

Abstract
Pancreatic ductal adenocarcinoma (PDAC) has few effective treatments. Immunotherapy, an attractive alternative strategy, remains challenging with the lack of knowledge on the tumor-infiltrating lymphocyte (TIL) landscape in PDAC. To generate a reference of T-cell subpopulations, we profiled 80,000 T cells from 57 PDAC, 22 uninvolved/normal samples, and cultured TIL using single-cell transcriptomic and T-cell receptor analysis. These data revealed 20 cell states and heterogeneous distributions of TIL populations. The CD8+ TIL contained a putative transitional GZMK+ population based on TCR clonotype sharing, and cell-state trajectory analysis showed similarity to a GZMB+PRF1+ cytotoxic and a CXCL13+ dysfunctional population. Statistical analysis suggested that certain TIL states, such as dysfunctional and inhibitory populations, often occurred together. Finally, analysis of cultured TIL revealed that high-frequency clones from effector populations were preferentially expanded. These data provide a framework for understanding the PDAC TIL landscape for future TIL use in immunotherapy for PDAC.
Keywords: single-cell sequencing, T-cell receptor sequencing, tumor-infiltrating lymphocytes, T cells, pancreatic cancer, TIL, TCR
Introduction
Pancreatic ductal adenocarcinoma (PDAC) accounts for ~85% of all diagnosed pancreatic cancers and, with a 5-year survival rate of only 9%, is the fourth-highest cause of cancer-related death in the United States(1,2). Chemotherapy and surgery remain the chief therapy options, providing curative potential. However, only 10–20% of patients are eligible for surgery, and 80% of those will experience relapse(3). The unmet clinical need of this patient population has spurred efforts to expand therapeutic options.
Immunotherapy represents an exciting new therapeutic strategy, in part due to the success of immune checkpoint blockade in melanoma, lung cancers, and renal cell carcinoma(4–7). Cellular immunotherapies also have found success. These include the adoptive transfer of T cells engineered to express chimeric antigen receptors (CAR-T) in several hematologic cancers(8,9) and tumor-infiltrating lymphocytes (TIL) in metastatic melanoma (10,11). However, immune checkpoint blockade and CAR-T have provided limited benefit in PDAC so far(12–14).
Due to the variable response to immunotherapy across cancer patients, it is important to understand the underlying immune landscape of the tumor microenvironment (TME) relevant to clinical response. Although the presence of TIL has been correlated with better prognosis in a variety of cancers, including PDAC(15–20), suppression by regulatory T cells (Tregs) or attenuation of cytotoxic function by entering a dysfunctional state have been identified as mechanisms of tumor immune escape(21–23). Conversely, TIL populations such as T-stem cell-like memory and tissue-resident memory-like T cells (TRM) have been identified as important for response or improved survival(24–26). These populations are often defined by a complex combination of surface markers and transcriptional states, and no consensus signature relevant for outcome has emerged yet. T-cell repertoire analysis can also provide insight on the anti-tumor response in the TME, given that expansion of T-cell clones at the tumor site can indicate an immune response and high-frequency clones have been found to be the tumor-reactive clones(27).
Single-cell RNA sequencing (scRNA-seq) has emerged as a powerful tool to interrogate the heterogeneity of complex cell populations such as TIL by combining the transcriptomic profile and T-cell receptor (TCR) repertoire in the same cells. Indeed, several scRNA-seq studies have revealed TIL diversity across multiple solid tumor types, identifying various states of T-cell dysfunction as well as potentially novel TIL populations associated with response and prognosis(24,28–35). The lack of response of PDAC to immunotherapy thus far, despite the presence of tumor-reactive TIL(36–38), and the dire need for improved therapy options makes PDAC an ideal candidate for deeper analysis using scRNA-seq.
Previous work by this group has shown the feasibility of ex vivo expansion of tumor reactive PDAC TIL for adoptive cell therapy (ACT)(38). Therefore, the purpose of this study was to characterize the PDAC TIL landscape with an eye toward informing TIL use in cellular immunotherapy of PDAC. To perform this study, freshly resected tumor and uninvolved tissue from PDAC patients were analyzed using combined single-cell RNA and TCR sequencing to interrogate the heterogeneous TIL populations present to better understand the tumor-infiltrating T cells. Additionally, cultured PDAC TIL from a subset of tumor samples were analyzed using similar methods to investigate the contribution of each initial TIL population to the composition of the final TIL product with its therapeutic potential in mind. Overall, seven CD8+ TIL cell states and five CD4+ TIL cell states in the fresh tissue were elucidated along with their inferred relationship based on transcriptomic and TCR data. Furthermore, transcriptomic and TCR analysis of the ex vivo expanded TIL product showed preferential expansion of select effector T-cell states originally identified in situ.
Results
Single T-cell transcriptome data generation and curation
To delineate TIL sub-states in PDAC, we performed scRNA-seq on seven primary, human PDAC samples and one uninvolved pancreas sample (hereafter referred to as “uninvolved”), focusing on the CD3+ TIL (Fig. 1A, MDA1 data set). To further increase the data sample size, CD3+ TIL scRNA-seq data from another cohort at the institution (MDA2 data set; 26 primary PDAC and 10 uninvolved) and from a previous study by Peng et al. (PUMCH data set; 24 PDAC and 11 normal pancreas) were combined(39). In total, 39,694 T cells were analyzed from 57 PDAC samples and 22 uninvolved/normal samples. Clustering of T cells from all samples identified 13 populations overall, with five CD4+ TIL cell states (CD4-FOXP3, CD4-CXCR4, CD4-CCR7, CD4-CXCL13, CD4-MX1), seven CD8+ TIL cell states (CD8-GZMK, CD8-CXCR6/IL7R, CD8-ZNF683, CD8-GZMB/PRF1, CD8-CXCL13, CD8-CCR7/IL7R, CD8-MX1), and a cycling sub-population (Fig. 1B). Cohort-specific and patient-specific batch effects were limited as evidenced by the even mixing between the cohorts and the patients as shown in UMAPs (Uniform Manifold Approximation and Projection) (Supplementary Fig. S1A and B respectively). These cell states were annotated after the identification of defining T-cell genes that are expressed within that cell state (Supplementary Fig. S1C). The presence of all CD4+ and CD8+ TIL cell states, with the exception of CD8-CXCR6/IL7R was validated in formalin-fixed paraffin-embedded primary PDAC tissues using RNA in situ hybridization (RNAScope) that enables visualization of single RNA molecules in individual cells (Supplementary Fig. S1D).
Figure 1. Clustering and differential gene expression analysis of pancreatic ductal adenocarcinoma (PDAC) tumor-infiltrating lymphocytes (TIL).
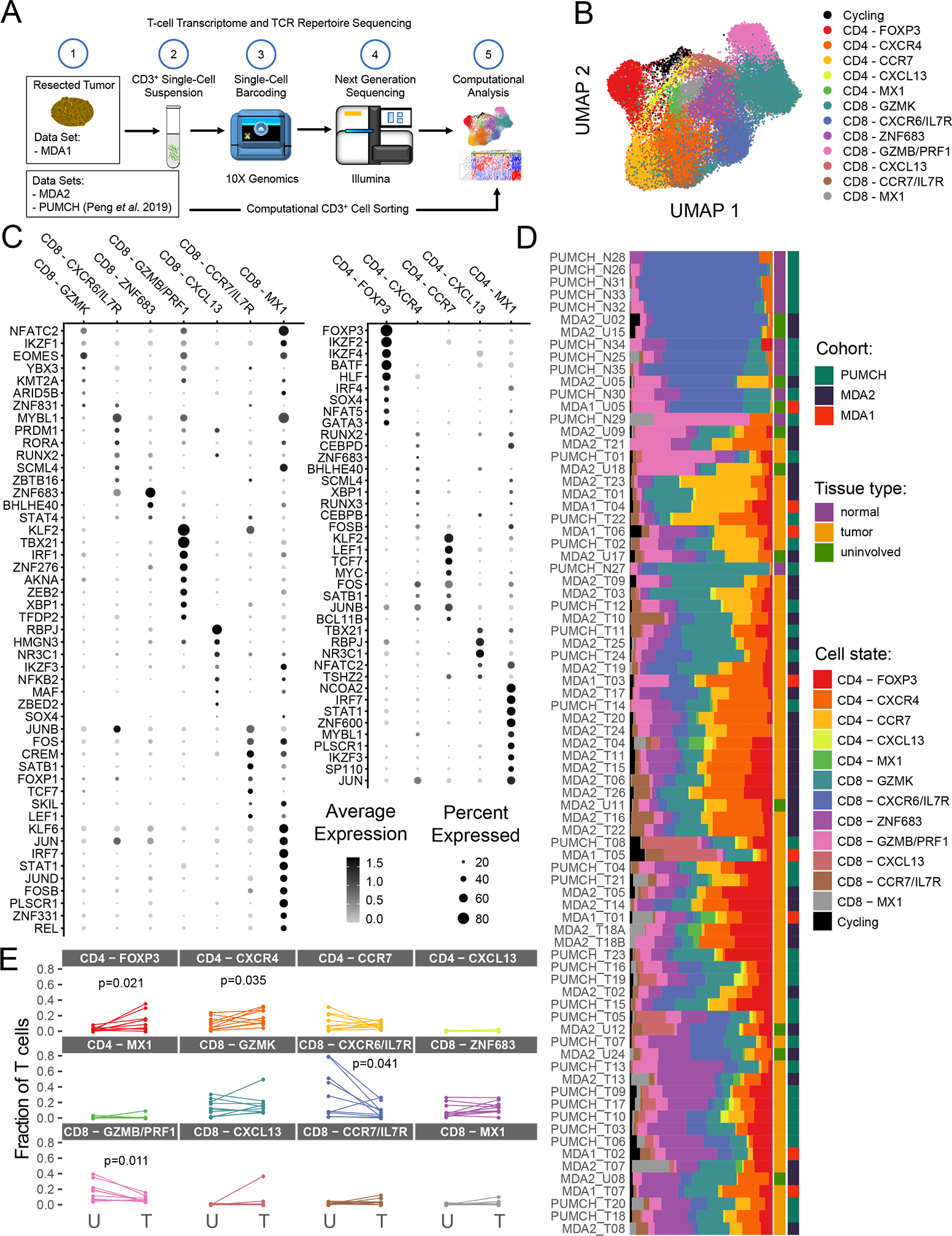
(A) Scheme of overall study in which TIL from three cohorts (two internal and one previously published) from PDAC and uninvolved/normal tissue underwent single-cell RNA sequencing analysis, and the data generated were used for transcriptomic and TCR sequencing analyses. (B) The uniform manifold approximation and projection (UMAP) projection of 39,694 single T cells from 57 PDAC samples and 22 uninvolved samples, showing seven CD8+ and five CD4+ cell state populations identified by shared nearest neighbor clustering. Each dot is a single cell and is colored according to transcriptomic state. (C) Top differentially expressed transcription factors for CD8+ and CD4+ TIL. Expression is marked by appearance of a circle, where greater the size indicates a greater percentage of cells express this gene and a color gradient that indicates higher level of expression as it moves from light gray to black. (D) A breakdown of the cell states within each patient sample is shown, where the samples are ordered by hierarchical tree. The fraction of T cells within each transcriptomic cell state is indicated by the size of the colored segments of each horizontal bar. Each horizontal bar represents an individual patient sample. Starting from the right side of the graph to the left side, the cohort, the tissue type, and cell state for each sample are indicated by colored squares. (E) Comparison of T-cell state frequencies between matched tumor and uninvolved samples (n=10). Only P values < 0.05 are displayed and were generated using a paired Wilcoxon statistical test, but no significant correlation was maintained after P value correction for multiple-testing.
Transcriptomic clustering defines TIL populations with functional T-cell markers
To understand the biological function of each T-cell cluster, the unbiased top differentially expressed genes were investigated as well as the expression of specific known transcription factors. This analysis revealed cluster-specific expression of key genes, indicating cell-state programming differences between the clusters (Fig. 1C and Supplementary Fig. S2A). While the TIL cell states were primarily composed of αβ T cells, marked by expression of the TCR alpha-chain gene TRAC, γδ T cells (expressing the TCR gamma-chain gene TRDC) were found to be a small component of the CD8-GZMB/PRF1 cell state (Supplementary Fig. S2B). In addition to the transcriptional states, there was a distinct cycling T-cell state with expression of MKI67 and other cell-cycle related genes (Fig. 1B and Supplementary Fig. S2C).
Regarding the potential functional identity of the TIL states, the CD4-FOXP3 cluster was classified as Tregs due to the expression of FOXP3, IL2RA (CD25), CTLA4 (Supplementary Fig. S2A), and Treg-associated transcription factors IKZF2 (Helios) and IKZF4 (Ikaros; Fig. 1C). An activated CD4+ helper T-cell population, CD4-CXCR4, was also identified based on CD69, GZMA, and CCL5 expression (Supplementary Fig. S2A). However, low expression of transcription factors TBX21 (TBET) and GATA3 made the classification as a Type 1 or Type 2 helper T-cell unclear (Fig. 1C). Within the CD8+ TIL population, the CD8-ZNF683 cluster appeared to be a TRM-like population as suggested by expression of the TRM transcription factor ZNF683, TRM-associated genes XCL1 and GNLY, and low expression of KLF2, as previously reported (Fig. 1C and Supplementary Fig. S2A)(29). Moreover, a cytotoxic CD8+ cell state was identified (CD8-GZMB/PRF1) by high expression of well-characterized effector cell markers GZMB and PRF1 (Supplementary Fig. S2A) and transcription factor TBX21 (Fig. 1C).
Beyond these unique cell states, the single-cell data showed CD8+ and CD4+ clusters that shared expression of several defining genes. One of these sets, CD4-CXCL13 and CD8-CXCL13, showed expression of transcription factor RBPJ and chemokine CXCL13, which are both identified as part of a reputed dysfunctional T-cell phenotype (Fig. 1C and Supplementary Fig. S1A)(35,40). These two cell states also showed expression of the well-known immune checkpoint marker genes CTLA4, PDCD1 (PD1), HAVCR2 (TIM3), TNFRSF18 (GITR), LAG3, and TIGIT (Supplementary Fig. S2D). Another set of cell states, CD4-CCR7 and CD8-CCR7/IL7R, shared expression of transcription factors LEF1 and TCF7 (Fig. 1C) and surface markers CCR7, IL7R, and SELL (CD62L; Supplementary Fig. S2A), consistent with a central-memory T-cell phenotype (TCM).
Across the cohorts, T-cell states were detected at varying frequencies (Fig. 1D and Supplementary Fig. S2E). For example, in tumor samples with ≥50 T cells detected, the CD8-CXCL13 population was observed in 43/53 PDAC samples, but only 7 patients had frequencies of this cluster ≥5% of their total TIL (Fig. 1D). This contrasts with the CD8-GZMK state that was identified in all tumors, and 50/53 patients had frequencies ≥5% of their total T cells. Additionally, uninvolved/normal samples predominantly clustered away from PDAC samples, suggesting global differences in their T-cell cluster composition (Fig. 1D). The CD8-CXCR6/IL7R cell state was significantly enriched in the uninvolved/normal tissue compared to PDAC samples (P < 0.001), while the reverse was found in the CD8-ZNF683 cell state (P < 0.001, permutation test, 1000 permutations). Furthermore, to establish if the cell states are tumor tissue-specific or reflecting the organ from which the tumor originated, the cell-state composition was compared in a subset of 10 patients that had matched tumor and uninvolved tissue samples (Fig. 1E). In these patients, it was found that the CD4-FOXP3 and CD4-CXCR4 cell states were enriched in the tumor over the uninvolved tissue (P = 0.021 and P = 0.035, respectively, two-sided, paired Wilcox test), whereas the CD8-CXCR6/IL7R and CD8-GZMB/PRF1 cell states were enriched in the uninvolved versus the tumor (P = 0.041 and P = 0.011, respectively, two-sided, paired Wilcox test). However, these changes were not statistically significant after P value correction for multiple testing.
To further verify if the cell states were tumor-specific or overlapping with cell states found in the periphery, their transcriptional profiles were compared to that of peripheral blood T cells (Supplementary Fig. S3). Publicly available sequencing data from healthy donors’ and PDAC patients’ peripheral blood mononuclear cells (PBMC) were combined to make the comparison (Supplementary Fig. S3A)(41). Shared Nearest Neighbors clustering revealed five CD4+ T cell states and four CD8+ T cell states as defined by their differentially expressed genes (Supplementary Fig. S3B and Supplementary Table S1). Comparing the top 20 differentially expressed genes (by p-value) of the tissue T-cell states and the PBMC cell states showed top-marker similarity only between three states, the CD8 TCM and the CD4 TCM cell states (both defined by CCR7), and the CD8 cytotoxic cell states (defined by GZMB and PRF1) (Supplementary Fig. S3C). Since CCR7 can identify less differentiated T cells and naïve T cells, it is not surprising that the two are found in both the tumor and the blood. Nonetheless, this highlights that the majority of the defined cell states are unique to the tissue.
Transition of TIL transcriptomic states revealed by trajectory mapping
Because T cells are known for their capacity to transition between different functional and differentiation states, we applied pseudotemporal inference to understand the relationship between the identified cell states. Because CD4+ and CD8+ T cells are not developmentally related beyond thymic selection, trajectory analysis on these major subsets was performed separately (Fig. 2A and 2B). Additionally, due to the high complexity of the T-cell development landscape, the lines displayed on the graph are weighted by the density of cells around each node. This density is proportional to the amount of data supporting the trajectory at that point, and therefore the confidence of that location in the graph.
Figure 2. Relationship between pancreatic ductal adenocarcinoma (PDAC) tumor-infiltrating lymphocyte (TIL) cell states inferred from pseudotime analysis and correlation of co-occurrence.
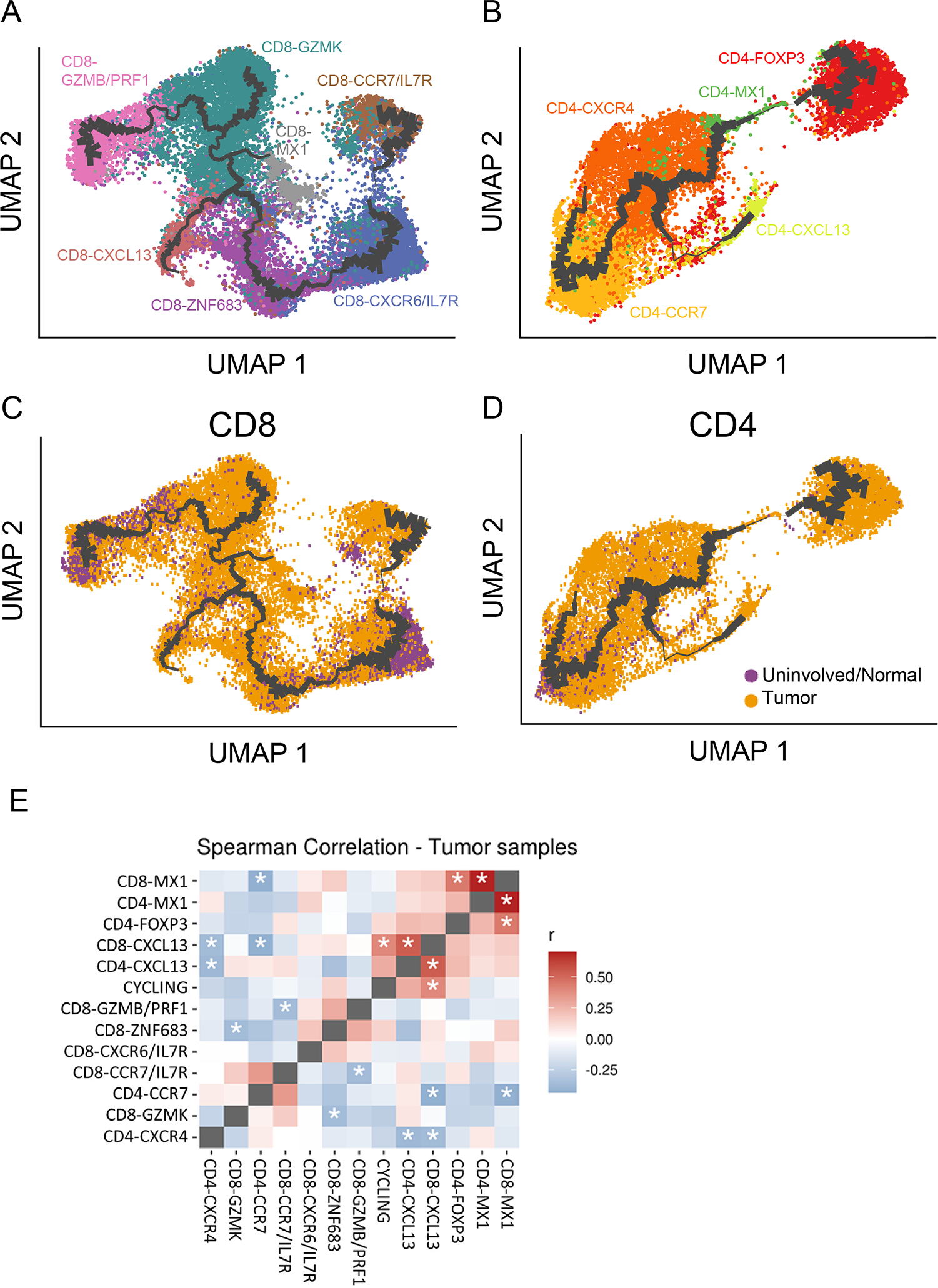
Monocle 3 pseudotime trajectory inference analysis for (A) CD8+ TIL and (B) CD4+ TIL. The thickness of the connecting lines is weighted based on the density of cells at that node, which is proportional to the amount of support for the trajectory. Each dot is a single cell and cell states are colored by transcriptional cell state. The same pseudotime plots are presented for (C) CD8+ TIL and (D) CD4+ TIL, again with the connecting lines weighted by cell density, but now colored by whether that cell came from a tumor sample or uninvolved/normal sample. (E) The degree and significance to which cell states co-occur with each other was computed using Spearman correlation. The matrix pits each cell state against every other transcriptomic state. Red squares indicate positive correlation, blue squares indicate negative correlation, and asterisks denote statistical significance (P < 0.05).
For the CD8+ TIL, the CD8-CCR7/IL7R TCM state and the CD8-CXCR6/IL7R state were inferred to be related in the trajectory. However, that portion of the graph had low cellular density support, suggesting the other CD8+ TIL states did not entirely arise from the TCM population detected in the tumor (Fig. 2A). In contrast, the transition of TIL through the other cell states was strongly supported by the trajectory analysis, based on the closeness on the path, and the thickness and length of their branches. For example, the CD8-CXCR6/IL7R and CD8-ZNF683 cell states occupied the same long branch, indicating a high degree of similarity (Fig. 2A). This contrasts with the latter part of the pseudotime trajectory beyond CD8-ZNF683, where the shorter branch lengths and multiple forks indicate that the CD8+ TIL can potentially enter distinct states. The trajectory also suggests that CD8-GZMK may be an intermediate cell state between the CD8-GZMB/PRF1 cytotoxic cell state and CD8-CXCL13 potentially dysfunctional state. The relationship of the CD8-MX1 IFN-I response cell state to the others appeared unclear from these trajectories, since they did not occur on one of the branches of the trajectory, suggesting they may be a separate state. Additionally, the contributions of CD8+ TIL from tumor and uninvolved samples overlapped, with the exception of CD8-CXCR6/IL7R (Fig. 2C). The inference of similarity and proximal state transition from the pseudotime analysis may suggest that a TIL population found outside the tumor (CD8-CXCR6/IL7R) could be contributing to a state inside the tumor (CD8-ZNF683).
Similar trajectory analysis was performed on the CD4+ TIL (Fig. 2B). These data showed that a CD4-CXCR4 cell state was adjacent to the TCM cluster, with the suggested transition having a much higher confidence (higher density of cells) for CD4+ TIL in contrast to what the data suggested for CD8+ TIL. The trajectory analysis suggested that a small subset of effector cells may be related to the CD4-CXCL13 dysfunctional cluster. However, this portion of the trajectory had a relatively low number of cells whose transcriptional data supported the transition (as displayed by the low cell density surrounding the purported transition on the graph), and therefore represents an unlikely cell state transition. Likewise, the CD4+ T-cell trajectory also tenuously projected a transition from an effector state to a final Treg phenotype. However, this transition is again weakly supported based on the distance between the CD4-FOXP3 cell state and the other CD4 cell states as well as the extremely sparse cellular density along the trajectory. This may indicate these were natural Tregs that infiltrated the tumor as opposed to induced Tregs that transitioned from another state in situ. Similar to the CD8-MX1 IFN-I cluster, the relationship between the CD4-MX1 IFN-I response population and the other clusters was unclear, likely due to its small sample size. Unlike the CD8+ TIL, there was homogeneity of the CD4+ TIL populations between tumor and the uninvolved normal (Fig. 2D).
PDAC TIL cell state co-occurrence analysis highlights different tumor immune milieus
To determine the degree to which T-cell states co-occur in the same PDAC sample, we performed Spearman correlations on cell state frequencies of all tumor samples in a patient-by-patient manner, similar to an analysis performed by Bassez et al. (42). A significantly positive correlation between two cell states indicates that if a patient has a higher fraction of cluster “A,” they also tended to have a higher fraction of cluster “B” (Fig. 2E). This analysis showed that the frequency of the highly related CD8-MX1 and CD4-MX1 IFN-I response cell states (r = 0.70, P = 8.17×10−8) were significantly correlated with each other. Although this cell state for both CD4 and CD8 shared many genes expressed by other cell states (Supplementary Fig. S2A), only CD8-MX1 significantly, positively correlated with CD4-FOXP3 (r = 0.45, P = 0.013), validating that these MX1 states stand on their own, but also that a TME rich with interferon, which influences these cell states, might be responsible for their significant correlation(43,44). Finally, the CD8-CXCL13 and CD4-CXCL13 cell states (r = 0.53, P = 6.71×10−4) were significantly correlated with each other, plus a significant correlation between the CD8-CXCL13 cell state and the cycling population of cells (r = 0.40, P = 0.028) was also observed, indicating that the CXCL13 cell state might be a proliferative population of TIL despite potentially being dysfunctional.
Inverse relationships were also observed for several T-cell states (Fig. 2E). A significant negative correlation was found for the CD8/CD4 CXCL13 populations and the CD4+ CXCR4 effector population (r = −0.37, P = 0.041 and r = −0.39, P = 0.031, respectively), suggesting that a TME with dysfunctional T cells has a lesser content of some but not all effector populations. On the other hand, when a richer content of the effector population CD8-GZMB/PRF1, it was associated with a scarcity of the CD8 TCM cell state (CD8-CCR7/IL7R; r = −0.36, P = 0.04). Another effector cell state, previously shown has the most prevalent effector population (Fig. 1D), CD8-GZMK, had a significant negative correlation with the TRM-associated CD8-ZNF683 cluster (r = −0.37, P = 0.04) (Fig. 2E). Generally, dysfunctional (CXCL13), interferon-response (MX-1) and suppressive (FOXP3) cell states clustered away from effector (ZNF683, GZMK, GZMB/PRF1) or less-differentiated cell states (CCR7/IL7R) (Figure 2E).
TCR clonotype distribution shows expanded TIL clones can occupy multiple cell states
To gain further insight into the potential function of T-cell states as well as the relationship between them, single-cell TCR sequencing and transcriptomic profiling on the same cells was performed on a subset of seven samples (the MDA1 cohort). As shown in Supplementary Figure S1A, the MDA1 cohort is comparable to the combined data set given the degree of overlap between data sets. The similarity was further verified by quantifying the overlap of genes between the MDA1 data set and the larger data set (Fig. 3A). Each cell state had at least 70% overlap of the top genes (P ≤ 0.05 and fold change ≥ 0.2), except for the CD4-CXCL13 cell state which was only composed a few cells in the MDA1 cohort. Furthermore, unsupervised clustering of the MDA1 TIL showed that the cell state markers in this subset were similar to the larger data set (Supplementary Fig. S4A and B). For example, the top markers of the CD8+ cell states included GZMK, IL7R, ZNF683, PRF1, CXCL13, and MX1, while top markers in the CD4+ TIL cell states included FOXP3, CD40LG, SELL, CXCL13, and MX1. UMAP embedding of the smaller MDA1 cohort suggested the robustness of the clustering, with the original cell state labels retaining co-localization in the UMAP projection for both CD8+ and CD4+ TIL (Fig. 3B and 3C). To assess the possibility of sampling bias in the MDA1 sample set, the contribution of each sample to the different cell states was analyzed (Fig. 3D and 3E). Apart from the CD8-CXCL13 cell state deriving predominantly from one sample (MDA1_T05), the cell states were not dominated by a single patient (Fig. 3D). Together, these data suggest that conclusions drawn from the smaller MDA1 cohort could potentially be extended to the larger data set of PDAC patients.
Figure 3. Features of multi-cohort data set are retained within subset of samples used to model the original data set.
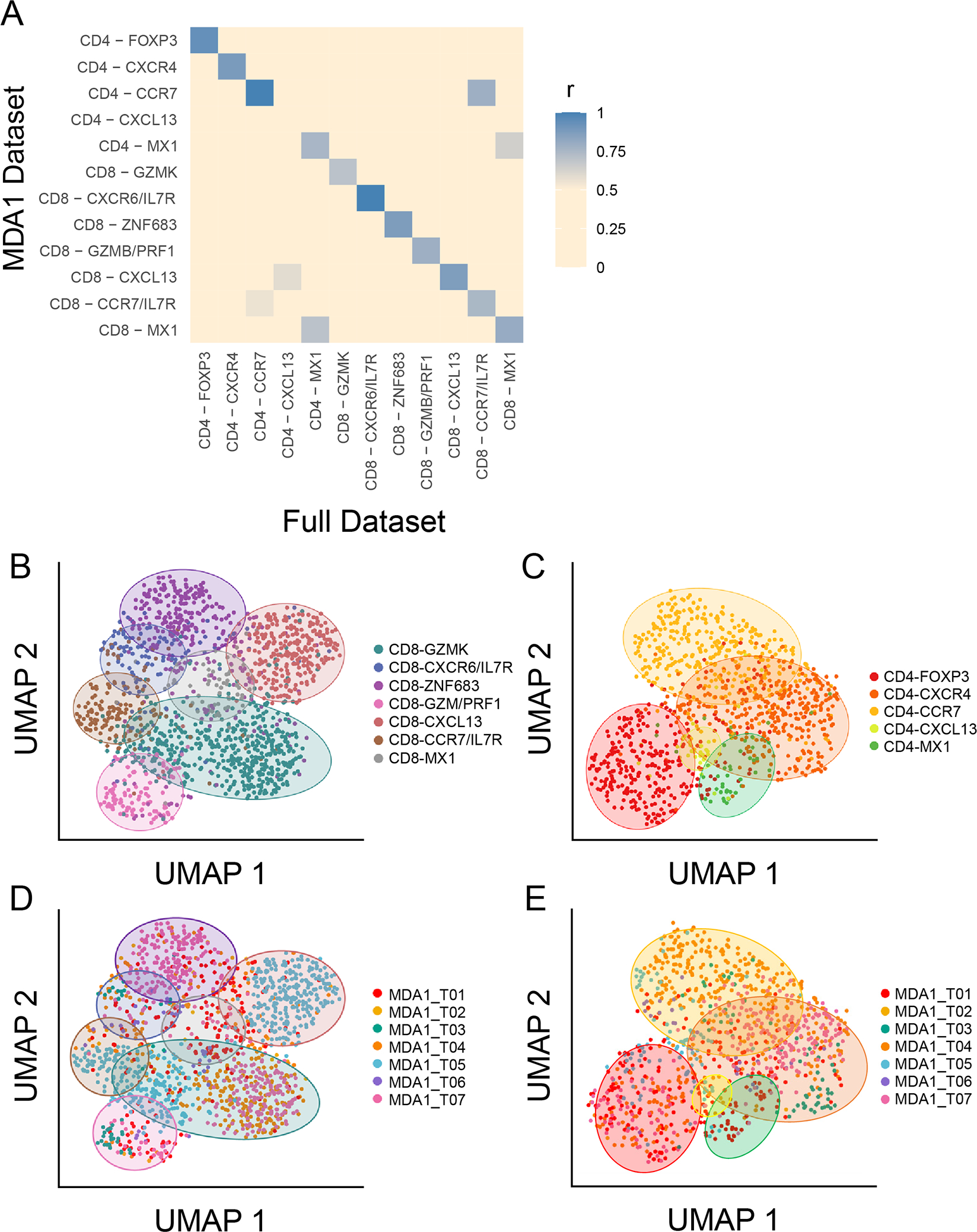
(A) The degree to which the top genes in the tumor-infiltrating lymphocyte (TIL) cell states in the full data set (MDA1, MDA2, and PUMCH) overlap with the those in the MDA1 data set are visualized in a gene overlap matrix. Blue indicates a high degree of overlap, and beige indicates a low degree. Each cell-state has at least 70% of the top (P ≤ 0.05 and fold change ≥ 0.2) genes overlapping, except for CD4-CXCL13, which was hardly detected in the MDA1 data set. (B-E) The seven-sample MDA1 data set is represented using uniform manifold approximation and projection (UMAP) plots. The (B) CD8+ TIL and (C) CD4+ TIL states are labeled and colored using the same scheme as the larger data set, showing that co-localization of the original cell states is maintained. Colored ovals are used to indicate the position of each cell state. (D) and (E) represent the same UMAP plot for CD8 and CD4, respectively, but now colored by patient sample. The same colored ovals from (B) and (C) are used to indicate in which cell states each patient falls.
Next, TCR sequences and transcriptomic information from the same single cells were analyzed to determine the relationship between cell states, as clonotypes found within multiple cell states (referred to here as cell state sharing) may indicate a transition between those states. Visualization of the combination of the transcriptional cell states with their clonal frequency for CD8+ TIL and CD4+ TIL showed that most clones were detected at low frequencies, suggesting limited proliferation at the tumor site (Fig. 4A and 4B). In Figures 4A and B, each bar of the Circos plots represents one T-cell clone where the height of the grey bar on the outer ring of the Circos plots is proportional to the number of cells bearing this TCR. The middle ring indicates the transcriptomic state(s) in which each TIL clonotype can be found, and the inner ring is colored by the patient to which this TCR belongs. When a clonotype is found in multiple transcriptomic clusters, the bar in the middle ring is comprised of several, smaller colored segments. Focusing on the CD8 transcriptomic states in Figure 4A, we found that the expanded TIL clonotypes (i.e., found in multiple copies in the tumor tissue) in the CD8-GZMK cell state (teal) had a high degree of cell state sharing (Fig. 4A, red trapezoidal segment), which is highlighted in the zoomed-in sections of the circular clonotype plots showing the contrast between expanded clones (Fig. 4C, left panel) and unexpanded clones (Fig. 4C, right panel). These observations were further emphasized by the upset plot showing the amount of cell state sharing and the number of times a specific intersection was detected (Fig. 4D and Supplementary Fig. S5). For the CD8-GZMK cell state, 18/40 TIL clonotypes (with ≥2 cells detected) were also assigned to at least one other cell state, such as the six clonotypes shared between CD8-GZMK and CD8-ZNF683 (Fig. 4D). Furthermore, the CD8-ZNF683 cell state also exhibited many expanded clonotypes (Fig. 4A, purple bars) and a high number of cell state sharing (Fig. 4D), with 23 of its 41 clonotypes detected in at least one other cell state, particularly the CD8-CXCR6/IL7R state (blue bars) with 7 instances of overlap. The CD8-GZMK and CD8-ZNF683 clusters also both exhibited sharing with the CD8-GZMB/PRF1 cell state (5 times and 3 times respectively). The TCR overlap between these cell states shows a relationship and further strengthens the cell state transitions depicted by the pseudotime trajectory analysis in Figure 2A. In contrast, the clones in the CD8-CXCL13 cell state (pinkish-red) were highly expanded (Fig. 4A) but were only detected once in other cell states, perhaps indicating a termination point on the trajectory (Fig 4D). In the CD4+ TIL, only very limited cluster sharing was identified, pointing to a lack of proliferation in the tumor (Fig. 4B, 4E, and Supplementary Fig. S6). This is particularly notable for the CD4-FOXP3 cell state (red), which was noted in the pseudotime plot (Fig. 2B) to have a very weak connection to the rest of the trajectory. This lack of TCR sharing between this cell state and other CD4 states further supports the idea that these are innate rather than induced Tregs.
Figure 4. Single-cell T-cell receptor (TCR) sequencing reveals certain T-cell clonotypes are shared among multiple transcriptional cell states.
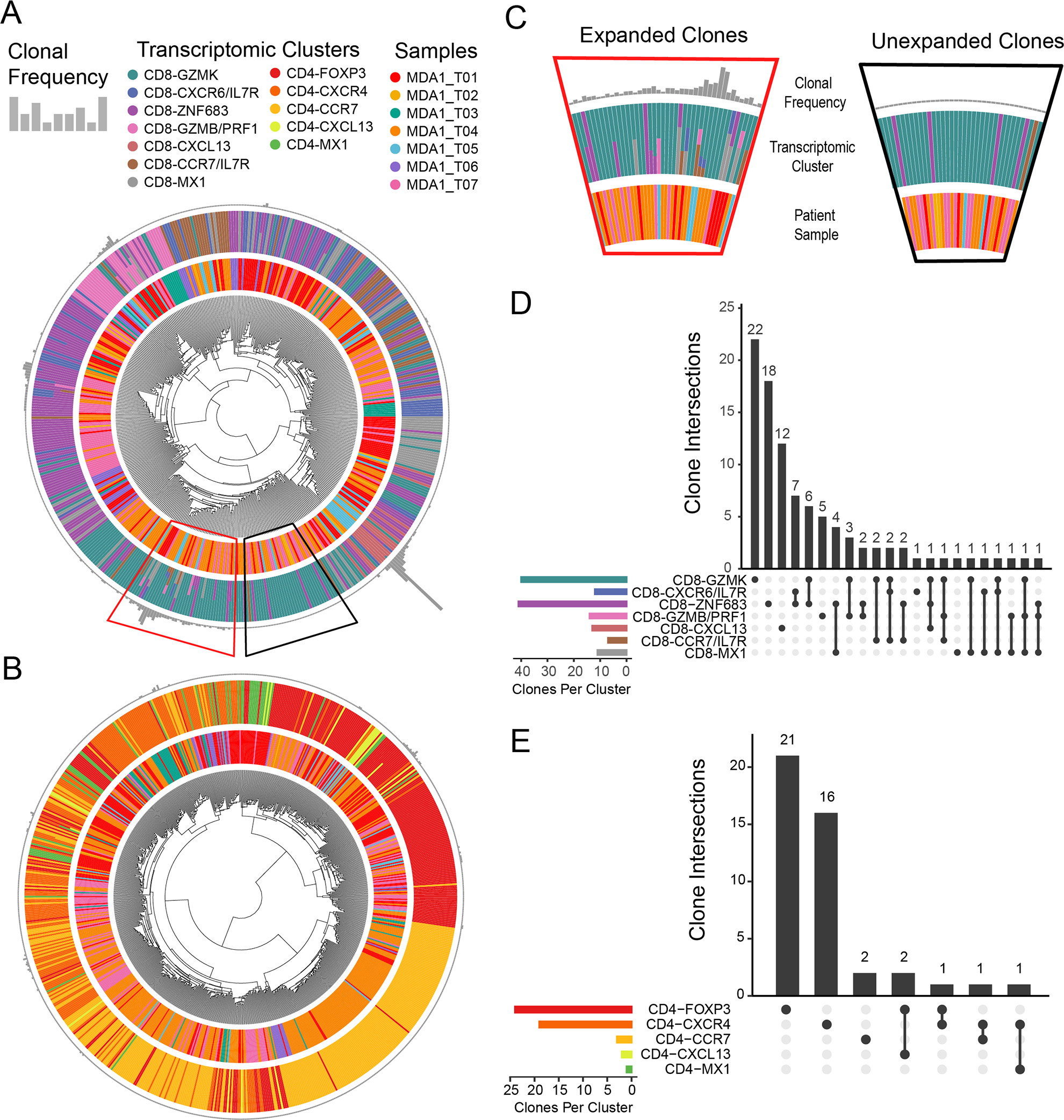
Circos plots are used for (A) CD8+ tumor-infiltrating lymphocytes (TIL) and (B) CD4+ TIL in order to combine the TCR sequencing data with the transcriptomic data. The outer ring constituted of gray bar graphs indicates TCR frequency, where each bar represents one TIL clonotype found in the tumor of a patient. The adjacent, middle ring is colored by the assigned transcriptomic states, where the corresponding TIL clonotype is found for that particular patient sample. When a clonotype is found in multiple transcriptomic cell states, the bar in the middle ring is comprised of several, smaller colored segments. Finally, the inner ring is colored by patient sample. Hence, one patient sample can encompass multiple clonotypes. (C) Trapezoidal segments of the CD8 Circos plot were selected and enlarged to show the detail of cell state sharing found in expanded clones, which is in contrast to that of the unexpanded clones. Upset plots for (D) CD8+ TIL and (E) CD4+ TIL show the degree to which TCR clones are shared between multiple cell states. The vertical, black bars indicate the number of times an intersection is detected. Underneath each bar, the cell states in which at least one expanded clonotype (>2 cells) is detected are symbolized by a solid, black circle. If clones are present in multiple cell states, a line is drawn between those states. If there is no sharing, only a single black dot is shown. The horizontal bars are colored by transcriptomic cell states and indicate the number of TCR clones per cell state.
Clonal expansion occurs within CD8+ TIL cell states and not CD4 cell states
Further information can be gleaned from the TCR-sequencing data by using clonal expansion and diversity of TIL clones at the tumor site as a surrogate for functional involvement in the anti-tumor response. In Figure 4A, the effector cell states of CD8-GZMK, CD8-ZNF683, CD8-GZMB/PRF1, and CD8-CXCL13 showed the highest levels of clonal expansion, with the potentially dysfunctional CD8-CXCL13 cell state having the highest levels of clonal expansion. Although the CD8-CXCL13 cell state was found in only one patient (MDA1_T05), the observed high clonal expansion matches what has been previously reported by several other studies (33,40,45,46). In contrast, very little CD4+ TIL expansion was observed, suggesting their involvement in the response to the tumor may have been suppressed (Fig. 4B). The lack of CD4+ TIL expansion reported here is also consistent with previous observations in the literature for other solid tumor types(29,33,34,40,47).
To quantify the degree of clonal expansion within each cluster, we measured clonal frequency evenness by computing the Gini index for CD4+ and CD8+ TIL across patients (Fig. 5A, left panel). The Gini coefficient is a measure of inequality, ranging from 0 to 1, where lower values indicate clonal frequency evenness and higher values signify more inequality in clonal expansion (i.e., expansion of only one or very few clones). With a median Gini coefficient value closer to 1, the CD8+ TIL population showed a higher degree of oligoclonality than the CD4+ TIL across patients (P = 0.0313, paired t-test). This observation was validated using the related clonal evenness metrics DE50 (P = 0.0313) and Pielou’s index (P = 0.0313), which both showed CD8+ TIL having a lower median value than CD4+ TIL (Supplemental Fig. S7A). When we evaluated the diversity of the repertoire using the diversity metric log(Chao1) index, we did not observe a significant difference between the CD4+ and CD8+ TIL, indicating the repertoires are composed of a similar number of clonotypes (P = 0.156; Fig. 5A, center panel). However, when we utilized Shannon’s diversity index, another diversity metric that takes into account both clonal evenness and clonotype abundance, we observed that overall the CD8+ TIL have a lower diversity than the CD4+ TIL (P = 0.0313; Fig 5A right panel). This observation was again validated using the related Simpson’s index (P = 0.0313; Supplemental Fig. S7A, right panel). Based on previous analyses of T-cell repertoire metrics presented by Chiffelle et al., we concluded that this indicated a greater number of clones comprise the expanded fraction of the CD8+ TIL repertoire than is the case for the CD4+ TIL repertoire(48).
Figure 5. CD8+ tumor-infiltrating lymphocytes (TIL) show a greater degree of oligoclonality than CD4+ TIL.
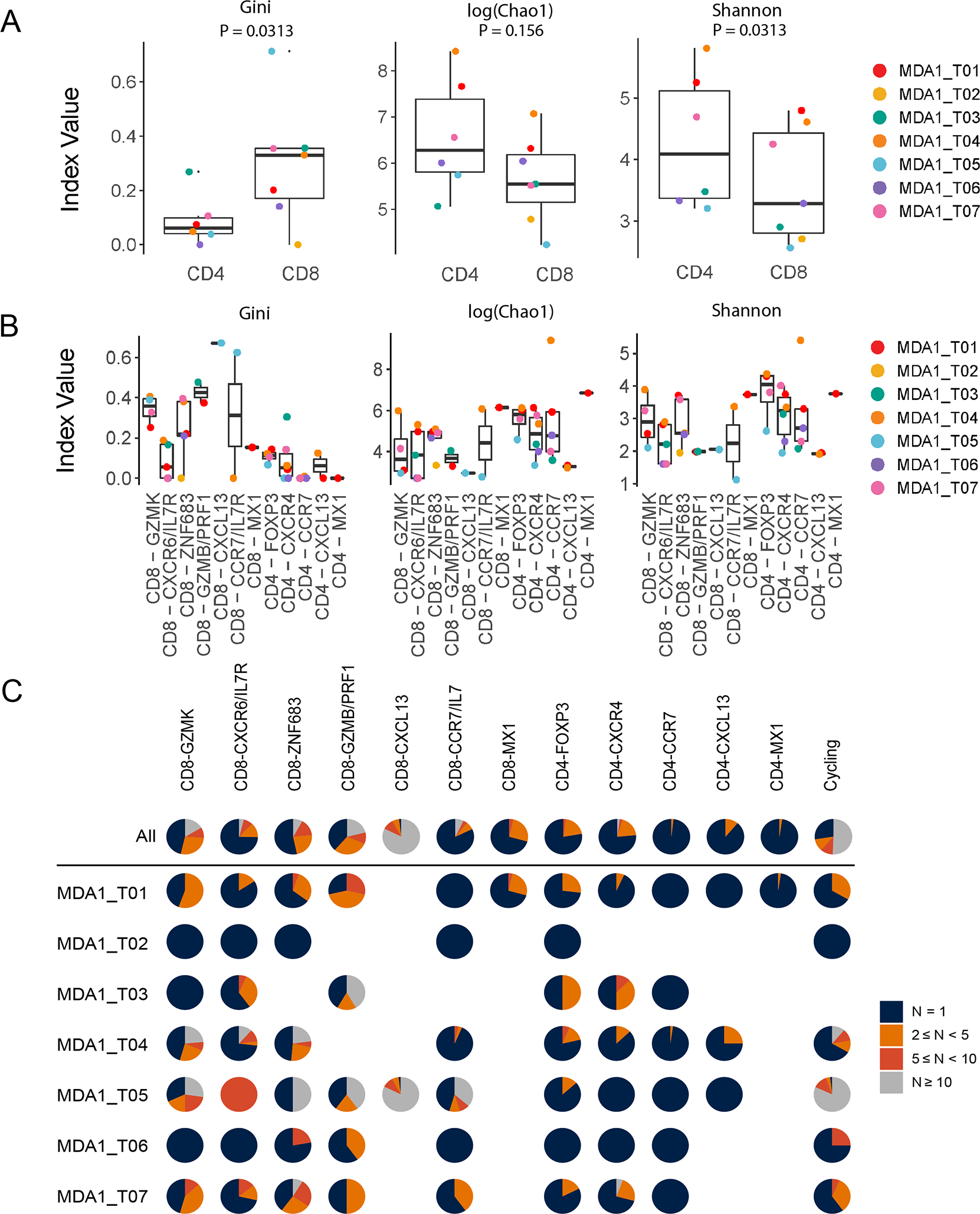
(A-B) The Gini index (left panel), Chao 1 index (middle pannel), and Shannon diversity index (right panel) were applied to measure clonal evenness and diversity within (A) the bulk CD4 and CD8 TIL for each patient and (B) for each cell state within the CD8 and CD4 populations for each patient. The p-values for the bulk comparisons in (A) were obtained following a paired Wilcox analysis. Only those samples with ≥5 cells detected for a cell state are displayed. (C) Pie charts showing the fraction of clonally expanded clonotypes per cell state along the top and broken down by patient underneath. The colors indicate how many times that clonotype was detected (i.e., the number of TIL with that TCR), where blue = 1, orange = 2 ≤ N < 5, red = 5 ≤ N < 10, and gray ≥ 10. Missing pie charts indicate that the cell state was not detected in that patient sample.
When breaking down the T-cell repertoire by cell states, we observed a similar pattern with all CD4+ TIL states exhibiting a lower clonality than the majority of CD8+ TIL states (Fig. 5B, left panel, and Supplemental Fig. S7B, left and center panel). Contributing the most to the higher clonality for the CD8+ TIL were the CD8-GZMK, CD8-GZMB/PRF1, and CD8-CXCL13 cell states. Notably, the median number of clones was lower for these oligoclonal CD8 cell states (Fig. 5B, center panel). However, this was not necessarily the case with the CD4+ TIL as the most oligoclonally expanded CD4 cell state (CD4-FOXP3) had nearly the highest value (i.e., number of clones in its repertoire) (Fig. 5B, left and center panels). Finally, the oligoclonal CD8-GZMB/PRF1 and CD8-CXCL13 cell states not only had lower clonotype richness, but the lowest median diversity as indicated by Shannon and Simpson’s index (Figure 5B, right panel and Supplementary Figure 7B, right panel). Taken altogether, this indicates expansion of select TIL clones in these cell states.
This degree of clonal expansion, and the difference between the CD4+ and CD8+ repertoires in each cell state is further illustrated in Figure 5C, now depicted by patient. The CD8-GZMK, CD8-ZNF683, CD8-GZMB/PRF1, and CD8-CXCL13 had the greatest proportion of clonally expanded cells within the TME, with at least half of the expanded (≥2) CD8+ TIL found ≥ 5 times, indicating a robust expansion. This contrasts with the CD8-CXCR6/IL7R cell states where μ of the clones were detected only once (Fig. 5C). In concordance with our earlier findings regarding the clonality and diversity of the TCR repertoire, CD4+ cell states presented a minority of clonally-expanded TIL as evidenced by the majority of the clones found in these cell states only being detected once (Fig. 5C). Furthermore, even within the expanded fraction, most CD4 clones were detected between 2–5 times. Taken together, these metrics indicate that although the CD8+ TIL population has a similar level of clonotype richness as the CD4+ TIL, the CD8+ effector populations are more clonal, suggesting participation of select TIL clones in the immune response to the tumor.
TIL transcriptomic profiling and TCR sequencing reveal their fate after ex vivo propagation
Concurrently, as the tissue samples from the MDA1 cohort were processed for scRNA-seq, a piece from each tissue was used for ex vivo TIL growth. The cultured TIL were then used for transcriptomic and TCR scRNA-seq (n = 40,065; Fig. 6A). The propagated TIL expression data revealed seven major cell states comprising five CD8+ states (G1-CD8-CD27, G2-CD8-GNLY, G3-CD8-ZNF683, G4-CD8-CCL3, G4-CD8-MKI67), one CD4+ states (G6-CD4-CD40L), and one γδ+ T-cell state (G7-γδΤ-TRDC; Fig. 6B). As expected, a large fraction of the cultured TIL were cycling cells. Cells initially classified as cycling were re-classified into the non-cycling clusters using a random forest classifier. CD8+ TIL states predominated the grown TIL products, which was expected because the culture method was optimized to enhance CD8+ TIL proliferation(49). The expression of signature genes suggested several activated CD8+ cell sates, a proliferating CD8+ state, an effector CD4+ state, and a γδ+ T-cell state (Fig. 6C and Supplementary Fig. S8). Cell states were labelled after key defining genes that were highly expressed (Fig. 6C).
Figure 6. Transcriptomic and T-cell receptor (TCR) sequencing of cultured tumor-infiltrating lymphocytes (TIL) shows preferential expansion of specific TIL states.
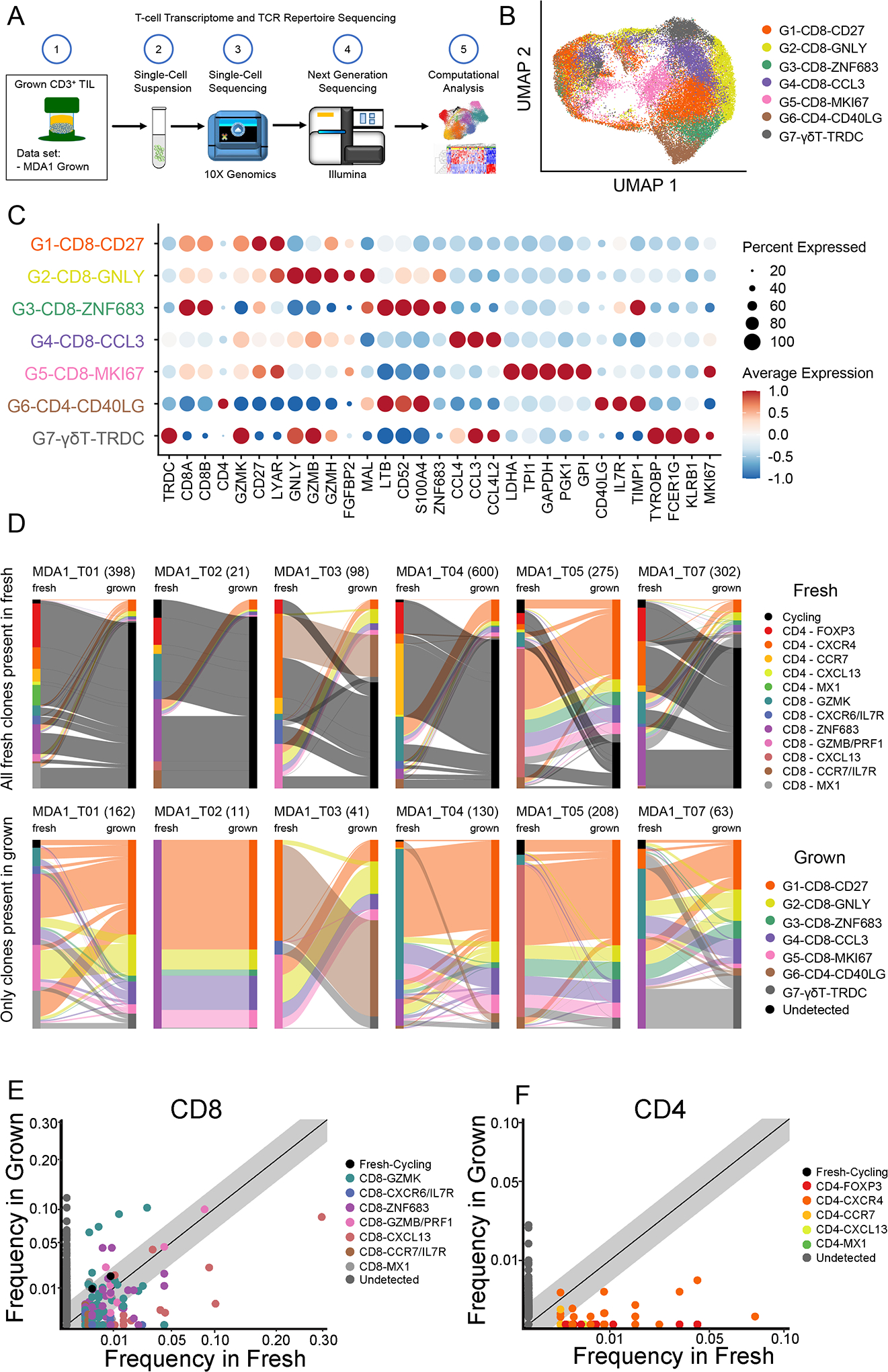
(A) From the same samples that make up the MDA1 data set, tissue was also used to generate TIL. The scheme is shown, in which TIL grown from these samples were sent for single-cell RNA sequencing to generate transcriptomic and TCR analyses. (B) The uniform manifold approximation and projection (UMAP) projection of 40,605 single T cells from six pancreatic ductal adenocarcinoma (PDAC) TIL cultures, showing five CD8, one CD4, and one γδ T-cell cell states. Each dot is a single cell and is colored according to transcriptomic state. (C) Top differentially expressed genes for each cell state are shown. Expression is marked by a circle, where greater size indicates a greater percentage of cells expressing this gene and the color gradient indicates a higher level of expression as it moves from blue to red. (D) Sankey plot visualization shows the transcriptomic cell state in which the TCR initially started in the tissue and where it finished in the culture (fresh tissue on left and grown TIL on right). Clonotypes are classified by the cell state the majority of that clonotype is found. For each sample/graph, two plots are shown with the upper row of graphs showing all clones detected in the fresh tissue and the bottom row showing only those clones detected in both the fresh tissue and grown TIL. Total number of clones are shown in parentheses. Scatter plot for (E) CD8+ TIL and (F) CD4+ TIL comparing the relative frequency of T-cell clones in the fresh pancreatic ductal adenocarcinoma (PDAC) tissue versus the grown TIL. Each dot is a single clonotype and is colored by the transcriptomic state from the fresh TIL. Clonotypes that were not detected in both the fresh tissue and grown culture are colored gray. The diagonal line indicates a 1:1 relative frequency, where clones above the line are found at greater relative frequency in the grown than in the fresh tissue and vice versa.
A major goal of the cellular-based cancer immunotherapy field has been to identify and select the best T-cell subtypes to provide the most efficacious adoptive cell therapy. To this end, we attempted to discern the fate of the different fresh TIL cell states identified in Figure 1 to determine whether the TIL culture process expanded subpopulations of interest. The individual TCRs were used to track TIL from the fresh samples to their corresponding grown TIL product. We found that regardless of their initial transcriptomic cell state in the tissue, the TIL clonotypes were identified in multiple grown cell states with a majority in the G1-CD8-CD27 subtype in the cultured product (Fig. 6D). As opposed to the trajectory inferred between the different cell states in the tissue in our pseudotime analyses in Figure 2 (where this cell state was not described), there was no apparent directionality from one specific fresh tissue T-cell state to a grown T-cell state. This difference can be attributed in part to the controlled activation environment provided by the in vitro expansion using the TIL 3.0 method, which is in contrast to the activation the TIL received in situ. Analysis of the TCR frequency of the grown TIL paired with their transcriptomic clusters showed a high degree of cluster sharing, where the same TCR sequence was found in multiple transcriptomic clusters (Supplementary Fig. S9 and S10A & B).
Despite the lack of connection between any final grown cell state and specific initial PDAC TIL populations, there was an indication that certain TIL populations from the tissue were preferentially grown over others (Fig. 6E and 6F). The T-cell clones originating from CD8-GZMK, CD8-ZNF683, and CD8-GZMB/PRF1 clusters were preferentially expanded ex vivo while those from CD8-CXCL13 populations decreased in frequency relative to their initial frequency in the tissue (Fig. 6E). The CD4+ TIL showed a similar pattern, in which mainly CD4+ effector cells expanded while Tregs did not (Fig. 6F). In both CD8+ and CD4+ TIL, the activated cell states expanded while cell states such as the potentially dysfunctional CXCL13+ populations had limited expansion. Finally, the frequency of TIL clones in the tumor can be an indication of their involvement in an immune response, where high-frequency clones are assumed to have expanded at the tumor site in response to the disease(27). In considering the use of TIL as a therapeutic modality, the expansion of high frequency clones from the tumor is highly desired. Thus, we investigated if the culture methodology employed expanded high frequency clones from the tissue. Comparing all TIL clones detected in fresh tissue and grown samples for six pairs, our data showed that the high-frequency clones detected in the fresh tissue were expanded, and they remained at a relatively high frequency in the grown product (Supplementary Fig. S11).
Discussion
Here, we used scRNA-seq to investigate CD8+ and CD4+ T cells and defined 13 cell states in PDAC and uninvolved/normal pancreas tissue samples plus seven cell states resulting from ex vivo culture. This work was executed using an initial cohort of seven patients treated at MD Anderson Cancer Center, and then further strengthened by the addition of another cohort of 26 patients from MD Anderson Cancer Center as well as a cohort of 24 patients from a previous study at Peking Union Medical College Hospital(39). Based on the transcriptional data from all three cohorts, and concurrent single-cell TCR sequencing in the initial seven patient cohort, we present a model for the organizational relationship and differentiation trajectory of PDAC TIL as well as their unrestricted expansion into each component population of the ex vivo culture (Fig. 7). The TIL cell states detected in this study are consistent with those reported in the literature for other cancer types and validates the lack of institutional and sample-size bias in the three data sets used in this study. For example, CD8+ TIL populations similar to the CD8-GZMK, CD8-ZNF683, and CD8-CXCL13 found here have been reported in other scRNA-seq studies in melanoma, triple-negative breast cancer, non-small cell lung cancer, colorectal cancer, and liver cancer(24,29,31,33,34).
Fig. 7. Model summary of pancreatic ductal adenocarcinoma (PDAC) tumor-infiltrating lymphocyte (TIL) states initially in patient-tissue samples and following in vitro cell culture.
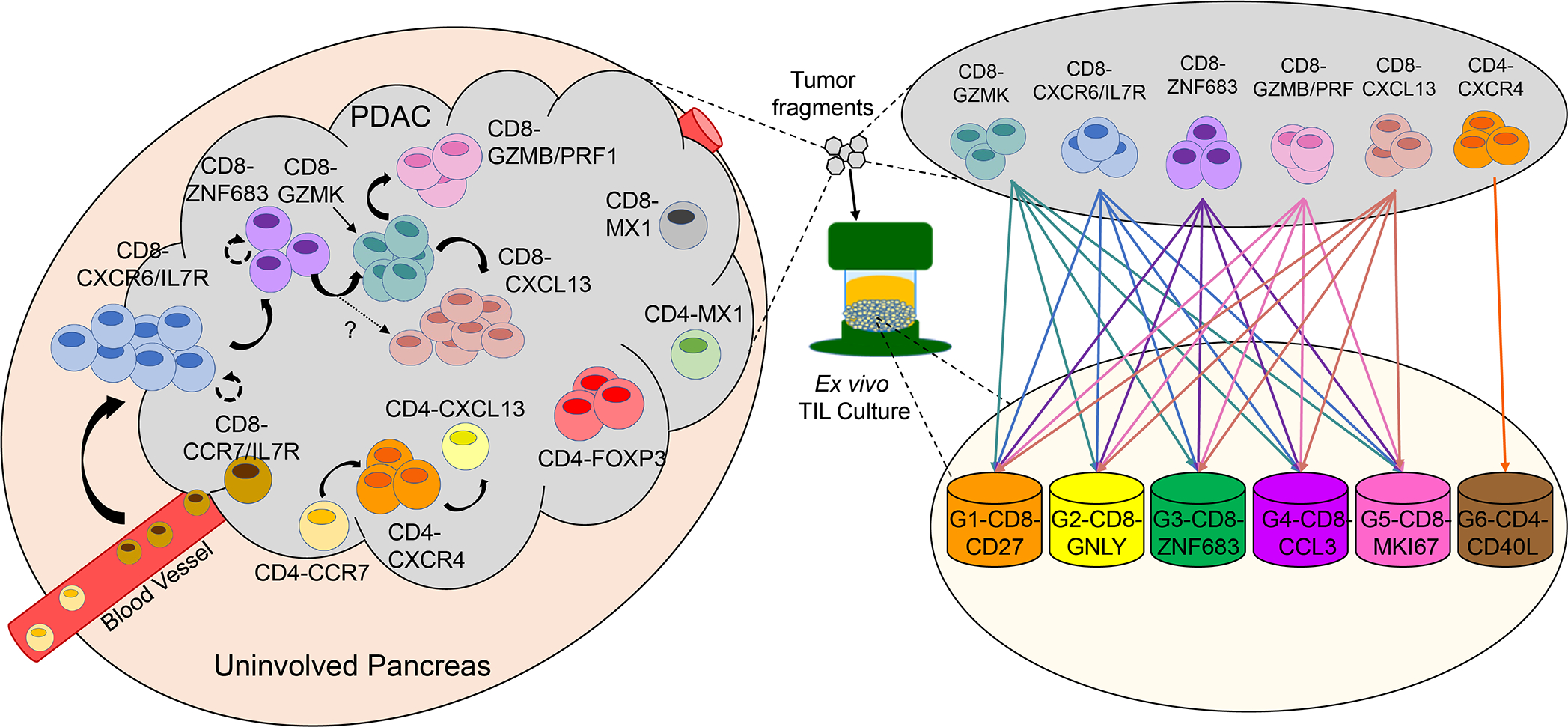
Based on data presented in this paper, this model depicts the TIL differentiation states in PDAC and the suggested transitional relationship between them in the tumor (left side) and following cell culture (right side).
In addition to GZMK, the CD8-GZMK cell state in our analysis showed expression of EOMES, which has notably been tied to a T-cell exhaustion phenotype when paired with expression of immune checkpoint molecules(50,51). Moreover, a recent study in pancreatic cancer reported an exhausted CD8+ T-cell population expressing GZMK, EOMES, and TIGIT(41). However, the CD8-GZMK population described in our study lacked expression of checkpoint molecules or other putative exhaustion markers. It’s position on the pseudotime trajectory along with it sharing TCRs with multiple other cell states indicates it could be an intermediate cell state. Indeed, the CD8+GZMK+ population is similar to those reported in several other studies that classified these cells as a transitional “pre-dysfunctional” population that gives rise to dysfunctional T cells(29,31,33,34,40,52). The CD8-GZMK cell state found here and in these studies are characterized by intermediate clonality, low/intermediate expression of checkpoint markers, and shared TCRs with their respective cytotoxic (i.e., CD8-GZMB/PRF1) and/or dysfunctional (i.e., CD8-CXCL13) populations.
CD8-ZNF683 may be a TRM-like cell state based on the expression of TRM transcription factor ZNF683(53). While this cell state did not express ITGAE (CD103) or have high expression of checkpoint markers as is commonly reported(54), this cell state was similar to a state reported by Guo et al. in non–small cell lung cancer(29). The TRM population is of particular interest due to its correlation with improved survival outcomes and increased cytotoxic activity in several epithelial cancers(25,31,55) and its involvement in response to checkpoint blockade therapy(26,56). However, it is unclear whether this population plays the same role in PDAC due to the lack of checkpoint marker expression observed in this study.
Another population of interest is the CD8-CXCL13 cell state, which has been reported by other groups to be an exhausted/dysfunctional population based on its high expression of checkpoint molecules. This population is also described as being highly clonally expanded, and able to retain proliferative potential at early stages of dysfunction(33,40,45,46). While we similarly report in PDAC that the CD8-CXCL13 cell state has high checkpoint expression and is clonally expanded, the population did not appear to retain great proliferative potential as it did not expand well in our TIL 3.0 culture method (simultaneous engagement of the TCR and 4–1BB combined with high-dose IL-2). This suggests they could be at a later stage of dysfunction and/or require stimulation beyond that provided by our culture method. Nonetheless, studies in melanoma, ovarian cancer, and lung cancer respectively show this population contains tumor reactive TIL(40,46,57). Moreover, the dysfunctional appearance of this population could be a consequence of its ability to react against the tumor repeatedly, and therefore there is value in leveraging this TIL population for therapeutic application.
One of the big questions that remains unanswered is what factor(s) of the pancreatic cancer TME might be causing TIL to move to certain cell states or might be responsible for the overall composition of TIL. One factor that could have a role is malignant progression from intraductal papillary mucinous neoplasm (IPMN) precursor lesions to PDAC. It was previously reported that as lesions progress from IPMN to PDAC, there is a reduction in cytotoxic CD8+ TIL and CD69+ CD4+ TIL(58). Likewise, the same trend was observed for the conventional dendritic cell (cDC) subset cDC2 which is a critical mediator of tumor antigen cross-presentation, an important process in the activation of CD8+ and CD4+ TIL(58). Because of the use of scRNA-seq on CD3+ TIL, we were still able to detect both a cytotoxic CD8+ TIL cell state (CD8-GZMB/PRF1) and an activated CD4+ TIL cell state (CD4-CXCR4) in our study, although with the limitation of not being able to follow their presence through the tumor progression.
Transforming growth factor beta (TGFβ) is a well-known factor with pleiotropic effects that is found at high levels in PDAC and is responsible for not only PDAC progression at late stage but can affect immune cell phenotype, which in turn can affect the progression(59). TGFβ is responsible for suppressing cytotoxic CD8+ and activated CD4+ T cells, either directly or indirectly through suppression of cDC2. TGFβ is also partly responsible for the accumulation of Tregs and myeloid-derived suppressor cells in PDAC, which the latter are responsible for cancer progression(60). While the focus of our study was not to characterize the myeloid compartment, we did observe a strong signature for activated Tregs which appeared to be natural Tregs based on their lack of TCR sharing with other cell states and position on the pseudotime trajectory.
TGFβ is also a known factor for the development of TRM, a population which closely matches with our CD8-ZNF683 cell state (54). In our study, this cell state and the CD8-CXCR6/IL7R cell state appear to be linked based on the pseudotime trajectory, their transcriptome, and TCR overlap. In fact, it has been reported in the literature that a CXCR6+ precursor cell can transition to a TRM phenotype after being activated(61). The occurrence of this transition is supported here in this study by the observation that the CD8-ZNF683 cell state is more clonally expanded than the CD8-CXCR6/IL7R one. Perhaps some of the TIL that comprise the CD8-CXCR6/IL7R cell state were activated, expanded and transitioned to the CD8-ZNF683 cell state. In contrast, the connection between the CD8-ZNF683 and the CD8-GZMK cell states that is suggested by the pseudotime and the shared TCRs is less clear. The relationship raises the question of could the CD8-ZNF683 cells be transitioning to the CD8-GZMK state, which is postulated to be a pre-dysfunctional state, after repeated antigen challenge (62).
Checkpoint blockade immunotherapy is a method that has been successful in altering the TME in some malignancies, but the benefit of this immunotherapy has not yet been demonstrated in PDAC patients. As a potential alternative approach, previous work by our group focused on developing ACT using PDAC TIL showed it is feasible to generate large amounts of tumor-reactive CD8+ TIL from primary and metastatic tumors(38). The TCR scRNA-seq data presented here show that the culture process expands high-frequency clones from potentially important CD8+ TIL cell states such as CD8-GZMK, CD8-ZNF683, CD8-CXCL13, and CD8-GZMB/PRF1 while avoiding the detrimental CD4-FOXP3 Tregs. Clonotypes from the CD8-GZMK and CD8-ZNF683 cell states expanded favorably (frequency in situ < frequency ex vivo) during culture, and exploration of these cell states following adoptive transfer may be of interest due to their memory status and proliferative potential. Recently, scRNA-seq analysis by Lu et al. found that ZNF683 was associated with improved TIL persistence after adoptive transfer in a patient with metastatic colorectal cancer(63). Furthermore, if CD8-ZNF683 is truly a TRM-like population, then it may have improved tissue-homing properties as previously reported for this type of T-cell(64). In contrast, clonotypes from the CD8-CXCL13 cell state did not expand favorably (frequency in situ > frequency ex vivo). Given the fact this population is reported to contain tumor-antigen reactive TIL, and therefore has therapeutic value, expansion techniques will need to be modified to take this into account as even the engagement of a powerful costimulatory molecule like 4–1BB used in this study did not optimally expand it.
Ultimately, regardless of the in situ cell states, the grown product consisted of several activated cell states with the largest cell state being the G1-CD8-CD27 cluster. This overall state of activation could be attributed in part to the controlled activation environment provided by the in vitro expansion using the TIL 3.0 method. Finally, our TIL culture process also expanded a population of γδ+ T cells that were present at low frequency in the fresh tissue. Although the culture process was designed to favor CD8+ αβ T cell expansion by using an agonistic 4–1BB antibody, γδ+ T cells can also express 4–1BB and respond to this stimulation. Even so, γδ+ T cells have anti-tumor potential, and thus their presence may add to the effectiveness of the TIL product(65).
In conclusion, this study defines the TIL cell states in PDAC patients, their inferred differentiation trajectory in situ, and their fate during ex vivo TIL culture for application in TIL ACT. Although the observations presented here are consistent with other cancer types, it is important to note that the conclusions presented here are based on a snapshot of a dynamic process. Future studies in the context of immunotherapy-treated PDAC patients with longitudinal combined transcriptomic and T-cell repertoire analysis will be needed to better understand the TIL cell states presented here and their clinical significance for this difficult cancer type. Finally, we expect that the granular knowledge gained from these data will help inform future T-cell targeted therapeutic strategies as well as manufacturing processes for TIL-based strategies.
Methods
Patient sample accrual
After providing written informed consent, 33 patients with primary pancreatic ductal adenocarcinoma underwent surgical resection (Supplementary Table S2). Patients are referred to by their de-identified number. Tissue from surgical resections was used under protocols (PA 15–0176 and PA17–0793) approved by the Institutional Review Board of The University of Texas MD Anderson Cancer Center. This study was conducted in accordance with the Declaration of Helsinki. For the MDA1 and MDA2 data sets, tissue from normal pancreas is referred to as “uninvolved” because it came from PDAC patients. For the PUMCH data set, tissue from control pancreases is referred to as “normal” because it came from patients without malignant pancreatic tumors.
Sample preparation for sequencing
MDA1 cohort:
Fresh tumor samples were cut into 1–3 mm3 fragments and disaggregated using a Medimachine (BD Biosciences, Franklin Lakes, NJ) to create a single-cell suspension in 1x PBS according to manufacturer’s instructions. After disaggregation, CD3+ T cells were isolated for sequencing via magnetic bead separation using the EasySep Release Human CD3 Positive Selection Kit (StemCell Technologies, Vancouver, Canada) according to the manufacturer’s instructions. The enriched cell suspension was collected in 1x PBS containing 2% FBS and 1mM EDTA, enumerated using an automatic cell counter, and sent for oil immersion on the 10X platform. For cultured TIL, previously cryopreserved samples were thawed in warm TIL-CM, washed with 1× PBS, and resuspended at 1×106 cells/mL in 1% bovine serum albumin in 1x PBS before being sent for oil immersion on the 10X platform.
MDA2 cohort:
PDAC tumors were washed with sterile PBS and minced with sterile scalpels in human wash media (DMEM/F12, 0.01M HEPES, 1X GlutaMAX supplement, 10% FBS) until tumor fragments were <1mm3. Tissue suspensions were centrifuged, the supernatant removed and resuspended in dissociation buffer (collagenase II [5mg/ml], dispase [1mg/ml] in human wash media). The samples were placed on a rotation plate in a 37°C incubator for 45 minutes. The single cell solution was then centrifuged and filtered. Single cells were collected as a pellet following centrifugation of the flow-through and resuspended in warm resuspension buffer (1% bovine serum albumin in 1X PBS). Cells were enumerated using a hemocytometer and sent for oil immersion on the 10X platform.
Single-cell RNA/TCR sequencing
Single-cell capture and library construction was performed with the 10x Genomics Chromium Single Cell 5’ kits v.1.0 (product codes 1000014, 1000020, and 1000151) with TCR enrichment (1000005) for cohort MDA1 and 3’ V.2 kits (product codes120237, 120236, 120262) for MDA2, according to the manufacturer’s instructions. Briefly, cells were loaded into the Chromium Single Cell Chip A for a recovery target of 10,000 cells. Reverse transcription was performed on a BioRad T100 thermal cycler, and the barcoded cDNA was purified with Dynabeads (Thermo Fisher Scientific, 37002D) prior to 14 cycles of cDNA amplification. Of this transcriptome cDNA, 2 μL was used for TCR enrichment and subsequent TCR library construction in the MDA1 cohort. Per the manufacturer’s protocol, up to 50 ng (or 20 μL) of transcriptome cDNA was used for single-cell library construction. Transcriptome libraries were sequenced on a HiSeq 400 (Read 1, 26 cycles; Index 1, 8 cycles; Read 2, 91 cycles), and TCR libraries were pooled and sequenced 150 cycles paired-end on a MiSeq (Illumina). For transcriptome libraries, median sequencing depth for each sample was targeted for a median of 30,000 reads/cell; for TCR libraries, 1,000 reads/cell. Read counts can be found in Supplementary Table S3.
Single-cell data processing and filtering
The 10x Genomics Cell Ranger pipeline (v. 3.1.0) was used to demultiplex and generate unique molecular identifier (UMI) matrices for all samples in the MDA1 cohort. UMI matrices for the MDA2 and PUMCH cohorts were also generated from the provided FASTQ files using this version of the Cell Ranger pipeline. The UMI data were then processed in R (v.3.5.3) using the Seurat package (V.3). Cells with more than 6000 genes (doublet removal), fewer than 200 genes, greater than 20% UMI in mitochondrial genes, or a UMI:gene ratio greater than 10 or less than 1.3 were filtered. To filter out non–T cells from the data sets, all samples were combined, the top 5000 variable genes were reduced to 50 top principal components and clustered using Seurat’s SNN clustering function. Clusters with significant average expression (Wilcoxon test) of CD3 genes (i.e., T cells) were then extracted for further analysis. This gene selection, principal component analysis, and clustering step was performed once more to further remove clusters not expressing CD3 genes.
Cohort data integration and dimension reduction
To account for library chemistry, and other possible unknown differences between cohorts, the T cells were integrated employing the methods as described in https://satijalab.org/seurat/v3.0/integration.html. Briefly, the top 5000 variable features were selected with TCR variable genes (“^TR[ABGD][VDJ]”) excluded. These genes were used to compute 20 “anchors” with the FindIntegrationAnchors function, and subsequently “integrated” by cohort with IntegrateData to produce a batch-corrected data set. The first 30 principal components of the integrated expression data were used for subsequent UMAP embedding.
CD4/CD8 classification, clustering, and marker identification
T cells with detectable expression of either CD4 or CD8 genes were assigned to their respective clusters. The first 30 principal components (excluding these genes) from a randomly selected training set of 75% of these cells were used to train a random forest classifier (RandomForest package in R) for CD4+ vs. CD8+ T cells. The remaining 25% were used as a validation set. This classifier was then used to classify T cells with no detectible CD4 or CD8 expression (likely due to gene dropout) as either CD4+ or CD8+ T cells and then further clustered separately. After CD4/CD8 classification and separation, the top 3000 variable features were re-selected for each group and principal components were computed. The top 30 principal components were used to perform clustering with Seurat’s FindNeighbors and FindClusters functions. Top marker genes for each cluster were identified with Seurat’s FindAllMarkers function, and P values were determined by the Wilcoxon test (Supplementary Tables S4 and S5).
RNA Scope materials and methods
We performed RNAscope on 5μM FFPE sections of PDAC and adjacent, uninvolved pancreas tissue using the ACD RNAscope Multiplex Fluorescent kit v2 and 4-Plex ancillary kit (CAT NO: 323100, CAT NO: 323120). Slides were prepared as described in the kit’s user guide (Doc. No. 323100-USM), briefly they were baked at 60˚C for 1 hour in an ACD HybEZ II oven (PN 321710/321720) and then deparaphanized in xylenes and 100% ethanol. Exogenous peroxides were blocked for 10 minutes, using the provided hydrigen peroxide and target retrieval was performed for 10 minutes in the provided retrieval buffer in a 5.5qt Hamilton Beach digital steamer (MODEL: 37530Z). A hydrophobic barrier was applied around the tissue sections, and they were treated with RNAscope protease plus for 30 minutes and washed in water. Probes sets were then applied, and the slides were probed for 2 hrs before washing (5 minutes x2) in the kit’s wash buffer and storage overnight in 5X SSC buffer. Hybridization Amplication, and 4 rounds of HRP development, Opal Dye conjugation, and blocking was performed per the kit’s instructions, washing twice, for 5 minutes with agitation in the provided wash buffer after each step. Slides were then stained with DAPI, washed in PBS, mounted in Invitrogen Prolong Diamond Antifade (Cat. P36965) and dried overnight in the dark. Imaging was performed on a Nikon Eclipse Ti2E microscope (PN MEA54000) using the NIS Elements AR 5.30.05 software as recommended by the manufacturer.
For samples with T cells detected on the initial screen, further sections were then stained for T-cell subpopulations with the following 8 panels: Panel 1 (CD4, CXCR4, CCR7, IFIT1); Panel 2 (CD4, CXCR4, FOXP3, CXCL13); Panel 3 (CD8A, CD4, GZMK, ZNF683); Panel 4 (CD8A, CXCR6, ZNF683, IL7R); Panel 5 (CD8A, GZMK, CXCL13, PRF1); Panel 6 (CD8A, CXCR6, CCR7, IL7R); Panel 7 (CD8A, GZMK, IFIT1, CXCL13); Tumor Panel (KRT19/MUC1, CD3E/CD3D/CD3G, CD8A, CD4). The panels were composed of the following probes from ACDBio: Hs-CD3-pool-C2 (426621-C2), Hs-CD4 (605601), Hs-CD4-C4 (605601-C4), Hs-CXCR4-C3 (310511-C3), Hs-CCR7-C2 (410721-C2), Hs-IFIT1 (415551), Hs-CXCL13-C4 (311321-C4), Hs-PRF1 (407381), Hs-GZMK (475901), Hs-GZMK-C2 (475901-C2), Hs-CXCR6 (468461), Hs-IL7R-C4 (408841-C4), Hs-ZNF683-C2 (586921-C2), Hs-MUC1 (310391), and Hs-KRT19 (310221). The probes were visualized using the following OPAL dye fluorophores from Akoya Biosciences: Opal 520 (FP1487001KT), filter: FITC, color: green, dilution 1:500; Opal 570 (FP1488001KT), filter: Cy3, color: cyan, dilution 1:750; Opal 620 (cat: FP1495001KT), filter: Texas Red, color: red, dilution 1:750; Opal 690 (FP1497001KT), filter: Cy5, color: white, dilution 1:750.
PBMC processing and clustering
Publicly available Single cell RNAseq data from PBMC s from healthy and PDAC patients were obtained from 10X genomics [10K PBMCs from Healthy Donor (v3, 28×91), Single Cell Immune Profiling Dataset by Cell Ranger 3.0.0, 10x Genomics, (2018, November 19th)] and Steele et al(41). The data was filtered and processed as described previously in the manuscript for the other single cell dataset. The T cells were then integrated by cohort (10X PBMCs, Steele-Healthy PBMCs, Steele-PDAC PBMCs) and clustered separately by CD4 and CD8 T cells as described for the other datasets in the manuscript. The top 20 cluster marking genes by p-value (Wilcox test) were used to compare to the similarly selected top marker genes from the tissue T cells.
Pseudotime trajectory analysis
Pseudotime analysis was performed on cells in the CD4 and CD8 populations separately using Monocle 3 (version 0.2.2.0; https://github.com/cole-trapnell-lab/monocle3). Expression data were UMAP embedded using the Monocle function “reduce_dimension” with the default parameters. The trajectory graph was inferred with the function “learn_graph” with the minimal branch length set to 15 and close_loop = FALSE. The CD4 and the CD8 trajectories were rooted in the clusters CD4-CCR7 and CD8-CCR7/IL7R, respectively.
Spearman Correlation of Cluster Frequencies
For all tumor samples with at least 50 T cells, cluster fractions were calculated by dividing the number of cells in a cluster by the total number of T cells in the sample. For each cluster, the cluster fractions across samples were then pairwise spearman correlated to all other cluster fractions using the cor() function in R. The spearman’s Rho value obtained where then hierarchically clustered with Ward.D2 linkage for the graph and tested for being significantly different from 0 using a t-test. P-values were corrected by the Benjamini-Hotchberg method for multiple testing.
TCR analysis and clonotype re-assignment
TCR calls on the single-cell TCR sequencing data were performed with the “cellranger vdj” function of the Cell Ranger software suite (v.3.1.0). Cells with only a TCR alpha or beta chain detectable whose TCR nucleotide sequence exactly matched that of another clonotype with both the alpha and beta chain detectable were re-assigned to that clonotype. Clonotype data was then attached to single cell metadata by exact matches in cell barcodes within a sample.
Visualizing TCR overlap between transcriptomic clusters using Circos plots and Upset plots
TCR Circos plots were created with code adapted from: https://github.com/aislyn/TCR_circos. For each clonotype (defined by unique CDR3 nucleotide sequence), the median expression was calculated across cells, and then hierarchically clustered using Euclidean distance between the medoids and ward.d2 linkage (using hclust function in R). The hierarchal graph is shown at the center of the plot with each leaf/spoke representing one clonotype. For each clonotype, the fraction of cells in that clonotype coming from each patient, and each cluster was calculated and plotted as the inner and outer solid rings, respectively. Finally, the number of cells detected in each clonotype was plotted on the outermost ring of the plot.
For the Upset plots, the number of TCRs existing in a single or multiple states was quantified by filtering for clonotypes with at least 2 cells detected (such that overlap was possible), and then the clonotype was classified by overlap state (e.g., CD8-GZMK only, CD8-ZNF683/CD8-CXCR6/IL7R overlap, etc.). The number of clonotypes observed in each overlapping state was then counted and plotted using the upsetR package in R. In order to quantify whether an overlap state existed more or less frequently than expected by chance (because high frequency clonotypes have more chances to overlap between states by chance than lower frequency ones), we performed a permutation test for our overlap counts by performing the same computation on permutated clonotypes and expression states within patients 1000 times.
TCR Repertoire Analysis
For each patient and cluster with at least 5 clonotypes present in the cluster, the number of cells in the clonotype were tallied and the following indexes were calculated as follows: chao1 species richness (computed with the Chao1 function from the ‘fossil’ package in R), Gini coefficient (computed with the Gini function from the ‘DescTools’ package in R), Diversity Evenness 50 (DE50) was the (frequency-ordered) number of clonotypes necessary to account for 50% of cells divided by the total number of clonotypes. Simpson’s Index was calculated as the sum of the clonotype fractions squared, Shannon Diversity Index was calculated as the negative sum of the clonotype fractions multiplied by their natural log, Pielou’s Evenness was calculated as the Shannon Index divided by the natural log of the number of clonotypes.
Expansion of TIL from tumor samples
Fresh tumor samples were cut into 1–3 mm3 fragments, and five fragments were placed in G-Rex10 flasks (Wilson Wolf, Saint Paul, MN) containing 20 mL of TIL culture media (TIL-CM: RPMI-1640 with GlutaMAX [Gibco/Invitrogen], 1× Pen-Strep [Gibco/Invitrogen], 50 μM 2-mercaptoethanol [Gibco/Invitrogen], 20 μg/mL gentamicin [Gibco/Invitrogen], and 1 mM sodium pyruvate [Gibco/Invitrogen]) with 6000 IU/mL IL-2, 10 μg/mL 4–1BB mAb, and 30 ng/mL anti-CD3 (OKT3) as previously described(49). Four to five days after culture initiation, 20 mL of additional TIL-CM with 6000 IU/mL IL-2 was added for a total volume of 40 mL. Half-media changes were done every 3–4 days with fresh TIL-CM containing 6000 IU/mL IL-2 for up to 35 days or until the cells formed a thick layer completely covering the bottom of the flask. The cell suspensions were collected and cryopreserved in FBS plus 10% DMSO.
TIL culture reagents
A purified, human IgG4 monoclonal antibody (mAb) against human CD137/4–1BB, Urelumab (663513), was kindly provided by Bristol-Myers Squibb (New York, NY). Human recombinant interleukin-2 (IL-2) (Proleukin) was generously provided by Prometheus Therapeutics & Diagnostics (San Diego, CA). GMP-grade soluble anti-CD3 antibody (OKT3 clone) was obtained from Miltenyi Biotec (Bergisch Gladbach, Germany).
Re-assignment in cycling cultured cells
Due to the (intentional) highly proliferating state of the cultured T-cell samples, a large fraction of the T cells formed a cluster of cycling cells. Non-cycling cell clusters were used to train a random forest classifier (similar to CD4/CD8 classification in the fresh T cells), which was used to re-classify cycling cells to the most similar non-cycling cluster.
Data Availability
The data generated in this study are publicly available on Sequence Read Archive (SRA) under BioProject ID PRJNA806978
Supplementary Material
Significance:
To improve the efficacy of immunotherapy in PDAC, there is a great need to understand the PDAC TIL landscape. This study represents a reference of PDAC TIL sub-populations and their relationships, and provides a foundation upon which to base future immunotherapeutic efforts.
Acknowledgments
The authors thank Bristol-Myers Squibb for their generous contribution of the agonistic anti-4–1BB antibody, Urelumab (BMS-663513). Human recombinant interleukin-2 (IL-2) (Proleukin) was generously provided by Prometheus Therapeutics & Diagnostics. Editorial support was provided by Bryan Tutt, Scientific Editor, Research Medical Library, MD Anderson Cancer Center. This paper is dedicated to the memory of Dr. Gauri Varadhachary, who is dearly missed by all her colleagues in the MD Anderson pancreatic cancer team.
Financial Support:
The study was supported by philanthropic contributions to The University of Texas MD Anderson Moon Shots Program and the Sheikh Khalifa bin Zayed Foundation. N.E. Navin is a Grady Saunders Distinguished Professor, an AAAS Fellow, AAAS Wachtel Scholar, Damon-Runyon Rachleff Innovator, Andrew Sabin Fellow and a Jack & Beverly Randall Innovator. This work was supported by grants to N.E. Navin from the NIH National Cancer Institute (RO1CA240526, RO1CA236864), the CPRIT Single Cell Genomics Center (RP180684) and the MD Anderson Sequencing Core Facility Grant (CA016672). This work also supported by grants to M.P Kim from the NIH National Cancer Institute (K08CA218690), the Ben and Rose Cole Foundation, and to T.G. Hughes from the NIH (T32 CA009599).
Footnotes
Conflict of Interest: A.M. receives royalties for a pancreatic cancer biomarker test from Cosmos Wisdom Biotechnology, and this financial relationship is managed and monitored by the UTMDACC Conflict of Interest Committee. A.M. also receives royalties as an inventor on a patent that has been licensed by Johns Hopkins University to Thrive Earlier Detection (an Exact Sciences Company).
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin 2019;69(1):7–34 doi 10.3322/caac.21551. [DOI] [PubMed] [Google Scholar]
- 2.Ryan DP, Hong TS, Bardeesy N. Pancreatic Adenocarcinoma. New England Journal of Medicine 2014;371(11):1039–49 doi 10.1056/NEJMra1404198. [DOI] [PubMed] [Google Scholar]
- 3.Kleeff J, Korc M, Apte M, La Vecchia C, Johnson CD, Biankin AV, et al. Pancreatic cancer. Nat Rev Dis Primers 2016;2:16022 doi 10.1038/nrdp.2016.22. [DOI] [PubMed] [Google Scholar]
- 4.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med 2015;373(1):23–34 doi 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Antonia S, Goldberg SB, Balmanoukian A, Chaft JE, Sanborn RE, Gupta A, et al. Safety and antitumour activity of durvalumab plus tremelimumab in non-small cell lung cancer: a multicentre, phase 1b study. The Lancet Oncology 2016;17(3):299–308 doi 10.1016/s1470-2045(15)00544-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hellmann MD, Rizvi NA, Goldman JW, Gettinger SN, Borghaei H, Brahmer JR, et al. Nivolumab plus ipilimumab as first-line treatment for advanced non-small-cell lung cancer (CheckMate 012): results of an open-label, phase 1, multicohort study. Lancet Oncol 2017;18(1):31–41 doi 10.1016/S1470-2045(16)30624-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med 2015;373(19):1803–13 doi 10.1056/NEJMoa1510665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med 2017;377(26):2531–44 doi 10.1056/NEJMoa1707447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N Engl J Med 2018;378(5):439–48 doi 10.1056/NEJMoa1709866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res 2011;17(13):4550–7 doi 10.1158/1078-0432.CCR-11-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Forget MA, Haymaker C, Hess KR, Meng YJ, Creasy C, Karpinets T, et al. Prospective Analysis of Adoptive TIL Therapy in Patients with Metastatic Melanoma: Response, Impact of Anti-CTLA4, and Biomarkers to Predict Clinical Outcome. Clin Cancer Res 2018;24(18):4416–28 doi 10.1158/1078-0432.CCR-17-3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Royal RE, Levy C, Turner K, Mathur A, Hughes M, Kammula US, et al. Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother 2010;33(8):828–33 doi 10.1097/CJI.0b013e3181eec14c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012;366(26):2455–65 doi 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beatty GL, O’Hara MH, Lacey SF, Torigian DA, Nazimuddin F, Chen F, et al. Activity of Mesothelin-Specific Chimeric Antigen Receptor T Cells Against Pancreatic Carcinoma Metastases in a Phase 1 Trial. Gastroenterology 2018;155(1):29–32 doi 10.1053/j.gastro.2018.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ino Y, Yamazaki-Itoh R, Shimada K, Iwasaki M, Kosuge T, Kanai Y, et al. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br J Cancer 2013;108(4):914–23 doi 10.1038/bjc.2013.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li J, Wang JY, Chen RF, Bai Y, Lu X. The prognostic value of tumor-infiltrating T lymphocytes in ovarian cancer. Oncotarget 2017;8(9):15621–31 doi 10.18632/oncotarget.14919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mlecnik B, Tosolini M, Kirilovsky A, Berger A, Bindea G, Meatchi T, et al. Histopathologic-based prognostic factors of colorectal cancers are associated with the state of the local immune reaction. J Clin Oncol 2011;29(6):610–8 doi 10.1200/JCO.2010.30.5425. [DOI] [PubMed] [Google Scholar]
- 18.Ibrahim EM, Al-Foheidi ME, Al-Mansour MM, Kazkaz GA. The prognostic value of tumor-infiltrating lymphocytes in triple-negative breast cancer: a meta-analysis. Breast Cancer Res Treat 2014;148(3):467–76 doi 10.1007/s10549-014-3185-2. [DOI] [PubMed] [Google Scholar]
- 19.Erdag G, Schaefer JT, Smolkin ME, Deacon DH, Shea SM, Dengel LT, et al. Immunotype and immunohistologic characteristics of tumor-infiltrating immune cells are associated with clinical outcome in metastatic melanoma. Cancer Res 2012;72(5):1070–80 doi 10.1158/0008-5472.CAN-11-3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Horne ZD, Jack R, Gray ZT, Siegfried JM, Wilson DO, Yousem SA, et al. Increased levels of tumor-infiltrating lymphocytes are associated with improved recurrence-free survival in stage 1A non-small-cell lung cancer. J Surg Res 2011;171(1):1–5 doi 10.1016/j.jss.2011.03.068. [DOI] [PubMed] [Google Scholar]
- 21.Togashi Y, Shitara K, Nishikawa H. Regulatory T cells in cancer immunosuppression - implications for anticancer therapy. Nat Rev Clin Oncol 2019;16(6):356–71 doi 10.1038/s41571-019-0175-7. [DOI] [PubMed] [Google Scholar]
- 22.Kurachi M CD8(+) T cell exhaustion. Semin Immunopathol 2019;41(3):327–37 doi 10.1007/s00281-019-00744-5. [DOI] [PubMed] [Google Scholar]
- 23.Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol 2015;15(8):486–99 doi 10.1038/nri3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, et al. Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell 2018;175(4):998–1013 e20 doi 10.1016/j.cell.2018.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Webb JR, Milne K, Watson P, Deleeuw RJ, Nelson BH. Tumor-infiltrating lymphocytes expressing the tissue resident memory marker CD103 are associated with increased survival in high-grade serous ovarian cancer. Clin Cancer Res 2014;20(2):434–44 doi 10.1158/1078-0432.CCR-13-1877. [DOI] [PubMed] [Google Scholar]
- 26.Edwards J, Wilmott JS, Madore J, Gide TN, Quek C, Tasker A, et al. CD103(+) Tumor-Resident CD8(+) T Cells Are Associated with Improved Survival in Immunotherapy-Naive Melanoma Patients and Expand Significantly During Anti-PD-1 Treatment. Clin Cancer Res 2018;24(13):3036–45 doi 10.1158/1078-0432.CCR-17-2257. [DOI] [PubMed] [Google Scholar]
- 27.Pasetto A, Gros A, Robbins PF, Deniger DC, Prickett TD, Matus-Nicodemos R, et al. Tumor- and Neoantigen-Reactive T-cell Receptors Can Be Identified Based on Their Frequency in Fresh Tumor. Cancer Immunol Res 2016;4(9):734–43 doi 10.1158/2326-6066.CIR-16-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Azizi E, Carr AJ, Plitas G, Cornish AE, Konopacki C, Prabhakaran S, et al. Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell 2018;174(5):1293–308 e36 doi 10.1016/j.cell.2018.05.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guo X, Zhang Y, Zheng L, Zheng C, Song J, Zhang Q, et al. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat Med 2018;24(7):978–85 doi 10.1038/s41591-018-0045-3. [DOI] [PubMed] [Google Scholar]
- 30.Lavin Y, Kobayashi S, Leader A, Amir ED, Elefant N, Bigenwald C, et al. Innate Immune Landscape in Early Lung Adenocarcinoma by Paired Single-Cell Analyses. Cell 2017;169(4):750–65 e17 doi 10.1016/j.cell.2017.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Savas P, Virassamy B, Ye C, Salim A, Mintoff CP, Caramia F, et al. Single-cell profiling of breast cancer T cells reveals a tissue-resident memory subset associated with improved prognosis. Nat Med 2018;24(7):986–93 doi 10.1038/s41591-018-0078-7. [DOI] [PubMed] [Google Scholar]
- 32.Tirosh I, Izar B, Prakadan SM, Wadsworth MH 2nd, Treacy D, Trombetta JJ, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016;352(6282):189–96 doi 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang L, Yu X, Zheng L, Zhang Y, Li Y, Fang Q, et al. Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature 2018;564(7735):268–72 doi 10.1038/s41586-018-0694-x. [DOI] [PubMed] [Google Scholar]
- 34.Zheng C, Zheng L, Yoo JK, Guo H, Zhang Y, Guo X, et al. Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell 2017;169(7):1342–56 e16 doi 10.1016/j.cell.2017.05.035. [DOI] [PubMed] [Google Scholar]
- 35.Thommen DS, Koelzer VH, Herzig P, Roller A, Trefny M, Dimeloe S, et al. A transcriptionally and functionally distinct PD-1(+) CD8(+) T cell pool with predictive potential in non-small-cell lung cancer treated with PD-1 blockade. Nat Med 2018;24(7):994–1004 doi 10.1038/s41591-018-0057-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meng Q, Liu Z, Rangelova E, Poiret T, Ambati A, Rane L, et al. Expansion of Tumor-reactive T Cells From Patients With Pancreatic Cancer. J Immunother 2016;39(2):81–9 doi 10.1097/CJI.0000000000000111. [DOI] [PubMed] [Google Scholar]
- 37.Poschke I, Faryna M, Bergmann F, Flossdorf M, Lauenstein C, Hermes J, et al. Identification of a tumor-reactive T-cell repertoire in the immune infiltrate of patients with resectable pancreatic ductal adenocarcinoma. Oncoimmunology 2016;5(12):e1240859 doi 10.1080/2162402X.2016.1240859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sakellariou-Thompson D, Forget MA, Creasy C, Bernard V, Zhao L, Kim YU, et al. 4–1BB Agonist Focuses CD8(+) Tumor-Infiltrating T-Cell Growth into a Distinct Repertoire Capable of Tumor Recognition in Pancreatic Cancer. Clin Cancer Res 2017;23(23):7263–75 doi 10.1158/1078-0432.CCR-17-0831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peng J, Sun BF, Chen CY, Zhou JY, Chen YS, Chen H, et al. Single-cell RNA-seq highlights intratumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res 2019;29(9):725–38 doi 10.1038/s41422-019-0195-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li H, van der Leun AM, Yofe I, Lubling Y, Gelbard-Solodkin D, van Akkooi ACJ, et al. Dysfunctional CD8 T Cells Form a Proliferative, Dynamically Regulated Compartment within Human Melanoma. Cell 2019;176(4):775–89 e18 doi 10.1016/j.cell.2018.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Steele NG, Carpenter ES, Kemp SB, Sirihorachai VR, The S, Delrosario L, et al. Multimodal mapping of the tumor and peripheral blood immune landscape in human pancreatic cancer. Nature Cancer 2020;1(11):1097–112 doi 10.1038/s43018-020-00121-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bassez A, Vos H, Van Dyck L, Floris G, Arijs I, Desmedt C, et al. A single-cell map of intratumoral changes during anti-PD1 treatment of patients with breast cancer. Nat Med 2021;27(5):820–32 doi 10.1038/s41591-021-01323-8. [DOI] [PubMed] [Google Scholar]
- 43.Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, et al. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci Transl Med 2013;5(200):200ra116 doi 10.1126/scitranslmed.3006504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Szabo PA, Levitin HM, Miron M, Snyder ME, Senda T, Yuan J, et al. 2019. doi 10.1101/555557. [DOI] [Google Scholar]
- 45.Clarke J, Panwar B, Madrigal A, Singh D, Gujar R, Wood O, et al. Single-cell transcriptomic analysis of tissue-resident memory T cells in human lung cancer. J Exp Med 2019;216(9):2128–49 doi 10.1084/jem.20190249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Laumont CM, Wouters MCA, Smazynski J, Gierc NS, Chavez EA, Chong LC, et al. Single-cell Profiles and Prognostic Impact of Tumor-Infiltrating Lymphocytes Coexpressing CD39, CD103, and PD-1 in Ovarian Cancer. Clin Cancer Res 2021;27(14):4089–100 doi 10.1158/1078-0432.CCR-20-4394. [DOI] [PubMed] [Google Scholar]
- 47.Reuben A, Zhang J, Chiou SH, Gittelman RM, Li J, Lee WC, et al. Comprehensive T cell repertoire characterization of non-small cell lung cancer. Nat Commun 2020;11(1):603 doi 10.1038/s41467-019-14273-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chiffelle J, Genolet R, Perez MA, Coukos G, Zoete V, Harari A. T-cell repertoire analysis and metrics of diversity and clonality. Curr Opin Biotechnol 2020;65:284–95 doi 10.1016/j.copbio.2020.07.010. [DOI] [PubMed] [Google Scholar]
- 49.Tavera RJ, Forget MA, Kim YU, Sakellariou-Thompson D, Creasy CA, Bhatta A, et al. Utilizing T-cell Activation Signals 1, 2, and 3 for Tumor-infiltrating Lymphocytes (TIL) Expansion: The Advantage Over the Sole Use of Interleukin-2 in Cutaneous and Uveal Melanoma. J Immunother 2018;41(9):399–405 doi 10.1097/CJI.0000000000000230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, Kalia V, et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 2007;27(4):670–84 doi 10.1016/j.immuni.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 51.Li J, He Y, Hao J, Ni L, Dong C. High Levels of Eomes Promote Exhaustion of Anti-tumor CD8(+) T Cells. Front Immunol 2018;9:2981 doi 10.3389/fimmu.2018.02981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yost KE, Satpathy AT, Wells DK, Qi Y, Wang C, Kageyama R, et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat Med 2019;25(8):1251–9 doi 10.1038/s41591-019-0522-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mackay LK, Minnich M, Kragten NA, Liao Y, Nota B, Seillet C, et al. Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science 2016;352(6284):459–63 doi 10.1126/science.aad2035. [DOI] [PubMed] [Google Scholar]
- 54.Smazynski J, Webb JR. Resident Memory-Like Tumor-Infiltrating Lymphocytes (TILRM): Latest Players in the Immuno-Oncology Repertoire. Front Immunol 2018;9:1741 doi 10.3389/fimmu.2018.01741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ganesan AP, Clarke J, Wood O, Garrido-Martin EM, Chee SJ, Mellows T, et al. Tissue-resident memory features are linked to the magnitude of cytotoxic T cell responses in human lung cancer. Nat Immunol 2017;18(8):940–50 doi 10.1038/ni.3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Boddupalli CS, Bar N, Kadaveru K, Krauthammer M, Pornputtapong N, Mai Z, et al. Interlesional diversity of T cell receptors in melanoma with immune checkpoints enriched in tissue-resident memory T cells. JCI Insight 2016;1(21):e88955 doi 10.1172/jci.insight.88955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hanada KI, Zhao C, Gil-Hoyos R, Gartner JJ, Chow-Parmer C, Lowery FJ, et al. A phenotypic signature that identifies neoantigen-reactive T cells in fresh human lung cancers. Cancer Cell 2022;40(5):479–93 e6 doi 10.1016/j.ccell.2022.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bernard V, Semaan A, Huang J, San Lucas FA, Mulu FC, Stephens BM, et al. Single-Cell Transcriptomics of Pancreatic Cancer Precursors Demonstrates Epithelial and Microenvironmental Heterogeneity as an Early Event in Neoplastic Progression. Clin Cancer Res 2019;25(7):2194–205 doi 10.1158/1078-0432.CCR-18-1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Korc M Pathways for aberrant angiogenesis in pancreatic cancer. Mol Cancer 2003;2:8 doi 10.1186/1476-4598-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ouzounova M, Lee E, Piranlioglu R, El Andaloussi A, Kolhe R, Demirci MF, et al. Monocytic and granulocytic myeloid derived suppressor cells differentially regulate spatiotemporal tumour plasticity during metastatic cascade. Nat Commun 2017;8:14979 doi 10.1038/ncomms14979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kok L, Dijkgraaf FE, Urbanus J, Bresser K, Vredevoogd DW, Cardoso RF, et al. A committed tissue-resident memory T cell precursor within the circulating CD8+ effector T cell pool. J Exp Med 2020;217(10):e20191711 doi 10.1084/jem.20191711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.van der Leun AM, Thommen DS, Schumacher TN. CD8(+) T cell states in human cancer: insights from single-cell analysis. Nat Rev Cancer 2020;20(4):218–32 doi 10.1038/s41568-019-0235-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lu YC, Jia L, Zheng Z, Tran E, Robbins PF, Rosenberg SA. Single-Cell Transcriptome Analysis Reveals Gene Signatures Associated with T-cell Persistence Following Adoptive Cell Therapy. Cancer Immunol Res 2019;7(11):1824–36 doi 10.1158/2326-6066.CIR-19-0299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fonseca R, Beura LK, Quarnstrom CF, Ghoneim HE, Fan Y, Zebley CC, et al. Developmental plasticity allows outside-in immune responses by resident memory T cells. Nat Immunol 2020;21(4):412–21 doi 10.1038/s41590-020-0607-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zou C, Zhao P, Xiao Z, Han X, Fu F, Fu L. gammadelta T cells in cancer immunotherapy. Oncotarget 2017;8(5):8900–9 doi 10.18632/oncotarget.13051. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated in this study are publicly available on Sequence Read Archive (SRA) under BioProject ID PRJNA806978