

SUMMARY
Breakdown of the blood-central nervous system barrier (BCNSB) is a hallmark of many neuroinflammatory disorders, such as multiple sclerosis (MS). Using a mouse model of MS, experimental autoimmune encephalomyelitis (EAE), we show that endothelial-to-mesenchymal transition (EndoMT) occurs in the CNS before the onset of clinical symptoms and plays a major role in the breakdown of BCNSB function. EndoMT can be induced by an IL-1β-stimulated signaling pathway in which activation of the small GTPase ADP ribosylation factor 6 (ARF6) leads to crosstalk with the activin receptor-like kinase (ALK)-SMAD1/5 pathway. Inhibiting the activation of ARF6 both prevents and reverses EndoMT, stabilizes BCNSB function, reduces demyelination, and attenuates symptoms even after the establishment of severe EAE, without immunocompromising the host. Pan-inhibition of ALKs also reduces disease severity in the EAE model. Therefore, multiple components of the IL-1β-ARF6-ALK-SMAD1/5 pathway could be targeted for the treatment of a variety of neuroinflammatory disorders.
In Brief
Loss of blood-CNS barrier (BCNSB) function is a hallmark of neuroinflammatory diseases, including MS. Sun et al. show that ARF6-mediated crosstalk between IL-1β and ALK signaling induces presymptomatic endothelial-to-mesenchymal transition (EndoMT), causing BCNSB disruption and disease progression in a mouse MS model. Inhibiting this pathway reverses EndoMT and disease severity, suggesting a new therapeutic strategy to treat MS.
INTRODUCTION
Neuroinflammatory disorders comprise many conditions in which the host’s immune response damages the nervous system. Multiple sclerosis (MS), an example of a neuroinflammatory disorder, is characterized by chronic inflammation and demyelination of the central nervous system (CNS) and is one of the most prevalent neurological disorders leading to long-term disability among young adults (Aharoni, 2014; Schampel et al., 2017). Current and emerging treatment options for MS target the host immune response and include natalizumab (Fletcher et al., 2010; Polman et al., 2006), which inhibits the penetration of immune T cells across the blood–central nervous system barrier (BCNSB) into the CNS parenchyma, and fingolimod (Choi et al., 2011) and siponimod (Behrangi et al., 2019), which hinder egression of immune cells from lymph nodes to the circulatory system. However, such immunosuppressive drugs can have serious side effects. For example, over 700 cases of often-fatal natalizumab-associated progressive multifocal leukoencephalopathy were reported in MS patients between its approval for clinical use in 2004 through December 2017 (Abbott et al., 2010; Faulkner, 2015; Fletcher et al., 2010; Schwab et al., 2015). Therefore, it is important to identify new molecular targets for the development of novel therapeutic strategies that do not immunocompromise the patient.
The BCNSB is a dynamic and complex cellular system composed of tight junction-forming endothelial cells, astrocytes, and pericytes that strictly controls the exchange of cells and molecules between the bloodstream and CNS parenchyma (Abbott et al., 2010; Kim et al., 2006; Palmer, 2010, 2013). However, in MS patients, this barrier exhibits increased permeability early in the disease process (Kermode et al., 1990a; Kermode et al., 1990b; Stone et al., 1995). Likewise, in mouse and marmoset experimental autoimmune encephalomyelitis (EAE) models (experimental models for MS), the BCNSB breakdown occurs prior to the appearance of inflammatory demyelinating lesions (Aubé et al., 2014; Maggi et al., 2014; Munji et al., 2019). BCNSB disruption leads to edema, disruption of metabolic function, excitotoxicity, and parenchymal entry of autoantibodies and plasma proteins (Chapouly et al., 2015; LeVine, 2016; Meinl, 2011; Oliveira et al., 2017) such as fibrinogen (Akassoglou et al., 2002) that contribute to a hostile CNS environment and promote demyelination both by inducing the recruitment of peripheral inflammatory macrophages/monocytes and T cells and by inhibiting oligodendrocyte-mediated remyelination (Adams et al., 2007; Akassoglou et al., 2002; Davalos et al., 2012; Petersen et al., 2017; Ryu et al., 2015). A growing body of evidence indicates that BCNSB disruption is not only one of the early pathophysiological hallmarks of MS but that it also plays a critical pathogenic role in the initial progression and amplification of disease (Argaw et al., 2012; Chapouly et al., 2015; Horng et al., 2017; Podjaski et al., 2015; Smith et al., 2015). Disease severity is enhanced by the loss of endothelial cell platelet/endothelial cell adhesion molecule-1 (PECAM-1/CD31) (Graesser et al., 2002; Privratsky and Newman, 2014) or activated leukocyte cell adhesion molecule (ALCAM/CD166) (Lécuyer et al., 2017), both of which promote optimal BCNSB function. Reduced clinical severity has been observed in acute EAE in endothelial-specific Il-1r (Li et al., 2011) or astrocyte-specific Vegfa knockout mice (Argaw et al., 2012) and in mice treated with the barrier-stabilizing factors netrin-1 (Podjaski et al., 2015) or angiopoietin (Ang-1) (Jiang et al., 2014). Thus, the BCNSB controls primary molecular and cellular mechanisms that are thought to play key roles in the etiology of MS.
Mesenchymal markers have been observed in the brain endothelium of MS patients, suggesting a potential role for endothelial-to-mesenchymal transition (EndoMT) in MS pathophysiology, possibly by disrupting BCNSB function (Derada Troletti et al., 2016; Derada Troletti et al., 2019). EndoMT is a process whereby an endothelial cell undergoes a series of molecularly-driven events leading it to assume a mesenchymal cell phenotype (Platel et al., 2019). The transforming growth factor β (TGF-β)/bone morphogenetic protein (BMP) signaling pathway, via either SMAD or non-SMAD signaling, plays a pivotal role in EndoMT by inducing the expression of transcription factors, such as Snail (SNAI1), Slug (SNAI2), or Twist, which increase the expression of mesenchymal markers, including N-cadherin, and repress the expression of endothelial cell adhesion molecules, such as vascular endothelial (VE)-cadherin or CD31 (Ma et al., 2020; Pardali et al., 2017). EndoMT leads to a reorganization of the microvasculature via cytoskeletal remodeling and an increase in endothelial permeability (Platel et al., 2019), a key player in the inflammatory process (Cho et al., 2018).
Our previous studies have demonstrated that the small GTPase ADP ribosylation factor 6 (ARF6) acts as a central convergence point for both vasculature-destabilizing and stabilizing pathways. Interleukin 1β (IL-1β) (Zhu et al., 2012) and VEGF (Zhu et al., 2017) disrupt endothelial integrity through the activation of ARF6 and subsequent disassembly of cell-cell junctions, while Slit2-Robo4 signaling promotes vascular stability by blocking ARF6 activity (Jones et al., 2009). We thus ask whether the activation of ARF6 might play a major role in the pathophysiology of EAE by inducing the breakdown of the BCNSB. In this study, we show that EndoMT occurs in the CNS prior to the onset of clinical symptoms and that it is likely a driver for the loss of BCNSB function. EndoMT can be induced by IL-1β-activated ARF6 via crosstalk with the activin receptor-like kinase (ALK)-SMAD1/5 pathway. We further show that inhibiting ARF6 activation can both prevent and reverse EndoMT, stabilize BCNSB function, reduce demyelination, and reverse severe clinical symptoms in the EAE model of MS. Pan-inhibition of ALK receptors also inhibits disease progression in the EAE model, thus suggesting a possible new approach for the treatment of not only MS but other neuroinflammatory disorders that exhibit a breakdown of the BCNSB (Davalos et al., 2019; Drouin-Ouellet et al., 2015; Jenkins et al., 2018; Rosenberg, 2014; Sweeney et al., 2018; Zlokovic, 2008).
RESULTS
Endothelial-specific Arf6 knockout blocks disease progression and reduces vascular leakage in an experimental model of MS
To examine whether ARF6 is involved in the establishment and progression of EAE, we generated whole-body knockout mice (Arf6f/−; Rosa26-CreERT), endothelial-specific Arf6 knockout mice (Arf6f/f; Cdh5-CreERT2), T-cell specific Arf6 knockout mice (Arf6f/−; Lck-Cre), and hematopoietic cell specific Arf6 knockout mice (Arf6f/−; Vav1-iCre). We immunized 9-week-old mice with myelin oligodendrocyte glycoprotein (MOG35–55) to induce EAE and assessed clinical scores and body weight over a 28-day period. Whole body or endothelial-specific knockout of Arf6 at postnatal day 1 (P1) significantly reduced the severity of clinical scores and limited body weight loss (Figures S1A, 1A, 1B, S1B, and S1C), while no reduction in clinical scores was observed in the other knockout mice (Figure S1B). H&E and Luxol fast blue (LFB) staining of the spinal cords revealed reduced T cell infiltration and reduced demyelination, respectively, in endothelial-specific Arf6 knockout mice (Figure 1C). The numbers of CD4+, CD4+IFNγ+, CD8+, and CD8+IFNγ+ cells in the spinal cord were reduced (although reduction in CD8+ cells was not significant) (Figures 1D and S1D and S1E), while no statistically significant differences in the numbers of these cell types were observed in the blood, spleen, or lymph nodes (Figures S1F–S1H). Brain and spinal cord permeability were markedly reduced in endothelial-specific Arf6 knockout mice (Figures 1E–1G, S1I–1K). Overall, our data reveal that endothelial-specific loss of ARF6 stabilizes the BCNSB and significantly inhibits the progression of EAE without affecting the systemic levels of T cells.
Figure 1. Endothelial-specific Arf6 knockout in mice blocks disease progression and reduces CNS vascular leak in the mouse EAE model.
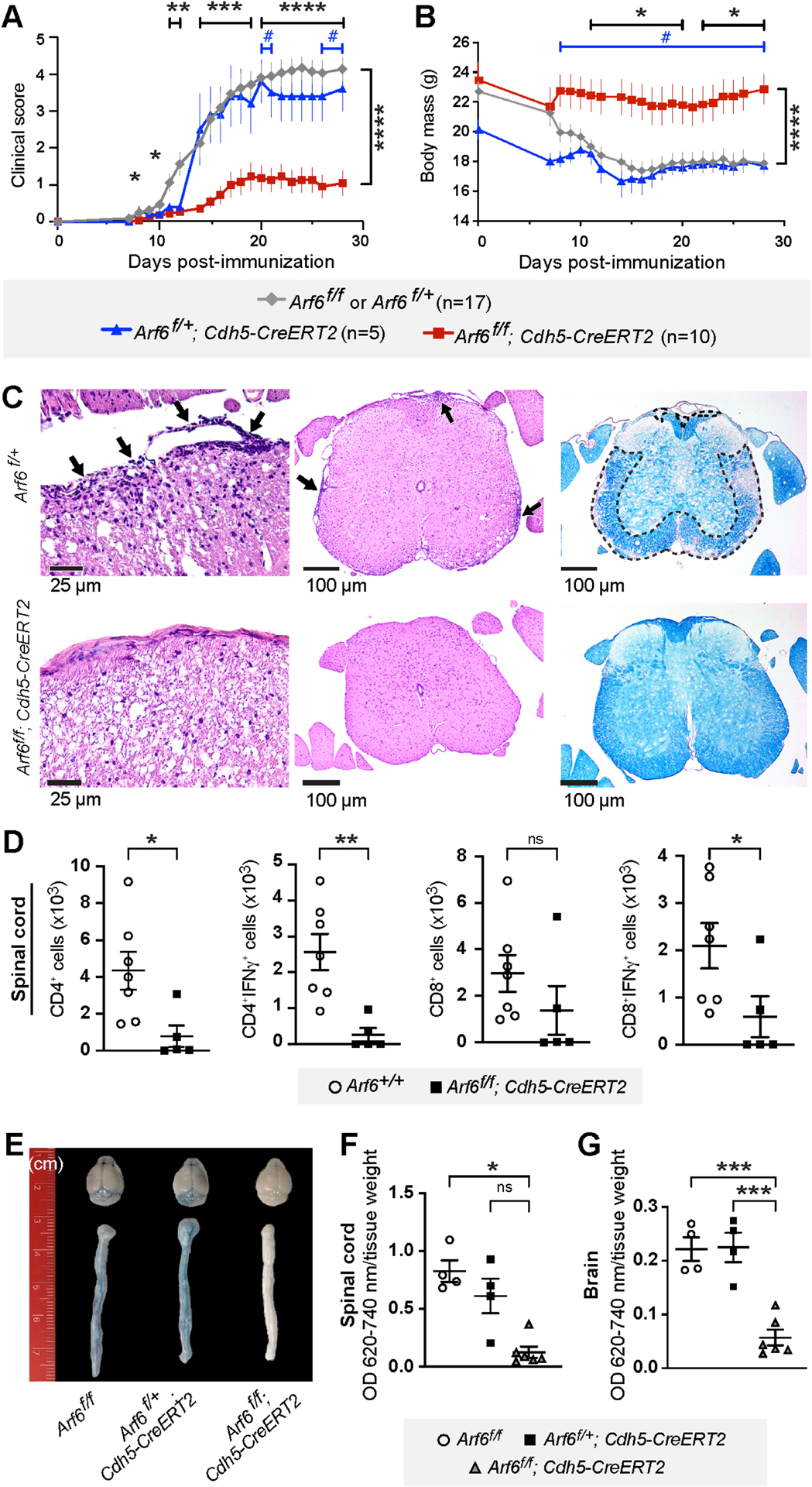
9-week-old endothelial-specific Arf6 knockout mice were immunized with myelin oligodendrocyte glycoprotein (MOG) to induce EAE. Results shown in panels A and B are a composite of two independent rounds of experiments.
(A) Clinical score of EAE induced in endothelial-specific Arf6 knockout mice (Arf6f/f; Cdh5-CreERT2) compared to littermate controls. # p<0.05, *p<0.05, **p<0.01, ****p<0.0001. *: Arf6f/f or Arf6f/+ vs Arf6f/f; Cdh5-CreERT2, #: Arf6f/+; Cdh5-CreERT2 vs Arf6f/f; Cdh5-CreERT2. Mixed-effects analysis with Tukey’s correction for multiple comparisons.
(B) Endothelial-specific Arf6 knockout prevents reduction of body weight in MOG-induced EAE model.
(C) Representative spinal cord sections. Left panels show that meningeal and parenchymal inflammation (arrows) in Arf6f/+ mice is absent in the Arf6f/f; Cdh5-CreERT2 mice, 400x H&E. Middle panels show H&E stain, 100x. Right panels show convergent foci of demyelinated white matter (outlined) of Arf6f/+ mice spinal cord. Normal myelination of Arf6f/f; Cdh5-CreERT2 mice. LFB stain, 100x.
(D) The total number of spinal cord CD4+, CD4+IFNγ+, CD8+, CD8+IFNγ+ T-cells in endothelial-specific Arf6 knockout EAE mice and their littermates was measured by flow cytometry on day 28 post-MOG immunization. Two-tailed Mann-Whitney test.
(E-G) ARF6 ablation by gene knockout in endothelial cells reduces brain and spinal cord vascular permeability as measured by Evans blue assay (14 days post-immunization). Kruskal-Wallis with Dunn’s multiple comparison test (F) and one-way ANOVA with Tukey’s correction for multiple comparisons (G).
Individual symbols shown in the graphs in panels D, F, and G represent a single mouse (n=4–7).
*p<0.05, **p<0.01, ***p<0.001, ns: not significant. All error bars represent the SEM.
See also Figure S1.
Pharmacologic inhibition of ARF6 is a potential therapeutic strategy for treating MS
Prophylactic treatment of wild-type C57BL/6J mice with NAV-2729 (daily intraperitoneal (IP) injection of 30 mg/kg), a small molecule ARF6 inhibitor (Yoo et al., 2016), reduced clinical scores, weight loss, and vascular leakage (Figures 2A–2C and S2A–2B). To determine whether inhibiting ARF6 might provide a potential therapeutic strategy for treating MS, we next treated EAE mice daily with NAV-2729 starting at 13 days post-EAE induction at a time point where clinical scores had nearly reached a maximum. NAV-2729 reduced clinical scores, prevented loss of body weight, and reduced T cell infiltration and demyelination (Figures 2D–2F). Flow cytometry revealed a significant reduction in CD4+, CD4+IFNγ+, CD8+, CD8+IFNγ+ cells infiltrating the spinal cord in the NAV-2729-treated group, while no statistically significant differences were observed in the different cell types in the blood, spleen, and lymph nodes (Figures 2G and S2C–S2F). One potential explanation for this phenotype is that NAV-2729 prevents the infiltration of immune cells into the CNS. In contrast to this idea, treatment with NAV-2729 did not significantly alter levels of total or virus-specific CD4+ T cells, total or virus-specific CD8+ T cells, or macrophages in the CNS of murine hepatitis virus strain JHMV-infected mice, nor did whole body knockout of Arf6 (Arf6f/−; Rosa26-CreERT) inhibit clearance of fecal bacteria following an intraperitoneal injection of 100 μL of cecal slurry (200 mg/mL) (Figure S3A–S3C). These results, combined with data from Figures S1F–S1H showing no change in the percentage of CD4+, CD4+IFNγ+, CD8+, CD8+IFNγ+ cells in the blood, spleen, or lymph nodes in endothelial cell-specific Arf6 knockout mice, indicate that blocking ARF6 activation using two approaches—Arf6 knockout and pharmacologic treatment—does not affect the host’s normal immune function, which is consistent with our previous published results (Gebremariam et al., 2020; Zhu et al., 2012). NAV-2729 treatment also inhibited EAE-induced vascular leakage in the brain and spinal cord (Figure 2H–2J). Taken together, these results suggest that blocking ARF6 activation stabilizes the BCNSB and reduces harmful proteins, such as fibrinogen, from entering the CNS parenchyma where they can cause additional damage and inflammation (Akassoglou et al., 2002; Chapouly et al., 2015; LeVine, 2016; Meinl, 2011; Oliveira et al., 2017). Reduction in damage and inflammation has the indirect effect of reducing immune cell infiltration, which otherwise would further damage neurons and exacerbate the disease. However, stabilizing the BCNSB does not appreciably inhibit immune cells from entering the CNS to fight infection presumably because the pathogen plays a major role in driving the initial immune response. These results suggest that pharmacologic inhibition of ARF6 could provide a novel approach for treating MS while still preserving an intact immune system for fighting viral reactivation.
Figure 2. Pharmacologic inhibition of ARF6 reverses symptoms and pathology in MOG-induced EAE mice.
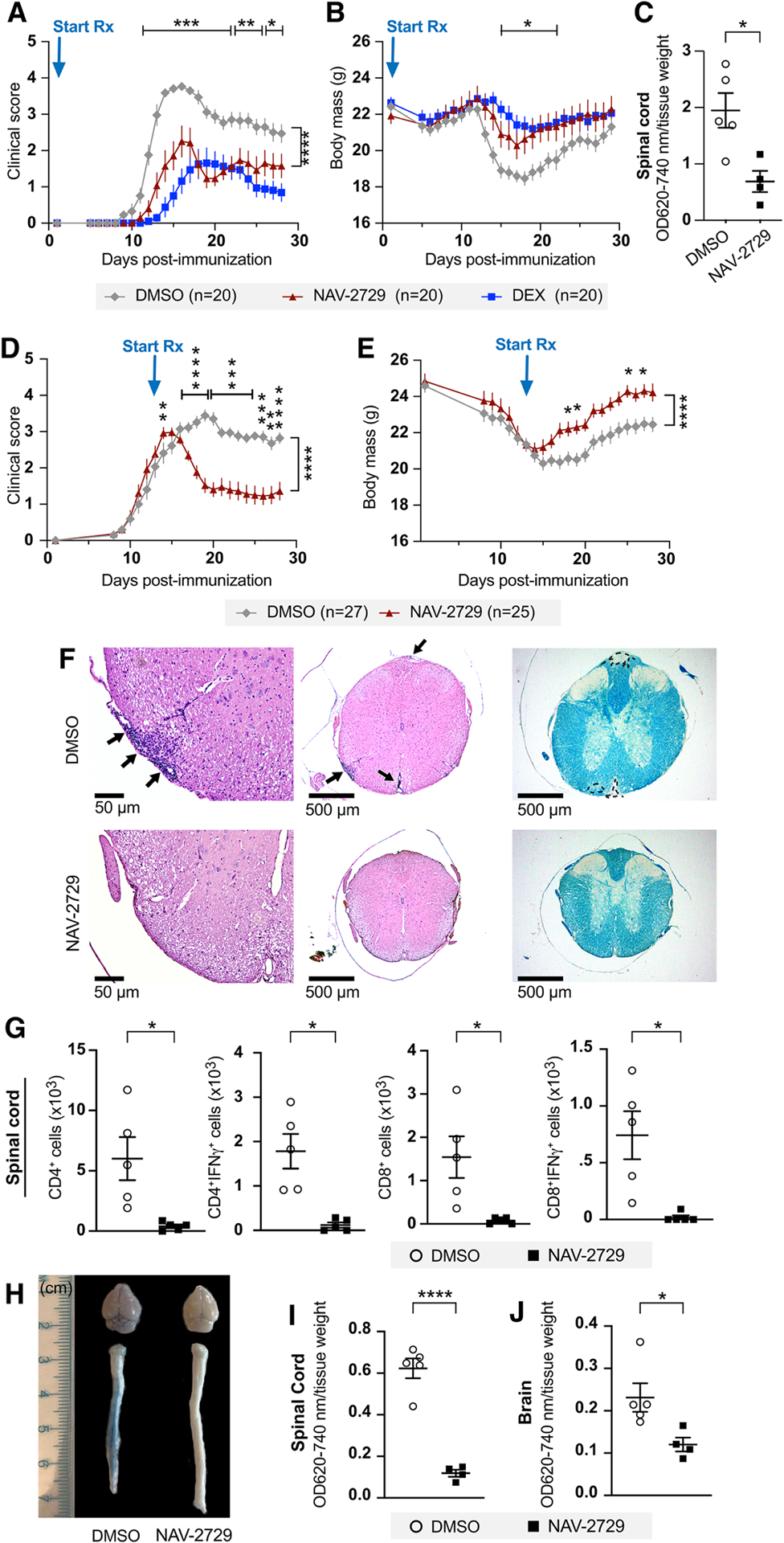
9-week-old C57BL/6J mice were immunized with MOG35–55 to induce EAE. Results shown in panels A and B are representative of two independent experiments and those shown in panels D-E are a composite of three independent rounds of experiments.
(A) Clinical scores in mice treated daily with intraperitoneal injection of the ARF6 inhibitor NAV-2729 (30 mg/kg) starting at day 1 post-MOG immunization.
(B) Changes in mouse body weight over the course of the experiment.
(C) Vascular permeability of spinal cords measured by Evans blue assay at 14 days post-immunization (daily treatments initiated on day 1).
Mice used for panels (D) through panel (J) were all treated daily with intraperitoneal injections of 30 mg/kg NAV-2729 starting at day 13 post-immunization.
(D, E) Clinical scores (D) and mouse body weight (E) over the course of the experiment. Two-way ANOVA with Šidák’s correction for multiple comparisons for panels A, B, D, and E.
(F) Representative spinal cord sections. Left panels show that meningeal and parenchymal inflammation (arrows) in DMSO control is reduced/absent in the NAV-2729-treated mice, 200x H&E. Middle panels show H&E stain, 40x. Right panels show convergent foci of demyelinated white matter (outlined) of spinal cord from DMSO-treated mice. Normal myelination of NAV-2729-treated mice. LFB stain, 40x.
(G) The total number of spinal cord CD4+, CD4+IFNγ+, CD8+, CD8+IFNγ+ T-cells in NAV-2729- and DMSO-treated mice were measured by flow cytometry on day 28 post-MOG immunization.
(H-J) Brain and spinal cord vascular permeability in NAV-2729-treated mice at day 21 post-immunization.
A two-tailed Welch’s t test was used for panel C and the first three graphs of panel G, a Mann-Whitney test was used for the right graph of panel G, a Student’s t test for panel I, and a Mann-Whitney test for panel J. *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001. All error bars represent the SEM.
Individual symbols shown in the graphs in panels C, G, I, and J represent a single mouse (n=4 or 5).
See also Figures S2 and S3.
Endothelial-specific loss of Arf6 or systemic inhibition of ARF6 reduces EndoMT in CNS blood vessels
To determine whether vascular leakage may play a role in the early progression of EAE that leads to clinical symptoms, we assessed vascular permeability in the brains and spinal cords of C57BL/6J EAE mice one day before clinical symptoms became apparent (day 6 post-EAE induction) using an Evans blue assay and found that indeed vascular permeability was increased at this presymptomatic time point with much of it occurring in the brain and in the caudal spinal cord (Figures 3A, S4A, and S4B). Permeability increased and spread to the entire spinal cord at days 14 and 21 post-immunization (Figures 3B, 3C, and S4C–S4F). Consistent with this result, we also found that vascular permeability is present by day 6 post-immunization in the brain and caudal region of spinal cord in Arf6f/f and Arf6f/+; Cdh5-CreERT2 mice but is reduced in Arf6f/f; Cdh5-CreERT2 mice and markedly increases by day 15 in Arf6f/f and Arf6f/+; Cdh5-CreERT2 mice, while remaining largely absent in Arf6f/f; Cdh5-CreERT2 mice (Figures S4G–S4I and 1E–1G). It should be noted that EAE symptoms were first manifested as impaired motor function in the tail on day 7, which correlates with the vascular leak first appearing in the caudal region of the spinal cord.
Figure 3. Endothelial-specific loss of Arf6 or systemic inhibition of ARF6 reduces EndoMT in microvessels isolated from spinal cords of EAE mice.
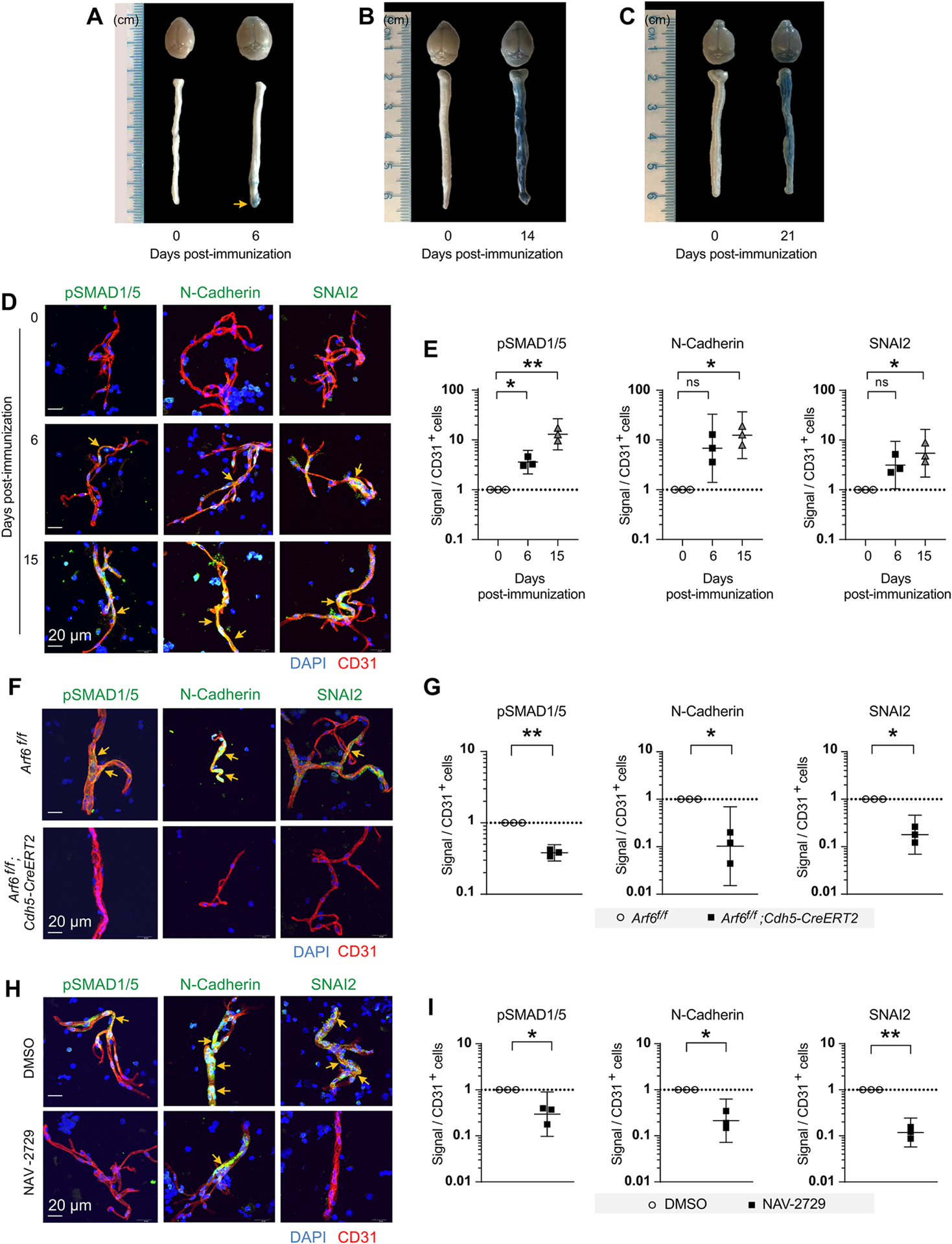
(A-C) Vascular permeability in the brains and spinal cords of C57BL/6J EAE mice was assessed one day before clinical symptoms became apparent (day 6 post-EAE induction) (A), at day 14 post-EAE induction (B), and at day 21 post-EAE induction (C) using an Evans blue assay.
(D, E) EndoMT in isolated spinal cord microvessels at 6 and 15 days post-EAE induction was assessed by examining pSMAD1/5, N-Cadherin, and SNAI2 expression using immunofluorescence.
(F, G) Phosphorylated SMAD1/5 and the EndoMT markers were assessed in the spinal cord microvessels of EAE-induced endothelial-specific Arf6 knockout mice. The tissues were harvested at day 28 post-EAE induction.
(H, I) Treatment with NAV-2729 (30 mg/kg IP daily) was initiated on day 13 post-EAE induction (a time when EndoMT markers were high) and continued daily through day 21. Phosphorylated SMAD1/5 and the EndoMT markers were assessed in the spinal cord microvessels of EAE mice.
A one-way randomized block ANOVA with Dunnett’s correction for multiple comparisons was used for panel E. A two-tailed ratio paired t test was used for panels G and I. *p<0.05 and **p<0.01. All error bars represent the SEM.
Individual symbols shown in the graphs in panels E, G, and I represent a single mouse (n=3).
See also Figures S4–S6.
A previous study showed that EndoMT can be detected in the brains of MS patients at autopsy, suggesting that EndoMT might play a role in MS pathophysiology possibly by inducing vascular permeability (Derada Troletti et al., 2016; Derada Troletti et al., 2019). Therefore, we hypothesized that EndoMT might be a driver of BCNSB disruption in the EAE model. To test this hypothesis, we isolated spinal cord microvessels at 6 and 15 days post-EAE induction and examined pSMAD1/5, N-Cadherin, and SNAI2 (which can be found in both the cytoplasm and nucleus) (Hill et al., 2019; Hultgren et al., 2020) expression using immunofluorescence and found evidence for EndoMT at day 6 (statistically significant for pSMAD1/5) with increased and statistically significant EndoMT signals for all markers at day 15 (Figures 3D, 3E, and S4J–S4L). Increased EndoMT signals persisted at 21- and 28-days post EAE-induction (Figure S5A). To further address whether EndoMT might be driving vascular leak, we searched fluorescently labeled brain and spinal cord tissue sections for colocalization of EndoMT lesions and vascular leakage and identified EndoMT sites adjacent to areas where Alexa fluor™ 647-labeled albumin had leaked into the CNS parenchyma (Figure S6). To assess whether ARF6 may be required for EndoMT, we isolated spinal cord microvessels from EAE-induced endothelial-specific Arf6 knockout mice and found that pSMAD1/5 and the EndoMT markers were greatly reduced compared to wild-type mice (Figures 3F, 3G, and S5B–S5D). Although EndoMT sites were more difficult to identify in CNS tissue sections and therefore quantification was not possible, our limited data from spinal cord tissue sections were consistent with the data obtained from isolated microvessels, showing a reduction in EndoMT in EAE-induced endothelial-specific Arf6 knockout mice (Figure S5E). Initiation of NAV-2729 administration (30 mg/kg IP daily for 7 days) at day 13 post-EAE induction (a time when EndoMT markers were high) and continued daily administration through day 21 significantly reduced the EndoMT markers pSMAD1/5, N-Cadherin, and SNAI2 (Figures 3H, 3I, S5F–S5H). Together, these results indicate that EndoMT is a catalyst for BCNSB dysfunction and that EAE pathophysiology can be reversed by ARF6 inhibition.
Crosstalk between IL-1β and ALK signaling induces EndoMT in human brain microvascular endothelial cells
Whole-body or endothelial-specific knockdown of Il-1r1 in mice reduces disease severity in the EAE mouse model (Li et al., 2011; Lukens et al., 2012). In addition, certain cytokines (e.g., IL-1β, TNFα) can induce EndoMT, resulting in the loss of endothelial barrier function (Cho et al., 2018), and ARF6 can be activated by IL-1β to increase endothelial permeability (Zhu et al., 2012). To determine whether IL-1β may be driving EndoMT and vascular leakage, we treated human brain microvascular endothelial cells (HBMECs) with IL-1β. IL-1β induced both morphological changes in endothelial cells (Figures 4A and S7A) and EndoMT markers at 12 h and 20 h with CD31 (an endothelial cell marker) being downregulated and Snail and N-cadherin (mesenchymal markers) being upregulated (Figures 4B and S7B–S7G). In addition, IL-1β activated pSMAD1/5 within 15 min (Figure 4C). Similarly, IL-1β also induced EndoMT in mouse brain microvascular endothelial cells (MBMECs) (Figure S7H–S7K). Blocking IL-1 receptor (IL-1R) signaling with Interleukin-1 receptor antagonist (IL-1RA) reduced phosphorylation of SMAD1/5 within the same time frame (Figures 4D, S7L, and S7M). We also blocked IL-1R in vivo in EAE mice using anakinra (a pharmaceutical drug version of IL-1RA) and observed a marked reduction in EndoMT at 14 days post-EAE induction (Figure S7N), suggesting that IL-1β signaling is important for EndoMT in the EAE model. We hypothesized that IL-1R agonism activated the SMAD pathway through crosstalk with signaling pathways activated by activin receptor-like kinases (ALKs), which are type I receptors for TGF-β/BMPs and are known to regulate SMADs. To test this idea, we treated IL-1β-stimulated HBMECs with the ALK inhibitors LDN193189 (Vogt et al., 2011), SB431542 (Maddaluno et al., 2013), or both LDN193189 and SB431542. When used in combination, these inhibitors block IL-1β-induced activation of SMAD1/5 (Figures 4E and S8A) and significantly upregulate CD31 and downregulate Snail and N-Cadherin (Figures 4F and S8B–S8D). With monotherapy, the changes in EndoMT markers were not as pronounced and, in most cases, not statistically significant (Figure S8E–S8L). These results are consistent with the known functions of various ALKs in endothelial cells and the inhibitory specificities of the inhibitors. LDN193189 inhibits ALK1, ALK2, ALK3, and ALK6, while SB431542 inhibits ALK4, ALK5, and ALK 7. In endothelial cells, both ALK1 and ALK5 have been shown to play a role in SMAD1/5 activation (Daly et al., 2008; Goumans et al., 2003a; Goumans et al., 2003b; Goumans et al., 2002); therefore, observing an enhanced inhibition of EndoMT in the presence of both inhibitors was not surprising. These results indicate that IL-1β can activate SMAD1/5 in an ALK-dependent manner, suggesting that crosstalk between IL-1β and ALK signaling induces EndoMT.
Figure 4. Crosstalk between IL-1β and ALK receptor signaling induces EndoMT in HBMECs.
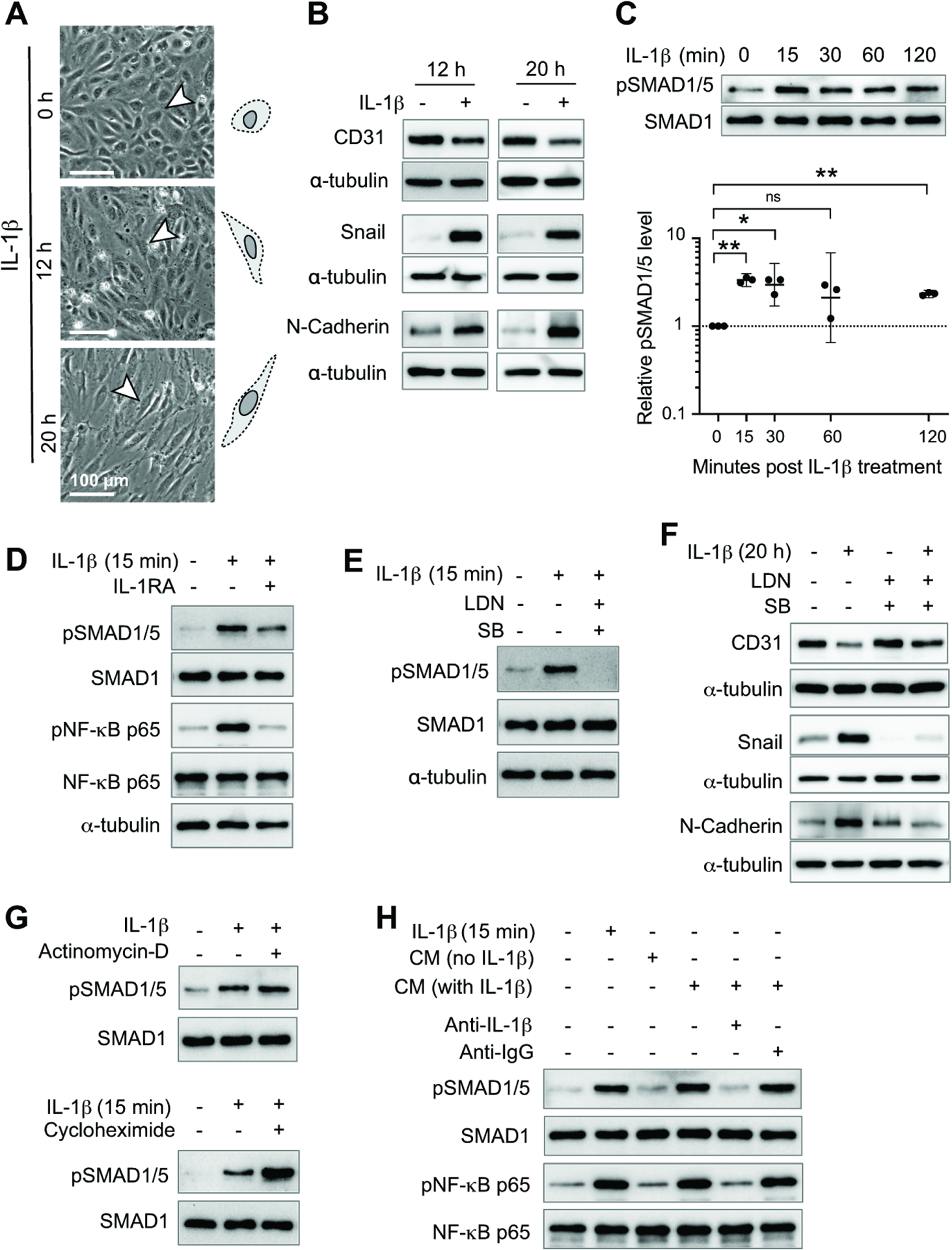
(A) Morphological changes in HBMECs after IL-1β treatment. Quantification of changes in cell morphology is shown in Figure S7A.
For panels B-H, western blotting was used to assess protein levels.
(B) Endothelial cell marker CD31 and mesenchymal markers Snail and N-cadherin were assessed in HBMECs treated with IL-1β for 12 or 20 h.
(C) Levels of pSMAD1/5, an important ALK receptor downstream signaling molecule, were assessed in HBMECs at different time points following IL-1β treatment. One-way randomized block ANOVA with Dunnett’s correction for multiple comparisons following log10 transformation. *p<0.05, **p<0.01, ns: not significant. Geometric means and 95% confidence intervals are shown.
(D) HBMECs treated with or without IL-1RA were stimulated with IL-1β for 15 min. The level of pSMAD1/5 and pNF-κB p65 were assessed.
(E) HBMECs treated with or without LDN193189 (LDN) and SB431542 (SB) were stimulated with IL-1β for 15 min and pSMAD1/5 levels were assessed.
(F) HBMECs treated with or without LDN and SB were stimulated with IL-1β for 20 h and the level of EndoMT markers were assessed.
(G) HBMECs treated with or without actinomycin-D or cycloheximide were stimulated with IL-1β for 15 min and the levels of pSMAD1/5 was assessed.
(H) Conditional media was collected from HBMECs treated with IL-1β for 15 min and incubated with anti-IL-1β neutralization antibody or IgG for 1 h. These conditional media were then used to treat starved HBMECs for 15 min and the level of pSMAD1/5 and pNF-κB p65 were then assessed.
Individual symbols shown in the graph in panel C represents data from a single western blot.
All other panels show representative western blots that are quantified in Figures S5 and S6. For each experiment, 3–5 independent western blots were generated.
See also Figures S7 and S8.
The rapid activation of SMAD1/5 by IL-1β suggests that the mechanism of activation may be independent of transcription and translation. To test this hypothesis directly, we treated HBMECs with IL-1β in the presence of either the transcription inhibitor actinomycin D or the translation inhibitor cycloheximide and observed no decrease in the activation of pSMAD1/5 (Figures 4G, S8M, and S8N). Transcription- and translation-independent activation of ALK (indicated by increased pSMAD1/5 levels) by IL-1β-activated ARF6 could occur by at least two different mechanisms: 1) intracellular activation or 2) secretion of pre-existing cytoplasmic pools of ALK receptor ligands. To distinguish between these two possible mechanisms, we collected conditioned media (CM) from HBMECs that had been treated with media containing either 0.1% BSA (starved) or IL-1β (20 ng/mL) in 0.1% BSA for 15 min after being starved in 0.5% BSA for 16 h. We then treated starved (16 h) HBMECs for 15 min with either these conditioned media or various control media as follows: 1) 0.1% BSA alone; 2) IL-1β in 0.1% BSA; 3) conditioned medium from starved cells lacking IL-1β; 4) conditioned medium from cells treated with IL-1β; 5) conditioned medium from cells treated with IL-1β and spiked with neutralizing IL-1β antibody; and 6) conditioned medium from cells treated with IL-1β and spiked with IgG. We observed SMAD1/5 activation only when IL-1β was present and not neutralized by its antibody, while cIL-1β and its neutralizing antibody functioned properly as indicated by appropriate NF-κB activation (Figures 4H, S8O, and S8P). These results demonstrate that the conditioned media did not contain enough ALK receptor ligands to activate SMAD1/5 and suggest that the activation of the ALK receptors was via an intracellular mechanism rather than secretion of pre-existing ALK ligands.
ARF6 is both necessary for IL-1β-induced EndoMT and sufficient to induce EndoMT in HBMECs
Our data indicate that ARF6 activation is required for the pathophysiology observed in EAE, including clinical symptoms, vascular leakage, and EndoMT and that IL-1β can induce ALK-dependent EndoMT in HBMECs. To explore whether ARF6 is required for IL-1β-driven EndoMT, we treated HBMECs with IL-1β and observed activation of ARF6 within 5 min (Figure 5A). Likewise, IL-1β decreased endothelial barrier function of HBMEC monolayers over an extended period of more than 26 h, while inhibition of ARF6 by dominant-negative ARF6T27N increased barrier function of IL-1β-treated monolayers (Figure 5B). ARF6T27N also inhibited the rapid activation of SMAD1/5 by IL-1β (15 and 30 min) without affecting the activation of NF-κB (Figures 5C, S9A, and S9B). At a later time point (20 h), ARF6T27N prevented IL-1β-induced loss of CD31 and decreased the expression levels of Snail and N-cadherin (Figures 5D and S9C–S9E). ARF6T27N also prevented IL-1β from inducing an elongated spindle-shaped cell morphology in HBMECs (Figures 5E and S9F). Constitutively active ARF6 (ARF6Q67L) activated SMAD1/5 and induced EndoMT (Figures 5F and S9G–S9J). ARF6Q67L also enhanced the phosphorylation of SMAD1/5, while the ALK inhibitors LDN193189 and SB431542 inhibited this activation in HBMECs (Figure 5G and S9K), suggesting either that ARF6 lies upstream of ALK or is part of a separate pathway that activates SMAD1/5 interdependently with the ALK pathway. These data indicate that ARF6 activation is both necessary for IL-1β-induced EndoMT and sufficient to induce EndoMT in HBMECs.
Figure 5. ARF6 is both necessary for IL-1β-induced EndoMT and sufficient to induce EndoMT in HBMECs.
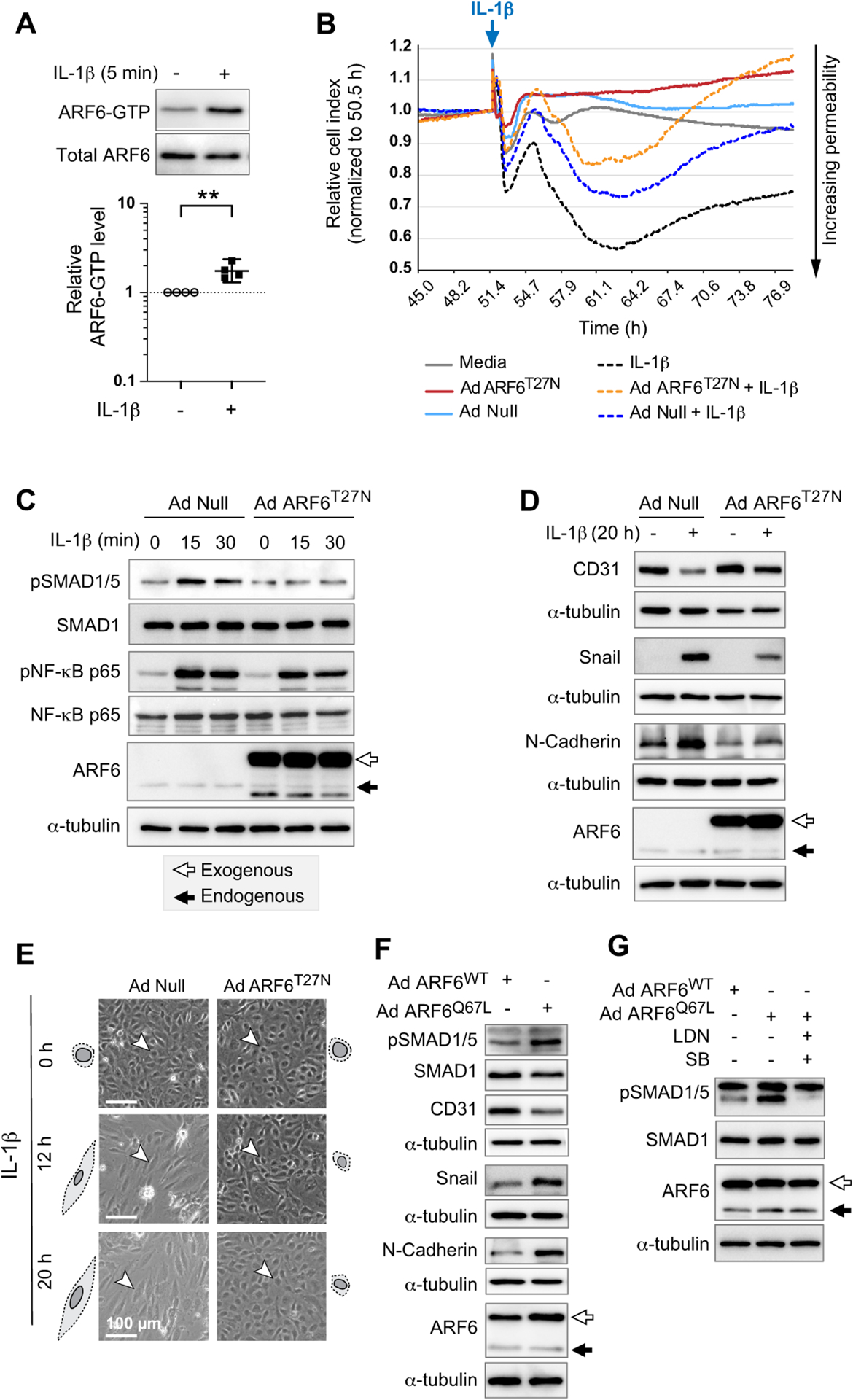
(A) IL-1β treated HBMECs were assayed for ARF6 activation using an ARF6-GTP pulldown assay. Two-tailed ratio paired t test. **p<0.01. Geometric mean and 95% confidence interval shown.
(B) AdNull-infected HBMEC monolayer stimulated with IL-1β causes a decrease in cell index (black dot data points) compared to control media treated cells (grey line data points) as measured by TEER. Ectopic expression of dominant-negative ARF6T27N reduces IL-1β-induced permeability (orange dot compared to blue dot data points). Representative of 2 independent experiments.
(C) HBMECs transduced with AdNull or AdARF6T27N were stimulated with IL-1β for 15 or 30 min. The levels of pSMAD1/5 and pNF-κB p65 were assessed by western blot.
(D) HBMECs transduced with AdNull or AdARF6T27N were stimulated with IL-1β for 20 h. The level of EndoMT markers were assessed by western blot.
(E) HBMECs transduced with AdNull or AdARF6T27N were stimulated with IL-1β. Changes in cell morphology were monitored at 12- and 20-h of treatment with IL-1β and are quantified in Figure S7F.
(F) The level of EndoMT markers in HBMECs transduced with AdARF6WT or AdARF6Q67L were determined by western blotting.
(G) HBMECs transduced with AdARF6WT or AdARF6Q67L were treated with LDN193189 (LDN) and SB431542 (SB). The level of pSMAD1/5 was assayed by western blot. A band larger than the expected size for pSMAD1/5 appears whenever cells are transduced with Ad ARF6WT or AdARF6Q67L (F, G). Therefore, quantification is done only on the band of the correct size.
Panels C, D, F, and G show representative western blots that are quantified in Figure S9. For each experiment, 3–5 independent western blots were generated.
See also Figure S9.
Inhibition of ALK receptors reduces vascular leakage and blocks the progression of EAE
Our in vivo and cellular data suggested that ARF6 plays an essential role in regulating the pathophysiology of EAE through its activation of ALKs, leading to EndoMT and the breakdown of the BCNSB and vascular leakage. This implies that blocking ALKs should reduce clinical symptoms, demyelination, EndoMT, and vascular leak in the EAE model of MS. Therefore, we treated EAE mice with the ALK inhibitors LDN193189 and SB431542 daily from day 1 to day 28 post-EAE induction. Inhibition of ALKs decreased clinical scores (Figure 6A), stabilized body weight (Figure 6B), and reduced T cell infiltration and demyelination (Figure 6C), EndoMT (Figures 6D, 6E, and S10A–S10C), and vascular permeability in the spinal cord and brain (Figure 6F and 6G). ALK inhibition did not appear to inhibit the phytohemagglutinin (PHA-L) activation of T cells as measured by mRNA levels of CD68, CD69, GATA3, and INFγ (Figure S10D) nor did we find any evidence indicating that in vivo ALK inhibition significantly altered the viability of T cells or the levels of the following EAE-associated T cells (Gartner et al., 2006; Grewal et al., 2001; Perrin et al., 1999): CD4+CD25+, CD4+CD28+,CD4+CD62L+CD44(Low)+, CD4+CD62L(Low)+CD44(High)+, CD8+CD25+, CD8+CD28+, CD8+CD62L+CD44(Low)+, and CD8+CD62L(Low)+CD44(High)+ (Figure S10E). These latter results suggest that the effects we observe with ALK inhibition are not due to systemic immune suppression. These data provide confirmatory evidence that the ARF6-ALK-SMAD1/5 pathway leads to EndoMT and vascular leak, which contribute to the pathophysiology of EAE.
Figure 6. Inhibition of ALK receptors reduces vascular leakage and blocks the progression of MS.
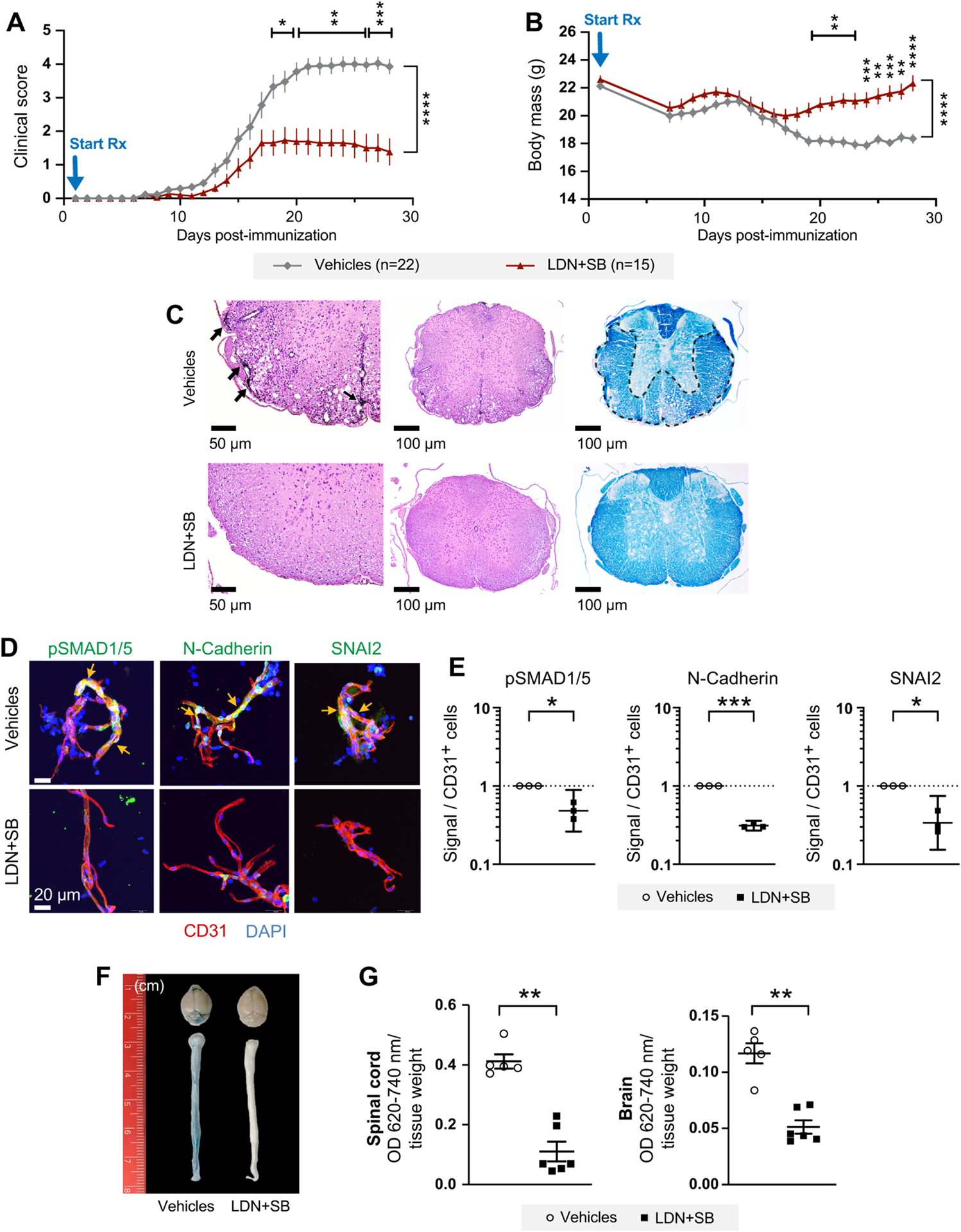
(A) Clinical scores in mice treated daily with intraperitoneal injections of LDN193189 and SB431542 (LDN+SB) starting at day 1 post-MOG immunization. Results shown in panels A and B are a composite of two independent rounds of experiments.
(B) LDN+SB improves body weight recovery in mice shown in panel A.
(C) Representative spinal cord sections. Left panels show that meningeal and parenchymal inflammation (arrows) in spinal cord from DMSO+H2O-treated mice is absent in the LDN+SB-treated mice, 200x H&E. Middle panels show H&E stain, 100x. Right panels show convergent foci of demyelinated white matter (outlined) of spinal cord from DMSO+H2O-treated mice. Normal myelination of LDN+SB-treated mice. LFB stain, 100x.
(D, E) Phosphorylated SMAD1/5 and the EndoMT markers were assessed in the spinal cord microvessels of EAE mice at day 28 post-immunization. Two-tailed ratio paired t tests.
(F, G) Vascular permeability was measured in spinal cords by the Evans blue assay at day 14 post-immunization.
*p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001. All error bars represent the SEM. Individual symbols shown in the graphs in panels E and G represent a single mouse (n=3–6).
See also Figure S10.
DISCUSSION
In this study, we show that blocking ARF6 activation by whole body or endothelial-specific Arf6 knockout or by pharmacologic treatment with an ARF6 inhibitor (NAV-2729) enhances BCNSB function and reduces disease progression in the MOG35–55-induced EAE model. However, Arf6 knockout in T-cell and hematopoietic lineages does not offer such protection. There appears to be a potential difference in the degree of protection that is offered against EAE by whole-body versus endothelial-specific knockout of Arf6 with the latter offering greater protection (Figures 1A and S1B). This apparent difference in protection could result from genuine biological differences, such as compensation resulting from loss of Arf6 in cell types other than endothelial cells, or it could be caused by a variety of experimental variables, including, but not limited to, differences in promoters used to drive Cre recombinase expression and Cre excision efficiency. Treatment with NAV-2729, even after the establishment of severe EAE, markedly reduces clinical symptoms, stabilizes the BCNSB, reverses EndoMT, and deceases demyelination without immunocompromising the host. These results indicate that ARF6 plays a critical role in BCNSB function in the EAE model and provide preclinical rationale for targeting ARF6 as a therapeutic approach to treat MS.
Reducing T cell infiltration into the CNS to inhibit neuroinflammation and demyelination has long been a primary goal in the development of therapies for MS. In fact, one of the most successful drugs for treating MS is natalizumab, a monoclonal antibody against the α4-integrin chain of VLA-4 that reduces T cell transmigration across the vascular endothelium and into the CNS parenchyma (Fletcher et al., 2010; Polman et al., 2006). Natalizumab is currently reserved for patients with highly active relapsing-remitting MS because of its strong immunosuppressive effects and potential for inducing the fatal complication progressive multifocal leukoencephalopathy during JC virus reactivation (Faulkner, 2015; Fletcher et al., 2010; Schwab et al., 2015). Our present study shows that in the EAE model, endothelial-specific loss of ARF6 or its systemic inhibition by NAV-2729 significantly reduces T cell infiltration and vascular leakage in the CNS (Figures 1C–1E and 2F–2J) but does not affect the number of T cells in the lymph node, blood, or spleen (Figures S1F–S1H and S2D–S2F). In contrast, NAV-2729 does not change the percentage of virus-specific CD4+ and CD8+ T-cells that have infiltrated the brain in the acute phase of a mouse hepatitis viral brain infection nor does whole-body knockout of Arf6 prevent bacterial clearance following infection (Figure S3). Together, these results suggest that ARF6 inhibition does not directly block the recruitment of immune cells into the CNS through the BCNSB nor does it cause immunosuppression; rather, it indirectly decreases the number of CNS immune cells during EAE by inhibiting the damage-induced neuroinflammation that is caused by infiltrating fluids and proteins (such as fibrinogen) (Cho et al., 2018; Davalos et al., 2019; Davalos et al., 2012) following breakdown of the BCNSB. Therefore, stabilizing the CNS vasculature and preventing fluid and protein leak via ARF6 inhibition should reduce T-cell-recruiting neuroinflammation caused by fluid and protein extravasation without blocking the ability of immune cells to clear acute viral infections in the CNS.
Previous studies have shown that proinflammatory signals, such as IL-1β and VEGF, activate ARF6, which switches off signaling pathways involved in the maintenance of the vascular endothelium and induces vascular destabilization and increased permeability by disrupting adherens junctions (Zhu et al., 2012; Zhu et al., 2017). These studies implied that ARF6 activation could be a major inducer of the vascular instability that plays a pivotal role in MS pathophysiology. However, we found that in the EAE model of MS, ARF6 can induce vascular permeability through an entirely different mechanism known as EndoMT. ARF6-dependent EndoMT occurs by day 6 post-EAE induction before clinical symptoms are present. Proinflammatory signals, such as IL-1β, activate ARF6, which in turn activates ALK through an unknown mechanism (Figure 7). One intriguing possibility is that ARF6 may promote the interaction between IL-1R and ALK, allowing crosstalk between these signaling pathways to occur. IL-1R can form a complex with TGFBR1 and TGFBR2 when stimulated with IL-1β (Lu et al., 2007), and we have previously shown that ARF6 can promote VEGF signaling by inducing the interaction between VEGFR2 and its coreceptor NRP1 (Zhu et al., 2017), thus making this potential mechanism an attractive hypothesis. Alternatively, ARF6 could be acting to promote interaction between other important components of the ALK signaling pathway, including interaction between SMAD1/5 and the ALKs that activate it. Once activated, the ALK signaling pathway promotes vascular leakage by inducing SMAD1/5 phosphorylation and EndoMT as indicated by downregulation of the endothelial marker CD31 and upregulation of the mesenchymal markers N-cadherin and Snail.
Figure 7. Schematic representation of the EndoMT in the BCNSB in response to inflammatory stimuli in MS.
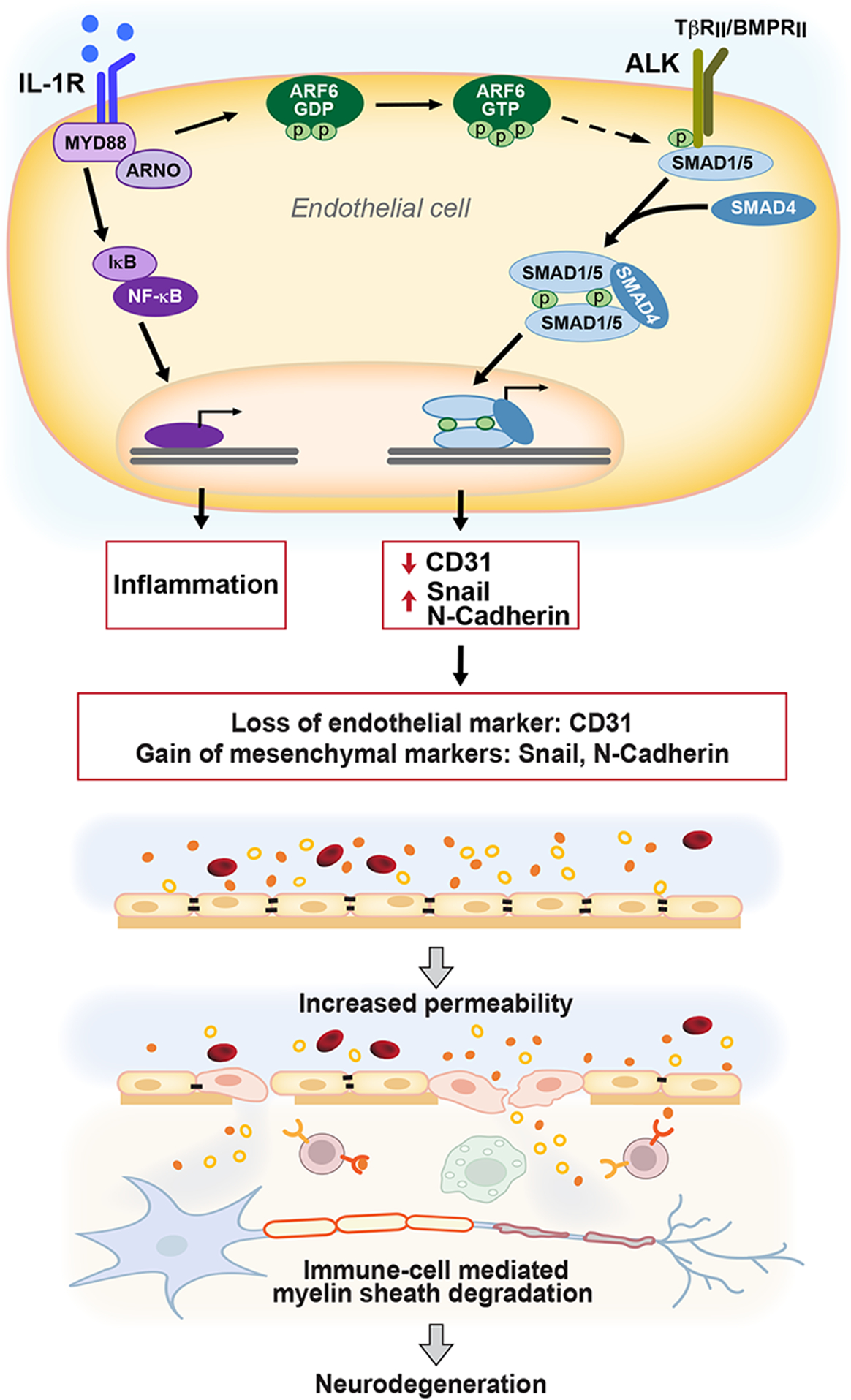
Under chronic inflammatory conditions, IL-1β induces ARF6 activation, promoting crosstalk with the ALK-SMAD1/5 pathway through a yet undetermined mechanism. The activation of SMAD1/5 stimulates the expression of transcriptional regulators, such as Snail, which results in loss of endothelial cell markers and acquisition of mesenchymal cell markers and contributes to vascular leakage of the soluble portion of the blood, including proteins (represented by small filled and open circular objects), but without hemorrhage (red blood cells shown as red discs). White blood cells are recruited to the sites of demyelination and inflammation in the CNS parenchyma. This process accelerates CNS inflammation and inhibits remyelination, which exacerbates symptoms and can lead to neurodegeneration. Although this study did not examine MYD88, ARNO, or SMAD4, previous studies have shown their importance in these signaling pathways (Watanabe et al., 2000; Zhu et al., 2012), and they are included in this diagram for completeness.
EndoMT has been implicated in brain endothelial cell dysfunction and brain pathologies (Evrard et al., 2016; Maddaluno et al., 2013). In EAE mice, we found evidence of both EndoMT (increased expression SNAI2 and N-Cadherin) and CNS vascular leakage at a very early time point (6 days post-EAE induction) prior to clinical manifestations of disease (Figures 3A, 3D, 3E, S4A, S4B, and S4G–S4I). Although other studies have shown that vascular leakage in the CNS occurs early in the disease process in the EAE model and that EndoMT can occur in MS patients at some point during the course of the disease (Argaw et al., 2012; Troletti et al., 2019), our in vivo data place EndoMT at the very early stages of the disease process in the EAE model before the onset of symptoms. Thus, EndoMT likely contributes to the early disruption of the BCNSB. We also show that EndoMT plays a critical role in the pathophysiology of EAE, given that reversing EndoMT by inhibiting ARF6 after the establishment of severe clinical symptoms not only restores BCNSB function but also reduces axon demyelination and significantly attenuates disease severity (Figures 2D–2J, 3H, 3I, and S5F–S5H). This latter finding is supported by our data demonstrating that pharmacologic inhibition of ALK also reduces EndoMT, vascular leak, axon demyelination, and clinical symptoms (Figures 6 and S10A–S10C), anakinra reduces EndoMT (Figure S7N), and ARF6 controls IL-1β-induced endothelial permeability and crosstalk between IL-1β and the EndoMT-driving ALK-SMAD1/5 pathway (Figure 5). These findings suggest that EndoMT is likely a major catalyst for the development of BCNSB dysfunction and neuroinflammation in MS pathophysiology.
Vascular leakage has also been implicated both in other neuroinflammatory disorders, including Alzheimer’s disease, Huntington’s disease, Parkinson’s disease (Drouin-Ouellet et al., 2015; Jenkins et al., 2018; Rosenberg, 2014; Sweeney et al., 2018; Zlokovic, 2008), and in chronic and acute inflammatory disorders, such as arthritis, diabetic retinopathy, and sepsis (Davis et al., 2014; Jones et al., 2009; Wallez and Huber, 2008; Zhu et al., 2012; Zhu et al., 2017). Although the mechanisms underlying vascular leakage in these other inflammatory disorders have largely been attributed to the breakdown of adherens junctions and tight junctions (Davis et al., 2014; Jones et al., 2009; Wallez and Huber, 2008; Zhu et al., 2012; Zhu et al., 2017), our data, coupled with a recent study (Glaser et al., 2020), suggest that EndoMT may also be a major contributor. Further investigation will be required to fully dissect the mechanisms that promote vascular leakage in all inflammatory disorders and the role EndoMT may play in this process. Regardless, the data we present here suggest that targeting ARF6 or components of its downstream signaling pathways may have broad implications for the treatment of many inflammatory disorders, including those primarily affecting the CNS.
STAR ☆ METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to the Lead Contact, Weiquan Zhu (weiquan.zhu@u2m2.utah.edu).
Materials availability
No new reagents were generated as a result of this study.
Data and code availability
All data reported in this paper will be shared by the Lead Contact upon request.
For this project, we used some custom code written in Matlab R2017b with the built-in image visualization toolbox. A simple GUI was used where images could be selected from the local machine and the amount of overlap between the staining with two separate antibodies was calculated. Use the attached GitHub link to read the description and see the custom code: https://github.com/learner97/CellImageAnalysis_Odelberg.
Any additional information required to reanalyze the data reported in this paper is available from the Lead Contact upon request.
EXPERIMENTAL MODELS AND SUBJECT DETAILS
All animal experiments were conducted following the NIH Guide for the Care and Use of Laboratory Animals (National Academies Press, 2011) and all animal protocols were approved by the University of Utah Institutional Animal Care and Use Committee (IACUC).
Mouse Strains
Arf6 was mutated by homologous recombination in mouse embryonic stem (ES) cells. Two alleles were generated. The conditional (f) allele contains 1 loxP site 5′ of the Arf6 gene at position 69,371,642 (UCSC genome browser) on chromosome 12 and a second loxP site 3′ of the Arf6 ORF at position 67,373,928. Whole-body knockout of Arf6 was achieved by crossing Arf6+/− mice with Arf6f/f; RosaCreERT mice and tamoxifen (50 μL at 1 mg/ml) induction at P1. P1 was chosen for the following reasons: 1) in our hands, Cre recombination occurs more efficiently at this time point than at later time points; 2) given that tamoxifen suppresses myelin antigen-specific T-cells, we needed to wait until it was cleared so we could avoid any residual effects of tamoxifen on the EAE disease course; and 3) whole-body loss of Arf6 after birth has no apparent effect on the development or longevity of mice. Endothelial-specific knockout of Arf6 was achieved by crossing Arf6f/f mice with Arf6f/f; Cdh5-CreERT2 mice and tamoxifen induction P1. T-cell specific knockout of Arf6 was generated by crossing Arf6f/f mice with Lck-Cre mice. Hematopoietic cell specific knockout of Arf6 was achieved by crossing Arf6f/f mice with Vav1-iCre mice. C57BL/6J mice were purchased from the Jackson Laboratory. All mice were bred on a standard 12:12 h light: dark cycle at the Comparative Medicine Center at the University of Utah, School of Medicine. Only adult (8–10 weeks old) male C57BL/6J mice were used for the NAV-2729 or LDN193189/SB431542 treatment experiments. In genetic experiments, where both adult (8–12 weeks old) female and male mice were used, we combined both sexes for reporting data and performing statistical analyses due to sample size constraints. However, we did analyze the two sexes separately whenever possible to identify any obvious outcome differences between them and found none.
METHOD DETAILS
Cells and compounds
HBMECs were cultured using the VascuLife® VEGF-Mv LifeFactors Kit and passaged for a maximum of six times. To assess the function of IL-1β/IL-1R signaling, cells were pretreated with the IL-1R antagonist IL-1RA (100 ng/ml) for 1 h. LDN193189 (1 μM), SB431542 (10 μM), or a combination of both compounds were added to the cell culture to inhibit ALKs for 2 h. In a separate experiment, HBMECs were treated for 30 min with either the transcription inhibitor actinomycin D (3 μg/mL) or the translation inhibitor cycloheximide (50 μg/mL). Following these treatments, HBMECs were stimulated with IL-1β (20 ng/mL) to test for SMAD1/5 activation (pSMAD 1/5) or the induction of EndoMT. In addition, MBMECs were treated with murine IL-1β (150 ng/mL) for 20 h to test for the induction of EndoMT.
Jurkat cells (a human T-cell leukemia cell line) were cultured with RPMI-1640 supplemented with 10% FBS, 2 mM L-glutamine, 10 mM HEPES, 1 mM sodium pyruvate, 4500 mg/L glucose, 1500 mg/L sodium bicarbonate, and 50 μM 2-ME. To access whether ALK inhibition would affect T cell activation, Jurkat cells were pre-incubated with LDN193189 and SB431542 for 2 h and then activated by Phytohemagglutinin-L (PHA-L, 5 μg/mL). After 24 h of culture, the cells were lysed, and total mRNA was extracted. CD68, CD69, GATA3 and IFNγ mRNA expression levels were accessed by quantitative RT-PCR. The expression of the housekeeping gene GAPDH was used for normalization.
Transduction with adenoviruses
Adenoviruses were diluted in medium, and the final working concentrations of Ad ARF6T27N, Ad ARF6Q67L, Ad ARF6WT, and Ad NULL were 1.4 × 107, 4.3 × 106, 4.3 × 106 and 4.3 × 106, respectively. HBMECs were plated and allowed to grow overnight, and then growth medium was replaced. Two days after the initial infection, the cells were treated with either IL-1β or 0.1% BSA for various lengths of time. For the short time frame (15 and 30 min), the HBMECs were starved with basal medium containing 0.5% BSA for 16 h before being treated with IL-1β, while cells were treated with IL-1β with no starvation for longer treatments (12 h and 20 h).
Western blotting
Whole-cell lysates were prepared in 1X assay/lysis buffer (Cell Biolabs) plus protease and phosphatase inhibitors. Protein concentrations were measured by Micro BCA Protein Assay (ThermoFisher Scientific), adjusted for equal loading into the wells of 8% or 10% polyacrylamide gels, and western blotting was performed using a BIO-RAD system with semi-dry transfer (0.2A, 90–120 min). Proteins were detected with the following primary and secondary antibodies to assess the activation of pSMAD1/5 and pNF-κB p65 and the induction of EndoMT: anti-pSMAD1/5 (1:1,000), anti-SMAD1(1:2,000), anti-pNF-κB p65 (1:2,000), anti-NF-κB p65 (1:2,000), anti-CD31 (1:4,000), anti-Snail (1:2,000), anti-N-Cadherin (1:2,000), anti- α -tubulin (1:10,000), and appropriate secondary antibody conjugated to horse-radish peroxidase (HRP) (1:10,000). Quantification was by ImageJ where changes were normalized to loading controls or input and represents an amalgamation of all independent experimental replicates (n ≥ 3 each). The limit of detection was considered to be 3 standard deviations above the mean of the blanks (Armbruster and Pry, 2008). Any western signal that fell below the limit of detection was assigned a value equal to the limit of detection for data analysis.
IL-1β neutralization assay
HBMECs were treated with 20 ng/mL IL-1β for 15 min. IL-1β-15 min conditional medium was collected for the following IL-1β neutralization. Protein A/G–Plus-Agarose beads were washed with medium containing 0.5% BSA for 5 min. Subsequently, beads were incubated with anti-IL-1β (2.5 μg) or anti-IgG (2.5 μg) for 1 h at 4°C. After incubation, the beads were washed with medium containing 0.5% BSA 3 times every 5 min. IL-1β-15 min conditional medium was incubated with the beads for 1 h at 4°C. Finally, the IL-1β neutralization conditioned medium was collected and the starved cells were treated with this conditioned medium for 15 min. Cell lysates were prepared, and western blotting was performed as described above.
ARF6-GTP–pulldown assays
ARF6-GTP–pulldown assays were performed as previously described (Zhu et al., 2012). Briefly, HBMECs were grown in VascuLife® VEGF-Mv medium in 10-cm dishes to 80% to 90% confluence and were treated with either IL-1β (20 ng/mL) or 0.1% BSA for 5 min after being starved in 0.5% BSA for 16 h. After treatment, media were aspirated, and cells were rinsed with chilled UltraSaline A (Lonza). The dishes were frozen on dry ice. Following thawing, ARF6-pulldown lysis buffer (50 mM Tris-HCl, 100 mM NaCl, 1 mM MgCl2, 1% NP-40, 10% glycerol, 1% protease inhibitors and phosphatase inhibitors) was added to the cells. Lysates were centrifuged, and supernatants were added to GGA3-conjugated beads and agitated at 4°C for 60 min. Beads were washed in ARF6-pulldown lysis buffer and resuspended in 2× Laemmli buffer prior to loading onto 15% SDS-PAGE gels. A fraction of the cell lysate was withheld for use as a measure of total ARF6 in each sample. Anti-ARF6 (1:2,000) and HRP-conjugated secondary antibody were used to determine ARF6-GTP levels.
Electric cell-substrate impedance sensing (ECIS)
HBMECs were cultured to 80% confluency in VascuLife® VEGF-Mv medium, treated with trypsin, and then collected for the permeability assay. The 96-well E-plate (ACEA Biosciences, Inc., San Diego, CA, USA) was coated with human fibronectin (10 μg/mL, 200 μL/well, 37°C for 1 h) before seeding the HBMECs. The HBMEC single cell suspension was diluted to 1 × 105 cells/mL, then seeded at 100 μL/well (10,000 cell/well) onto a 96-well E-plate. The 96-well E-plate was loaded onto xCELLigence RTCA System (ACEA Biosciences, Inc.) inside a tissue culture incubator (37°C, 5% CO2). Cell growth and monolayer formation were monitored in real-time. After 12 h, the culture media was replaced with fresh, complete media containing Ad ARF6T27N or Ad Null. After another 48 h, the culture media was again replaced with fresh complete media with IL-1β (20 ng/mL) or 0.1% BSA. Changes in impedance were monitored for another 48 h. The xCELLigence RTCA software converted the impedance values to a cell index by normalizing the impedance to the time point before treatment, also known as “the relative cell index”.
MOG35–55-induced EAE
EAE was induced in mice using MOG35–55 as described by Bittner et al. (Bittner et al., 2014). Briefly, 8-week-old endothelial-specific Arf6 knockout mice (Arf6f/f; Cdh5-CreERT2) and their control sibling littermates or C57BL/6J mice were immunized with MOG35–55 in CFA emulsion followed by administration of pertussis toxin (PTX) in PBS at day 0 and again at day 2. Clinical Scores for EAE were obtained daily by individuals blinded to the genotype starting day 7 post-immunization and continuing to day 28. For some experiments, we monitored clinical scores from day 1 post-immunization. The following grading scale was used: Grade 0 = no clinical disease; grade 1 = loss of tail tonicity; grade 2 = mild hind leg paresis grade; grade 3 = moderate hind leg paralysis grade; grade 4 = complete paraplegia; grade 5 = quadriplegia, moribund state, or death. Body weight was measured daily.
In vivo pharmacologic treatment of EAE mice
C57BL/6J mice were immunized with MOG35–55 to induce EAE and randomized into different groups. Once randomized, the mice were daily administered NAV-2729 (30 mg/kg), LDN193189 (3 mg/kg) + SB431542 (10 mg/kg), anakinra (100 mg/kg), or vehicle intraperitoneally starting at day 1 post-immunization. A different set of EAE mice were daily administered 30 mg/kg NAV-2729 or vehicle intraperitoneally starting at day 13 post-immunization to determine whether inhibiting ARF6 might provide a potential therapeutic strategy for treating MS. Clinical scoring was conducted over the course of the experiment and the mice were tested for permeability or euthanized on days 14, 21, or 28. Brains and spinal cords were harvested from euthanized mice for histology, flow cytometry, or immunofluorescence studies.
Brain/Spinal cord vascular leakage
Quantification of brain and spinal cord vascular leakage was obtained as previously described (Scheppke et al., 2008; Zhu et al., 2017). Briefly, 6, 14, or 21 days post-EAE immunization, mice were anesthetized with ketamine-HCl (100 mg/kg) and xylazine (13 mg/kg) and then injected into the tail vein with 50 μL of 60 mg/mL Evans blue solution (MilliporeSigma) in UltraSaline A. After 5 h, the mice were euthanized and perfused with PBS to eliminate the remaining Evans blue dye from the circulation, and the spinal cords and brains were collected and weighed. The dye was eluted in 1 mL formamide for 18 h at 70°C. The extract was filtered with a 40 μm strainer, and absorbances at 620 nm and 740 nm were measured with the absorbance at 740 nm being subtracted out. Finally, the absorbances were normalized to the tissue weight.
To determine the brain or spinal cord to plasma ratio (Michalicova et al., 2017), two groups of mice (Arf6f/f; Cdh5-CreERT2 EAE mice, Day 21; and NAV-2729 treated EAE mice, Day 14) were injected with 50 μL of 60 mg/mL Evans blue, and after 5 h, blood was drawn just prior perfusion. The plasma was collected following centrifugation, diluted 1:100 in PBS, and absorbance was measured at 620 nm and 740 nm with the 740 nm absorbance subtracted out as described above. The Evans blue absorbance in the diluted plasma was multiplied by the dilution factor (100), and the spinal cord or brain to plasma ratio was then obtained by dividing the Evans blue absorbance in the brain or spinal cord by the absorbance in the plasma.
Cecal slurry model of multi-bacterial infection
Cecal slurry was batch prepared from adult wildtype C57BL/6J mice housed in specific pathogen free environment, based on an established protocol (Starr et al., 2014). Briefly, whole cecum from each euthanized mouse was dissected and the cecal contents were collected using sterile tools. The collected cecal contents were weighed and mixed with sterile water at a ratio of 0.5 mL of water to 100 mg of cecal content. This cecal slurry was filtered through 70 μm strainer and mixed with an equal volume of 30% glycerol in PBS. The resulting cecal slurry stock was aliquoted and stored at −80°C. 100 μL of cecal slurry was IP injected into experimental mice indicated in Figure S3B and S3C. After indicated time periods, mice were euthanized, and peripheral blood was immediately collected via cardiac puncture using sterile syringes. Peritoneal lavage fluid was collected following injection of 5 mL sterile PBS into unopened peritoneal cavity. 100 μL of diluted or undiluted peripheral blood or peritoneal lavage fluid was spread onto LB agar plates, and colony forming units of aerobic and facultative anaerobic bacteria were counted after overnight incubation at 37 °C.
JHMV Infection
For intracranial (IC) injections, age-matched (5–7 weeks) C57BL/6 mice were anesthetized with an IP injection of 200 μL of a mixture of ketamine (Hospira, Lake Forest, IL, USA) and xylazine (Phoenix Pharmaceutical, Saint Joseph, MO, USA) in Hank’s balanced salt solution (HBSS). Mice were injected intracranially with 200 plaque-forming units (PFU) of JHMV (strain V34) suspended in 30 μL HBSS. Beginning 24 h post-infection, ARF6 inhibitor NAV-2729 was IP injected every other day for 10 days. Mice were euthanized 14 days post infection. Immunophenotyping of immune cells present within brains of JHMV-infected mice was accomplished by homogenizing isolated tissue and generating single-cell suspensions for analysis by flow cytometry. In brief, isolated cells were stained with the following antibodies: APC-conjugated rat anti-mouse CD4 and a PE-conjugated tetramer specific for the CD4 immunodominant epitope present within the JHMV matrix (M) glycoprotein spanning amino acids 133–147 (M133–147 tetramer) to determine total and virus-specific CD4+ cells, respectively (Chen et al., 2014); APC-conjugated rat anti-mouse CD8a and a PE-conjugated tetramer specific for the CD8 immunodominant epitope present in the spike (S) glycoprotein spanning amino acids 510–518 (S510–518) to identify total and virus-specific CD8+ cells, respectively; and APC-conjugated rat anti-mouse CD45 and FITC-conjugated anti-F4/80 to identify macrophages. Samples were analyzed using a BD LSR Fortessa X-20 flow cytometer and FloJo software.
Mouse lung endothelial cells isolation
Mouse lung endothelial cells were isolated following the protocol described by Wang et al. (Wang et al., 2019). Briefly, the lungs were harvested and digested by collagenase I, followed by sequential sorting and isolation with PECAM1- and ICAM2-coated Dynabeads. For this purification procedure, we used 4 mice (~ 4 weeks old) for each genotype (Arf6f/f or Arf6f/f; Cdh5-CreERT2). After the 2nd round of sorting and isolation, the mouse lung endothelial cells were harvested and lysed in 1X assay/lysis buffer (Cell Biolabs) for western blotting using anti-ARF6 to check knockout efficiency.
Flow cytometry
For the T cell infiltration assay, leukocytes were obtained from EAE mouse spinal cords, lymph nodes, blood, or spleens by homogenization and centrifugation. Cells were incubated with viability dye (1:500, eBioscience™ Fixable Viability Dye eFluor™ 780), antibodies (Anti-CD45, 1:400; CD8, 1:400; CD4, 1:400 and CD3, 1:200), and their respective isotype controls (at same dilution) at room temperature for 30 min after Fc blocking and then fixed with 4% paraformaldehyde for 15 min before washing. Intracellular staining (Anti IFNγ, 1:30) was carried out according to the BD Fixation/Permeabilization Solution Kit protocol. Stained cells were then analyzed on a flow cytometer.
To test whether globally blocking the ALK receptors would suppress immune activation, leukocytes were obtained from spleens of LDN193189 (3 mg/kg) + SB431542 (10 mg/kg)-treated EAE mice by homogenization and centrifugation. Cells were then incubated with viability dye (1:1,000, LIVE/DEAD™ Fixable Aqua Dead Cell Stain Kit), antibodies (anti-CD62L, 1:200; CD44, 1:200; CD28, 1:200; CD25, 1:200; CD8, 1:400; CD4, 1:400; and CD3, 1:200), and their respective isotype controls at room temperature for 30 min after Fc blocking and then fixed with 4% paraformaldehyde for 15 min before washing. Stained cells were then analyzed by flow cytometry.
Histology
C57BL/6J or Arf6f/f; Cdh5-CreERT2 mice were anesthetized with isoflurane and the spinal cords were perfused with 4% paraformaldehyde (PFA) as described according to Maeda et al. (Maeda et al., 2019). After fixation in 4% PFA for 24 h, the lumbar cords were carefully removed from the surrounding tissues and processed for paraffin embedding. Samples were then cut into 8 μm sections, followed by hematoxylin and eosin (H&E) staining and Luxol fast blue (LFB) staining. All stained slides were interpreted by a board-certificated anatomic pathologist who provided representative histologic figures and conclusions based on the analysis.
To check for the colocalization of EndoMT and vascular leakage, the EAE mice at day 15 were anesthetized, then injected into the tail vein with 10 μL/g of 1 mg/mL Albumin from Bovine Serum (BSA), Alexa Fluor™ 647 Conjugate. After 5 h, the mice were euthanized with no perfusion, and the spinal cords were collected and fixed with 4% PFA at 4°C overnight. Samples were embedded in OCT for cryosection in the next day, and then cut into 5 μm sections, followed by immunofluorescence analysis.
Immunofluorescence studies
Microvessels from EAE mouse spinal cords were isolated and stained following the protocol described by Lee et al. (Lee et al., 2019). Antibodies against CD31 were used to label the endothelial cells (1:200). Antibodies against pSMAD1/5, SNAI2, and N-cadherin were applied at 1:50 dilution. Sectioned EAE mouse spinal cords were also stained with CD31 (1:200), pSMAD1/5 (1:50), SNAIL (1:50), and N-cadherin (1:50). DAPI was used to label nuclear DNA (1:1000). Images are maximum-Z-projections of confocal images acquired at 0.38 μm z-section intervals. Acquisition settings were identical for all images. Images are representative of at least 3 independent experiments, and we took about 8–12 independent images for each experiment. Quantification was performed on images from single optical slices (0.38 μm) using Matlab R2017b to obtain intensity of total fluorescence signal for each specific marker (pSMAD1/5, SNAI2, or N-cadherin) per area of image exhibiting CD31+ fluorescence signals. Data are presented as mean ± SEM of at least three independent experiments.
Quantification and statistical analysis
Statistical analyses were performed using GraphPad Prism 9.0 (GraphPad Software), and individual data points were graphed using scatter plots as recommended by Weissgerber et al. (Weissgerber et al., 2015). Extreme outliers were removed from the data sets using a very stringent ROUT analysis at Q = 0.1% prior to statistical analysis. One-way ANOVA with Tukey’s or Dunnett’s multiple comparisons test was used to assess statistical significance when more than two groups were being compared, the data were consistent with a normal distribution, standard deviations were similar between groups, and pairing of groups within an individual experiment was not warranted. When the standard deviations varied between the groups, Welch’s one-way ANOVA was used with Dunnett’s T3 multiple comparisons test. A Kruskal-Wallis test with Dunn’s multiple comparisons test was used when more than two groups were being compared and the data did not appear to be normally distributed. A two-tailed Student’s t test was used when the experiment involved only two groups, the data were normally distributed, and the variation was similar in the two groups. When the variation between groups was not similar, a two-tailed Welch’s t test was used. A two-tailed Mann-Whitney test was used when the data were not normally distributed. For assays that exhibited greater variability between the controls of independent experiments, such as western blotting and immunofluorescence imaging of spinal cord blood vessels, we normalized experimental groups of each independent experiment to their own control and either used a two-tailed ratio paired t test (if only two groups were being compared) or a one-way randomized block ANOVA with Tukey’s or Dunnett’s multiple comparisons tests following log10 transformation of the data when more than two groups were being compared. For these data, semi-logarithmic graphs showing geometric means and 95% confidence intervals were used. For clinical scores and body mass data, a two-way ANOVA (when no data points were missing) or mixed effects analysis (if some data points were missing) was used in conjunction with Tukey’s or Šidák’s multiple comparisons test for individual time points and overall differences between treatment or genotype curves were corrected for multiple comparisons using the Holm-Šidák method. Quantification for data shown in Figures 4 and 5 are presented in Figures S7–S9. Adjusted p-values of less than 0.05 were considered statistically significant. Sample sizes are provided in the figure legends.
Supplementary Material
KEY RESOURCES TABLE
| Reagents | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| APC/Cyanine7 Rat IgG2a, κ Isotype Ctrl Antibody | BioLegend | Cat. #: 400523 |
| ARF6 | Cell Signaling | Cat. :5740S; RRID: AB_10694539 |
| CD25 | BioLegend | Cat. #: 102005 |
| CD28 | BioLegend | Cat. #: 102105 |
| CD3 | BioLegend | Cat. #: 100218 |
| CD31 | Abcam | Cat. #: Ab28364; RRID: AB_726362 |
| CD31 | BD Pharmingen | Cat. #:553370; RRID: AB_396660 |
| CD4 | BioLegend | Cat. #: 100406 |
| CD4 | Invitrogen | Cat. #: 25-0041-82; RRID: AB_469576 |
| CD44 | Invitrogen | Cat. #: 17-0441-82; RRID: AB_469390 |
| CD45 | BioLegend | Cat. #: 103126 |
| CD62L | Invitrogen | Cat. #: 47-0621-82; RRID: AB_1603256 |
| CD8 | BioLegend | Cat. #: 100714 |
| CD8 | BioLegend | Cat. #: 100708 |
| Donkey anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | ThermoFisher Scientific | Cat. #: A21206 RRID: AB_2535792 |
| Donkey anti-Rat IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 594 | ThermoFisher Scientific | Cat. #: A21209 RRID: AB_2535795 |
| FITC Rat IgG1, λ Isotype Ctrl Antibody | BioLegend | Cat. #: 401913 |
| FITC Rat IgG2b, κ Isotype Ctrl Antibody | BioLegend | Cat. #:400634 |
| GAPDH | Cell Signaling | Cat. #:5174S; RRID: AB_10622025 |
| Human IL-1β/IL-1F2 antibody | R&D Systems | Cat. #: MAB201-100 |
| IFNγ | ThermoFisher Scientific | Cat. #: 50-7311-82 RRID: AB_11217680 |
| IgG | Santa Cruz | Cat. #:SC2025; RRID: AB_737182 |
| N-cadherin | ThermoFisher Scientific | Cat. #:MA5-32088; RRID: AB_2800542 |
| NF-κB p65 | Cell Signaling | Cat. #:8242S; RRID: AB_10859369 |
| Pacific Blue™ Rat IgG2b, κ Isotype Ctrl Antibody | BioLegend | Cat. #:400627 |
| PE Rat IgG2a, κ Isotype Ctrl Antibody | BioLegend | Cat. #:400508 |
| PE Syrian Hamster IgG Isotype Ctrl Antibody | BioLegend | Cat. #: 402008 |
| PerCP/Cyanine5.5 Rat IgG2b, κ Isotype Ctrl Antibody | BioLegend | Cat. #:400631 |
| pNF-κB p65, | Cell Signaling | Cat. #:3033S; RRID: AB_331284 |
| pSMAD1/5 | Cell Signaling | Cat. #: 9516S; RRID: AB_491015 |
| pSMAD1/5 | ThermoFisher Scientific | Cat. #: MA5-15124 RRID: AB_11000599 |
| Rat IgG1 kappa Isotype Control (eBRG1), eFIuor™ 660, eBioscience™ | Invitrogen | Cat. #: 50-4301-82 RRID: AB_10598505 |
| Rat IgG2a kappa Isotype Control (eBR2a), eFluor 450, eBioscience™ | Invitrogen | Cat. #: 48-4321-80 RRID: AB_1272002 |
| Rat IgG2b kappa Isotype Control (eB149/10H5), APC, eBioscience™ | Invitrogen | Cat. #: 17-4031-81 RRID: AB_470175 |
| Rat IgG2b kappa Isotype Control (eB149/10H5), PE-Cyanine7, eBioscience™ | Invitrogen | Cat. #: 25-4031-82 RRID: AB_891624 |
| Rat-anti-mouse ICAM2 | BD Biosciences | Cat. #:553325; RRID: AB_394783 |
| Rat-anti-mouse Pecam-1 |
BD Biosciences | Cat. #: 553370 RRID: AB_394816 |
| Slug (SNAI2) | Cell Signaling | Cat. #:9585S; RRID: AB_2239535 |
| SMAD1 | Cell Signaling | Cat. #:9743S; RRID: AB_2107780 |
| Snail | Cell Signaling | Cat. #:3879S; RRID: AB_2255011 |
| SNAIL | Invitrogen | Cat. # PA5-85493; RRID: AB_2792633 |
| α-tubulin | Cell Signaling | Cat. #:3873S; RRID: AB_1904178 |
| Chemicals, Peptides, Proteins | ||
| Animal-Free Human IL-1beta | PEPROTECH | Cat. #: AF-200-01B |
| 2-Mercaptoethanol | Sigma-Aldrich | Cat. #: M3148 |
| 5X Assay/Lysis Buffer | Cell Biolabs | Cat. #:240102 |
| Actinomycin D | Sigma-Aldrich | Cat. #: A1410 |
| Albumin From Bovine serum (BSA), Alexa Fluor™ 647 Conjugate | Invitrogen | Cat. #: A34785 |
| Anakinra | SOBI | NDC: 66658-234-07 |
| Animal-Free Recombinant Murine IL-1β | PEPROTECH | Cat. #: AF-211-11B |
| Bordetella Pertussis toxin | List Biological Labs | Cat. #: 180 |
| BSA | Sigma | Cat. #: A-8806 |
| Collagenase Type I | Worthington Biochemical Corp | Cat. #:LS004196 |
| Complete Mouse Endothelial Cell Medium /w Kit | Cell Biologics | Cat. #: M1168 |
| Cycloheximide | Sigma-Aldrich | Cat. #: 01810 |
| DAPI | ThermoFisher Scientific | Cat. #: D1306 |
| Dulbecco’s Modified Eagle’s Medium | ATCC | Cat. #:30-2002 |
| Dxtran-70 | TCI | Cat. #: D1449 |
| Dynabeads | ThermoFisher Scientific | Cat. #:11035 |
| eBioscience Cell Stimulation Cocktail | ThermoFisher Scientific | Cat. #:00-4975-93 |
| eBioscience™ Fixable Viability Dye eFIuor™ 780 | ThermoFisher Scientific | Cat. #:65-0865-14 |
| Evans blue | MilliporeSigma | Cat. #: E2129 |
| Formamide | Millipore | Cat. #: S4117 |
| Halt™ Protease and Phosphatase Inhibitor Cocktail (100X) | ThermoFisher Scientific | Cat. #: 78446 |
| Human BD Fc Block | BD Biosciences | Cat. #: 564220 |
| Human Fibronectin | Promocell | Cat. #:43060 |
| IL-1RA | Abcam | Cat. #: Ab229516 |
| Incomplete Freund’s adjuvant | MP Bio | Cat. #:642851 |
| LDN193189 | STEMCELL | Cat. #: 72147 |
| MCDB 131 | Corning | Cat. #:15-100-cv |
| Myelin Oligodendrocyte Glycoprotein Peptide (35-55) | GenScript | Cat. #: CP0005 |
| NAV-2729 | Navigen, Inc | N/A |
| Paraformaldehyde | MP Biomedicals | Cat. #: 150146 |
| Phytohemagglutinin-L | Roche | Cat. #:11249738001 |
| Protein A/G PLUS-Agarose | Santa Cruz | Cat. #:sc-2003 |
| RPMI-1640 | ATCC | Cat. #: 30-2001 |
| SB431542 | Selleckchem | Cat. #: S1067 |
| UltraSaline A | Lonza | Cat. #:12-747F |
| VascuLife® Basal Medium | Lifeline cell technology | Cat. #: LM-0002 |
| Commercial Assays | ||
| ARF6-GTP-pulldown kits | Cell Biolabs | Cat. #: STA-419 |
| Fixation/Permeabilizati on Solution Kit | BD Biosciences | Cat. #: 554714 |
| LIVE/DEAD™ Fixable Aqua Dead Cell Stain Kit, for 405 nm excitation | Invitrogen™ | Cat. #: L34965 |
| Micro BCA™ protein assay kit | ThermoFisher Scientific | Cat. #: 23235 |
| VascuLife® VEGF-Mv LifeFactors Kit | Lifeline Cell Technology | Cat. #: LS-1029 |
| Experimental Models: Cell line | ||
| C57BL/6 Mouse Primary Brain Microvascular Endothelial Cells | Cell Biologics | Cat. #: C57-6023 |
| Human brain microvascular endothelial cells | Cell Systems | Cat. #: ACBRI 376 |
| Jurkat, Clone E6-1 | ATCC | Cat. #: TIB-152 |
| Experimental Models: Organisms/Strains | ||
| Arf6 floxed mice | Zhu et al. (2017) | N/A |
| C57BL/6 | The Jackson Laboratory | Strain: 000664 |
| Cdh5-CreERT2 mice | Wang et al. (2010); Gift from Dr. Ralf Adams, Max Planck Institute for Molecular Biomedicine, Münster, Germany | N/A |
| Lck-Cre mice | The Jackson Laboratory | Strain: 003802 |
| Rosa-CreERT mice | The Jackson Laboratory | Strain: 004847 |
| Vav1-iCre mice | The Jackson Laboratory | Strain: 008610 |
| Oligonucleotides | ||
| GGAAATGCCACGGTTCATCCA | PrimerBank | CD68_F |
| TGGGGTTCAGTACAGAGATGC | PrimerBank | CD68_R |
| ATTGTCCAGGCCAATACACATT | PrimerBank | CD69_F |
| CCTCTCTACCTGCGTATCGTTTT | PrimerBank | CD69_R |
| GCCCCTCATTAAGCCCAAG | PrimerBank | GATA3_F |
| TTGTGGTGGTCTGACAGTTCG) | PrimerBank | GATA3_R |
| TCGGTAACTGACTTGAATGTCCA | PrimerBank | IFNγ_F |
| TCGCTTCCCTGTTTTAGCTGC | PrimerBank | IFNγ_R |
| GAGTCAACGGATTTGGTCGT | In this paper | GAPDH_F |
| TTGATTTTGGAGGGATCTC | In this paper | GAPDH_R |
| Software and Algorithms | ||
| FlowJo v10.6.1 | FlowJo, LLC | https://www.flowjo.com |
| ImageJ | NIH | https://imagej.net |
| Matlab R2017b | MathWorks | https://www.mathworks.com |
| CellImageAnalysis_Odelberg | This paper | https://github.com/learner97/CellImageAnalysis_Odelberg |
| Photoshop | Adobe | https://www.adobe.com/products/photoshop.html |
| Prism 9 | GraphPad | https://www.graphpad.com/scientificsoftware/prism/ |
Highlights.
EndoMT occurs in the CNS before the onset of symptoms in a mouse model of MS
EndoMT helps drive the loss of BCNSB function and is induced by ARF6 activation
ARF6 activation mediates crosstalk between IL-1β and ALK-SMAD1/5 signaling pathways
Inhibiting the IL-1β-ARF6-ALK-SMAD1/5 pathway reverses EndoMT and disease severity
Acknowledgements:
We thank D. Lim for preparation of the figures, comments, and consultation regarding responsible and effective display of the images. We thank R.A. Campbell for reading the manuscript. We thank the University of Utah Flow Cytometry Facility and ARUP Laboratories Research Histology and Anatomic Pathology. We thank Jiajia Wang and Xuefeng Cao for assisting in data collection.
Funding:
This work was funded by grants to W.Z. from the National Institutes of Health (R01HL077671 and R01EY025342) and from the National Multiple Sclerosis Society (RG-1907-34532) and to S.J.O. from the National Institutes of Health (R01HL130541 and R01AR064788).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests: The University of Utah has filed intellectual property concerning ARF6 pathways. The authors declare competing financial interests: The University of Utah has licensed technology to Navigen, a biotechnology company owned in part by the University of Utah Research Foundation. A.L.M., is employee of Navigen.
REFERENCES
- Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, and Begley DJ (2010). Structure and function of the blood-brain barrier. Neurobiol. Dis 37, 13–25. [DOI] [PubMed] [Google Scholar]
- Adams RA, Bauer J, Flick MJ, Sikorski SL, Nuriel T, Lassmann H, Degen JL, and Akassoglou K (2007). The fibrin-derived γ377–395 peptide inhibits microglia activation and suppresses relapsing paralysis in central nervous system autoimmune disease. J. Exp. Med 204, 571–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aharoni R (2014). Immunomodulation neuroprotection and remyelination–The fundamental therapeutic effects of glatiramer acetate: A critical review. J. Autoimmun 54, 81–92. [DOI] [PubMed] [Google Scholar]
- Akassoglou K, Yu WM, Akpinar P, and Strickland S (2002). Fibrin inhibits peripheral nerve remyelination by regulating Schwann cell differentiation. Neuron 33, 861–875. [DOI] [PubMed] [Google Scholar]
- Argaw AT, Asp L, Zhang J, Navrazhina K, Pham T, Mariani JN, Mahase S, Dutta DJ, Seto J, and Kramer EG (2012). Astrocyte-derived VEGF-A drives blood-brain barrier disruption in CNS inflammatory disease. J. Clin. Invest 122, 2454–2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armbruster DA, and Pry T (2008). Limit of blank, limit of detection and limit of quantitation. Clin. Biochem. Rev 29 Suppl 1, S49–52. [PMC free article] [PubMed] [Google Scholar]
- Aubé B, Lévesque SA, Paré A, Chamma É, Kébir H, Gorina R, Lécuyer M-A, Alvarez JI, De Koninck Y, and Engelhardt B (2014). Neutrophils mediate blood–spinal cord barrier disruption in demyelinating neuroinflammatory diseases. J. Immunol 193, 2438–2454. [DOI] [PubMed] [Google Scholar]
- Behrangi N, Fischbach F, and Kipp M (2019). Mechanism of Siponimod: Anti-Inflammatory and Neuroprotective Mode of Action. Cells 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittner S, Afzali AM, Wiendl H, and Meuth SG (2014). Myelin oligodendrocyte glycoprotein (MOG35–55) induced experimental autoimmune encephalomyelitis (EAE) in C57BL/6 mice. J. Vis. Exp 86, 51275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapouly C, Tadesse Argaw A, Horng S, Castro K, Zhang J, Asp L, Loo H, Laitman BM, Mariani JN, and Straus Farber R (2015). Astrocytic TYMP and VEGFA drive blood–brain barrier opening in inflammatory central nervous system lesions. Brain 138, 1548–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Coleman R, Leang R, Tran H, Kopf A, Walsh CM, Sears-Kraxberger I, Steward O, Macklin WB, Loring JF, et al. (2014). Human neural precursor cells promote neurologic recovery in a viral model of multiple sclerosis. Stem Cell Rep. 2, 825–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho JG, Lee A, Chang W, Lee MS, and Kim J (2018). Endothelial to Mesenchymal Transition Represents a Key Link in the Interaction between Inflammation and Endothelial Dysfunction. Front. Immunol 9, 294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JW, Gardell SE, Herr DR, Rivera R, Lee C-W, Noguchi K, Teo ST, Yung YC, Lu M, and Kennedy G (2011). FTY720 (fingolimod) efficacy in an animal model of multiple sclerosis requires astrocyte sphingosine 1-phosphate receptor 1 (S1P1) modulation. Proc. Natl. Acad. Sci. USA 108, 751–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly AC, Randall RA, and Hill CS (2008). Transforming growth factor β-induced Smad1/5 phosphorylation in epithelial cells is mediated by novel receptor complexes and is essential for anchorage-independent growth. Mol. Cell. Biol 28, 6889–6902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos D, Mahajan KR, and Trapp BD (2019). Brain fibrinogen deposition plays a key role in MS pathophysiology - Yes. Multiple Sclerosis J. 25, 1434–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos D, Ryu JK, Merlini M, Baeten KM, Le Moan N, Petersen MA, Deerinck TJ, Smirnoff DS, Bedard C, Hakozaki H, et al. (2012). Fibrinogen-induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation. Nat. Commun 3, 1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis CT, Zhu W, Gibson CC, Bowman-Kirigin JA, Sorensen L, Ling J, Sun H, Navankasattusas S, and Li DY (2014). ARF6 inhibition stabilizes the vasculature and enhances survival during endotoxic shock. J. Immunol 192, 6045–6052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derada Troletti C, de Goede P, Kamermans A, and de Vries HE (2016). Molecular alterations of the blood-brain barrier under inflammatory conditions: The role of endothelial to mesenchymal transition. Biochim. Biophys. Acta 1862, 452–460. [DOI] [PubMed] [Google Scholar]
- Derada Troletti C, Fontijn RD, Gowing E, Charabati M, van Het Hof B, Didouh I, van der Pol SMA, Geerts D, Prat A, van Horssen J, et al. (2019). Inflammation-induced endothelial to mesenchymal transition promotes brain endothelial cell dysfunction and occurs during multiple sclerosis pathophysiology. Cell Death Dis. 10, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drouin-Ouellet J, Sawiak SJ, Cisbani G, Lagace M, Kuan WL, Saint-Pierre M, Dury RJ, Alata W, St-Amour I, Mason SL, et al. (2015). Cerebrovascular and blood-brain barrier impairments in Huntington’s disease: Potential implications for its pathophysiology. Ann. Neurol 78, 160–177. [DOI] [PubMed] [Google Scholar]
- Evrard SM, Lecce L, Michelis KC, Nomura-Kitabayashi A, Pandey G, Purushothaman K-R, d’Escamard V, Li JR, Hadri L, and Fujitani K (2016). Endothelial to mesenchymal transition is common in atherosclerotic lesions and is associated with plaque instability. Nat. Commun 7, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faulkner M (2015). Risk of progressive multifocal leukoencephalopathy in patients with multiple sclerosis. Expert Opin Drug Saf 14, 1737–1748. [DOI] [PubMed] [Google Scholar]
- Fletcher JM, Lalor SJ, Sweeney CM, Tubridy N, and Mills KH (2010). T cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clin. Exp. Immunol 162, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gartner D, Hoff H, Gimsa U, Burmester GR, and Brunner-Weinzierl MC (2006). CD25 regulatory T cells determine secondary but not primary remission in EAE: impact on long-term disease progression. J. Neuroimmunol 172, 73–84. [DOI] [PubMed] [Google Scholar]
- Gebremariam T, Zhang L, Alkhazraji S, Gu Y, Youssef EG, Tong Z, Kish-Trier E, Bajji A, de Araujo CV, Rich B, et al. (2020). Preserving Vascular Integrity Protects Mice against Multidrug-Resistant Gram-Negative Bacterial Infection. Antimicrob. Agents Chemother 64, e00303–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser SF, Heumuller AW, Tombor L, Hofmann P, Muhly-Reinholz M, Fischer A, Gunther S, Kokot KE, Hassel D, Kumar S, et al. (2020). The histone demethylase JMJD2B regulates endothelial-to-mesenchymal transition. Proc. Natl. Acad. Sci. USA 117, 4180–4187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goumans MJ, Lebrin F, and Valdimarsdottir G (2003a). Controlling the angiogenic switch: a balance between two distinct TGF-b receptor signaling pathways. Trends Cardiovasc. Med 13, 301–307. [DOI] [PubMed] [Google Scholar]
- Goumans MJ, Valdimarsdottir G, Itoh S, Lebrin F, Larsson J, Mummery C, Karlsson S, and ten Dijke P (2003b). Activin receptor-like kinase (ALK)1 is an antagonistic mediator of lateral TGFbeta/ALK5 signaling. Mol. Cell 12, 817–828. [DOI] [PubMed] [Google Scholar]
- Goumans MJ, Valdimarsdottir G, Itoh S, Rosendahl A, Sideras P, and ten Dijke P (2002). Balancing the activation state of the endothelium via two distinct TGF-beta type I receptors. EMBO J. 21, 1743–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graesser D, Solowiej A, Bruckner M, Osterweil E, Juedes A, Davis S, Ruddle NH, Engelhardt B, and Madri JA (2002). Altered vascular permeability and early onset of experimental autoimmune encephalomyelitis in PECAM-1-deficient mice. J. Clin. Invest 109, 383–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grewal IS, Foellmer HG, Grewal KD, Wang H, Lee WP, Tumas D, Janeway CA Jr., and Flavell RA (2001). CD62L is required on effector cells for local interactions in the CNS to cause myelin damage in experimental allergic encephalomyelitis. Immunity 14, 291–302. [DOI] [PubMed] [Google Scholar]
- Hill C, Li J, Liu D, Conforti F, Brereton CJ, Yao L, Zhou Y, Alzetani A, Chee SJ, Marshall BG, et al. (2019). Autophagy inhibition-mediated epithelial-mesenchymal transition augments local myofibroblast differentiation in pulmonary fibrosis. Cell Death Dis. 10, 591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horng S, Therattil A, Moyon S, Gordon A, Kim K, Argaw AT, Hara Y, Mariani JN, Sawai S, and Flodby P (2017). Astrocytic tight junctions control inflammatory CNS lesion pathogenesis. J. Clin. Invest 127, 3136–3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hultgren NW, Fang JS, Ziegler ME, Ramirez RN, Phan DTT, Hatch MMS, Welch-Reardon KM, Paniagua AE, Kim LS, Shon NN, et al. (2020). Slug regulates the Dll4-Notch-VEGFR2 axis to control endothelial cell activation and angiogenesis. Nat. Commun 11, 5400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins DR, Craner MJ, Esiri MM, and DeLuca GC (2018). Contribution of Fibrinogen to Inflammation and Neuronal Density in Human Traumatic Brain Injury. J. Neurotrauma 35, 2259–2271. [DOI] [PubMed] [Google Scholar]
- Jiang H, Zhang F, Yang J, and Han S (2014). Angiopoietin-1 ameliorates inflammation-induced vascular leakage and improves functional impairment in a rat model of acute experimental autoimmune encephalomyelitis. Exp. Neurol 261, 245–257. [DOI] [PubMed] [Google Scholar]
- Jones CA, Nishiya N, London NR, Zhu W, Sorensen LK, Chan AC, Lim CJ, Chen H, Zhang Q, Schultz PG, et al. (2009). Slit2-Robo4 signalling promotes vascular stability by blocking Arf6 activity. Nat. Cell Biol 11, 1325–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kermode A, Thompson A, Tofts P, MacManus D, Kendall B, Kingsley D, Moseley I, Rudge P, and McDonald W (1990a). Breakdown of the blood-brain barrier precedes symptoms and other MRI signs of new lesions in multiple sclerosis: pathogenetic and clinical implications. Brain 113, 1477–1489. [DOI] [PubMed] [Google Scholar]
- Kermode A, Tofts P, Thompson A, MacManus D, Rudge P, Kendall B, Kingsley D, Moseley I, Du Boulay E, and McDonald W (1990b). Heterogeneity of blood-brain barrier changes in multiple sclerosis: an MRI study with gadolinium-DTPA enhancement. Neurology 40, 229–229. [DOI] [PubMed] [Google Scholar]
- Kim J-H, Kim J-H, Park J, Lee S-W, Kim W-J, Yu Y-S, and Kim K-W (2006). Blood-neural barrier: intercellular communication at glio-vascular interface. BMB Rep 39, 339–345. [DOI] [PubMed] [Google Scholar]
- Lécuyer M-A, Saint-Laurent O, Bourbonnière L, Larouche S, Larochelle C, Michel L, Charabati M, Abadier M, Zandee S, and Jahromi NH (2017). Dual role of ALCAM in neuroinflammation and blood–brain barrier homeostasis. Proc. Natl. Acad. Sci. USA 114, E524–E533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y-K, Uchida H, Smith H, Ito A, and Sanchez T (2019). The isolation and molecular characterization of cerebral microvessels. Nat. Protoc 14, 3059–3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeVine SM (2016). Albumin and multiple sclerosis. BMC Neurol. 16, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Powell N, Zhang H, Belevych N, Ching S, Chen Q, Sheridan J, Whitacre C, and Quan N (2011). Endothelial IL-1R1 is a critical mediator of EAE pathogenesis. Brain Behav. Immun 25, 160–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu T, Tian L, Han Y, Vogelbaum M, and Stark GR (2007). Dose-dependent cross-talk between the transforming growth factor-beta and interleukin-1 signaling pathways. Proc. Natl. Acad. Sci. USA 104, 4365–4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukens JR, Barr MJ, Chaplin DD, Chi H, and Kanneganti TD (2012). Inflammasome-derived IL-1beta regulates the production of GM-CSF by CD4(+) T cells and gammadelta T cells. J. Immunol 188, 3107–3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Sanchez-Duffhues G, Goumans MJ, and Ten Dijke P (2020). TGF-β-Induced Endothelial to Mesenchymal Transition in Disease and Tissue Engineering. Front. Cell Dev. Biol 8, 260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddaluno L, Rudini N, Cuttano R, Bravi L, Giampietro C, Corada M, Ferrarini L, Orsenigo F, Papa E, and Boulday G (2013). EndMT contributes to the onset and progression of cerebral cavernous malformations. Nature 498, 492–496. [DOI] [PubMed] [Google Scholar]
- Maeda Y, Nakagomi N, Nakano-Doi A, Ishikawa H, Tatsumi Y, Bando Y, Yoshikawa H, Matsuyama T, Gomi F, and Nakagomi T (2019). Potential of adult endogenous neural stem/progenitor cells in the spinal cord to contribute to remyelination in experimental autoimmune encephalomyelitis. Cells 8, 1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maggi P, Macri SMC, Gaitán MI, Leibovitch E, Wholer JE, Knight HL, Ellis M, Wu T, Silva AC, and Massacesi L (2014). The formation of inflammatory demyelinated lesions in cerebral white matter. Ann. Neurol 76, 594–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinl E (2011). Untapped targets in multiple sclerosis. J Neurol Sci 311, S12–S15. [DOI] [PubMed] [Google Scholar]
- Michalicova A, Galba J, Novak M, and Kovac A (2017). Determination of Evans blue as a blood–brain barrier integrity tracer in plasma and brain tissue by UHPLC/UV method. J. Liq. Chromatogr. Relat. Technol 40, 442–448. [Google Scholar]
- Munji RN, Soung AL, Weiner GA, Sohet F, Semple BD, Trivedi A, Gimlin K, Kotoda M, Korai M, Aydin S, et al. (2019). Profiling the mouse brain endothelial transcriptome in health and disease models reveals a core blood-brain barrier dysfunction module. Nat. Neurosci 22, 1892–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira SR, Kallaur AP, Reiche EM, Kaimen-Maciel DR, Panis C, Lozovoy MAB, Morimoto HK, Maes M, and Dichi I (2017). Albumin and protein oxidation are predictors that differentiate relapsing-remitting from progressive clinical forms of multiple sclerosis. Mol. Neurobiol 54, 2961–2968. [DOI] [PubMed] [Google Scholar]
- Palmer AM (2010). The role of the blood–CNS barrier in CNS disorders and their treatment. Neurobiol. Dis 37, 3–12. [DOI] [PubMed] [Google Scholar]
- Palmer AM (2013). Multiple sclerosis and the blood-central nervous system barrier. Cardiovasc. Psychiatry Neurol. 2013, 530356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardali E, Sanchez-Duffhues G, Gomez-Puerto MC, and Ten Dijke P (2017). TGF-β-Induced Endothelial-Mesenchymal Transition in Fibrotic Diseases. Int. J. Mol. Sci 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrin PJ, June CH, Maldonado JH, Ratts RB, and Racke MK (1999). Blockade of CD28 during in vitro activation of encephalitogenic T cells or after disease onset ameliorates experimental autoimmune encephalomyelitis. J. Immunol 163, 1704–1710. [PubMed] [Google Scholar]
- Petersen MA, Ryu JK, Chang K-J, Etxeberria A, Bardehle S, Mendiola AS, Kamau-Devers W, Fancy SP, Thor A, and Bushong EA (2017). Fibrinogen activates BMP signaling in oligodendrocyte progenitor cells and inhibits remyelination after vascular damage. Neuron 96, 1003–1012. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platel V, Faure S, Corre I, and Clere N (2019). Endothelial-to-Mesenchymal Transition (EndoMT): Roles in Tumorigenesis, Metastatic Extravasation and Therapy Resistance. J. Oncol 2019, 8361945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podjaski C, Alvarez JI, Bourbonniere L, Larouche S, Terouz S, Bin JM, Lecuyer M-A, Saint-Laurent O, Larochelle C, and Darlington PJ (2015). Netrin 1 regulates blood–brain barrier function and neuroinflammation. Brain 138, 1598–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polman CH, O’Connor PW, Havrdova E, Hutchinson M, Kappos L, Miller DH, Phillips JT, Lublin FD, Giovannoni G, Wajgt A, et al. (2006). A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N. Engl. J. Med 354, 899–910. [DOI] [PubMed] [Google Scholar]
- Privratsky JR, and Newman PJ (2014). PECAM-1: regulator of endothelial junctional integrity. Cell Tissue Res. 355, 607–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg GA (2014). Blood-Brain Barrier Permeability in Aging and Alzheimer’s Disease. J. Prev. Alzheimers Dis 1, 138–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu JK, Petersen MA, Murray SG, Baeten KM, Meyer-Franke A, Chan JP, Vagena E, Bedard C, Machado MR, Rios Coronado PE, et al. (2015). Blood coagulation protein fibrinogen promotes autoimmunity and demyelination via chemokine release and antigen presentation. Nat. Commun 6, 8164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schampel A, Volovitch O, Koeniger T, Scholz C-J, Jörg S, Linker RA, Wischmeyer E, Wunsch M, Hell JW, and Ergün S (2017). Nimodipine fosters remyelination in a mouse model of multiple sclerosis and induces microglia-specific apoptosis. Proc. Natl. Acad. Sci. USA 114, E3295–E3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheppke L, Aguilar E, Gariano RF, Jacobson R, Hood J, Doukas J, Cao J, Noronha G, Yee S, and Weis S (2008). Retinal vascular permeability suppression by topical application of a novel VEGFR2/Src kinase inhibitor in mice and rabbits. J. Clin. Invest 118, 2337–2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab N, Schneider-Hohendorf T, and Wiendl H (2015). Therapeutic uses of anti-alpha4-integrin (anti-VLA-4) antibodies in multiple sclerosis. Int. Immunol 27, 47–53. [DOI] [PubMed] [Google Scholar]
- Smith ES, Jonason A, Reilly C, Veeraraghavan J, Fisher T, Doherty M, Klimatcheva E, Mallow C, Cornelius C, and Leonard JE (2015). SEMA4D compromises blood–brain barrier, activates microglia, and inhibits remyelination in neurodegenerative disease. Neurobiol. Dis 73, 254–268. [DOI] [PubMed] [Google Scholar]
- Starr ME, Steele AM, Saito M, Hacker BJ, Evers BM, and Saito H (2014). A new cecal slurry preparation protocol with improved long-term reproducibility for animal models of sepsis. PLoS One 9, e115705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone L, Smith M, Albert P, Bash C, Maloni H, Frank J, and McFarland H (1995). Blood-brain barrier disruption on contrast-enhanced MRI in patients with mild relapsing-remitting multiple sclerosis: relationship to course, gender, and age. Neurology 45, 1122–1126. [DOI] [PubMed] [Google Scholar]
- Sweeney MD, Sagare AP, and Zlokovic BV (2018). Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol 14, 133–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troletti CD, Fontijn RD, Gowing E, Charabati M, van Het Hof B, Didouh I, van der Pol SM, Geerts D, Prat A, and van Horssen J (2019). Inflammation-induced endothelial to mesenchymal transition promotes brain endothelial cell dysfunction and occurs during multiple sclerosis pathophysiology. Cell Death Dis. 10, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt J, Traynor R, and Sapkota GP (2011). The specificities of small molecule inhibitors of the TGFss and BMP pathways. Cell. Signal 23, 1831–1842. [DOI] [PubMed] [Google Scholar]
- Wallez Y, and Huber P (2008). Endothelial adherens and tight junctions in vascular homeostasis, inflammation and angiogenesis. Biochim. Biophys. Acta 1778, 794–809. [DOI] [PubMed] [Google Scholar]
- Wang J, Niu N, Xu S, and Jin ZG (2019). A simple protocol for isolating mouse lung endothelial cells. Sci. Rep 9, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Nakayama M, Pitulescu ME, Schmidt TS, Bochenek ML, Sakakibara A, Adams S, Davy A, Deutsch U, Luthi U, et al. (2010). Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature 465, 483–486. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Masuyama N, Fukuda M, and Nishida E (2000). Regulation of intracellular dynamics of Smad4 by its leucine-rich nuclear export signal. EMBO Rep. 1, 176–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissgerber TL, Milic NM, Winham SJ, and Garovic VD (2015). Beyond bar and line graphs: time for a new data presentation paradigm. PLoS Biol. 13, e1002128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo JH, Shi DS, Grossmann AH, Sorensen LK, Tong Z, Mleynek TM, Rogers A, Zhu W, Richards JR, Winter JM, et al. (2016). ARF6 Is an Actionable Node that Orchestrates Oncogenic GNAQ Signaling in Uveal Melanoma. Cancer Cell 29, 889–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, London NR, Gibson CC, Davis CT, Tong Z, Sorensen LK, Shi DS, Guo J, Smith MC, Grossmann AH, et al. (2012). Interleukin receptor activates a MYD88-ARNO-ARF6 cascade to disrupt vascular stability. Nature 492, 252–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Shi DS, Winter JM, Rich BE, Tong Z, Sorensen LK, Zhao H, Huang Y, Tai Z, Mleynek TM, et al. (2017). Small GTPase ARF6 controls VEGFR2 trafficking and signaling in diabetic retinopathy. J. Clin. Invest 127, 4569–4582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlokovic BV (2008). The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 57, 178–201. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the Lead Contact upon request.
For this project, we used some custom code written in Matlab R2017b with the built-in image visualization toolbox. A simple GUI was used where images could be selected from the local machine and the amount of overlap between the staining with two separate antibodies was calculated. Use the attached GitHub link to read the description and see the custom code: https://github.com/learner97/CellImageAnalysis_Odelberg.
Any additional information required to reanalyze the data reported in this paper is available from the Lead Contact upon request.