

Abstract
We have recently shown that co-administration of monoclonal antibodies (mAb) with anti-idiotypic distribution enhancers (AIDEs) that inhibit mAb binding to tumor antigens enabled increased intra-tumoral mAb distribution and increased efficacy of an antibody-drug conjugate (trastuzumab emtansine, T-DM1). In the present work, a PK/PD model was applied to predict the impact of this optimization strategy on the within-tumor distribution and anti-tumor efficacy of trastuzumab-gelonin, where the released payload (gelonin) is expected to exhibit negligible bystander activity. Immunofluorescence histology was used to investigate trastuzumab-gelonin distribution in solid tumors following dosing with or without co-administration of anti-trastuzumab AIDEs. Anti-tumor efficacy of trastuzumab-gelonin, with or without co-administration of AIDEs, was also evaluated in tumor-bearing mice. Trastuzumab-gelonin efficiently induced cytotoxicity when applied to NCI-N87 cells in culture (IC50: 0.224±0.079 nM). PK/PD simulations predicted that anti-idiotypic single-domain antibodies AIDEs with dissociation rate constants between 0.03-0.2 hour−1 would provide optimal enhancement of trastuzumab-gelonin efficacy. LE8 and 1HE, anti-trastuzumab AIDEs, were selected for evaluation in vivo. Co-administration of trastuzumab-gelonin with the inhibitors increased the portion of tumor area that stained positive for trastuzumab-gelonin by 58% (p=0.0059). Additionally, LE8 or 1HE co-administration improved trastuzumab-gelonin efficacy in NCI-N87 xenograft bearing mice by increasing the percent increase in life span (%ILS) from 27.8% (for trastuzumab-gelonin administered alone) to 62.5% when administered with LE8 (p=0.0007) or 83.3% (p=0.0007) when administered with 1HE. These findings support the hypothesis that transient, competitive inhibition of mAb-tumor binding can improve the intra-tumoral distribution and efficacy of immunotoxins when applied for treatment of solid tumors.
Keywords: Binding site barrier, Antibody distribution, Immunotoxins, Trastuzumab, Tumor PK-PD model
INTRODUCTION
There is substantial interest in the development of immunoconjugates for targeted cancer treatment, including antibody-drug conjugates (ADCs), radioimmunoconjugates, and immunotoxins (1, 2). ADCs, which are designed to deliver small molecule chemotherapeutic agents, have been particularly successful with 11 ADCs receiving Food and Drug Administration approval, and with more than 100 ADCs under current clinical development (3, 4). However, ADCs often exhibit substantial off-site off-target toxicity, which may be a function of off-site exposure to the released (i.e., free or unconjugated) small-molecule payloads (5, 6). Due to their small size and high membrane permeability, released payloads may easily diffuse across the plasma membranes of non-targeted cells, entering the cytoplasm and mediating off-site off-target toxicity.
Immunotoxins contain a targeting domain, such as an antibody or antibody fragment, that is either chemically conjugated or recombinantly fused to a highly cytotoxic protein toxin (7). One potential advantage for immunotoxins, in comparison to ADCs, is that the large size and high polarity of protein toxins may minimize risk for off-site toxicity due to inefficient cellular entry of released payload via off-target mechanisms (e.g., membrane diffusion). For example, recombinant gelonin, which has been evaluated as the payload for several immunotoxins that have undergone preclinical evaluation, is a 29 kDa type-I single-chain ribosome-inactivating protein (RIP) that irreversibly inhibits ribosome translation on the 28S rRNA unit (8). Gelonin has no cell entry domain and is unable to cross the plasma membrane efficiently. Consequently, extracellular unconjugated gelonin is essentially non-toxic with IC50 values > 1 ¼M (9–11) when applied to cells in culture, and with a very high (>40 mg/kg) median lethal dose when administered to mice (12).
In general, antibody-based therapies have shown more success for hematological cancers than for the treatment of solid tumors (13, 14). Sub-optimal efficacy in solid tumors has been explained by limited intra-tumoral penetration and heterogeneous distribution, which have been attributed to many factors (15–17). Following extravasation, high-affinity monoclonal antibodies (mAb) often bind tightly to targeted antigens on cancer cells, limiting the extent of mAb distribution within tumors. This phenomenon is known as the binding site barrier (BSB). The BSB may be overcome by saturating tumor antigens via administration of large doses of mAbs (18, 19). However, saturating doses of ADCs or immunotoxins are often not feasible due to dose-limiting toxicities (20). The BSB is a direct function of antibody affinity for tumor antigens, and it is possible to improve within-tumor distribution with use of low affinity mAb, but the benefit of this approach would be expected to be offset by decreased tumor selectivity (21).
Recently, our lab has proposed a new and perhaps non-intuitive strategy to improve antibody efficacy in the treatment of solid tumors. We proposed that co-administration of mAb with competitive inhibitors of mAb-tumor binding would enable mAb to bypass the BSB, improving within tumor distribution and improving anti-cancer efficacy. We demonstrated the feasibility of this strategy with use of a high-affinity anti-trastuzumab sdAb (1HE), as a model anti-idiotypic distribution enhancer (AIDE) that enables transient, competitive inhibition of trastuzumab binding to tumor antigens (22). Co-administration of 1HE with trastuzumab significantly increased the distribution of the mAb in solid tumors and significantly increased the efficacy of trastuzumab emtansine (T-DM1) in mice bearing NCI-N87 xenograft tumors.
Although AIDEs may have utility in enhancing the intra-tumoral distribution of any high affinity mAb-based therapy, we project that benefit will be highly dependent on the significance of bystander activity. For ADCs, the significance of the BSB is, to some extent, reduced by the diffusion of payload molecules from sites of payload release within targeted cells to neighboring cells. Substantial within-tumor distribution of released payload, and substantial bystander activity, may compensate for poor intra-tumoral antibody distribution (23). For example, Tsumura et. al. compared the efficacy of high and low affinity antibody conjugates with the bystander payload MMAE. Despite the dramatically greater tumor penetration of the low affinity antibody conjugates, the increase in therapeutic effect of the low affinity conjugates relative to the high affinity conjugates was modest (24). A similar observation was made by Singh et. al, in which naked antibody co-administration synergistically increased the therapeutic effect of the non-bystander ADC trastuzumab-DM1 in NCI-N87 tumors whereas a less than additive effect was observed for trastuzumab-MMAE co-administered with naked antibody (25). For immunotoxins, where the payload is a protein toxin with low membrane permeability, there is much less opportunity for bystander activity (2, 13) and, therefore, application of AIDEs may be particularly beneficial.
Pharmacokinetic/pharmacodynamic (PK/PD) modeling uses a series of mathematical equations to simulate changes in drug concentration (or mass) over time and to predict drug effects over time (e.g., tumor regression following chemotherapeutic administration). PK/PD modeling is an important component of drug development and optimization, as it helps to identify critical drug attributes (e.g., binding affinity) or dosing protocols to achieve a desired therapeutic outcome. As the AIDEs co-administration approach is non-intuitive and dependent on many factors, mechanistic PK/PD modeling and simulation is useful to identify ideal AIDE characteristics (e.g., optimal dissociation rate constants). In this paper, we have evaluated the utility of the transient competitive inhibition approach for enhancing the intra-tumoral distribution and efficacy of trastuzumab-gelonin, as a model immunotoxin. Based on PK/PD simulation results, two sdAb AIDEs, 1HE and LE8, were selected for in vivo evaluation to assess effects on the intra-tumoral distribution and anti-cancer efficacy of trastuzumab-gelonin with use of NCI-N87 xenograft-bearing mice.
MATERIALS AND METHODS
Preparation of Trastuzumab-Gelonin Conjugate
Recombinant gelonin amino acid sequence was obtained from UniProt (ID# P33186). As described in our prior work, gelonin was expressed in E. Coli and purified using a Ni2+-NTA column (Thermo Fisher Scientific, Waltham, MA, 88226) followed by a Bio-Scale mini CHT ceramic hydroxyapatite multimodal chromatography type I cartridge (BioRad, Hercules, CA, 7324324) (10). Trastuzumab was purchased through Millard Fillmore Memorial Hospital (Williamsville, NY). Trastuzumab was conjugated to gelonin via an SPDP linker as shown in Figure 1A and purified by protein G chromatography (10, 26). The trastuzumab-gelonin conjugate was evaluated using a validated anti-gelonin capture antibody previously described (27).
Figure 1: Generation and functional analysis of trastuzumab-gelonin:
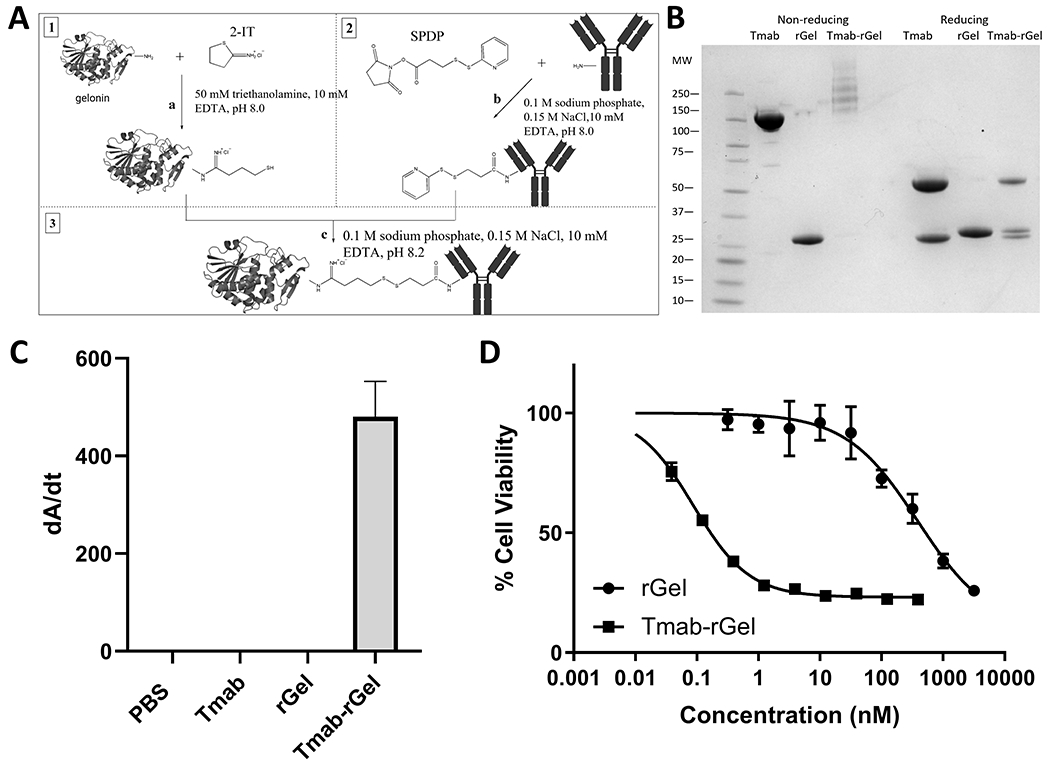
(A) Schematic diagram for generation of trastuzumab-gelonin conjugate. Free gelonin amine groups are modified with 2-IT on ice under a nitrogen blanket. Trastuzumab was reacted with SPDP at room temperature. After 30 minutes reaction, both reactions were stopped by buffer exchange. Then amine-modified gelonin and SPDP-linked trastuzumab were combined and allowed to react overnight on ice under a nitrogen blanket. Iodoacetamide was added to block unreacted sulfhydryl groups. Protein G purification was performed to remove excess gelonin. (B) Shown is an image of an SDS-PAGE gel with trastuzumab (Tmab), gelonin (rGel) and purified trastuzumab-gelonin (Tmab-rGel). Under non-reducing conditions, the trastuzumab-gelonin conjugate presents as several bands at a higher molecular weight than trastuzumab (~150 kDa). Under reducing conditions, the purified trastuzumab-gelonin conjugate lane has three bands that correspond to the light and heavy chains of trastuzumab in addition to free gelonin. The averaged DAR was 1.84 determined by SDS-PAGE. (C) An ELISA plate was coated with anti-gelonin antibody, incubated with PBS, trastuzumab, gelonin and trastuzumab-gelonin and subsequently incubated with an anti-human Fab AP conjugated secondary antibody. No signal is observed for PBS, trastuzumab or gelonin, whereas trastuzumab-gelonin treated wells were observed to have a high positive signal, consistent with a successful conjugation of trastuzumab to gelonin. (D) Shown is the cell viability of NCI-N87 cells treated with gelonin (circles) or trastuzumab-gelonin (squares) for 72 hours. Points represent the mean of triplicate wells with standard deviation error bars.
Cell Viability Assay
NCI-N87 cells were seeded in a 96-well microtiter plate at a density of 2500 cells/well. After 24 hours, culture media was replaced with fresh media containing gelonin or trastuzumab-gelonin. After a 72-hour incubation, 25 μL of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (TGI, Chuo-ku, Tokyo, D0801) solution (5 mg/mL in DPBS) was added and incubated for 4 hours. After MTT reduction, 100 μL of 10% SDS/0.01 M HCl was added and cells were incubated overnight to solubilize the formazan crystals. Solubilized formazan dye was measured at 590 nm and 640 nm. Half-maximal inhibitory concentration values (IC50s) were determined by fitting a four-parameter inhibition model with variable slope using GraphPad Prism 8.2.1 (GraphPad, San Diego, CA).
PK/PD Modeling and Simulation
A hybrid mechanistic pharmacokinetic model was adapted from a spherical tumor model that was developed by our laboratory (28, 29) to identify optimal inhibitor dissociation rates, and to predict the impact of AIDEs on trastuzumab-gelonin tumor disposition and on the tumor growth of NCI-N87 xenografts. Systemic concentrations of trastuzumab-gelonin, trastuzumab and inhibitor were modeled with a 2-compartment model. The pharmacokinetic parameters of trastuzumab-gelonin were fit from the pharmacokinetic study conducted by Cao et al. (11) using a two-compartment model and the maximum likelihood estimation method in ADAPT 5 (BMSR, CA) (30); parameters fits are provided in Table S1. The solid tumor pharmacokinetic model is comprised of a collection of concentric spheres. Each sphere is divided into regions A-E, which represent 5 well-mixed sub-compartments with the same width and different volumes. Region A is the point of extravasation for antibody or immunotoxin through a single tumor blood vessel into the tumor space. Region E represents the most distant region from vasculature and accounts for 49% of the whole sphere volume and is the primary metric for evaluating intratumor distribution. Within each tumor sub-compartment region, trastuzumab-gelonin and trastuzumab-gelonin with only one Fab arm binding to the inhibitor, can bind cellular HER2, bind-free 1HE, or diffuse to adjacent tumor regions. Trastuzumab-gelonin that has both Fab arms bound to inhibitor, is unable to bind to free HER2 and can freely diffuse through tumor regions A-E.
In each individual tumor region, the internalized number of trastuzumab-gelonin per cell drives a cell killing function, with a killing rate constant (Kkill), half-maximal killing constant (TN50) and a slope ɣ (9). The values of the three parameters were estimated by fitting observed in-vitro cellular cytotoxicity data using the weighted least squares (WLS) estimation method in ADAPT 5 (BMSR, CA). All the other PK/PD parameter values can be found in our prior publication (29).
All simulations were conducted in Berkley-Madonna Version 9 (University of California at Berkeley, CA), using a mouse weight of 25 grams and a trastuzumab-gelonin dose of 2.5 mg/kg. Model equations and initial conditions are provided in the supplement files.
Production and Characterization of Competitive sdAb Inhibitors
The sequence of a high-affinity anti-idiotypic anti-trastuzumab sdAb 1HE was taken from Alvarez-Rueda et al (31) and was synthesized by GeneArt. LE8 is a 1HE mutant identified by phage display (29). The DNA of LE8 was isolated from phagemid by colony PCR and ligated into the plasmid pET22b(+), and transformed into the E. coli strain Shuffle T7. 1HE or LE8 were expressed and purified by IMAC affinity chromatography and CHT chromatography as described for trastuzumab gelonin. The trastuzumab dissociation rate constants for 1HE and LE8 were estimated using a dissociation ELISA assay that we have described previously (29).
Animal Xenograft Model
Male athymic nude mice aged 4 to 6 weeks were purchased from the Jackson Laboratory (Bar Harbor, ME). NCI-N87 cells (CRL-5822, a gift from Dr. Dhaval Shah) were cultured following cell line specific ATCC recommendations. NCI-N87 cells were authenticated by STR profiling and tested negative for mycoplasma in January of 2021. NU/J mice were injected subcutaneously into the right flank with 5 million cells/mouse. Tumor growth was monitored using digital vernier calipers, and tumor volume was calculated using the following equation: TV = 0.5∙l∙w2, where “l” represents the longest diameter of the tumor and “w” represents the diameter perpendicular to “l”. Mice were euthanized if tumor diameter exceeded 20 mm, weight loss >15%, or if mice displayed signs of pain or distress.
Animals
All animal studies were approved by the University at Buffalo’s Institutional Animal Care and Use Committee (IACUC). All animal experiments were carried out in accordance with the standards and regulations set by the IACUC.
Immunofluorescence Histology
Mice bearing NCI-N87 xenografts at a volume of 250-300 mm3 were intravenously injected with 2.5 mg/kg trastuzumab-gelonin. Subsequently PBS (n=3) or 1HE in a 3:1 molar ratio (n=2) was immediately intravenously administered through retro-orbital injection. Mice were sacrificed 24 hours post-dosing. The protocol for tumor processing, fluorescence staining and image acquisition is provided in our prior work (22).
Trastuzumab-Gelonin Efficacy Study
Once the tumor volume of NCI-N87 xenograft mice reached 100 mm3, mice were intravenously injected with PBS, trastuzumab-gelonin, or trastuzumab-gelonin with LE8 or 1HE administered at a ten-fold molar excess. PBS or AIDEs were administered immediately after trastuzumab-gelonin injection. Mice were split into four groups of PBS vehicle (n=5), 2.5 mg/kg trastuzumab-gelonin (n=6), 2.5 mg/kg trastuzumab-gelonin + 10:1 LE8 (n=6) and 2.5 mg/kg trastuzumab-gelonin + 10:1 1HE (n=6). Mice body weight and tumor size were monitored using digital calipers. Mice were sacrificed if tumor diameter exceeded 20 mm or weight loss >15%. The log-rank test was used to evaluate the Kaplan-Meier survival curves created in GraphPad Prism 8.2.1 (GraphPad, San Diego, CA). Percent increase in life span (%ILF) was calculated as 100*(median survival time of treatment group - median survival time of control group)/ median survival time of treatment group.
Data Availability
The data generated in this study are available within the article and its supplementary data files.
RESULTS
Conjugation and Cytotoxicity of Trastuzumab-Gelonin Immunoconjugates
Recombinant gelonin (rGel) was expressed in E.Coli and purified with high purity (Figure 1B). Trastuzumab-gelonin immunotoxin was then conjugated through a disulfide-based SPDP linker and purified using protein G chromatography to remove excess gelonin. Both SDS-PAGE and ELISA confirmed the successful conjugation (Figure 1B & 1C). The cytotoxicity of unconjugated rGel and trastuzumab-gelonin were evaluated in NCI-N87 cancer cells and the MTT assay demonstrated IC50 values of 555 ± 69.6 nM and 0.224 ± 0.079 nM, respectively (Figure 1D).
Mathematical Modeling of Cellular Toxicity
In the PD model, the internalized number of trastuzumab-gelonin per cell drives a cell killing function in individual regions of the growth model (Figure 2A). The values of PD parameters Kkill, TN50 and ɣ were estimated by fitting the observed cell viability data of NCI-N87 cells treated with various concentrations of trastuzumab-gelonin conjugates. Figure 2B shows the observed and model predicted cell viability of NCI-N87 cells that were incubated with varying concentrations of trastuzumab-gelonin for 72 hours. The PD model accurately captured the observed cellular cytotoxicity profiles with low coefficients of variation (CV%) for the estimated PD parameters (Figure 2B & Figure S1). Equations are provided in supplement files and the fit values for Kkill, TN50 and ɣ are listed in Table S1.
Figure 2: PK/PD model structure and model fitting results for trastuzumab gelonin cell killing:
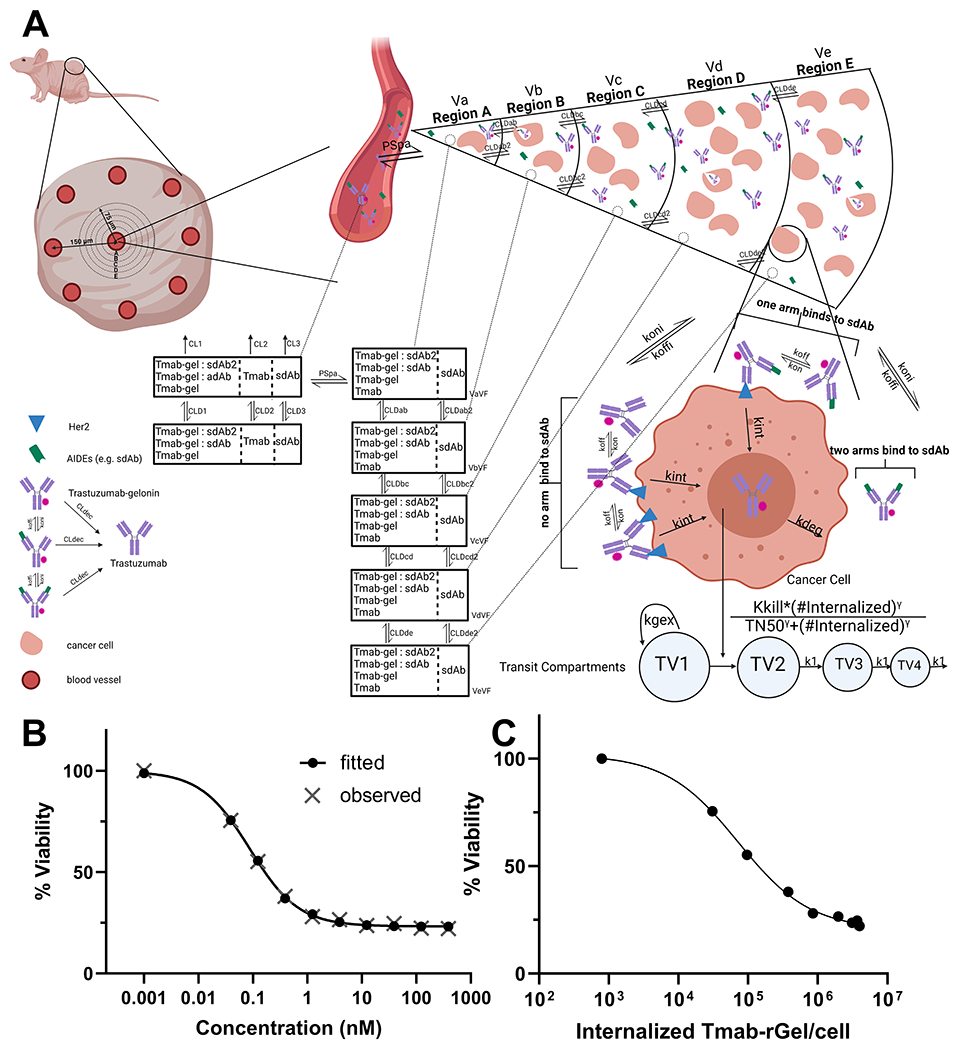
(A) A graphic representation of the sphere PK/PD model structure is shown. A description of the model structure, parameters and assumptions can be found in the methods section, supplementary table 1 and in our prior work (29). (B) Provided is the observed (grey X) and model fitted (black closed circles) cell viability for NCI-N87 cells after treatment with various concentrations of trastuzumab-gelonin. (C) Shown are the model predictions for the number of internalized trastuzumab-gelonin molecules per NCI-N87 cell following treatment with different concentrations of trastuzumab-gelonin.
Simulations Predicting Ideal Inhibitor Koff for Improving Within-Tumor Distribution and Anti-Tumor Efficacy
The sphere pharmacokinetic model was combined with the PD model of trastuzumab-gelonin intoxication process, and the model structure is shown as Figure 2A. The optimal inhibitor dissociation rate constant for improving tumor distribution was identified by simulation. The area under the curve (AUC) of HER2-bound to trastuzumab-gelonin in region E, or fractional HER2 occupancy in tumor region E, were evaluated over a mAb-inhibitor dissociation rate constant (koff) range of 0.001-100 hour−1 (Figure 3A & 3B). Both simulations predict that an inhibitor-mAb dissociation rate (koff) of 0.03~1 hour−1 is optimal for increasing the HER2-bound trastuzumab-gelonin AUC or fractional occupancy of region E, with lower-affinity inhibitors dissociating too rapidly to significantly improve distribution and higher-affinity inhibitors out-competing tumor antigen for mAb binding.
Figure 3: Simulated effect of AIDE co-administration on the tumor distribution of trastuzumab-gelonin:
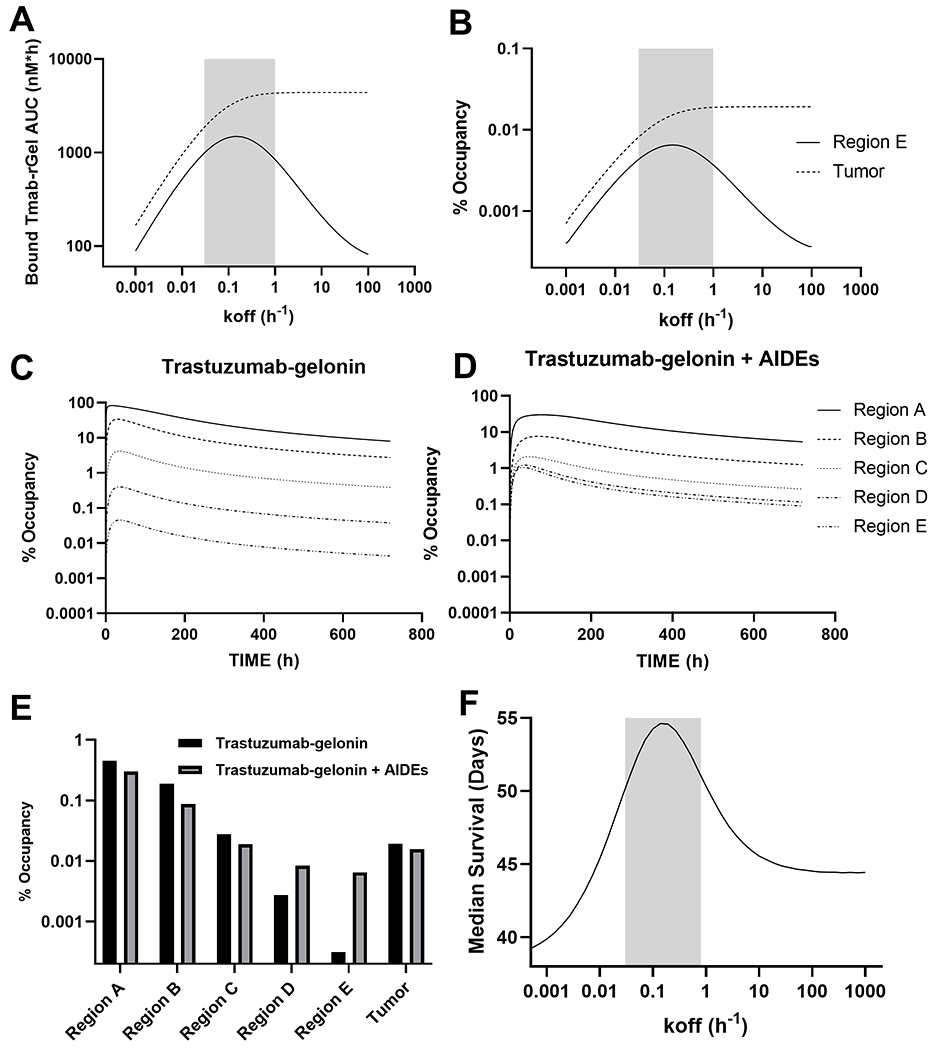
Shown are model simulations evaluating the relationship between the AIDE dissociation rate constant (koff) and the (A) AUC of HER2-bound by trastuzumab-gelonin and the (B) time averaged percent of HER2 bound by trastuzumab-gelonin for the whole tumor (average of regions A-E) and for tumor region E. The percent occupancy of HER2 overtime is provided for all tumor regions following trastuzumab-gelonin injection (C) or following trastuzumab-gelonin administered with a model AIDE (koff: 0.15 h−1) (D). (E) Provided is the time averaged percent of HER2 bound by trastuzumab-gelonin administered with (grey) and without (black) AIDE co-administration. (F) The PK/PD model predictions for the relationship between the AIDE dissociation rate constant (koff) and the response of NCI-N87 xenografts to a single 2.5 mg/kg dose of trastuzumab-gelonin are shown. The simulated median survival time is the time for the tumor profile to reach a tumor volume of 1200 mm3. The grey shaded regions represent an estimated range for a suitable AIDE dissociation rate constant to use in combination of trastuzumab-gelonin.
Using the optimal inhibitor dissociation rate constant of 0.15 hour−1 simulations were conducted to evaluate the impact of inhibitor co-administration on the within-tumor distribution of trastuzumab-gelonin. Trastuzumab-gelonin administered alone leads to a maximum percent occupancy of 84.0%, 40.9%, 6.74%, 0.780% and 0.108% for tumor regions A-E (Figure 3C), indicating trastuzumab-gelonin administered alone is predicted to be restricted to the peri-vasculature (region A), with ~6-fold stepwise decreases for subsequent sub-compartments. Maximum percent occupancy for trastuzumab administered with inhibitor are 25.1%, 8.08%, 2.58%, 1.60% and 1.41% for regions A-E (Figure 3D). The time averaged fractional occupancy of HER2 bound by trastuzumab-gelonin for trastuzumab-gelonin with or without inhibitor are shown in figure 3E. Fractional occupancy values for trastuzumab-gelonin / trastuzumab-gelonin:1HE respectively are region A: 0.411%/0.246%, region B: 0.208%/0.091%, region C: 0.041%/0.024%, region D: 0.005%/0.011%, region E: 0.0007%/0.0088%, whole tumor: 0.023%/0.018%. The dramatic increase in the fractional occupancy of HER2-bound by trastuzumab-gelonin for regions D and E, with a small decrease in total tumor exposure, demonstrate that co-administration of AIDEs is predicted to improve the intra-tumoral distribution trastuzumab-gelonin.
Simulations with the PK/PD model were conducted across a koff range from 0.001–10 hour−1 to determine the optimal inhibitor koff to enhance the efficacy of trastuzumab-gelonin in NCI-N87 xenografts (Figure 3F). Simulations predict that competitive inhibitors with a koff between 0.03 and 0.2 hour−1 result in the greatest extension in median survival days (>50 days) relative to trastuzumab-gelonin administered alone (~45 days). The median survival time is equal to the time it takes for the simulated tumor volume to exceed 1200 mm3.
Production and Assessment of Competitive sdAb Inhibitors
Model AIDEs, 1HE or LE8, were recombinantly expressed and purified with good purity (Figure S2A), and dissociation rate constants were estimated by ELISA with monoexponential decline fittings (Figure S2B). Dissociation rate constants for LE8 and 1HE were 0.151 ± 0.025 hour−1 and 0.0418 ± 0.0052 hour−1, which are within the optimal range (0.03~0.2 hour−1) predicted for improving trastuzumab-gelonin tumor distribution and efficacy. Therefore, LE8 and 1HE were selected for in vivo evaluation using the NCI-N87 xenograft model.
Impact of 1HE Co-Administration on Trastuzumab-Gelonin Distribution in NCI-N87 Xenografts
To experimentally investigate the impact of 1HE co-administration on trastuzumab-gelonin distribution in NCI-N87 xenografts, NCI-N87 xenograft mice were administered 2.5 mg/kg trastuzumab-gelonin with or without a 3-fold molar excess of 1HE. After 24 hours, tumors were excised, sectioned, and fluorescently stained to detect trastuzumab-gelonin (green) and tumor vasculature (red). Trastuzumab-gelonin administered alone is observed to penetrate a short distance from vasculature (Figure 4A). However, 1HE co-administration increased trastuzumab-gelonin tumor penetration with a more homogeneous distribution in trastuzumab-gelonin staining (Figure 4B). The fluorescence images corresponding to the trastuzumab color channel (Figure S3A & S3B) were converted to a binary black and white image to evaluate the percent of the tumor area that stained positive for trastuzumab (Figure S3C & S3D). Compared to tumors treated with trastuzumab-gelonin alone, 1HE co-administration significantly increased the fraction of the tumor that stained positive for trastuzumab-gelonin from 21.6 ± 8.1 % to 34.0 ± 7.4 % (p=0.0059) (Figure 4C).
Figure 4: 1HE co-administration increases the within tumor distribution of trastuzumab-gelonin:
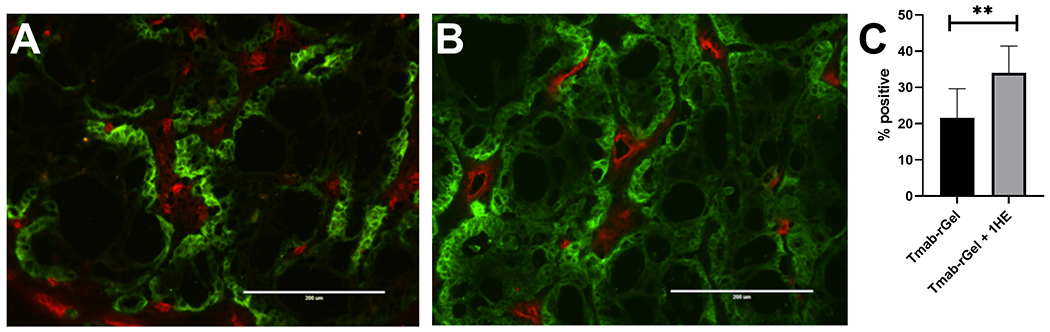
Shown are representative fluorescence histology images of NCI-N87 xenograft tumor sections that were obtained from mice that were treated with (A) Trastuzumab-gelonin or (B) Trastuzumab-gelonin+1HE. (C) Whole tumor sections were imaged and analyzed to evaluate the percent of the tumor area that stained positive for trastuzumab. Data is presented as the group mean (n=6 Tmab-rGel, 4 Tmab-rGel+1HE) with standard deviation error bars.
Impact of Inhibitors Co-Administration on Trastuzumab-Gelonin Efficacy in NCI-N87 Xenografts
NCI-N87 xenograft bearing NU/J mice were used to evaluate the anti-cancer efficacy of trastuzumab-gelonin. Mice were intravenously injected with single doses of 2.5 mg/kg trastuzumab-gelonin, with or without 1HE or LE8. Observed tumor volumes are shown in Figure 5A and Kaplan-Meier survival curves are provided in Figure 5B. Trastuzumab-gelonin in combination with LE8 and 1HE significantly extended the median survival time to 58.5 days (p=0.0007) and 66 days (p=0.0007), respectively, with a corresponding %ILS of 62.5% and 83.3%. Both 1HE and LE8 co-administered with trastuzumab-gelonin significantly extended the median survival relative to trastuzumab-gelonin administered alone (p=0.0013). There was no significant difference between the LE8 and 1HE groups (p=0.736). Administration of 2.5 mg/kg trastuzumab-gelonin with or without the inhibitors did not significantly decrease mean body weight (Figure 5C) or result in any signs of toxicity.
Figure 5: AIDE co-administration improves the response of NCI-N87 xenografts to trastuzumab-gelonin.
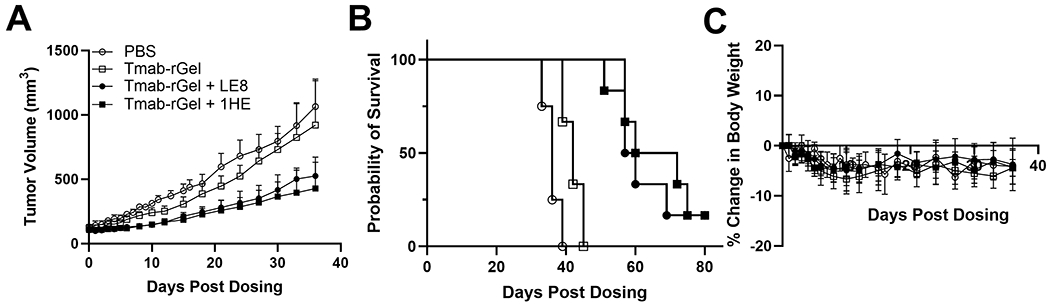
Provided is the (A) tumor volume over time, (B) Kaplan-Meier survival curves and (C) change in body weight of NCI-N87 xenograft bearing mice following a single intravenous injection of PBS (open circles), 2.5 mg/kg trastuzumab-gelonin (open squares), 2.5 mg/kg trastuzumab-gelonin+LE8 (closed circles) and 2.5 mg/kg trastuzumab-gelonin+1HE (closed squares). Data is presented as the group mean (n=5 for the PBS group, 6 for trastuzumab gelonin treated groups) with standard deviation error bars.
Comparison of Tumor Volume Observations and Simulations
PK/PD model simulations were compared to the observed NCI-N87 tumor volumes for mice treated with PBS, trastuzumab-gelonin, trastuzumab-gelonin with LE8 or with 1HE. The simulations for the four groups captured the observed tumor volumes accurately, except for several xenografts in the trastuzumab-gelonin + 1HE group showed superior delays in tumor growth relative to model predictions at time points greater than 20-days after injection (Figure 6A–D).
Figure 6: The PK/PD model accurately predicts the tumor response to trastuzumab gelonin treatment:
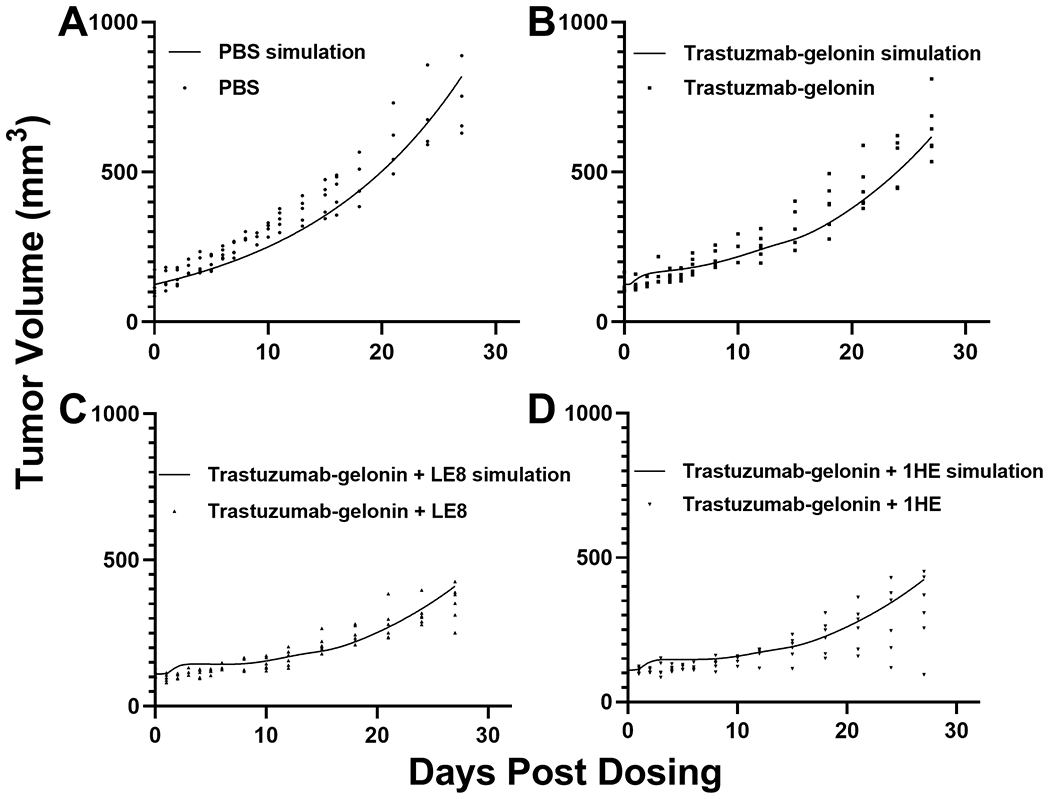
Simulated tumor profiles following treatment with (A) PBS, (B) 2.5 mg/kg trastuzumab-gelonin, (C) 2.5 mg/kg trastuzumab-gelonin+LE8, and (D) 2.5 mg/kg trastuzumab-gelonin+1HE are shown as solid lines. Solid symbols represent observed NCI-N87 tumor volumes for individual xenografts at different timepoints.
Simulations were also conducted to determine if greater enhancements in trastuzumab-gelonin efficacy could be achieved with multiple doses. The simulations used an inhibitor koff of 0.087 h−1, which was predicted to be the optimal for delaying tumor growth (Figure 3F). Simulations predicted that trastuzumab-gelonin, administered alone with regimens of 2.5 mg/kg every 4 days for eight doses (Q4Dx8) or every seven days for five doses (Q7Dx5) will lead to limited anti-cancer efficacy with a median predicted survival time of 49.5 or 44.4 days (Figure S4A & S4B). However, co-dosing of trastuzumab-gelonin, via the same protocols, with AIDE therapy was predicted to lead to dramatically increased tumor response with median survival times of 88.96 or 66.5 days, respectively (Figure S4C & S4D).
DISCUSSION
The development of immunotoxins as anti-cancer therapeutics has been pursued for over 40 years. Two immunotoxins, Lumoxiti and Elzonris, are approved by the FDA for the treatment of refractory hairy cell leukemia and blastic plasmacytoid dendritic cell neoplasm, with both agents requiring “black-box” warnings relating to capillary leak syndrome (32, 33). The narrow therapeutic window of immunotoxins limits the immunotoxin dose below what is required to eradicate cancer. In comparison to pseudomonas exotoxin (PE) payload which is used in Lumoxiti, gelonin has a better safety profile, with a median lethal dose of >40 mg/kg in mice (12), and an observed in-vitro IC50 value of more than 1 µM (9–11). The observed IC50 value for untargeted gelonin is ~1,000-fold higher than untargeted PE38 (34) and 10,000-fold higher than the ADC payload monomethyl auristatin E (35). Due to the favorable safety profile of unconjugated gelonin, and due to the high potency of this toxin when delivered to the cytoplasm of targeted cells, there has been substantial interest in the development of gelonin-mAb conjugates for many cancer indications (10, 11, 36). Clinical development of HUM195-rGEL, the only gelonin based immunotoxin that advanced to clinical trials, was halted due to modest efficacy (37). Although many factors contribute to the disappointing efficacy of immunotoxin therapies, such as the inefficient cytosolic translocation, immunogenicity and dose-limiting toxicities (7), a key contributor to limited immunotoxin efficacy is the poor uptake and penetration of immunotoxins into and within solid tumors (2, 7, 17). Effective delivery of immunotoxins to solid tumors is hampered by abnormal tumor vasculature, high tumor pressure and the overdevelopment of the tumor interstitial matrix (15–17, 38). Following entry into the tumor tissue, poor immunotoxin distribution within the tumor may occur due to the BSB. A binding site barrier for high affinity antibodies against tumor associated antigens was predicted by theoretical mathematical modeling in 1990 by Fujimori et. al. (39) and was subsequently supported by experimental observations (40).
Several groups have investigated approaches that specifically focused on enhancing the tumor penetration of immunotoxin conjugates. Mazor et. al. compared the tumor uptake and efficacy of an intact antibody PE38 conjugate with a MW of 200 kDa to a scFv PE38 recombinant immunotoxin conjugate (RIT) with a MW of 69 kDa (41). Despite the expected penetration advantage, the intact immunoconjugate exhibited a greater anti-tumor effect than the scFv RIT. The poorer efficacy of the scFv conjugate was attributed to the increased rate of elimination and decreased tumor retention relative to the intact conjugate (41). Cao et al. developed recombinant anti-HER2 scFv RIT conjugates with an anti-HER2 equilibrium dissociation constant (KD) range of 10−8 to 10−11 M. However, a direct comparison of the anti-tumor effect in BT474 xenograft bearing mice was unable to be demonstrated as the highest affinity scFv, with a KD of 0.013 nM, formed immune complexes with shed HER2, resulting in liver toxicity. However, greater anti-tumor efficacy was observed for the scFv with a KD of 0.12 nM relative to the scFv with a KD of 16 nM (42). The results by Cao et al. were consistent with the observations of Adams et al., where high affinity antigen binding is required for scFv tumor retention (43). Benefits relating to increased tumor penetration for low and intermediate affinity constructs were offset by poor tumor selectivity (43–45). Using mathematical modeling, Pak et al. predicted that shed mesothelin may improve the tumor penetration of an anti-mesothelin PE immunoconjugate, resulting in an enhanced anti-tumor effect (46). Unfortunately, experimental observations contradicted the model predictions, as immunotoxin efficacy was observed to be lower when applied in the treatment of tumors with high mesothelin shedding cell-lines relative tumors developed with cell lines with low rates of mesothelin shedding (47). The impact of AIDE coadministration on shed antigen mediated immunoconjugate disposition is unknown. AIDEs co-administration may decrease immunoconjugate binding to shed antigen in circulation. The impact of the AIDE strategy on overcoming shed antigen is likely to depend on the rate of shed antigen production and elimination relative to the rate of immunoconjugate elimination. The mathematical model that is presented in the current work may be adapted to predict if there is a potential benefit of AIDE co-administration on overcoming shed antigen barriers to immunoconjugate tumor uptake. The concentration of shed HER2 in NCI-N87 xenografts has been reported to be ~5 pM (48), therefore, it is likely that shed antigen had minimal impact on our evaluations of trastuzumab-gelonin efficacy.
In prior work, we took steps to extend the AIDEs approach through generation of trastuzumab competitive inhibitors with trastuzumab dissociation half-lives ranging from 1 hour to 107 hours (29). We expect that there is an ideal AIDE competitive inhibition window for specific cytotoxin conjugates. To rationally select an optimal AIDE, we developed a mechanistic PK/PD mathematical model to evaluate the relationship between AIDE binding kinetics and the tumor uptake and efficacy of trastuzumab cytotoxin conjugates. Model sensitivity analysis predict that antibody uptake into region E is most sensitive to the tumor inter-vessel radius, tumor antigen concentration, mAb diffusion rate constant, and mAb-antigen association rate constant (kon), while the uptake into layer E is insensitive to the clearance rate of the mAb conjugate, mAb-antigen dissociation rate (koff), or AIDE clearance, vascular permeability and interstitial diffusion (29). In the present work, the sphere model was updated with trastuzumab-gelonin specific plasma pharmacokinetics obtained in mice by Cao et al. who used the same SPDP linker conjugation method (11). The estimated clearance value for trastuzumab-gelonin is 6.6-fold faster than the clearance rate for trastuzumab (49). Consistent with the prior sensitivity analysis the prototype inhibitor 1HE, which was predicted to be optimal for improving T-DM1, was predicted by model simulations to be within the optimal inhibition range for improving trastuzumab-gelonin penetration and efficacy, despite the faster plasma clearance rate. Although 1HE is predicted to be optimal for improving the penetration of intact antibody conjugates, model simulations are required for application of AIDEs to additional immunotoxin formats. For example, prior model simulations predict that the within-tumor distribution of an scFv based RIT, with a >100-fold faster clearance rate than trastuzumab, would be most improved by co-administration of an AIDE with an inhibition half-life of ~1 hour. Co-administration of 1HE (dissociation half-life of 15 hours) would decrease the total tumor uptake and efficacy of an scFv based RIT (29).
Simulations predicted that AIDE co-administration may increase the uptake of trastuzumab-gelonin into tumor region E by ~30-fold. Consistent with the model predictions, the immunofluorescence signal of trastuzumab-gelonin at regions distant from the vasculature was greatly increased with coadministration of the AIDE 1HE. In our prior work we developed a quantitative image analysis algorithm to evaluate trastuzumab distribution in SKOV3 xenografts. 1HE increased the fluorescence intensity of trastuzumab at tumor regions between 50-100 µm from vasculature by >2-fold (22). Direct comparison of image analysis results to the PK model predictions is difficult as there is an undefined relationship between fluorescence signal and antibody concentration. Additionally, useful quantitative analysis of fluorescence images is complicated by background fluorescence, photobleaching, quenching and non-functional vasculature staining. Due to the limitations of immunofluorescence, we did not attempt to compare the distribution of trastuzumab-gelonin following administration of LE8 to 1HE. Both AIDEs were predicted to fall within the optimal range for enhancing the efficacy of trastuzumab-gelonin and were chosen to be compared in the NCI-N87 xenograft efficacy study. 1HE and LE8 were administered separately from trastuzumab gelonin in both the immunofluorescence and efficacy studies. In our prior work, we have administered trastuzumab in-complex with 1HE and observed similar effects as the co-dosing results observed here (22). Due to the high affinity binding, AIDEs that are administered separately rapidly bind to trastuzumab in circulation and model simulations predict that there is no difference between pre-mixing in equimolar ratios or co-dosing with an excess of AIDEs. Both administration approaches have unique advantages, for example, premixing requires less AIDEs and ensures complex formation prior to administration whereas the separation administration of AIDEs would not require approved therapies, such as T-DM1, to have a new formulation approved for clinical use. In the immunofluorescence study, a 3-fold excess of 1HE was administered while a 10-fold excess was used for LE8 and 1HE in the efficacy study. A 10-fold excess was used to control for a decrease in the association rate constant for LE8 to trastuzumab and as a result, a slower rate of complex formation in systemic circulation.
Improved tumor penetration of trastuzumab gelonin was expected to increase therapeutic efficacy similar to the results observed with T-DM1. However, it is possible that application of AIDEs may increase tumor penetration and decrease efficacy, due to dilution of trastuzumab-gelonin to subtherapeutic concentrations in some tumor regions. To improve our ability to predict the impact of AIDE co-administration on the effect of trastuzumab-gelonin we incorporated a pharmacodynamic component into the sphere model. The anti-tumor component of the pharmacodynamic model is driven by the internalized quantity of trastuzumab-gelonin immunotoxins, in individual tumor regions. Parameter values for trastuzumab-gelonin killing were estimated based on our observed in-vitro data (Figure 1D). Prior modeling work published by the Murphy lab estimated that for every 1 million internalized gelonin immunotoxin molecules, only one reaches the cytoplasm (50). The Wittrup group modeled data for many gelonin immunotoxins and concluded that cell killing required internalization of ~4.7 million gelonin molecules, independent of: the antigen targeted, immunotoxin binding affinity, internalization rate, or the external immunotoxin concentration (9). The estimated TN50 from our in-vitro cell cytotoxicity experiments was 1.3 × 105 molecules/cell, which is 36-fold lower than the 4.7 million estimate from the Wittrup group. Notably, the observed IC50 for trastuzumab gelonin in our cell cytotoxicity assays is consistent with prior reports of the cytotoxicity of trastuzumab-gelonin using the same conjugation method when applied to NCI-N87 cells (11). The mechanism for the increased potency for trastuzumab-gelonin relative to the immunoconjugates used by the Wittrup group is unknown. It is possible there are physiochemical characteristics of immunoconjugates or antigen specific dynamics that result in the higher potency. Interestingly, within the published data by Wittrup, the C7rGel conjugate also appears to deviate from the 4.7 million TN50 value with ~2 × 105 internalized C7rGel molecules leading to a ~50% reduction in HT29 cell viability (9). Therefore, it will be of interest to evaluate if there are key immunotoxin attributes that result in enhanced potency. Pharmacodynamic parameters, estimated from in vitro data and implemented into the full PK/PD model, enabled accurate prediction of the anti-tumor effect of trastuzumab- gelonin with and without the co-administration of the AIDEs, supporting the current model structure and values of parameters employed.
IgG-gelonin conjugates are reported to have LD50 values ranging from 2–25 mg/kg (12). In pilot investigations, we observed significant adverse effects (>15% weight loss) following administration of two doses of 5 mg/kg trastuzumab-gelonin four days apart. As a result of the toxicity observed at 5 mg/kg we evaluated the impact of AIDE co-administration following a single dose of 2.5 mg/kg trastuzumab-gelonin. Our work demonstrated that single-dose co-administration of AIDEs with trastuzumab-gelonin significantly enhanced tumor growth inhibition in NCI-N87 xenograft mice. However, it is important to note that tumor regression was neither predicted nor observed for single doses of the combination therapy. The PK/PD model does predict greater efficacy for multiple doses of trastuzumab-gelonin when administered with AIDEs; however, multiple dose therapy with immunotoxins leads to high risk for the development of host anti-drug antibodies that can neutralize the immunotoxin, potentially resulting in the accelerated clearance and decreased efficacy (7). Notably 67 of 80 patients that were treated with Lumoxiti developed neutralizing antibodies after multiple dosing with Lumoxiti (i.e., 40 µg/kg on days 1, 3, and 5 of each 28-day cycle for up to 6 cycles) (51). To avoid the impact of immunogenicity, it is critical to obtain the greatest therapeutic effect in the fewest number of treatment cycles possible. Therefore, approaches to further increase the potency of trastuzumab immunotoxin conjugates are of particular interest. Aside from poor tumor penetration, insufficient in vivo efficacy of trastuzumab-gelonin is attributed to the inefficient cytosolic delivery of gelonin. Relative to other proteins toxins, such as PE, gelonin has no translocation domain and inefficiently escapes endosomes following antigen mediated internalization. To overcome this barrier and enhance potency, our laboratory recently evaluated co-treatment of a gelonin immunotoxin with an antibody-targeted endosomal escape peptide (H6CM18). Co-treatment of an anti-CEA antibody gelonin conjugate with an anti-CEA H6CM18 antibody conjugate resulted in a 1,000-10,000-fold increase in vitro potency and increased the median survival of mice bearing CEA+ LS174T xenografts by 69% (relative to results observed for animals treated with anti-CEA gelonin alone) (10). We have not evaluated the endosomal escape peptide coadministration approach with trastuzumab-gelonin; however, it is possible that further increases in trastuzumab-gelonin efficacy may be observed if the AIDEs strategy and endosomal escape approach are combined.
In summary, the present work was centered on the hypothesis that AIDEs would enable transient, competitive inhibition of immunotoxin binding to tumor antigens, allowing increased intra-tumoral distribution and increased anti-tumor efficacy. To pursue this strategy, a mechanistic PK/PD model was developed and applied to assist in the identification of optimal attributes for AIDEs. We developed and characterized a model immunotoxin, trastuzumab-gelonin, and model AIDEs (1HE and LE8). Consistent with our hypothesis, and also with PK/PD model predictions, AIDEs co-administration was shown to increase trastuzumab gelonin intra-tumoral distribution and efficacy when applied to mice bearing NCI-N87 xenograft tumors. This optimization strategy may find utility in enhancing the efficacy of a wide range of high-affinity, targeted therapies, including agents that are already in clinical development or in clinical use.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by the National Cancer Institute of the National Institutes of Health (R01CA204192, R01CA246785), and by the Center for Protein Therapeutics. JPB was supported through R01CA204192 and R01CA246785. BMB was supported through R01CA246785. PC and YZ were supported through funding provided by the Center for Protein Therapeutics.
Conflict of Interest Disclosure Statement:
J.P.B. serves as the Director of the University at Buffalo Center for Protein Therapeutics, which is supported by AbbVie, Amgen, AstraZeneca, CSL-Behring, Eli Lilly, Genentech, GSK, Janssen, Merck, Roche, Seagen, and Sanofi. During the course of this work, J.P.B. has received consulting fees from companies involved with the development of cancer therapies, including Abbvie, Amgen, Janssen, Eli Lilly, Merck, Sanofi, and Pfizer.
Data Availability Statements
The data generated in this study are available within the article and its supplementary data files.
REFERENCES
- 1.Subbiah V Optimizing anti-body drug conjugates and radiopharmaceuticals for precision therapy: The next frontier in precision oncology. Curr Probl Cancer. 2021;45(5):100799. [DOI] [PubMed] [Google Scholar]
- 2.Hassan R, Alewine C, Pastan I. New Life for Immunotoxin Cancer Therapy. Clinical cancer research : an official journal of the American Association for Cancer Research. 2016;22(5):1055–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.do Pazo C, Nawaz K, Webster RM. The oncology market for antibody-drug conjugates. Nature reviews Drug discovery. 2021;20(8):583–4. [DOI] [PubMed] [Google Scholar]
- 4.Lee A Loncastuximab Tesirine: First Approval. Drugs. 2021;81(10):1229–33. [DOI] [PubMed] [Google Scholar]
- 5.Saber H, Leighton JK. An FDA oncology analysis of antibody-drug conjugates. Regulatory toxicology and pharmacology : RTP. 2015;71(3):444–52. [DOI] [PubMed] [Google Scholar]
- 6.Masters JC, Nickens DJ, Xuan D, Shazer RL, Amantea M. Clinical toxicity of antibody drug conjugates: a meta-analysis of payloads. Invest New Drugs. 2018;36(1):121–35. [DOI] [PubMed] [Google Scholar]
- 7.Kim J-S, Jun S-Y, Kim Y-S. Critical Issues in the Development of Immunotoxins for Anticancer Therapy. Journal of pharmaceutical sciences. 2020;109(1):104–15. [DOI] [PubMed] [Google Scholar]
- 8.Endo Y, Tsurugi K. RNA N-glycosidase activity of ricin A-chain. Mechanism of action of the toxic lectin ricin on eukaryotic ribosomes. J Biol Chem. 1987;262(17):8128–30. [PubMed] [Google Scholar]
- 9.Pirie CM, Hackel BJ, Rosenblum MG, Wittrup KD. Convergent potency of internalized gelonin immunotoxins across varied cell lines, antigens, and targeting moieties. J Biol Chem. 2011;286(6):4165–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Polli JR, Chen P, Bordeau BM, Balthasar JP. Targeted delivery of endosomal escape peptides to enhance immunotoxin potency and anti-cancer efficacy. Aaps j. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cao Y, Marks JW, Liu Z, Cheung LH, Hittelman WN, Rosenblum MG. Design optimization and characterization of Her2/neu-targeted immunotoxins: comparative in vitro and in vivo efficacy studies. Oncogene. 2014;33(4):429–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lambert JM, Blättler WA, McIntyre GD, Goldmacher VS, Scott CF, Jr. Immunotoxins containing single-chain ribosome-inactivating proteins. Cancer Treat Res. 1988;37:175–209. [DOI] [PubMed] [Google Scholar]
- 13.FitzGerald DJ, Wayne AS, Kreitman RJ, Pastan I. Treatment of hematologic malignancies with immunotoxins and antibody-drug conjugates. Cancer research. 2011;71(20):6300–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cruz E, Kayser V. Monoclonal antibody therapy of solid tumors: clinical limitations and novel strategies to enhance treatment efficacy. Biologics : targets & therapy. 2019;13:33–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jain RK. Physiological barriers to delivery of monoclonal antibodies and other macromolecules in tumors. Cancer Res. 1990;50(3 Suppl):814s–9s. [PubMed] [Google Scholar]
- 16.Cruz E, Kayser V. Monoclonal antibody therapy of solid tumors: clinical limitations and novel strategies to enhance treatment efficacy. Biologics. 2019;13:33–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bordeau BM, Balthasar JP. Strategies to enhance monoclonal antibody uptake and distribution in solid tumors. Cancer Biol Med. 2021;18(3):649–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thurber GM, Zajic SC, Wittrup KD. Theoretic criteria for antibody penetration into solid tumors and micrometastases. J Nucl Med. 2007;48(6):995–9. [DOI] [PubMed] [Google Scholar]
- 19.Cilliers C, Menezes B, Nessler I, Linderman J, Thurber GM. Improved Tumor Penetration and Single-Cell Targeting of Antibody-Drug Conjugates Increases Anticancer Efficacy and Host Survival. Cancer Res. 2018;78(3):758–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krop IE, Beeram M, Modi S, Jones SF, Holden SN, Yu W, et al. Phase I study of trastuzumab-DM1, an HER2 antibody-drug conjugate, given every 3 weeks to patients with HER2-positive metastatic breast cancer. J Clin Oncol. 2010;28(16):2698–704. [DOI] [PubMed] [Google Scholar]
- 21.Adams GP, Schier R, McCall AM, Simmons HH, Horak EM, Alpaugh RK, et al. High affinity restricts the localization and tumor penetration of single-chain fv antibody molecules. Cancer Res. 2001;61(12):4750–5. [PubMed] [Google Scholar]
- 22.Bordeau BM, Yang Y, Balthasar JP. Transient Competitive Inhibition Bypasses the Binding Site Barrier to Improve Tumor Penetration of Trastuzumab and Enhance T-DM1 Efficacy. Cancer Res. 2021;81(15):4145–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Khera E, Cilliers C, Bhatnagar S, Thurber GM. Computational transport analysis of antibody-drug conjugate bystander effects and payload tumoral distribution: implications for therapy. Molecular Systems Design & Engineering. 2018;3(1):73–88. [Google Scholar]
- 24.Tsumura R, Manabe S, Takashima H, Koga Y, Yasunaga M, Matsumura Y. Influence of the dissociation rate constant on the intra-tumor distribution of antibody-drug conjugate against tissue factor. J Control Release. 2018;284:49–56. [DOI] [PubMed] [Google Scholar]
- 25.Singh AP, Guo L, Verma A, Wong GG, Thurber GM, Shah DK. Antibody Coadministration as a Strategy to Overcome Binding-Site Barrier for ADCs: a Quantitative Investigation. Aaps j 2020;22(2):28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rosenblum MG, Shawver LK, Marks JW, Brink J, Cheung L, Langton-Webster B. Recombinant Immunotoxins Directed against the c-erb-2/HER2/neu Oncogene Product: In Vitro Cytotoxicity, Pharmacokinetics, and in Vivo Efficacy Studies in Xenograft Models1. Clinical Cancer Research. 1999;5(4):865–74. [PubMed] [Google Scholar]
- 27.Engler FA, Balthasar JP. Development and validation of an enzyme-linked immunosorbent assay for the quantification of gelonin in mouse plasma. J Immunoassay Immunochem. 2016;37(6):611–22. [DOI] [PubMed] [Google Scholar]
- 28.Shah DK, Shin BS, Veith J, Tóth K, Bernacki RJ, Balthasar JP. Use of an anti-vascular endothelial growth factor antibody in a pharmacokinetic strategy to increase the efficacy of intraperitoneal chemotherapy. The Journal of pharmacology and experimental therapeutics. 2009;329(2):580–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bordeau BM, Abuqayyas L, Nguyen TD, Chen P, Balthasar JP. Development and Evaluation of Competitive Inhibitors of Trastuzumab-HER2 Binding to Bypass the Binding-Site Barrier. Frontiers in Pharmacology. 2022;13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.D’Argenio DZ SA, Wang X. . ADAPT 5 User’s Guide: Pharmacokinetic/Pharmacodynamic Systems Analysis Software. Biomedical Simulations Resource, Los Angeles; 2009. [Google Scholar]
- 31.Alvarez-Rueda N, Ladjemi MZ, Béhar G, Corgnac S, Pugnière M, Roquet F, et al. A llama single domain anti-idiotypic antibody mimicking HER2 as a vaccine: Immunogenicity and efficacy. Vaccine. 2009;27(35):4826–33. [DOI] [PubMed] [Google Scholar]
- 32.Janus A, Robak T. Moxetumomab pasudotox for the treatment of hairy cell leukemia. Expert Opin Biol Ther. 2019;19(6):501–8. [DOI] [PubMed] [Google Scholar]
- 33.Syed YY. Tagraxofusp: First Global Approval. Drugs. 2019;79(5):579–83. [DOI] [PubMed] [Google Scholar]
- 34.Onda M, Nagata S, FitzGerald DJ, Beers R, Fisher RJ, Vincent JJ, et al. Characterization of the B Cell Epitopes Associated with a Truncated Form of Pseudomonas Exotoxin (PE38) Used to Make Immunotoxins for the Treatment of Cancer Patients. The Journal of Immunology. 2006;177(12):8822–34. [DOI] [PubMed] [Google Scholar]
- 35.Abuhay M, Kato J, Tuscano E, Barisone GA, Sidhu RS, O Donnell RT, et al. The HB22.W vcMMAE antibody drug conjugate has efficacy against non-Hodgkin lymphoma mouse xenografts with minimal systemic toxicity. Cancer Immunology, Immunotherapy. 2016;65:1169–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shin MC, Zhang J, Ah Min K, Lee K, Moon C, Balthasar JP, et al. Combination of antibody targeting and PTD-mediated intracellular toxin delivery for colorectal cancer therapy. Journal of controlled release : official journal of the Controlled Release Society. 2014;194:197–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Borthakur G, Rosenblum MG, Talpaz M, Daver N, Ravandi F, Faderl S, et al. Phase 1 study of an anti-CD33 immunotoxin, humanized monoclonal antibody M195 conjugated to recombinant gelonin (HUM-195/rGEL), in patients with advanced myeloid malignancies. Haematologica. 2013;98(2):217–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jain RK, Baxter LT. Mechanisms of heterogeneous distribution of monoclonal antibodies and other macromolecules in tumors: significance of elevated interstitial pressure. Cancer Res. 1988;48(24 Pt 1):7022–32. [PubMed] [Google Scholar]
- 39.Fujimori K, Covell DG, Fletcher JE, Weinstein JN. A modeling analysis of monoclonal antibody percolation through tumors: a binding-site barrier. J Nucl Med. 1990;31(7):1191–8. [PubMed] [Google Scholar]
- 40.Juweid M, Neumann R, Paik C, Perez-Bacete MJ, Sato J, van Osdol W, et al. Micropharmacology of monoclonal antibodies in solid tumors: direct experimental evidence for a binding site barrier. Cancer Res. 1992;52(19):5144–53. [PubMed] [Google Scholar]
- 41.Mazor Y, Noy R, Wels WS, Benhar I. chFRP5-ZZ-PE38, a large IgG-toxin immunoconjugate outperforms the corresponding smaller FRP5(Fv)-ETA immunotoxin in eradicating ErbB2-expressing tumor xenografts. Cancer Letters. 2007;257(1):124–35. [DOI] [PubMed] [Google Scholar]
- 42.Cao Y, Marks JD, Huang Q, Rudnick SI, Xiong C, Hittelman WN, et al. Single-Chain Antibody-Based Immunotoxins Targeting Her2/neu: Design Optimization and Impact of Affinity on Antitumor Efficacy and Off-Target Toxicity. Molecular Cancer Therapeutics. 2012;11(1):143–53. [DOI] [PubMed] [Google Scholar]
- 43.Adams GP, Schier R, Marshall K, Wolf EJ, McCall AM, Marks JD, et al. Increased affinity leads to improved selective tumor delivery of single-chain Fv antibodies. Cancer Res. 1998;58(3):485–90. [PubMed] [Google Scholar]
- 44.Thurber GM, Zajic SC, Wittrup KD. Theoretic Criteria for Antibody Penetration into Solid Tumors and Micrometastases. Journal of Nuclear Medicine. 2007;48(6):995. [DOI] [PubMed] [Google Scholar]
- 45.Thurber GM, Schmidt MM, Wittrup KD. Antibody tumor penetration: transport opposed by systemic and antigen-mediated clearance. Adv Drug Deliv Rev. 2008;60(12):1421–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pak Y, Zhang Y, Pastan I, Lee B. Antigen Shedding May Improve Efficiencies for Delivery of Antibody-Based Anticancer Agents in Solid Tumors. Cancer Research. 2012;72(13):3143–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Awuah P, Bera TK, Folivi M, Chertov O, Pastan I. Reduced Shedding of Surface Mesothelin Improves Efficacy of Mesothelin-Targeting Recombinant Immunotoxins. Mol Cancer Ther. 2016;15(7):1648–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pereira PMR, Sharma SK, Carter LM, Edwards KJ, Pourat J, Ragupathi A, et al. Caveolin-1 mediates cellular distribution of HER2 and affects trastuzumab binding and therapeutic efficacy. Nature Communications. 2018;9(1):5137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Abuqayyas L Evaluation of the Mechanistic Determinants for IgG Exposure in Tissues, Pharmaceutical Sciences. Buffalo, NY: University at Buffalo[Google Scholar]. 2012. [Google Scholar]
- 50.Yazdi PT, Murphy RM. Quantitative analysis of protein synthesis inhibition by transferrin-toxin conjugates. Cancer Res. 1994;54(24):6387–94. [PubMed] [Google Scholar]
- 51.Kreitman RJ, Dearden C, Zinzani PL, Delgado J, Karlin L, Robak T, et al. Moxetumomab pasudotox in relapsed/refractory hairy cell leukemia. Leukemia. 2018;32(8):1768–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated in this study are available within the article and its supplementary data files.
The data generated in this study are available within the article and its supplementary data files.