germline variants per tumor type
Figure S9. Pathogenic germline variants with therapy options
Figure S10. Cascade strategy for molecular diagnostics
Table S1. Genes for which pathogenic germlines were analyzed
Table S2. NGS modified Ampliseq panel
Table S3. Archer Fusionplex – Lung v1.0
Table S4. Archer Fusionplex – (Expanded) Sarcoma v2.0
Table S5. Information per WGS attempt
Table S6. Additional variants detected by WGS
Table S7. Clinical validity
Table S8. Pathogenic germline variants detected by WGS
Table S9. Correlation signatures in BRCA and Lynch patients
Data Availability Statement
The researchers are willing to share with external qualified researchers access to patient‐level data and supporting clinical documents. These requests are reviewed and approved by an independent review committee on the basis of scientific merit. All data provided are anonymized to respect the privacy of patients who have participated in the study, in line with applicable laws and regulations. Data can be requested via https://www.hartwigmedicalfoundation.nl/applying‐for‐data/ or g.meijer@nki.nl.