

Spinal meningiomas (SM) comprise 5–10% of primary meningiomas and up to 30% of spinal intradural tumors. SMs are usually sporadic, but rarely, they can develop in association with genetic diseases like neurofibromatosis type 2 or schwannomatosis [2, 4, 6]. While the mutational landscape of intracranial meningiomas has been extensively studied [3, 5, 11, 14], our understanding of the molecular profile of SM remains incomplete. To date, genomic studies in SMs have been underpowered to make significant conclusions about the correlations between main genomic driver alterations and clinical features of these tumors. Here, we sought to assess the mutational profile of WHO grade 1 SM and to investigate the clinical characteristics that correlate with the genomic status.
Targeted next-generation sequencing was performed using assays covering frequently mutated genes in meningiomas as previously described [8] and online resource. Moreover, we correlated clinical and imaging data with the molecular tumor status.
Our study cohort consisted of 50 patients with newly diagnosed SM WHO grade 1 (Table 1). Thirty-eight patients were female and 12 were male (female:male ratio of 3.2:1). The median age at diagnosis was 66 years (range 28–84 years). Cohort patients were included if they had suspected sporadic meningioma based on lack of family history of neurofibromatosis type 2, schwannomatosis, and/or lack of other meningiomas or CNS tumors. The mean follow-up time of our SM cohort was 60 months (range 6–288 months).
Table 1.
Comparison between patient’s demographics and tumor features of AKT1- and NF2-mutant spinal meningiomas
| All tumors (n = 50) |
AKT1-mutant (n = 15) |
NF2-mutant (n = 32) |
P value | |
|---|---|---|---|---|
| Median age (years) | 66 | 71 | 65 | 0.032 |
| Female sex | 38 | 7/15 (46.6%) | 30/32 (94%) | 0.0006 |
| Thoracic spine location | 28 | 4/15 (26.6%) | 24/32 (75%) | 0.0034 |
| Cervical spine location | 21 | 11/15 (73.3%) | 7/32 (21.8%) | 0.0012 |
| Ventral or ventro-lateral location to spinal cord | 28 | 13/15 (87%) | 14/32 (40.6%) | 0.010 |
| Dorsal or dorso-lateral location to spinal cord | 22 | 2/15 (13.3%) | 19/32 (59.3%) | 0.0043 |
| Meningothelial histology | 24 | 14/15 (93.3%) | 7/32 (21.8%) | 0.0001 |
| Tumor calcification | 17 | None | 17/32 (53.2%) | 0.0002 |
Bold indicates significance (P < 0.05)
Two predominant recurrent mutations were observed: AKT1E17K mutations were detected in 15 (30%, 7 females and 8 males) and NF2 mutations in 32 (64%, 30 females and 2 males) patients. Both mutations were mutually exclusive. In three cases (6%, one female and two males), no known driver mutation was found. All detected AKT1E17K mutations were confirmed using Sanger sequencing, as described above. Meningiomas with an AKT1 E17K mutation harbored additionally ATRX (n = 2), ARID1A (n = 2), TRAF7 (n = 1) and POLR2A (n = 1). Co-mutations in NF2-mutant meningiomas included SMARCB1 (n = 3) and PTEN (n = 1) mutations (Supplementary Tables 1 and 2, online resource).
Upon examination of the clinical features of patient harboring these two mutations, distinct cohorts emerged. The median age of patients with AKT1E17K mutations (71 years old) was significantly higher when compared with patients with NF2-mutant meningiomas (65.5 years-old, p = 0.032). Notably, NF2-mutant meningiomas had a near-complete female predominance (n = 30/32, 94%) when compared to the balanced female–male incidence of AKT1-mutant tumors (n = 7/15, p = 0.0006, Table 1). A tumor location in the thoracic spine was significantly more common in NF2-mutant meningiomas (75%) than in their AKT1-mutant counterparts (26.6%) (p = 0.0034). In contrast, meningiomas harboring an AKT1 mutation were predominantly located in the cervical spine (73.3%) and 87% of AKT1-mutant meningiomas (n = 13) (compared to 43.75% of NF2-mutant meningiomas, n = 14) arose ventrally or ventro-laterally to the spinal cord (p = 0.010). In contrast to AKT1-mutant meningiomas, a substantial proportion of NF2-mutant meningiomas developed in the dorsal or dorso-lateral location to the spinal cord (59.3%, p = 0.0043, Fig. 1 and Supplementary Fig. 1, online resource). Consistent with intracranial meningiomas, the histologic subtype of NF2-mutant meningiomas was variable (7 meningothelial, 16 psammomatous, 4 transitional, 5 fibrous), while all but 1 AKT1-mutant meningioma showed a meningothelial histology (93.3%, p = 0.0001). None of the AKT1-mutant meningiomas showed calcifications in the preoperative MRI or CT scan, whereas all calcified meningiomas (n = 17) harbored a NF2 mutation (p = 0.0002). Several prior reports have discussed tumor calcification as a potential risk for permanent neurological deterioration due to difficult surgical removal [9, 12]. In our series, four patients (8%) experienced local tumor recurrence with three of these cases harboring a NF2 mutation and with tumor calcifications.
Fig. 1.
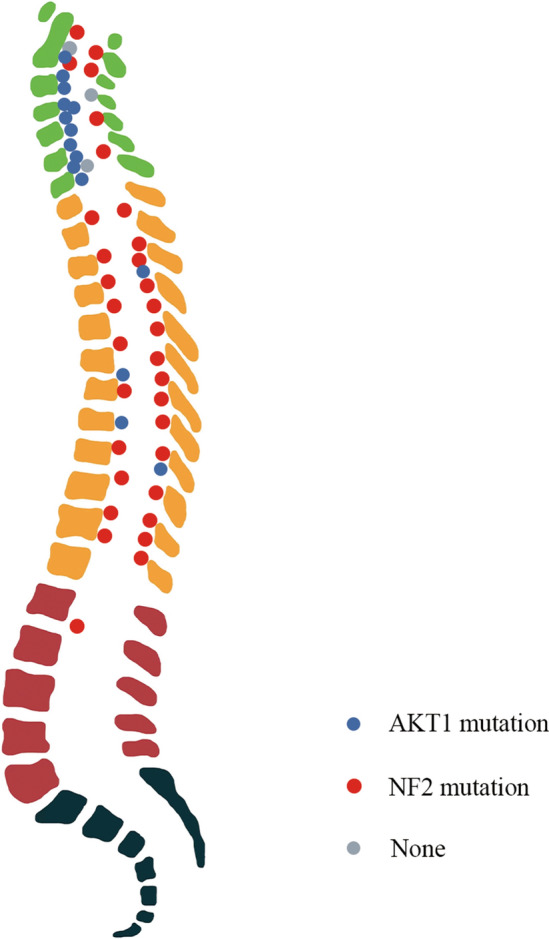
Anatomic distribution of AKT1- and NF2-mutant meningiomas along the spinal column and in relation to the spinal cord
Our data demonstrate the primary role of NF2 and AKT1 alterations as driver events in WHO grade 1 SM, as 94% of the cases harbored mutations in these genes. AKT1 and NF2 mutations presented in a mutually exclusive pattern, identifying unique clinical cohorts. In concordance with our findings, Arslantas et al. investigated 16 SM samples and described NF2 mutations in 8 cases[1]. Remarkably, all NF2-mutant cases in the aforementioned study were located in the thoracic spine and six of them were female patients. Subsequently, Sahm et al. screened 1437 tumors for AKT1 mutations and found 65 mutant cases, including 6 AKT1-mutant cases in 57 SMs [10], and described a strong association between AKT1E17K mutations and spinal tumor localization.
In contrast to AKT1-mutant intracranial meningiomas, where TRAF7 mutations are found to co-occur very frequently [5], we observed only a single AKT1-mutant SM case with a TRAF7 co-mutation. Combining our data with these prior reports, focusing on WHO grade 1 meningiomas only, Clark et al. found TRAF7 co-mutations in 50/68 AKT1-mutant intracranial meningiomas (15 were AKT1 “isolated”, and 3 were co-mutant for AKT1/NF2), while we observed only 1 of 15 co-mutant tumors. Therefore, there appears to be a strong locational difference (p < 0.0001), suggesting that AKT1-mutant meningiomas arising from the ventral cervical spinal arachnoid are genetically distinct from their intracranial counterparts, based on the relative absence of TRAF7 co-mutation. We speculate that the absence of TRAF7 co-mutations in these tumors may potentially correlate with the more indolent clinical behavior that has been observed in AKT1-mutant SM compared to their intracranial counterparts.
Furthermore, we found two SMARCB1 mutations that co-occurred in NF2-mutant meningiomas. SMARCB1 mutations have been associated with the development of SM [2, 7]. Of note, our study included WHO grade 1 SMs exclusively and thus, did not include meningiomas with clear cell histology. This selection criterion may explain the absence of SMARCE1 mutations in our study, which are known driver events in clear cell meningiomas [13]. Nevertheless, the presence of cryptic inactivation of SMARCB1, NF2 or other genes cannot be definitely excluded in the three cases in our series that did not show known hot spot mutations in driver genes.
In summary, we have identified two predominant molecular subgroups in WHO grade 1 SM, characterized by AKT1E17K and NF2 mutations. Both mutations are mutually exclusive and are associated with distinct patient characteristics and tumor features. AKT1-mutant meningiomas originate in the cervical spine ventrally to the spinal cord, are almost exclusively associated with meningothelial histology and exhibit no calcifications on imaging. In contrast, NF2-mutant meningiomas show strong female gender predominance, arise with a wider anatomic distribution, although most frequently in the thoracic spine dorsally to the spinal cord, and can be calcified while displaying variable histologic subtypes (Supplementary Figs. 2 and 3, online resource).
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
Priscilla Brastianos has consulted for Angiochem, Genentech-Roche, Lilly, Tesaro, ElevateBio, Axiom Healthcare Strategies, Pfizer (Array), Dantari, SK Life Sciences, Advise Connect Inspire (ICI), Voyager Therapeutics and Sintetica, and has received grant/research support to MGH from Merck, BMS, Mirati, Kinnate and Lilli and honoraria from Merck, Genentech-Roche, Pfizer and Lilly. The following for-profit companies have supported clinical trials conducted by PKB with payments made to the institution: Merck, Pfizer, Lilly, Bristol-Myers Squibb, AstraZeneca, and Genentech-Roche.
Funding
Open Access funding enabled and organized by Projekt DEAL. Sibylle Assmus Foundation, 2021 Neurooncology Award, to Tareq A. Juratli
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Lingyang Hua, Email: lyhua15@fudan.edu.cn.
Tareq A. Juratli, Email: Tareq.Juratli@ukdd.de
References
- 1.Arslantas A, et al. Detection of chromosomal imbalances in spinal meningiomas by comparative genomic hybridization. Neurol Med Chir (Tokyo) 2003;43(1):12–18. doi: 10.2176/nmc.43.12. [DOI] [PubMed] [Google Scholar]
- 2.Bacci C, et al. Schwannomatosis associated with multiple meningiomas due to a familial SMARCB1 mutation. Neurogenetics. 2010;11(1):73–80. doi: 10.1007/s10048-009-0204-2. [DOI] [PubMed] [Google Scholar]
- 3.Brastianos PK, et al. Genomic sequencing of meningiomas identifies oncogenic SMO and AKT1 mutations. Nat Genet. 2013;45(3):285–289. doi: 10.1038/ng.2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Champeaux-Depond C, Weller J, Resche-Rigon M. Neurofibromatosis type 2: a nationwide population-based study focused on survival after meningioma surgery. Clin Neurol Neurosurg. 2020;198:106236. doi: 10.1016/j.clineuro.2020.106236. [DOI] [PubMed] [Google Scholar]
- 5.Clark VE, et al. Genomic analysis of non-NF2 meningiomas reveals mutations in TRAF7, KLF4, AKT1, and SMO. Science. 2013;339(6123):1077–1080. doi: 10.1126/science.1233009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hua L, et al. Clinical and prognostic features of spinal meningioma: a thorough analysis from a single neurosurgical center. J Neurooncol. 2018;140(3):639–647. doi: 10.1007/s11060-018-2993-3. [DOI] [PubMed] [Google Scholar]
- 7.Hulsebos TJ, et al. Germline mutation of INI1/SMARCB1 in familial schwannomatosis. Am J Hum Genet. 2007;80(4):805–810. doi: 10.1086/513207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Juratli TA, et al. Sporadic multiple meningiomas harbor distinct driver mutations. Acta Neuropathol Commun. 2021;9(1):8. doi: 10.1186/s40478-020-01113-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Levy WJ, Bay J, Dohn D. Spinal cord meningioma. J Neurosurg. 1982;57(6):804–812. doi: 10.3171/jns.1982.57.6.0804. [DOI] [PubMed] [Google Scholar]
- 10.Sahm F, et al. AKT1E17K mutations cluster with meningothelial and transitional meningiomas and can be detected by SFRP1 immunohistochemistry. Acta Neuropathol. 2013;126(5):757–762. doi: 10.1007/s00401-013-1187-5. [DOI] [PubMed] [Google Scholar]
- 11.Sahm F, et al. DNA methylation-based classification and grading system for meningioma: a multicentre, retrospective analysis. Lancet Oncol. 2017;18(5):682–694. doi: 10.1016/S1470-2045(17)30155-9. [DOI] [PubMed] [Google Scholar]
- 12.Sandalcioglu IE, et al. Spinal meningiomas: critical review of 131 surgically treated patients. Eur Spine J. 2008;17(8):1035–1041. doi: 10.1007/s00586-008-0685-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smith MJ, et al. Germline SMARCE1 mutations predispose to both spinal and cranial clear cell meningiomas. J Pathol. 2014;234(4):436–440. doi: 10.1002/path.4427. [DOI] [PubMed] [Google Scholar]
- 14.Williams EA, et al. Distinct genomic subclasses of high-grade/progressive meningiomas: NF2-associated, NF2-exclusive, and NF2-agnostic. Acta Neuropathol Commun. 2020;8(1):171. doi: 10.1186/s40478-020-01040-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.