

Abstract
Apolipoprotein A-I (ApoA-I) is elevated in the plasma of a subgroup of trauma patients with systemic hyperfibrinolysis. We hypothesize that apoA-I inhibits platelet activation and clot formation. The effects of apoA-I on human platelet activation and clot formation were assessed by whole blood thrombelastography (TEG), platelet aggregometry, P-selectin surface expression, microfluidic adhesion, and Akt phosphorylation. Mouse models of carotid artery thrombosis and pulmonary embolism were used to assess the effects of apoA-I in vivo. The ApoA-1 receptor was investigated with transgenic mice knockouts (KO) for the scavenger receptor class B member 1 (SR-BI). Compared to controls, exogenous human apoA-I inhibited arachidonic acid and collagen-mediated human and mouse platelet aggregation, decreased P-selectin surface expression and Akt activation, resulting in diminished clot strength and increased clot lysis by TEG. ApoA-I also decreased platelet aggregate size formed on a collagen surface under flow. In vivo, apoA-I delayed vessel occlusion in an arterial thrombosis model and conferred a survival advantage in a pulmonary embolism model. SR-BI KO mice significantly reduced apoA-I inhibition of platelet aggregation versus wild type platelets. Exogenous human apoA-I inhibits platelet activation, decreases clot strength and stability, and protects mice from arterial and venous thrombosis via the SR-BI receptor.
Keywords: Thrombelastography, platelet inhibition, microfluidics, SR-B1 receptor, hyperfibrinolysis
Introduction
Trauma-induced coagulopathy (TIC) presents in approximately 25% of severely injured patients, new injury severity scores (NISS) >15 and lead to increased morbidity and mortality.1–5 Mechanistically TIC has been attributed to altered thrombin generation, endothelial dysfunction, reductions in serpins and clotting factors by activated protein C, circulating membrane vesicles, hypoperfusion and ischemia resulting in increases in tissue plasminogen activator (tPA), and of importance to the presented work, alterations in platelet activity.3,6–13 The pathogenesis of TIC is likely directly affected by circulating intracellular proteins released by traumatic tissue injury.3,6–13 Such mediators typically do not affect coagulation or fibrinolytic activity because they are not normally present in the plasma or circulate at higher concentrations following injury and may become moonlighting mediators that directly affect coagulation and fibrinolysis.14–17
Apolipoprotein A-I (ApoA-I) is a 28-kDa protein product of the APOA1 gene synthesized in the liver and small intestine, is the primary protein moiety in high-density lipoprotein (HDL) complexes, along with cholesterol and phospholipids.18,19 ApoA-I plays a central role in reverse cholesterol transport, with a plasma concentration of 96–183 mg/dL in healthy humans.18–20 Infusion of human apoA-I into wild-type (WT) mice decreases the risk for venous thrombus formation, and HDL-mediated inhibition of platelet activation is primarily due to the binding of oxidized HDL to the scavenger receptor class B member 1 (SR-BI) on vascular endothelium, increasing endothelial nitric oxide synthase (eNOS) activity.21–24 However, recent platelet aggregation data from eNOS−/−, AKT1−/−, and AKT2−/− mice suggest that HDL directly inhibits platelet activity independent of the eNOS/Akt pathway.25–29 We hypothesize that apoA-I circulates at elevated concentrations in severely injured patients and directly inhibits platelet function, predisposing patients to systemic hyperfibrinolysis, a lethal form of TIC.9,12,13.
Materials and Methods
Materials.
ApoA-I was purchased from Calbiochem (San Diego, CA) or PromoCell (Pittsburgh, PA). Collagen, arachidonic acid (AA), ristocetin, adenosine triphosphate (ATP) standard, Chrono-Lume® and the Chrono-Log 700 aggregometer were all obtained from CHRONO-LOG Corporation (Havertown, PA). HDL isolated from human plasma was purchased from Athens Research and Technology (Athens, GA). Recombinant human SR-BI protein and anti–SR-BI antibody were obtained from Novus (Littleton, CO). Rabbit IgG was purchased from Thermo Scientific (Waltham, MA). Murine platelet aggregations were performed on the Biodata Platelet Aggregation Profiler PAP 8 v 2.0 (Horsham, PA). Antibodies for flow cytometry: CD41, PAC-1, and CD62P were obtained from BD Biosciences (San Jose, CA) and an FC-500 flow cytometer was purchased from Beckman-Coulter (Brea, CA). Thrombelastography (TEG) was completed on a TEG 5000 Thrombelastograph (Haemonetics Inc., Niles, IL) and TEG supplies: kaolin, Platelet Mapping® and Functional Fibrinogen reagents were gifted by Haemonetics Corporation (Braintree, MA). ApoA-I ELISA assays were obtained from Abcam (Cambridge, MA).
Blood Samples and Platelet Concentrates.
Human whole blood (WB) was collected by venipuncture from healthy volunteers after obtaining informed consent under the auspices of the Colorado Multiple Institutional Review Board (COMIRB #09-0816), Aurora, CO. Washed platelets (WP) and platelet-rich plasma (PRP) were prepared as described.14,30–32 Citrated plasma samples from WB samples of injured patients were collected at the scene or immediately upon arrival to the Emergency Department at Denver Health Medical Center (DHMC) an urban level-1 trauma center from 2010–2017 under waiver of consent as approved, COMIRB #13-3087.14,33 Basic demographic data and clinical findings, including: injury pattern, shock, and outcome were also gathered.
TEG/Platelet Mapping/Citrated Functional Fibrinogen and ApoA-I Levels.
Citrated kaolin-TEGs (CK-TEG) were completed as described, with the addition of apoA-I [50–300 μg/dl] or normal saline (NS) prior to activation.34,35 The TEG parameters measured included the activated clotting time (ACT) (min), angle (degrees), maximum amplitude (MA, mm), clot strength (G, derived from the MA) and clot lysis at 30 minutes following the MA (LY30, percent).34,35 The maximum amplitude (MA) is the maximum width of the TEG tracing and corresponds with maximum clot strength. G (dynes/cm2) is a measure of shear elastic modulus strength derived according to the formula G=(5,000*MA)/(100-MA).36,37 Citrated Functional Fibrinogen was evaluated with abciximab (a GPIIb/IIIa monoclonal blocking antibody) to eliminate platelet function and examine fibrin-based clot formation.38,39 This assay has been previously demonstrated to closely correlate with von Clauss and Clauss fibrinogen assays. 38 Platelet mapping studies were completed employing reptilase and factor XIII to activate fibrin with arachidonic acid (AA) and adenosine diphosphate (ADP) to activate platelets ± pre-incubation with apoA-I [300 μg/ml] or vehicle control.32 ApoA-I levels were measured in four consecutive severely injured patients with systemic hyperfibrinolysis because of their clot instability using a commercial enzyme-linked immunosorbent assay (ELISA).
Platelet Aggregations.
Platelet aggregations were performed on both washed platelets (WP) and platelet-rich plasma (PRP) via standard light-transmission aggregometry.30 The samples were incubated with NS or apoA-I [50–300 μg/ml] for 5 minutes at 37°C and then activated with collagen [1μg/mL] or ristocetin [1.25 mg/mL]. Platelet samples were also pre-incubated with human plasma-derived HDL, recombinant human SR-BI protein, or anti–SR-BI antibody or non-immune isotype control antibody for 5 minutes, followed by incubation with apoA-I [300 μg/ml] or vehicle for five minutes at 37°C and stimulated with collagen [1 μg/mL]. Platelet aggregation was also performed with WP from wild type (WT) mice and SR-BI knockout mice after pre-treatment with either apoA-I or NS (control) for 5 minutes at 37°C.
Flow Cytometry.
Human WP were prepared and diluted to a platelet concentration of 200–250 × 103/μL.30 Samples were then incubated with NS or apoA-I [300 μg/ml] for 5 minutes at 37°C. The platelets were then activated with AA [0.5 mM] or convulxin [10 ng/mL] for 10 minutes at 37°C, incubated with antibodies to CD41, PAC-1, or P-selectin for 5 minutes at 37°C, and fixed with cold 2% paraformaldehyde.30
Microfluidic Flow Assay.
Whole blood was incubated with NS or apoA-I [300 μg/ml] for 5 minutes at room temperature (RT) and perfused through a polydimethylsiloxane (PDMS) microfluidic channel flow device (W 500 μm × H 50 μm channels) by syringe pump at a wall shear rate of 600 s−1 over type I fibrillar equine collagen [0.25 mg/mL].30,31 Platelet adhesion was captured with a CCD camera (Hamamatsu Orca-R2) using relief contrast microscopy (40X, NA 0.6) on an inverted microscope (Olympus IX81) at a frame rate of 1 s−1. Platelet aggregates were counted by hand, and grouped by number of platelets per aggregate, using the brightfield image at the end of the run
Akt Phosphorylation.
Human WP were pre-incubated with NS or apoA-I [300 μg/ml] for 5 minutes at RT and 37°C as a temperature control.30 Platelets were then activated with convulxin [10 ng/mL], incubated for 1–10 minutes, lysis buffer was added, the proteins separated by polyacrylamide gel electrophoresis, transferred to nitrocellulose, and incubated with an antibody to phosphorylated Akt.30 Densitometry was completed using ImageJ software.40
Murine Model of FeCl3-Induced Carotid Artery Thrombosis.
All animal procedures were approved by the University of Colorado Institutional Animal Care and Use Committee (IACUC #00501). Briefly, twelve-week-old, male C57BL/6 mice (The Jackson Laboratory, Bar Harbor, ME) were anesthetized, intubated, mechanically ventilated under a dissecting microscope, and maintained at a rectal temperature of 37±1°C.30 The mice were given NS or apoA-I [300 μg/ml]FINAL and incubated for 10 min, and an ultrasound probe was attached to the exposed carotid artery which was injured with 6% FeCl3.30 Mean blood flow was monitored for 30 minutes past the time of first vessel occlusion, the experiment was terminated, and the mice euthanized.30,41–43
Murine Model of Pulmonary Embolism.
Twelve-week old C57BL/6 mice were anesthetized, NS or apoA-I [300 μg/ml]FINAL were injected and allowed to circulate for 10 minutes, followed by injection of collagen [0.28mg/kg] and epinephrine [0.029 mg/kg]. 30,41–44 The time to breathing cessation was measured for 30 minutes, and mice were euthanized following the procedure.30,41–43 All animal studies were reviewed and approved by Institutional Animal Care and Use Committees.
Statistical Analysis.
Data are expressed as the mean±the standard error of the mean (SEM) or the median±the interquartile. All statistical analyses were done using either StatView 5.0 (SAS, Cary, NC USA) or GraphPad Prism version 7.05 (GraphPad Software, La Jolla, CA USA). Comparisons were completed using a paired analysis of variance (ANOVA) followed by a Bonferroni/Dunn test for multiple comparisons (based upon the equality of variance), or, if appropriate, via a paired t-test; p<0.05 denoted statistical significance unless otherwise stated. Platelet aggregation was evaluated by surface area coverage and aggregate size, compared with a paired t-test and chi-square test, respectively. Surface area coverage and platelet aggregate size in the microfluidic platelet aggregation analysis were compared using ImageJ software with a paired t-test and chi-square test used for comparisons, respectively.40 Because of the distribution of TEG values and the apoA-I plasma concentrations, comparisons were completed using a Mann-Whitney test; p<0.01 denoted statistical significance.
Results
Severe injury caused increased ApoA-I and a decrease in hemostatic potential.
ApoA-I levels were significantly elevated in 4 severely injured (NISS 27–50), male trauma patients in the DHMC Trauma Activation Protocol (TAP) study as compared to 121 healthy male controls (median 248 μg/ml, range 237–268 μg/ml vs. 128 μg/ml, range 125–133 μg/ml, p=.005) (Table 1).14 All 4 patients had delayed clot formation with a prolonged activating clotting times and systemic hyperfibrinolysis (Table 1).45–47 Three of these patients had suffered blunt trauma and one a penetrating gunshot wound. Of the blunt trauma patients, one died of traumatic brain injury and the other two developed pneumonia 4 days post-injury. Seventy five percent (3/4) of these patients had decreased MA, a measure of clot stability, and all had normal angles (Table 1).
Table 1.
Traumatically-injured patients with increased ApoA-I plasma concentrations.
| Ethnicity& Age (years) | NISS | ACT (sec) | Angle (°) | MA (mm) | Ly30 (%) | Mechanism | Hosp/ICU/Vent Days | Outcome |
|---|---|---|---|---|---|---|---|---|
| W 28.3 | 38 | 238 | 49.4 | 47.5 | 41.6 | Blunt Auto-Ped | 1/1/1 | Died of TBI |
| WH 48.9 | 27 | 136 | 70.2 | 59.4 | 7.8 | Blunt MVC occupant | 23/14/11 | Pneumonia Discharged |
| W 28.3 | 50 | 160 | 72.7 | 56 | 11.6 | Blunt Auto-Ped | 18/8/7 | α-hemolytic strep Pneumonia Discharged |
| NA 24.5 | 27 | 136 | 71.1 | 54.5 | 3.6 | Penetrating GSW | 7/4/3 | Discharged |
| Controls 21–54 (n=121) | 117 (113–121) | 71.4 (68.0–74.2) | 61.5 (58.2–64.8) | 2.6 (1.6–3.5) |
W = White, H = Hispanic, NA = Native American, NISS = New Injury Severity Score, ACT = activated clotting time, MA = maximal amplitude, Ly30 = lysis time 30 after achieving MA, auto = automobile, Ped = pedestrian, MVC = motor vehicle crash, GSW = gunshot wound, Hosp = Hospital days, ICU = Intensive Care Unit days, Vent = ventilator days, TBI = traumatic brain injury. Controls are healthy, male volunteers on no medications that would inhibit platelet function.
ApoA-I Inhibits Clot Formation and Increases Clot Instability.
Whole blood samples from healthy controls were pre-treated with apoA-I [300 μg/ml] or NS and a Kaolin-TEG was performed. An apoA-I concentration of 300 μg/mL caused a significant reduction in G (n=20) and a similar decrease in MA (n=20) and increased LY30 (n=20) (Figs.1A–B). Importantly, this apoA-I concentration is similar to concentrations seen in the 4 injured patients’ plasma with systemic hyperfibrinolysis: 300 μg/ml vs. 237–268 μg/ml. The ApoA1 concentrations of the healthy controls was 74±7 (range 34–101) μg/ml, and all WB samples had normal K-TEG values (Fig.1A–B).
Figure 1: Clot Strength and Stability.
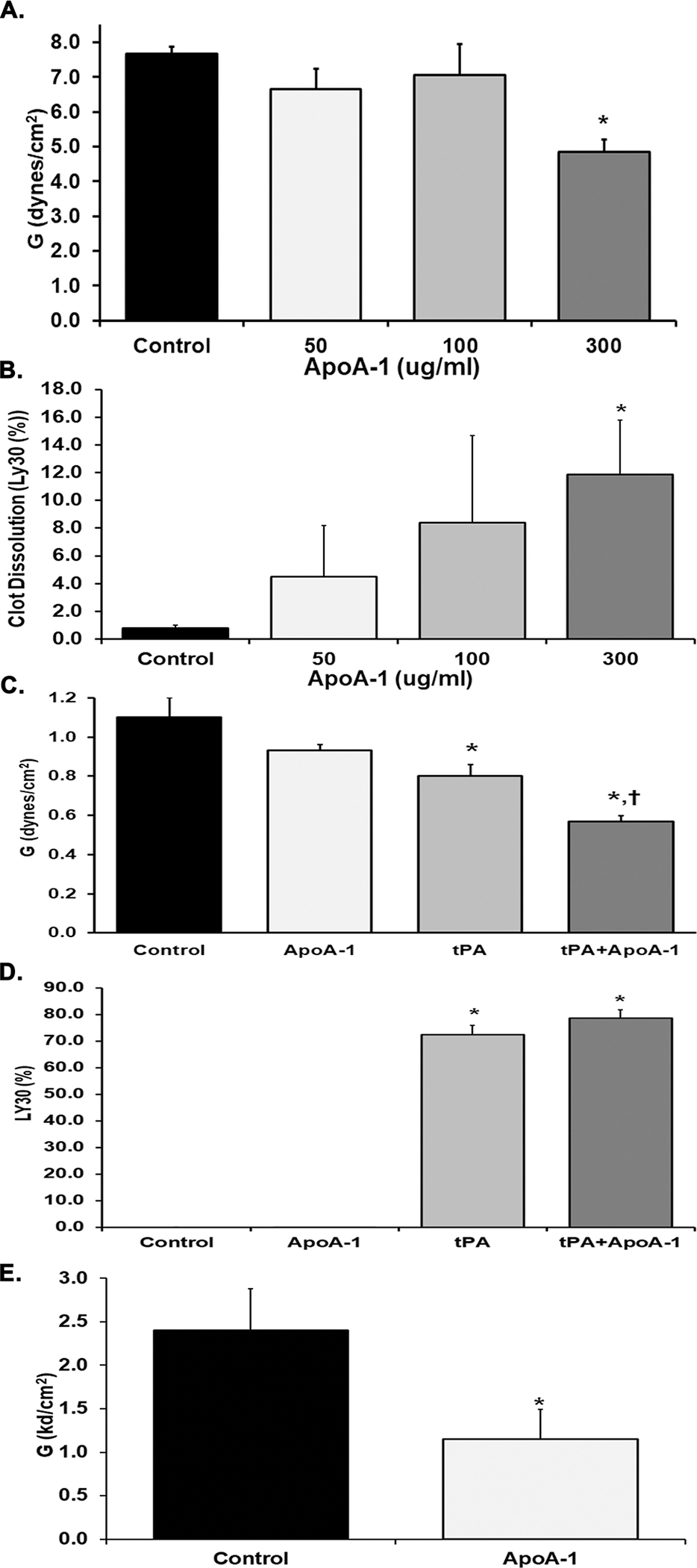
Evaluation of thrombus strength and stability in 3.2% citrate-treated human whole blood (A-D) or heparinized (17 U/mL) human whole blood (E), demonstrating median +/− interquartile range. A. Clot strength, G (Dynes/cm2), as determined by Kaolin-TEG after pre-treatment with vehicle control (black bar) or increasing concentrations of apoA-I (white, gray; and charcoal gray bars). Pretreatment with 300 mg/ml of ApoA-1 significantly decreased clot strength vs. NS-treated controls (*=p<0.01). B. Clot dissolution after 30 minutes, Ly30 (%), as determined by Kaolin-TEG following pre-treatment with vehicle control (black) or increasing concentrations of apoA-I ((white, gray; and charcoal gray bars). Preincubation with apoA-I at 300 mg/ml significantly increased Ly30 versus controls (*=p<0.01). C. Clot strength (Dynes/cm2) was evaluated by the TEG citrated functional fibrinogen (CFF) assay in samples pre-treated with vehicle control (black), 300 μg/ml apoA-I (white), 100 ng/mL tPA (gray), or combination of tPA and apoA-I (charcoal gray). Preincubation with tPA inhibited formation of the functional fibrinogen clot vs. controls and apoA-I (*=p<0.01) and pre-treatment with the combination of apoA-I and tPA caused significant decrease in the functional fibrinogen clots versus both NS-treated controls and pre-incubation with apoA-I (*=p<0.01 vs. NS; †=P<.01 vs. apoA-I). D. Clot dissolution (%) was evaluated by the TEG CFF assay in samples pre-treated with vehicle control (black, no detectable value), 300 μg/ml apoA-I (clear, no detectable value), 100 ng/mL tPA (gray), or combination of tPA and apoA-I (charcoal gray) Both tPA and tPA+ apoA-I treatment induced significant amounts of CFF clot lysis as compared to NS-apoA-I treated samples (*=p<0.01). E. Clot strength (Dynes/cm2) was evaluated by thrombin-independent platelet mapping studies using factor XIIIa, reptilase, and arachidonic acid as agonists in vehicle controls (black) compared to samples treated with apoA-I (white). ApoA-I inhibited the clot strength in the platelet mapping assay (*=P<0.01). Statistical significance was determined by Mann Whitney; *=p<0.01.
Fibrin Formation is not affected by ApoA-I.
The addition of apoA-I [300 μg/ml] did not affect fibrin formation in the citrated functional fibrinogen (CFF) TEG assay,38 which measures fibrin formation independent from platelet activities contribution (n=6) (Fig.1C). The addition of tPA in the CK-TEGs induced an expected significant decrease in G whether ApoA1 was present or not (n=6). Moreover, apoA-I alone did not alter the LY30 (Fig.1D). As expected, tPA added to the CK–TEG assay resulted in a dramatic increase in Ly30 (n=6), while the addition of both tPA + apoA-I did not cause a significant increase in Ly30 versus tPA alone (n=6).
ApoA-I Abrogates TxA2-Induced Platelet Clot Formation.
TEG platelet mapping, traditionally used to evaluate inhibitory contribution of various anti-platelet agents, was employed to delineate the specific effect of apoA-I on the platelet component of clot formation.32,39 ApoA-I [300 μg/ml] significantly inhibited the TxA2-induced clot strength of human WB samples (n=6) (Fig.1E).
ApoA-I Inhibits Platelet Aggregation and Dense Granule Release.
ApoA-I [300 μg/ml] significantly decreased collagen-mediated [1μg/mL] maximal aggregation of PRP compared to NS-treated controls (n=5) (Fig.2A). Neither pre-treatment with native HDL nor recombinant human SR-BI protein induced any changes in collagen-induced aggregation compared to NS-treated PRP, HDL, or SR-BI protein (n=5 for both). Addition of apoA-I to the SR-BI protein significantly decreased collagen-induced aggregation compared to SR-BI pretreatment alone (n=5) (Fig.2A). Samples pretreated with an anti-SR-BI antibody alone (n=3) and the SR=BI antibody + apoA-I (n=3) significantly decreased collagen-induced aggregation compared to NS-treated controls (n=5). The addition of apoA-I to the anti-SR-BI antibody did not significantly alter the collagen-induced aggregation versus PRP pretreated with anti-SR-BI antibody alone (p>0.1) (Fig.2A).
Figure 2. Platelet aggregation and granule release are inhibited by ApoA-I.
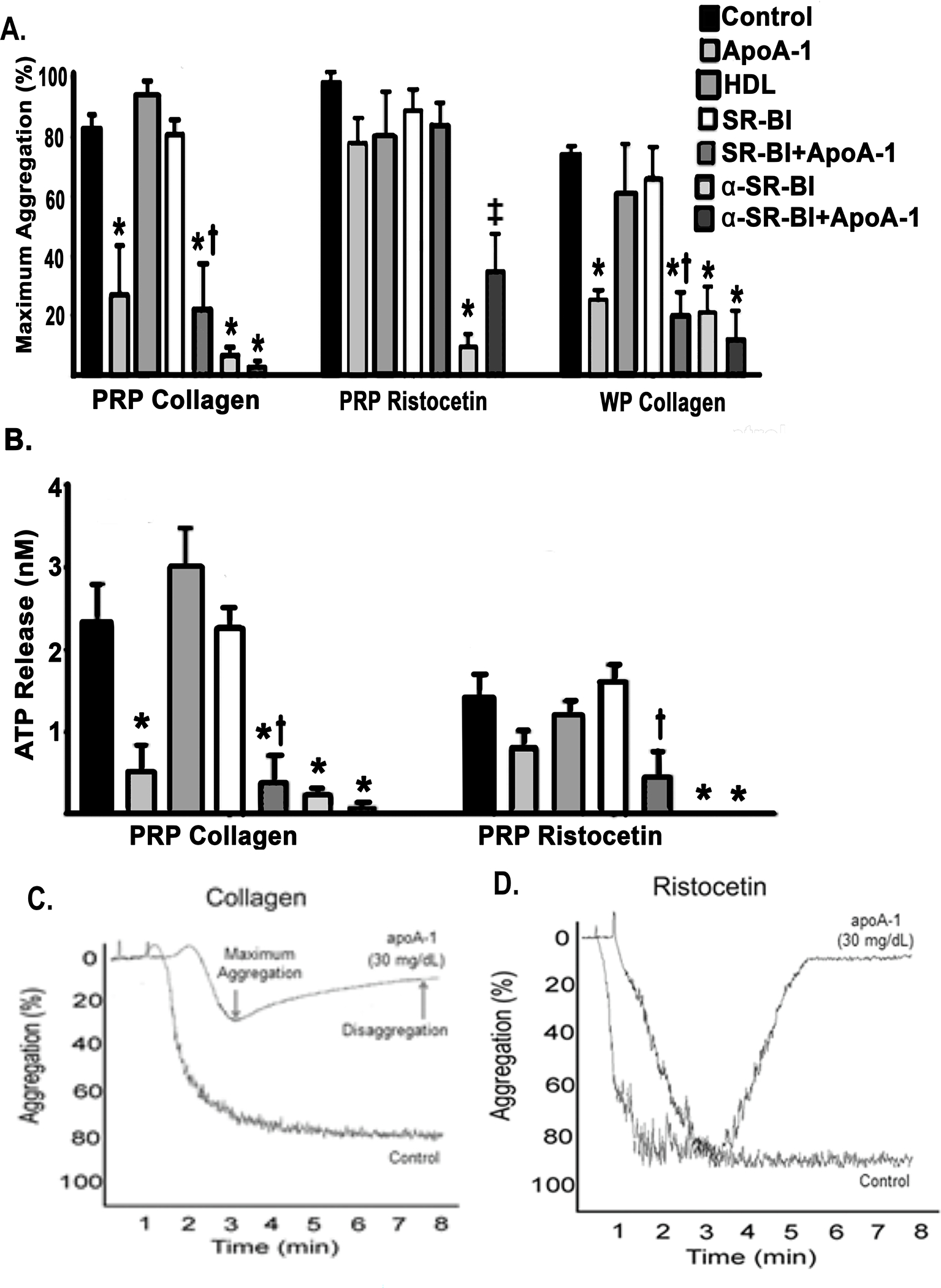
A. The maximum light transmission aggregometry (percent) of PRP or WP stimulated with collagen (1 μg/mL) or ristocetin (1.25 mg/mL) after pre-treatment with vehicle control (NS, n=5), apoA-I (n=5), HDL (n=5), SR-BI±apoA-I (n=5), and anti-SR-BI (α-SR-BI) antibody±apoA-I (n=3) expressed as the mean±SEM. ApoA-I, SR-BI + apoA-I, α-SR-BI and α-SR-BI+ apoA-I pretreatments inhibited PRP platelet aggregation to collagen as compared to NS-treated controls (*=p<0.05). Pretreatment of platelets from PRP with α-SR-BI inhibited aggregation to ristocetin vs. controls (*=p<0.05). This inhibition by α-SR-BI inhibition was partially ameliorated by the addition of apoA-I and was significantly increased (‡=p<0.05 vs. α-SR-BI). Preincubation with ApoA-1, SR-BI+apoA-I, α-SR-BI, and α-SR-BI+apoA-I of washed platelets (right) stimulated with collagen showed a significant inhibition vs. controls (*=p<0.05). SB-RI+apoA-I pre-incubation inhibited collagen-induced platelet aggregation compared to SR-BI preincubation alone (†=p<0.05). B. ATP release is shown as the mean±SEM (nM) from PRP samples stimulated with collagen (1μg/mL, left) or ristocetin (1.25 mg/mL, right) after pre-treatment with control (saline, n=5, black bars), apoA-I [300 μg/ml] (n=5, light gray bars), HDL (n=5, darker gray bars), SR-BI +/− apoA-I (n=5, white and darkest gray bars). In response to collagen apoA-I inhibited ATP release as compared to controls (*=p<0.05), SR-BI +apoA-I together inhibited platelet ATP release vs. SAR-BI treated platelets alone (†=p<0.05), and anti-SR-BI antibody-treated platelets released significantly less ATP than control platelets. Ristocetin activation demonstrated that only platelet treated with SR-BI+ ApoA-1 released less ATP than SR-BI-treated platelets alone (†=p<0.05) and antibodies to SR-BI + ApoA-1 released less ATP than controls (*=p<0.05). Statistical significance was determined by Kruskal-Wallis one-way ANOVA with Dunn’s multiple corrections. C. and D. Representative light-transmission aggregation tracings demonstrate reaction after addition of collagen or ristocetin, respectively, of apoA-I treated human samples compared to vehicle treated controls. PRP, platelet rich plasma; WP, washed platelets.
Similar results were seen with PRP activated by 1.25 mg/mL ristocetin demonstrating a trend towards decreased agglutination in samples pre-treated with apoA-I compared to NS-treated controls (n=5) (Fig.2A). Neither native HDL nor recombinant human SR-BI pretreatment of PRP samples, was associated with a significant difference in ristocetin-induced agglutination when compared to vehicle controls (n=5). As opposed to the decrease noted in collagen-induced aggregation described in the preceding paragraph, addition of apoA-I to the SR-BI protein was not associated with a significant decrease in ristocetin-induced agglutination compared to SR-BI pretreatment alone: apoA-I + SRBI: 84.2±7.3% vs. SRBI: 89.0±6.6% (p>0.1, n=5). Samples pretreated with anti-SR-BI antibody alone exhibited significantly decreased ristocetin-induced agglutination compared to vehicle-treated controls.(n=5). Addition of apoA-I to the anti-SR-BI antibody did not significantly alter the ristocetin-induced agglutination compared to samples pretreated with anti-SR-BI antibody alone (p>0.1, n=3).
In WP, apoA-I significantly inhibited collagen-elicited aggregation [1μg/mL] as compared to vehicle controls apoA-I (n=5) (Fig.2A). Neither native HDL nor recombinant human SR-BI protein pretreatment affected collagen-induced WP aggregation when compared to controls (n=5) (Fig.2A). However, the addition of apoA-I to the SR-BI protein significant decreased in collagen-mediated aggregation compared to SR-BI alone (n=5) (Fig.2A). Pretreatment of WP with SR-BI antibodies or the SR-BI antibodies + apoA-I significantly decreased collagen-induced aggregation compared to vehicle-treated controls (n=3) (Fig.2A). Similar to the results obtained with PRP, pretreatment with both apoA-I and SR-BI antibodies did not further in decrease collagen-induced WP aggregation compared to pretreatment with SR-BI antibodies alone (Fig.2A).
In PRP apoA-I pretreatment also inhibited collagen-induced dense granule mobilization, ATP release, as measured by mean (± SEM) ATP release, was significantly decreased in apoA-I-treated samples compared to vehicle-treated controls (n=5) (Fig.2B). Neither native HDL nor recombinant human SR-BI protein pretreatment of PRP affected collagen-induced ATP release vs. controls (n=5) (Fig.2B). Addition of apoA-I to the SR-BI protein significantly decreased collagen-induced ATP release versus SR-BI pretreatment alone (n=5) (Fig.2B). Pretreatment with SR-BI antibodies exhibited significantly decreased collagen-induced ATP release compared to controls (n=5). Pretreatment with both apoA-I and SR-BI antibodies did not significantly alter the collagen-induced ATP release compared to pretreatment with SR-BI antibodies alone (n=3) (Fig.2B).
ApoA-I also inhibited ristocetin-mediated ATP release in PRP as compared to vehicle-treated controls (n=5) (Fig.2B). Neither pretreatment with native HDL nor recombinant human SR-BI protein pretreatment of PRP samples, was associated with a difference in ristocetin-induced ATP release when compared to controls (n=5) (Fig.2B). Preincubation with both apoA-I and the SR-BI protein resulted in a significant decrease in ristocetin-induced ATP release compared to SR-BI pretreatment alone (n=5) (Fig.2B).
ApoA-I Inhibits Granule Release but Does Not Affect Integrin αIIbβ3 Activation.
ApoA-I significantly inhibited both convulxin [10 ng/ml]- and arachidonic acid (AA, 0.5 mM)-elicited α-granule release as measured by the mean fluorescence intensity (MFI) of P-selectin surface expression by flow cytometry versus vehicle-treated controls (n=4) (Fig.3A). However, apoA-I pre-incubation did not affect convulxin- or AA-induced PAC-1 binding to the activated αIIbβ3 integrin in comparison to controls (p>0.1, n=4 for both) (Fig.3B).
Figure 3. ApoA-I inhibits human the surface expression P-selectin surface expression but not that of αiibβiii-integrin in washed human platelets.
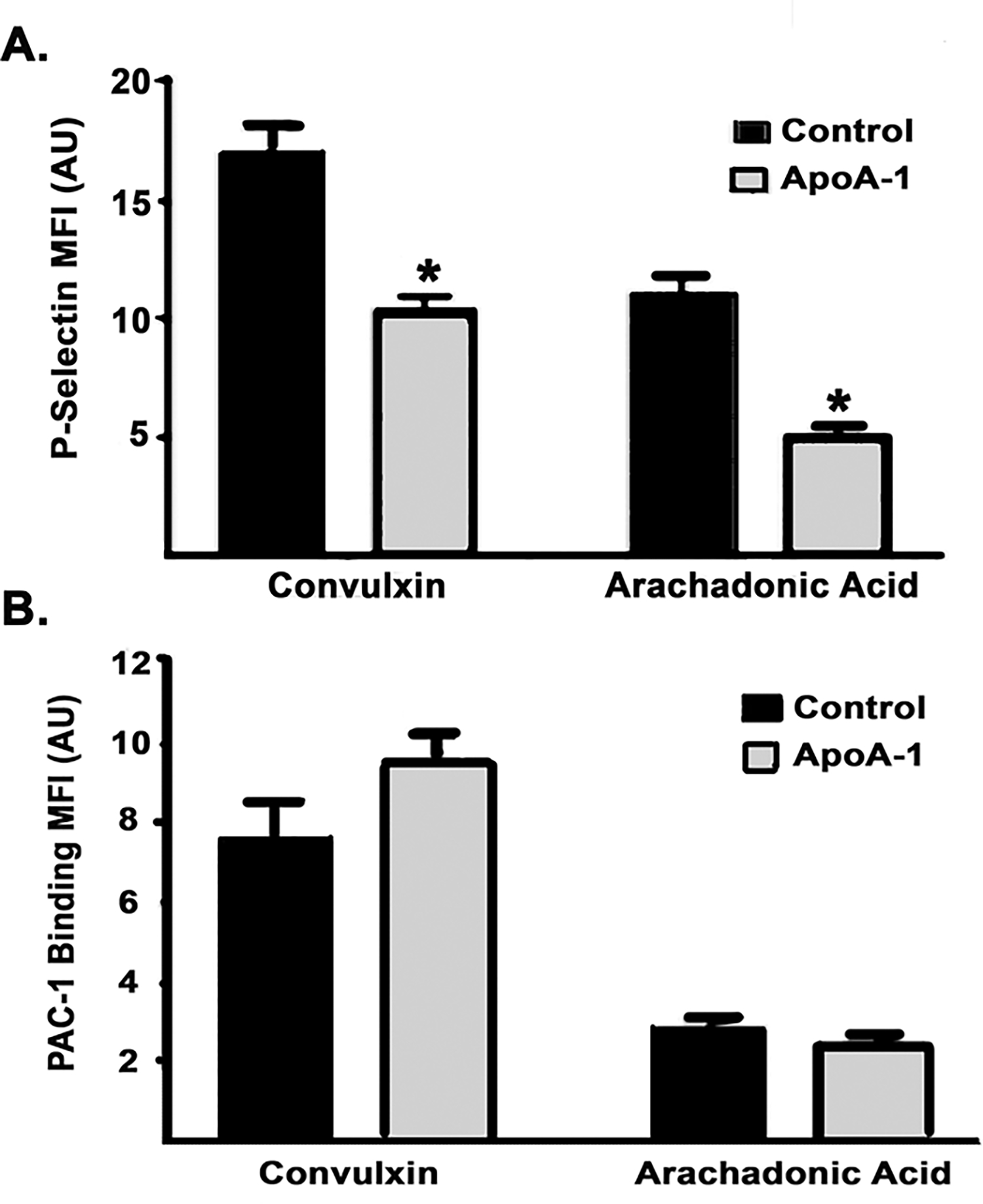
A. P-selectin surface expression quantified by Mean fluorescence intensity (MFI)±SEM of anti-P-selectin antibody (arbitrary units, AU) on platelets stimulated with convulxin or AA after pre-treatment with NS (black bar) or apoA-I (n=4, charcoal gray bar). ApoA-I pretreatment inhibited the convulxin- or AA-induced increase in P-selectin surface expression (*=p<0.05). B. αiibβiii-integrin (PAC-1) surface expression (MFI) expressed as the mean±SEM on platelets stimulated with convulxin or AA after pre-treatment with normal saline (black) or apoA-I (charcoal gray) apoA-I preincubation did not alter PAC-1 surface expression to either convulxin or AA.
ApoA-I Limits Platelet Spreading on Collagen Fibers, Decreases the Size of Platelet Aggregates Bound to Collagen under Flow, and inhibits Akt phosphorylation.
Figure 4A demonstrates a qualitative difference in the human platelets pretreated with apoA-I (resting, round) compared to those pretreated with vehicle control (stimulated, flattened and spread). Microfluidic assays were employed to study apoA-I-mediated functional changes under physiologic flow (Fig.4B–C). Preincubation of human WB with apoA-I [300 μg/ml] did not affect initial platelet adhesion to collagen at a wall shear rate of 650 s−1 but did confer a significant decrease in the formation of intermediate and large platelet aggregates in favor of small aggregates compared to vehicle-treated WB (Fig.4B–C). Small aggregates (3–10 platelets/aggregate) formed by vehicle-treated platelets comprised 31.8±3.1% of total aggregates, while small aggregates of apoA-I-treated platelets represented a significantly higher proportion (n=4), of the total (Fig.4B). ApoA-I significantly inhibited the formation of Intermediate (51–100 platelets/aggregate) and large (>100 platelets/aggregate) platelet aggregates versus vehicle controls (n=4). Lastly, compared to NS-treated controls, apoA-I [300 μg/ml] decreased the convulxin-induced phosphorylation of Akt in WP with quantification by densitometry (Fig.5).
Figure 4. ApoA-I inhibits platelet spreading on fibrillary collagen, and limits the formation of large platelet aggregates on collagen under physiologic flow conditions.
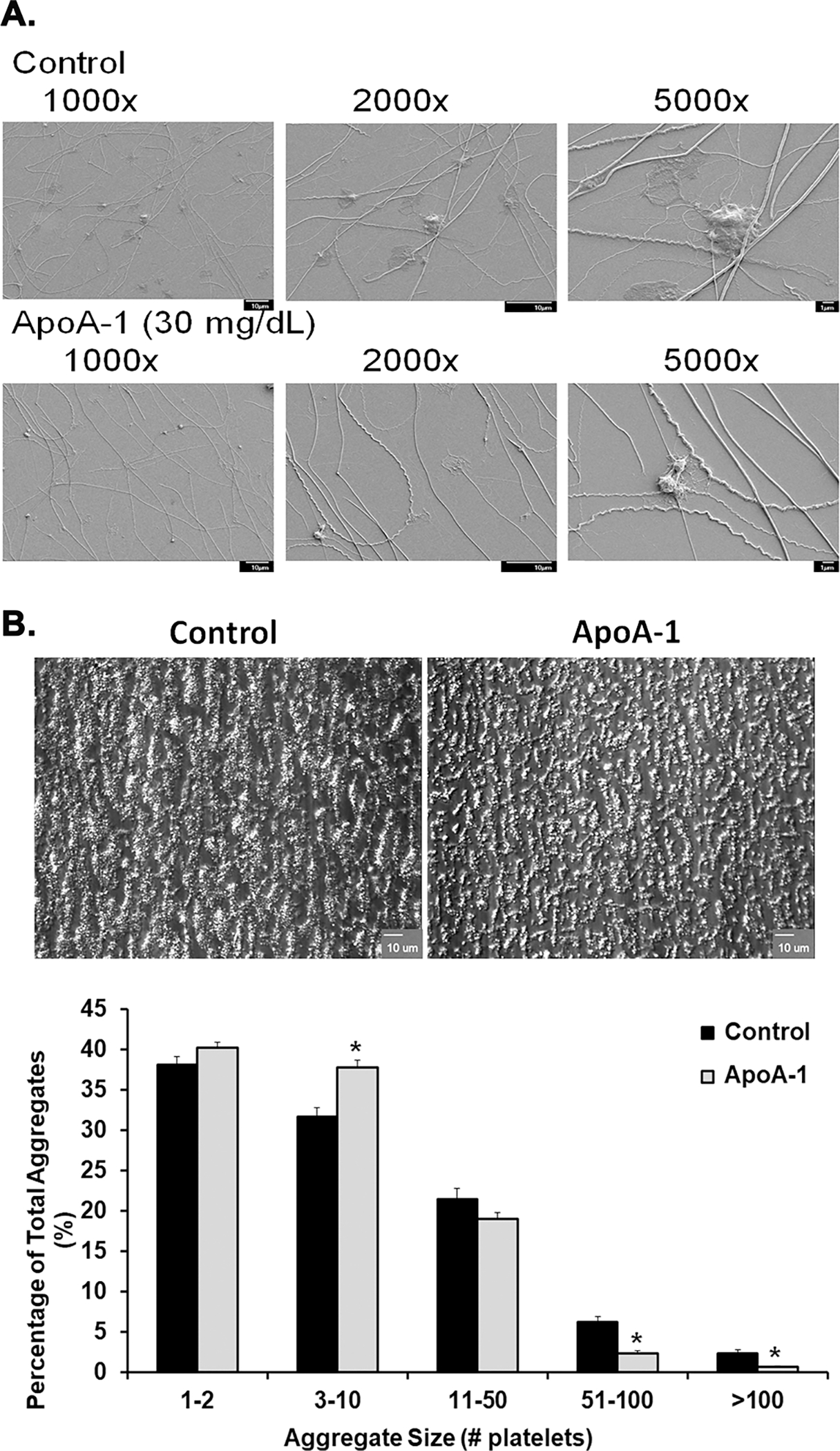
Panel A: representative scanning electron microscopy of NS- (top row of micrographs) or apoA-I-pretreated (bottom row of micrographs) platelets forming aggregates on fibrillar collagen at different magnifications: 1,000X, 2,000X, and 5,000X. ApoA-I visually reduced the size of the human platelet aggregates on collagen fibrils versus the NS-treated controls. Panel B: two representative bright-field microscopic images of platelet aggregates: NS (vehicle)-treated controls on the left and apoA-I [300 μg/ml]-treated platelets on the right (100X). The ApoA-1 pretreatment diminished the size of the aggregates vs. the NS-treated controls. Panel C: Percentage of the total aggregates represented by the different size groups of various platelets per aggregate. Controls (black bars) and apoA-I-pre-treated samples (light gray bars) are divided into groups of various aggregate sizes along the x-axis, and corresponding proportions of the surface area covered by aggregates of each size (percent) shown on the y-axis. ApoA-I caused an increased in platelet aggregates of 3–10 and decreased the large aggregates 51–100 and >100 platelets on collagen versus NS-pretreated controls (*=p<0.05).
Figure 5. ApoA-I inhibits Akt phosphorylation.
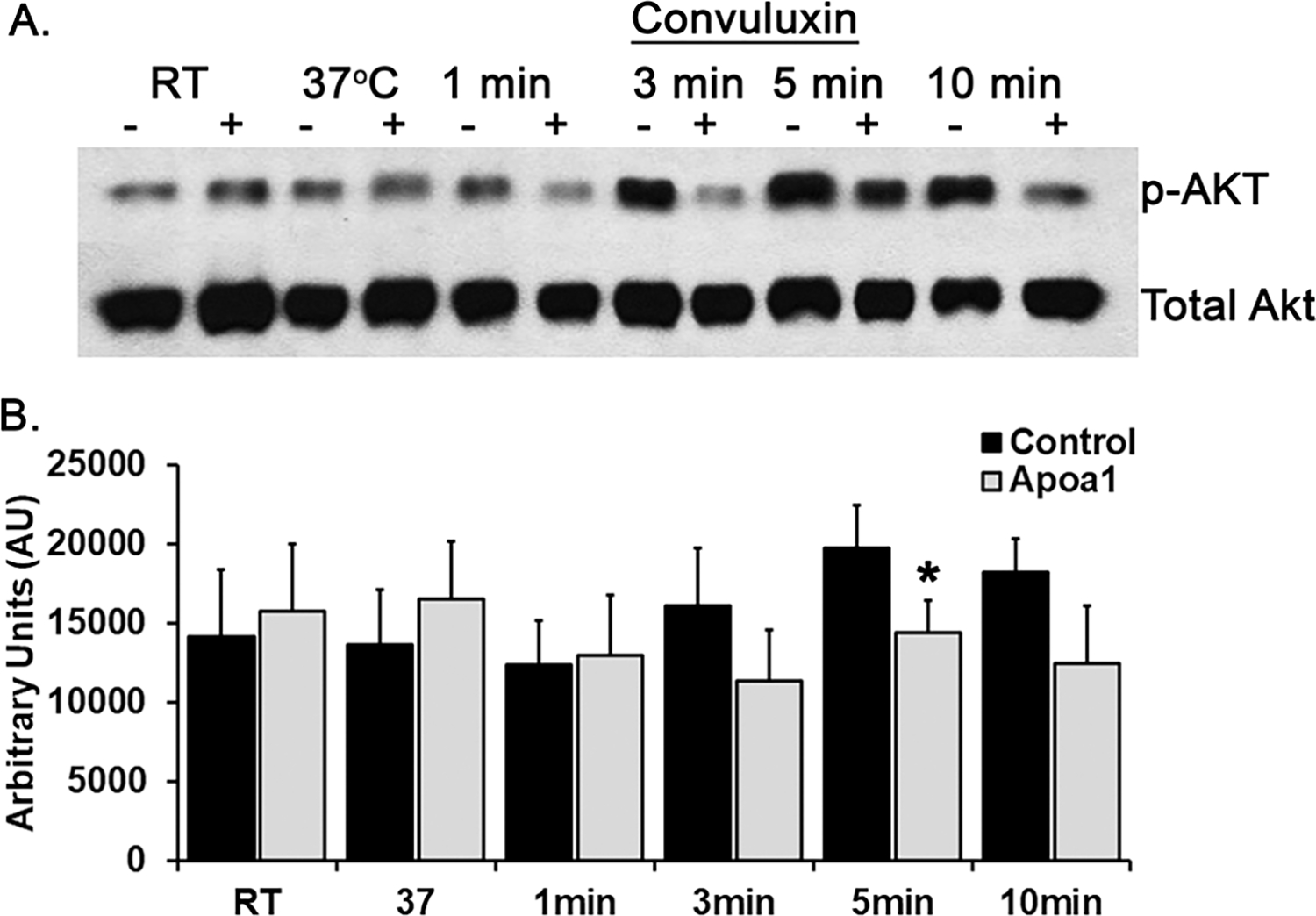
A representative western blot comparing phosphorylated Akt from NS-pre-treated human platelet controls (−) to platelets pre-treated with ApoA-1 (+) for 5 min at 37°C and activated with convulxin [10 ng/mL] over a time course of 1–10 min at 37°C. Apolipoprotein A-I pretreatment decreased the amounts of Akt phosphorylation over the entire time course as seen both visually (upper panel) and by densitometry (lower panel) with statistical significance between total and phosphorylated Akt present at 5 min (*=p<0.05, n=3).
ApoA-I inhibits thrombus formation in vivo.
In a 6% ferric chloride (FeCl3) carotid artery injury, apoA-I [300 μg/ml] preincubation significantly delayed the time to vessel occlusion in mice compared to NS-treated control animals (n=5, Fig.6A).30,41–43 Following initial vessel occlusion, reperfusion was not noted in the controls, but was occasionally seen in apoA-I treated animals (Fig 6A). A second murine model of venous thrombosis was employed in which the venous injection of collagen [0.28 mg/kg] and epinephrine [0.029 mg/kg] causes venous thrombosis leading to pulmonary embolus (PE) with a 30 minute experimental observation time, during which no reperfusion or resumption of breathing was noted after the time of initial breathing cessation was noted.44 Incubation of mice with apoA-I [300 μg/ml] completely inhibited PE formation and all apoA-I treated mice survived as compared to saline-injected controls that all died (n=5, Fig.6B).30,41–43
Figure 6. ApoA-I inhibits thrombosis in mice and SR_BI is necessary for apoA-I inhibition.
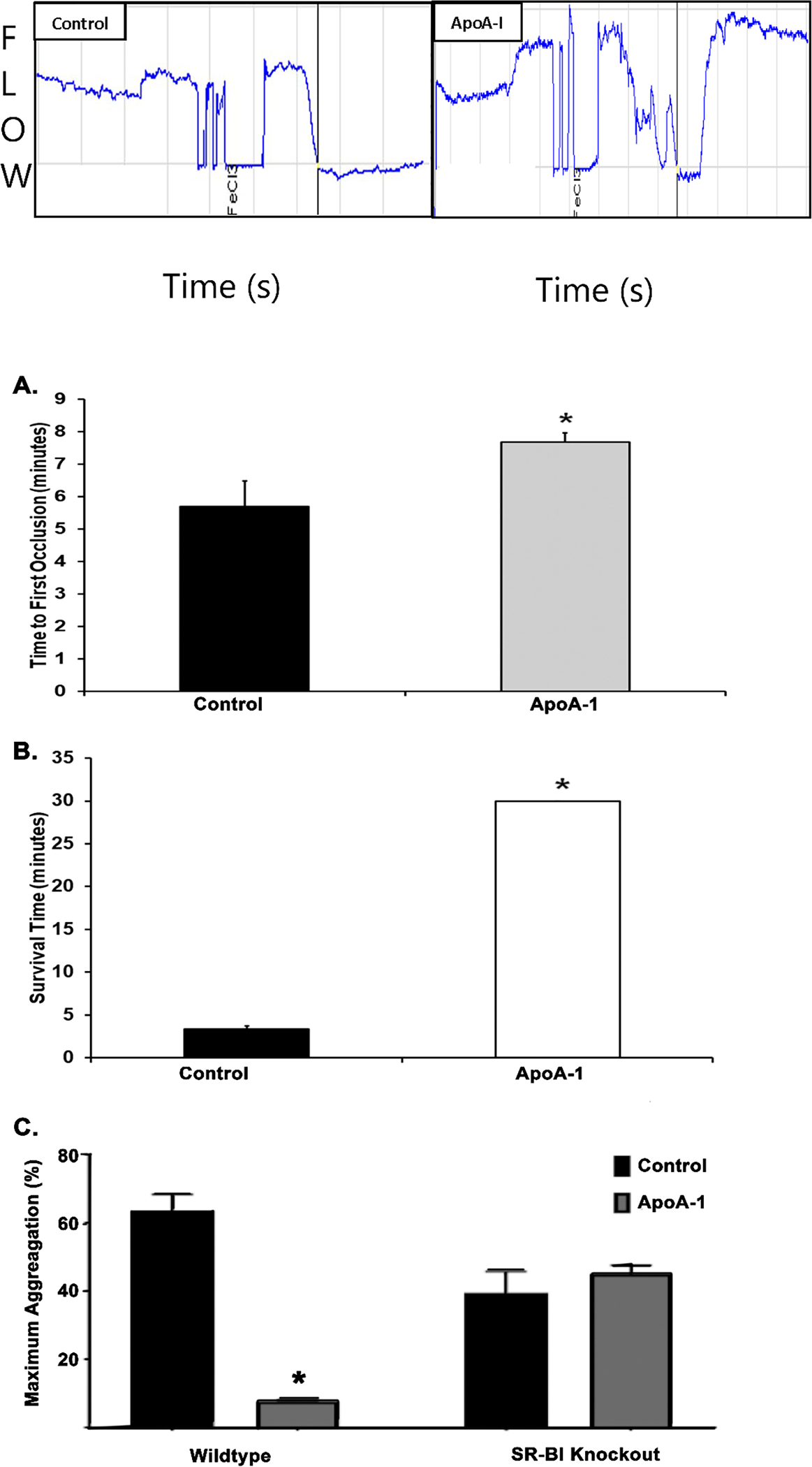
A. Time (minutes) to vessel occlusion in a 6% ferric chloride-induced carotid artery injury model with vehicle control-treated WT mice (black bar, n=5) compared to those treated with apoA-I (gray bar). ApoA-I preincubation significantly prolonged the time to vessel occlusion versus NS-pretreated control mice (n=5; *=p<0.05). B. Survival time (minutes) following pulmonary embolism induced by intravenous injection of collagen and epinephrine in mice treated with vehicle control (black, n=5) or apoA-I (red, n=5). All the NS-treated controls died via a pulmonary embolus within 5 minutes following injury. ApoA-I pre-treated mice survived and did not develop pulmonary emboli during the observation period. Statistical significance determined by Mann-Whitney test; *=p<0.05. C. Maximal aggregation (%) was compared between experimental groups with no difference seen in SR-BI KO mice treated with apoA-I (n=3) or vehicle (normal saline, n=3), but significant decrease in aggregation seen in WT mice treated with apoA-I (n=4) compared to controls treated with vehicle only (n=7, *=p<0.05).
The SR-B1 receptor is Essential for the Inhibitory Effect of ApoA-I in vivo.
Preincubation with apoA-I [300 μg/ml] of WP from WT mice significantly inhibited collagen-elicited aggregation as compared to NS-treated controls (n=4) (Fig.6C). When the identical experiments were performed in SR-BI receptor knockout mice, apoA-I did not affect collagen-induced platelet aggregation (n=3) (Fig.6C).
Discussion
Severe injury (NISS=27–50) caused increased circulating concentrations of apoA-I with delayed clot formation, increased ACT values, and systemic hyperfibrinolysis: increased Ly30 values with insufficient clot strength. Using the blood from healthy volunteers, who had normal K-TEGs, addition of exogenous apoA-I caused significant increases in LY30 and decreases in G, a measure of clot strength derived from the MA, without affecting fibrin formation via CFF-TEGs. Apolipoprotein A-I significantly inhibited TxA2-induced clot strength in the TEG platelet mapping assay. Furthermore, apoA-I directly diminished platelet activity by significantly decreasing 1) platelet aggregation to collagen and 2) ATP and P-selectin release from both dense and α-granules, respectively, to both ristocetin and collagen without affecting αIIbβ3-integrin activity. Under physiologic flow rates apoA-I significantly decreased the formation of intermediate and large platelet aggregates without affecting the initial collagen binding. Apolipoprotein AI activity in vitro was mimicked by antibodies to the SR-BI receptor (which acted as activating ligands, rather than inhibitors), and apoA-I also decreased convulxin-induced Akt phosphorylation. In vivo apoA-I delayed the time to carotid artery occlusion after ferric chloride-induced vessel injury and inhibited the cessation of breathing in WT mice following venous injection of collagen/epinephrine, but did not inhibit aggregation in platelets from the SR-BI receptor KO mice, suggesting apoA-I inhibition requires the SR-BI receptor.
Clots formed in the presence of apoA-I [300 μg/ml] were less stable and more susceptible to lysis, which are consistent with the inverse correlation of apoA-I with lysis time, measured by spectrophotometry.48 Importantly, apoA-I did not affect fibrin clot formation and did not significantly enhance plasmin activity induced by the addition of exogenous tPA. The dose-dependent increase in Ly30 is likely due to apoA-I-mediated reduction in clot strength resulting in an increased susceptibility to plasmin cleavage due to its direct inhibition of platelet activity. However, maximal agglutination induced by 1.25 mg/mL ristocetin was not different in PRP treated with apoA-I compared to vehicle-treated controls.
Formation of a stable thrombus in vivo depends on inside-out signaling to activate the αIIbβ3-integrin and outside-in signaling, occurring after ligand-occupied integrins form clusters.49–51 Receptor-specific platelet activation signaling pathways converge into common signaling events that induce the inside-out signaling process, leading to activation of the ligand-binding function of the integrin, platelet shape change, and granule secretion.50 Upon ligand binding, αIIbβ3 integrins cluster and trigger outside-in signaling that stabilizes the aggregate and supports responses, resulting in platelet spreading, additional granule secretion, stabilization of platelet adhesion and aggregation, and clot retraction.50–52 ApoA-I interrupts outside-in signaling and blocks platelet granule release as seen in the decreased ATP release from dense granules and the decreased P-selectin expression from α-granules. Interestingly, PAC-I binding, a marker of integrin αIIbβ3 activation, was not inhibited, suggesting apoA-I does not completely block inside-out signaling. This pattern is consistent with previous data in which apoA-I decreased the storage lesion and extracellular vesicle release in stored platelet concentrates.53
Intracellular signaling is vital for platelet activation, and the phosphorylation of Akt is a key signaling step for multiple agonists, including collagen and AA.54–56 Activated platelets release agonists, like TxA2, ADP, and serotonin, further amplifying their responsiveness, and recruiting circulating platelets.54–56 These agonists activate platelets via G-protein-coupled receptors (GPCRs) that also promote outside-in signaling.50 These GPCRs involve multiple feedback loops and crosstalk between different pathways, including the phosphorylation of Akt.56 Apolipoprotein A-I demonstrated partial inhibition of Akt phosphorylation in platelets, which may explain the inhibitory effect on granule release without concomitant blockage of αIIbβ3 activation.
The apoA-I induced inhibition of platelet activation was consistent irrespective of the different agonists employed and was documented in both isolated platelets and WB and resulted in the formation of an unstable clot (a dose-dependent increased LY30). This increased LY30 is not due to enhanced plasmin activity, but rather to diminished clot strength because apoA-I did not enhance tPA-induced fibrinolysis in either WB or fibrin clots. Diminished clot strength is also the likely result of apoA-I inhibition of large and intermediate platelet aggregates under physiologic flow. These data are consistent with the inhibitory effect of HDL and apoA-I on the self-association of von Willebrand Factor (VWF) and decreased platelet adhesion to VWF fibers, limiting aggregate size.57 Thus, platelets treated with exogenous apoA-I are still able to adhere to exposed collagen through GPVI or α2β1 receptors. ApoA-I also inhibits platelet function by decreasing autocrine/paracrine stimulation through the reduction in granule release, decreased P-selectin surface expression. ApoA-I also had inhibitory effects on thrombus formation in vivo, using two commonly accepted murine models (ferric chloride-induced carotid artery thrombosis, and cessation of breathing following venous injection of collagen/epinephrine), which are consistent with a role of apoA-I as an inhibitor of platelet aggregation and possibly a blocker of VWF self-association.57
Oxidized HDL binds to the SR-BI receptor on platelets and inhibits platelet activation, which was thought to be mediated by a different pathway not involving the eNOS/Akt pathway, based on platelet aggregation data from eNOS−/−, AKT1−/−, and AKT2−/− mice.27,28 Conversely, exogenous apoA-I partially inhibited Akt phosphorylation and may account for the differences in HDL and apoA-I inhibitory activity. The lack of apoA-I-mediated aggregation antagonism in platelets from SR-BI KO mice revealed the necessity of this receptor for apoA-I inhibition.
ApoA-I, within the HDL molecule, inhibits platelets; however, prior studies attributed this inhibition to the entire HDL molecule and its lipid content.24,57,58 ApoA-I Milano, a rare naturally occurring mutant apoA-I in which arginine 173 is substituted by cysteine, causes decreased arterial thrombus formation in rats without a concomitant increase in HDL or total cholesterol levels.59 Congruent with the presented data, the direct anti-platelet effect of apoA-I Milano may represent the inhibitory effects of the native protein. Additionally, apoA-I immune complexes have been associated with adverse cardiovascular events in humans.60 Null mutations of apoA-I in human populations are also associated with early coronary heart disease (CHD); whereas; overexpression of apoA-I in transgenic mice protects against atherogenesis.61,62 Thus, the apoA-I subunit of HDL may be a key driver in preventing the formation of a stable thrombus by partial antagonism of Akt inhibiting platelet granule release leading to the platelet dysfunction associated with TIC.32 Although we cannot rule out that there is an independent mechanism for the apoA-1 observed effect, the fact that the deletion of the SR-BI receptor (which has been shown to be a receptor for HDL/apoA-1), abrogates the inhibitory effect of apoA-1 strongly suggests an SR-BI dependent mechanism.
One limitation of our study is the use of normal saline (the solvent for apoA-I in our study) as a negative control, rather than a different non-apoA-I protein. The number of different static and dynamic platelet activation assays in which the difference between apoA-I and saline vehicle is clearly demonstrated, however, which reduces the likelihood that nonspecific platelet binding would produce the consistent effect we observed across these various experiments. This decision is similar to that made in other related papers, such as the work by Barret, et al, in which the authors demonstrate apoA-I promotes atherosclerosis regression in diabetic mice by suppressing mhelopoiesis and plaque inflammation, using vehicle controls.63 Another limitation is the relatively low number of patients in whom apoA-I levels were measured, with only four consecutive severely injured patients having levels measured because of their clot instability. Most TAP patients at our institution are not severely injured and samples on the severely injured with systemic hyperfibrinolysis are not easily found, however, which led to the decision to use these samples. A final limitation is the fact that necropsies were not performed on the mice used in the in vivo experiments to demonstrate that the mice who ceased breathing during the experiment definitively had larger thrombus burden following the venous injection of collagen and epinephrine compared to those mice that survived the initial experimental period. This model has been previously described as decreasing systemic platelet count and leading to pulmonary embolism,43,64 and has been used by our group before, with the initial studies including such histologic confirmation following necropsy.30,44 While the injection of collagen/epinephrine induces systemic clotting, when taken together with the other data presented herein, it is more likely that the apoA-I treated mice demonstrated increased embolization (leading to the increased rate of breathing cessation) due to decreased clot strength, rather than generating a lower initial thrombus burden.
We believe the full length HDL likely does not, itself, inhibit platelet activation due to steric hindrance of the interaction between apoA-I and platelets by lipid moieties. ApoA-I may lack this steric hindrance, thereby executing its inhibitory effect directly on to platelets. Indeed, many proteins circulate in injured patients in forms that are not normally present in the plasma, such as intracellular proteins, enzymes, and constituents of circulating complexes like HDL that have been radically changed by tissue injury and/or hemorrhagic shock. Further work must be completed to define the possible role of ApoA1-induced platelet inhibition resulting in the hyperfibrinolysis form of TIC and establish methods to reverse or modulate such platelet inhibition.
The direct interaction between apoA-I and platelets may have additional clinical implications beyond TIC in cardiac pathophysiology.65 Increased HDL levels correspond to decreased thromboembolic events, while low HDL levels are associated with increased CHD risk.58,66–70 HDL levels are also a predictor of platelet-dependent thrombus formation, independent of platelet aggregation and circulating prothrombotic factors.71 Data to delineate the specific role of apoA-I in the reduction of thrombosis are required.
Acknowledgments
WLJ, CRR, and BRB performed all experiments. WLJ and CRR wrote the initial drafts of the manuscript and BRB performed the bulk of writing as well as planning and helping to design the experiments. EEM provided data analyses and clinical correlation with injured patients. KCH performed experiments and identified apoA-I. JC and MK performed experiments and analyzed the TEG data. KBN assisted with experiments and reviewed/edited the manuscript. CCS synthesized and provided data from trauma patient samples, contributed to overall conceptualization of the project, and reviewed/edited the manuscript. AB contributed to overall conceptualization of the project and reviewed/edited the manuscript. JDP provided equipment, supplies, and reagents, as well as contributing to overall conceptualization of the project, experiment planning, and reviewing/editing the manuscript. All authors have reviewed, and approved, the manuscript for submission.
Funding Details
Branchford: HRSA/MCHB H30MC24049
Jones, Ramos, Coleman: NIGMS, NIH: T32-GM008315.
Moore, Silliman, Hansen, Banerjee: NIGMS, NIH: grants P50 GM49222 and 1RM1GM131968, NHLBI, NIH: UM1 HL120877 and Department of Defense: W81XWH-12-2-08.
Di Paola: HRSA/MCHB H30MC24049, University of Colorado School of Medicine Postle Endowed Chair in Cancer and Blood Disorders, and NHLBI, NIH: R01 HL120728, R01 HL139825
Footnotes
Disclosure Statement
No authors have relevant conflicts of interest to declare.
References
- 1.Chang R, Cardenas JC, Wade CE, Holcomb JB. Advances in the understanding of trauma-induced coagulopathy. Blood. 2016;128(8):1043–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gonzalez E, Moore EE, Moore HB, Chapman MP, Silliman CC, Banerjee A. Trauma-Induced Coagulopathy: An Institution’s 35 Year Perspective on Practice and Research. Scand J Surg. 2014;103(2):89–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moore EE, Moore HB, Kornblith LZ, et al. Trauma-induced coagulopathy. Nat Rev Dis Primers. 2021;7(1):30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neal MD, Moore HB, Moore EE, et al. Clinical assessment of trauma-induced coagulopathy and its contribution to postinjury mortality: A TACTIC proposal. J Trauma Acute Care Surg. 2015;79(3):490–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sumislawski JJ, Christie SA, Kornblith LZ, et al. Discrepancies between conventional and viscoelastic assays in identifying trauma-induced coagulopathy. Am J Surg. 2019;217(6):1037–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bohm JK, Schafer N, Maegele M, Stumpges B, Bauerfeind U, Caspers M. Plasmatic and cell-based enhancement by microparticles originated from platelets and endothelial cells under simulated in vitro conditions of a dilutional coagulopathy. Scand J Trauma Resusc Emerg Med. 2021;29(1):38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brohi K, Cohen MJ, Davenport RA. Acute coagulopathy of trauma: mechanism, identification and effect. Curr Opin Crit Care. 2007;13(6):680–685. [DOI] [PubMed] [Google Scholar]
- 8.Brohi K, Cohen MJ, Ganter MT, et al. Acute coagulopathy of trauma: hypoperfusion induces systemic anticoagulation and hyperfibrinolysis. J Trauma. 2008;64(5):1211–1217. [DOI] [PubMed] [Google Scholar]
- 9.Cotton BA, Harvin JA, Kostousouv V, et al. Hyperfibrinolysis at admission is an uncommon but highly lethal event associated with shock and prehospital fluid administration. J Trauma Acute Care Surg. 2012;73(2):365–370. [DOI] [PubMed] [Google Scholar]
- 10.Davenport RA, Brohi K. Cause of trauma-induced coagulopathy. Curr Opin Anaesthesiol. 2016;29(2):212–219. [DOI] [PubMed] [Google Scholar]
- 11.Gando S, Shiraishi A, Wada T, et al. A multicenter prospective validation study on disseminated intravascular coagulation in trauma-induced coagulopathy. J Thromb Haemost. 2020;18(9):2232–2244. [DOI] [PubMed] [Google Scholar]
- 12.Ives C, Inaba K, Branco BC, et al. Hyperfibrinolysis elicited via thromboelastography predicts mortality in trauma. J Am Coll Surg. 2012;215(4):496–502. [DOI] [PubMed] [Google Scholar]
- 13.Schochl H, Frietsch T, Pavelka M, Jambor C. Hyperfibrinolysis after major trauma: differential diagnosis of lysis patterns and prognostic value of thrombelastometry. J Trauma. 2009;67(1):125–131. [DOI] [PubMed] [Google Scholar]
- 14.Banerjee A, Silliman CC, Moore EE, et al. Systemic Hyperfibrinolysis after Trauma: A Pilot study of Targeted Proteomic Analysis of Superposed Mechanisms in Patient Plasma. J Trauma Acute Care Surg. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.D’Alessandro A, Moore HB, Moore EE, et al. Plasma succinate is a predictor of mortality in critically injured patients. J Trauma Acute Care Surg. 2017;83(3):491–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.D’Alessandro A, Nemkov T, Moore HB, et al. Metabolomics of trauma-associated death: shared and fluid-specific features of human plasma vs lymph. Blood Transfus. 2016;14(2):185–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nunns GR, Vigneshwar N, Kelher MR, et al. Succinate Activation of SUCNR1 Predisposes Severely Injured Patients to Neutrophil-Mediated ARDS. Ann Surg. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Albers JJ, Wahl PW, Cabana VG, Hazzard WR, Hoover JJ. Quantitation of apolipoprotein A-I of human plasma high density lipoprotein. Metabolism. 1976;25(6):633–644. [DOI] [PubMed] [Google Scholar]
- 19.Fisher EA, Feig JE, Hewing B, Hazen SL, Smith JD. High-density lipoprotein function, dysfunction, and reverse cholesterol transport. Arterioscler Thromb Vasc Biol. 2012;32(12):2813–2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bashtovyy D, Jones MK, Anantharamaiah GM, Segrest JP. Sequence conservation of apolipoprotein A-I affords novel insights into HDL structure-function. J Lipid Res. 2011;52(3):435–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Assanasen C, Mineo C, Seetharam D, et al. Cholesterol binding, efflux, and a PDZ-interacting domain of scavenger receptor-BI mediate HDL-initiated signaling. J Clin Invest. 2005;115(4):969–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brill A, Yesilaltay A, De Meyer SF, et al. Extrahepatic high-density lipoprotein receptor SR-BI and apoA-I protect against deep vein thrombosis in mice. Arterioscler Thromb Vasc Biol. 2012;32(8):1841–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saddar S, Mineo C, Shaul PW. Signaling by the high-affinity HDL receptor scavenger receptor B type I. Arterioscler Thromb Vasc Biol. 2010;30(2):144–150. [DOI] [PubMed] [Google Scholar]
- 24.van der Stoep M, Korporaal SJ, Van Eck M. High-density lipoprotein as a modulator of platelet and coagulation responses. Cardiovasc Res. 2014;103(3):362–371. [DOI] [PubMed] [Google Scholar]
- 25.Brodde MF, Korporaal SJ, Herminghaus G, et al. Native high-density lipoproteins inhibit platelet activation via scavenger receptor BI: role of negatively charged phospholipids. Atherosclerosis. 2011;215(2):374–382. [DOI] [PubMed] [Google Scholar]
- 26.Calkin AC, Drew BG, Ono A, et al. Reconstituted high-density lipoprotein attenuates platelet function in individuals with type 2 diabetes mellitus by promoting cholesterol efflux. Circulation. 2009;120(21):2095–2104. [DOI] [PubMed] [Google Scholar]
- 27.Calzada C, Vericel E, Colas R, et al. Inhibitory effects of in vivo oxidized high-density lipoproteins on platelet aggregation: evidence from patients with abetalipoproteinemia. FASEB J. 2013;27(7):2855–2861. [DOI] [PubMed] [Google Scholar]
- 28.Valiyaveettil M, Kar N, Ashraf MZ, Byzova TV, Febbraio M, Podrez EA. Oxidized high-density lipoprotein inhibits platelet activation and aggregation via scavenger receptor BI. Blood. 2008;111(4):1962–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang N, Tall AR. Cholesterol in platelet biogenesis and activation. Blood. 2016;127(16):1949–1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Branchford BR, Stalker TJ, Law L, et al. The small-molecule MERTK inhibitor UNC2025 decreases platelet activation and prevents thrombosis. J Thromb Haemost. 2018;16(2):352–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Neeves KB, Onasoga AA, Hansen RR, et al. Sources of variability in platelet accumulation on type 1 fibrillar collagen in microfluidic flow assays. PLoS One. 2013;8(1):e54680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wohlauer MV, Moore EE, Thomas S, et al. Early platelet dysfunction: an unrecognized role in the acute coagulopathy of trauma. J Am Coll Surg. 2012;214(5):739–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chapman MP, Moore EE, Chin TL, et al. Combat: Initial Experience with a Randomized Clinical Trial of Plasma-Based Resuscitation in the Field for Traumatic Hemorrhagic Shock. Shock. 2015;44 Suppl 1:63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moore HB, Moore EE, Chapman MP, et al. Viscoelastic measurements of platelet function, not fibrinogen function, predicts sensitivity to tissue-type plasminogen activator in trauma patients. J Thromb Haemost. 2015;13(10):1878–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moore HB, Moore EE, Chin TL, et al. Activated clotting time of thrombelastography (T-ACT) predicts early postinjury blood component transfusion beyond plasma. Surgery. 2014;156(3):564–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang RS, McDonald MM, Wetzel JS, et al. Clot Strength as Measured by Thrombelastography Correlates with Platelet Reactivity in Stroke Patients. Ann Clin Lab Sci. 2015;45(3):301–307. [PubMed] [Google Scholar]
- 37.Lu D, Owens J, Kreutz RP. Plasma and whole blood clot strength measured by thrombelastography in patients treated with clopidogrel during acute coronary syndromes. Thromb Res. 2013;132(2):e94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harr JN, Moore EE, Ghasabyan A, et al. Functional fibrinogen assay indicates that fibrinogen is critical in correcting abnormal clot strength following trauma. Shock. 2013;39(1):45–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Collyer TC, Gray DJ, Sandhu R, Berridge J, Lyons G. Assessment of platelet inhibition secondary to clopidogrel and aspirin therapy in preoperative acute surgical patients measured by Thrombelastography Platelet Mapping. Br J Anaesth. 2009;102(4):492–498. [DOI] [PubMed] [Google Scholar]
- 40.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9(7):671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Naik MU, Stalker TJ, Brass LF, Naik UP. JAM-A protects from thrombosis by suppressing integrin alphaIIbbeta3-dependent outside-in signaling in platelets. Blood. 2012;119(14):3352–3360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.O’Brien KA, Stojanovic-Terpo A, Hay N, Du X. An important role for Akt3 in platelet activation and thrombosis. Blood. 2011;118(15):4215–4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Westrick RJ, Winn ME, Eitzman DT. Murine models of vascular thrombosis (Eitzman series). Arterioscler Thromb Vasc Biol. 2007;27(10):2079–2093. [DOI] [PubMed] [Google Scholar]
- 44.Sather S, Kenyon KD, Lefkowitz JB, et al. A soluble form of the Mer receptor tyrosine kinase inhibits macrophage clearance of apoptotic cells and platelet aggregation. Blood. 2007;109(3):1026–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chapman MP, Moore EE, Moore HB, et al. The “Death Diamond”: Rapid thrombelastography identifies lethal hyperfibrinolysis. J Trauma Acute Care Surg. 2015;79(6):925–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chapman MP, Moore EE, Ramos CR, et al. Fibrinolysis greater than 3% is the critical value for initiation of antifibrinolytic therapy. J Trauma Acute Care Surg. 2013;75(6):961–967; discussion 967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gonzalez E, Moore EE, Moore HB, et al. Goal-directed Hemostatic Resuscitation of Trauma-induced Coagulopathy: A Pragmatic Randomized Clinical Trial Comparing a Viscoelastic Assay to Conventional Coagulation Assays. Ann Surg. 2016;263(6):1051–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zabczyk M, Hondo L, Krzek M, Undas A. High-density cholesterol and apolipoprotein AI as modifiers of plasma fibrin clot properties in apparently healthy individuals. Blood Coagul Fibrinolysis. 2013;24(1):50–54. [DOI] [PubMed] [Google Scholar]
- 49.Durrant TN, van den Bosch MT, Hers I. Integrin alphaIIbbeta3 outside-in signaling. Blood. 2017;130(14):1607–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li Z, Delaney MK, O’Brien KA, Du X. Signaling during platelet adhesion and activation. Arterioscler Thromb Vasc Biol. 2010;30(12):2341–2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zou Z, Chen H, Schmaier AA, Hynes RO, Kahn ML. Structure-function analysis reveals discrete beta3 integrin inside-out and outside-in signaling pathways in platelets. Blood. 2007;109(8):3284–3290. [DOI] [PubMed] [Google Scholar]
- 52.Ginsberg MH, Du X, Plow EF. Inside-out integrin signalling. Curr Opin Cell Biol. 1992;4(5):766–771. [DOI] [PubMed] [Google Scholar]
- 53.Pienimaeki-Roemer A, Fischer A, Tafelmeier M, et al. High-density lipoprotein 3 and apolipoprotein A-I alleviate platelet storage lesion and release of platelet extracellular vesicles. Transfusion. 2014;54(9):2301–2314. [DOI] [PubMed] [Google Scholar]
- 54.Barry FA, Gibbins JM. Protein kinase B is regulated in platelets by the collagen receptor glycoprotein VI. J Biol Chem. 2002;277(15):12874–12878. [DOI] [PubMed] [Google Scholar]
- 55.Cho MJ, Pestina TI, Steward SA, Lowell CA, Jackson CW, Gartner TK. Role of the Src family kinase Lyn in TxA2 production, adenosine diphosphate secretion, Akt phosphorylation, and irreversible aggregation in platelets stimulated with gamma-thrombin. Blood. 2002;99(7):2442–2447. [DOI] [PubMed] [Google Scholar]
- 56.Kim S, Jin J, Kunapuli SP. Akt activation in platelets depends on Gi signaling pathways. J Biol Chem. 2004;279(6):4186–4195. [DOI] [PubMed] [Google Scholar]
- 57.Chung DW, Chen J, Ling M, et al. High-density lipoprotein modulates thrombosis by preventing von Willebrand factor self-association and subsequent platelet adhesion. Blood. 2016;127(5):637–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Deguchi H, Pecheniuk NM, Elias DJ, Averell PM, Griffin JH. High-density lipoprotein deficiency and dyslipoproteinemia associated with venous thrombosis in men. Circulation. 2005;112(6):893–899. [DOI] [PubMed] [Google Scholar]
- 59.Li D, Weng S, Yang B, et al. Inhibition of arterial thrombus formation by ApoA1 Milano. Arterioscler Thromb Vasc Biol. 1999;19(2):378–383. [DOI] [PubMed] [Google Scholar]
- 60.Henson D, Tahhan AS, Nardo D, Quyyumi AA, Venditto VJ. Association Between ApoA-I (Apolipoprotein A-I) Immune Complexes and Adverse Cardiovascular Events-Brief Report. Arterioscler Thromb Vasc Biol. 2019;39(9):1884–1892. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 61.Ng DS, Vezina C, Wolever TS, Kuksis A, Hegele RA, Connelly PW. Apolipoprotein A-I deficiency. Biochemical and metabolic characteristics. Arterioscler Thromb Vasc Biol. 1995;15(12):2157–2164. [DOI] [PubMed] [Google Scholar]
- 62.Rubin EM, Krauss RM, Spangler EA, Verstuyft JG, Clift SM. Inhibition of early atherogenesis in transgenic mice by human apolipoprotein AI. Nature. 1991;353(6341):265–267. [DOI] [PubMed] [Google Scholar]
- 63.Barrett TJ, Distel E, Murphy AJ, et al. Apolipoprotein AI) Promotes Atherosclerosis Regression in Diabetic Mice by Suppressing Myelopoiesis and Plaque Inflammation. Circulation. 2019;140(14):1170–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Beviglia L, Poggi A, Rossi C, et al. Mouse antithrombotic assay. Inhibition of platelet thromboembolism by disintegrins. Thromb Res. 1993;71(4):301–315. [DOI] [PubMed] [Google Scholar]
- 65.Benjamin EJ, Blaha MJ, Chiuve SE, et al. Heart Disease and Stroke Statistics-2017 Update: A Report From the American Heart Association. Circulation. 2017;135(10):e146–e603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ageno W, Becattini C, Brighton T, Selby R, Kamphuisen PW. Cardiovascular risk factors and venous thromboembolism: a meta-analysis. Circulation. 2008;117(1):93–102. [DOI] [PubMed] [Google Scholar]
- 67.Doggen CJ, Smith NL, Lemaitre RN, Heckbert SR, Rosendaal FR, Psaty BM. Serum lipid levels and the risk of venous thrombosis. Arterioscler Thromb Vasc Biol. 2004;24(10):1970–1975. [DOI] [PubMed] [Google Scholar]
- 68.Eichinger S, Pecheniuk NM, Hron G, et al. High-density lipoprotein and the risk of recurrent venous thromboembolism. Circulation. 2007;115(12):1609–1614. [DOI] [PubMed] [Google Scholar]
- 69.Sacks FM, Rudel LL, Conner A, et al. Selective delipidation of plasma HDL enhances reverse cholesterol transport in vivo. J Lipid Res. 2009;50(5):894–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yamashita S, Tsubakio-Yamamoto K, Ohama T, Nakagawa-Toyama Y, Nishida M. Molecular mechanisms of HDL-cholesterol elevation by statins and its effects on HDL functions. J Atheroscler Thromb. 2010;17(5):436–451. [DOI] [PubMed] [Google Scholar]
- 71.Naqvi TZ, Shah PK, Ivey PA, et al. Evidence that high-density lipoprotein cholesterol is an independent predictor of acute platelet-dependent thrombus formation. Am J Cardiol. 1999;84(9):1011–1017. [DOI] [PubMed] [Google Scholar]