

Abstract
Mast cells (MCs) are widely recognized as central effector cells during type 2 inflammatory thought to also play a role in innate immune responses, wound healing, and potentially cancer. Circulating progenitor cells mature to MCs in peripheral tissues, where they exhibit phenotypic and functional heterogeneity. This diversity likely originates from differences in MC development imprinted by microenvironmental signals. The advent of single-cell transcriptomics reveals MC diversity beyond differences in proteases that were classically used to identify MC phenotypes. Here, we provide an overview of the current knowledge on MC progenitor differentiation and characteristics, and MC heterogeneity seen in health versus disease that are drastically advanced through single-cell profiling technologies. This powerful approach can provide detailed cellular maps of tissues to decipher the complex cellular functions and interactions that may lead to identifying candidate factors to target in therapies.
Keywords: Mast cell progenitors, Mast cell heterogeneity, single-cell RNA-sequencing, Type 2 inflammation
Background
Mast cells (MCs) are histochemically distinctive tissue resident effector cells derived from the hematopoietic system. Although best known as principal effector cells involved in IgE-driven type 1 hypersensitivity that underlies many allergic disorders, MCs are an ancient component of the immune system, with MC-like cells detected in tunicates, predating adaptive immunity by hundreds of millions of years (1, 2). MCs almost certainly serve diverse additional functions in inflammation, innate and adaptive host defense, wound healing, regulation of vasomotor tone, and cancer. Such diverse functions in turn require the capacity for context-specific modifications of MC effector properties. While MC functional diversification has long been suspected based on histochemical markers, the exclusive tissue residence of mature MCs presents significant challenges to understanding the true range of MC diversity and how this diversity is regulated based on functional studies. The recent application of single cell genomics to the MC field has begun to overcome some of these limitations and reveals a much broader range of diversification than predicted by histochemistry alone. Moreover, these studies have also permitted the identification of receptors and transcriptional systems that are promising targets for the development of therapeutics for diseases associated with or driven by MC-dependent effector systems. Here, we provide an overview of insights gained on MC progenitor (MCp) characteristics and MC heterogeneity across tissue and diseases using single-cell transcriptomic analysis and discuss the findings and remaining questions to unravel the underlying drivers of their infiltration and diversity in health and disease.
Insights into MC development from murine models
MCs arise from circulating hematopoietic MCps that mature following recruitment to peripheral tissues (3). Recent murine studies have indicated that MC populations found in peripheral connective tissues arise from a series of developmentally discrete waves of MCps during and following embryogenesis. In the first wave, early erythro-myeloid progenitor cells (EMPs) from yolk sac give rise to MCs expressing CD117, T1/ST2, and containing avidin-binding granules. These early MCs are then replaced by a second wave of late EMP-derived MCps that give rise to transcriptionally distinct connective tissue MCs (CTMCs) that are maintained independently of bone marrow (BM) through adulthood, indicating that progenitor origin and developmental stage of the host likely play roles in shaping MC identity (4, 5). In contrast, MCs in the intestinal mucosa arise from MCps derived from hematopoietic stem cells (HSCs), first from aorta-gonad-mesonephros (AGM) hemogenic endothelium and later from the fetal liver and BM (4, 6, 7). MCs across peripheral connective tissues share a core transcriptional signature, consistent with a shared developmental origin, yet also show tissue-specific differences in transcriptional profile and cell surface protein expression, indicating a role for their tissue microenvironment in shaping MC identity (8). A recent study from our laboratory examined the transcriptional distinction between constitutive and inflammation-induced MCs within murine lung tissue (6). Through this approach, we found that the resident lung CTMCs were significantly enriched for a transcriptional signature shared by CTMCs across five tissues in naïve mice relative to recruited BM-derived MCs, even following allergic lung inflammation (6, 8). Notably, the BM-derived MCs infiltrating the pulmonary tissue during inflammation exhibit enrichment for TGF-β signature genes along with inflammation-associated superimposed signals (6). These studies suggest that MCp origin, microenvironmentally-derived tissue signals, and tissue inflammatory state all influence the ultimate phenotype of MC subsets. Although peripheral CTMCs have minimal transcriptional overlap with basophils beyond components of the high affinity IgE receptor FcɛR1 and histamine biosynthetic components, numerous studies have suggested the existence of a bi-potent basophil/MC progenitor within the mouse BM and spleen (9–11). Supporting this concept, we determined that Mcpt8, previously identified as a basophil-specific transcript, is expressed by BM-derived MCs in vivo, a finding also reported by Hamey and colleagues (6, 12). While these findings are consistent with the derivation of MCs and basophils from a common progenitor, there has been substantial debate within the field as to where exactly the MC and basophil progenitor fits within broader hematopoiesis.
Historically, hematopoietic progenitors have been identified based on surface marker expression and characterized based on their progeny in clonal expansion assays and following transplantation (13, 14). Classical model of hematopoiesis proposes that multipotent progenitors (MPPs) give rise to common myeloid progenitors (CMPs) with an immunophenotype distinct from common lymphoid progenitors (CLPs). In this model, CMPs give rise to both granulocyte-monocyte progenitors (GMPs) and megakaryocyte-erythroid progenitors (MEP) (13, 15). Adolfsson et al. identified lymphoid-primed multipotent progenitors (LMPPs) with the capacity to give rise to both lymphocytes and granulocyte/macrophages but not the megakaryocyte-erythroid lineage, establishing that MPP branch into to CMPs and LMPPs, with both possessing the potential to develop to GMPs (16). Studies based on clonal assays and transplantation suggested that GMPs, MEPs, and MPPs all may have the potential to differentiate into MCs (9, 17–19). Franco and colleagues were among the first to combine single-cell cloning- and transcriptomics-based approaches to understand mouse granulocyte differentiation, finding that MCps are distinct from the GMP population that gives rise to monocytes and neutrophils, instead sharing expression of a set of transcription factors (Gata2, Gata1) and surface receptors (T1/ST2 and erythropoietin receptor) with the MEP pool (20). Drissen et al. similarly identified Gata1 expression as an early point of lineage divergence through single-cell RNA-sequencing (scRNA-seq), generating a reporter mouse and discovering that Gata1+ cells in the immunophenotypically-defined pre-GMP and GMP pools had megakaryocyte, erythrocyte, MC, and eosinophil differentiation capacity but minimal monocyte and neutrophil potential (21). While LMPP, Gata1− GMPs and pre-GMPs were able to give rise to monocytes and neutrophils, they were unable to differentiate into MCs. These findings were consistent with prior lineage tracing approaches supporting early bifurcation of myeloid and erythroid lineages within a heterogenous MPP pool (22). Subsequent studies using scRNA-seq supported this bifurcation and further found that the MPP trajectory towards defined lineages progresses in a continuum rather than through distinct subpopulations (23, 24). Tusi and colleagues similarly characterized a linked developmental trajectory for MCs and basophils that was closely associated with the megakaryocyte and erythroid lineages and separate from the neutrophil and monocyte (25). This linked trajectory was confirmed by Dahlin and colleagues, who used a massively parallel scRNA-seq-based approach to analyze BM of WT and W41/W41 Kit mutant mice (26). While Kit mutant mice had intact MEP and basophil differentiation trajectories, the MC progenitor branch from the MC/Basophil bipotent progenitor was notably absent, indicating a requirement for Kit signaling for MCp development in vivo. Although eosinophils were identified through sequencing in this study, their developmental relationship to MC and basophils was unclear. The same dataset was later used by Wanet et al. to identify E-cadherin as a surface marker capable of identifying both basophil and MC progenitors within the BM, confirming a link between these cells, eosinophils, and the MEP lineage, but also identifying rare clones capable of giving rise to monocytes or neutrophils in addition to basophils and/or eosinophils (27).
Clarification of human MC and basophil development through scRNA-seq
In humans, progenitor cells from both the AGM and the yolk sac contribute to the fetal liver progenitor pool that seeds peripheral tissues prior to the emergence of BM-derived progenitors (28). MCs develop in yolk sac prior to emergence of HSCs in non-lymphoid tissues, suggesting the potential for multiple waves of MCs in humans as in mouse, although this has not been experimentally confirmed (29). Several studies suggest that MC differentiation in BM is also similar between mouse and human. As in mouse, the classical model of hematopoiesis in human was challenged due to the potential of both LMPPs and CMPs to give rise to myeloid cells and granulocytes (30, 31). Using a sort-based approach, Görgens and colleagues found that the eosinophil and basophil lineages originated from a common EMP that segregated from GMPs based on surface expression of CD133, while neutrophils arose from GMPs. Macrophages also developed from EMP-cultured cells, albeit within colonies forming a mixed population of erythroid and myeloid cells (CFU-MIX) that were assumed to be transient CD133low CMPs which lacked the potential to develop into neutrophils (32). Their findings challenged the existence of a true CMP and proposed a model where MPPs branched to LMPPs and EMPs, the latter lacking GMP potential. The advent of transcriptomics contributed to resolving the relationship between the progenitors and dynamic of progression from primitive progenitor cells to more defined lineages. Velten et al. integrated cell indexing and scRNA-seq approaches to illustrate that early stem and progenitor cells gradually progress in a continuum into more defined lineages expressing CD38 (33). This finding indicated that traditional use of a limited number of surface markers creates subpopulations that, although immunophenotypically distinct, exhibit transcriptional overlap and potentially share differentiation trajectories. The progression to the basophil/eosinophil/MC lineages coincided with simultaneous induction and suppression of shared modules of transcripts where these progenitors displayed an immunophenotype that was similar to the MEP trajectory but distinct from classic GMP commitment. A similar relationship was observed in umbilical cord blood by Zheng and colleagues, who identified a close link between the basophil/eosinophil/MC lineages and MEP, and determined that human MCp strongly express the transferrin receptor CD71 (TFRC), a classical marker of erythroid precursors (34). They further identified expression of MC-associated transcripts in prior bulk transcriptomics studies of hematopoiesis that used CD71 as a marker for the MEP lineage. Drissen et al. similarly used transcriptomic analysis to show that differential expression of CD131 and CD114 separates the Bas/Eos/MC lineages from neutrophil/monocyte progenitors, respectively (35). Thus, scRNA-seq based approaches have proven highly useful identifying markers and modulators of each lineage. These important studies clarified the lineage relationship between MCs, basophils, and eosinophils in humans, suggesting that all three major granulocytes involved in type 2 (T2) inflammation may have evolved from a common precursor.
Within peripheral blood, Dahlin and colleagues used flow cytometry to identify the existence of a Lin- CD34+ CD117+ FcɛR1+ committed MCp (36). Ex vivo culture assays suggested that these MCps lack the multi-lineage capacity of MC/Basophil/eosinophil progenitors within the bone marrow or cord blood. More recently, Wu and colleagues used scRNA-seq to define MCps within the context of other circulating CD34+ CD117+ cells, finding that while some MC-associated transcripts (TPSAB1, HDC) were tightly linked to the MCp, others associated with FcɛR1α were more broadly expressed (37). They further found that IL-3 drove the proliferation of the MCp population, while IL-5 supported its survival. However, identification of human MCp within peripheral tissue has proven more elusive, in part because CD34 is inconsistently detected on the surface of MCs in inflamed tissue (38, 39).
The differentiation potential of the progenitor cells into various lineages as evaluated in vitro using a cocktail of cytokines and a layer of feeder cells may not recapitulate their actual fate potential in vivo due to a lack of critical microenvironmental signals or the simultaneous administration of cytokines that may not be present within the same niche in vivo. Transcriptomic studies suggest that HSCs progress through intermediate progenitor stages, gradually losing their potential to develop into some lineages while increasing their propensity to become others following fate decision points along the pathway (Fig, 1). It is therefore likely that some intermediate populations retain the capacity to develop into more than one cell type depending on the growth factors in the microenvironment and the degree to which they have progressed down a differentiation pathway to a particular lineage. An example is the derivation of macrophages from eosinophil, basophil common progenitor prior to commitment to the latter cell types (32). Moreover, while scRNA-seq approaches have substantially increased our understanding of how MCs fit within the broader hematopoietic landscape, several important questions remain. In particular, whether and how MCp characteristics may change in disease and the potential contribution of other less committed progenitors to driving MC expansion in health and disease have yet to be explored.
Fig 1.
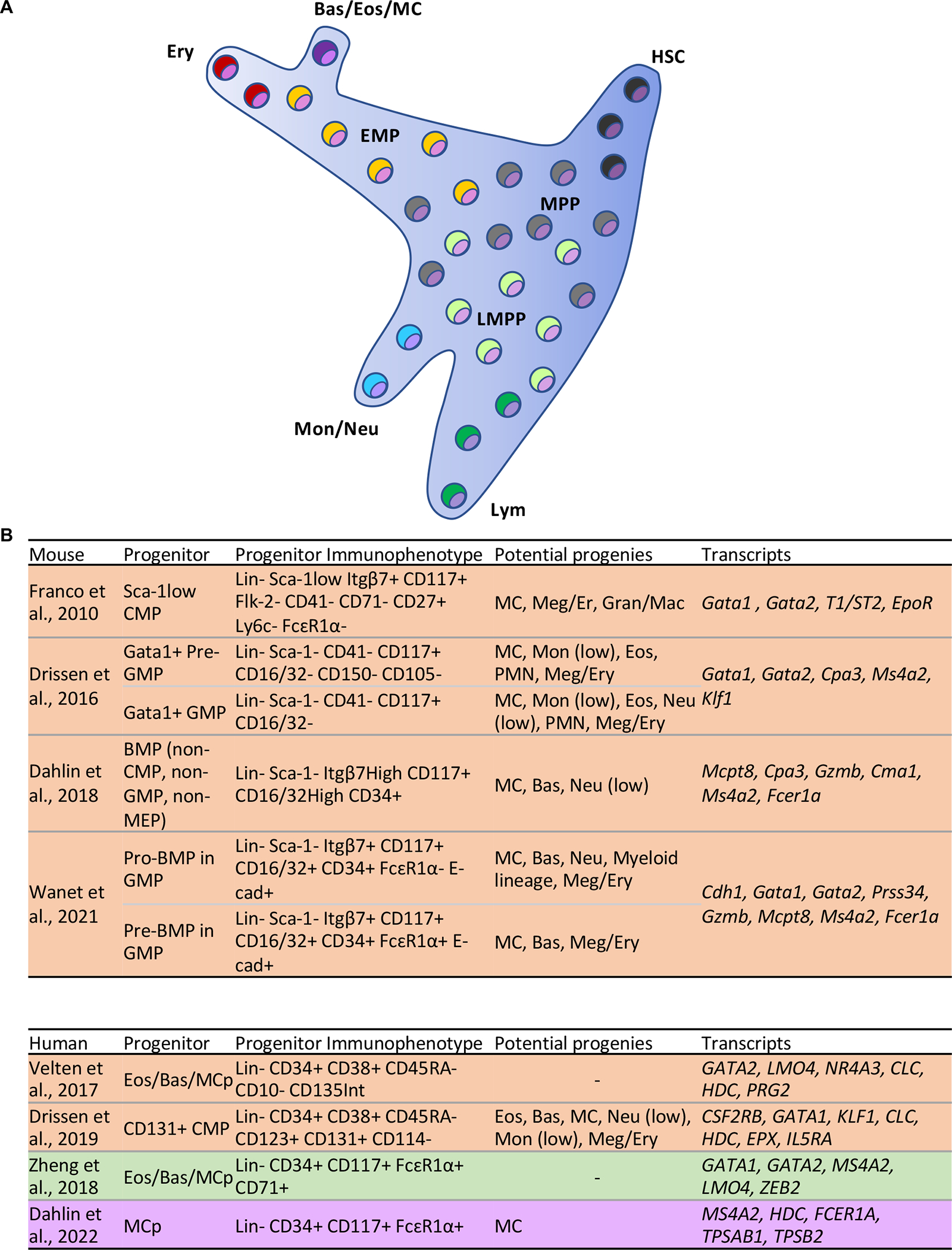
MC differentiation and immunophenotype of MC progenitors. (A) In human, hematopoietic stem cell/multipotent progenitor cells (HSC/MPP) in a continuum progress to erythro myeloid progenitors (EMP) or lymphoid-primed multipotent progenitors (LMMP) that give rise to different lineages (33, 34). Basophil/eosinophil/MC lineages arise from a common precursor that in turn differentiates from a progenitor shared with megakaryocytes and erythrocytes. (B) MC progenitors in mouse and human have been defined through a series of studies and can be identified using the indicated markers expressed on their surface (20, 21, 26, 27, 33–35, 37). These progenitor cells have the potential to differentiate to MCs alone or MCs and other lineages. A few of the transcripts expressed by the progenitors are also listed. Colors indicate tissue origin for each study, including bone marrow (orange), cord blood (green), and peripheral blood (purple). Lin: markers used to label defined lineages such as lymphocytes and monocytes, which may vary across studies; BMP: Bipotent MC basophil progenitor; Meg/Ery: Megakaryocyte/Erythrocyte; Gran/Mac: Granulocyte/Macrophage; Bas: Basophil; Neu: Neutrophil; Eos: Eosinophil; PMN: Polymorphonuclear; Mon: Monocyte; Lym: Lymphocyte.
Identification of disease-associated human MC clusters in whole tissue scRNA-seq studies
As noted previously, both the scarcity of MCs and their exclusive tissue residence presents challenges to their study in humans. Traditionally, MCs in human tissues have therefore been studied using histologic approaches. Collectively these studies have contributed substantially to our understanding of the correlation of MC burden in tissues to disease outcomes, such as in allergic rhinitis and severe asthma, and identified disease-associated differences in MC protease expression, such as the induced expression of carboxypeptidase A3 by intraepithelial MCs arising in several T2 inflammatory diseases (40–43). While functional studies performed on MCs derived by in vitro culture techniques have identified the capacity of MCs to generate a broad range of pro-inflammatory mediators, the relationship between what MCs have the capacity to make in vitro and what they actually make in vivo remains unclear (44). Whole tissue transcriptomic studies have long hinted at a prominent role for MCs in T2 disease, with MC-specific transcripts appearing among the most differentially regulated transcripts between health and disease in eosinophilic esophagitis (EoE) and asthma (41, 45). However, we still have little understanding of how and whether these mediators differ among MC subsets, and how mediator production varies across disease states.
With the advent of scRNA-seq, our understanding of mature tissue MCs in health and disease has improved substantially (46). MCs often form a distinct cluster within scRNA-seq datasets, likely due at least in part to their robust expression of protease-encoding transcripts that are often high on the list of differentially expressed genes. Interestingly, while scRNA-seq-based studies of hematopoiesis have posited a developmental link between MC, basophils, and eosinophils, the transcriptomes of the latter two have proven frustratingly elusive in peripheral tissues. Eosinophil clusters are rarely detected, even within inflammatory conditions under which they comprise the majority of the immune infiltrate (47–49). This may be due to a number of factors, ranging from expression of endogenous RNAses to mechanical instability of eosinophils and basophils. The lack of basophil representation contributes to a lack of knowledge regarding the transcriptional distinctions between induced MCs and basophils. While human peripheral basophils are transcriptionally distinct from MCs within the skin, they may either be transcriptionally similar enough to intraepithelial MCs in the lung and sinonasal tissue to appear indistinguishable at commonly used clustering resolution or, as with eosinophils, resistant to detection via scRNA-seq (8, 50).
Defining roles for MCs in upper airway inflammation
While few tissue-level studies have examined MC heterogeneity, these studies have provided significant insight into effector programs in vivo. A number of studies have focused on understanding MC functionality in nasal polyposis, a disease associated with large-scale MC hyperplasia and that is routinely treated via surgical resection. In a study of chronic rhinosinusitis (CRS) using the SeqWell platform, similar to previous studies that used histology (42), we found that MCs were significantly expanded in CRS with nasal polyps (CRSwNP) relative to CRS without nasal polyps (CRSsNP) (47). We identified MCs as a potentially major source of T2-associated cytokine transcripts (IL5, IL13) and amphiregulin (AREG), all of which were upregulated in MCs in CRSwNP relative to CRSsNP tissue (47). Stevens et al. also evaluated MCs in nasal polyps with a focus on understanding distinctions between nasal polyp of patients with aspirin exacerbated respiratory disease (AERD), a severe endotype of CRSwNP associated with asthma, rapid polyp regrowth, and respiratory responses to aspirin AERD (48). In agreement with our studies, they identified MCs as a dominant site of expression for transcripts encoding a number of eicosanoid-associated biosynthetic proteins (LTC4S, PTGS1, PTGS2, ALOX5, ALOX5AP, TBXAS, HPGD). They further identified a link between MC expression of HPGD (encoding 15-keto prostaglandin dehydrogenase) and an accumulation of arachidonic acid products driven by 15-lipoxygenase (15-LO) in AERD, specifically 15-oxo-ETE, a potent eosinophil chemoattractant that forms from HPGD-mediated conversion of 15-LO-derived 15-hydroxytetraenoic acid (15-HETE). ALOX15 (encoding 15-LO) was highly upregulated in the epithelium of nasal polyps from AERD subjects and correlated with MC burden and with HPGD expression. Bangert and colleagues separately evaluated differences between MCs in AERD and CRSwNP tissue, identifying enhanced MC expression of HPGD in AERD MCs, as well as elevations in transcripts encoding cytokines and growth factors (IL13, VEGFA, CSF1), histidine decarboxylase (HDC), and protease-encoding transcripts (CPA3, TPSD1, PRSS57) (49). Thus, MC effector programs are differentially regulated both between nasal polyps and controls and across nasal polyp endotypes. Collectively, these studies support a major role for MCs as drivers of the immunopathology of CRSwNP and AERD through both cytokines and lipid mediators.
Defining roles for MCs in lung inflammation
Several recent scRNA-seq-based studies provide fresh insight into MC function in the lung across a number of human diseases. In a study comparing lung tissue of subjects with asthma relative to healthy controls, Vierra-Braga identified a prominent MC cluster that was enriched in asthma, which was notable for a lack of CMA1 (encoding MC-specific chymase) expression (51), likely reflecting the chymase-negative intraepithelial MCs that previously shown to infiltrate the tissue in T2 high asthma based on immunohistology (52). MCs were the only identified cell type to express entire biosynthetic pathway for prostaglandin (PG)D2 pathway in asthma (51), similar to our findings in CRSwNP tissue MCs. Given the ability of PGD2 to selectively recruit and activate eosinophils, basophils, Th2 cells, and group 2 innate lymphoid cells, these findings suggest another important pathway for respiratory MCs in coordinating T2 inflammation. An analysis of asthma GWAS-associated transcripts identified MCs as the dominant cell type expressing IL1RL1 and GATA2 within the lung, as well as a surprising site of IKZF3 expression (encoding the transcription factor IKAROS Family Zinc Finger 3), with expression levels comparable to that observed in lymphocytes. Sauler and colleagues noted a significant increase in MC concentration within the lung tissues of subjects with COPD relative to healthy control tissue using RNAseq (53), potentially reflecting the increase in MC density within smooth muscle of small airways and alveolar wall of patients with centrilobular emphysema reported previously based on histology (54). While Li et al. did not identify MC expansion, in a separate, smaller-scale scRNA-seq based study of COPD, they found that the MC cluster was one of only four cell populations (together with proliferating macrophages, monocytes, and FBPB4+ macrophages) that exhibited significant enrichment for a COPD gene signature generated using a dataset from the Lung Tissue Research Consortium (55). Together, these findings support a major role for MCs in the pathobiology of multiple lung diseases, and again indicate that their transcriptomes (and corresponding effector programs) are highly disease- and tissue-specific.
MCs have also been identified in several scRNA-seq-based studies of lung tissue during COVID-19 infection, which together have suggested the possibility for MCs to actively participate in disease progression. Delorey et al. generated a COVID-19 atlas using tissue from the lung, kidney, liver and heart of patients post-mortem. Within lung tissue, they noted a significant correlation between MC concentration and viral burden, the strongest such correlation observed among immune cells (56). They further noted the presence of SARS-CoV-2 RNA within several cells in the MC cluster, despite the absence of expression of the hypothesized entry receptor ACE2. Filbin and colleagues collected plasma from COVID-19 patients for proteomics analysis on the day of hospital admission and three days afterwards (57). Using a previously generated scRNAseq dataset of brocheoalveolar lavage fluid from COVID-19 patients (58), they characterized potential communication axes between lung cells that they compared with differentially detected proteins at each time point. They observed a dramatic increase in mediators potentially indicative of MC interaction with CD4+ T cells, CD8+ T cells, and epithelial progenitors, along with an increase in tryptase in samples from patients with severe disease. Whether these findings indicate a role for MCs in the protective antiviral immune response, in the virally-induced lung injury, or in both processes remains to be determined.
Characterizing MCs in skin disease
MCs are strikingly abundant in scRNA-seq-based studies of human skin, appearing across a range of human disease states (59), although they are notably absent from the epidermis and can only be detected when full-thickness biopsies are examined (60). In psoriasis, histologic and flow cytometric studies have identified MCs as a source of interferon (IFN)-γ, IL-22, and IL-17 (61, 62), while histology-based studies have come to opposite conclusions regarding MC concentration in psoriatic lesions (63, 64). In an scRNA-seq based comparative study of 31 donors across a range of disease states, Liu and colleagues identified increased AREG expression (encoding amphiregulin) in MCs in psoriasis compared with healthy control skin (65). They further identified a cluster of proliferating MCs, but did not observe any concentration differences in either cluster across disease states. In a separate study of psoriasis, Gao and colleagues confirmed the restriction of MCs to the dermis and identified two clusters of MCs, designated MastC1 and MastC2 (66). They observed that MastC1 decreased in psoriasis while MastC2 increased but did not further characterize the two populations, and thus it is unclear whether these represent discrete clusters or differences in activation state.
Characterizing MCs in cancer
MC expansion within tumors, first described by Paul Ehrlich in the 19th century, has been long appreciated (67). However, defining the roles of MCs in cancer has proven challenging. More recently, MC clusters have been identified across a range of tumor-focused scRNA-seq studies, including breast cancer (68), prostate cancer (69), colorectal cancer (70), non-small cell lung cancer (71), and lung adenocarcimona (72). MCs were found to be specifically enriched in adenocarcinoma in the latter study, where they were hypothesized to promote tumor angiogenesis. This concept was further supported by a meta-analysis published by Cheng and colleagues, who combined previously published and newly generated scRNA-seq datasets encompassing 15 cancer types across 210 patients to examine similarities and distinctions between myeloid lineage cells (73). Through this approach, they found that MC infiltration was beneficial in some tumor types, but detrimental in others. Specifically, they found that MCs increased patient survival in tumors where they showed upregulation of TNFA (encoding the proinflammatory cytokine tumor necrosis factor (TNF)-α) relative to MCs in the surrounding tissue, but decreased survival when they instead upregulated VEGF, encoding the angiogenic cytokine vascular endothelial growth factor. It will be important for future studies to determine if this distinction is associated with the infiltration of different MC phenotypes, or if they are instead responding to different activating stimuli within the tumor microenvironment.
Re-defining MC heterogeneity through scRNA-seq analysis
Human MCs are classically categorized as either co-expressing tryptase and chymase (MCTC), localizing within the skin, peripheral connective tissues, and the mucosal subepithelium, or expressing tryptase in the absence of chymase (MCT), found predominantly within mucosal epithelium (74). To better understand the distinction between these MC subsets, we recently conducted a focused analysis of MC heterogeneity in CRSwNP by flow cytometrically sorting MCs for scRNA-seq analysis, examining 7,355 MCs derived from the nasal polyps of 6 donors (38). We identified polyp MC clusters reflecting MCT and MCTC expressing respective discrete gene expression profiles, differing by 282 transcripts. Immunohistochemical studies had previously suggested that these MC subsets differ in their profiles of expressed cytokines (75). Consistent with these differences, we found that the MCTC cluster was specifically enriched for chemokines (CCL2, CCL3, CCL4) and growth factors (CSF1, CSF2) potentially important for coordinating monocyte responses, and also strongly expressed IL13. MCT, in contrast, were enriched for TRAIL, encoded by TNFSF10, and FGL2, implying a potential regulatory role within the epithelium. MCT were additionally enriched for IL17RB, encoding the IL-25 receptor, likely indicating sensitivity to this epithelial-derived cytokine. Ex vivo studies suggest that it is at least partially regulated by IL-4 (which notably increased IL17RB expression), suggesting that it represents a distinct T2 inflammation-driven MC phenotype. Re-clustering MCs in datasets from several previously generated scRNA-seq studies of pulmonary fibrosis (76–78) and asthma (51) identified a similar IL17RB-expressing MCT cluster. Similar populations were recently confirmed by Bangert et al, who further noted enhanced expression of IL17RB in MCT from AERD polyps (49), potentially suggesting enhanced IL-4 signaling in AERD polyps.
In contrast to the dichotomous MCT and MCTC populations identified by immunohistology of human tissues, we found unexpectedly that within the nasal polyp MCT and MCTC appeared to be two ends of a transcriptional gradient rather than discrete populations, suggesting that MC differentiation within this tissue is the result of polarization in response to tissue microenvironmental signals. Supporting this hypothesis, we identified a transcriptionally intermediate population of transitional MCs that expressed a core set of MC genes, including IL1RL1 and HDC, but lacked enrichment for genes associated with polarized MCT and MCTC. We additionally identified a cluster of MCs expressing proliferation-associated genes, suggesting a role for in-situ proliferation in MC expansion. We were able to further use the transcriptomes associated with MCT, MCTC, and transitional MCs to develop cell surface marker panels capable of distinguishing MCT (CD38 high CD117 low), MCTC (CD38low CD117 high), and transitional MCs (CD38 high CD117high) via flow cytometry, and confirmed that MC proliferation was predominantly associated with the transitional MC pool. Notably, MCTC within the nasal polyp had little transcriptional similarity to MCTC from human skin, notably lacking expression of both MRGPRX2 and C5AR1, markers previously linked to MCTC. These observations indicate that the classical definitions of MC subsets based on protease markers vastly underestimate their true functional diversity, and that tissue- and context-specific factors further modify the functions underlying the immunohistochemically identified subsets. This led us to propose re-classifying these subsets as airway inflammatory MCT and MCTC (iMCT, iMCTC) (Fig. 2).
Fig. 2.
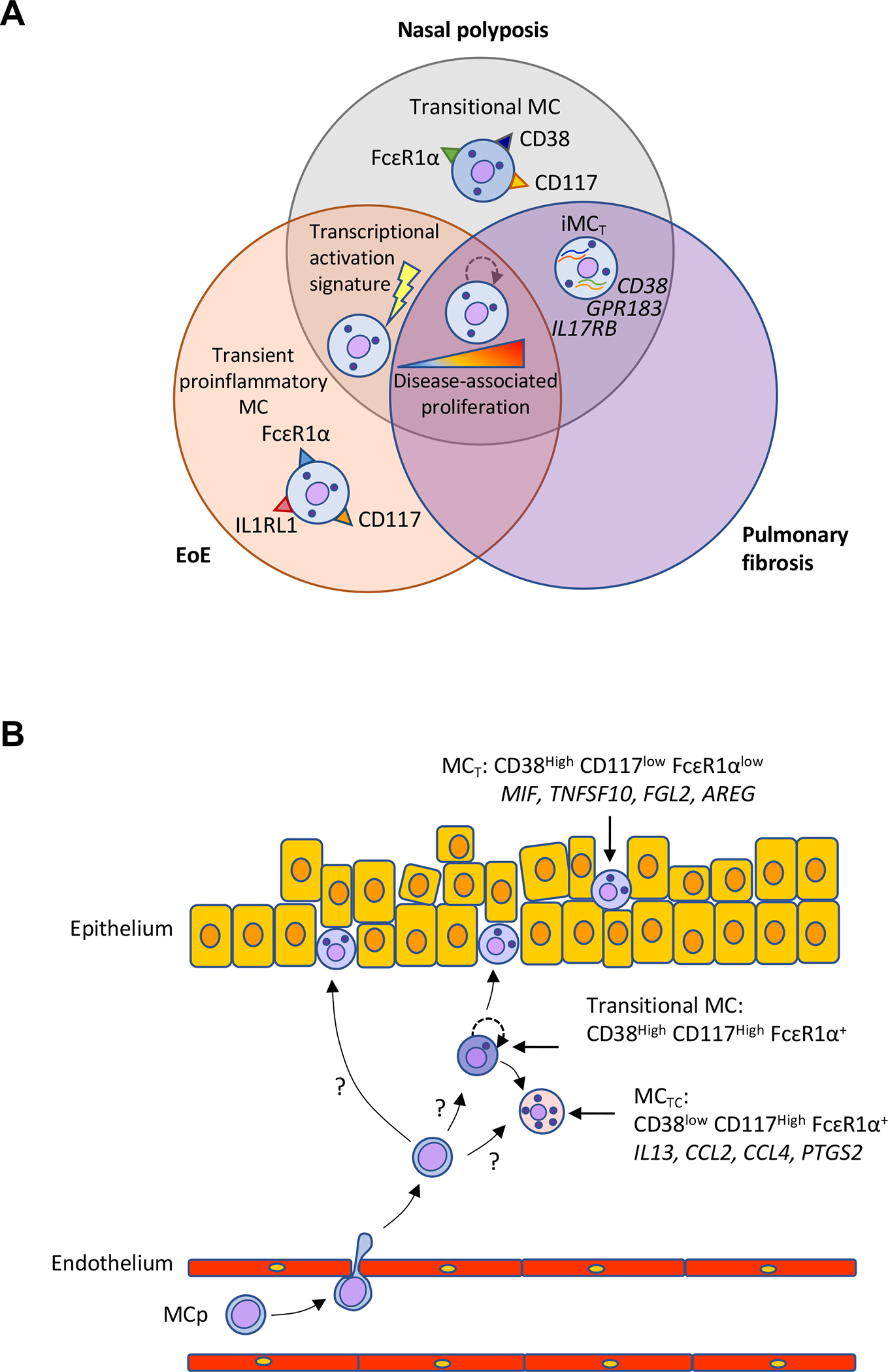
MCs exhibit specific features across tissue and diseases. (A) Characterization of scRNA-seq defined features identified in MCs across nasal polyposis, EoE, and pulmonary fibrosis. MC expansion is a common feature across all three diseases and is associated with disease severity. In both nasal polyposis and EoE, a MC activation-associated gene signature is enriched in MCs, while in nasal polyposis and pulmonary fibrosis, intraepithelial MCs expressing distinct cassette of genes including CD38, IL17RB, and GPR183 appear during inflammation (iMCTs). (B) Schematic indicating a recently defined model for development of phenotypically heterogeneous MCs in nasal polyposis (38). Three phenotype of MCs, MCTC, MCT, and transitional MCs can be identified through differential expression of CD117, FcɛR1α, and CD38 on their surface. MCTC and MCTs may arise directly from MC progenitors recruited to the tissue or from transitional MCs with elevated proliferation potential that develop from MC progenitors and expand within tissue. Each phenotype has the potential to contribute to disease through expression of pro-inflammatory mediators.
While fewer tissue-level studies have focused on heterogeneity within MC transcriptional clusters, distinctions can be readily observed given sufficient cell numbers and reclustering. Within the colon mucosa, Smillie and colleagues identified two clusters of MCs separated by CD69 expression in a scRNA-seq based study of colitis, with the CD69+ cluster enriched in diseased tissue compared with healthy controls (79). While differences in protease expression between these clusters were not examined, the authors noted that the CD69− MC cluster was among the few tissue-resident immune cell populations found that localized to the epithelium. Thus, it is likely that this cluster is at least enriched for MCs associated with the intraepithelial MCT phenotype. MC clusters have also been identified by multiple labs using scRNA-seq to study EoE (80, 81). In one of these studies, Morgenstern and colleagues re-clustered the MC population to conduct a more in-depth characterization of MC heterogeneity across healthy control donors, and both EoE patients with active disease and those in remission. Through this approach, they identified a TPSAB1high AREGhigh cluster present across all conditions, a cluster expressing transcripts encoding MCTC-associated proteases present in both disease and remission, and a pro-inflammatory KIThigh IL1RL1high FCER1Ahigh population restricted to disease. They found that even the constitutive cluster present across active disease, remission, and control donors exhibited transcriptional differences associated with disease severity, and identified MCs as a prominent source of the therapeutic target IL13 in disease. Moreover, this study also identified a proliferating cluster of MCs that persists even in remission. It seems possible that MCs in EoE may be poised to drive disease recurrences when therapy is withdrawn.
Conclusion
In summary, single cell genomics has rapidly and dramatically increased our knowledge of MC developmental mechanisms, heterogeneity, and effector properties. The lineage relationship to eosinophils and basophils suggests evolutionary pressures to diversify effector cell populations for helminth expulsion and other functions. It is tempting to speculate that at least a fraction of the CD34+ cells in the bone marrow, blood, and sputum of human subjects with allergic disease, identified as eosinophil/basophil bipotent progenitors based on their expressions of CD34 and IL-5Rα, might also have the capacity to develop into tissue MCs in the appropriate tissue milieu (82–84). The capacity of MCs to proliferate in situ suggests that MC hyperplasia during inflammatory diseases likely occurs largely through in situ proliferation, a unique feature among granulocytes. Identifying the factors responsible for driving MC proliferation carries substantial pathobiologic implications. Finally, while all MCs share common transcriptional features, the wide diversity identified in studies of different tissues and in health versus disease will likely expand further as more and more tissues and diseases are examined. With the increasing prevalence of newer transcriptomics-based technologies, such as Cellular Indexing of Transcriptomes and Epitopes (CITE-seq), allowing for the simultaneous evaluation surface protein expression and transcript expressions, allowing for the improved identification of specific markers for each MC phenotype, and spatial transcriptomics, linking MC phenotypes to their microenvironmental context, our understanding of MC heterogeneity derived by their tissue milieu will drastically evolve. While harnessing this knowledge for prognostic and therapeutic purposes represents a major challenge for the MC field, a deeper understanding of how MCs participate in human disease will aid in both the development of strategies to specifically target MCs and defining situations where this would be therapeutically beneficial.
Funding
This work was supported by NIH grants U19 AI095219 and K22 AI146281, and by generous contributions from the Vinik family.
Abbreviations:
- MC
Mast cell
- MCp
MC progenitor
- EMP
Erythro-myeloid progenitor cell
- CTMC
Mouse connective tissue MC
- BM
Bone marrow
- HSC
Hematopoietic stem cell
- AGM
Aorta-gonad-mesonephros
- MPP
Multipotent progenitor
- CMP
Common myeloid progenitor
- CLP
Common lymphoid progenitor
- LMPP
lymphoid-primed multipotent progenitor
- GMPs
Granulocyte-monocyte progenitors
- MEP
Megakaryocyte-erythroid progenitors
- scRNA-seq
Single-cell RNA-sequencing
- CRS
Chronic rhinosinusitis
- CRSsNP
CRS without nasal polyps
- CRSwNP
CRS with nasal polyps
- EoE
Eosinophilic esophagitis
- AERD
Aspirin exacerbated respiratory disease
- T2
Type 2
- MCTC
Human MCs expressing tryptase and chymase
- MCT
Human MCs expressing tryptase
- iMCT
Inflammatory MCT
- iMCTC
Inflammatory MCTC
- PGD2
Prostaglandin D2
Footnotes
Disclosure Statement
The authors declare that they have no relevant conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cavalcante MC, de Andrade LR, Du Bocage Santos-Pinto C, Straus AH, Takahashi HK, Allodi S, et al. Colocalization of heparin and histamine in the intracellular granules of test cells from the invertebrate Styela plicata (Chordata-Tunicata). J Struct Biol. 2002;137(3):313–21. [DOI] [PubMed] [Google Scholar]
- 2.Voehringer D Protective and pathological roles of mast cells and basophils. Nat Rev Immunol. 2013;13(5):362–75. [DOI] [PubMed] [Google Scholar]
- 3.Gurish MF, Austen KF. Developmental origin and functional specialization of mast cell subsets. Immunity. 2012;37(1):25–33. [DOI] [PubMed] [Google Scholar]
- 4.Li Z, Liu S, Xu J, Zhang X, Han D, Liu J, et al. Adult Connective Tissue-Resident Mast Cells Originate from Late Erythro-Myeloid Progenitors. Immunity. 2018;49(4):640–53.e5. [DOI] [PubMed] [Google Scholar]
- 5.Gentek R, Ghigo C, Hoeffel G, Bulle MJ, Msallam R, Gautier G, et al. Hemogenic Endothelial Fate Mapping Reveals Dual Developmental Origin of Mast Cells. Immunity. 2018;48(6):1160–71.e5. [DOI] [PubMed] [Google Scholar]
- 6.Derakhshan T, Samuchiwal SK, Hallen N, Bankova LG, Boyce JA, Barrett NA, et al. Lineage-specific regulation of inducible and constitutive mast cells in allergic airway inflammation. Journal of Experimental Medicine. 2020;218(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kitamura Y, Shimada M, Hatanaka K, Miyano Y. Development of mast cells from grafted bone marrow cells in irradiated mice. Nature. 1977;268(5619):442–3. [DOI] [PubMed] [Google Scholar]
- 8.Dwyer DF, Barrett NA, Austen KF. Expression profiling of constitutive mast cells reveals a unique identity within the immune system. Nat Immunol. 2016;17(7):878–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arinobu Y, Iwasaki H, Gurish MF, Mizuno S-i, Shigematsu H, Ozawa H, et al. Developmental checkpoints of the basophil/mast cell lineages in adult murine hematopoiesis. Proceedings of the National Academy of Sciences. 2005;102(50):18105–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qi X, Hong J, Chaves L, Zhuang Y, Chen Y, Wang D, et al. Antagonistic Regulation by the Transcription Factors C/EBPα and MITF Specifies Basophil and Mast Cell Fates. Immunity. 2013;39(1):97–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iwasaki H, Mizuno S, Arinobu Y, Ozawa H, Mori Y, Shigematsu H, et al. The order of expression of transcription factors directs hierarchical specification of hematopoietic lineages. Genes Dev. 2006;20(21):3010–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hamey FK, Lau WWY, Kucinski I, Wang X, Diamanti E, Wilson NK, et al. Single-cell molecular profiling provides a high-resolution map of basophil and mast cell development. Allergy. 2021;76(6):1731–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Akashi K, Traver D, Miyamoto T, Weissman IL. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature. 2000;404(6774):193–7. [DOI] [PubMed] [Google Scholar]
- 14.Osawa M, Hanada K-i, Hamada H, Nakauchi H. Long-Term Lymphohematopoietic Reconstitution by a Single CD34-Low/Negative Hematopoietic Stem Cell. Science. 1996;273(5272):242–5. [DOI] [PubMed] [Google Scholar]
- 15.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414(6859):105–11. [DOI] [PubMed] [Google Scholar]
- 16.Adolfsson J, Månsson R, Buza-Vidas N, Hultquist A, Liuba K, Jensen CT, et al. Identification of Flt3+ Lympho-Myeloid Stem Cells Lacking Erythro-Megakaryocytic Potential: A Revised Road Map for Adult Blood Lineage Commitment. Cell. 2005;121(2):295–306. [DOI] [PubMed] [Google Scholar]
- 17.Suda T, Suda J, Ogawa M. Single-cell origin of mouse hemopoietic colonies expressing multiple lineages in variable combinations. Proceedings of the National Academy of Sciences. 1983;80(21):6689–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen C-C, Grimbaldeston MA, Tsai M, Weissman IL, Galli SJ. Identification of mast cell progenitors in adult mice. Proceedings of the National Academy of Sciences. 2005;102(32):11408–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martin DI, Zon LI, Mutter G, Orkin SH. Expression of an erythroid transcription factor in megakaryocytic and mast cell lineages. Nature. 1990;344(6265):444–7. [DOI] [PubMed] [Google Scholar]
- 20.Franco CB, Chen C-C, Drukker M, Weissman IL, Galli SJ. Distinguishing Mast Cell and Granulocyte Differentiation at the Single-Cell Level. Cell Stem Cell. 2010;6(4):361–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Drissen R, Buza-Vidas N, Woll P, Thongjuea S, Gambardella A, Giustacchini A, et al. Distinct myeloid progenitor–differentiation pathways identified through single-cell RNA sequencing. Nature Immunology. 2016;17(6):666–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perié L, Duffy KR, Kok L, de Boer RJ, Schumacher TN. The Branching Point in Erythro-Myeloid Differentiation. Cell. 2015;163(7):1655–62. [DOI] [PubMed] [Google Scholar]
- 23.Paul F, Arkin Y, Giladi A, Jaitin DA, Kenigsberg E, Keren-Shaul H, et al. Transcriptional Heterogeneity and Lineage Commitment in Myeloid Progenitors. Cell. 2015;163(7):1663–77. [DOI] [PubMed] [Google Scholar]
- 24.Nestorowa S, Hamey FK, Pijuan Sala B, Diamanti E, Shepherd M, Laurenti E, et al. A single-cell resolution map of mouse hematopoietic stem and progenitor cell differentiation. Blood. 2016;128(8):e20–e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tusi BK, Wolock SL, Weinreb C, Hwang Y, Hidalgo D, Zilionis R, et al. Population snapshots predict early haematopoietic and erythroid hierarchies. Nature. 2018;555(7694):54–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dahlin JS, Hamey FK, Pijuan-Sala B, Shepherd M, Lau WWY, Nestorowa S, et al. A single-cell hematopoietic landscape resolves 8 lineage trajectories and defects in Kit mutant mice. Blood. 2018;131(21):e1–e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wanet A, Bassal MA, Patel SB, Marchi F, Mariani SA, Ahmed N, et al. E-cadherin is regulated by GATA-2 and marks the early commitment of mouse hematopoietic progenitors to the basophil and mast cell fates. Science Immunology. 2021;6(56):eaba0178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Holt PG, Jones CA. The development of the immune system during pregnancy and early life. Allergy. 2000;55(8):688–97. [DOI] [PubMed] [Google Scholar]
- 29.Popescu D-M, Botting RA, Stephenson E, Green K, Webb S, Jardine L, et al. Decoding human fetal liver haematopoiesis. Nature. 2019;574(7778):365–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Doulatov S, Notta F, Eppert K, Nguyen LT, Ohashi PS, Dick JE. Revised map of the human progenitor hierarchy shows the origin of macrophages and dendritic cells in early lymphoid development. Nature Immunology. 2010;11(7):585–93. [DOI] [PubMed] [Google Scholar]
- 31.Goardon N, Marchi E, Atzberger A, Quek L, Schuh A, Soneji S, et al. Coexistence of LMPP-like and GMP-like leukemia stem cells in acute myeloid leukemia. Cancer Cell. 2011;19(1):138–52. [DOI] [PubMed] [Google Scholar]
- 32.Görgens A, Radtke S, Möllmann M, Cross M, Dürig J, Horn PA, et al. Revision of the human hematopoietic tree: granulocyte subtypes derive from distinct hematopoietic lineages. Cell Rep. 2013;3(5):1539–52. [DOI] [PubMed] [Google Scholar]
- 33.Velten L, Haas SF, Raffel S, Blaszkiewicz S, Islam S, Hennig BP, et al. Human haematopoietic stem cell lineage commitment is a continuous process. Nat Cell Biol. 2017;19(4):271–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zheng S, Papalexi E, Butler A, Stephenson W, Satija R. Molecular transitions in early progenitors during human cord blood hematopoiesis. Mol Syst Biol. 2018;14(3):e8041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Drissen R, Thongjuea S, Theilgaard-Mönch K, Nerlov C. Identification of two distinct pathways of human myelopoiesis. Sci Immunol. 2019;4(35). [DOI] [PubMed] [Google Scholar]
- 36.Dahlin JS, Malinovschi A, Öhrvik H, Sandelin M, Janson C, Alving K, et al. Lin− CD34hi CD117int/hi FcεRI+ cells in human blood constitute a rare population of mast cell progenitors. Blood. 2016;127(4):383–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu C, Boey D, Bril O, Grootens J, Vijayabaskar MS, Sorini C, et al. Single-cell transcriptomics reveals the identity and regulators of human mast cell progenitors. Blood Adv. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dwyer DF, Ordovas-Montanes J, Allon SJ, Buchheit KM, Vukovic M, Derakhshan T, et al. Human airway mast cells proliferate and acquire distinct inflammation-driven phenotypes during type 2 inflammation. Science Immunology. 2021;6(56):eabb7221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Andersson CK, Shikhagaie M, Mori M, Al-Garawi A, Reed JL, Humbles AA, et al. Distal respiratory tract viral infections in young children trigger a marked increase in alveolar mast cells. ERJ Open Research. 2018;4(4):00038–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bentley AM, Jacobson MR, Cumberworth V, Barkans JR, Moqbel R, Schwartz LB, et al. Immunohistology of the nasal mucosa in seasonal allergic rhinitis: Increases in activated eosinophils and epithelial mast cells. Journal of Allergy and Clinical Immunology. 1992;89(4):877–83. [DOI] [PubMed] [Google Scholar]
- 41.Abonia JP, Blanchard C, Butz BB, Rainey HF, Collins MH, Stringer K, et al. Involvement of mast cells in eosinophilic esophagitis. J Allergy Clin Immunol. 2010;126(1):140–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takabayashi T, Kato A, Peters AT, Suh LA, Carter R, Norton J, et al. Glandular mast cells with distinct phenotype are highly elevated in chronic rhinosinusitis with nasal polyps. J Allergy Clin Immunol. 2012;130(2):410–20.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Balzar S, Fajt ML, Comhair SA, Erzurum SC, Bleecker E, Busse WW, et al. Mast cell phenotype, location, and activation in severe asthma. Data from the Severe Asthma Research Program. Am J Respir Crit Care Med. 2011;183(3):299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mukai K, Tsai M, Saito H, Galli SJ. Mast cells as sources of cytokines, chemokines, and growth factors. Immunol Rev. 2018;282(1):121–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Woodruff PG, Boushey HA, Dolganov GM, Barker CS, Yang YH, Donnelly S, et al. Genome-wide profiling identifies epithelial cell genes associated with asthma and with treatment response to corticosteroids. Proc Natl Acad Sci U S A. 2007;104(40):15858–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cildir G, Yip KH, Pant H, Tergaonkar V, Lopez AF, Tumes DJ. Understanding mast cell heterogeneity at single cell resolution. Trends Immunol. 2021;42(6):523–35. [DOI] [PubMed] [Google Scholar]
- 47.Ordovas-Montanes J, Dwyer DF, Nyquist SK, Buchheit KM, Vukovic M, Deb C, et al. Allergic inflammatory memory in human respiratory epithelial progenitor cells. Nature. 2018;560(7720):649–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stevens WW, Staudacher AG, Hulse KE, Carter RG, Winter DR, Abdala-Valencia H, et al. Activation of the 15-lipoxygenase pathway in aspirin-exacerbated respiratory disease. J Allergy Clin Immunol. 2021;147(2):600–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bangert C, Villazala-Merino S, Fahrenberger M, Krausgruber T, Bauer WM, Stanek V, et al. Comprehensive Analysis of Nasal Polyps Reveals a More Pronounced Type 2 Transcriptomic Profile of Epithelial Cells and Mast Cells in Aspirin-Exacerbated Respiratory Disease. Front Immunol. 2022;13:850494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Travaglini KJ, Nabhan AN, Penland L, Sinha R, Gillich A, Sit RV, et al. A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature. 2020;587(7835):619–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vieira Braga FA, Kar G, Berg M, Carpaij OA, Polanski K, Simon LM, et al. A cellular census of human lungs identifies novel cell states in health and in asthma. Nat Med. 2019;25(7):1153–63. [DOI] [PubMed] [Google Scholar]
- 52.Dougherty RH, Sidhu SS, Raman K, Solon M, Solberg OD, Caughey GH, et al. Accumulation of intraepithelial mast cells with a unique protease phenotype in T(H)2-high asthma. The Journal of allergy and clinical immunology. 2010;125(5):1046–53.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sauler M, McDonough JE, Adams TS, Kothapalli N, Barnthaler T, Werder RB, et al. Characterization of the COPD alveolar niche using single-cell RNA sequencing. Nat Commun. 2022;13(1):494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ballarin A, Bazzan E, Zenteno RH, Turato G, Baraldo S, Zanovello D, et al. Mast cell infiltration discriminates between histopathological phenotypes of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2012;186(3):233–9. [DOI] [PubMed] [Google Scholar]
- 55.Li X, Noell G, Tabib T, Gregory AD, Trejo Bittar HE, Vats R, et al. Single cell RNA sequencing identifies IGFBP5 and QKI as ciliated epithelial cell genes associated with severe COPD. Respir Res. 2021;22(1):100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Delorey TM, Ziegler CGK, Heimberg G, Normand R, Yang Y, Segerstolpe A, et al. COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets. Nature. 2021;595(7865):107–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Filbin MR, Mehta A, Schneider AM, Kays KR, Guess JR, Gentili M, et al. Longitudinal proteomic analysis of severe COVID-19 reveals survival-associated signatures, tissue-specific cell death, and cell-cell interactions. Cell Rep Med. 2021;2(5):100287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bost P, Giladi A, Liu Y, Bendjelal Y, Xu G, David E, et al. Host-Viral Infection Maps Reveal Signatures of Severe COVID-19 Patients. Cell. 2020;181(7):1475–88 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hughes TK, Wadsworth MH 2nd, Gierahn TM, Do T, Weiss D, Andrade PR, et al. Second-Strand Synthesis-Based Massively Parallel scRNA-Seq Reveals Cellular States and Molecular Features of Human Inflammatory Skin Pathologies. Immunity. 2020;53(4):878–94 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rojahn TB, Vorstandlechner V, Krausgruber T, Bauer WM, Alkon N, Bangert C, et al. Single-cell transcriptomics combined with interstitial fluid proteomics defines cell type-specific immune regulation in atopic dermatitis. J Allergy Clin Immunol. 2020;146(5):1056–69. [DOI] [PubMed] [Google Scholar]
- 61.Ackermann L, Harvima IT, Pelkonen J, Ritamäki-Salo V, Naukkarinen A, Harvima RJ, et al. Mast cells in psoriatic skin are strongly positive for interferon-gamma. Br J Dermatol. 1999;140(4):624–33. [DOI] [PubMed] [Google Scholar]
- 62.Mashiko S, Bouguermouh S, Rubio M, Baba N, Bissonnette R, Sarfati M. Human mast cells are major IL-22 producers in patients with psoriasis and atopic dermatitis. J Allergy Clin Immunol. 2015;136(2):351–9.e1. [DOI] [PubMed] [Google Scholar]
- 63.Jiang WY, Chattedee AD, Raychaudhuri SP, Raychaudhuri SK, Farber EM. Mast cell density and IL-8 expression in nonlesional and lesional psoriatic skin. International Journal of Dermatology. 2001;40(11):699–703. [DOI] [PubMed] [Google Scholar]
- 64.Zhang Y, Shi Y, Lin J, Li X, Yang B, Zhou J. Immune Cell Infiltration Analysis Demonstrates Excessive Mast Cell Activation in Psoriasis. Front Immunol. 2021;12:773280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu Y, Wang H, Taylor M, Cook C, Martinez-Berdeja A, North JP, et al. Classification of human chronic inflammatory skin disease based on single-cell immune profiling. Sci Immunol. 2022;7(70):eabl9165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gao Y, Yao X, Zhai Y, Li L, Li H, Sun X, et al. Single cell transcriptional zonation of human psoriasis skin identifies an alternative immunoregulatory axis conducted by skin resident cells. Cell Death Dis. 2021;12(5):450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Crivellato E, Beltrami C, Mallardi F, Ribatti D. Paul Ehrlich’s doctoral thesis: a milestone in the study of mast cells. Br J Haematol. 2003;123(1):19–21. [DOI] [PubMed] [Google Scholar]
- 68.Azizi E, Carr AJ, Plitas G, Cornish AE, Konopacki C, Prabhakaran S, et al. Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell. 2018;174(5):1293–308 e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ma X, Guo J, Liu K, Chen L, Liu D, Dong S, et al. Identification of a distinct luminal subgroup diagnosing and stratifying early stage prostate cancer by tissue-based single-cell RNA sequencing. Mol Cancer. 2020;19(1):147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pelka K, Hofree M, Chen JH, Sarkizova S, Pirl JD, Jorgji V, et al. Spatially organized multicellular immune hubs in human colorectal cancer. Cell. 2021;184(18):4734–52 e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wu F, Fan J, He Y, Xiong A, Yu J, Li Y, et al. Single-cell profiling of tumor heterogeneity and the microenvironment in advanced non-small cell lung cancer. Nat Commun. 2021;12(1):2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lu T, Yang X, Shi Y, Zhao M, Bi G, Liang J, et al. Single-cell transcriptome atlas of lung adenocarcinoma featured with ground glass nodules. Cell Discov. 2020;6:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cheng S, Li Z, Gao R, Xing B, Gao Y, Yang Y, et al. A pan-cancer single-cell transcriptional atlas of tumor infiltrating myeloid cells. Cell. 2021;184(3):792–809 e23. [DOI] [PubMed] [Google Scholar]
- 74.Irani AA, Schechter NM, Craig SS, DeBlois G, Schwartz LB. Two types of human mast cells that have distinct neutral protease compositions. Proceedings Of The National Academy Of Sciences Of The United States Of America. 1986;83(12):4464–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bradding P, Okayama Y, Howarth PH, Church MK, Holgate ST. Heterogeneity of human mast cells based on cytokine content. Journal Of Immunology (Baltimore, Md: 1950). 1995;155(1):297–307. [PubMed] [Google Scholar]
- 76.Adams TS, Schupp JC, Poli S, Ayaub EA, Neumark N, Ahangari F, et al. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci Adv. 2020;6(28):eaba1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Habermann AC, Gutierrez AJ, Bui LT, Yahn SL, Winters NI, Calvi CL, et al. Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis. Sci Adv. 2020;6(28):eaba1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Morse C, Tabib T, Sembrat J, Buschur KL, Bittar HT, Valenzi E, et al. Proliferating SPP1/MERTK-expressing macrophages in idiopathic pulmonary fibrosis. Eur Respir J. 2019;54(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Smillie CS, Biton M, Ordovas-Montanes J, Sullivan KM, Burgin G, Graham DB, et al. Intra- and Inter-cellular Rewiring of the Human Colon during Ulcerative Colitis. Cell. 2019;178(3):714–30.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Morgan DM, Ruiter B, Smith NP, Tu AA, Monian B, Stone BE, et al. Clonally expanded, GPR15-expressing pathogenic effector TH2 cells are associated with eosinophilic esophagitis. Sci Immunol. 2021;6(62). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ben-Baruch Morgenstern N, Ballaban AY, Wen T, Shoda T, Caldwell JM, Kliewer K, et al. Single-cell RNA sequencing of mast cells in eosinophilic esophagitis reveals heterogeneity, local proliferation, and activation that persists in remission. J Allergy Clin Immunol. 2022;149(6):2062–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wood LJ, Inman MD, Watson RM, Foley R, Denburg JA, O’Byrne PM. Changes in bone marrow inflammatory cell progenitors after inhaled allergen in asthmatic subjects. Am J Respir Crit Care Med. 1998;157(1):99–105. [DOI] [PubMed] [Google Scholar]
- 83.Sehmi R, Wood LJ, Watson R, Foley R, Hamid Q, O’Byrne PM, et al. Allergen-induced increases in IL-5 receptor alpha-subunit expression on bone marrow-derived CD34+ cells from asthmatic subjects. A novel marker of progenitor cell commitment towards eosinophilic differentiation. J Clin Invest. 1997;100(10):2466–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dorman SC, Efthimiadis A, Babirad I, Watson RM, Denburg JA, Hargreave FE, et al. Sputum CD34+IL-5Ralpha+ cells increase after allergen: evidence for in situ eosinophilopoiesis. Am J Respir Crit Care Med. 2004;169(5):573–7. [DOI] [PubMed] [Google Scholar]