

Abstract
Background
Keloid scarring is a fibroproliferative disease caused by aberrant genetic activation with an unclear underlying mechanism. Genetic predisposition, aberrant cellular responses to environmental factors, increased inflammatory cytokines and epithelial–mesenchymal transition (EMT) phenomena are known as major contributors. In this study, we aimed to identify the molecular drivers that initiate keloid pathogenesis.
Methods
Bulk tissue RNA sequencing analyses of keloid and normal tissues along with ex vivo and in vitro tests were performed to identify the contributing genes to keloid pathogenesis. An animal model of inflammatory keloid scarring was reproduced by replication of a skin fibrosis model with intradermal bleomycin injection in C57BL/6 mice.
Results
Gene set enrichment analysis revealed upregulation of Wnt family member 5A (WNT5A) expression and genes associated with EMT in keloid tissues. Consistently, human keloid tissues and the bleomycin-induced skin fibrosis animal model showed significantly increased expression of WNT5A and EMT markers. Increased activation of the interleukin (IL)-6/Janus kinase (JAK)/signal transducer and activator of transcription (STAT) pathway and subsequent elevation of EMT markers was also observed in keratinocytes co-cultured with WNT5A-activated fibroblasts or keloid fibroblasts. Furthermore, WNT5A silencing and the blockage of IL-6 secretion via neutralizing IL-6 antibody reversed hyperactivation of the STAT pathway and EMT markers in keratinocytes. Lastly, STAT3 silencing significantly reduced the EMT-like phenotypes in both keratinocytes and IL-6-stimulated keratinocytes.
Conclusions
Intercellular communication via the WNT5A and STAT pathways possibly underlies a partial mechanism of EMT-like phenomena in keloid pathogenesis. IL-6 secreted from WNT5A-activated fibroblasts or keloid fibroblasts activates the JAK/STAT signaling pathway in adjacent keratinocytes which in turn express EMT markers. A better understanding of keloid development and the role of WNT5A in EMT will promote the development of next-generation targeted treatments for keloid scars.
Keywords: Keloid; Wnt family member 5A; Interleukin-6; JAK/STAT pathway; Epithelial–mesenchymal transition; Scar, Epithelial mesenchymal transition
Highlights.
This study shows that keloid scarring is associated with activation of the WNT5A gene and the IL-6/JAK/STAT pathway.
Intercellular communication via the WNT5A and STAT pathways possibly underlies the EMT-like phenomenon in keloid pathogenesis
IL-6 secreted from WNT5A-activated fibroblasts or keloid fibroblasts might be a factor inducing adjacent keratinocytes to express EMT-like phenotype markers in keloid scars.
Background
During epithelial–mesenchymal transition (EMT), epithelial cells can acquire remarkable cell plasticity and differentiate into various mesenchymal cells [1]. Keloid scars are benign dermal fibroproliferative lesions that are unique to humans [2]; however, their locally invasive behavior somewhat resembles that of neoplastic tumors [3]. It has been suggested that their recurrence and invasiveness may involve aberrant genetic activation of EMT during the abnormal wound-healing process [4–6]. Upon EMT activation, epithelial cells acquire a spindle-shaped morphology and increase the expression of mesenchymal markers, such as neural cadherin (N-cadherin), slug, vimentin, α-smooth muscle actin (α-SMA) and fibronectin [7]. This phenomenon has been observed in many physiological processes, including embryogenesis, inflammation and wound healing [8]. During pathologic transition of epithelial cells into mesenchymal cells, dissolution of cell–cell junctions, loss of apical–basal polarity, reorganization of cytoskeletal architecture and the degradation of extracellular matrix (ECM) proteins that enables invasive behavior can occur [9]. Numerous studies have suggested that abnormal EMT-like phenotypes are observed in the development of keloid scars [3,6,8,10]. Because keloid scars result from abnormal fibrosis that extends beyond the original skin-injury site, EMT might play a role in their development and aggravation [11,12].
Zhang et al. [13] have previously described a population of self-renewing keloid-derived precursor cells expressing mesenchymal and embryonic stem cell markers driven by interleukin (IL)-6/IL-17-mediated inflammation. The expression of IL-6, a pro-tumorigenic cytokine, is frequently elevated in keloid fibroblasts (KFs) [14]. Interestingly, not only IL-6 but also its second messengers, Janus kinase (JAK)/signal transducer and activator of transcription (STAT)3, are elevated in keloids and potentially contribute to tumorigenesis [15,16]. The IL-6/JAK/STAT3 pathway is a key mechanism in EMT pathogenesis seen in other organ systems. IL-6-dependent STAT3 activation positively regulates transforming growth factor (TGF)-β1-induced EMT and invasion in hepatocellular carcinoma. Furthermore, IL-6 promotes EMT by activating the JAK/STAT pathway, thereby causing fibrotic injury of the peritoneal membrane [17]. A recent transcriptome and open-chromatin analysis suggested that the STAT3 and WNT signaling pathways are both independently involved in keloid pathogenesis [18].
Wnt family member 5A (WNT5A) is a secreted growth factor in the non-canonical Wnt family. In liver myofibroblasts, WNT5A knockdown reduces production of IL-1β and IL-6 and the matrix proteins collagen types I and III, whereas its overexpression increases the expression of these factors [19]. WNT5A is substantially upregulated in activated liver myofibroblasts [20] and its expression during fibrosis is associated with myofibroblast proliferation and migration and ECM protein production [21]. Moreover, a previous study showed that WNT5A mRNA and protein levels are increased in KFs relative to those in normal fibroblasts [22]. In the present study, we examine the intricate relationship between WNT5A signaling and the JAK/STAT pathway in keloids and propose potential treatment targets for suppressing pathogenic fibrosis of the skin.
Methods
Patient samples
Keloid tissues from 11 patients with active-stage keloids and undergoing surgical excision, and three normal skins from remnant tissues after breast reconstruction surgery at the plastic and reconstructive surgery department in Yonsei University College of Medicine, were harvested after obtaining informed consent according to the protocol approved by the Institutional Review Board of Yonsei University Hospital (IRB No. 4–2017-0259, 4–2021-0262; Table 1). The keloid patients enrolled in the study had either a family history of keloid scars or a past history of keloid formation after trauma; their scar types were clinically evaluated by both the plastic surgeon and the dermatologist to be defined as keloids, which showed extension into normal adjacent scars, and prolonged inflammation of the epidermis without resolution of the scar proliferation and inflammation within 1 year. The scars were identified as in their ‘active stage’, since all patients complained of itching, stinging and painful sensation of the lesion; the scars also had either spread in size or were accompanied by new adjacent keloid scar formation. All experiments involving humans adhered to the Declaration of Helsinki. Tissue specimens from patients who underwent scar treatments within the previous 6 months were excluded from the study.
Table 1.
Clinical information of keloids and normal skin tissues
| Sample ID | Cohort | Gender | Race | Site | Experiment type |
|---|---|---|---|---|---|
| Healthy 1 | Public #1 | Female | Caucasian | Lower back | RNAseq |
| Healthy 2 | Public #1 | Male | Caucasian | Lower back | RNAseq |
| Healthy 3 | Public #1 | Male | Caucasian | Lower back | RNAseq |
| Healthy 4 | Public #1 | Female | Caucasian | Lower back | RNAseq |
| Healthy 5 | public #1 | Male | Caucasian | Lower back | RNAseq |
| Healthy 6 | Public #1 | Female | Caucasian | Lower back | RNAseq |
| Healthy 7 | Public #1 | Male | Caucasian | Lower back | RNAseq |
| Keloid 1 | Yonsei | Female | Asian | Neck | RNAseq, IHC, IF |
| Keloid 2 | Yonsei | Female | Asian | Lower abdomen | RNAseq, IHC, qRT-PCR |
| Keloid 3 | Yonsei | Female | Asian | Earlobe | RNAseq, IHC, qRT-PCR |
| Keloid 4 | Yonsei | Female | Asian | Anterior chest | RNAseq, qRT-PCR |
| Keloid 5 | Yonsei | Female | Asian | Lower abdomen | RNAseq |
| Keloid 6 | Yonsei | Female | Asian | Earlobe | RNAseq |
| Keloid 7 | Yonsei | Female | Asian | Earlobe | RNAseq |
| Keloid 8 | Yonsei | Female | Asian | Forearm | RNAseq |
| Keloid 9 | Yonsei | Female | Asian | Upper arm | RNAseq |
| Keloid 10 | Yonsei | Male | Asian | Anterior chest | Cell isolation |
| Keloid 11 | Yonsei | Female | Asian | Earlobe | Cell isolation |
| Normal 1 | Yonsei | Female | Asian | Anterior chest | IHC, qRT-PCR, IF |
| Normal 2 | Yonsei | Female | Asian | Anterior chest | IHC, qRT-PCR |
| Normal 3 | Yonsei | Female | Asian | Anterior chest | IHC, qRT-PCR |
Information for a publicly available normal skin cohort (public #1) along with the in-house-generated keloid tissues and normal tissues are provided. Detailed patients’ demographic information for public #2 cohort (normal skin tissues) was not provided by the authors to protect patients’ identities. The raw data are available in the ArrayExpress database (Accession number E-MTAB-5678). RNAseq RNA sequencing, IHC immunohistochemistry, IF immunofluorescence
Sample selection and RNA sequencing
We analyzed a newly generated in-house dataset for 9 Asian keloid tissues (Sample ID: Keloid 1–9; Table 1) and two publicly controlled datasets from the Sequence Read Archive containing data for 11 normal skin samples. To address the collinearity problem from utilizing multiple datasets, we modified the scBatch method [23] to correct the quantile distribution from distinct batch blocks in a sample-distance matrix. Sample distances were measured as a correlation with the expression matrix of housekeeping genes. Genes that differed significantly between keloids and normal tissues were identified using DESeq2 version 1.26.0. Pathway analysis using gene set enrichment analysis (GSEA; v2.07; Broad Institute, Cambridge, MA, USA) was performed. The in-house data set for the keloid tissue samples is publicly available under accession number GSE173900.
Sequencing was performed by the Genomics Core at Avison Biomedical Research Center, Yonsei University. Total RNA was isolated from the 9 human keloid tissue samples using the RNeasy mini kit (Qiagen, Hilden, Germany) according to a standard extraction protocol. Tissue samples for analysis were obtained from the margin of the lesion via 3-mm-diameter skin-punch biopsy. RNA sequencing (RNA-seq) libraries were constructed using a SureSelect RNA-seq library prep kit (Agilent Technologies) and sequenced in 100-bp paired-end mode on a NextSeq550 platform (Illumina). Two public data sets were assessed from the SRA database to establish a dataset of 11 normal tissue samples as a control. Raw data (FASTQ files) from 7 healthy skin biopsies sequenced on the NextSeq500-platform (generating 800 million total reads) were downloaded from the SRA database (accession no. SRP227263). Another raw dataset (FASTQ files) was assessed from 4 normal skin tissues sequenced on the Hiseq2000-platform (accession no. ERP022968). Multiple normal tissue datasets were collected and used as control data to reduce selection bias from using a single public dataset.
Total RNA sequencing analysis and modified batch effect removal
RNA-seq data were analyzed by the Bioinformatics Collaboration Unit, Yonsei University College of Medicine. The in-house keloid dataset and the public control datasets were analyzed using an identical RNA-seq analysis pipeline. All FASTQ reads were trimmed for quality and adapter content using Trimmomatic v.0.32 [24], and the data were aligned to the human reference genome (assembly: hg38) using HISAT2 v.2.1.0 [25]. To minimize misalignment with the remaining rRNA reads, we also removed ribosomal RNA (rRNA) reads using RSeQC v.2.6.1 [26]. Data were then quantified as transcripts per million using StringTie v.2.0.6 [27], and raw read counts retrieved using the Python script available from StringTie were used as input into the DESeq2 R package.
To address the collinearity problem from utilizing multiple datasets, we modified the scBatch method [23], an assumption-free batch-effect correction algorithm, to correct the quantile distribution from the distinct batch blocks in the sample-distance matrix. Sample distances were measured using correlation with the expression matrix of the housekeeping genes, which characteristically maintained constant expression levels under all biological conditions. To adjust for batch effects, we used 98 housekeeping genes compiled in a previous study [28].
Differentially expressed gene analysis on DESeq2
Genes that differed significantly in their expression between the two groups (keloid scars and normal tissues) were identified using DESeq2 V.1.26.0 from Bioconductor. The count matrix corrected for batch effect was entered as an input to DESeq2, and genes with non-zero expression values in at least three samples from each dataset were extracted. Only genes exhibiting logarithmic changes (log2) in expression of >0.5-fold and an adjusted p < 0.01 using the Benjamini–Hochberg procedure were considered differentially expressed genes (DEGs).
Cell culture and transfection
Experiments were conducted using human primary epidermal keratinocytes (KCs) (HEKa; ATCC, Manassas, VA, USA), human dermal fibroblasts (HDFs; ATCC PCS-201-012) and KFs (ATCC CRL-1762) at passages 2–4. The isolation of patient-derived keloid KCs (KKs) and KFs was conducted as previously described [29]. All patient-derived cells (Sample ID: Keloid 10–11; Table 1) were used up to 2–3 passages for qRT-PCR and enzyme-linked immunosorbent assay (ELISA). HDFs (5 × 105) were treated with WNT5A (250 ng/mL; 645-WN-010; R&D Systems, Minneapolis, MN, USA) for the times indicated. HEKs (5 × 105) were treated with recombinant human IL-6 (50 ng/mL; 206-IL-010; R&D Systems). Small-interfering (si) RNAs targeting WNT5A (siWNT5A) and STAT3 (siSTAT3) were applied for gene silencing. Cells were transfected either with control siRNA (D-001810-10-05; Dharmacon, Lafayette, CO, USA), siWNT5A (L-003939-00-0005; Dharmacon) or siSTAT3 (6582S; Cell Signaling Technology, Danvers, MA, USA) for 24 h using Lipofectamine (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. Neutralizing monoclonal antibody against human IL-6 (anti-hIL-6-IgG, mabg-hil6–3; InvivoGen) was used to block the effect of IL-6 secretion on KCs.
Co-culture of HDFs or KFs with KCs
Primary KCs (5 × 105) were seeded into the lower chamber and 5 × 105 HDFs or 5 × 105 KFs were seeded into the upper chamber of a 6.5-mm diameter Transwell separated by a 0.4-μm pore-sized membrane (Corning, Inc., Corning, NY, USA) and cultured in DMEM. Co-cultures were incubated with or without WNT5A (250 ng/mL) for the times indicated. The supernatant of each stimulation experiment with or without WNT5A was collected. KCs co-cultured with HDFs or KFs from the lower chamber were washed three times with phosphate-buffered saline (PBS) and collected for further experiments.
Animals and replication of a bleomycin-induced skin fibrosis model
Specific-pathogen-free female C57BL/6 mice (6 weeks old; 20–25 g) were purchased from the ORIENT BIO Animal Center (Seongnam-si, Korea). Before the experiment, mice underwent a 1-week acclimation period. Mice that were 7–12-weeks old were used in all experiments. Injection of 100 μL of 1 mg/mL bleomycin sulfate (S1214; Selleckchem, Houston, TX, USA) or PBS was performed on 1-cm2 shaved patches on the back skins of the mice five times weekly for 3 weeks, as previously described in a systemic sclerosis skin fibrosis model [30]. All experiments were conducted in accordance with the Guide for the Care and Use of Laboratory Animals provided by the Animal Laboratory Ethics Committee.
RNA isolation and real-time PCR analysis
Total RNA was extracted from cells using RNAiso plus reagent (#9109; Takara Biotechnology, Shiga, Japan) and purified using a RNeasy Plus mini kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. RNA was reverse transcribed into cDNA using an RNA to cDNA EcoDry premix kit (Takara Biotechnology). Quantitative PCR was performed using a QuantStudio 3 real-time PCR system (Applied Biosystems, Foster City, CA, USA) in 20-μL reactions containing SYBR Green master mix (Promega, Madison, WI, USA) and specific primer pairs (Macrogen, Seoul, Korea). The primer sequences are listed in Table 2. mRNA levels were normalized to that of the housekeeping genes glyceraldehyde 3-phosphate dehydrogenase (GAPDH) or beta-actin as indicated in each Figure. Three independent experiments as technical replicates were performed for statistical analyses.
Table 2.
Primers used in this study
| Target gene | Primer sequences (5′–3′) |
|---|---|
| WNT5A | Forward: TACGAGAGTGCTCGCATCCTCA |
| Reverse: TGTCTTCAGGCTACATGAGCCG | |
| VIM (vimentin) | Forward: AGGCAAAGCAGGAGTCCACTGA |
| Reverse: ATCTGGCGTTCCAGGGACTCAT | |
| SNAI2 (slug) | Forward: ATCTGCGGCAAGGCGTTTTCCA |
| Reverse: GAGCCCTCAGATTTGACCTGTC | |
| CDH2 (N-cadherin) | Forward: CCTCCAGAGTTTACTGCCATGAC |
| Reverse: GTAGGATCTCCGCCACTGATTC | |
| CDH1 (E-cadherin) | Forward: GCCTCCTGAAAAGAGAGTGGAAG |
| Reverse: TGGCAGTGTCTCTCCAAATCCG | |
| COL1A1 | Forward: GATTCCCTGGACCTAAAGGTGC |
| Reverse: AGCCTCTCCATCTTTGCCAGCA | |
| α-SMA | Forward: GCACCCCTGAACCCCAAGGC |
| Reverse: GCACGATGCCAGTTGTGCCT | |
| IL-6 | Forward: AGACAGCCACTCACCTCTTCAG |
| Reverse: TTCTGCCAGTGCCTCTTTGCTG | |
| GAPDH | Forward: GTCTCCTCTGACTTCAACAGCG |
| Reverse: ACCACCCTGTTGCTGTAGCCAA |
WNT5A Wnt family member 5A, COL1A1 collagen type 1 α1, α-SMA smooth muscle actin, IL interleukin, GAPDH glyceraldehyde 3-phosphate dehydrogenase, E-cadherin epithelial cadherin
Tissue handling and immunohistochemical analysis
The samples were stained with hematoxylin and eosin (H&E) and Masson’s trichrome (MT) staining kit (ab150686; Abcam, Cambridge, UK) according to the manufacturer’s instructions. Immunohistochemical (IHC) staining was performed on deparaffinized, formalin-fixed tissue sections (3–5 μm thickness) with the antibodies listed in Table 3. Protein expression levels on IHC-stained keloid and normal tissue sections were quantified using ImageJ software (National Institutes of Health, Bethesda, MD, USA). All analyses were performed on stained images obtained from three adjacent tissue sections of each study group, and the average data were statistically analyzed.
Table 3.
Immunohistochemistry staining protocols
| Target marker | Antibody source | Antibody dilution |
|---|---|---|
| WNT5A | Rabbit polyclonal IgG antibody (ab229220; Abcam, Cambridge, UK) | 1:100 |
| N-Cadherin | Rabbit monoclonal IgG antibody (#13116S; Cell Signaling Technology, Danvers, MA, USA) | 1:125 |
| Vimentin | Mouse monoclonal IgG antibody (ab8978; Abcam) | 1:100 |
| α-SMA | Mouse monoclonal IgG antibody (ab7817l; Abcam) | 1:100 |
| E-Cadherin | Mouse monoclonal IgG antibody (#14472S; Cell Signaling Technology) | 1:100 |
WNT5A Wnt family member 5A, α-SMA smooth muscle actin
Immunofluorescence
Double-immunofluorescence staining of WNT5A (1:200; ab229220; Abcam) with cytokeratin 14 (1:200; ab9220; Abcam), CD31 (1:800; #3528; Cell Signaling Technology) and α-SMA (1:100; 14–9760-82; Invitrogen) was performed to determine cell localization and differential expression of WNT5A in keloid and normal tissues. Sections were observed using fluorescence microscopy (confocal LSM 700; Zeiss, Oberkochen, Germany).
Proteome profiler human cytokine array and ELISA
The cytokine profile assay was performed using the Proteome Profiler Human Cytokine Array (#ARY0005B; R&D Systems) according to the manufacturer’s instructions. A pre-coated human ELISA kit (IL-6; PeproTech, Rocky Hill, NJ, USA) was used to quantify the IL-6 secretion level in culture medium. Human protein WNT5A ELISA kit (CSB-EL026138HU; Cusabio, Houston, TX, USA) was used to quantify WNT5A secretion from HDFs and KFs.
Western blot analysis
Protein levels were normalized to that of GAPDH or the ratio of phosphorylated and total isoforms. Antibodies against WNT5A (ab229200), vimentin (ab8978), α-SMA (ab7817), collagen type 1 α1 (COL1A; ab260043), STAT3 (ab68153) and phosphorylated (p)-STAT3 (ab76315) were purchased from Abcam. Antibodies against slug (C79G7), N-cadherin (#13116), E-cadherin (#14472) and GAPDH (#2118) were purchased from Cell Signaling Technology. Antibodies against β-actin (sc-47 778) were purchased from Santa Cruz Biotechnology (Dallas, TX, USA). The optical densities of bands on the developed film were analyzed using ImageJ software.
Statistical analysis
Data are represented as the mean ± standard deviation. Differences between groups of normally distributed data were analyzed with independent t tests for two-group comparisons, and non-parametric variables were compared using the Mann–Whitney U test. Bleomycin-induced fibrosis assays were analyzed by repeated-measure analysis of variance, followed by post hoc tests via Bonferroni correction. A p < 0.05 was considered statistically significant. SPSS (v.25.0; IBM Corp., Armonk, NY, USA) was used for all statistical analyses.
Results
RNA sequencing analysis revealed differential expression of WNT5A and EMT markers between keloid and normal tissues
We utilized 9 in-house-generated sets of keloid Asian RNA-seq data and two independent public datasets that included 11 normal skin samples as control data (Table 1). After removing batch effects, principal component analysis showed that the two independent public datasets generated from different batches could be combined. Keloid and normal samples showed significant dispersion, demonstrating meaningful biological differences (Supplementary Figure 1a-b, see online supplementary material). Genes differing significantly between keloid and normal tissues were identified, with 1939 and 1610 upregulated and downregulated genes, respectively, selected as DEGs (Figure 1a). Among the various candidates, WNT5A transcripts were significantly upregulated in keloids by a log2 fold-change of 1.72 before removing batch effects (adjusted p = 7.6 × 10−10) and 0.637 after removing batch effects (adjusted p = 2.7 × 10−7). Additionally, GSEA allowed visualization of the top 10 pathways with the highest normalized enrichment scores (NESs) (Supplementary Figure 2, see online supplementary material). Notably, the HALLMARK EMT pathway gene set showed the highest NES, containing the most significantly enriched genes (NES = 3.06; p = 1.8 × 10−4) and emphasizing upregulation of EMT-related genes in keloid samples (Figure 1b). The HALLMARK IL-6/JAK/STAT pathway gene set revealed a significant NES (NES = 1.98, p = 0.002) (Figure 1c).
Figure 1.

Human keloid tissues show increased expression of WNT5A and EMT markers. (a) Bulk RNA-seq shows upregulation of WNT5A and EMT phenotype markers in keloid samples. GSEA of RNA-seq reveals significantly enriched gene sets in the (b) EMT and (c) IL-6/JAK/STAT3 pathways in keloid samples. Keloid tissues express (d) increased N-cadherin and decreased E-cadherin in the epidermis [scale bars, 100 μm (low-power field, LPF) and 50 μm (high-power field, HPF)]. Expression of (e) vimentin and α-SMA mRNA increases in keloid tissues including the reticular dermis [scale bars, 2 mm (LPF) and 250 μm (HPF)]. Expression of (f) WNT5A is significantly increased in keloid tissues [scale bars, 200 μm (LPF) and 50 μm (HPF)]. The tissue analyses were performed from three different keloid and normal tissues, with three independent experiments performed as technical replicates. *p < 0.05, **p < 0.01, ***p < 0.005, Mann–Whitney U test. EMT epithelial-mesenchymal transition, GSEA gene set enrichment analysis, LPF low-power field, HPF high-power field
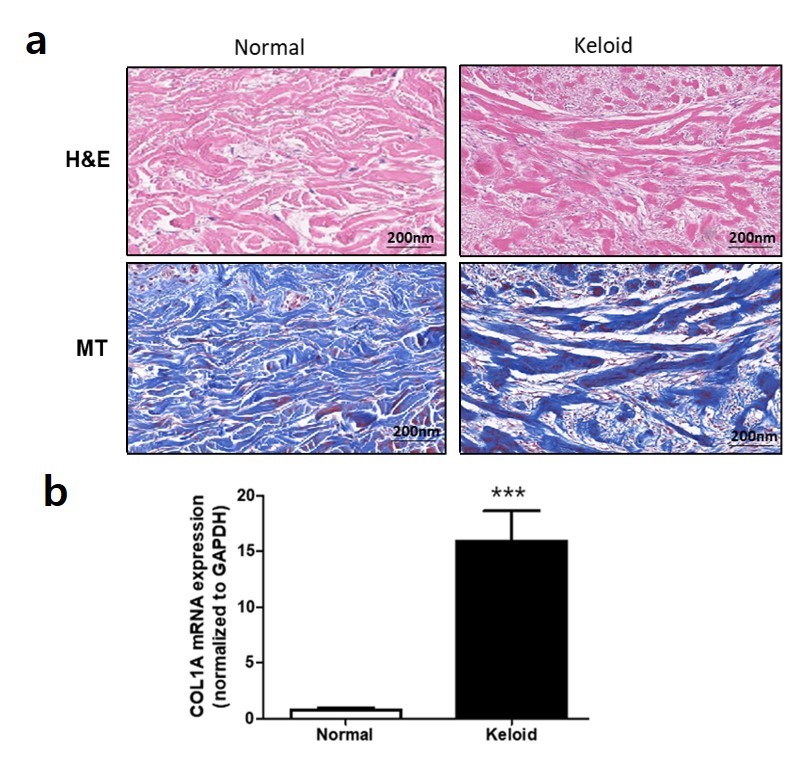
Elevated levels of EMT-like phenotype markers and WNT5A in human keloid tissue
H&E and MT staining of human keloid tissues showed excessive and thick bundles of collagen fibers and increased dermal thickness relative to normal skin (Supplementary Figure 3a, see online supplementary material). Additionally, COL1A1 mRNA level was significantly elevated in keloids (Supplementary Figure 3b). However, IHC staining of N-cadherin revealed significantly higher expression in keloid epidermis, whereas E-cadherin showed decreased expression relative to normal epidermis (p < 0.01) (Figure 1d). IHC staining of vimentin, as well as α-SMA mRNA analysis, revealed significantly increased levels of both in keloids (p < 0.01) (Figure 1e). Furthermore, IHC staining of WNT5A showed significantly upregulated expression in keloid tissues, particularly in the dermis, which agreed with upregulated levels of WNT5A mRNA expression in keloids (p < 0.05, p < 0.01, respectively) (Figure 1f).
A bleomycin-induced skin fibrosis mouse model shows a similar increase in EMT markers and WNT5A expression as observed in human keloid tissue
We generated an animal model for inflammatory skin fibrosis by repeated intradermal injection of bleomycin sulfate into C57BL/6 mice. H&E and MT staining showed sufficient formation of dermal fibrosis at week 3 accompanied by an abnormally thick connective tissue layer and increased collagen (Figure 2a). Additionally, IHC staining showed significantly increased vimentin expression in bleomycin-injected fibrotic skin relative to PBS-injected skin (p < 0.005) (Figure 2b), and qRT-PCR of tissue samples revealed increased mRNA levels of vimentin, slug and N-cadherin (p < 0.005) (Figure 2b, c). These results confirmed that bleomycin-induced dermal fibrosis mice expressed increased EMT markers similar to human keloid tissues. Moreover, we observed significantly upregulated levels of WNT5A mRNA and protein following bleomycin injection, particularly throughout the dermis (p < 0.005) (Figure 2d).
Figure 2.

Replication of a bleomycin (BLM)-induced fibrosis model in C57BL/6 mice. Daily injection of BLM induces (a) significantly increased dermal thickness and collagen [scale bars, 200 μm from hematoxylin and eosin (H&E) staining and Masson’s trichrome staining (MTS)]. Induction of dermal fibrosis via BLM injection for 3 weeks causes significantly increased (b) vimentin and (c) slug and N-cadherin expression. (d) WNT5A expression is significantly increased in the dermis after BLM injection (scale bars, 100 μm from immunohistochemical staining). Data are from three independent experiments of at least six mice per group. *p < 0.05, ***p < 0.01, ***p < 0.005, repeated-measure analysis of variance with Bonferroni’s correction for multiple comparisons. LPF Low-power field, WNT5A Wnt family member 5A, PBS phosphatebuffered saline, N-CAD N-cadherin
KFs express increased WNT5A and pro-inflammatory cytokine levels including IL-6
To determine the skin cell type with the highest differential expression of WNT5A in keloids relative to normal tissues, we performed double-immunofluorescence staining of WNT5A and markers of KCs (cytokeratin 14), endothelial cells (CD31) and fibroblasts (α-SMA). Although the differential expression of WNT5A between normal and keloid epidermis was not significant, WNT5A expression within the dermis was most significantly upregulated in KFs relative to normal tissues (Figure 3a, Supplementary Figure 4 see online supplementary material). Subsequent immunofluorescence staining showed notably higher expression of WNT5A in KFs relative to HDFs (Figure 3b). qRT-PCR revealed significantly increased WNT5A gene expression level of KFs compared to HDFs; ELISA also showed significantly increased secretion level of WNT5A from KFs compared to HDFs (p < 0.05, p < 0.005, respectively) (Figure 3b). The subsequent human cytokine profile array showed that KFs produced elevated levels of multiple pro-inflammatory cytokines, including C–C motif chemokine ligand 2 (CCL2), IL-8, C–X–C chemokine ligand 1 (CXCL1) and IL-6, relative to those in HDFs (Figure 3c).
Figure 3.

KFs express higher levels of WNT5A and pro-inflammatory cytokines relative to normal fibroblasts. (a) Double-immunofluorescence staining of keloid tissues show elevated WNT5A expression in α-SMA-positive cells in the dermis relative to normal tissues (scale bars, 50 μm). In vitro analysis shows (b) increased gene and protein expression of WNT5A in KFs relative to HDFs (scale bars, 20 μm). *p < 0.01, ***p < 0.005; Mann–Whitney U test. Data are from three independent experiments. Human cytokine profile array shows (c) increased CCL-2/MCP-1, IL-6, IL-8 and CXCL-1 production from KFs relative to HDFs. KF keloid fibroblasts, HDF human dermal fibroblasts, α-SMA α-smooth muscle actin, CCL2 C–C motif chemokine ligand 2, CXCL-1 C–X–C chemokine ligand 1, IL interleukin, WNT5A Wnt family member 5A
IL-6 secreted by WNT5A-stimulated fibroblasts can increase EMT-like phenotype markers in co-cultured KCs via the JAK/STAT pathway
The ELISA revealed significantly increased secretion of IL-6 from HDFs treated with WNT5A for 24 and 48 h (p < 0.01) (Figure 4a). To evaluate the role of IL-6 in EMT, we cultured KCs with or without recombinant IL-6. As the result, Western blot analysis revealed that IL-6 treatment for 24 h increased p-STAT, N-cadherin and slug levels in KCs, whereas E-cadherin level was decreased (Figure 4b). The gene expression levels in KCs treated with IL-6 for 24 h also showed a significant reduction in E-cadherin level (p < 0.005), whereas N-cadherin and vimentin levels were significantly elevated following IL-6 treatment for 24 h (p < 0.005) (Figure 4c).
Figure 4.

Production of IL-6 from WNT5A-activated fibroblasts could indirectly cause KCs to undergo EMT. (a) HDFs treated with WNT5A for 6, 12, 24 or 48 h produce significantly increased level of IL-6. (b) Western blot analysis of KCs treated with IL-6 for 24 h confirms increased expression of p-STAT3, STAT3, N-cadherin and slug and decreased expression of E-cadherin. (c) Treatment of KCs with IL-6 decreases mRNA levels of E-cadherin and increases those of N-cadherin and vimentin after 24 h. Data are from three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.005; Mann–Whitney U test. EMT Epithelial–mesenchymal transition, IL interleukin, KF keloid fibroblasts, HDF human dermal fibroblasts, KC keratinocytes
To determine the role of WNT5A in keloid formation, we designed a co-culture system of KCs and HDFs (Figure 5a). KCs co-cultured with HDFs expressed significantly higher levels of EMT-like phenotype markers (vimentin, slug and N-cadherin) when treated with WNT5A for 24 h, whereas KCs cultured without HDFs did not respond significantly to WNT5A stimulation (p < 0.01) (Figure 5b). On the other hand, patient-derived KKs from two different keloid tissues showed significantly elevated gene expression levels of vimentin compared to the normal KCs (p < 0.005) (Figure 5c). Additional western blot analysis revealed significantly increased levels of p-STAT3/T-STAT3 ratio, vimentin and slug, and decreased E-cadherin levels in KCs co-cultured with WNT5A-activated HDFs (p < 0.05, p < 0.01, respectively) (Figure 5d). Notably, KCs did not express p-STAT3 with or without stimulation with WNT5A when cultured without HDFs.
Figure 5.
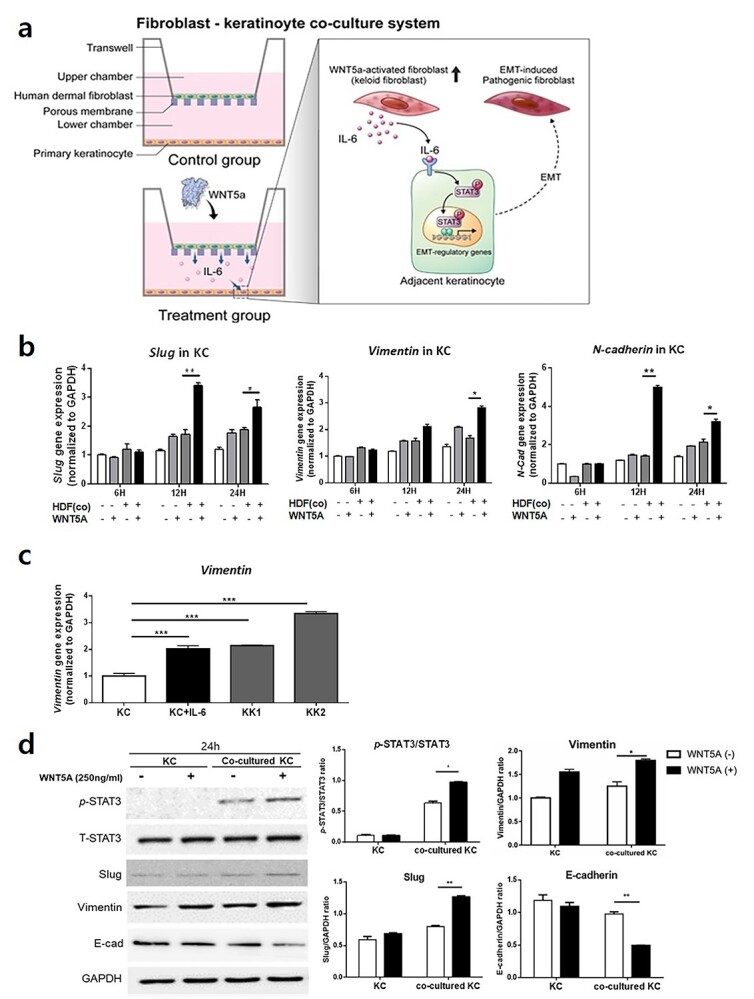
KCs express elevated levels of EMT markers during co-culture with WNT5A-activated HDFs or IL-6 stimulation. (a) Illustration of the co-culture system involving KCs and HDFs. (b) Increased levels of slug, vimentin and N-cadherin following co-culture of KCs with WNT5A-activated HDFs. (c) IL-6-stimulated KCs as well as patient-derived keloid keratinocytes from two different keloid tissues (KK1, KK2) show elevated gene expression levels of vimentin compared to the normal keratinocytes. (d) Western blot analysis of KCs co-cultured for 24 h with WNT5A-treated HDFs shows significantly increased levels of p-STAT3, slug and vimentin and decreased level of E-cadherin. Data are from three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.005; Mann–Whitney U test. KC keratinocytes, KK keloid keratinocytes, HDF human dermal fibroblasts, EMT epithelial-mesenchymal transitio, WNT5A Wnt family member 5A, IL interleukin
WNT5A knockdown decreases IL-6 production by KFs and reduces levels of STAT pathway as well as EMT phenotype markers in co-cultured KCs
We silenced WNT5A to confirm its role in excessive production of IL-6 in KFs and subsequent EMT activation by adjacent KCs. Indeed, KFs secreted significantly higher levels of IL-6 relative to HDFs (p < 0.005); following siWNT5A transfection, KFs showed significantly reduced IL-6 production (p < 0.01) (Figure 6a). qRT-PCR analysis of KCs co-cultured with fibroblasts revealed that KCs co-cultured with WNT5A-silenced KFs expressed significantly reduced levels of EMT phenotype markers (α-SMA, vimentin and N-cadherin) relative to KCs co-cultured with KFs treated with control siRNA (p < 0.005) (Figure 6b–d). Western blot analysis of KCs also showed significantly decreased levels of p-STAT3/T-STAT3, α-SMA, N-cadherin and vimentin when co-cultured with WNT5A-knockdown KFs compared to control siRNA-treated KFs (p < 0.005) (Figure 6e).
Figure 6.

Effect of WNT5A silencing on IL-6 production and levels of EMT markers. (a) Control KFs produce significantly increased levels of IL-6 compared to control HDFs, while KFs treated with siWNT5A produce significantly decreased levels of IL-6 compared to siWNT5A-treated KFs. mRNA levels of (b) α-SMA, (c) vimentin, and (d) N-cadherin and (e) Western blot analysis of KCs co-cultured with HDFs, KFs, KFs treated with control siRNA and KFs treated with siWNT5A. (f) Western blot analysis of KCs co-cultured with KFs treated with or without IL-6 neutralizing antibody (αIL-6). Data are from three independent experiments. *p < 0.05, ***p < 0.05, ***p < 0.005; Mann–Whitney U test. HDF human dermal fibroblasts, KF keloid fibroblasts, KC keratinocytes, EMT epithelial–mesenchymal transition, IL interleukin, α-SMA α-smooth muscle actin
In order to confirm the role of IL-6 in initiating the EMT phenomenon in KCs via the JAK/STAT pathway, we performed 24 h IL-6 blocking treatment on co-cultured KFs (1 × 105) and KCs (2 × 105) using 100 ng/ml neutralizing monoclonal antibody against IL-6. Western blot analysis showed significantly decreased levels of p-STAT3/T-STAT3 and vimentin in KCs co-cultured with IL-6 neutralizing antibody (p < 0.05) (Figure 6f). KCs cultured without fibroblasts did not express p-STAT, similar to our previous experiment with HDFs with or without WNT5A stimulation.
STAT3 silencing decreases levels of EMT phenotype markers in KCs cultured with or without IL-6 stimulation
We further silenced STAT3 to confirm the mechanism involving the JAK/STAT pathway in KCs undergoing EMT-like phenotype changes due to IL-6 stimulation. Compared to the control, KCs (2 × 105) following siSTAT3 transfection for 24 h showed significantly reduced STAT3 levels (p < 0.005); stimulation of KCs with 50 ng/mL human recombinant IL-6 significantly increased the expression level of STAT3 in KCs compared to the control, but it again decreased significantly upon siSTAT3 transfection (p < 0.005) (Figure 7a). The subsequent analyses of EMT markers from KCs transfected with siSTAT3 revealed significantly decreased gene expression levels of slug, α-SMA and N-cadherin compared to the control; these EMT phenotype markers again decreased when treated with siSTAT3 after stimulation with IL-6 (p < 0.005) (Figure 7b–d).
Figure 7.

STAT3 silencing decreases the level of EMT phenotype markers in KCs. Gene expression levels of (a) STAT3, (b) α-SMA, (c) slug and (d) N-cadherin after silencing of STAT3 in KCs reveals decreased expression of each gene compared to the control group. The increased STAT3, a-SMA, slug and N-cadherin levels after stimulation with 50 ng/ml IL-6 again decreased significantly when treated with siSTAT3. Data are from three independent experiments. *p < 0.05, ***p < 0.005; Mann–Whitney U test. KC Keratinocytes, EMT epithelial–mesenchymal transition, α-SMA α-smooth muscle actin
Discussion
Although the molecular drivers of keloid pathogenesis remain unclear, in various fibrotic diseases, such as keloid scarring, disease-specific triggers initiate local site inflammation, which activates a distinct cell population that drives fibrosis in susceptible individuals [31]. Here, we analyzed DEGs using next-generation RNA-seq of keloid and normal tissues to identify genes contributing to keloid formation and aggravation. Notably, GSEA from tissue RNA-seq revealed that upregulated genes in keloid tissues were most highly enriched in the pathway of EMT gene signatures. Additionally, representative genes related to EMT, including vimentin, slug, N-cadherin, and α-SMA and WNT5A were simultaneously upregulated in keloid tissues.
Keloids only occur in humans, making it difficult to study disease pathogenesis [32]. Currently available potential keloid animal models involve keloid reproduction either by chemical-induced abnormal fibrosis or implantation of human keloid tissues [33]. In the present study, we adopted an in vivo skin fibrosis animal model to reproduce keloid inflammatory conditions by intradermal bleomycin injection in C57BL/6 mice, as previously described [34]. Bleomycin triggers the production of reactive oxygen species, which can damage surrounding cells, ultimately recruiting inflammatory cells and causing aberrant activation of resident fibroblasts [35]. In the literature, a study on targeting IL-6 in a bleomycin-induced dermal fibrosis mouse model via the introduction of monoclonal anti-IL-6R antibody has shown decreased dermal thickness as well as hydroxyproline content [36]. Another in vivo study on the intradermal bleomycin-induced dermal fibrosis mouse model revealed that the administration of a JAK inhibitor, Tofacitinib, significantly alleviated fibrosis of the skin; it also suppressed IL-6-producing effector B cells and gene expression levels of extracellular proteins and fibrogenic cytokines [37]. A study on idiopathic pulmonary fibrosis generated a mouse model of bleomycin-induced lung fibrosis and demonstrated that WNT5A knock-out in airway smooth muscle leads to an improved clinical phenotype [38]. In this study, our intradermal bleomycin-induced dermal fibrosis model showed significantly increased expression of IL-6, EMT markers and WNT5A, consistent with results from human keloid tissue. These results suggest its successful reproduction of the inflammatory fibrosis condition of keloids and the possible presence of the WNT5A/IL-6/JAK/STAT pathway in the bleomycin-induced dermal fibrosis model, further indicating a potential therapeutic outcome via blocking the pathway.
We observed that WNT5A transcription was highly upregulated in keloid tissues. WNT5A activates non-canonical Wnt pathways, and its overexpression is observed in tumorigenic progression by promoting cell motility, EMT and metastasis [39]. The correlation of WNT5A with increased EMT in cancer development has been reported previously [40–42]. Additionally, increasing evidence indicates a central role for Wnt signaling in driving fibrotic responses, including liver, kidney and lung fibrosis [43–45]. Previous studies have also suggested a relationship between WNT5A and the fibrogenic factor TGF-β, necessitating further studies of the functional role of WNT5A in fibrotic diseases [21,46,47]. Here, the expression of WNT5A and EMT markers as well as activation of IL-6/JAK/STAT3 signaling were notably upregulated in keloid scars. Moreover, KFs expressed higher levels of WNT5A relative to HDFs.
The role of WNT5A in stimulating the production of pro-inflammatory cytokines, which drive tumor invasion, has gained increased attention in the literature. Cytokines upregulated by WNT5A include IL-1, CXCL8 (IL-8), CCL2 (monocyte chemoattractant protein-1; MCP-1) and IL-6 [39], of which IL-6 overexpression by tumor-associated fibroblasts was reported in invasive skin lesions, such as melanoma [48,49]. We also observed increased expression of CCL-2/MCP-1, CXCL8 and IL-6 in media cultured with KFs relative to that in media cultured with HDFs. Furthermore, IL-6 and one of its downstream mediators, the JAK/STAT3 pathway, are known to be aberrantly hyperactivated in several cancer types and drive tumor cell proliferation, invasiveness and EMT [50,51]. Gao et al. [52] also demonstrated that treatment with IL-6 induces EMT through the JAK/STAT3 pathway in hepatocellular carcinoma. In the present study, activation of the JAK/STAT3 pathway and subsequent elevation of EMT-like phenotype markers were observed in KCs co-cultured with WNT5A-activated fibroblasts or KFs. In addition, both WNT5A silencing and IL-6 neutralizing antibody treatment in KFs decreased IL-6 production and suppressed levels of EMT markers in co-cultured KCs. Furthermore, KKs as well as IL-6-stimulated KCs showed increased expressions of EMT-phenotype markers compared to normal KCs. These results might indicate a partial role for WNT5A in activating fibroblasts, which abnormally increase the secretion of IL-6 to adjacent KCs to promote EMT-like phenomena. Our subsequent siSTAT3 transfection assay showed significantly reduced expression levels of EMT markers on KCs treated with IL-6 when STAT3 gene expression was silenced. Hence, these results propose a possible mechanism involving WNT5A stimulating the production of IL-6 in KFs, and the successive activation of the JAK/STAT3 pathway in IL-6-stimulated KCs expressing EMT-like phenotype markers in keloid scars.
Our study has several limitations. First, although WNT5A in the skin is known to be primarily expressed in fibroblasts, it is also expressed in Langerhans cells, endothelial cells, basal KCs and T-cells [22,53]. Although our immunofluorescence study revealed that the difference in the degree of WNT5A expression between normal and keloid tissues differed most prominently in dermal fibroblasts, further WNT5A expression studies on various other types of patient-derived cells would be informative. Secondly, although the need for an in vivo animal model of a keloid scar is obvious, keloids are known to occur exclusively in humans [33]. No other animal species has been found to naturally develop scar tissue that can be compared to that of human keloids, making keloid research difficult. Nonetheless, there have been several attempts to understand the pathophysiology of keloid scars via the application of alternative in vivo animal models for studying abnormal tissue fibrosis. For instance, the bleomycin-induced murine dermal fibrosis model, which is a well-known animal model for scleroderma, had shown advantages for studying early inflammatory components of fibrosis [54]. As seen in many fibrosis cases, including keloids, there is an inflammatory component that drives fibrosis in early scar development [55], and the bleomycin-induced model for skin fibrosis has been previously utilized to recreate abnormal scar formation and wound healing [54,56–58]. We adapted the scleroderma animal model to understand the pro-inflammatory process between fibroblasts and adjacent epidermal KCs in keloid scars, which in turn display EMT-like phenotypes in gene expression. As the bleomycin animal model does not fully encounter several other factors of abnormal wound healing, including hypoxia, genetic susceptibility and trauma, further research on developing an improved inherent keloid animal model is necessary [54]. Lastly, the proposed mechanism of the intercellular communication via the WNT5A and IL-6/JAK/STAT pathways between KFs and KKs might only be a partial story in promoting EMT. Further in vitro studies on the interaction between various other types of cells in keloid dermis and epidermis in both mono- and co-culture systems also need to be performed for a complete understanding of the mechanism involving EMT-like phenomena in keloid scars.
Conclusions
To our knowledge, this is the first description of the use of RNA-seq in human keloid tissue to describe keloid pathogenesis in a Korean population. Because the need for effective anti-fibrotic therapies remains high, understanding keloid pathogenesis and developing targeted therapies is necessary. This study identified the gene responsible for keloid pathogenesis (WNT5A) and its possible role in assisting aberrant activation of dermal fibroblasts and EMT-like phenotype changes in adjacent KCs via the IL-6/JAK/STAT3 pathway. Further investigations are warranted to evaluate its potential role as a keloid biomarker and develop new treatment strategies.
Authors’ contributions
Conceptualization: YIL and JHL; data curation: JES and KHN; methodology: YIL and JES; formal analysis: YIL, JK, and JWK; funding acquisition: YIL and JHL; investigation: YIL, WJL and JHL; writing–original draft: YIL; writing–review and editing: YIL and JHL; supervision: JHL. All author reviewed and approved the final version of the manuscript for publication.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
The authors thank Medical Illustration & Design, part of the Medical Research Support Services of Yonsei University College of Medicine, for the artistic support related to this work. We also thank Bioinformatics Collaboration Unit (BiCU) at the Department of Biomedical Systems Informatics, Yonsei University College of Medicine, for RNA-seq and downstream data analysis.
Contributor Information
Young In Lee, Department of Dermatology, Severance Hospital, Cutaneous Biology Research Institute, Yonsei University College of Medicine, Seoul 03722, Korea; Scar Laser and Plastic Surgery Center, Yonsei Cancer Hospital, Yonsei University College of Medicine, Seoul 03722, Korea.
Jung Eun Shim, Bioinformatics Collaboration Unit, Yonsei University College of Medicine, Seoul 03722, Korea.
Jihee Kim, Department of Dermatology, Severance Hospital, Cutaneous Biology Research Institute, Yonsei University College of Medicine, Seoul 03722, Korea; Scar Laser and Plastic Surgery Center, Yonsei Cancer Hospital, Yonsei University College of Medicine, Seoul 03722, Korea.
Won Jai Lee, Department of Plastic and Reconstructive Surgery, Severance Hospital, Yonsei University College of Medicine, Seoul 03722, Korea; Scar Laser and Plastic Surgery Center, Yonsei Cancer Hospital, Yonsei University College of Medicine, Seoul 03722, Korea.
Jae Woo Kim, Department of Biochemistry and Molecular Biology, Chronic Intractable Disease for Systems Medicine Research Center, Yonsei University College of Medicine, Seoul 03722, Korea.
Kee Hyun Nam, Department of Surgery, Severance Hospital, Yonsei University College of Medicine, Seoul, Korea.
Ju Hee Lee, Department of Dermatology, Severance Hospital, Cutaneous Biology Research Institute, Yonsei University College of Medicine, Seoul 03722, Korea; Scar Laser and Plastic Surgery Center, Yonsei Cancer Hospital, Yonsei University College of Medicine, Seoul 03722, Korea.
Abbreviations
N-Cadherin: Neural cadherin; CCL2: C–C motif chemokine ligand 2; COL1A: Collagen type 1 α1; CXCL1: C–X–C chemokine ligand 1; DEG: Differentially expressed gene; ECM: Extracellular matrix; ELISA: Enzyme-linked immunosorbent assay; EMT: Epithelial–mesenchymal transition; GAPDH: Glyceraldehyde 3-phosphate dehydrogenase; GSEA: Gene set enrichment analysis; H&E: Hematoxylin and eosin; HDF: Human dermal fibroblasts; IHC: Immunohistochemical; KC: Keratinocytes; KK: Keloid keratinocytes; IL: Interleukin; JAK: Janus kinase; KF: Keloid fibroblasts; MT: Masson’s trichrome; NES: Normalized enrichment score; PBS: Phosphate-buffered saline; α-SMA: α-Smooth muscle actin; STAT: Signal transducer and activator of transcription; pSTAT3: Phosphorylated STAT3; TGF: Transforming growth factor; WNT5A: Wnt family member 5A.
Funding
This research was supported by the Health Fellowship Foundation, Seoul, Korea.
Ethics approval and consent to participate
The experiments involving human participants were reviewed and approved by approved by the appropriate Review Board (IRB No. 4–2017-0259), Yonsei University College of Medicine. The patients provided their written informed consents to participate in this study.
Consent for publication
All presentations of case reports in this manuscript have consent for publication.
Data availability
The original contributions presented in the study are publicly available. The data can be found at https://www.ncbi.nlm.nih.gov/geo with accession number GSE173900.
Competing interests
None declared.
References
- 1. Wesseling M, Sakkers TR, de Jager SCA, Pasterkamp G, Goumans MJ. The morphological and molecular mechanisms of epithelial/endothelial-to-mesenchymal transition and its involvement in atherosclerosis. Vasc Pharmacol. 2018;106:1–8. [DOI] [PubMed] [Google Scholar]
- 2. Kang Y, Roh MR, Rajadurai S, Rajadurai A, Kumar R, Njauw CN, et al. Hypoxia and HIF-1alpha Regulate Collagen Production in Keloids. J Invest Dermatol. 2020;140:2157–65. [DOI] [PubMed] [Google Scholar]
- 3. Tan S, Khumalo N, Bayat A. Understanding Keloid Pathobiology From a Quasi-Neoplastic Perspective: Less of a Scar and More of a Chronic Inflammatory Disease With Cancer-Like Tendencies. Front Immunol. 2019;10:1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hahn JM, McFarland KL, Combs KA, Supp DM. Partial epithelial-mesenchymal transition in keloid scars: regulation of keloid keratinocyte gene expression by transforming growth factor-beta1. Burns Trauma. 2016;4:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yan L, Cao R, Wang L, Liu Y, Pan B, Yin Y, et al. Epithelial-mesenchymal transition in keloid tissues and TGF-beta1-induced hair follicle outer root sheath keratinocytes. Wound Repair Regen. 2015;23:601–10. [DOI] [PubMed] [Google Scholar]
- 6. Yuan FL, Sun ZL, Feng Y, Liu SY, Du Y, Yu S, et al. Epithelial-mesenchymal transition in the formation of hypertrophic scars and keloids. J Cell Physiol. 2019;234:21662–9. [DOI] [PubMed] [Google Scholar]
- 7. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Satish L, Evdokiou A, Geletu E, Hahn JM, Supp DM. Pirfenidone inhibits epithelial-mesenchymal transition in keloid keratinocytes. Burns Trauma. 2020;8:tkz007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15:178–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yang CE, Moon SJ, Kim SJ, Lee JH, Yun CO, Lew DH, et al. Epithelial-mesenchymal transition in keloid tissue. Arch Plast Surg. 2018;45:600–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nakamura M, Tokura Y. Epithelial-mesenchymal transition in the skin. J Dermatol Sci. 2011;61:7–13. [DOI] [PubMed] [Google Scholar]
- 12. Kuwahara H, Tosa M, Egawa S, Murakami M, Mohammad G, Ogawa R. Examination of Epithelial Mesenchymal Transition in Keloid Tissues and Possibility of Keloid Therapy Target. Plast Reconstr Surg Glob Open. 2016;4:e1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang Q, Yamaza T, Kelly AP, Shi S, Wang S, Brown J, et al. Tumor-like stem cells derived from human keloid are governed by the inflammatory niche driven by IL-17/IL-6 axis. PLoS One. 2009;4:e7798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bussard KM, Mutkus L, Stumpf K, Gomez-Manzano C, Marini FC. Tumor-associated stromal cells as key contributors to the tumor microenvironment. Breast Cancer Res. 2016;18:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lim CP, Phan TT, Lim IJ, Cao X. Stat3 contributes to keloid pathogenesis via promoting collagen production, cell proliferation and migration. Oncogene. 2006;25:5416–25. [DOI] [PubMed] [Google Scholar]
- 16. Chang Q, Bournazou E, Sansone P, Berishaj M, Gao SP, Daly L, et al. The IL-6/JAK/Stat3 feed-forward loop drives tumorigenesis and metastasis. Neoplasia. 2013;15:848–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xiao J, Gong Y, Chen Y, Yu D, Wang X, Zhang X, et al. IL-6 promotes epithelial-to-mesenchymal transition of human peritoneal mesothelial cells possibly through the JAK2/STAT3 signaling pathway. Am J Physiol Renal Physiol. 2017;313:F310–8. [DOI] [PubMed] [Google Scholar]
- 18. Lee YS, Liang YC, Wu P, Kulber DA, Tanabe K, Chuong CM, et al. STAT3 signalling pathway is implicated in keloid pathogenesis by preliminary transcriptome and open chromatin analyses. Exp Dermatol. 2019;28:480–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dong S, Wu C, Hu J, Wang Q, Chen S, Wang Z, et al. Wnt5a Promotes Cytokines Production and Cell Proliferation in Human Hepatic Stellate Cells Independent of Canonical Wnt Pathway. Clin Lab. 2015;61:537–47. [DOI] [PubMed] [Google Scholar]
- 20. Rashid ST, Humphries JD, Byron A, Dhar A, Askari JA, Selley JN, et al. Proteomic analysis of extracellular matrix from the hepatic stellate cell line LX-2 identifies CYR61 and Wnt-5a as novel constituents of fibrotic liver. J Proteome Res. 2012;11:4052–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Beljaars L, Daliri S, Dijkhuizen C, Poelstra K, Gosens R. WNT-5A regulates TGF-beta-related activities in liver fibrosis. Am J Physiol Gastrointest Liver Physiol. 2017;312:G219–27. [DOI] [PubMed] [Google Scholar]
- 22. Igota S, Tosa M, Murakami M, Egawa S, Shimizu H, Hyakusoku H, et al. Identification and characterization of Wnt signaling pathway in keloid pathogenesis. Int J Med Sci. 2013;10:344–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fei T, Yu T. scBatch: batch-effect correction of RNA-seq data through sample distance matrix adjustment. Bioinformatics. 2020;36:3115–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol. 2019;37:907–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang L, Wang S, Li W. RSeQC: quality control of RNA-seq experiments. Bioinformatics. 2012;28:2184–5. [DOI] [PubMed] [Google Scholar]
- 27. Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33:290–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tirosh I, Izar B, Prakadan SM, Wadsworth MH, 2nd, Treacy D, Trombetta JJ, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352:189–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lim IJ, Phan TT, Song C, Tan WT, Longaker MT. Investigation of the influence of keloid-derived keratinocytes on fibroblast growth and proliferation in vitro. Plast Reconstr Surg. 2001;107:797–808. [DOI] [PubMed] [Google Scholar]
- 30. Sekiguchi A, Motegi SI, Fujiwara C, Yamazaki S, Inoue Y, Uchiyama A, et al. Inhibitory effect of kaempferol on skin fibrosis in systemic sclerosis by the suppression of oxidative stress. J Dermatol Sci. 2019;96:8–17. [DOI] [PubMed] [Google Scholar]
- 31. Distler JHW, Gyorfi AH, Ramanujam M, Whitfield ML, Konigshoff M, Lafyatis R. Shared and distinct mechanisms of fibrosis. Nat Rev Rheumatol. 2019;15:705–30. [DOI] [PubMed] [Google Scholar]
- 32. Sharma JR, Lebeko M, Kidzeru EB, Khumalo NP, Bayat A. In Vitro and Ex Vivo Models for Functional Testing of Therapeutic Anti-scarring Drug Targets in Keloids. Adv Wound Care (New Rochelle). 2019;8:655–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Limandjaja GC, Niessen FB, Scheper RJ, Gibbs S. The Keloid Disorder: Heterogeneity, Histopathology, Mechanisms and Models. Front Cell Dev Biol. 2020;8:360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Do NN, Eming SA. Skin fibrosis: Models and mechanisms. Curr Res Transl Med. 2016;64:185–93. [DOI] [PubMed] [Google Scholar]
- 35. Batteux F, Kavian N, Servettaz A. New insights on chemically induced animal models of systemic sclerosis. Curr Opin Rheumatol. 2011;23:511–8. [DOI] [PubMed] [Google Scholar]
- 36. Desallais L, Avouac J, Frechet M, Elhai M, Ratsimandresy R, Montes M, et al. Targeting IL-6 by both passive or active immunization strategies prevents bleomycin-induced skin fibrosis. Arthritis Res Ther. 2014;16:R157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Aung WW, Wang C, Xibei J, Horii M, Mizumaki K, Kano M, et al. Immunomodulating role of the JAKs inhibitor tofacitinib in a mouse model of bleomycin-induced scleroderma. J Dermatol Sci. 2021;101:174–84. [DOI] [PubMed] [Google Scholar]
- 38. Carmo-Fernandes A, Puschkarow M, Peters K, Gnipp S, Peters M. The Pathogenic Role of Smooth Muscle Cell-Derived Wnt5a in a Murine Model of Lung Fibrosis. Pharmaceuticals (Basel). 2021;14:755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lopez-Bergami P, Barbero G. The emerging role of Wnt5a in the promotion of a pro-inflammatory and immunosuppressive tumor microenvironment. Cancer Metastasis Rev. 2020;39:933–52. [DOI] [PubMed] [Google Scholar]
- 40. Wang B, Tang Z, Gong H, Zhu L, Liu X. Wnt5a promotes epithelial-to-mesenchymal transition and metastasis in non-small-cell lung cancer. Biosci Rep. 2017;37:BSR20171092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Qi H, Sun B, Zhao X, Du J, Gu Q, Liu Y, et al. Wnt5a promotes vasculogenic mimicry and epithelial-mesenchymal transition via protein kinase Calpha in epithelial ovarian cancer. Oncol Rep. 2014;32:771–9. [DOI] [PubMed] [Google Scholar]
- 42. Bo H, Zhang S, Gao L, Chen Y, Zhang J, Chang X, et al. Upregulation of Wnt5a promotes epithelial-to-mesenchymal transition and metastasis of pancreatic cancer cells. BMC Cancer. 2013;13:496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jiang F, Parsons CJ, Stefanovic B. Gene expression profile of quiescent and activated rat hepatic stellate cells implicates Wnt signaling pathway in activation. J Hepatol. 2006;45:401–9. [DOI] [PubMed] [Google Scholar]
- 44. Li X, Yamagata K, Nishita M, Endo M, Arfian N, Rikitake Y, et al. Activation of Wnt5a-Ror2 signaling associated with epithelial-to-mesenchymal transition of tubular epithelial cells during renal fibrosis. Genes Cells. 2013;18:608–19. [DOI] [PubMed] [Google Scholar]
- 45. Vuga LJ, Ben-Yehudah A, Kovkarova-Naumovski E, Oriss T, Gibson KF, Feghali-Bostwick C, et al. WNT5A is a regulator of fibroblast proliferation and resistance to apoptosis. Am J Respir Cell Mol Biol. 2009;41:583–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kumawat K, Menzen MH, Slegtenhorst RM, Halayko AJ, Schmidt M, Gosens R. TGF-beta-activated kinase 1 (TAK1) signaling regulates TGF-beta-induced WNT-5A expression in airway smooth muscle cells via Sp1 and beta-catenin. PLoS One. 2014;9:e94801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kumawat K, Menzen MH, Bos IS, Baarsma HA, Borger P, Roth M, et al. Noncanonical WNT-5A signaling regulates TGF-beta-induced extracellular matrix production by airway smooth muscle cells. FASEB J. 2013;27:1631–43. [DOI] [PubMed] [Google Scholar]
- 48. Tsukamoto H, Fujieda K, Hirayama M, Ikeda T, Yuno A, Matsumura K, et al. Soluble IL6R Expressed by Myeloid Cells Reduces Tumor-Specific Th1 Differentiation and Drives Tumor Progression. Cancer Res. 2017;77:2279–91. [DOI] [PubMed] [Google Scholar]
- 49. Hoejberg L, Bastholt L, Schmidt H. Interleukin-6 and melanoma. Melanoma Res. 2012;22:327–33. [DOI] [PubMed] [Google Scholar]
- 50. Johnson DE, O'Keefe RA, Grandis JR. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat Rev Clin Oncol. 2018;15:234–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sullivan NJ, Sasser AK, Axel AE, Vesuna F, Raman V, Ramirez N, et al. Interleukin-6 induces an epithelial-mesenchymal transition phenotype in human breast cancer cells. Oncogene. 2009;28:2940–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gao Y, Li W, Liu R, Guo Q, Li J, Bao Y, et al. Norcantharidin inhibits IL-6-induced epithelialmesenchymal transition via the JAK2/STAT3/TWIST signaling pathway in hepatocellular carcinoma cells. Oncol Rep. 2017;38:1224–32. [DOI] [PubMed] [Google Scholar]
- 53. Sole-Boldo L, Raddatz G, Schutz S, Mallm JP, Rippe K, Lonsdorf AS, et al. Single-cell transcriptomes of the human skin reveal age-related loss of fibroblast priming. Commun Biol. 2020;3:188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Marttala J, Andrews JP, Rosenbloom J, Uitto J. Keloids: Animal models and pathologic equivalents to study tissue fibrosis. Matrix Biol. 2016;51:47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bagabir R, Byers RJ, Chaudhry IH, Muller W, Paus R, Bayat A. Site-specific immunophenotyping of keloid disease demonstrates immune upregulation and the presence of lymphoid aggregates. Br J Dermatol. 2012;167:1053–66. [DOI] [PubMed] [Google Scholar]
- 56. Cameron AM, Adams DH, Greenwood JE, Anderson PJ, Cowin AJ. A novel murine model of hypertrophic scarring using subcutaneous infusion of bleomycin. Plast Reconstr Surg. 2014;133:69–78. [DOI] [PubMed] [Google Scholar]
- 57. Padmanabhan J, Maan ZN, Kwon SH, Kosaraju R, Bonham CA, Gurtner GC. In Vivo Models for the Study of Fibrosis. Adv Wound Care (New Rochelle). 2019;8:645–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Seo BF, Lee JY, Jung SN. Models of abnormal scarring. Biomed Res Int. 2013;2013:423147. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The original contributions presented in the study are publicly available. The data can be found at https://www.ncbi.nlm.nih.gov/geo with accession number GSE173900.