

ABSTRACT
Background
Hypermanganesemia with dystonia 1 and 2 (HMNDYT1 and 2) are rare, inherited disorders of manganese transport.
Objectives
We aimed to describe clinical, laboratory features, and outcomes among children with HMNDYT.
Methods
We conducted a retrospective multicenter study involving tertiary centers across India. We enrolled children between 1 month to 18 years of age with genetically confirmed/clinically probable HMNDYT. Clinical, laboratory profile, genetic testing, treatment details, and outcomes scored by treating physicians on a Likert scale were recorded.
Results
We enrolled 27 children (19 girls). Fourteen harbored SLC30A10 mutations; nine had SLC39A14 mutations. The SLC39A14 cohort had lower median age at onset (1.3 [interquartile range (IQR), 0.7–5.5] years) versus SLC30A10 cohort (2.0 [IQR, 1.5–5.1] years). The most frequent neurological features were dystonia (100%; n = 27), gait abnormality (77.7%; n = 21), falls (66.7%; n = 18), and parkinsonism (59.3%; n = 16). Median serum manganese (Mn) levels among SLC39A14 (44.9 [IQR, 27.3–147.7] mcg/L) cohort were higher than SLC30A10 (29.4 [17.1–42.0] mcg/L); median hemoglobin was higher in SLC30A10 (16.3 [IQR, 15.2–17.5] g/dL) versus SLC39A14 cohort (12.5 [8.8–13.2] g/dL). Hepatic involvement and polycythaemia were observed exclusively in SLC30A10 variants. A total of 26/27 children underwent chelation with disodium calcium edetate. Nine demonstrated some improvement, three stabilized, two had marked improvement, and one had normalization. Children with SLC39A14 mutations had poorer response. Two children died and nine were lost to follow‐up.
Conclusions
We found female predominance. Children with SLC39A14 mutations presented at younger age and responded less favorably to chelation compared to SLC30A10 mutations. There is emerging need to better define management strategies, especially in low resource settings.
Keywords: manganese, dystonia, parkinsonism, cirrhosis
Hypermanganesemia with dystonia (HMNDYT) is a recently described neurogenetic disorder caused by abnormalities in intracellular manganese (Mn) transport. 1 , 2 , 3 , 4 Pathogenic variants in two Mn transporter genes have been reported. HMNDYT1 resulting from pathogenic variants in SLC30A10 gene manifests typically with dystonia‐parkinsonism, hepatic involvement, and polycythemia with depleted iron stores. Patients affected with SLC39A14 mutations (HMNDYT2) develop rapidly progressive dystonia‐parkinsonism with onset during early childhood, but lack hepatic involvement and polycythemia. In both conditions, pathognomonic magnetic resonance imaging (MRI) features include T1‐weighted hyperintensities in basal ganglia, midbrain, dorsal pons, and medulla with characteristic sparing of the ventral pons. 2 , 3 , 5 , 6 , 7 , 8 Forty‐five cases with SLC30A10 4 , 9 , 10 , 11 , 12 and 18 with SLC39A14 mutations have been described. 4 , 13 , 14
Treatment comprises chelation with disodium calcium edetate (Na2CaEDTA) and iron supplementation. However, Na2CaEDTA is not widely available, incurs recurrent expenditure, monthly hospitalization and intravenous access, and therefore, presents several barriers in resource‐constrained settings. Although treatment has been reported to be of benefit in case reports and case series, there is paucity of data on long term treatment outcomes.
In this multicentric retrospective study, we describe clinical, genetic, and radiological features and outcomes among Indian children with IH‐Mn.
Methods
This was a retrospective, multicenter study across tertiary care hospitals in India with pediatric neurology expertise.
Participants
Children age 1 month to 18 years with genetically proven HMNDYT1/2 (ie, with mutations in SLC30A10 or SLC39A14 genes) diagnosed before March 1, 2019 were enrolled. We also included patients in who genetic analysis was not available, but the following features were present: extrapyramidal symptoms in the form of dystonia and/or parkinsonism; MRI brain showing T1 hyperintensities in basal ganglia; elevated blood/urine Mn levels; and absence of risk factors for acquired manganism like prolonged parenteral nutrition or environmental exposure. Children who had any other concurrent neurometabolic/neurodegenerative disorder or acquired neurological injury such as traumatic brain injury or meningoencephalitis were excluded.
Procedure
Details of clinical presentation, investigations, treatment regimen, and response were collected via a predesigned proforma. Clinical details included age at onset, presence of dystonia/parkinsonism, other movement disorders, cognitive impairment, seizures, vision, or hearing impairment.
MRI findings were reviewed by a neuroradiologist (K.M.). Because Mn is a paramagnetic element, magnetic susceptibility was expected on T1 and fluid attenuated inversion recovery (FLAIR) weighted sequences. 15 , 16 T1 shortening was graded on a qualitative scale as mild = 1+, moderate = 2+, strong = 3+.
Molecular genetic analysis was reviewed by two geneticists (U.S. and M.F.). A total of 23 patients had undergone next generation sequencing (NGS)/Sanger sequencing to identify underlying defects (additional data are listed in Table S1).
Treatment details included drugs used and duration of chelation, and supportive care for neurological and extra‐neurological features. Treating clinicians rated treatment response in neurological features on a Likert scale (no change, somewhat improved, marked improvement, normalization, mild worsening, moderate worsening, and severe worsening). Results of available follow‐up investigations such as liver function tests, hemogram, and serum/urinary Mn levels were noted.
Statistical Analysis
Data were recorded on an Excel spreadsheet and analyzed with SPSS version 22.0 (IBM group, Armonk, NY). Results were expressed as frequency (%), mean ± standard deviation or median (interquartile range).
Data Sharing
The data that support the findings of this study are available from the corresponding author on reasonable request.
Results
We enrolled 27 children with HMNDYT1/2 from 20 families (19 girls) (Tables 1 and 2). These included eight previously reported children. 5 , 9 , 17 , 18 Among these, 14 harbored SLC30A10 mutations and nine had SLC39A14 mutation (additional data are listed in Tables S1 and S2). Genetic confirmation could not be performed for four children, but all fulfilled clinical, radiological, and laboratory features of HMNDYT.
TABLE 1.
Detailed clinical and laboratory characteristics and treatment outcomes of individual patients with SLC30A10 mutations
| Subject no. | Subject | Sex | Age (y) | C | Onset age (y) | Dystonia | Other features | I | H | Hb (g/dL) | LFT | Mn (mcg/L) | Variant identified | Treatment outcomes |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Children with SLC30A10 mutations | ||||||||||||||
| 1 | F118 | F | 8 | + | 5 | + (LL only) | Spasticity, brisk reflexes, recurrent falls | − | − | 15.4 | A | 42.0 |
chr1:220101323G>A c.460C>T; p.Gln154* |
Initial improvement on Na2CaEDTA, but suboptimal. D‐penicillamine added leading to further improvement. LFT normalized on treatment. She had severe polycythemia despite chelation and required four sessions of plasma exchange. |
| 2 | F2‐C118 | F | 9 | + | 2 | + (b/l feet) | Central hypotonia, chorea | − | + | 15.6 | A | 23.9 |
chr1:220101291_220101291delG c.492delC; p.Gly165Alafs*27 |
Marked neurological improvement noted. LFT normalized. Hepatomegaly regressed. |
| 3 | F2‐C2 | F | 2.5 | + | 2.0 | + (bilateral feet) | Choreiform and athetoid movements of hands and feet | − | − | 17.5 | A | 29.2 |
chr1:220101291_220101291delG c.492delC; p.Gly165Alafs*27 |
Some neurological improvement. LFT normalized. |
| 4 | F2‐C318 | F | 7 | + | 2 | + (b/l toes) | − | − | − | 11.4 | N | 29.5 |
chr1:220101291_220101291delG c.492delC; p.Gly165Alafs*27 |
Some neurological improvement although could not ambulate independently. LFT normalized. |
| 5 | F2‐C4 | F | 2 | + | 1.5 | + (b/l feet and hands) | − | − | − | 15.6 | N | 34.0 |
chr1:220101291_220101291delG c.492delC; p.Gly165Alafs*27 |
Patient showed initial improvement on Na2CaEDTA, but suboptimal. D‐penicillamine was added leading to further improvement from baseline. LFT normalized on treatment. |
| 6 | F2‐C5 | F | 2.5 | + | 1.5 | + (b/l hands, feet) | Spasticity, brisk reflexes, tongue dyskinesia | − | − | 15 | N | 42.0 |
chr1:220101291_220101291delG c.492delC; p.Gly165Alafs*27 |
Initial improvement on Na2CaEDTA, but suboptimal. D‐penicillamine was added leading to further improvement. LFT normalized on treatment. |
| 7 | F218 | M | 4 | + | 3 | + (b/l LL) | Central hypotonia | − | − | 16.3 | A | 19.5 |
chr1:220101291_220101291delG c.492delC; p.Gly165Alafs*27 |
Died in a road traffic accident. |
| 8 | F39 | M | 4.5 | + | 2 | + (b/l feet) | Parkinsonism | − | + | 10.2 to >15.7 | A | 186.0 |
chr1:220101764_220101765insA; c.19_20insT (p.Lys7Ilefs*106 |
Initial improvement, then moderate worsening despite Na2CaEDTA. Became non‐ambulatory and developed progressive polycythemia. |
| 9 | F4 | F | 16 | − | 8 | + (b/l feet) | Spasticity, brisk reflexes | + | + | 17.5 | A | 16.3 | Chr1:220100449A>G; g.31541A>G; c.641‐2A > G [Splice site] | Some neurological improvement noted. Hepatomegaly persistent. |
| 10 | F5 | M | 10.5 | + | 1.5 | + (all limbs) | Parkinsonism | − | − | 17.0 | N | 10.0 |
chr1:220089243C>T; c.1006C>T; p.His336Tyr |
Lost to follow‐up. |
| 11 | F6 | M | 16 | + | 12 | + | Bradykinesis, hypomimia, facial dystonia | + | − | 16.6 | A | UrineMn++ |
chr1:220089150C>T; c.1099C>T; p.Arg367* |
Some improvement noted in gait, dystonia, cognitive functions. |
| 12 | F82 | F | 5.5 | + | 5.1 | + (b/l feet) | Parkinsonism | − | + | 21.4 | A | 130.0 | chr1:220089203 T>C; c.1046 T>C;p.Leu349Pro | Some neurological improvement noted. Only three cycles of chelation given at the time of enrolment. |
| 13 | F918 | F | 15 | + | 3 | + Gen. | Dysarthria, dysphagia | − | + | 19.0 | A | 9.8 |
chr1:220101230_220101288del58 c.496_553del58 p.Ala166Glnfs*7 |
She received trihexyphenidyl, levodopa/carbidopa and oral iron supplementation. Because of unavailability, chelation therapy could not be offered. She required blood‐letting twice. |
| Clinical exome sequencing negative | ||||||||||||||
| 14 | F7 | F | 2.5 | + | 1.4 | + (b/l LL) | Spasticity, brisk reflexes | + | − | 19.2 | A | Serum and urine Mn++ | No exonic pathogenic variations identified; UTR not covered | No improvement but no further deterioration noted. Partial exchange done for polycythemia. |
Abbreviations: A, abnormal; AFO, ankle foot orthoses; ALT, alanine aminotransferase; AST, aspartate aminotransferase; b/l, bilateral; Br, bilirubin; C, consanguinity; F, female; Gen., generalized; H, hepatomegaly; I, incoordination; LFT, liver function test; LL, lower limb; M, male; Mn, manganese; N, normal; N/A, not available; S, sex; UL, upper limb.
TABLE 2.
Detailed clinical and laboratory characteristics and treatment outcomes of individual patients with SLC39A14 mutations and those with genetics unknown
| Subject no. | Subject | S | Age (y) | C | Onset age (y) | Dystonia | Other features | I | H | Hb (g/dL) | LFT | Serum Mn (mcg/L) | Variant identified | Treatment outcomes |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Children with SLC39A14 mutations | ||||||||||||||
| 1 | F10 | M | 5 | + | 3 | + Gen. | Parkinsonism, hyperreflexia | − | − | 12.5 | N | 110.0 |
chr8:22265856A>G c.304A>G; p.Asn102Asp |
Lost to follow‐up. |
| 2 | F11 | F | 0.75 | + | 0.75 | + (trunk, b/l LL) | Intermittent opisthotonos | − | − | 11.7 | N | 260.9 |
chr8:22262399 T>A c.176 T>A; p.Leu59Gln |
No improvement, but no further deterioration noted. |
| 3 | F12 | M | 2 | − | 1.9 | + Gen. | − | − | − | 12.8 | N | 60.5 | Deletion of Exon4 to Exon9 | Some improvement noted neurologically. Developed anemia on treatment. |
| 4 | F1317 | F | 1 | − | 0.75 | + Gen. | Brisk reflexes | − | − | 12.5 | N | 200 (blood) |
chr8:22265934C>T c.382C>T; p.Arg128Trp |
Lost to follow‐up. |
| 5 | F14 | F | 12 | − | 11 | + Gen. | Brisk reflexes, parkinsonism | + | − | 11.7 | N | 28.5 |
chr8:22265874C>T c.322C>T; p.Arg108Trp |
Lost to follow‐up. |
| 6 | F14‐C1 | F | 9 | − | 8 | + b/l LL | Brisk reflexes | − | − | 11.4 | N | 29.3 |
chr8:22265874C>T c.322C>T; p.Arg108Trp |
Lost to follow‐up. |
| 7 | F15 | F | 2.75 | − | 0.33 | + LL>UL | Brisk reflexes, parkinsonism | + | + | 8.8 | N | 23.5 |
chr8:22275310G>C; c.1294G>C; p.Ala432Pro |
Lost to follow‐up. |
| 8 | F16 | F | 2.6 | + | 1.25 | + (b/l ankles) | Lower limb hyperreflexia | + | − | 14.5 | N | Not done |
chr8:22275329A>G; c.1313A>G; p.Tyr438Cys |
Some improvement noted: stands with support, unbuttons shirt, tells stories, dry by night. |
| 9 | F17 | F | 1.2 | + | 0.67 | + Gen. | Brisk reflexes | − | − | 8.8 | N | ↑ urine | chr8:22262404C>T c.181C>T; p.Gln61* | Some improvement noted. |
| Genetics not available | ||||||||||||||
| 1 | F18 | F | 6 | + | 2 | + Gen. | Quadruped gait | − | − | 10.1 | N | 20.8 | N/A | Lost to follow‐up. |
| 2 | F19 | M | 9.5 | + | 9 | + b/l UL | − | − | + | 12.8 | A | 38.7 | N/A | Died because of complications of liver disease. |
| 3 | F20 | F | 5.1 | + | 4 | + b/l feet | Parkinsonism, spasticity | − | − | 12.5 | A | 72.9 | N/A | Marked improvement: walks independently with AFO. |
| 4 | F21 | M | 2.75 | + | 1.5 | + (UL) | Central hypotonia, chorea‐athetosis | − | + | 9.8 | A | ↑ blood, urine | N/A | Lost to follow‐up. |
Abbreviations: AFO, ankle foot orthoses; ALT, alanine aminotransferase; AST, aspartate aminotransferase; b/l, bilateral; Br, bilirubin; C, consanguinity; F, female; Gen., generalized; H, hepatomegaly; I, incoordination; LFT, liver function test; LL, lower limb; M, male; Mn, manganese; N, normal; N/A, not available; S, sex; UL, upper limb.
Median age at enrolment was 4.3 years (IQR, 2.5–9.0 years). Median age at onset for SLC39A14 cohort was 1.3 years (IQR, 0.7–5.5 years), whereas for the SLC30A10 cohort was 2.0 years (IQR, 1.5–5.1 years). Eighteen children were born to third‐degree consanguineous and three to second‐degree consanguineous parentage. The remaining six children had non‐consanguineous parentage. Pedigree chart of the largest family affected is provided in Figure 1. Family history of dystonia was present in the largest family with HMNDYT1 (F2, F2‐C1, F2‐C2, F2‐C3, F2‐C4, and F2‐C5). None of the other children had family history of dystonia.
FIG. 1.
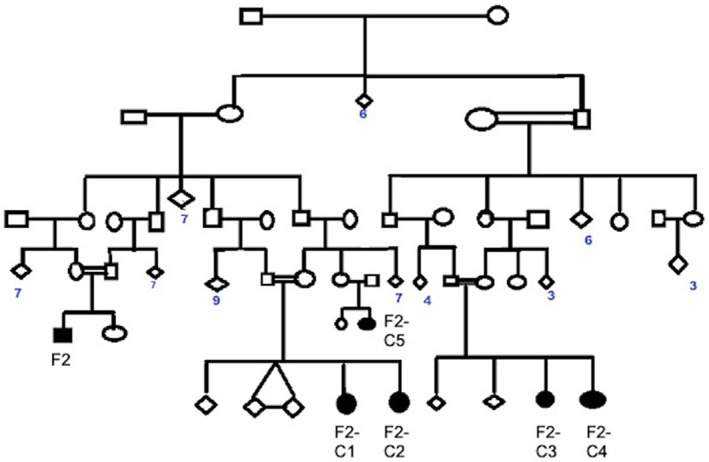
Pedigree chart of the largest family affected in our cohort.
Clinical Phenotype
The most frequent presenting complaint was gait abnormality (21/27; 77.7%) (Table 3). This varied from dystonic gait with toe‐walking to the typical “cock‐walk” gait. One patient developed a peculiar gait with partial walking on all fours, reminiscent of the gait in Uner Tan syndrome. Recurrent falls were common (18, 66.7%). Three children presented with abdominal complaints (jaundice/abdominal distention) preceding neurological complaints by 2 to 4 weeks. All three had SLC30A10 mutations. Two children were non‐ambulatory at presentation; both had SLC39A14 mutations. Two children with SLC30A10 mutations developed loss of ambulation during advanced stage (mean disease duration, 11 months); one while on chelation. Seizures were observed in two children (F16 and F20). In F16, who had HMNDYT2, onset of seizure was at 15 months of age, with sudden onset of up‐rolling of eyes and loss of consciousness. This child developed three more similar episodes over the next 1.5 years. Electroencephalography (EEG) was normal. Levetiracetam was used for control of seizures. In F20, who was genetically uncharacterized, two episodes of generalized seizures occurred at 1.5 years of age, 2 weeks apart. EEG was normal. The child was initiated on valproate, which was eventually tapered and stopped after a seizure‐free period of 2 years. Postural tremors involving both upper limbs, and chorea‐athetosis were noted in five children each. Features of incoordination were present in four: dysdiadochokinesia (2), gaze evoked nystagmus (1), and gait ataxia (1). Eight had abnormal abdominal examination with clinically appreciable hepatic and/or splenic enlargement. Twelve children (44.4%) had spasticity involving limbs.
TABLE 3.
Clinical features and laboratory evaluation of patients with HMNDYT
| Clinical features | Total (n = 27) | SLC30A10 mutations (n = 14) | SLC39A14 mutations (n = 9) |
|---|---|---|---|
| Median age (y) (IQR) | 4.3 (2.5–9.0) | 5.5 (2.7–9.8) | 2.3 (1.0–6.4) |
| Sex | |||
| Female (%) | 19 (70.4) | 10 (71.4) | 7 (77.8) |
| Median age at onset (y) (IQR) | 2.0 (1.5–5.0) | 2.0 (1.5–5.1) | 1.3 (0.7–5.5) |
| Prominent complaints | |||
| Gait abnormality/Falls | 21/18 | 12/14 | 4/− |
| Dystonia | 27 | 14 | 9 |
| Regression of milestones | 3 | 1 | 2 |
| Dysarthria | 3 | 2 | – |
| Abdominal a | 3 | 2 a | – |
| Seizures | 2 | – | 1 |
| Tremors | 5 | 2 | 1 |
| Non‐ambulatory at presentation | 2 | – | 2 |
| Consanguinity | |||
| Second degree/third degree | 3/18 | 1/11 | −/5 |
| Examination findings | |||
| Cognitive dysfunction | 3 | 1 | 1 |
| Motor system | |||
| Spasticity | 12 | 5 | 7 |
| Extrapyramidal system | |||
| Dystonia (generalized) | 27 (21) | 13 (12) | 10 (8) |
| Tremor | 5 | 2 | 1 |
| Parkinsonism | 16 | 11 | 4 |
| “Cock‐walk” gait | 8 | 6 | 2 |
| Chorea‐athetosis | 5 | 2 | 2 |
| Dysarthria | 8 | 6 | 2 |
| Incoordination | 4 | 2 | 2 |
| Abdominal examination | |||
| Hepatomegaly/Splenomegaly | 8 | 4 | 1 |
| Laboratory parameter | |||
| Hb (g/dL) | |||
| Mean | 12.8 | 16.1 | 11.7 |
| SD | 3.3 | 2.9 | 2.3 |
| Median | 13.8 | 16.3 | 12.5 |
| IQR | 11.4–16.4 | 15.2–17.5 | 8.8–13.2 |
| PCV | |||
| Mean | 42.1 | 48.5 | 36.2 |
| SD | 9.5 | 8.4 | 6.3 |
| Median | 40.8 | 49.5 | 37.5 |
| IQR | 35.5–49.9 | 46.1–55.6 | 29.5–41.7 |
| Liver function test (LFT) | |||
| Normal | 13 | 3 | 9 |
| Abnormal | 14 | 11 | – |
| Hyperbilirubinemia | 6 | 6 (all mild) | – |
| Elevated SGOT | 8 | 6 | – |
| Elevated SGPT | 5 | 6 | – |
| Elevated ALP | 5 | 5 | – |
| Hypoalbuminemia | 4 | 3 | – |
| Abdominal imaging b | |||
| Normal | 15 | 6 | 9 |
| Hepatomegaly with increased echotexture | 3 | 2 | – |
| Hepatomegaly with normal echotexture | 5 | 5 | – |
| Splenomegaly | 1 | – | – |
| Others c | 1 | ||
| Serum manganese level (0.3–1.8 mcg/L) | n = 21 | n = 12 | n = 6 |
| Mean | 58.1 | 47.7 | 85.4 |
| SD | 63.9 | 53.9 | 91.9 |
| Median | 32.5 | 29.4 | 44.9 |
| IQR | 22.2–66.7 | 17.1–42.0 | 27.3–147.7 |
| Thyroid function test (n = 18) | Normal | ||
In one child, the abdominal complaint (jaundice) accompanied the neurological complaints. In the second child, abdominal complaints preceded neurological complaints be 2 weeks and in the third by 4 weeks.
Ultrasonography abdomen: 22; MRI abdomen = 1; CT abdomen = 1.
Splitting of the pelvi‐calyceal system noted.
Abbreviations: IQR, interquartile range; PCV, packed cell volume; SD, standard deviation; SGOT, serum glutamic oxaloacetate transaminase; SGPT, serum glutamic pyruvic transaminase.
Laboratory Evaluation
Median blood Mn level among children with SLC39A14 variants was 44.9 mcg/L (IQR, 27.3–147.7 mcg/L) and among those with SLC30A10 variants was 29.4 (17.1–42.0 mcg/L) (Table 3). Among patients with SLC30A10 variants, 13 of 14 (92.3%) children had polycythemia. Of 14 who had abnormal liver function test (LFT), 10 belonged to the SLC30A10 cohort and the other four had not undergone genetic testing, although presence of liver dysfunction in three indicated likelihood of underlying SLC30A10 mutations. Thyroid function, tested in 18 children, was normal in all.
Genetic Abnormalities
Genetic analysis identified eight probands with SLC30A10 variants and eight probands with SLC39A14 variants. Importantly, no variant could be identified either in SLC30A10 nor SLC39A14 for one proband (F7) despite characteristic HMNDYT1 phenotype. Moreover, all genetic variants identified in either SLC30A10 or SLC39A14 genes existed in homozygous state. None of the patients carried compound heterozygous mutations. These variants were mostly not reported in gnomAD and 1KGP (1000 genome project) demonstrating their absence in the general population, except one variant in SLC30A10 reported in ExAC in heterozygous state.
Mutations in SLC30A10 included missense variants along with small indels in three cases (F2, F3, and F9) (additional data are listed in Tables S1 and S2). Of eight mutations, three were novel variants, a frameshift (p. Lys7Ilefs*106 in F3), a splice site change (c.641‐2A>G in F4), and a nonsense variation (p.Arg367* in F6) reported as likely pathogenic variations through ACMG guidelines (VARSOME; https://varsome.com). 19
Mutations observed in the SLC39A14 gene in our patient cohort were mostly missense variants with only one patient (F12) harboring a major deletion encompassing several exons (Ex4–Ex9) (additional data are listed in Tables S1 and S2). All variants in SLC39A14 were novel except one, which is a reported pathogenic variation (p.Arg128Trp). 17 Among the novel variants, a large deletion of Exon4‐Exon9 (F12) is reported to be pathogenic, whereas a nonsense change p.Gln61* (F17) and missense variants like p.Ala432Pro and p.Tyr438Cys (F15, F16), respectively, were likely pathogenic The remaining missense mutations, p.Asn102Asp, p.Leu59Gln, p.Arg108Trp were classified as variant of unknown significance (VUS) following the American College of Medical Genetics and Genomics (ACMG) guidelines (VARSOME; https://varsome.com). 19
However, location of these VUS in SLC39A14 gene was confined to important functional domains of the SLC39A14 protein. p.Arg108Trp (shared by F14 and F14‐C1 subjects), p.Asn102Asp (F10) fell in the extracellular N terminal domain, whereas p.Leu59Gln (F11) in the transmembrane domain 1 of SLC39A14, similar mutations in these domains are known to alter function. 5 The p.Arg108Trp variation is predicted benign (DEOGEN2, EIGEN, FATHMM‐MKL, MVP, MutationTaster, PrimateAI, and REVEL), and pathogenic (DANN, LIST‐S2, M‐CAP, MutationAssessor, and SIFT) within an evolutionary non‐conserved site, whereas p.Leu59Gln has transmembrane domain location and predicted majorly as pathogenic by prediction tools, BayesDel_addAF, DANN, EIGEN, FATHMM‐MKL, LIST‐S2, M‐CAP, MutationAssessor, MutationTaster, and SIFT. The functional consequence of nonsense mutations and indels revealed that the occurrence of a premature stop codon either results in formation of truncated protein or complete loss of functional protein because of nonsense mediated decay of the transcript. Missense variants in both SLC30A10 (F5, F8) and SLC39A14 (F10, F11, F13, F14, F15, and F16) were mostly present at transmembrane domains, involved in binding and trafficking of metal ions, therefore, likely hindering protein function.
Loss of functional genomic variants in either SLC30A10 or SLC39A14 did not result in higher Mn levels in all patients. Henceforth, a genotype–phenotype correlation could not be observed. However, Mn levels were found to be more elevated in patients carrying SLC39A14 variants than SLC30A10 carriers. In SLC30A10, only two cases showed substantial increase in Mn levels; p.Lys7*(F3), which is predicted to truncate almost the full length of the protein, and in case F8, a nonsynonymous p.Leu349Pro variant in regulatory cytoplasmic C‐terminal domain (CTD). This domain has a role in structure and stability of SLC30A10 protein.
Radiology
Marked T1‐shortening was demonstrated in all cases (additional data are listed in Table S3). Within the basal ganglia, globus pallidus was most strongly involved, followed by subthalamic nucleus, putamen, substantia nigra, and caudate nucleus. The thalamus was characteristically spared in majority. We also observed a generally increased T1‐weighted white matter signal also in periventricular and deep white matter of the cerebral hemispheres, as well as within the cerebellar peduncles in the majority of cases. We also noted involvement of the dentate nuclei. Parenchymal atrophy was not noted.
Six patients underwent post‐treatment MRI. We observed dramatic improvement in the extent of T1 shortening, with residual changes mostly in the globus pallidus, internal capsule and or subthalamic nucleus. In three cases, T2‐weighted sequence showed central gliosis within the globus pallidus, especially on post treatment follow‐up (Fig. 2).
FIG. 2.
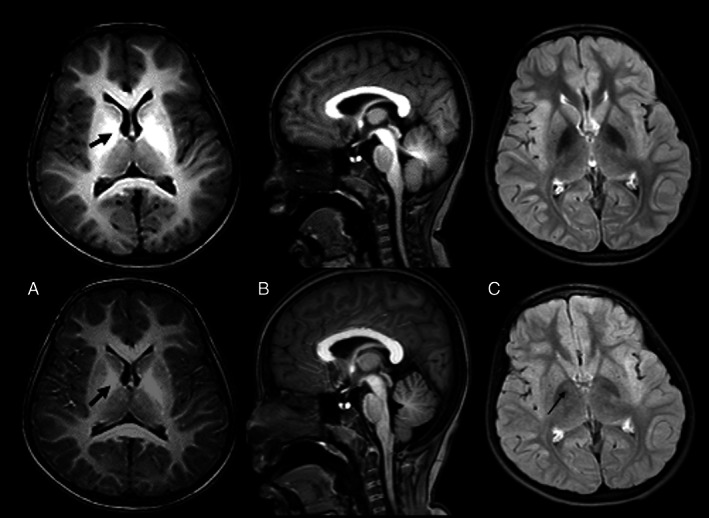
Case F20. Top panel (a = axial T1, B = sagittal T1, C = FLAIR) shows presentation scans. Bottom panel shows corresponding post treatment images. Note the reduction in swelling and T1 shortening globally within the white matter, particularly within the globus pallidi (arrows) and the tegmentum and superior cerebellar peduncles. The axial FLAIR images show concomitant reduction in the hypointensity within the globus pallidi, but with some increased central focal hyperintensity (C‐arrow), which would be consistent with mild central pallidal gliosis.
Among children with SLC30A10 mutations, 8/14 children demonstrated hepatomegaly on ultrasonography with or without increased liver echotexture. Interestingly, hepatomegaly with normal liver echotexture was also reported in one child with SLC39A14 mutations. All these children also had clinically appreciable liver enlargement. MRI abdomen, done in one patient with SLC30A10 mutations, showed increased T1 and low T2 signal in the liver.
Treatment Outcomes
Among the 27 children, 26 were treated with Na2CaEDTA (regimen: 20 mg/kg per dose, given twice daily, for 5 days. Solution prepared in 250 mL 0.9% sodium chloride, and administered over 1 hour). In addition, iron was supplemented orally in 22 and oral zinc in 16 children. D‐penicillamine was used in two children. Several drugs for symptomatic treatment including trihexyphenidyl, clonazepam, levodopa/carbidopa, baclofen, and botulinum toxin were also used.
In our series, 16 children continued to be on regular follow‐up, for a median duration of 2.0 (IQR, 0.7–3.0) years; nine were lost to follow‐up (Table 4). Two children died, one because of complications of chronic liver disease and one in a road traffic accident. Neurological outcomes on treatment were scored by treating physicians on a Likert scale. Nine of 16 children had some improvement and three had stabilization with no further deterioration (additional data are listed in Table S4 and Video 1). Only two children had marked improvement, and one had normalization. One child had moderate worsening after some initial improvement. Among nine children with some improvement, two had suboptimal improvement on chelation with Na2CaEDTA and were placed on additive d‐penicillamine, following which they showed some improvement. On comparing children with some to marked improvement (n = 12) versus no improvement to worsening (n = 4) (Table S4), it was observed that the latter group had later median age at presentation and shorter duration of chelation compared to children with improvement. Of four children with SLC39A14 variants with available follow‐up, one stabilized and three showed some improvement.
TABLE 4.
Treatment outcomes among children with HMNDYT
| Total | SLC30A10 mutations | SLC39A14 mutations | |
|---|---|---|---|
| Patients continuing to be on follow‐up | 16 | 11 | 4 |
| Lost to follow‐up | 9 | 2 | 5 |
| Died a | 2 | 1 | – |
| Duration of follow‐up available (yrs) | n = 16 | ||
| Median | 2.0 | ||
| IQR | 0.7–3.0 | ||
| Neurological course on treatment | |||
| Stabilized b | 3 | 2 | 1 |
| Some improvement b | 9 | 6 | 3 |
| Marked improvement | 2 | 1 | – |
| Normalization | 1 | 1 | – |
| Mild worsening | – | – | – |
| Moderate worsening c | 1 | 1 | – |
| Severe worsening | – | – | – |
Died in a road traffic accident = 1; died because of liver dysfunction = 1.
In 3 patients, because of suboptimal response with Na2CaEDTA, d‐penicillamine was added leading to some improvement from baseline in two and stabilization of the disease in one patient.
The patient showed initial improvement followed by moderate worsening.
VIDEO 1.
Pre‐chelation (segment A) (at age 4 years) and post‐chelation (segment B) (at age 9 years) videos of a child with SLC3010 mutation (F2‐C1), demonstrating abdominal distention (hepatomegaly) and dystonic gait pre‐chelation. The same child demonstrates regression of abdominal distention and improvement of lower limb dystonia after 4 years of chelation therapy.
Of 15 children with abnormal liver function tests, follow‐up reports were available for 10 children; among these, hepatic parameters normalized for seven and remained stable for three children without further progression. Among eight children with hepatomegaly, one showed regression in liver size with treatment. Hepatomegaly stabilized among the remaining six children. Three children continued to have worsening polycythemia despite chelation (F1, F3, and F8) of whom one child required partial exchange for hyperviscosity syndrome (F1) (Table 1). One child (F9) required phlebotomy twice for hyperviscosity, but she had been off chelation because of non‐availability of Na2CaEDTA at the time. None of these children had clinical features of hyperviscosity and a hematocrit cut‐off of 70% and above prompted phlebotomy.
Follow‐up urinary and serum Mn levels could be measured in only four children who demonstrated increase in urinary Mn excretion during chelation therapy: F1, 99.3 to 699 mcg/L, serum 42 to 17.8 mcg/L; F2, 8.33 to 253 mcg/L, serum 19.5 to 11.2 mcg/L; F2‐C1, 3.43 to 81.5 mcg/L, serum 23.9 to 16.2 mcg/L; F2‐C3, 5.66 to 285 mcg/L, serum 29.5 to 18.2 mcg/L. Repeat serum manganese levels persisted to be high despite 12 to 18 months of chelation therapy.
Discussion
To the best of our knowledge, this is the largest case series of children with HMNDYT. Patients with SLC39A14 mutations were younger at symptom onset (50% had onset in infancy) and at presentation, compared to patients with SLC30A10 mutations. This is consistent with original descriptions of SLC39A14 mutations‐related HMNDYT among 10 children with mean age at onset being 15.8 (7–36) months compared to SLC30A10 with mean age at onset of 7.1 (1–57) years. 6 We also noted a strong female predominance, with female:male ratio being 2.25:1. Although female preponderance has been noted in previous reports, we found a higher female proportion than previously observed. 6
Neurological presentation was dominated by extrapyramidal features including dystonia, parkinsonism, chorea‐athetosis, and tremors, although we also found mild cerebellar features in some children. The most common presenting complaint was gait abnormality, which ranged from dystonic gait with toe‐walking to the typical “cock‐walk” gait.
We evaluated phenotypic association of each of the reported and identified novel variants. Overall, locations of each of the missense variants in both SLC30A10 (F5, F8) and SLC39A14 (F10, F11, F13, F14, F15, and F16) were confined to functional domains like transmembrane domains, involved in binding and trafficking of metal ions, and hence, may hinder protein function and lead to pathogenic effects. Missense protein truncating variants were identified as well. There was lack of overall understanding of genotype–phenotype correlation for each of the variants; however, some of the observations are described for SLC30A10 and SLC39A14 overall.
Children with SLC39A14 variants seemed to have more severe neurological disease. Most had generalized dystonia, with two being non‐ambulatory and one requiring support to walk at presentation. In contrast, most children with SLC30A10 mutations had lower limb (foot/toe) dystonia, but were independently ambulatory despite recurrent falls until advanced stage.
Liver dysfunction and hepatomegaly were observed exclusively among children with SLC30A10 mutations. The lack of hepatic Mn deposition in SLC39A14 mutations has been partly attributed to impaired hepatic Mn uptake, believed to be a function of the SLC39A14 protein. 5 In knockout (KO) mouse models with deficient SLC30A10 and SLC39A14 transporters individually as well as together (double KOs), elevated Mn levels could be demonstrated in all three models in blood and brain, but increased hepatic Mn was demonstrable only in the SLC30A10 model. 20 , 21 Another interesting observation in our study was that with chelation, LFTs normalized in several patients, suggesting reversibility in early stages.
Presence of polycythemia was seen exclusively with SLC30A10 mutations, in line with previous reports, with some children demonstrating severe polycythemia. However, the proportion of polycythemic children is much higher in our series than noted previously. 6 This is interesting, considering that children in our resource‐constrained settings often exhibit anemia, and hence, normal Hb values usually raise concerns of relative polycythemia. Mn may mimic effects of hypoxia, stabilize hypoxia‐inducible factor (HIF), and induce genetic expression of erythropoietin (Epo) leading to increase in Hb, 22 , 23 supported by raised Epo levels in some patients. Elevated Mn levels activate HIF‐1 and 2, to upregulate SLC30A10, which reduces cellular Mn levels. This mechanism and subsequent polycythaemia may be absent in HMNDYT2 because of lack of hepatic involvement. 24
Apart from brain and liver, thyroid dysfunction, presumably because of accumulation of Mn in thyroid tissue, has been demonstrated in SLC30A10 KO mice, 21 , 25 with severe hypothyroidism because of inhibition of thyroxine hormone by Mn. However, we did not observe any thyroid dysfunction.
Neuroimaging is useful not only for diagnosis, but also to monitor therapeutic response. We found classic T1 shortening of basal ganglia in all children. Post‐chelation scans demonstrated dramatic improvement in the extent of T1 shortening, with residual changes mostly in the globus pallidus, internal capsule, and/or subthalamic nucleus. This is in line with the few case reports where repeat imaging has been described. 8 , 26 , 27 Hence, MRI scans can be used to monitor treatment response. However, limitations of cost and need for sedation in young children need consideration. Optimal timing for follow‐up scans needs to be determined as well.
Treatment outcomes on chelation with Na2CaEDTA among our patients were variable. Although most patients on follow‐up had some alleviation of neurological symptoms, marked improvement/ normalization was achieved in very few. Children with deterioration had shorter duration of chelation and presented somewhat later than children who showed improvement, which may be important influencers of outcome. However, these results must be interpreted with caution owing to small patient numbers, and predictors of outcome require careful evaluation in larger cohorts. Long‐term treatment outcomes on chelation have been reported in a few patients. A 10‐year follow‐up of a patient with SLC30A10 variant observed good clinical and biochemical response with chelation therapy with Na2CaEDTA, but reduction in MRI T1 signal change occurred only after 4 years. 8 The patient showed clinical deterioration when d‐penicillamine was substituted, because of lack of availability of Na2CaEDTA. In another adult patient with SLC30A10 variant, a 2‐year follow‐up revealed significant improvement in parkinsonism and hepatic steatosis. 28
We also noted a large proportion of loss to follow‐up, particularly in children with SLC39A14 mutations. This may be because of suboptimal response after the first few cycles of chelation, as well as recurrent need for hospitalization. Apart from limited availability of Na2CaEDTA in India, its use necessitates hospitalization and monthly intravenous infusion for 5 days. This demanding regime leads to parental fatigue, loss of work, and school days and consequent loss to follow‐up. Additionally, potential side effects of chelation therapy include hypocalcemia (that may develop during infusion), trace metal and vitamin deficiency, low platelets and leucopenia, and nephrotoxicity. Therefore, related monitoring needs to be done regularly, including renal function, complete blood count, calcium/phosphate, and trace metal (copper, zinc).
Previously, the use of oral 2,3‐dimercaptosuccininc acid as well as d‐penicillamine has been trialed in some patients with varying benefits. 7 , 8 , 26 , 27 , 29 D‐penicillamine as an add‐on agent in two of our patients yielded appreciable results. D‐penicillamine may serve as a more feasible agent in our settings, because it is orally administered, widely available, and inexpensive.
Limitations of our study include its retrospective nature and loss to follow‐up of a large proportion of patients. Repeat serum/urinary manganese levels and neuroimaging were not available in most children. These tests are very expensive and not easily available in India. There are no guidelines for using serum/urinary manganese levels to titrate chelation. Furthermore, we could not apply functional scales to assess baseline and post‐treatment disease severity and used a Likert scale instead. It will be useful to design severity scales and functional scoring in future studies.
Conclusions
We report clinical presentation and short term outcomes of children with HMNDYT in a low‐resource setting. We found female preponderance in both genetic subtypes, younger age at onset and more severe clinical phenotype in children with SLC39A14 mutations, and moderate response to Na2CaEDTA chelation therapy overall. Our study raises concerns regarding treatment options in these patients, inviting the possibility of exploring further therapeutic options such as d‐penicillamine in the future.
Author Roles
(1) Research Project: A. Conception, B. Organization, C. Execution; (2) Statistical Analysis: A. Design, B. Execution, C. Review and Critique; (3) Manuscript Preparation: A. Writing of the First Draft, B. Review and Critique.
D.G.: 1A, 1B, 1C; 2A, 2B, 2C; 3A, 3B
S.Y.: 1A, 1B, 2C, 3C
M.F.: 1A, 1B, 2C, 3C
U.S.: 1A, 1B, 2C, 3C
K.M.: 1A, 1B, 2C, 3C
P.G.: 1A, 1B, 2C, 3C
V.B.: 1A, 1B, 2C, 3C
A.B.: 1A, 1B, 2C, 3C
U.K.: 1A, 1B, 2C, 3C
A.G.S.: 1A, 1B, 2C, 3C
N.S.: 1A, 1B, 2C, 3C
K.S.: 1A, 1B, 2C, 3C
V.K.G.: 1A, 1B, 2C, 3C
M.J.: 1A, 1B, 2C, 3C
M.K.: 1A, 1B, 2C, 3C
H.P.: 1A, 1B, 2C, 3C
D.P.: 1A, 1B, 2C, 3C
S. Pa.: 1A, 1B, 2C, 3C
V.U.: 1A, 1B, 2C, 3C
A.K.: 1A, 1B, 2C, 3C
S.P.: 1A, 1B, 2C, 3C
M.T.: 1A, 1B, 2C, 3C
S.D.: 1A, 1B, 2C, 3C
A.Sh.: 1A, 1B, 2C, 3C
A.S.: 1A, 1B, 2C, 3C
H.P: 1A, 1B, 2C, 3C
V.S: 1A, 1B, 2C, 3C
S.S.: 1A, 1B, 1C; 2A, 2B, 2C; 3B, 3C.
Disclosures
Ethical Compliance Statement: Ethical clearance for this study was obtained from the Ethics Committee for Human Research of Lady Hardinge Medical College (Reference LHMC/IEC/2019/31R) and other participating institutes as necessary by their local ethics committees. Informed consent was obtained from the participating parents, and assent from the child when applicable. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Funding Sources and Conflicts of Interest: This study has not received any funding support. The authors declare that there are no conflicts of interest relevant to this work.
Financial Disclosures for the Previous 12 Months: V.B receives salary from the Erasmus MC Rotterdam, The Netherlands; He received research grants from the Stichting ParkinsonFonds (The Netherlands) and from the Stichting Alzheimer Nederland; He received honoraria from Elsevier, for serving as co‐Editor‐in‐Chief of Parkinsonism and Related Disorders; he also received honoraria from the International Parkinson and Movement Disorders Society, and as Chair of the MDS International Congress Program Committee 2020‐2021. N.S. has received research support from PTC Therapeutics for a Multicentric RCT. N.S. has received research support from Institute Grant, PGIMER, (Government Entity). V.U. has received personal compensation in the range of $0–$499 for serving on a Scientific Advisory or Data Safety Monitoring board for Sanofi Pharmaceuticals. V.U. has received personal compensation in the range of $0–$499 for serving on a Scientific Advisory or Data Safety Monitoring board for Abbott Pharmaceuticals. V.U. has received personal compensation in the range of $0–$499 for serving on a Scientific Advisory or Data Safety Monitoring board for Dr. Reddys Laboratory. S.S. has received personal compensation in the range of $0–$499 for serving as an Editor, Associate Editor, or Editorial Advisory Board Member for Jaypee publishers.
Supporting information
Table S1. Variants identified in patients with associated genetic and clinical interpretation.
Table S2. Methodology deployed for genetic screening.
Table S3. MRI features among children with inherited hypermanganesemia.
Table S4. Differences between children with no improvement/worsening versus some/marked improvement (n = 16).
Acknowledgments
We thank the patients and their families for participation in the study.
Relevant disclosures and conflict of interest are listed at the end of this article.
References
- 1. Tuschl K, Mills PB, Parsons H, et al. Hepatic cirrhosis, dystonia, polycythaemia and hypermanganesaemia—a new metabolic disorder. J Inherit Metab Dis 2008;31(2):151–163. 10.1007/s10545-008-0813-1. [DOI] [PubMed] [Google Scholar]
- 2. Tuschl K, Clayton PT, Gospe SM, et al. Syndrome of hepatic cirrhosis, dystonia, polycythemia, and hypermanganesemia caused by mutations in SLC30A10, a manganese transporter in man. Am J Hum Genet 2012;90(3):457–466. 10.1016/j.ajhg.2012.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Quadri M, Federico A, Zhao T, et al. Mutations in SLC30A10 cause parkinsonism and dystonia with Hypermanganesemia, polycythemia, and chronic liver disease. Am J Hum Genet 2012;90(3):467–477. 10.1016/j.ajhg.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Anagianni S, Tuschl K. Genetic disorders of manganese metabolism. Curr Neurol Neurosci Rep 2019;19(6):33. 10.1007/s11910-019-0942-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tuschl K, Meyer E, Valdivia LE, et al. Mutations in SLC39A14 disrupt manganese homeostasis and cause childhood‐onset parkinsonism–dystonia. Nat Commun 2016;7(1):11601. 10.1038/ncomms11601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Marti‐Sanchez L, Ortigoza‐Escobar JD, Darling A, et al. Hypermanganesemia due to mutations in SLC39A14: further insights into Mn deposition in the central nervous system. Orphanet J Rare Dis 2018;13:28. 10.1186/s13023-018-0758-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mukhtiar K, Ibrahim S, Tuschl K, Mills P. Hypermanganesemia with dystonia, polycythemia and cirrhosis (HMDPC) due to mutation in the SLC30A10 gene. Brain and Development 2016;38(9):862–865. 10.1016/j.braindev.2016.04.005. [DOI] [PubMed] [Google Scholar]
- 8. Stamelou M, Tuschl K, Chong WK, Burroughs AK, Mills PB, Bhatia KP, Clayton PT. Dystonia with brain manganese accumulation resulting from SLC30A10 mutations: a new treatable disorder. Mov Disord 2012;27(10):1317–1322. 10.1002/mds.25138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Padmanabha H, Krishnamurthy S, Sharath Kumar GG, Chikkanayakana I, Sethuraman A, Mathew T. Teaching NeuroImages: an imaging clue for treatable early childhood‐onset dystonia: manganism. Neurology 2019;92(6):e628–e629. 10.1212/WNL.0000000000006881. [DOI] [PubMed] [Google Scholar]
- 10. Lambrianides S, Nicolaou P, Michaelidou M, et al. A novel SLC30A10 missense variant associated with parkinsonism and dystonia without hypermanganesemia. J Neurol Sci 2020;418:117101. 10.1016/j.jns.2020.117101. [DOI] [PubMed] [Google Scholar]
- 11. Yapici Z, Tuschl K, Eraksoy M. Hypermanganesemia with dystonia 1: a novel mutation and response to iron supplementation. Mov Disord Clin Pract 2020;7(1):94–96. 10.1002/mdc3.12861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tavasoli A, Arjmandi Rafsanjani K, Hemmati S, Mojbafan M, Zarei E, Hosseini S. A case of dystonia with polycythemia and hypermanganesemia caused by SLC30A10 mutation: a treatable inborn error of manganese metabolism. BMC Pediatr 2019;19:229. 10.1186/s12887-019-1611-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zeglam A, Abugrara A, Kabuka M. Autosomal‐recessive iron deficiency anemia, dystonia and hypermanganesemia caused by new variant mutation of the manganese transporter gene SLC39A14. Acta Neurol Belg 2019;119(3):379–384. 10.1007/s13760-018-1024-7. [DOI] [PubMed] [Google Scholar]
- 14. Namnah M, Bauer M, Mor‐Shaked H, Bressman SB, Raymond D, Ozelius LJ, Arkadir D. Benign SLC39A14 course of dystonia‐parkinsonism secondary to inherited manganese accumulation. Mov Disord Clin Pract 2020;7(5):569–570. 10.1002/mdc3.12947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Josephs KA, Ahlskog JE, Klos KJ, Kumar N, Fealey RD, Trenerry MR, Cowl CT. Neurologic manifestations in welders with pallidal MRI T1 hyperintensity. Neurology 2005;64(12):2033–2039. 10.1212/01.WNL.0000167411.93483.A1. [DOI] [PubMed] [Google Scholar]
- 16. Sadek AH, Rauch R, Schulz PE. Parkinsonism due to manganism in a welder. Int J Toxicol 2003;22(5):393–401. 10.1177/109158180302200511. [DOI] [PubMed] [Google Scholar]
- 17. Juneja M, Shamim U, Joshi A, et al. A novel mutation in SLC39A14 causing hypermanganesemia associated with infantile onset dystonia. J Gene Med 2018;20(4):e3012. 10.1002/jgm.3012. [DOI] [PubMed] [Google Scholar]
- 18. Quadri M, Kamate M, Sharma S, et al. Manganese transport disorder: novel SLC30A10 mutations and early phenotypes. Mov Disord 2015;30(7):996–1001. 10.1002/mds.26202. [DOI] [PubMed] [Google Scholar]
- 19. Kopanos C, Tsiolkas V, Kouris A, Chapple CE, Albarca Aguilera M, Meyer R, Massouras A. VarSome: the human genomic variant search engine. Bioinforma Oxf Engl 2019;35(11):1978–1980. 10.1093/bioinformatics/bty897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xin Y, Gao H, Wang J, et al. Manganese transporter Slc39a14 deficiency revealed its key role in maintaining manganese homeostasis in mice. Cell Discov 2017;3:17025. 10.1038/celldisc.2017.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hutchens S, Liu C, Jursa T, et al. Deficiency in the manganese eff1lux transporter SLC30A10 induces severe hypothyroidism in mice. J Biol Chem 2017;292(23):9760–9773. 10.1074/jbc.M117.783605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zogzas CE, Mukhopadhyay S. Inherited disorders of manganese metabolism. Adv Neurobiol 2017;18:35–49. 10.1007/978-3-319-60189-2_3. [DOI] [PubMed] [Google Scholar]
- 23. Ebert BL, Bunn HF. Regulation of the erythropoietin gene. Blood 1999;94(6):1864–1877. [PubMed] [Google Scholar]
- 24. Liu C, Jursa T, Aschner M, Smith DR, Mukhopadhyay S. Up‐regulation of the manganese transporter SLC30A10 by hypoxia‐inducible factors defines a homeostatic response to manganese toxicity. Proc Natl Acad Sci U S A 2021;118(35):e2107673118. 10.1073/pnas.2107673118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu C, Hutchens S, Jursa T, et al. Hypothyroidism induced by loss of the manganese efflux transporter SLC30A10 may be explained by reduced thyroxine production. J Biol Chem 2017;292(40):16605–16615. 10.1074/jbc.M117.804989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zaki MS, Issa MY, Elbendary HM, et al. Hypermanganesemia with dystonia, polycythemia and cirrhosis in 10 patients: six novel SLC30A10 mutations and further phenotype delineation. Clin Genet 2018;93(4):905–912. 10.1111/cge.13184. [DOI] [PubMed] [Google Scholar]
- 27. Dutta A, Majumdar R, Dubey S, Pandit A. Penicillamine for hypermanganesemia with dystonia, polycythemia, and cirrhosis in 2 sisters. Neurology 2020;2:123–125. 10.1212/WNL.0000000000011296. [DOI] [PubMed] [Google Scholar]
- 28. Di Toro ML, Mignarri A, Battisti C, et al. Two‐year follow‐up after chelating therapy in a patient with adult‐onset parkinsonism and hypermanganesaemia due to SLC30A10 mutations. J Neurol 2014;261(1):227–228. 10.1007/s00415-013-7187-5. [DOI] [PubMed] [Google Scholar]
- 29. Rodan LH, Hauptman M, D'Gama AM, et al. Novel founder intronic variant in SLC39A14 in two families causing manganism and potential treatment strategies. Mol Genet Metab 2018;124(2):161–167. 10.1016/j.ymgme.2018.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Variants identified in patients with associated genetic and clinical interpretation.
Table S2. Methodology deployed for genetic screening.
Table S3. MRI features among children with inherited hypermanganesemia.
Table S4. Differences between children with no improvement/worsening versus some/marked improvement (n = 16).