

SUMMARY
The hippocampal CA2 region, an area important for social memory, has been suspected to play a role in temporal lobe epilepsy (TLE) because of its resistance to degeneration observed in neighboring CA1 and CA3 regions in both humans and rodent models of TLE. However, little is known about whether alterations in CA2 properties promote seizure generation or propagation. Here we addressed the role of CA2 using the pilocarpine-induced status epilepticus model of TLE. Ex vivo electrophysiological recordings from acute hippocampal slices revealed a set of coordinated changes that enhance CA2 PC intrinsic excitability, reduce CA2 inhibitory input, and increase CA2 excitatory output to its major CA1 synaptic target. Moreover, selective chemogenetic silencing of CA2 pyramidal cells caused a significant decrease in the frequency of spontaneous seizures. These findings provide the first evidence that CA2 actively contributes to TLE seizure activity and may thus be a promising therapeutic target.
Keywords: Designer receptors activated by designer drugs (DREADDs), CA2, electroencephalogram (EEG), hippocampal slices, patch clamp, optogenetics
IN BRIEF
Whitebirch et al. find increased intrinsic excitability and net synaptic excitation of CA2 PCs in hippocampal slices from epileptic mice. Chemogenetic inhibition of CA2 PCs in vivo reduces the frequency of spontaneous recurring seizures. Together these data suggest pathological hyperexcitability in CA2 circuitry may contribute to seizure activity in epilepsy.
INTRODUCTION
Temporal lobe epilepsy (TLE) is among the most prevalent neurological disorders, with approximately one third of patients experiencing seizures refractory to medication (P. Kwan & Sander, 2004; Patrick Kwan & Brodie, 2000). The identification of new therapeutic targets therefore requires deeper insights into the mechanisms of seizure generation. Anatomical and functional studies have suggested that the relatively unexplored CA2 region of the hippocampus may play an important role in seizure generation. Thus, autopsy specimens from human TLE patients display a characteristic pattern of hippocampal neurodegeneration termed mesial temporal sclerosis (MTS), with a substantial loss of neurons in the hilus of the dentate gyrus hilus and in the CA3 and CA1 pyramidal cell layer. In contrast, there is a relative sparing of dentate gyrus granule cells (GCs) and CA2 pyramidal cells (PCs) (Blümcke et al., 2013; Steve, Jirsch, & Gross, 2014).
Although relatively small, the CA2 subfield is of particular interest as it forms the nexus of a powerfully excitatory disynaptic circuit that directly links cortical input to hippocampal output (Chevaleyre & Siegelbaum, 2010; Srinivas et al., 2017), thereby regulating in vivo hippocampal network excitability and population activity (Boehringer et al., 2017; He et al., 2021; Lehr et al., 2021; Oliva, Fernández-Ruiz, Buzsáki, & Berényi, 2016; Oliva, Fernández-Ruiz, Leroy, & Siegelbaum, 2020). Examination of both surgically-resected human epileptic tissue and rodent models of TLE has revealed alterations in CA2, including synaptic reorganization of dentate gyrus granule cell mossy fiber axons (Freiman et al., 2021; Häussler, Rinas, Kilias, Egert, & Haas, 2016), decreases in parvalbumin expression (Andrioli & Arellano, 2007; Wittner et al., 2009), and the presence of spontaneous interictal-like spikes in recordings from surgically-resected tissue from patients with refractory TLE (Wittner et al., 2009). Altogether, this evidence suggests that CA2 may be an important component of an epileptic network in TLE. To date, however, there has been no direct test of the hypothesis that CA2 contributes to seizure activity in chronic epilepsy.
Here we examined the role of CA2 in seizure activity using the pilocarpine-induced status epilepticus (PILO-SE) model of TLE. We took advantage of the Amigo2-Cre mouse line, which enables the relatively selective targeting of CA2 using Cre-dependent viral vectors (Hitti & Siegelbaum, 2014). By expressing chemogenetic and optogenetic probes in CA2 pyramidal cells (PCs), we found that chemogenetic silencing of CA2 significantly reduced seizure frequency in PILO-SE mice. Moreover, ex vivo recordings from acute hippocampal slices revealed that PILO-SE enhanced the intrinsic excitability of surviving CA2 neurons, decreased CA2 synaptic inhibition, increased synaptic excitation of CA2 by its mossy fiber inputs, and increased CA2 excitatory output onto its downstream CA1 targets. Thus, our results point to an important role of CA2 in seizure activity that makes it an attractive target for novel therapeutic interventions.
RESULTS
We performed both ex vivo electrophysiological slice recordings and in vivo chronic EEG recordings from mice that had been treated with systemic injections of pilocarpine to induce status epilepticus (PILO-SE mice) and age-matched controls. PILO-SE mice developed spontaneous recurring seizures (epilepsy) 4–6 weeks after the induction of SE, at which time our recordings were performed. The mice developed a pattern of hippocampal neurodegeneration with preferential loss of CA1 and CA3 PCs and relative sparing of dentate gyrus (DG) granule cells and CA2 PCs, similar to hippocampal sclerosis in human TLE (Blümcke et al., 2013; Sastri et al., 2014; Steve et al., 2014; Thom et al., 2010), and consistent with prior work in rodents (Buckmaster, Abrams, & Wen, 2017; Jain, LaFrancois, Botterill, Alcantara-Gonzalez, & Scharfman, 2019; Mazzuferi, Kumar, Rospo, & Kaminski, 2012; Winawer et al., 2007). Although we observed a range of severity of neurodegeneration following PILO-SE, the relative survival of neurons in the CA2 subfield and the dentate gyrus granule cell layer were consistent features across animals (Figure S1).
We first report results of experiments in acute hippocampal slices to determine whether CA2 PCs from PILO-SE mice underwent changes in intrinsic excitability, excitatory or inhibitory synaptic input, or synaptic output. We then report results from in vivo experiments using chronic EEG recordings and video monitoring of behavioral seizures to determine whether chemogenetic inhibition of CA2 PC activity altered seizure frequency or seizure duration.
PILO-SE treatment enhances CA2 PC intrinsic excitability
We first performed whole-cell current clamp recordings to examine intrinsic electrophysiological properties of CA2 PCs from PILO-SE and control mice. Injection of depolarizing current steps (one-second square pulses from 100 to 1000 pA) revealed a significant increase in excitability of CA2 PCs from PILO-SE mice compared to control animals. Thus, the action potential firing rate versus current curve was shifted to higher firing rates in PILO-SE mice relative to controls (Figure 1B, C; two-way ANOVA; ****P < 0.0001; n = 92 cells from 48 control mice, 138 cells from 56 PILO-SE mice), with a significant increase in maximum firing rate (Figure 1D; Mann-Whitney; PILO-SE vs control, ****P < 0.0001; n = 92 cells from 48 control mice, 138 cells from 56 PILO-SE mice). We also observed increased action potential firing in CA2 PCs from PILO-treated mice that did not enter status epilepticus (PILO non-SE mice; two-way ANOVA; *P = 0.0138; n = 92 cells from 48 control mice, 45 cells from 22 PILO non-SE mice).
Figure 1. CA2 pyramidal cells (PCs) in slices from pilocarpine-treated mice had increased intrinsic excitability.
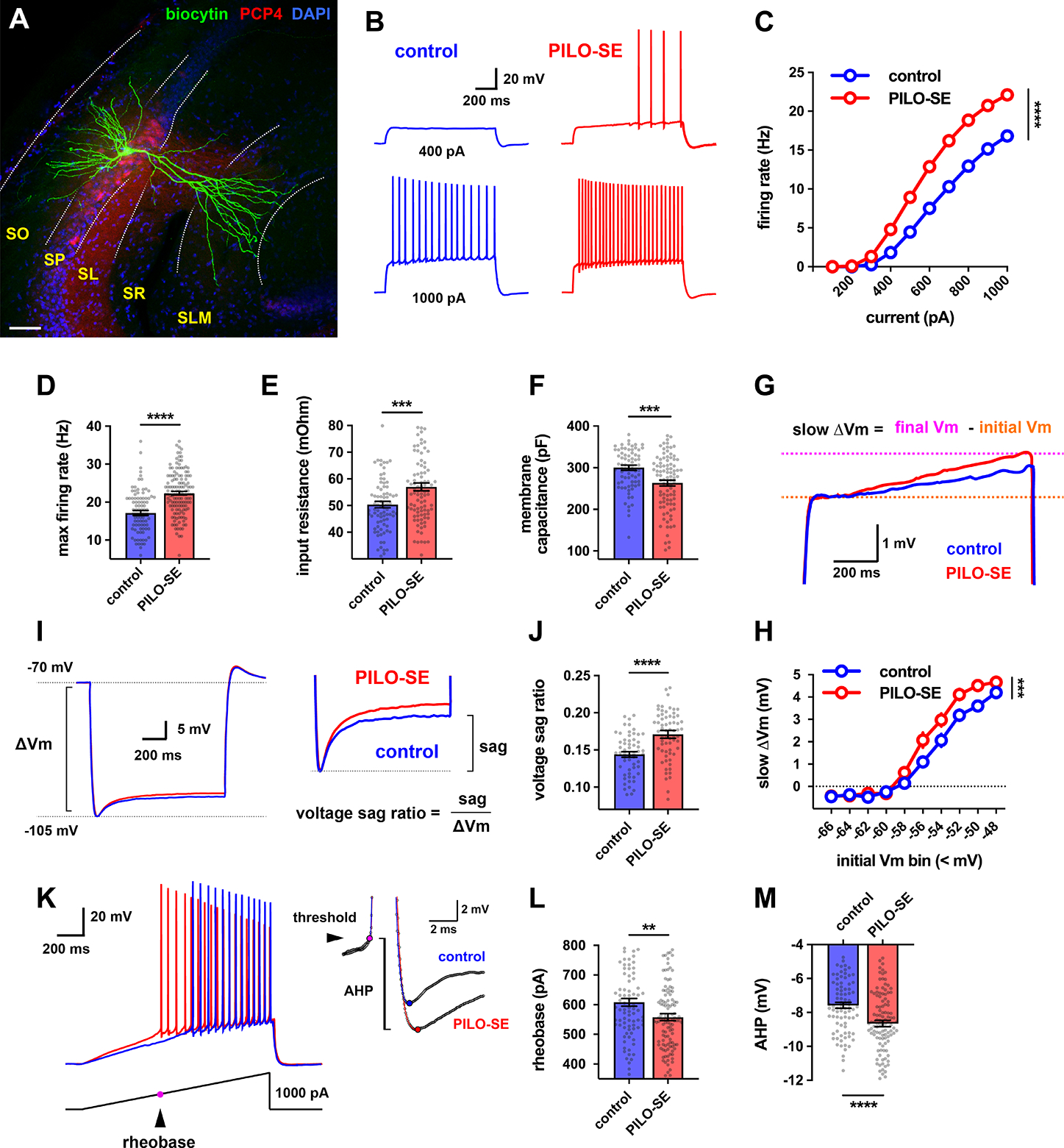
(A) A representative hippocampal slice stained for Nissl (blue) and PCP4, which labels CA2 PCs (red), with a single CA2 PC filled with intracellular biocytin (green). The hippocampal layers in this and all subsequent images are labeled as follows: stratum oriens (SO), stratum pyramidale (SP), stratum lucidum (SL), stratum radiatum (SR), stratum lacunosum moleculare (SLM). Scale bar, 60 μm. (B) Representative traces of membrane depolarization and action potential firing patterns in control (blue) and PILO-SE (red) CA2 PCs in response to a 1 second current step. (C) CA2 PCs in slices from PILO-SE mice fire a greater number of action potentials in response to depolarizing current steps. (D) The maximum firing rate was increased in CA2 PCs from PILO-SE mice. (E) Input resistance was increased in CA2 PCs from PILO-SE mice. (F) Membrane capacitance was reduced in CA2 PCs from PILO-SE mice. (G) Representative averaged traces of the slow ramping depolarization exhibited by CA2 PCs near action potential threshold. (H) Cells in slices from PILO-SE mice exhibited a larger slow depolarization. (I) Representative averaged traces from control and PILO-SE CA2 PCs showing hyperpolarization-induced membrane voltage sag. (J) The voltage sag ratio was greater in CA2 PCs from PILO-SE mice. (K) Representative membrane voltage responses from control (blue) and PILO-SE (red) CA2 PCs to a ramp of applied current. (L) The rheobase current was reduced in CA2 PCs from PILO-SE mice. (M) The amplitude of the AHP was significantly increased in CA2 PCs from PILO-SE mice. See also Supplemental Table 1.
We found no difference in resting potential between CA2 PCs from control or PILO-SE mice (Mann-Whitney; P = 0.3439; n = 120 cells from 58 control mice, 143 cells from 59 PILO-SE mice). In contrast, there was a significant increase in input resistance in cells from PILO-SE mice relative to controls (Figure 1E; Mann-Whitney; ***P = 0.0009; n = 81 cells from 48 control mice, 94 cells from 46 PILO-SE mice). We also observed a reduction in membrane capacitance in cells from PILO-SE mice (Figure 1F; Mann-Whitney; ***P = 0.0005; n = 78 cells from 47 control mice, 92 cells from 45 PILO-SE mice), suggesting a decreased membrane surface area. In addition, a slow ramping depolarization seen during current steps that is characteristic of CA2 PCs (Kohara et al., 2014) was increased in amplitude in PILO-SE mice relative to controls (Figure 1G, H; mixed-effects model; ***P = 0.0001; n = 110 cells from 56 control mice, 129 cells from 52 PILO-SE mice). Furthermore, we found an increase in voltage sag in response to hyperpolarizing current steps (Figure 1I, J; Mann-Whitney; ****P < 0.0001; n = 57 cells from 37 control mice, 67 cells from 34 PILO-SE mice), indicative of an increased hyperpolarization-activated HCN channel current (Srinivas et al., 2017).
We next examined action potential properties by applying a 1-second depolarizing current ramp (Figure 1K). The minimal current needed to elicit an action potential (rheobase) was significantly reduced in cells from PILO-SE mice (Figure 1L; Mann-Whitney; **P = 0.0072; n = 88 cells from 49 control mice, 119 cells from 54 PILO-SE mice), consistent with the increased action potential firing during current steps. However, there was no change in action potential voltage threshold, amplitude, half-width, maximum rate of rise, or maximum rate of descent (Supplemental Table 1), suggesting that the increase in spike firing may be due to the increase in input resistance, rather than to a change in voltage-gated channels. Although action potential parameters were largely unchanged, the fast afterhyperpolarization (AHP) was significantly larger in amplitude in CA2 PCs from PILO-SE animals relative to cells from control mice (Figure 1K, M; Mann-Whitney; ****P < 0.0001; n = 88 cells from 49 control mice, 119 cells from 54 PILO-SE mice). These results are summarized in Supplemental Table 1.
PILO-SE reduces synaptic inhibition but not excitation of CA2 PCs by their CA3 inputs, with no change in synaptic response to CA2 cortical inputs
CA2 receives its main excitatory inputs from the entorhinal cortical perforant path axons and CA3 PC Schaffer collaterals (Chevaleyre & Siegelbaum, 2010). In addition, CA2 receives recurrent collateral excitatory synapses from other CA2 neurons and weaker excitatory input from the mossy fibers of dentate gyrus granule cells (Kohara et al., 2014; Okamoto & Ikegaya, 2018). Most excitatory inputs also recruit strong feedforward inhibition of CA2 PCs, mediated by a diverse and widespread population of GABAergic interneurons (Kaoutsar Nasrallah et al., 2019; Sun et al., 2017). We thus systematically evaluated whether the PILO-SE model of TLE affects the strength of these excitatory and inhibitory synapses.
We first examined the effects of electrical stimulation of local synaptic inputs using a stimulating electrode in nearby stratum radiatum (SR), which recruits the excitatory Schaffer collateral inputs and inhibitory inputs from local interneurons (Figure 2A).
Figure 2. Inhibition of CA2 PCs recruited by stimulation of CA2/CA3 axons was diminished in slices from PILO-SE mice.
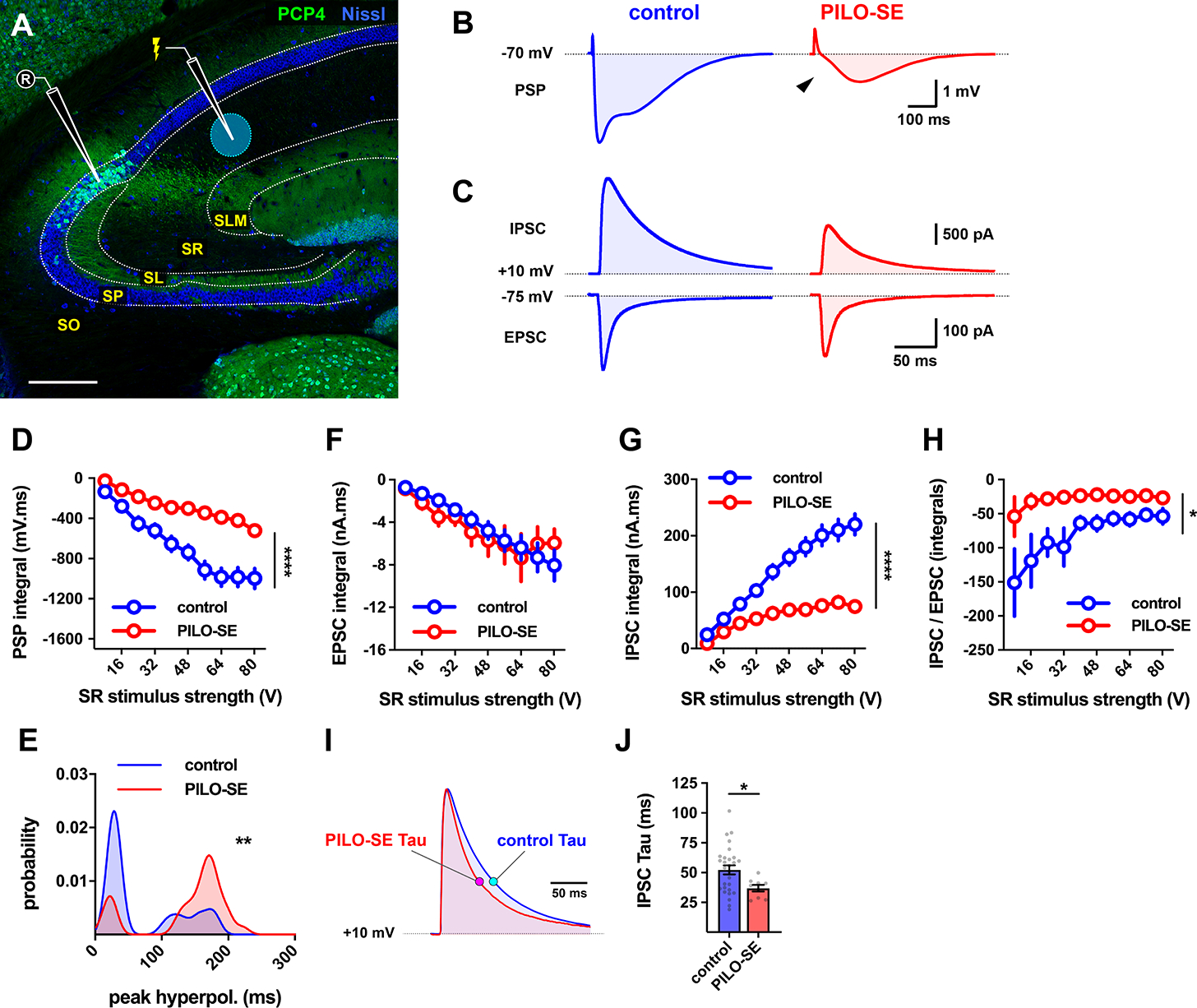
(A) Representative hippocampal section stained for Nissl (blue) and PCP4 (green) to illustrate the configuration used to measure synaptic input to CA2 PCs from the CA2/3 local collaterals in the SR. Scale bar is 250 μm. (B) Representative averaged PSPs from control and PILO-SE CA2 PCs in response to SR stimulation. (C) Top, representative averaged SR-evoked IPSCs in voltage clamp configuration (with intracellular Cs+) from CA2 PCs voltage-clamped at +10 mV. Below, SR-evoked EPSC from CA2 PCs voltage-clamped at −75 mV. (D) The integral of the SR-evoked postsynaptic potential was significantly less negative in CA2 PCs from PILO-SE mice. (E) Probability density functions constructed from the measured latencies between stimulation and the peak hyperpolarization of SR-evoked PSPs. (F - H) Input-output curves of the integral of the SR-evoked EPSC (F), the integral of the SR-evoked IPSC (G), and the ratio between the integrals of the IPSC and EPSC (H). (I) Representative averaged traces illustrating the time course of SR-evoked IPSCs with the exponential time constant of decay, tau, indicated for the currents from cells from control and PILO-SE mice. (J) The time constant of the SR-evoked IPSC was faster in CA2 PCs from PILO-SE mice. See also Figure S2.
In current clamp recordings from CA2 PCs from control mice, SR stimulation evoked a triphasic postsynaptic potential (PSP), consisting of a small initial depolarization followed by a larger hyperpolarization (Figure 2B, left). In CA2 PCs from epileptic mice, we observed a striking decrease in the magnitude of the prolonged hyperpolarization (Figure 2B, right). As a result, the net integral of the synaptic voltage response was significantly smaller in cells from PILO-SE mice compared to controls (Figure 2D), suggesting a reduction in inhibition (mixed-effects model; ****P < 0.0001; n = 36 cells from 23 control mice, 35 cells from 21 PILO-SE mice).
The triphasic PSP observed in CA2 PCs reflects the sum of monosynaptic excitation, from CA2 and CA3 associational collaterals, and inhibition from interneurons, which consists of both fast and slow hyperpolarizing components mediated by GABAA and GABAB receptors, respectively. We observed a pronounced loss of fast inhibition in PILO-SE mice relative to controls (Figure 2B, arrowhead). This altered the dynamics of synaptic inhibition, resulting in a significant shift in the latency to peak inhibition distribution to longer times (Figure 2E; Kolmogorov-Smirnov; **P = 0.0018; n = 25 cells from 18 control mice, 30 cells from 20 PILO-SE cells).
We next performed voltage clamp recordings to isolate the SR-evoked excitatory and inhibitory postsynaptic currents (EPSCs and IPSCs). There was little difference in the peak amplitude of the EPSC (recorded at a holding potential of −75 mV, near the reversal potential of the IPSC) in PILO-SE mice compared to controls (Figure S2A). However, the peak amplitude of the IPSC (recorded with the membrane held near the reversal potential of the EPSC, +10 mV), was markedly reduced in PILO-SE animals compared to controls (Figure 2C, Figure S2B; mixed-effects model; ***P = 0.0007; n = 32 cells from 15 control mice, 15 cells from 9 PILO-SE mice).
Measurement of the integral of the synaptic currents (Figure 2F, G) confirmed a significant decrease in the total charge carried by the IPSC (mixed-effects model; ****P < 0.0001; n = 32 cells from 15 control mice, 15 cells from 9 PILO-SE mice) with no change in EPSC charge (mixed-effects model; P = 0.7856; n = 30 cells from 15 control mice, 15 cells from 10 PILO-SE mice), resulting in a pronounced decrease in the IPSC/EPSC ratio (Figure 2H; mixed-effects model; *P = 0.0321; n = 30 cells from 15 control mice, 14 cells from 9 PILO-SE mice). Consistent with the above findings, the PILO-SE mice showed no change in the maximal slope of the EPSC (mixed-effects model; P = 0.6809; n = 30 cells from 15 control mice, 15 cells from 10 PILO-SE mice), compared to a significant decrease in the IPSC maximal slope (mixed-effects model; **P = 0.0064; n = 32 cells from 15 control mice, 15 cells from 9 PILO-SE mice; at a stimulus intensity of 64 V, the SR-evoked IPSC slope was 1.419 ± 0.199 nA/ms in control cells and 0.583 ± 0.094 nA/ms in cells from PILO-SE mice). Finally, the IPSC decayed significantly faster in PILO-SE mice compared to controls, as quantified by a decrease in the exponential decay time constant tau (Figure 2I, J; Mann-Whitney; *P = 0.0403; n = 28 cells from 13 control mice, 9 cells from 7 PILO-SE mice).
In contrast to the reduced synaptic inhibition in response to electrical stimulation of the SR inputs to CA2, PILO-SE caused no significant change, relative to controls, in either synaptic inhibition or excitation evoked by stimulation of the direct entorhinal cortical projections to CA2, using a stimulating electrode in stratum lacunosum moleculare (SLM) (Figure S3).
PILO-SE reduces inhibition but not excitation evoked by optogenetic activation of CA2 recurrent collaterals
Next we examined whether PILO-SE altered synaptic responses evoked by optogenetic activation of CA2 PC recurrent connections. We stereotactically injected Cre-dependent adenoassociated virus (AAV) in the dorsal hippocampus of Amigo2-Cre mice to drive expression of channelrhodopsin-2 (ChR2-eYFP) selectively in CA2 PCs (see Methods, Figure S4). In acute hippocampal slices photostimulation (2 ms pulses) effectively triggered CA2 action potential output (Figure S4B), which evoked mixed excitatory-inhibitory synaptic responses recorded from CA2 PCs not expressing ChR2 (Figure 3A - C).
Figure 3. The inhibitory-excitatory balance of the CA2 → CA2 recurrent circuit was reduced in slices from PILO-SE.
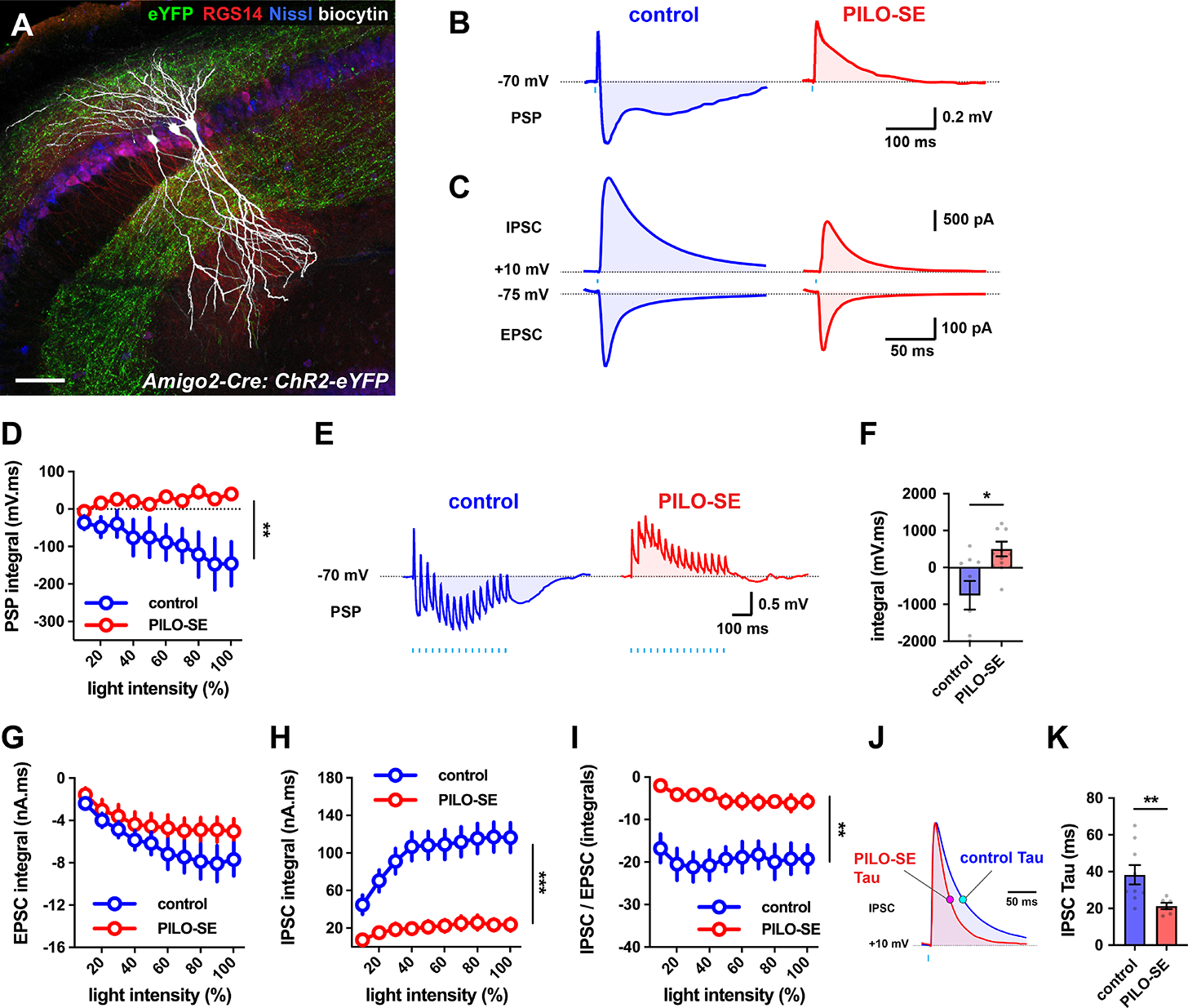
(A) Biocytin-filled CA2 PCs (white) in a slice from intermediate hippocampus, with ChR2-eYFP-expressing CA2 PC axons (green) visible in SO and SR. CA2 PCs were labeled for RGS14 (red) and neuronal somata were visualized with Nissl stain (blue). Scale bar, 80 μm. (B) Representative averaged light-evoked PSPs from CA2 PCs from control and PILO-SE mice. (C) Representative averaged light-evoked EPSCs and IPSCs from control and PILO-SE CA2 PCs. (D) The integral of the light-evoked PSP was significantly more positive in CA2 PCs from PILO-SE mice. (E) Representative averaged PSPs evoked by 15 pulses of light delivered at 30 Hz in cells from control and PILO-SE mice. (F) The integral of the train-evoked PSP is significantly more positive in cells from PILO-SE mice. (G - I) Input-output curves of the integral of the light-evoked EPSC, the integral of the light-evoked IPSC, and the ratio of the integrals of the light-evoked IPSC and EPSC. (J) Representative averaged light-evoked IPSCs from CA2 PCs with the time constant indicated with magenta and cyan markers on the control and PILO-SE currents, respectively. (K) The time constant of the light-evoked IPSC was significantly shorter in cells from PILO-SE mice. See also Figure S2.
In control slices, optogenetic stimulation evoked a triphasic PSP, with an initial fast EPSP followed by discernable fast and slow IPSPs (Figure 3B). PILO-SE reduced markedly the inhibitory component of the PSP (Figure 3B), similar to its effect on the SR-evoked PSP. The integral of the light-evoked PSP revealed a net hyperpolarization in control mice that was transformed into a net depolarization in PILO-SE mice (Figure 3D; mixed-effects model; **P = 0.0096; n = 12 cells from 10 control mice, 16 cells from 12 PILO-SE mice). The change from net inhibition to net excitation was also observed in the PSPs evoked by a train of 15 photostimulation pulses at 30 Hz (Figure 3E, F; Mann-Whitney; *P = 0.0208; n = 9 cells from 9 control mice, 13 cells from 10 PILO-SE mice). In PILO-SE, the depolarizing PSPs summated to exceed the depolarizing response to the first light pulse (one sample t test; *P = 0.0362; 13 cells from 10 PILO-SE mice).
Voltage-clamp recordings confirmed the current clamp results (Figure 3C, G – K). Thus, PILO-SE greatly reduced the IPSC integral (Figure 3H; mixed-effects model; ***P = 0.0001; n = 16 cells from 9 control mice, 11 cells from 4 PILO-SE mice), with no change in EPSC integral (Figure 3G; mixed-effects model; P = 0.2108; n = 15 cells from 8 control mice, 11 cells from 5 PILO-SE mice), leading to a large decrease in the IPSC/EPSC integral ratio (Figure 3I; mixed-effects model; **P = 0.0013; n = 14 cells from 8 control mice, 9 cells from 4 PILO-SE mice).
PILO-SE also caused a profound decrease in the light-evoked IPSC peak amplitude (Figure S2E; mixed-effects model; **P = 0.0052; n = 16 cells from 9 control mice, 11 cells from 4 PILO-SE mice), maximal slope (IPSC slope at 80% maximal intensity = 1.22 ± 0.19 nA/ms in control [n = 16 cells from 9 mice] and 0.49 ± 0.15 nA/ms in PILO-SE [n = 11 cells from 4 mice]; mixed-effects model; **P = 0.0034), and IPSC duration (Figure 3J, K; Mann-Whitney; **P = 0.0076; n = 9 cells from 8 control mice, 6 cells from 3 PILO-SE mice). In contrast, PILO-SE had no effect on EPSC amplitude (Figure S2D; two-way ANOVA; P = 0.8888; n = 15 cells from 8 control mice, 11 cells from 5 PILO-SE mice), slope (two-way ANOVA; P = 0.5812; n = 15 cells from 8 control mice, 11 cells from 5 PILO-SE mice), or duration (Mann-Whitney; P = 0.4378; n = 13 cells from 7 control mice, 7 cells from 3 PILO-SE mice). Thus, PILO-SE caused a selective loss of inhibition in the CA2 recurrent network, similar to that seen with electrical stimulation of the CA3 inputs to CA2.
PILO-SE increases synaptic excitation of CA2 PCs by their mossy fiber inputs from DG
Next we used an optogenetic approach to examine whether PILO-SE altered the DG mossy fiber input to CA2 PCs given that mossy fiber sprouting is seen in both human TLE (Freiman et al., 2021) and rodent models (Häussler et al., 2016). To express ChR2 in selectively in DG granule cells, we crossed the proopiomelanocortin (POMC)-Cre mouse line (McHugh et al., 2007), in which Cre is transiently expressed in newly-born DG granule cells, with a Cre-dependent ChR2-eYFP reporter line (Madisen et al., 2012). ChR2-eYFP+ mossy fiber axons colocalized with the proximal apical dendrites of CA2 PCs, whose lack of thorny excrescences distinguishes them from CA3 PCs (Figure 4A–C, Figure S5).
Figure 4. The inhibitory-excitatory balance of the dentate gyrus granule cell mossy fiber pathway to CA2 was reduced after PILO-SE.
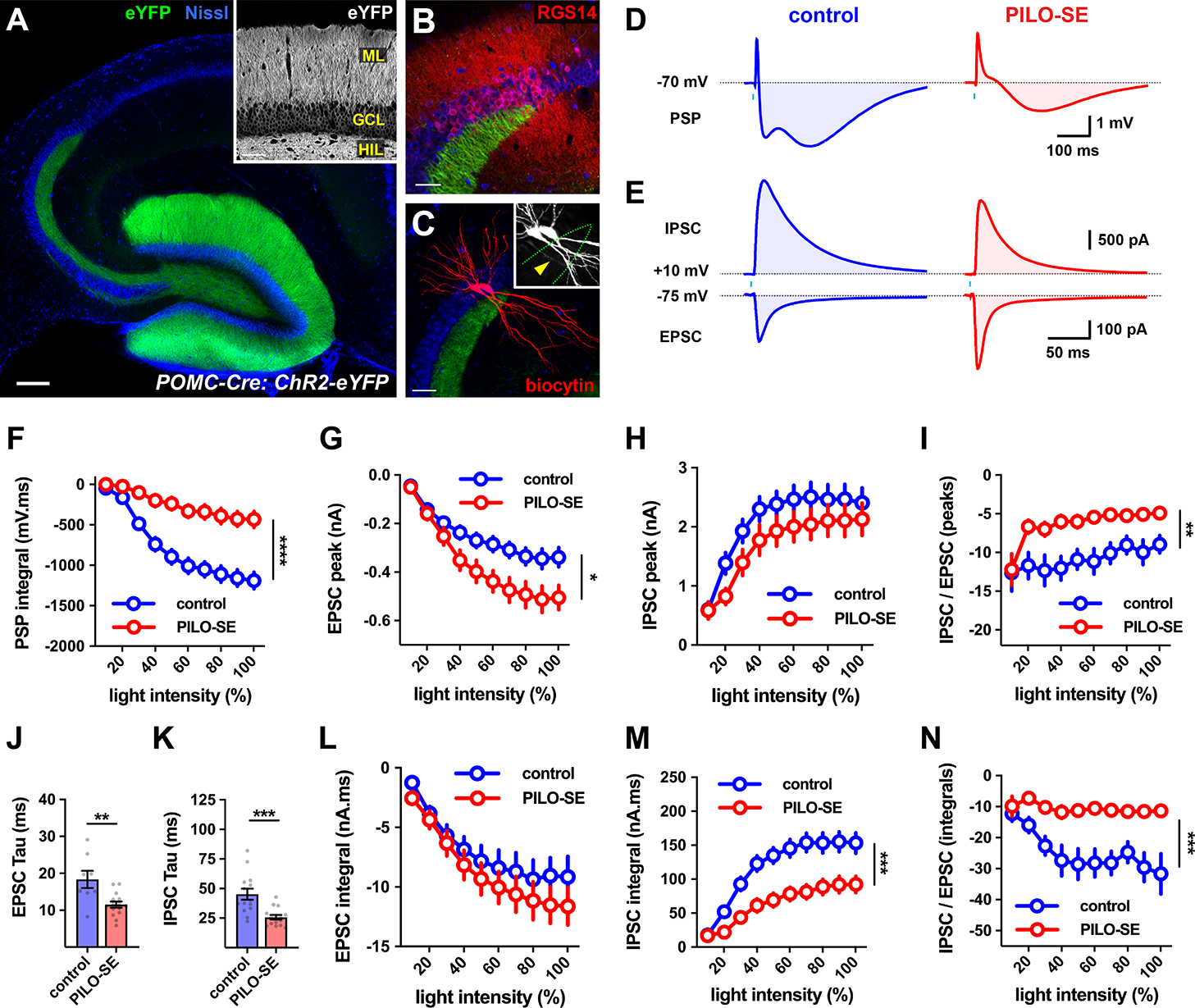
(A) A representative section from a POMC-Cre mouse expressing ChR2-eYFP (green) in DG granule cells, with neuronal somata stained for Nissl substance (blue). Scale bar is 200 μm. Inset, ChR2-eYFP expression in the granule cell layer (GCL), the apical dendrites in the molecular layer (ML), and the mossy fiber axons in the hilus (HIL). Scale bar, 60 μm. (B) The CA2 region (defined by RSG14, red) straddles the end of the mossy fiber projection (Dudek, Alexander, & Farris, 2016). Scale bar, 60 μm. (C) Two CA2 PCs (filled with biocytin, red) showing a lack of thorny excrescences (inset, arrowhead, biocytin, white). Scale bar, 80 μm. (D) Representative averaged light-evoked PSPs in CA2 PCs from control and PILO-SE mice. (E) Representative averaged light-evoked EPSCs and IPSCs in cells from control and PILO-SE mice. (F) The input-output curve of the integral of the light-evoked PSP in CA2 from control and PILO-SE mice. (G - I) Input-output curves of the peak amplitude of the light-evoked EPSC in CA2 PCs, the peak amplitude of the light-evoked IPSC, and the ratio of the peak amplitudes of the light-evoked IPSC and EPSC. (J, K) The time constants (tau) of the light-evoked EPSC and IPSC were shorter in cells from PILO-SE mice. (L - N) Input-output curves of the integral of the light-evoked EPSC, the integral of the light-evoked IPSC, and the ratio between the integrals of the light-evoked IPSC and the EPSC. See also Figure S5 and Figure S6.
In control mice, photostimulation of mossy fiber axons evoked a triphasic PSP in CA2 PCs (Figure 4D, left). PILO-SE caused a significant decrease in both the magnitude of the peak hyperpolarization during the light-evoked PSP (Figure 4D, right) and its integral (Figure 4F; mixed-effects model; control vs PILO-SE, ****P < 0.0001; n = 21 cells from 9 control mice, 26 cells from 9 PILO-SE mice).
In contrast to the lack of change in excitatory synaptic transmission at the CA3 Schaffer collateral or recurrent CA2 inputs to CA2 PCs, PILO-SE produced a significant increase in the peak amplitude of the mossy fiber-evoked EPSCs (Figure 4E, G; mixed-effects model; *P = 0.0334; n = 23 cells from 9 control mice, 29 cells from 7 PILO-SE mice) and in the EPSC slope (mixed-effects model; *P = 0.0282; n = 23 cells from 9 control mice, 29 cells from 7 PILO-SE mice; at a photostimulation intensity of 80% the EPSC slope was −0.111 ± 0.017 nA/ms for control cells and −0.183 ± 0.025 nA/ms in cells from PILO-SE mice).
Surprisingly, despite the clear decrease in the hyperpolarizing phase of the mossy fiber PSP, under voltage clamp, there was no decrease in either the peak amplitude of the IPSC (Figure 4H; mixed-effects model; P = 0.1820; n = 23 cells from 9 control mice, 28 cells from 7 PILO-SE mice) or in its slope (mixed-effects model; P = 0.7331; n = 23 cells from 9 control mice, 28 cells from 7 PILO-SE mice). However, because of the increased EPSC amplitude, the ratio of the peak IPSC to peak EPSC amplitude was significantly reduced in the epileptic mice (Figure 4I; mixed-effects model; **P = 0.0029; n = 21 cells from 9 control mice, 28 cells from 7 PILO-SE mice).
Although PILO-SE altered the EPSC but not IPSC amplitude, this treatment significantly speeded the time course of decay of both the IPSCs and EPSCs (Figure 4J, K; Mann-Whitney; EPSC tau: **P = 0.0067, n = 8 cells from 5 control mice and 14 cells from 7 PILO-SE mice; IPSC tau: ***P = 0.0003, n = 14 cells from 8 control mice, 15 cells from 7 PILO-SE mice). As a result of the offsetting effects of the faster EPSC decay with the increased EPSC amplitude, EPSC integral was unchanged by PILO-SE (Figure 4L; mixed effects model; P = 0.3705; n = 23 cells from 9 control mice, 29 cells from 7 PILO-SE mice). In contrast, as a result of the speeding of the IPSC decay with no change in its peak amplitude, the integral of the IPSC was significantly reduced in CA2 PCs from PILO-SE mice (Figure 4M; mixed-effects model; ***P = 0.0003; n = 23 cells from 9 control mice, 28 cells from 7 PILO-SE mice). This effect likely accounts for the decrease in the hyperpolarizing phase of the PSP seen under current clamp. The net effect of the reduction in IPSC integral and unchanged EPSC integral led to a marked net decrease in the ratio of IPSC/EPSC integrals (Figure 4N; mixed-effects model; ***P=0.0002; n = 21 cells from 9 control mice, 28 cells from 7 PILO-SE mice). These changes to the mossy fiber-evoked EPSC and IPSC in PILO-SE greatly increases the net excitatory drive from DG to CA2.
As previous studies in TLE models found an increased synaptic depression at mossy fiber synapses during a train of stimuli (Hendricks, Chen, Bensen, Westbrook, & Schnell, 2017; Hendricks, Westbrook, & Schnell, 2019), we examined mossy fiber to CA2 synaptic responses using a 500 ms long train of 30 Hz photostimulation. In control mice, the light-evoked EPSCs exhibited a small but significant short-term depression (Figure S6A, B), in which the EPSC amplitude at the end of the train was reduced to 55.8 ± 8.5% of its initial size (n = 14 cells from 8 control mice). The EPSC amplitude rapidly recovered following the end of the train, reaching 109.9 ± 9.5% of its initial value with a test stimulus delivered after 500 ms (Figure S6A, B). PILO-SE dramatically enhanced short-term depression in CA2 PCs during the train of photostimulation (Figure S6A, B), with the amplitude of the final EPSC reduced to 12.2 ± 1.8% of its initial level (control vs PILO-SE, two-way ANOVA; ****P < 0.0001; n = 14 cells from 8 control mice, 27 cells from 7 PILO-SE mice). Additionally, the depression was longer lasting in PILO-SE mice as the EPSC did not fully recover after the 500 ms interval, returning to only 45.8 ± 4.4% of its initial amplitude (n = 14 cells from 8 control mice, 27 cells from 7 PILO-SE mice; Figure S6A, B).
Because of the offsetting effects of the increased mossy fiber EPSC amplitude in response to a single stimulus and the increased synaptic depression during a train of stimuli, the net integral of the compound PSP evoked by the photostimulation train was not significantly altered by PILO-SE (Figure S6C, D; Mann-Whitney; P = 0.2279; n = 16 cells from 6 control mice and 23 cells from 9 PILO-SE mice). In contrast, the maximal hyperpolarization was reduced significantly (Figure S6C, E; Mann-Whitney; **P = 0.0016; n = 16 cells from 6 control mice and 23 cells from 9 PILO-SE mice).
PILO-SE strengthens CA2 excitation of CA1
To determine whether PILO-SE altered the synaptic responses elicited by optogenetic activation of CA2 excitatory inputs in their CA1 PC targets, we expressed ChR2 selectively in CA2 PCs using targeted injections of AAV-DIO-ChR2-eYFP into dorsal CA2 of Amigo2-Cre mice (Figure S4). We recorded from CA1 PCs in the deep pyramidal cell layer (Figure 5A - C) as these cells normally receive stronger input from CA2 than do superficial-layer CA1 PCs (Kohara et al., 2014; Valero et al., 2015). We further focused on the CA1c subfield (also termed proximal CA1), near the CA2 border, as this region was more resistant to neurodegeneration that other CA1 subfields in the PILO-SE mice.
Figure 5. PILO-SE strengthened CA2 excitation of CA1.
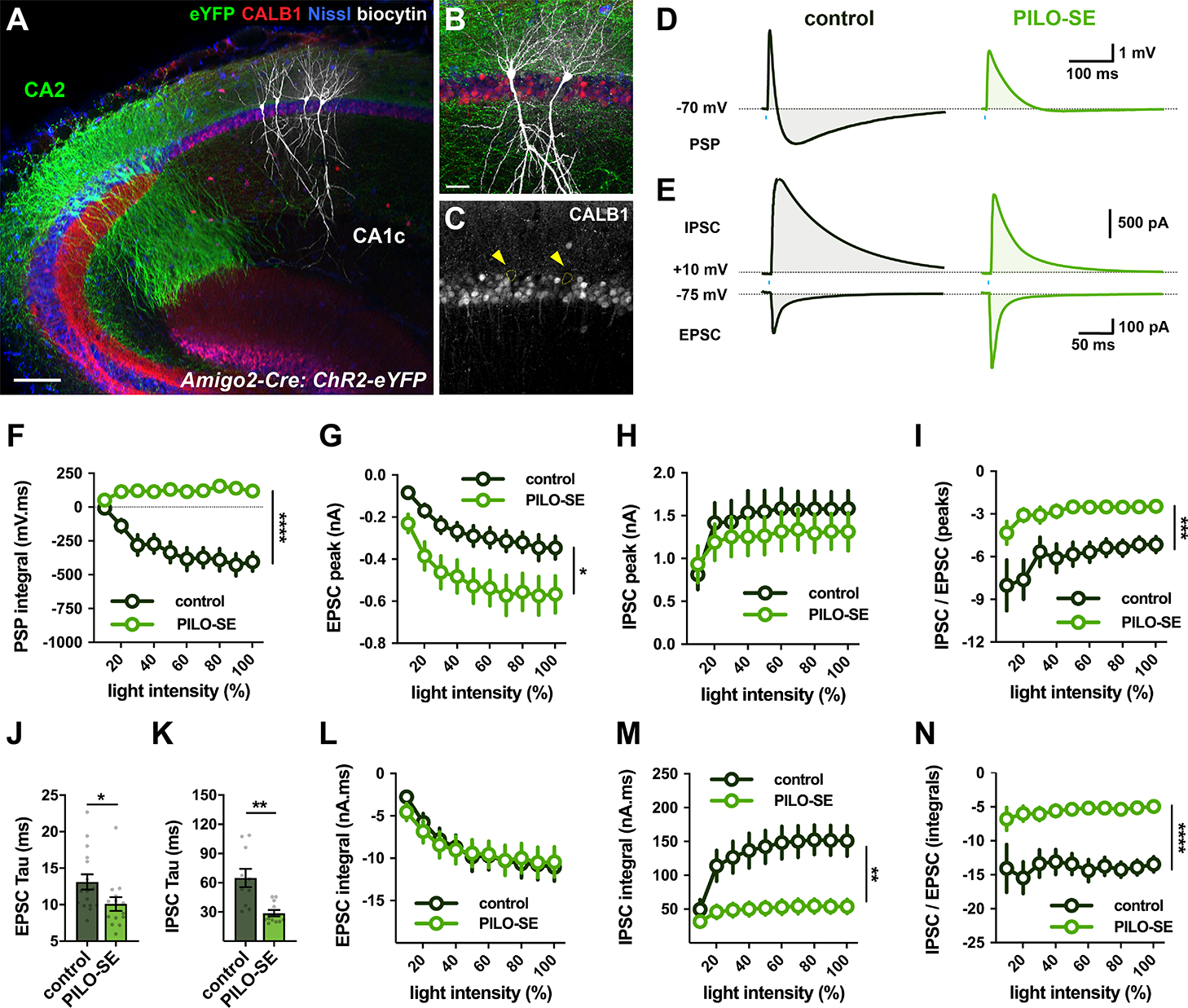
(A - C) A hippocampal slice with biocytin-filled CA1c PCs (white) located in the deep sublayer of SP, adjacent to SO. ChR2-eYFP-expressing CA2 PC dendrites and axonal projections (green) visible throughout SR and SO. Neuronal somata labeled with Nissl stain (blue); Calbindin-1 stain (CALB1, red) delineates superficial CA1 PCs (Lee et al., 2014). Scale bars, 150 μm in A and 40 μm in B. (D) Representative averaged light-evoked PSPs from deep CA1c PCs in slices from control (left, dark green) and PILO-SE (right, bright green) mice. (E) Representative averaged light-evoked EPSCs and IPSCs from control and PILO-SE CA1c PCs. (F) The integral of the light-evoked PSP was significantly more positive in CA1 PCs from PILO-SE mice. (G - I) Input-output curves of the light-evoked EPSC amplitude, IPSC amplitude, and the ratio of the IPSC and EPSC peak amplitudes. (J, K) The time constants of the light-evoked EPSC and IPSC were significantly shorter in cells from PILO-SE mice. (L - N) Input-output curves of the integral of the light-evoked EPSC, the integral of the IPSC, and the ratio of the IPSC and EPSC integrals.
Current clamp recordings from CA1 PCs of control mice revealed a biphasic PSP in response to optogenetic stimulation of CA2 inputs with a single light pulse, with an initial brief depolarization followed by a large hyperpolarization, resulting in a net negative PSP integral (Figure 5D, left). Similar to our findings in recordings from CA2 PCs, the net hyperpolarization was greatly reduced in PILO-SE mice, resulting in a net positive PSP integral (Figure 5D, F; mixed-effects model; ****P < 0.0001; n = 19 cells from 5 control mice, 24 cells from 7 PILO-SE mice). Moreover, whereas a brief train of light pulses evoked summating hyperpolarizing responses that often produced a net negative integral in CA1 PCs from control mice, the stimuli elicited a net depolarization and positive integral in cells from epileptic animals. (Figure 6A, B; Mann-Whitney; ****P < 0.0001; n = 16 cells from 5 control mice, 23 cells from 7 PILO-SE mice).
Figure 6. PILO-SE enhanced the ability of CA2 PCs to drive action potential output from CA1 PCs.
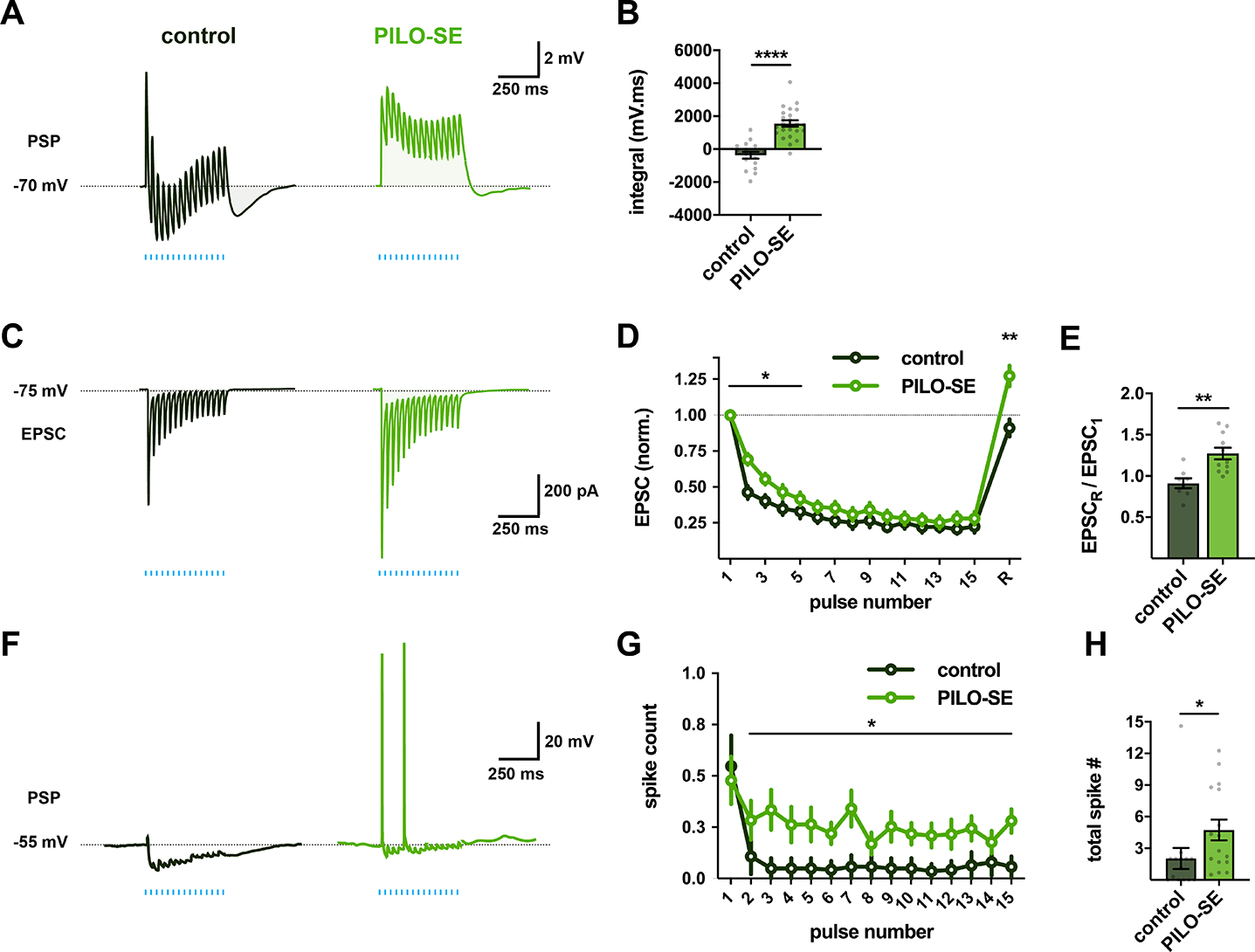
(A) Representative averaged PSPs from control and PILO-SE mice evoked by 15 lights pulses delivered at a frequency of 30 Hz for 500 ms. (B) The integral of the train-evoked PSP was significantly larger in deep CA1c PCs from PILO-SE mice. (C) Representative averaged EPSCs evoked by 15 light pulses delivered at a frequency of 30 Hz (blue lines) across 500 ms, recorded from control (dark green) and PILO-SE (bright green) PCs. (D) Normalized EPSC amplitudes evoked by a train of photostimulation, with 15 pulses delivered at 30 Hz, followed after 500 ms by a single recovery pulse. (E) The normalized amplitude of the recovery pulse EPSC was larger in cells from PILO-SE mice than in controls. (F) Representative photostimulation train-evoked PSPs from CA1 PCs held at an initial potential of −55 mV. (G) The mean number of action potentials evoked following the first pulse of the photostimulation train was increased in PILO-SE mice. (H) The mean total number of action potentials evoked by the 30 Hz photostimulation protocol was significantly increased in PCs from PILO-SE mice.
Voltage-clamp recordings from CA1 PCs during optogenetic activation of CA2 inputs (Figure 5E, left) revealed that PILO-SE enhanced significantly the EPSC amplitude (Figure 5G; two-way ANOVA; *P = 0.0184; n = 22 cells from 4 control mice, 23 cells from 8 PILO-SE mice) without changing the IPSC size (Figure 5H; mixed-effects model; P = 0.5439; n = 20 cells from 4 control mice, 20 cells from 8 PILO-SE mice). As a result, the ratio of the peak IPSC to the peak EPSC amplitudes was smaller in CA1 PCs from PILO-SE mice than from controls (Figure 5I; mixed-effects model; ***P = 0.0008; n = 20 cells from 4 control mice, 20 cells from 8 PILO-SE mice). A similar effect of PILO-SE was seen in an increased maximal EPSC slope (at a photostimulation intensity of 80%, EPSC slope was −0.13 ± 0.02 nA/ms in control cells and −0.28 ± 0.05 nA/ms in cells from PILO-SE mice; two-way ANOVA; **P = 0.0092; n = 22 cells from 4 control mice, 23 cells from 8 PILO-SE mice) and unchanged IPSC slope (mixed-effects model; P = 0.8502; n = 20 cells from 4 control mice, 20 cells from 8 PILO-SE mice). We also observed a significant speeding of the time course of decay of both EPSCs (Figure 5J; Mann-Whitney; *P = 0.0372; n = 16 cells from 4 control mice, 14 cells from 5 PILO-SE mice) and IPSCs (Figure 5K; Mann-Whitney; **P = 0.0011; n = 10 cells from 4 control mice, 11 cells from 4 PILO-SE mice). As a result of the offsetting effects of the increase in EPSC amplitude and speeding of EPSC decay, there was no net change in the EPSC integral in PILO-SE mice (Figure 5L; two-way ANOVA; P = 0.9400; n = 22 cells from 4 control mice, 23 cells from 8 PILO-SE mice). In contrast, the speeding of the IPSC decay in the face of a constant IPSC amplitude produced a marked decrease in IPSC integral in PILO-SE mice compared to controls, (Figure 5M; mixed-effects model; **P = 0.0025; n = 20 cells from 4 control mice, 20 cells from 8 PILO-SE mice). Consequently, the ratio of the IPSC/EPSC integral was greatly reduced in cells from the epileptic mice (Figure 5N; mixed-effects model; ****P < 0.0001; n = 20 cells from 4 control mice, 20 cells from 8 PILO-SE mice).
In contrast to the effect of PILO-SE to enhance synaptic depression of the mossy fiber to CA2 EPSC, this treatment reduced the synaptic depression observed in recordings from CA1 PCs (Figure 6C, D). This effect was also seen in the significant decrease in paired-pulse depression of the first two EPSCs of the train (Mann-Whitney; **P = 0.0018; n = 8 cells from 3 control mice, 11 cells from 5 PILO-SE mice). The decreased depression combined with the increased peak EPSC amplitude led to a synergistic increase in the excitation of CA1 PCs in response to a train of CA2 inputs in PILO-SE mice relative to that in control animals (Figure 6C, D; two-way ANOVA; first 5 EPSCs; *P = 0.0259; n = 8 cells from 3 control mice, 11 cells from 5 PILO-SE mice), Furthermore, a test stimulus to CA2 delivered 500 ms after the train revealed a short-term potentiation of the EPSC in CA1 PCs from the PILO-SE but not control mice (Figure 6D, E; one sample t test; **P = 0.0034; n = 8 cells from 3 control mice, 11 cells from 5 PILO-SE mice).
In addition to their altered CA2 synaptic input, CA1 PCs from PILO-SE mice, like CA2 PCs, showed an increased intrinsic excitability, firing action potentials at a higher rate (25.4 ± 1.5 Hz) than controls (17.7 ± 1.4 Hz) in response to 500 pA current injections (mixed-effects model; ****P <0.0001; n = 32 cells from 9 control mice, 48 cells from 13 PILO-SE mice). To determine how PILO-SE impacted the ability of CA2 inputs to drive CA1 PC action potential output, we delivered a train of CA2 optogenetic stimuli with the CA1 PCs held under current clamp at a membrane potential near threshold (−55 mV), similar to the in vivo membrane potentials observed in hippocampal neurons (Figure 6F). In slices from control mice, the train of photostimulation evoked only sparse firing in CA1 PCs, with postsynaptic responses dominated by inhibition (Figure 6F, G). In contrast, optogenetic stimulation was much more effective in exciting CA1c PCs from PILO-SE mice, with an increase in spike probability following the first stimulus of the train (Figure 6G; two-way ANOVA; *P = 0.0232; n = 14 cells from 4 control mice, 16 cells from 4 PILO-SE mice) and an increase in the total number of spikes elicited per train (Figure 6H; Mann-Whitney; *P = 0.0133; n = 14 cells from 4 control mice, 16 cells from 4 PILO-SE mice).
CA2 PCs also send an excitatory output to CA3 PCs (Figure S4C, D, G1 – G3), although the synaptic response in CA3 is dominated by strong feedforward inhibition (Kohara et al., 2014; Boehringer et al., 2017). In contrast to the increased net synaptic excitation of CA1 PCs by their CA2 inputs in epileptic mice, PILO-SE caused a decrease in the integrals of both the EPSCs and IPSCs recorded from CA3 PCs in response to optogenetic activation of their CA2 PC inputs, with no change in the ratio of inhibition to excitation (Figure S7B - E). Delivery of a 30 Hz photostimulation train revealed a decrease in the short-term depression of the light-evoked EPSCs (Figure S7F, G).
Chemogenetic inhibition of CA2 reduces the frequency of spontaneous seizures.
The above results indicate that PILO-SE leads to a series of changes in the intrinsic excitability, synaptic input to, and synaptic output from CA2 PCs that increases excitation through the cortico-hippocampal circuit and may therefore enhance seizure activity (Figure S8). These results led us to test this hypothesis using a chemogenetic approach to selectively suppress CA2 PC activity in Amigo2-Cre PILO-SE mice experiencing spontaneous recurrent seizures. We asked whether inhibition of CA2 activity had any effect on the frequency, severity, or other features of seizures (Figure 7A, see Methods) (Figure 7B). To achieve selective silencing of CA2, Amigo2-Cre mice were injected in CA2 with a Cre-dependent AAV (AAV-DIO-hM4Di-mCherry) expressing the hM4Di inhibitory DREADD (iDREADD; Figure 7C). Continuous video EEG recordings began 4 weeks after PILO-SE treatment and continued 24 hr/day, 7 days/week for at least 4 weeks. One group of mice received the DREADD ligand CNO in their drinking water for 2–3 weeks, followed by 2–3 weeks of water without CNO; in a separate group we reversed the order of treatment (Figure 7A). In some mice this schedule was then repeated with similar results. After video EEG recordings, mice were perfusion-fixed and brain tissue examined to determine PILO-SE induced pathology and confirm selective expression of virus in CA2 (Figure 7D1, D2). In some experiments, mice were perfusion-fixed at earlier times to confirm CA2-selective DREADD expression throughout the recordings. As noted above, we observed a classic pattern of neuropathology in PILO-SE mice consistent with human MTS, with substantial neuronal loss in the hilus, CA3 and CA1 (Figure S1) and relative survival of CA2.
Figure 7. Chemogenetic silencing of CA2 reduced convulsive seizure frequency.
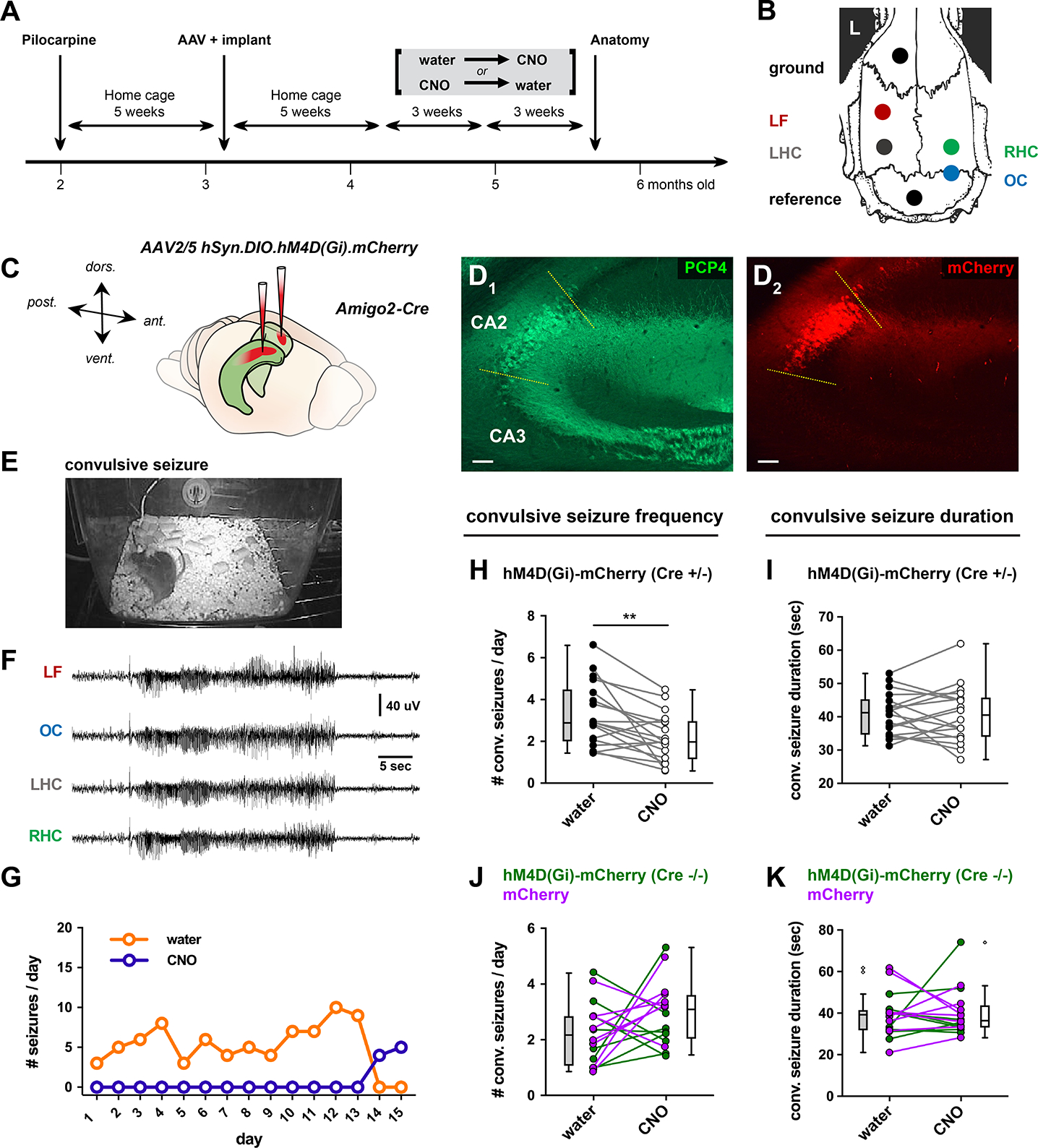
(A) Timeline of experiments. 5 weeks after PILO-SE, Cre-dependent AAV expressing hM4Di-mCherry or mCherry alone were injected in CA2 of Amigo2-Cre mice or wild-type controls. EEG electrodes were implanted at same time. CNO was either present or absent from drinking water for 3-week periods of continuous video EEG recording. Order of CNO delivery was randomized. (B) Locations of electrodes for EEG. LF, left frontal; LHC, left hippocampus; RHC, right hippocampus; OC, occipital cortex. (C) Locations of bilateral AAV injections in dorsal CA2. (D) Representative immunohistochemistry micrographs for CA2 marker PCP4 (D1) and AAV-mediated expression of hM4D(Gi)-mCherry (D2). Scale bars, 100 μm. (E, F) Frame from the continuous video (E) and corresponding EEG (F) during a convulsive seizure in an Amigo2-Cre mouse injected with hM4D(Gi)-mCherry. (G) Daily seizure counts in one mouse expressing hM4D(Gi)-mCherry in CA2 during 15 days in the absence of CNO (orange) followed by 15 days with CNO in drinking water (purple). (H) Paired analysis of convulsive seizure frequency in the presence of CNO compared with the absence of CNO (water control) in the same Amigo2-Cre mice expressing hM4D(Gi)-mCherry in CA2. CNO reduced convulsive seizure frequency. (I) CNO did not alter convulsive seizure duration in Amigo2-Cre mice expressing hM4D(Gi)-mCherry in CA2 (paired t-test; t = 0.0804, df = 17; P = 0.9368; n = 18 mice). (J) CNO did not alter convulsive seizure frequency in two groups of control mice: Amigo2-Cre mice expressing mCherry in CA2 (magenta symbols and lines) and wild-type mice (Cre−/−) injected with AAV2/5 hSyn.DIO.hM4D(Gi).mCherry (paired t-test; t = 1.573, df = 15; P = 0.1366; n = 16 mice). (K) CNO did not alter convulsive seizure duration in control mice (paired t-test; t = 0.3446, df = 15; P = 0.7352; n = 16 mice). See also Figure S9.
PILO-SE treatment reliably induced spontaneous seizures (9/9 males and 5/9 females; Fisher’s Exact test, p=0.0824), with >90% of seizures associated with stage 4–5 convulsions using the Racine scale (Racine, 1972). EEG activity was similar to seizures in human TLE, with large amplitude, high frequency, rhythmic activity that continued for over 20 sec and was generalized across four electrodes (Figure 7E, F). All mice included in the study had frequent convulsive seizures, with total numbers of seizures often exceeding 10 per week (the mean total number of convulsive seizures was 68.94 ± 7.56 over three weeks of recordings, Figure 7). In addition, much like human TLE (Baud et al., 2018), seizures could show clustering (Figure S9). Most seizures terminated in a prominent postictal depression of the EEG, another characteristic of robust seizures (Figure 7F). These properties were observed both during periods when CNO was absent from the drinking water and during periods when CNO was delivered. However, the frequency and total number of seizures declined significantly with CNO, as delineated below.
Periods with CNO treatment showed a striking decrease in convulsive seizure frequency compared with seizure frequency during periods when the drinking water did not contain CNO (exemplified in Figure 7G). This decrease was statistically significant when we compared seizure frequency from all mice with and without CNO (Figure 7H; paired t-test; t = 3.58, df = 17; **P = 0.0023; n = 18 mice). The decrease in seizure frequency was not caused by off-target effects of CNO as CNO treatment had no significant effect on seizure frequency in two groups of control mice that did not express iDREADD: Amigo2-Cre−/− mice injected with AAV-DIO-hM4Di-mCherry (which do not express iDREADD), and Amigo2-Cre+/− or Cre−/− mice injected with AAV-hSyn-DIO-mCherry (Figure 7J).
In contrast to the effect on seizure frequency, CNO treatment had no effect on seizure duration, in either iDREADD-expressing PILO-SE mice (Figure 7I) or in the two PILO-SE control groups (Figure 7K). Moreover, CNO treatment did not alter seizure severity or the tendency of seizures to occur in clusters (Figure S9). Consistent with the decreased convulsive seizure frequency during CNO treatment, the total number of convulsive seizures was reduced compared to periods without CNO (paired t-test; **P = 0.0023; n = 18 mice). There was no significant sex difference in either baseline seizure frequency or in the effect of CA2 silencing. For seizure frequency, a two-way ANOVA with treatment and sex as factors showed an effect of CNO treatment (F(3,48)63.57, p<0.0001), but not sex (F(1,16)0.2305, p=0.6377), and there was no interaction ((F(3,48)1.200, p=0.3197). Thus, results from male and female mice were pooled in the above analyses.
In addition to the convulsive seizures, a minority of seizures recorded in the EEG were not associated with behavioral convulsions (Figure 8A, B). Similar to its effect on convulsive seizures, CNO treatment of iDREADD-expressing mice also decreased non-convulsive seizure frequency relative to mice that were not treated (Figure 8C, E; paired t-test; t = 2.352, df = 16; *P = 0.0318; n = 17 mice). There was no effect of CNO on non-convulsive seizure duration (Figure 8D), and no effect of CNO in the two control groups described above (Figure 8E, F). As mentioned above, there were few nonconvulsive seizures compared to convulsive seizures (the mean total number of nonconvulsive seizures was 5.00 ± 2.54 over three weeks of recordings). In fact, several mice lacked nonconvulsive seizures entirely (Figure 8C, E).
Figure 8. CA2 silencing reduced nonconvulsive seizure frequency.
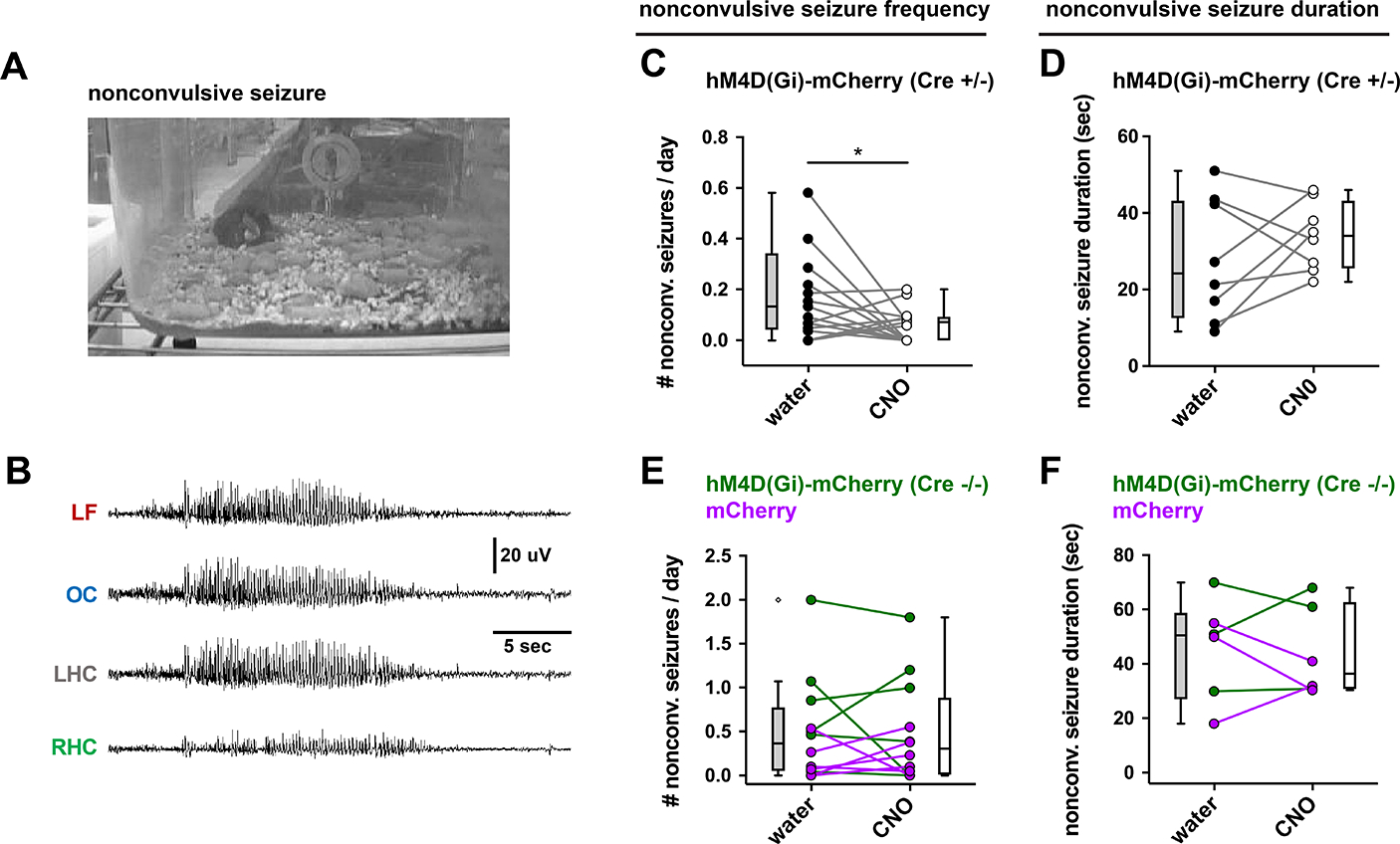
(A, B) A representative example of a nonconvulsive seizure in an Amigo2-Cre mouse expressing hM4D(Gi)-mCherry in CA2. Despite the absence of behavioral indicators (A), EEG revealed significant seizure activity in all electrodes (B). (C) CNO reduced nonconvulsive seizure frequency. (D) CNO did not alter seizure duration (paired t-test; t = 0.6743, df = 6; P = 0.5252; n = 8 mice). (E) CNO did not reduce nonconvulsive seizure frequency in two control groups of mice described in the previous figure (paired t-test; t = 0.1333, df = 11; P = 0.8963; n = 12 mice). (F) CNO did not reduce nonconvulsive seizure duration in control groups (paired t-test; t = 0.2927, df = 5; P = 0.7815; n = 6 mice).
These results indicate that CA2 PC activity contributes to seizure frequency in the PILO-SE model of TLE. Might CA2 also be important for the initial induction of status epilepticus in response to pilocarpine injection? To address this question, we injected AAV-DIO-hM4Di-mCherry in CA2 of Amigo2-Cre+/− mice and Amigo2-Cre−/− control mice, as described above, and injected CNO systemically into both groups of mice 30 min prior to injection of pilocarpine. We found that acute inhibition of CA2 PCs had no significant effect on acute seizures or SE induced by PILO. Thus, the number of convulsive seizures was comparable between Amigo2-Cre+/− mice (2.70 ± 0.27 seizures, n=24) and Amigo2-Cre−/− mice (2.33 ± 0.40 seizures, n=12; t-test; P = 0.53). CA2 inhibition also had no significant effect on either the latency to the first seizure (t-test; P = 0.97; Cre+/−, 52.6 ± 5.6 mins, n=19; Cre−/−, 52.3 ± 4.1 mins, n=12) or the latency to SE (t-test; P = 0.3954; Cre+/−, 77.9± 6.9 mins, n=19; Cre−/−, 68.7 ± 6.4 mins, n=10).
DISCUSSION
Here we provide direct evidence that CA2 PCs play an important role in recurring seizures, based on our findings that chemogenetic silencing of CA2 reduces chronic, spontaneous seizure frequency in the PILO-SE model of temporal lobe epilepsy. Because CA2 PCs do not survive in the most severe cases of MTS (Thom, 2014), CA2 is not absolutely necessary for seizures in TLE. However, in TLE with surviving CA2 PCs, our results suggest that CA2 makes a significant contribution to seizure activity. In principle, surviving CA2 PCs could play a relatively passive role in seizure activity by providing a pathway for propagation of seizures arising from abnormal activity in upstream regions. However, our findings that surviving CA2 PCs have increased intrinsic excitability, receive less synaptic inhibition, and exert a stronger excitatory synaptic output suggest that CA2 plays an active role in seizure generation and/or propagation in TLE. This view is consistent with findings from human TLE specimens, showing both a decrease in CA2 PC synaptic inhibition (Williamson and Spencer, 1994) and increased excitability (Wittner et al., 2009).
A DG to CA2 circuit is poised to contribute to seizure activity in TLE
The finding that DG granule cells are highly resistant to the cell death characteristic of mesial temporal sclerosis (MTS) suggests that DG functions as a critical network node in TLE that gates excitatory input from the entorhinal cortex to downstream hippocampal neuronal populations (Krook-Magnuson et al., 2015; Scharfman, 2019). However, it has been unclear how the DG participates in seizures since its major synaptic target, the CA3 PCs, undergo marked degeneration in TLE accompanied by MTS (Blümcke et al., 2013; Steve et al., 2014; Winawer et al., 2007). A number of anatomical studies have found that TLE is associated with the sprouting of mossy fibers, including in CA2 (Freiman et al., 2021; Häussler et al., 2016). However, the functional effects of such sprouting has been unclear, as the mossy fiber pathway normally provides relatively weak direct excitatory input to CA2 (Kohara et al., 2014; Sun et al., 2017). Our finding that TLE leads to enhanced excitation of CA2 by its DG inputs, together with increased CA2 action potential firing and enhanced excitation of CA1 PCs by CA2, suggests a novel pathway supporting the propagation of activity through the hippocampal network. Thus, the coordinated changes to CA2 we see in PILO-SE mice support the idea that CA2 plays an active role in seizure generation or propagation.
Our results do not provide a direct causal link between the enhanced excitability of CA2 that we observe in acute hippocampal slices and the ability of CA2 PCs to promote the frequency of spontaneous seizures in vivo. However, our finding that acute chemogenetic inhibition of CA2 in normal mice did not alter acute seizure generation or status epilepticus in response to pilocarpine, despite the ability of chemogenetic inhibition of CA2 to markedly reduce spontaneous seizure frequency in PILO-SE mice, suggests that the changes to CA2 we observe in response to PILO-SE may enhance its contribution to hippocampal hyperexcitability and seizure activity. Our finding that CA2 inhibition altered seizure frequency but not seizure duration or severity suggests that CA2 may play a more important role in the initiation of seizures within the hippocampus, and that once initiated the seizures proceed independently of CA2. As data from both the PILO-SE model and clinical seizure recordings suggest that seizures in TLE are generated from several areas, not only the hippocampus (Ogren et al., 2009; Spencer, 2002; Wyeth, Nagendran, & Buckmaster, 2020), CA2 may only contribute to a subset of seizures.
CA2 promotion of seizure activity in epileptic mice may reflect its function in the healthy hippocampus
Our findings that CA2 may play an important role in generating or propagating seizure activity is consistent with recent investigations into the contribution of CA2 to synchronized hippocampal network activity in healthy animals (Kay & Frank, 2019; Lehr et al., 2021). CA2 PC recurrent excitatory connections (Okamoto & Ikegaya, 2018) may contribute to the role of CA2 PCs in generating sharp wave ripples (SWRs) (Oliva et al., 2016), brief periods of synchronous firing observed during immobility or slow wave sleep. Although acute optogenetic inhibition of CA2 decreases the frequency of SWR occurrence (Oliva et al., 2020), more prolonged inhibition of CA2 using chemogenetics can trigger a paradoxical increase in SWRs (Alexander et al., 2018), consistent with findings that CA2 backprojections recruit powerful inhibition of CA3 PCs (Boehringer et al., 2017; Kohara et al., 2014), whose recurrent connections also contribute to SWRs. Thus, prolonged CA2 silencing may produce compensatory changes that enhance the excitability of the CA3 recurrent network through reduced inhibition (Boehringer et al., 2017). We speculate that in TLE, when synaptic inhibition is compromised, CA2 cannot constrain CA3 excitability so that the CA2 and CA3 subfields together act as a hyperexcitable hub. Importantly, in resected human TLE tissue CA2 generates epileptiform activity in parallel to, and independently from, the subiculum (Wittner et al., 2009). There is also a large body of literature suggesting that the DG generates seizures in the PILO-SE model (reviewed in Scharfman, 2019), and in cases where the CA3 subfield exhibits neurodegeneration, such seizures would depend on passage to CA2 to propagate from the hippocampus and develop into a prolonged (>20 sec) generalized seizure.
Do the changes in CA2 PC intrinsic and synaptic properties reflect a maladaptive or homeostatic response?
The coordinated changes we observe in CA2 with PILO-SE suggest that TLE may result, in part, from maladaptive responses that enhance overall CA2 excitation, leading to increased hippocampal network activity. Alternatively, the changes we observe might, in principle, represent a homeostatic response, restoring normal levels of CA2 activation in response to neurodegeneration of the CA3 inputs to CA2. Although it is difficult to provide a direct answer to such a question, several results suggest that the effects we observe are maladaptive, contributing to seizures, rather than homeostatic.
One important piece of evidence comes from our finding that chemogenetic inhibition of CA2 significantly decreases seizure activity in the PILO-SE mouse model of TLE. Second, although CA3 PCs form extensive excitatory connections with CA2 PCs and PILO-SE animals often show degeneration of CA3 PCs, activation of the CA3 Schaffer collaterals normally produces a net inhibitory response in CA2 PCs, due to the recruitment of very strong feedforward inhibition (Chevaleyre & Siegelbaum, 2010; K. Nasrallah, Piskorowski, & Chevaleyre, 2015). Thus, loss of CA3 excitatory input would be expected to enhance net CA2 activity, so that any homeostatic adaptation should decrease CA2 excitation and action potential firing, rather than to increase it as we observed in PILO-SE mice.
A final result arguing against a homeostatic change is based on the fact that the main excitatory drive to CA2 PCs is provided by the direct perforant path connections from Layer II (LII) neurons of entorhinal cortex (Chevaleyre & Siegelbaum, 2010; Sun, Srinivas, Sotayo, & Siegelbaum, 2014), which also provide the behaviorally relevant information that supports the role of CA2 PCs in social memory (Lopez Rojas et al., 2022). In contrast to the loss of CA3 PCs, there is little degeneration of EC LII neurons in TLE in either humans or mouse models (F. Du, Eid, Lothman, Kohler, & Schwarcz, 1995; Fu Du et al., 1993; Janz et al., 2017) and we found that the synaptic response of CA2 PCs to activation of their cortical inputs is unchanged following PILO-SE.
What underlies the increase in CA2 intrinsic excitability with PILO-SE?
Presently the mechanism responsible for the increase in CA2 intrinsic excitability after PILO-SE is unknown, although the increase in input resistance is likely to play an important role. As PILO-SE caused a decrease in membrane capacitance, the increase in input resistance could reflect a loss of membrane area. However, any loss of membrane area is unlikely to reflect a general degeneration of CA2 dendrites since there was no change in the magnitude of the synaptic response evoked by stimulation of the direct cortical perforant path inputs to CA2, which should be most sensitive to any loss of dendritic branches as these inputs target the most distal CA2 dendrites. Another possibility is that there is a downregulation of resting K+ conductance. One potential candidate is the TREK-1 resting K+ channel, which is normally highly enriched in CA2 (Talley, Solórzano, Lei, Kim, & Bayliss, 2001). Upregulation of this channel is thought to contribute to a membrane hyperpolarization and reduction in firing of CA2 PCs in a mouse model of the human 22q11.2 microdeletion (Donegan et al., 2020; Piskorowski et al., 2016), a genetic syndrome strongly linked to schizophrenia (Karayiorgou, Simon, & Gogos, 2010). As a result, we suggest that the downregulation of the same channel may contribute to the enhanced CA2 excitability in TLE.
Although the lack of change in voltage threshold with PILO-SE indicates a lack of direct effect on fast voltage-gated Na+ channels, we did observe a number of changes in active conductances that could also contribute to increased excitability. Thus, the increase in the fast afterhyperpolarization (AHP) that follows a single action potential (Figure 1M) may facilitate repetitive high-frequency firing by increasing the rate of sodium channel recovery from inactivation (Jaffe & Brenner, 2018). We also found an increase in the amplitude of the hyperpolarization-activated voltage sag (Figure 1I, J), suggesting an enhancement of currents mediated by hyperpolarization-activated cyclic nucleotide-gated (HCN) channels in CA2 PCs following PILO-SE (Srinivas et al., 2017). Such an enhancement can promote a rapid recovery or even overshoot of membrane voltage following an inhibitory synaptic potential. The increased sag stands in sharp contrast to numerous reports of HCN downregulation in CA1 PCs from epileptic rodents (Arnold, Mcmurray, Gray, & Johnston, 2019; Wolfart & Laker, 2015), although upregulation of HCN currents has been observed in DG granule cells in surgically resected human TLE tissue (Stegen et al., 2012). This wide array of alterations in CA2 intrinsic properties in epileptic mice (Figure 1, Supplemental Table 1) are likely to contribute to an increased firing of CA2 PCs in vivo.
In addition to finding an increased excitability in the PILO-SE mice, we also found an increased excitability of CA2 PCs from PILO-treated mice that did not experience SE. This result is intriguing as these mice only experienced minor (often nonconvulsive) acute seizures following PILO administration and showed no evidence of hippocampal neurodegeneration. This suggests that the cellular changes leading to increased action potential firing in CA2 PCs from epileptic mice may not be a response to hippocampal degeneration but may reflect a maladaptive response to the abnormal levels of activity induced by PILO treatment.
Mechanisms responsible for the alterations in inhibitory synaptic transmission in TLE
What might account for the dramatic loss of feedforward inhibition observed in CA2 during activation of the CA3 Schaffer collateral inputs? One simple explanation is that the degeneration of CA3 PCs reduces excitation of local inhibitory neurons in response to Schaffer collateral stimulation. However, we did not see a decrease in the peak amplitude of the Schaffer collateral-evoked EPSC in CA2 PCs. One clue as to mechanism comes from the increased rate of decay of the fast IPSC observed in both CA2 and CA1 PCs, which is compatible with either a change in expression of postsynaptic GABAA receptor subunit subtype and/or a preferential loss of inhibitory input from a class of interneurons that produces synaptic responses with slower kinetics. Given the marked heterogeneity in hippocampal inhibitory neurons, a future goal is to delineate how the IPSCs mediated by specific interneuron classes are modified in PILO-SE.
Increased synaptic excitation of and synaptic output from CA2 PCs in epileptic mice
We observed two prominent changes in excitatory synaptic transmission to and output from CA2 PCs. Thus, there was an increase in the amplitude of the mossy fiber-evoked EPSC in CA2 PCs and an increase in the EPSC in CA1 PCs in response to activation of their CA2 inputs. The increase in mossy fiber synaptic excitation is consistent with well-established reports of mossy fiber sprouting in TLE (Schmeiser, Zentner, Prinz, Brandt, & Freiman, 2017), including increased anatomical inputs to CA2 in both human TLE (Freiman et al., 2021) and a mouse model of MTS-like neurodegeneration (Häussler et al., 2016). However, there are also functional changes in the properties of mossy fiber synapses onto CA2 neurons, reflected by the substantial increase in short-term depression. This could result from elevated presynaptic release probability (Figure S6), as reported previously at other sprouted mossy fiber synapses (Hendricks et al., 2017, 2019).
The mechanism contributing to the increased excitation of CA1 PCs by their CA2 inputs is also unclear. Again, there may be an increased number of CA2 synapses, perhaps due to CA2 sprouting as a result of the loss of CA3 inputs to CA1. However, we also found functional changes at the CA2 to CA1 synapses after PILO-SE, reflected by an increased short-term synaptic potentiation compared to controls (Figure 6C - E).
Conclusion
In summary, our data provide compelling support for the hypothesis that CA2 circuits become pathologically hyperexcitable following PILO-SE as a result of changes in both intrinsic and extrinsic excitability, summarized in Figure S8. Together, our data suggest that in epileptic mice intrinsically hyperexcitable CA2 PCs are poised to convey excitatory activity along the alternate trisynaptic circuit from DG → CA2 → CA1. Importantly, as CA2 PC axons project along both the transverse and longitudinal axes of the hippocampus (Figure S4; see Meira et al., 2018; Okuyama, Kitamura, Roy, Itohara, & Tonegawa, 2016; Tamamaki, Abe, & Nojyo, 1988) and target numerous extrahippocampal structures (Cui, Gerfen, & Young, 2013), CA2 hyperexcitability may potentially facilitate the emergence of seizures through both hippocampal and extrahippocampal projections. However, it is important to note that a direct causal link between the alteration in CA2 intrinsic and synaptic properties and the generation of spontaneous seizure activity remains to be demonstrated. Nonetheless, our findings suggest that the surviving CA2 PC population may function as a resilient network node within the cortico-hippocampal loop that supports seizure generation and propagation, making CA2 an important novel target for the treatment of drug-resistant seizures in TLE.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Helen Scharfman (Helen.Scharfman@nki.rfmh.org).
Materials availability
This study did not generate new unique reagents.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
All original code has been deposited at Zenodo (see DOI in the Key Resources Table) and is publicly available as of the date of publication.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Cresyl Violet Acetate | Sigma-Aldrich | #C5042 |
| Rabbit anti-PCP4 | Sigma-Aldrich | #HPA005792; RRID:AB_1855086 |
| Mouse anti-RGS14 (IgG2a) | Neuromab | #75-170; RRID:AB_2179931 |
| Mouse anti-STEP (IgG1) | Cell Signaling Technology | #4396; RRID:AB_1904101 |
| Chicken anti-GFP | Aves | #GFP-1020; RRID:AB_10000240 |
| Rabbit anti-CALB1 | Abcam | #ab11426; RRID:AB_298031 |
| NeuroTrace 435/455 fluorescent dye | Invitrogen | #N21479 |
| Streptavidin-Alexa Fluor 647 | Invitrogen | #S21374; RRID:AB_2336066 |
| Bacterial and virus strains | ||
| rAAV5-EF1a-DIO-hChR2(E123T/T159C)-eYFP | UNC Vector Core | Lot AV4828b |
| AAV2/5 hSyn.DIO.hM4D(Gi)-mCherry | Krashes et al J Clin Invest. 2011 Apr;121(4):1424–8. doi: 10.1172/JCI46229 | Addgene: 44362-AAV5 |
| Chemicals, peptides, and recombinant proteins | ||
| Scopolamine methyl nitrate | Sigma-Aldrich | #S2250 |
| Terbutaline hemisulfate | Sigma-Aldrich | #T2528 |
| Ethosuximide | Sigma-Aldrich | #E7138 |
| Pilocarpine hydrochloride | Sigma-Aldrich | #P6503 |
| Diazepam | Hospira | NDC: 0409-3213-12 |
| Methylatropine bromide | Millipore Sigma | #M1300000 |
| Levetiracetam | West-Ward | NDC: 0143-9673-10 |
| Lactated Ringer’s solution | Aspen Veterinary Resources | NDC: 0409-7953-03 |
| Clozapine N-oxide | Sigma-Aldrich | #C0832 |
| Isoflurane | Covetrus | NDC: 11695-6777-2 |
| Betadine | Purdue Products | NDC: 67618-155-01 |
| Buprenorphine | Reckitt Benckiser | NDC: 12496-0757-1 |
| Carprofen | Zoetis Inc. | NADA #141-199 |
| Lubricant Ophthalmic Ointment | Akorn | NDC: 59399-162-35 |
| MediGel CPF | ClearH2O | CPF-74-05-5022 |
| Rodent MDs Rimadyl Tablets | Bio-Serv | #MD150-2 |
| Sucrose | Sigma-Aldrich | #S9378-1KG |
| D-Glucose anhydrous | Fisher Chemical | #D16-1 |
| Sodium chloride | VWR Chemicals | #BDH9286-2.5KG |
| Sodium bicarbonate | Fisher Chemical | #S233-500 |
| Potassium chloride | Sigma-Aldrich | #P3911-500G |
| Sodium phosphate monobasic monohydrate | Fisher Chemical | #S369-500 |
| Sodium pyruvate | Fisher Chemical | #BP356-100 |
| Ascorbic acid | Alfa Aesar | #36237 |
| Calcium chloride | G-Biosciences | #R040 |
| Magnesium chloride | Quality Biological | Cat# 351-033-721 |
| Agar | Difco | Ref# 214530 |
| Potassium gluconate | USP Reference Standard | Cat# 1550001 |
| Ethylene glycol-bis(2-aminoethylether)-N,N,N’,N’-tetraacetic acid | Sigma-Aldrich | #03777-10G |
| HEPES | Sigma-Aldrich | #H3375-25G |
| Adenosine 5’-triphosphate magnesium salt | Sigma-Aldrich | #A9187-500MG |
| Guanosine 5’-triphosphate sodium salt | Sigma-Aldrich | #G8877-250MG |
| Phosphacreatine disodium salt hydrate | Sigma-Aldrich | #P7936-1G |
| Biocytin | Sigma-Aldrich | #B4261-250MG |
| Cesium methanesulfonate | Sigma-Aldrich | #C1426-5G |
| Paraformaldehyde 16% aqueous solution | Electron Microscopy Sciences | Cat# 15710 |
| Triton X-100 | Sigma-Aldrich | #T-9284 |
| Critical Commercial Assays | ||
| New York University Mouse Genotyping Core facility | New York University | https://med.nyu.edu/research/scientific-cores-shared-resources/genotyping-core-laboratory |
| GeneTyper Mouse Genotyping Service | GeneTyper | https://www.genetyper.com/ |
| Experimental models: Organisms/strains | ||
| Amigo2-Cre mice | The Jackson Laboratory | Cat# 030215; RRID:IMSR_JAX:030215 |
| POMC-Cre mice | The Jackson Laboratory | Cat# 010714; RRID:IMSR_JAX:010714 |
| 129S1/SvlmJ mice | The Jackson Laboratory | Cat# 002448; RRID:IMSR_JAX:002448 |
| C57BL/6NTac mice | Taconic | # B6-F / B6-M; RRID:IMSR_TAC:b6 |
| C57BL/6J mice | The Jackson Laboratory | Cat# 000664; RRID:IMSR_JAX:000664 |
| B6;129S-Gt(ROSA)26Sortm32(CAG-COP4*H134R/EYFP)Hze/J mice | The Jackson Laboratory | Cat# 012569; RRID:IMSR_JAX:012569 |
| Software and algorithms | ||
| Sirenia Acquisition | Pinnacle Technology | https://www.pinnaclet.com/sirenia-download.html |
| Sirenia Seizure Pro 1.7.9 | Pinnacle Technology | https://www.pinnaclet.com/seizurePRO.html |
| MATLAB R2019a | MathWorks | https://www.mathworks.com/products/matlab.html |
| Prism 8.4.3 | GraphPad Software | https://www.graphpad.com/ |
| AxoGraph 1.6.4 | AxoGraph | https://axograph.com |
| ZEN Black 2.3 | Zeiss | https://www.micro-shop.zeiss.com/en/us/softwarefinder/software-categories/zen-black/ |
| FIJI / ImageJ 2.0.0 | ImageJ | https://imagej.net/software/fiji/ |
| Original MATLAB code | N/A | https://doi.org/10.5281/zenodo.6835900 |
| Other | ||
| Water Jacked Warmer Intensive Care Unit | ThermoCare | #FW-1 |
| Rodent stereotaxic apparatus | World Precision Instruments | Model #502063 |
| Surgical drill | Grobert | Model C300 |
| 33-gauge infusion needle | Plastics One | #C315I-SPC |
| 0.10” Stainless steel jeweler’s screws | Pinnacle Technology | #8209 |
| Infrared LED camera | Apex CCTV | #AP-DCS100W |
| Tethered four-channel EEG/EMG system | Pinnacle Technology | #8400-K1 |
| Nanoject II | Drummond | #3-000-204 |
| Constant voltage isolated stimulator | Digitimer Ltd. | Model DS2A-Mk.II |
| High-power 1-channel LED driver with pulse modulation | ThorLabs | Model DC2100 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
All procedures were performed in accordance with the Nathan Kline Research Institute for Psychiatric Research (NKI) and Columbia University Institutional Animal Care and Use Committees (IACUC). Adult male and female mice (8–12 weeks-old) were housed in a temperature and humidity-controlled environment with a 12-hour light/dark cycle with food and water provided ad libitum. For in vivo studies, Amigo2-Cre+/− mice from the Siegelbaum laboratory (C57BL/6J background) were initially bred at NKI with wild-type (WT) C57BL/6N mice (Taconic) and backcrossed with the C57BL/6N line for several generations before use. For ex vivo studies at Columbia University, the Amigo2-Cre+/− mice from the Siegelbaum laboratory were F1 generation hybrids resulting from a cross between Amigo2-Cre mice with a C57BL/6J genetic background and 129S1/SvlmJ mice (stock # 002448; Jackson Laboratory). The distribution of Cre activity and Cre-dependent protein expression has been characterized in detail in our previous publications (Hitti & Siegelbaum, 2014; Leroy et al., 2018; Meira et al., 2018; Oliva et al., 2020), which indicate that there is near complete overlap in expression of Cre and endogenous CA2 markers PCP4, RGS14, and STEP in the dorsal hippocampus. For both past publications and the present study, we estimate that in our viral injections we typically target at least 50% of CA2 PCs in the dorsal half of the hippocampus. In juvenile animals, the Amigo2 gene is widely expressed and as such a transgenic approach would not yield selective expression in CA2 neurons. Therefore, we performed stereotactic injection of AAV targeted to the CA2 region in adult animals (at least 8 weeks old), by which point Cre is expressed less broadly. In contrast, in experiments examining dentate gyrus granule cell mossy fiber input to CA2 PCs we utilized the POMC-Cre mouse line (stock # 010714; Jackson Laboratory) in a transgenic strategy. The POMC gene is only expressed in immature neurons, and therefore cannot be used to drive Cre-dependent expression via AAVs in adult mice. Rather, POMC-Cre mice with a C57BL/6J genetic background were initially crossed to Ai32 mice (stock # 012569; Jackson Laboratory) for Cre-driven expression of ChR2-eYFP, and then the progeny were subsequently crossed to 129S1/SvlmJ mice to generate F1 generation hybrids carrying both the POMC-Cre and the ChR2-eYFP transgenes. Thus, the distinct strategies used in the Amigo2-Cre and POMC-Cre experiments reflect the spatiotemporal expression patterns of the Amigo2 and POMC genes. Genotyping was performed using tail tip samples sent to the New York University Mouse Genotyping Core facility (NKI) or GeneTyper (Columbia University). In a small cohort at the outset of the study it was confirmed that genotypes of mice were the same using the two genotyping facilities.
METHOD DETAILS
I. Induction of epilepsy
Epileptic mice were generated after pilocarpine-induced SE (PILO-SE) for in vivo and ex vivo studies. In vivo studies used hM4Di (inhibitory DREADDs; iDREADDs) to selectively silence CA2 PCs during chronic seizures. Ex vivo studies used hippocampal slices and associated techniques to clarify potential mechanisms underlying the role of CA2 in epileptic conditions. Initial studies using identical mice and methods at NKI and Columbia University led to greater mortality at Columbia University, so Columbia University modified methods to reduce mortality. This led to slightly different methods but each location generated robust epileptic mice. One change that was made was Columbia University used a slightly different background strain to reduce mortality, a F1 generation hybrid resulting from a cross of the C57BL/6J strain (high mortality) and 129S1/SvlmJ mice (lower mortality). However, these mice did require a higher dose of pilocarpine (see below for dose). A higher dose was also likely to be a result of a higher body weight in the mixed background strain.
A. PILO-SE induction for in vivo experiments
Induction of SE with the convulsant pilocarpine was done in cohorts of 2–4 mice with the experimenter blinded to the experimental group. Baseline vEEG recordings were acquired for at least 1 hr to capture a wide range of EEG signals associated with various behavioral states (e.g., exploration, grooming, and rest). Following the baseline period, mice were injected with the peripheral muscarinic antagonist scopolamine methyl nitrate (1 mg/kg s.c.; #S2250, Sigma Aldrich) to reduce the peripheral effects of pilocarpine. The β2-adrenergic agonist terbutaline hemisulfate (1 mg/kg s.c.; #T2528, Sigma Aldrich) was also administered to support respiration. Ethosuximide (150 mg/kg s.c.; #E7138, Sigma Aldrich) was administered to reduce the occurrence of brainstem seizures which can lead to mortality (Iyengar et al., 2015). PILO-SE was induced by injecting pilocarpine hydrochloride (250 mg/kg s.c., #P6503, Sigma Aldrich). All mice were injected with diazepam (5 mg/kg, s.c.; NDC: 0409–3213–12, Hospira) 2 hrs after the pilocarpine injection to reduce the severity of SE, which appears to prevent morbidity and mortality after SE (Goodkin & Kapur, 2009; Iyengar et al., 2015). Mice were injected with 1 mL (s.c.) of lactated Ringer’s solution (Aspen Veterinary Resources) at this time to support hydration. Our previous studies suggest that SE is most intense for several hours after the pilocarpine injection, but there is continued spiking in the EEG overnight (Iyengar et al., 2015; Jain et al., 2019). For all in vivo experiments, stock solutions were freshly prepared in 0.9% NaCl in dH2O (saline; i.e., CNO, scopolamine, terbutaline) or phosphate buffered saline (i.e., ethosuximide).
B. PILO-SE induction for ex vivo experiments
All drugs were administered intraperitoneally (i.p.). Mice were first administered methylatropine bromide (5 mg/kg, i.p.; Millipore Sigma #M1300000) to suppress peripheral cholinergic activation from pilocarpine hydrochloride. Pilocarpine was administered 30 mins later (350 mg/kg, i.p.; Sigma Aldrich #P6503) and mice were closely and continually monitored for behavioral indicators of seizures. The onset of SE typically occurred between 30 and 60 mins following pilocarpine treatment and was defined as a convulsive seizure (stage 3, 4, or 5 on the Racine seizure scale (Racine, 1972)) that lasted continually for at least 5 mins and did not fully subside for several hours. Diazepam (5 mg/kg, i.p.; Hospira NDC: 0409–3213–12) was administered 1 hr after SE onset to curtail seizures. If mice did not exhibit SE following pilocarpine injection (non-SE) they were administered diazepam at a similar delay after pilocarpine as SE mice, 2 hrs after pilocarpine. In all cases diazepam was followed 20 mins later by levetiracetam (100 mg/kg, i.p.; West-Ward NDC: 0143–9673–10). Control mice were given an identical course of drug treatment to non-SE mice, except they were not administered pilocarpine. Thus, control animals received methylatropine bromide, then after 150 mins were administered diazepam and at 170 mins were administered levetiracetam. Immediately after levetiracetam SE and non-SE mice were transferred to a heated and humidified veterinary intensive care unit (ThermoCare #FW-1), where they were closely monitored, provided with dietary supplements, and given subcutaneous hydration (lactated Ringer’s solution; ICU Medical NDC: 0409–7953–03) until they showed normal locomotion and feeding (typically within 1–3 days). After recovery all mice were kept in standard group housing, except where aggression between cage-mates was observed in which cases aggressors were removed. In the cohort of mice used for ex vivo experiments, pilocarpine treatment resulted in three outcomes: acute mortality due to generalized tonic-clonic seizure leading to tonic hindlimb extension and death in 102/367 mice (27.8%), minor isolated seizures but without SE in 110/367 mice (30%), or SE in 155/367 mice (42.2%).
II. In vivo experiments with epileptic mice
A. Stereotaxic injection of AAV
Expression of the inhibitory designer receptor exclusively activated by designer drug (iDREADD) was achieved by stereotaxic injection of AAV2/5 hSyn.DIO.hM4D(Gi)-mCherry (Addgene 44362-AAV5) in the dorsal hippocampus. Mice were initially anesthetized with 5% isoflurane (Aerrane, Henry Schein). The mice were then immediately secured in a rodent stereotaxic apparatus (Model #502063, World Precision Instruments). A homeothermic blanket system maintained body temperature at 37 °C (Harvard Apparatus). Isoflurane (1–2%) was mixed with oxygen and delivered through a nose cone attached to the stereotaxic apparatus. Buprenex (Buprenorphine, 0.1 mg/kg, s.c.) was delivered prior to any surgical manipulations to reduce discomfort. The scalp of each mouse was then shaved and swabbed with Betadine (Purdue Products). Lubricating gel was applied to the eyes to prevent dehydration (Patterson Veterinary).
After a midline scalp incision, a surgical drill (Model C300, Grobert) was used to make two craniotomies for viral injections. All stereotaxic coordinates for craniotomies are described in anterior-posterior (AP) and medial-lateral (ML) coordinates (in reference to Bregma). Craniotomies were made over dorsal CA2 bilaterally (−1.9 mm AP, ± 1.4 mm ML). This injection site was chosen to maximize specific expression in CA2.
A 33-gauge infusion needle (#C315I-SPC, Plastics One) attached to a 0.5 μl Hamilton syringe was lowered from the skull surface 1.5 mm into each relatively anterior site in the hippocampus to target dorsal CA2. Each site was injected with 150 nL of virus at a rate of 40 nL/min. The needle remained in place for an additional 5 min after each injection to allow for diffusion of the virus and then it was slowly removed.
Next, subdural screw electrodes (0.10” length stainless steel jeweler’s screws #8209, Pinnacle Technology) were secured in the two anterior craniotomies positioned over the left and right dorsal hippocampus. A subdural screw electrode was also secured in the right occipital cortex (−3.5 mm AP, +2.0 ML). In addition, a subdural screw electrode was secured in a craniotomy made over the left frontal cortex (0.0 mm AP, −2.6 mm ML). Last, screw electrodes were secured in craniotomies made over the right olfactory bulb (+2.3 mm AP, +1.8 mm ML) which served as a ground and the cerebellum (−5.7 mm AP, −0.5 mm ML) which served as a reference electrode. The subdural screw electrodes were attached to an 8-pin connector that was centered over the skull and secured with dental cement. Mice were transferred to a clean cage at the end of surgery and placed on a heating blanket (37 °C) until fully ambulatory.
B. Video-EEG and CNO treatment
Mice were housed individually in a standard laboratory cage within a room where the video-EEG (vEEG) equipment was housed so they would acclimate to the recording environment. After the recovery period, each mouse was individually placed into a 21 cm × 19 cm transparent cage comparable to a standard laboratory cage. A pre-amplifier was inserted into the 8-pin headcap and connected to a multichannel commutator (Pinnacle Technology). This tethered EEG system allowed for free range of movement throughout the entire recording cage. EEG signals were acquired at 500 Hz and bandpass filtered at 1–100 Hz in Sirenia Acquisition software (Pinnacle Technology). Simultaneous video recordings synchronized with the EEG record were captured using an infrared LED camera (#AP-DCS100W, Apex CCTV) for offline analyses.
CNO (Sigma-Aldrich) was added to the drinking water at a 0.4 mg/ml concentration. For a 20 mg mouse the water delivered approximately 10 mg/kg per day, given an average volume of water consumption of 6 mL per day. The CNO stock solution (1 mg/ml dissolved in water) was made every 3 days and refrigerated between uses. We selected a 10 mg/kg/day dose of CNO because this dose has been widely used in the literature to inhibit diverse cell types throughout the brain with minimal to no off-target effects (Mahler & Aston-Jones, 2018; Smith, Benison, Bercum, Dudek, & Barth, 2018). Importantly, we controlled for potential off-target effects of CNO by also injecting Cre−/− mice with the same dose of CNO as iDREADD-expressing mice (MacLaren et al., 2016). In prior studies we found no evidence of non-specific effects of the 10 mg/kg dose of CNO (Botterill et al., 2019, 2021).
All vEEG recordings were analyzed offline using Sirenia Seizure Pro (v. 1.7.9, Pinnacle Technology). Seizures were defined as rapid and rhythmic (>3 Hz) deflections in all EEG channels that lasted >5 sec (Botterill et al., 2019; Iyengar et al., 2015; Jain et al., 2019) and were at least 3 standard deviations above the baseline root mean square (RMS) amplitude (Iyengar et al., 2015). Seizures were considered convulsive if the video record showed behaviors consistent with stages 3–5 on the Racine scale (stage 3, unilateral forelimb clonus; stage 4, bilateral forelimb clonus with rearing; stage 5, stage 4 followed by loss of posture; (Racine, 1972). Seizures were considered non-convulsive if the EEG criteria were met, but no stage 3–5 behaviors were detected in the video record. Nonconvulsive seizures were occasionally accompanied by small movements but often occurred during sleep or immobility. SE was defined as the first seizure that was severe (large amplitude EEG deflections appearing in all 4 electrodes simultaneously, stage 3–5) with the seizure activity in the EEG persisting continuously for >5 min, and seizure-associated behavior persisting for >5 min, a standard definition (Goodkin & Kapur, 2009; Iyengar et al., 2015). Five min was chosen because if the abnormal activity persistent for 5 min it lasted for several hours. Note that during SE the seizure-associated behavior included up to 10 stage 3–5 convulsions. Between convulsions behavior included twitching and abnormal body movement. Cessation of convulsions was indicated by a return to grooming and feeding although animals showed persistent EEG spiking, seizure activity and convulsions intermittently for the next 24 hours (Botterill et al. 2019 cell rep).
C. Immunohistochemistry
Following deep anesthesia by isoflurane inhalation and a terminal dose of urethane (2.5 g/kg, i.p.), the abdominal cavity was opened and a 26 g butterfly needle inserted into the ventricle. After cutting the left atria, 0.9% NaCl (30 mL) was perfused into the heart with a peristaltic pump (Gilson) on a high setting (1 mL/min). Fixative (4% paraformaldehyde in 0.1 M Tris buffer, pH 7.6; 30 mL) was perfused immediately thereafter. After storing the body at 4° C for over 30 min, the brain was removed and postfixed in paraformaldehyde for at least 24 hrs. Sections (50-μm thick) were cut with a vibratome (Leica 1200; Leica Biosystems) in the coronal plane and alternate sections were stained using Cresyl violet (Scharfman, 2002) or with immunohistochemical procedures using markers of CA2 (PCP4, RSG14; described further below). Sections were coverslipped with Permount (Fisher) and viewed with an upright microscope (BX61; Olympus of America) using a digital camera (Infinity3–6URC; Lumenera) and associated software (Infinity; Lumenara).
III. Ex vivo experiments with epileptic mice
A. Stereotaxic viral injections
Surgeries were performed at least three weeks after pilocarpine treatment. To drive Cre-dependent expression of channelrhodopsin2 (ChR2) in CA2 PCs for optogenetic experiments in acute hippocampal slices, we performed stereotactic injection (Nanoject II, Drummond #3–000-204) of Cre-dependent adenoassociated virus (rAAV5-EF1a-DIO-hChR2(E123T/T159C)-eYFP; UNC, lot AV4828b) targeted to CA1c, adjacent to the dorsal CA2 subfield in control and pilocarpine-treated Amigo2-Cre+/− mice (male and female F1 generation progeny from a C57BL/6J × 129S1/SvlmJ cross). The stereotactic coordinates used were measured in millimeters relative to bregma (for anterior-posterior and medial-lateral coordinates, A-P and M-L) and relative to the surface of the brain (for dorsal-ventral coordinates, D-V) and were as follows: A-P −2.0, M-L ± 1.80, D-V −1.20.
For surgery, carprofen was given as an analgesic agent (5 mg/kg, subcutaneous, s.c.), and postoperative analgesia was carprofen-supplemented food (either 60 mL of MediGel CPF-74–05–5022 food gel, or Rodent MDs #MD150–2 2 mg carprofen tablets, each estimated to be a dose of approximately 5 mg/kg over 24 hours of consumption). Before the commencement of surgery animals were weighed to record baseline body weight and to calculate carprofen dose. Anesthesia was achieved with isoflurane (Covetrus, NDC: 11695–6777–2), with an induction concentration of 4% and a maintenance concentration of approximately 2%. Surgical procedures were performed under sterile conditions as approved by the Columbia University IACUC. Following surgery mice were group housed and allowed to recover for a minimum of two weeks to allow for viral expression before preparation of acute hippocampal slices or processing of brains for immunohistochemistry.
B. Ex vivo slice electrophysiology and data analysis
1. Hippocampal slice preparation
Ex vivo electrophysiology was performed in the chronic phase of PILO-induced epilepsy at least 6 weeks after status epilepticus, when mice were 14 – 18 weeks old. Acute hippocampal slices were prepared from both Amigo2-Cre mice and their wild-type littermates. The Amigo2-Cre line was used to drive channelrhodopsin2-eYFP expression in CA2 PCs for the purpose of measuring synaptic input from CA2 to PCs located in CA1, CA2, and CA3 (Figure 3, Figure 5, Figure 6, Figure S7). Importantly, the intrinsic physiological properties of CA2 PCs from Amigo2-Cre+/− mice or wild-type (Amigo2-Cre−/−) mice are equivalent: we found no significant difference in resting membrane potential (Mann-Whitney; P = 0.7440; n = 51 cells from 27 Amigo2-Cre+/− mice, 51 cells from 24 Amigo2-Cre−/− mice) or action potential firing rates (two-way ANOVA; P = 0.4255; n = 43 cells from 24 Amigo2-Cre+/− mice, 37 cells from 19 Amigo2-Cre−/− mice).
Acute hippocampal slices were prepared using sucrose-substituted artificial cerebrospinal fluid (referred to as sucrose solution) and standard ACSF. ACSF and sucrose solutions were made with purified water that had been filtered through a 0.22 μm filter. ACSF contained (concentration expressed as millimolar, mM): 22.5 glucose, 125 NaCl, 25 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 3 sodium pyruvate, 1 ascorbic acid, 2 CaCl2, and 1 MgCl2. Sucrose solution used for slice preparation contained, in mM: 195 sucrose, 10 glucose, 25 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 2 sodium pyruvate, 0.5 CaCl2, 7 MgCl2. ACSF and sucrose cutting solution were prepared fresh before each experiment and the osmolarity was consistently measured to be between 315 and 325 mOsm. Sucrose solution was chilled on ice and bubbled with carbogen gas (95% O2 / 5% CO2) for at least 30 mins before slice preparation. A recovery beaker was prepared with a 50:50 mixture of ACSF and sucrose solution and warmed to approximately 32° C. Mice were deeply anesthetized by isoflurane inhalation and immediate incisions were made to sever the diaphragm and access the heart. Transcardial perfusion was performed with ice-cold carbogenated sucrose cutting solution for approximately 30 – 45 s. The mouse was decapitated and the brain quickly removed, at which point the hippocampi were dissected free, placed into an agar block, and secured to a vibratome slicing platform with cyanoacrylate adhesive. Hippocampal slices were cut at a thickness of 400 μm, parallel to the transverse plane. Slices were collected from the dorsal and intermediate hippocampus, as CA2 circuits have primarily been characterized within the dorsal hippocampus (Chevaleyre & Siegelbaum, 2010; Hitti & Siegelbaum, 2014; Kohara et al., 2014). Slices were transferred to the warmed recovery beaker and allowed to recover for 30 m, after which the beaker was allowed to come to room temperature and left for an additional 90 m.
2. Whole-cell recordings.
Recording and stimulation pipettes were prepared from borosilicate glass capillaries using a heated-filament puller programmed to produce pipettes with a resistance of approximately 3 – 5 mOhm. Stimulation pipettes were filled with 1 M NaCl. Recording pipettes were filled with intracellular solution composed of, in mM: 135 potassium gluconate (C6H11KO7), 5 KCl, 0.2 EGTA (C14H24N2O10), 10 HEPES (C8H18N2O4S), 2 NaCl, 5 MgATP (C10H16N5O13P3 · Mg2+), 0.4 Na2GTP (C10H16N5O14P3 · Na+), 10 Na2 phosphocreatine (C4H8N3O5PNa2 · H2O), and biocytin (0.2% by weight). Intracellular solution was prepared on ice, the osmolarity was adjusted to approximately 295 mOsm, and the pH titrated to approximately 7.2. For voltage clamp recordings, intracellular solution instead contained cesium (Cs+) methanesulfonate (CH3O3SCs). Hippocampal slices were individually transferred to the recording chamber of an electrophysiology station where ACSF, warmed to 32° C and continually bubbled with carbogen, was perfused through the chamber at a rate of approximately 1 – 3 mL/min. Whole-cell recordings from CA2 and CA3 PCs was accomplished primarily through a “blind patching” approach, whereas CA1c PCs were targeted by visual guidance to ensure cell position in the deep PCL. CA2 PCs were targeted using the end of the stratum lucidum (SL) as an anatomical landmark and cell identity was confirmed post-hoc based on cellular morphology as revealed by biocytin and colocalization with known CA2 markers, such as PCP4 (Figure 1A, Figure 2A). As previously reported (Chevaleyre & Siegelbaum, 2010; Hitti & Siegelbaum, 2014; Kohara et al., 2014), several key intrinsic physiological properties distinguished CA2 PCs from their neighbors in CA1 and CA3, including a relatively small input resistance, delayed action potential firing preceded by a gradual slow depolarizing ramp upon depolarizing current injection, a relatively small-amplitude voltage sag upon hyperpolarization, a relatively high rheobase current, and a large membrane capacitance. Moreover, the cellular morphology and intrinsic physiological properties of CA2 pyramidal cells, in particular their action potential firing patterns, are clearly distinguishable from those of fast-spiking interneurons, even in slices from epileptic mice. For example, the elevated firing rate of CA2 PCs from epileptic mice reaches a maximum of approximately 22 Hz with 1000 pA of injected current, whereas a fast-spiking interneuron fires at approximately 80 Hz to the same stimulus. Fast-spiking interneurons also exhibit a significantly larger action potential afterhyperpolarization and a larger input resistance as compared to CA2 PCs, even when compared to PCs from epileptic mice. Cells were left unperturbed for several minutes before any recordings to ensure the membrane potential and seal were stable, and cells with a resting membrane potential more depolarized than −50 mV or a series resistance greater than 25 MΩ were discarded.
3. Intrinsic properties.
The resting membrane potential was measured by recording at least one minute in the current clamp configuration, without any injected current, and averaging the membrane potential. Current was then applied as needed to hold cells at −70 mV. Neuronal identity was assessed according to several physiological characteristics in the current clamp configuration: action potential firing patterns upon current injection, a low input resistance, and the absence of large spontaneous postsynaptic potentials clearly distinguished CA2 PCs from nearby CA3a PCs. Cell identity was confirmed by post-hoc microscopy (see below). Action potential firing patterns and a current-firing rate curve were measured using ten current steps, from +100 pA to +1000 pA (in 100 pA increments). Subthreshold membrane properties, namely the input resistance and membrane capacitance, were assessed using eleven current steps from −100 pA to +100 pA (in 20 pA increments). Voltage sag was measured using a series of negative current steps, and sag measured from whichever current step produced an initial hyperpolarization to between −100 mV and −105 mV. Voltage sag was expressed as a ratio between the peak hyperpolarization at the beginning of the pulse (ΔVmpeak) and the difference between ΔVmpeak and the steady-state hyperpolarization (ΔVmss) at the end of the current step: sag ratio = (ΔVmpeak − ΔVmss) / ΔVmpeak.
Rheobase current, action potential voltage threshold, afterhyperpolarization (AHP), and other action potential characteristics were assessed with a current ramp that reached 1000 pA over one second. The first action potential to result from the current ramp was isolated and a phase-plane plot constructed from the action potential waveform and the derivative of the membrane potential during the spike. Action potential onset was defined as the timepoint where the rate of depolarization exceeded 5 mV/ms. This timepoint was used to measure the rheobase current and the action potential voltage threshold. The phase-plane plot was used to measure the maximum rate of rise and rate of descent during the action potential. A gridded data interpolation was constructed from the raw action potential waveform, and this interpolant was used to measure the action potential half-width and the AHP. The AHP was defined as the difference between the action potential voltage threshold and the negative peak membrane potential occurring just after the action potential.
The voltage-dependent ramp of slow depolarization in CA2 PCs (slow ΔVm) was measured from membrane potential responses produced by the ten +100 pA current steps (the same membrane potential responses described above, used to measure the current-firing rate curve). For every cell, membrane potential was measured near the onset of the current step (initial Vm) and at the end of the current step (final Vm). The difference of the final and initial Vm was measured as the slow depolarization (slow ΔVm) that occurred over the one second duration of the current step. The slow ΔVm values from each cell were binned based on the corresponding initial Vm values into ten bins ranging from −68 mV to −48 mV, in two mV increments. In other words, the first bin collected slow ΔVm values from responses with an initial depolarization between −68 mV and −66 mV, the second bin from −66 mV to −64 mV, and so on. Membrane potential responses with an initial Vm more negative than −68 mV (or more positive than −48 mV) were discarded. In this way the amplitude of the slow depolarization could be directly compared across groups while controlling for the voltage dependence of the response. Analysis of intrinsic properties was performed in MATLAB and AxoGraph.
4. Synaptic responses and optogenetics.
Electrical stimulation was delivered as a single 0.2 ms pulse (Digitimer Ltd. Constant voltage isolated stimulator, model DS2A-Mk.II) and photostimulation was delivered as a 2 ms pulse of 470 nm light produced by an LED light source (ThorLabs High-power 1-channel LED driver with pulse modulation, model DC2100). For input-output curves both electrical stimulation and photostimulation were delivered in ten steps of intensity, ranging from 8 V to 80 V (in 8 V increments) or from 10% to 100% light intensity (in 10% increments). Photostimulation trains consisted of fifteen 2 ms pulses delivered at 30 Hz, followed after 500 ms by an additional single 2 ms pulse. Postsynaptic potentials were measured in current clamp from an initial holding potential of −70 mV (except where specified as −55 mV). In the voltage clamp configuration cells were held at −75 mV (for EPSCs) or +10 mV (for IPSCs). The exponential time constant of decay tau was measured at the time point at which postsynaptic currents decayed from their peak amplitude to 36.8% of their peak (an approximation of 1/e). For each cell, only postsynaptic currents within a set range of amplitudes were pooled and their tau values averaged to yield one time constant per cell, such that comparisons of time constants between the control and PILO-SE groups utilized postsynaptic currents of comparable amplitudes. In recordings examining the DG input to CA2 PCs, optogenetic activation of granule cell mossy fiber axons occasionally resulted in polysynaptic responses (likely involving recruitment of CA3 PCs). Therefore cells were excluded from analysis if light-evoked postsynaptic responses occurred at a polysynaptic latency or involved multiple compounding potentials.
C. Immunohistochemistry
For immunohistochemistry mice were placed under deep isoflurane-induced anesthesia and transcardially perfused, first with 0.9% NaCl and then with 4% PFA. Brains were extracted and immersed whole in 4% PFA overnight at 4° C. The next day, brains were washed in 0.3% glycine in phosphate buffered saline (PBS) for one hour at room temperature on a shaker and were then rinsed three times briefly with PBS. Coronal or horizontal sections were prepared at a thickness of 60 μm using a vibratome. Post-hoc immunohistochemistry was performed by immersing 400-μm thick acute hippocampal slices in 4% paraformaldehyde (PFA) at the conclusion of recordings and fixing them overnight on a shaker at 4° C. Both 60 μm brain sections and 400 μm hippocampal slices were incubated for four hours at room temperature in PBS with 0.5% Triton-X. This solution was then exchanged for PBS containing primary antibodies and 0.1% Triton-X. The following antibodies were used: chicken anti-GFP (Aves #GFP-1020), rabbit anti-PCP4 (Sigma-Aldrich #HPA005792), mouse anti-RGS14 IgG2a (Neuromab #75–170), mouse anti-STEP IgG1 (Cell Signaling Technology #4396), and rabbit anti-CALB1 (Abcam #ab11426). Neuronal somata were visualized with a stain against the Nissl substance using the NeuroTrace 435/455 fluorescent dye (Invitrogen #N21479). Biocytin was visualized using streptavidin conjugated to Alexa Fluor 647 (Invitrogen #S21374). The anatomical definition of the CA2 subfield in this study is informed by prior work from our lab and others (Chevaleyre & Siegelbaum, 2010; Meira et al., 2018)(Hitti & Siegelbaum, 2014; Kohara et al., 2014). In mice, the end of the stratum lucidum falls within the bounds of the CA2 subfield: the distal border of CA2 (adjacent to CA1) is approximately 50 to 100 μm past the end of the stratum lucidum, and the proximal border of CA2 (adjacent to CA3) is typically located approximately 200 – 300 μm before the end of the mossy fiber projection. In other words, the total length of CA2 is approximately 300 – 400 μm in the dorsal hippocampus, and approximately three-quarters of this length overlaps with the stratum lucidum. This delineation of CA2 is clearly seen in the staining patterns of established markers for CA2 pyramidal cells PCP4 (Figures 1 and 2) and RGS14 (Figure 4, Figure S4). All images were acquired with a confocal microscope and processed using FIJI software.
QUANTIFICATION AND DATA ANALYSIS
All statistical tests were performed in GraphPad Prism and results of statistical tests can be found in the Results section and the figure legends. Values are given as the mean ± the standard error of the mean (SEM). In all cases the cutoff for significance was set to a P value of 0.05. For parametric data, t-tests and ANOVA followed by Tukey-Kramer post hoc tests were used. For nonparametric data, Mann-Whitney U tests and Kruskal-Wallis followed by Dunn’s multiple comparisons test were used instead. Normality tests were conducted in Prism to confirm parametric statistics could be performed. Where variances were unequal, all data were log-transformed prior to analysis. In some cases of two-way ANOVA, the Geisser-Greenhouse correction and Holm’s or Sidak’s multiple comparisons test were used. Note that in cases where datasets had missing values, Prism applied a comparable mixed-effects model, which utilizes a compound symmetry covariance matrix fit using Restricted Maximum Likelihood (REML) with the Holm’s multiple comparisons test. Sample sizes were determined based on power analysis of pilot data using Instat2 (GraphPad). Video-EEG acquisition and seizure analysis were performed in a blinded fashion, while ex vivo experiments and analysis were unblinded.
Supplementary Material
HIGHLIGHTS.
CA2 pyramidal cell (PC) intrinsic excitability is increased in an epilepsy model.
Inhibitory synaptic input to CA2 PCs is diminished in slices from epileptic mice.
Excitation of CA2 PCs by DG granule cells, and of CA1 PCs by CA2 PCs, is enhanced.
Chemogenetic inhibition of CA2 PCs reduces the frequency of spontaneous seizures.
Acknowledgements
This work was supported by grants R01NS106983 (PIs, H.E.S. and S.A.S.) and F31NS113466 (A.C.W.) from NIH, the American Epilepsy Society, and the New York State Office of Mental Health.
Footnotes
DECLARATION OF INTERESTS:
The authors declare no competing interests.
INCLUSION AND DIVERSITY
We worked to ensure sex balance in the selection of non-human subjects. One or more of the authors of this paper self-identifies as living with a disability. While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Alexander GM, Brown LY, Farris S, Lustberg D, Pantazis C, Gloss B, ... Dudek SM (2018). CA2 neuronal activity controls hippocampal low gamma and ripple oscillations. ELife, 7, 1–25. 10.7554/eLife.38052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrioli A, & Arellano JI (2007). Quantitative analysis of parvalbumin-immunoreactive cells in the human epileptic hippocampus. Neuroscience, 149, 131–143. 10.1016/j.neuroscience.2007.07.029 [DOI] [PubMed] [Google Scholar]
- Arnold EC, Mcmurray C, Gray R, & Johnston D (2019). Epilepsy-induced reduction in HCN channel expression contributes to an increased excitability in dorsal, but not ventral, hippocampal CA1 neurons. ENeuro, 6(April), 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baud MO, Kleen JK, Mirro EA, Andrechak JC, King-Stephens D, Chang EF, & Rao VR (2018). Multi-day rhythms modulate seizure risk in epilepsy. Nature Communications, 9(1), 1–10. 10.1038/s41467-017-02577-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blümcke I, Thom M, Aronica E, Armstrong DD, Bartolomei F, Bernasconi A, ... Spreafico R (2013). International consensus classification of hippocampal sclerosis in temporal lobe epilepsy: A task force report from the ILAE commission on diagnostic methods. Epilepsia, 54(7), 1315–1329. 10.1111/epi.12220 [DOI] [PubMed] [Google Scholar]
- Boehringer R, Polygalov D, Huang AJY, Piskorowski RA, Chevaleyre V, Mchugh TJ, ... Mchugh TJ (2017). Chronic loss of CA2 transmission leads to hippocampal hyperexcitability. Neuron, 94(3), 642–655.e9. 10.1016/j.neuron.2017.04.014 [DOI] [PubMed] [Google Scholar]
- Botterill JJ, Lu YL, LaFrancois JJ, Bernstein HL, Alcantara-Gonzalez D, Jain S, ... Scharfman HE (2019). An excitatory and epileptogenic effect of dentate gyrus mossy cells in a mouse model of epilepsy. Cell Reports, 29(9), 2875–2889.e6. 10.1016/j.celrep.2019.10.100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botterill JJ, Vinod KY, Gerencer KJ, Teixeira CM, LaFrancois JJ, & Scharfman HE (2021). Bidirectional regulation of cognitive and anxiety-like behaviors by dentate gyrus mossy cells in male and female mice. Journal of Neuroscience, 41(11), 2475–2495. 10.1523/JNEUROSCI.1724-20.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckmaster PS, Abrams E, & Wen X (2017). Seizure frequency correlates with loss of dentate gyrus GABAergic neurons in a mouse model of temporal lobe epilepsy. Journal of Comparative Neurology, 525(February), 2592–2610. 10.1002/cne.24226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevaleyre V, & Siegelbaum S. a. (2010). Strong CA2 pyramidal neuron synapses define a powerful disynaptic cortico-hippocampal loop. Neuron, 66(4), 560–572. 10.1016/j.neuron.2010.04.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Z, Gerfen CR, & Young WS (2013). Hypothalamic and other connections with dorsal CA2 area of the mouse hippocampus. Journal of Comparative Neurology, 521(8), 1844–1866. 10.1002/cne.23263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donegan ML, Stefanini F, Meira T, Gordon JA, Fusi S, & Siegelbaum SA (2020). Coding of social novelty in the hippocampal CA2 region and its disruption and rescue in a 22q11.2 microdeletion mouse model. Nature Neuroscience, 23, 1365–1375. 10.1038/s41593-020-00720-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du F, Eid T, Lothman EW, Kohler C, & Schwarcz R (1995). Preferential neuronal loss in layer III of the medial entorhinal cortex in rat models of temporal lobe epilepsy. Journal of Neuroscience, 15(10), 6301–6313. 10.1523/jneurosci.15-10-06301.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Fu, Whetsell WO, Abou-Khalil B, Blumenkopf B, Lothman EW, & Schwarcz R (1993). Preferential neuronal loss in layer III of the entorhinal cortex in patients with temporal lobe epilepsy. Epilepsy Research, 16(3), 223–233. 10.1016/0920-1211(93)90083-J [DOI] [PubMed] [Google Scholar]
- Dudek SM, Alexander GM, & Farris S (2016). Rediscovering area CA2: unique properties and functions. Nature Reviews Neuroscience, 17(2), 89–102. 10.1038/nrn.2015.22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freiman TM, Häussler U, Zentner J, Doostkam S, Beck J, Scheiwe C, ... Puhahn-schmeiser B (2021). Mossy fiber sprouting into the hippocampal region CA2 in patients with temporal lobe epilepsy. Hippocampus, 31(6), 1–13. 10.1002/hipo.23323 [DOI] [PubMed] [Google Scholar]
- Goodkin HP, & Kapur J (2009). The impact of diazepam’s discovery on the treatment and understanding of status epilepticus. Epilepsia, 50(9), 2011–2018. 10.1111/j.1528-1167.2009.02257.x [DOI] [PubMed] [Google Scholar]
- Häussler U, Rinas K, Kilias A, Egert U, & Haas CA (2016). Mossy fiber sprouting and pyramidal cell dispersion in the hippocampal CA2 region in a mouse model of temporal lobe epilepsy. Hippocampus, 26(5), 577–588. 10.1002/hipo.22543 [DOI] [PubMed] [Google Scholar]
- He H, Boehringer R, Huang AJY, Overton ETN, Polygalov D, Okanoya K, & Mchugh TJ (2021). CA2 inhibition reduces the precision of hippocampal assembly reactivation. Neuron, 109, 1–14. 10.1016/j.neuron.2021.08.034 [DOI] [PubMed] [Google Scholar]
- Hendricks WD, Chen Y, Bensen AL, Westbrook GL, & Schnell E (2017). Short-term depression of sprouted mossy fiber synapses from adult-born granule cells. Journal of Neuroscience, 37(23), 5722–5735. 10.1523/JNEUROSCI.0761-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendricks WD, Westbrook GL, & Schnell E (2019). Early detonation by sprouted mossy fibers enables aberrant dentate network activity. Proceedings of the National Academy of Sciences of the United States of America, 166(22), 10994–10999. 10.1073/pnas.1821227116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitti FL, & Siegelbaum S. a. (2014). The hippocampal CA2 region is essential for social memory. Nature, 508(7494), 88–92. 10.1038/nature13028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyengar SS, LaFrancois JJ, Friedman D, Drew LJ, Denny CA, Burghardt NS, ... Scharfman HE (2015). Suppression of adult neurogenesis increases the acute effects of kainic acid. Experimental Neurology, 264, 135–149. 10.1016/j.expneurol.2014.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe DB, & Brenner R (2018). A computational model for how the fast afterhyperpolarization paradoxically increases gain in regularly firing neurons. Journal of Neurophysiology, 119(4), 1506–1520. 10.1152/jn.00385.2017 [DOI] [PubMed] [Google Scholar]
- Jain S, LaFrancois JJ, Botterill JJ, Alcantara-Gonzalez D, & Scharfman HE (2019). Adult neurogenesis in the mouse dentate gyrus protects the hippocampus from neuronal injury following severe seizures. Hippocampus, 29(8), 1–27. 10.1002/hipo.23062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janz P, Savanthrapadian S, Häussler U, Kilias A, Nestel S, Kretz O, ... Haas CA (2017). Synaptic remodeling of entorhinal input contributes to an aberrant hippocampal network in temporal lobe epilepsy. Cerebral Cortex, 27(3), 2348–2364. 10.1093/cercor/bhw093 [DOI] [PubMed] [Google Scholar]
- Karayiorgou M, Simon TJ, & Gogos JA (2010). 22q11.2 microdeletions: linking DNA structural variation to brain dysfunction and schizophrenia. Nature Reviews Neuroscience, 11(6), 402–416. 10.1038/nrn2841.2q11.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay K, & Frank LM (2019). Three brain states in the hippocampus and cortex. Hippocampus, 29(3), 184–238. 10.1002/hipo.22956 [DOI] [PubMed] [Google Scholar]
- Kohara K, Pignatelli M, Rivest AJ, Jung H-Y, Kitamura T, Suh J, ... Tonegawa S (2014). Cell type-specific genetic and optogenetic tools reveal hippocampal CA2 circuits. Nature Neuroscience, 17(2), 269–279. 10.1038/nn.3614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krook-Magnuson E, Armstrong C, Bui A, Lew S, Oijala M, & Soltesz I (2015). In vivo evaluation of the dentate gate theory in epilepsy. The Journal of Physiology, 10, 2379–2388. 10.1113/JP270056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan Patrick, & Brodie MJ (2000). Early identification of refractory epilepsy. The New England Journal of Medicine, 342(5), 314–319. [DOI] [PubMed] [Google Scholar]
- Kwan P, & Sander JW (2004). The natural history of epilepsy: An epidemiological view. Journal of Neurology, Neurosurgery and Psychiatry, 75(10), 1376–1381. 10.1136/jnnp.2004.045690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Marchionni I, Bezaire M, Varga C, Danielson N, Lovett-Barron M, ... Soltesz I (2014). Parvalbumin-positive basket cells differentiate among hippocampal pyramidal cells. Neuron, 82(5), 1129–1144. 10.1016/j.neuron.2014.03.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehr AB, Kumar A, Tetzlaff C, Hafting T, Fyhn M, & St TM (2021). CA2 beyond social memory : Evidence for a fundamental role in hippocampal information processing. Neuroscience and Biobehavioral Reviews, 126(March), 398–412. 10.1016/j.neubiorev.2021.03.020 [DOI] [PubMed] [Google Scholar]
- Leroy F, Park J, Asok A, Brann DH, Meira T, Boyle LM, ... Siegelbaum SA (2018). A circuit from hippocampal CA2 to lateral septum disinhibits social aggression. Nature, 564, 213–218. 10.1038/s41586-018-0772-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLaren DAA, Browne RW, Shaw JK, Radhakrishnan SK, Khare P, España RA, & Clark SD (2016). Clozapine N-oxide administration produces behavioral effects in long-evans rats: Implications for designing DREADD experiments. ENeuro, 3(5). 10.1523/ENEURO.0219-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Mao T, Koch H, Zhuo JM, Berenyi A, Fujisawa S, ... Zeng H (2012). A toolbox of Cre-dependent optogenetic transgenic mice for light-induced activation and silencing. Nature Neuroscience, 15(5), 793–802. 10.1038/nn.3078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahler SV, & Aston-Jones G (2018). CNO evil? Considerations for the use of DREADDs in behavioral neuroscience. Neuropsychopharmacology, 43(5), 934–936. 10.1038/npp.2017.299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzuferi M, Kumar G, Rospo C, & Kaminski RM (2012). Rapid epileptogenesis in the mouse pilocarpine model : Video-EEG , pharmacokinetic and histopathological characterization. Experimental Neurology, 238(2), 156–167. 10.1016/j.expneurol.2012.08.022 [DOI] [PubMed] [Google Scholar]
- McHugh TJ, Jones MW, Quinn JJ, Balthasar N, Coppari R, Elmquist JK, ... Tonegawa S (2007). Dentate gyrus NMDA receptors mediate rapid pattern separation in the hippocampal network. Science, 317(July), 94–99. 10.4324/9781315657455-44 [DOI] [PubMed] [Google Scholar]
- Meira T, Leroy F, Buss EW, Oliva A, Park J, & Siegelbaum SA (2018). A hippocampal circuit linking dorsal CA2 to ventral CA1 critical for social memory dynamics. Nature Communications, 9(4163), 1–14. 10.1038/s41467-018-06501-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasrallah K, Piskorowski R. a., & Chevaleyre V (2015). Inhibitory plasticity permits the recruitment of CA2 pyramidal neurons by CA3. ENeuro, 2(August), 1–12. 10.1523/ENEURO.0049-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasrallah Kaoutsar, Therreau L, Robert V, Huang AJY, Mchugh TJ, Piskorowski RA, ... Mchugh TJ (2019). Routing hippocampal information flow through parvalbumin interneuron plasticity in area CA2. Cell Reports, 27(1), 86–98.e3. 10.1016/j.celrep.2019.03.014 [DOI] [PubMed] [Google Scholar]
- Ogren JA, Bragin A, Wilson CL, Hoftman GD, Lin JJ, Dutton RA, ... Staba RJ (2009). Three-dimensional hippocampal atrophy maps distinguish two common temporal lobe seizure-onset patterns. Epilepsia, 50(6), 1361–1370. 10.1111/j.1528-1167.2008.01881.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K, & Ikegaya Y (2018). Recurrent connections between CA2 pyramidal cells. Hippocampus, (May), 321513. 10.1101/321513 [DOI] [PubMed] [Google Scholar]
- Okuyama T, Kitamura T, Roy DS, Itohara S, & Tonegawa S (2016). Ventral CA1 neurons store social memory. Science, 353(6307), 1536–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliva A, Fernández-Ruiz A, Buzsáki G, & Berényi A (2016). Role of hippocampal CA2 region in triggering sharp-wave ripples. Neuron, 91, 1–14. 10.1016/j.neuron.2016.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliva A, Fernández-Ruiz A, Leroy F, & Siegelbaum SA (2020). Hippocampal CA2 sharp-wave ripples reactivate and promote social memory. Nature, (November 2019). 10.1101/2020.03.03.975599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piskorowski RA, Nasrallah K, Diamantopoulou A, Mukai J, Hassan SI, Siegelbaum SA, ... Chevaleyre V (2016). Age-dependent specific changes in area CA2 of the hippocampus and social memory deficit in a mouse model of the 22q11.2 deletion syndrome. Neuron, 89(1), 163–176. 10.1016/j.neuron.2015.11.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racine RJ (1972). Modification of seizure activity by electrical stimulation: II. Motor seizure. Electroencephalography and Clinical Neurophysiology, 32, 281–294. [DOI] [PubMed] [Google Scholar]
- Sastri BVS, Arivazhagan A, Sinha S, Mahadevan A, Bharath RD, Saini J, ... Satishchandra P (2014). Clinico-pathological factors influencing surgical outcome in drug resistant epilepsy secondary to mesial temporal sclerosis. Journal of the Neurological Sciences, 340(1–2), 183–190. 10.1016/j.jns.2014.03.026 [DOI] [PubMed] [Google Scholar]
- Scharfman HE (2019). The dentate gyrus and temporal lobe epilepsy: An “exciting” era. Epilepsy Currents, 19(4), 1–7. 10.1177/1535759719855952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmeiser B, Zentner J, Prinz M, Brandt A, & Freiman TM (2017). Extent of mossy fiber sprouting in patients with mesiotemporal lobe epilepsy correlates with neuronal cell loss and granule cell dispersion. Epilepsy Research, 129, 51–58. 10.1016/j.eplepsyres.2016.11.011 [DOI] [PubMed] [Google Scholar]
- Smith ZZ, Benison AM, Bercum FM, Dudek FE, & Barth DS (2018). Progression of convulsive and nonconvulsive seizures during epileptogenesis after pilocarpine-induced status epilepticus. Journal of Neurophysiology, 119, 1818–1835. 10.1152/jn.00721.2017 [DOI] [PubMed] [Google Scholar]
- Spencer SS (2002). Neural networks in human epilepsy: Evidence of and implications for treatment. Epilepsia, 43(3), 219–227. 10.1046/j.1528-1157.2002.26901.x [DOI] [PubMed] [Google Scholar]
- Srinivas KV, Buss EW, Sun XQ, Santoro B, Takahashi H, Nicholson DA, & Siegelbaum SA (2017). The dendrites of CA2 and CA1 pyramidal neurons differentially regulate information flow in the cortico-hippocampal circuit. Journal of Neuroscience, 37(12), 3276–3293. 10.1523/JNEUROSCI.2219-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegen M, Kirchheim F, Hanuschkin A, Staszewski O, Veh RW, & Wolfart J (2012). Adaptive intrinsic plasticity in human dentate gyrus granule cells during temporal lobe epilepsy. Cerebral Cortex, 22(9), 2087–2101. 10.1093/cercor/bhr294 [DOI] [PubMed] [Google Scholar]
- Steve TA, Jirsch JD, & Gross DW (2014). Quantification of subfield pathology in hippocampal sclerosis: A systematic review and meta-analysis. Epilepsy Research, 108(8), 1279–1285. 10.1016/j.eplepsyres.2014.07.003 [DOI] [PubMed] [Google Scholar]
- Sun Q, Sotayo A, Cazzulino AS, Snyder AM, Denny CA, Siegelbaum SA, ... Denny CA (2017). Proximodistal heterogeneity of hippocampal CA3 pyramidal neuron intrinsic properties, connectivity, and reactivation during memory recall. Neuron, 95(3), 656–672.e3. 10.1016/j.neuron.2017.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q, Srinivas KV, Sotayo A, & Siegelbaum S. a. (2014). Dendritic Na(+) spikes enable cortical input to drive action potential output from hippocampal CA2 pyramidal neurons. ELife, 3, 1–24. 10.7554/eLife.04551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talley EM, Solórzano G, Lei Q, Kim D, & Bayliss DA (2001). CNS distribution of members of the two-pore-domain (KCNK) potassium channel family. Journal of Neuroscience, 21(19), 7491–7505. 10.1523/jneurosci.21-19-07491.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamamaki N, Abe K, & Nojyo Y (1988). Three-dimensional analysis of the whole axonal arbors originating from single CA2 pyramidal neurons in the rat hippocampus with the aid of a computer graphic technique. Brain Research, 452(1–2), 255–272. 10.1016/0006-8993(88)90030-3 [DOI] [PubMed] [Google Scholar]
- Thom M (2014). Review: Hippocampal sclerosis in epilepsy: A neuropathology review. Neuropathology and Applied Neurobiology, 40(5), 520–543. 10.1111/nan.12150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thom M, Liagkouras I, Elliot KJ, Martinian L, Harkness W, McEvoy A, ... Sisodiya SM (2010). Reliability of patterns of hippocampal sclerosis as predictors of postsurgical outcome. Epilepsia, 51(9), 1801–1808. 10.1111/j.1528-1167.2010.02681.x [DOI] [PubMed] [Google Scholar]
- Valero M, Cid E, Averkin RG, Aguilar J, Sanchez-Aguilera A, Viney TJ, ... de la Prida LM (2015). Determinants of different deep and superficial CA1 pyramidal cell dynamics during sharp-wave ripples. Nature Neuroscience, 18(9), 1281–1290. 10.1038/nn.4074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winawer MR, Makarenko N, McCloskey DP, Hintz TM, Nair N, Palmer AA, & Scharfman HE (2007). Acute and chronic responses to the convulsant pilocarpine in DBA/2J and A/J mice. Neuroscience, 149(2), 465–475. 10.1016/j.neuroscience.2007.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittner L, Huberfeld G, Clemenceau S, Eross L, Dezamis E, Entz L, ... Miles R (2009). The epileptic human hippocampal cornu ammonis 2 region generates spontaneous interictal-like activity in vitro. Brain, 132(11), 3032–3046. 10.1093/brain/awp238 [DOI] [PubMed] [Google Scholar]
- Wolfart J, & Laker D (2015). Homeostasis or channelopathy? Acquired cell type-specific ion channel changes in temporal lobe epilepsy and their antiepileptic potential. Frontiers in Physiology, 6(June), 1–23. 10.3389/fphys.2015.00168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyeth M, Nagendran M, & Buckmaster PS (2020). Ictal onset sites and γ-aminobutyric acidergic neuron loss in epileptic pilocarpine-treated rats. Epilepsia, 61(5), 856–867. 10.1111/epi.16490 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
All original code has been deposited at Zenodo (see DOI in the Key Resources Table) and is publicly available as of the date of publication.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Cresyl Violet Acetate | Sigma-Aldrich | #C5042 |
| Rabbit anti-PCP4 | Sigma-Aldrich | #HPA005792; RRID:AB_1855086 |
| Mouse anti-RGS14 (IgG2a) | Neuromab | #75-170; RRID:AB_2179931 |
| Mouse anti-STEP (IgG1) | Cell Signaling Technology | #4396; RRID:AB_1904101 |
| Chicken anti-GFP | Aves | #GFP-1020; RRID:AB_10000240 |
| Rabbit anti-CALB1 | Abcam | #ab11426; RRID:AB_298031 |
| NeuroTrace 435/455 fluorescent dye | Invitrogen | #N21479 |
| Streptavidin-Alexa Fluor 647 | Invitrogen | #S21374; RRID:AB_2336066 |
| Bacterial and virus strains | ||
| rAAV5-EF1a-DIO-hChR2(E123T/T159C)-eYFP | UNC Vector Core | Lot AV4828b |
| AAV2/5 hSyn.DIO.hM4D(Gi)-mCherry | Krashes et al J Clin Invest. 2011 Apr;121(4):1424–8. doi: 10.1172/JCI46229 | Addgene: 44362-AAV5 |
| Chemicals, peptides, and recombinant proteins | ||
| Scopolamine methyl nitrate | Sigma-Aldrich | #S2250 |
| Terbutaline hemisulfate | Sigma-Aldrich | #T2528 |
| Ethosuximide | Sigma-Aldrich | #E7138 |
| Pilocarpine hydrochloride | Sigma-Aldrich | #P6503 |
| Diazepam | Hospira | NDC: 0409-3213-12 |
| Methylatropine bromide | Millipore Sigma | #M1300000 |
| Levetiracetam | West-Ward | NDC: 0143-9673-10 |
| Lactated Ringer’s solution | Aspen Veterinary Resources | NDC: 0409-7953-03 |
| Clozapine N-oxide | Sigma-Aldrich | #C0832 |
| Isoflurane | Covetrus | NDC: 11695-6777-2 |
| Betadine | Purdue Products | NDC: 67618-155-01 |
| Buprenorphine | Reckitt Benckiser | NDC: 12496-0757-1 |
| Carprofen | Zoetis Inc. | NADA #141-199 |
| Lubricant Ophthalmic Ointment | Akorn | NDC: 59399-162-35 |
| MediGel CPF | ClearH2O | CPF-74-05-5022 |
| Rodent MDs Rimadyl Tablets | Bio-Serv | #MD150-2 |
| Sucrose | Sigma-Aldrich | #S9378-1KG |
| D-Glucose anhydrous | Fisher Chemical | #D16-1 |
| Sodium chloride | VWR Chemicals | #BDH9286-2.5KG |
| Sodium bicarbonate | Fisher Chemical | #S233-500 |
| Potassium chloride | Sigma-Aldrich | #P3911-500G |
| Sodium phosphate monobasic monohydrate | Fisher Chemical | #S369-500 |
| Sodium pyruvate | Fisher Chemical | #BP356-100 |
| Ascorbic acid | Alfa Aesar | #36237 |
| Calcium chloride | G-Biosciences | #R040 |
| Magnesium chloride | Quality Biological | Cat# 351-033-721 |
| Agar | Difco | Ref# 214530 |
| Potassium gluconate | USP Reference Standard | Cat# 1550001 |
| Ethylene glycol-bis(2-aminoethylether)-N,N,N’,N’-tetraacetic acid | Sigma-Aldrich | #03777-10G |
| HEPES | Sigma-Aldrich | #H3375-25G |
| Adenosine 5’-triphosphate magnesium salt | Sigma-Aldrich | #A9187-500MG |
| Guanosine 5’-triphosphate sodium salt | Sigma-Aldrich | #G8877-250MG |
| Phosphacreatine disodium salt hydrate | Sigma-Aldrich | #P7936-1G |
| Biocytin | Sigma-Aldrich | #B4261-250MG |
| Cesium methanesulfonate | Sigma-Aldrich | #C1426-5G |
| Paraformaldehyde 16% aqueous solution | Electron Microscopy Sciences | Cat# 15710 |
| Triton X-100 | Sigma-Aldrich | #T-9284 |
| Critical Commercial Assays | ||
| New York University Mouse Genotyping Core facility | New York University | https://med.nyu.edu/research/scientific-cores-shared-resources/genotyping-core-laboratory |
| GeneTyper Mouse Genotyping Service | GeneTyper | https://www.genetyper.com/ |
| Experimental models: Organisms/strains | ||
| Amigo2-Cre mice | The Jackson Laboratory | Cat# 030215; RRID:IMSR_JAX:030215 |
| POMC-Cre mice | The Jackson Laboratory | Cat# 010714; RRID:IMSR_JAX:010714 |
| 129S1/SvlmJ mice | The Jackson Laboratory | Cat# 002448; RRID:IMSR_JAX:002448 |
| C57BL/6NTac mice | Taconic | # B6-F / B6-M; RRID:IMSR_TAC:b6 |
| C57BL/6J mice | The Jackson Laboratory | Cat# 000664; RRID:IMSR_JAX:000664 |
| B6;129S-Gt(ROSA)26Sortm32(CAG-COP4*H134R/EYFP)Hze/J mice | The Jackson Laboratory | Cat# 012569; RRID:IMSR_JAX:012569 |
| Software and algorithms | ||
| Sirenia Acquisition | Pinnacle Technology | https://www.pinnaclet.com/sirenia-download.html |
| Sirenia Seizure Pro 1.7.9 | Pinnacle Technology | https://www.pinnaclet.com/seizurePRO.html |
| MATLAB R2019a | MathWorks | https://www.mathworks.com/products/matlab.html |
| Prism 8.4.3 | GraphPad Software | https://www.graphpad.com/ |
| AxoGraph 1.6.4 | AxoGraph | https://axograph.com |
| ZEN Black 2.3 | Zeiss | https://www.micro-shop.zeiss.com/en/us/softwarefinder/software-categories/zen-black/ |
| FIJI / ImageJ 2.0.0 | ImageJ | https://imagej.net/software/fiji/ |
| Original MATLAB code | N/A | https://doi.org/10.5281/zenodo.6835900 |
| Other | ||
| Water Jacked Warmer Intensive Care Unit | ThermoCare | #FW-1 |
| Rodent stereotaxic apparatus | World Precision Instruments | Model #502063 |
| Surgical drill | Grobert | Model C300 |
| 33-gauge infusion needle | Plastics One | #C315I-SPC |
| 0.10” Stainless steel jeweler’s screws | Pinnacle Technology | #8209 |
| Infrared LED camera | Apex CCTV | #AP-DCS100W |
| Tethered four-channel EEG/EMG system | Pinnacle Technology | #8400-K1 |
| Nanoject II | Drummond | #3-000-204 |
| Constant voltage isolated stimulator | Digitimer Ltd. | Model DS2A-Mk.II |
| High-power 1-channel LED driver with pulse modulation | ThorLabs | Model DC2100 |