

Abstract
Lysosomes are key degradative compartments of the cell. Transport to lysosomes relies on GlcNAc-1-phosphotransferase-mediated tagging of soluble enzymes with mannose 6-phosphate (M6P). GlcNAc-1-phosphotransferase deficiency leads to the severe lysosomal storage disorder mucolipidosis II (MLII). Several viruses require lysosomal cathepsins to cleave structural proteins and thus depend on functional GlcNAc-1-phosphotransferase. Here, we used genome-scale CRISPR screens to identify Lysosomal Enzyme Trafficking factor (LYSET) as essential for infection by cathepsin-dependent viruses including SARS-CoV-2. LYSET deficiency resulted in global loss of M6P tagging and mislocalization of GlcNAc-1-phosphotransferase from the Golgi complex to lysosomes. Lyset knockout mice exhibited MLII-like phenotypes and human pathogenic LYSET alleles failed to restore lysosomal sorting defects. Thus, LYSET is required for correct functioning of the M6P trafficking machinery, and mutations in LYSET can explain the phenotype of the associated disorder.
One-Sentence Summary:
Genome-scale CRISPR screens identify a key protein in lysosome biogenesis relevant for inherited disease and viral infection.
Viruses have evolved to hijack the cellular endocytic machinery to enter the cell (1). Lysosomal cathepsin proteases mediate the stepwise proteolytic disassembly of reovirus particles, which is essential for infection (2). Cathepsins also cleave viral glycoproteins of several enveloped viruses during viral entry, triggering productive infection. This includes highly pathogenic viruses including the filovirus family member Ebola virus (EBOV) and the SARS-CoV-1, MERS and SARS-CoV-2 coronaviruses (3–6). These viruses are thus sensitive probes of lysosomal function.
LYSET is a cellular factor essential for infection by diverse viruses including SARS-CoV-2
To identify genes critical for reovirus infection, we performed a genome-scale CRISPR–Cas9 screen in human glioblastoma cells (U87MG). In these cells, productive viral infection leads to cell death. After CRISPR-Cas9 mutagenesis with the Brunello library (7), which covers 19,114 genes, cells were infected with reovirus type 3D (ReoT3D). To identify protective gene mutations, we retrieved the guide RNA (gRNA) sequences present in the resistant population and compared them with the unselected population (fig. S1A). In line with its essential role in reovirus entry, the gene encoding cathepsin L (CTSL) was highly enriched in the resistant population (Fig. 1A, and table S1) (8). Consistent with the importance of M6P-dependent lysosomal transport for cathepsin activity (9), genes encoding the α/β (GNPTAB (10)) and γ (GNPTG (11)) subunits of GlcNAc-1-phosphotransferase scored highly, as did site-1 protease (S1P), a Golgi-localized protein required for the activation of GlcNAc-1-phosphotransferase (12). We did not identify M6P receptors (MPR), likely owing to functional overlap between the two M6P receptor types, MPRD and MPRI (13). The second highest hit was TMEM251, a gene encoding a largely uncharacterized transmembrane protein. Based on the results outlined below, we renamed this gene Lysosomal Enzyme Trafficking factor, LYSET. A separate genome-scale CRISPR screen in eHAP cells also identified deletion of GNPTAB and LYSET as highly protective against reovirus infection, corroborating the essentiality of M6P-mediated lysosomal protein sorting and LYSET for reovirus infection (fig. S1B, and table S2). To validate and characterize the role of LYSET, GNPTAB, and S1P in viral infection, we generated clonal knock out (KO) cell lines in U87MG and 293FT cells using CRISPR/Cas9 (fig. S2). Knock out of LYSET, GNPTAB, and S1P resulted in strong protection against cell death following reovirus infection in both cell types (Fig. 1B). Intracellular viral RNA levels and virus production were severely reduced suggesting an early block in viral entry or replication (Fig. 1, C and D). Because GNPTAB, GNPTG and S1P have known roles in the sorting of lysosomal cathepsins, we reasoned that LYSET might act similarly. We first tested whether LYSET deficiency would prevent viral entry of other cathepsin-dependent viruses (3). As a faithful model of Ebola virus (EBOV) entry (14) we used a GFP-encoding vesicular stomatitis virus pseudotyped with the EBOV glycoprotein (VSV-EBOV-GP). While parental 293FT cells were susceptible to infection with VSV-EBOV-GP, as evidenced by a time-dependent increase in GFP fluorescence, LYSET KO cells were highly refractory to infection (Fig. 1E; fig. S3A; and movies S1 and S2). Similar results were obtained using clonal HAP1 LYSET KO cells and pooled knock outs in human skin fibroblasts (fig. S3B–D; and movie S3–S6). SARS-CoV-2 requires activation of its Spike protein during viral entry, which can be mediated by active endosomal/lysosomal cathepsins or by the transmembrane serine protease TMPRSS2 (5). In cells with very low TMPRSS2 expression, cathepsins become essential for SARS-CoV-2 entry. While parental A549-ACE2 cells were highly susceptible to infection by VSV pseudotyped with the SARS-CoV-2 Spike protein, deletion of LYSET strongly reduced infection levels during a time course of infection (fig. S3E, F and movie S7 and S8). To validate these results in the context of the authentic virus, we used an infectious molecular clone of SARS-CoV-2 that expresses the mNeonGreen fluorescent protein (15). Consistent with the results of the pseudotyped viruses, we observed a significant decrease in infection (Fig. 1F, and fig. S3G). In cells expressing TMPRSS2, most SARS-CoV-2 variants preferentially utilize this route of entry (16). However, the recently emerging omicron variant is less efficiently cleaved by TMPRSS2 and more dependent on cathepsin-mediated entry than other variants of concern such as the delta variant (16, 17). We thus tested lentiviral particles pseudotyped with SARS-CoV-2 S variants in cells that expressed or did not express TMPRSS2. Consistent with our results with Spike corresponding to the early Wuhan strain, LYSET KO resulted in decreased entry of both delta and omicron variant pseudotyped viruses in cells that did not express TMPRSS2 (Fig. 1G). In cells expressing TMPRSS2, the delta variant was only moderately affected by LYSET KO while the omicron variant was still strongly dependent on LYSET (Fig. 1G). Thus, our genetic screens identified LYSET as a host factor essential for a diverse group of viruses that depend on endosomal protease activation during their entry.
Fig. 1. LYSET is a critical host factor for multiple viruses that utilize activated cathepsins for entry.
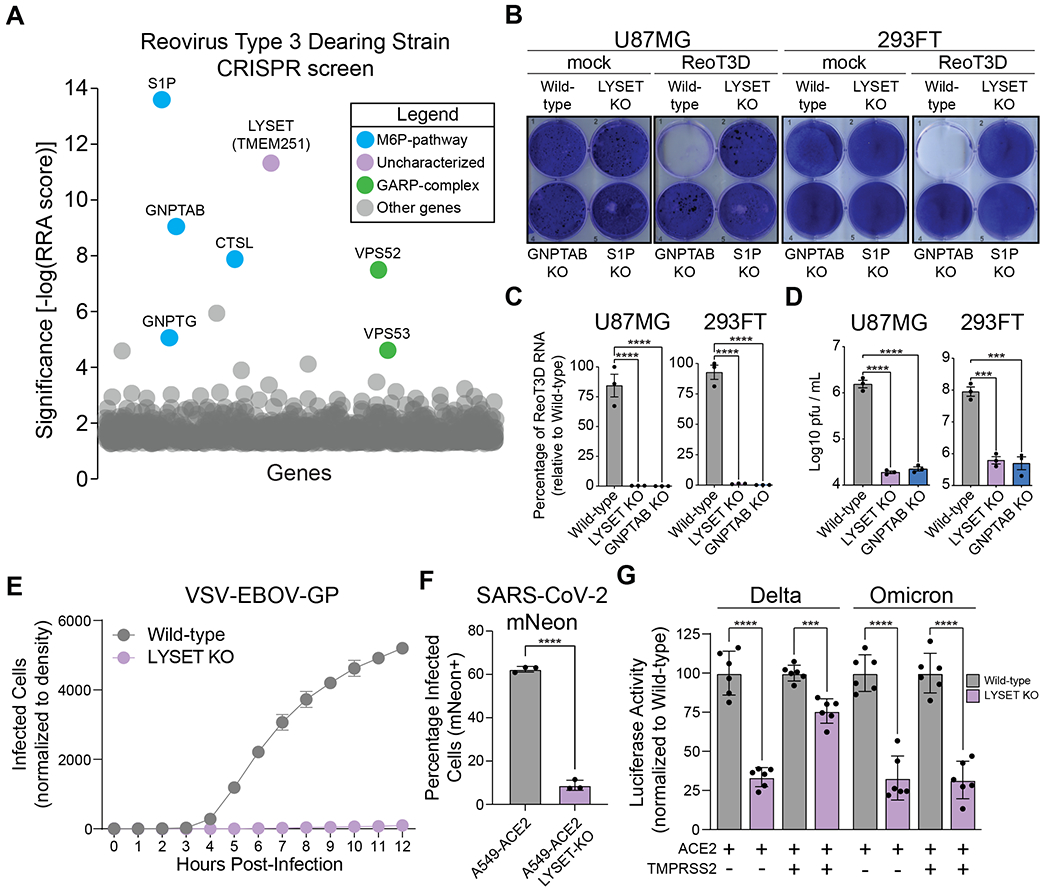
(A) CRISPR screen for reovirus T3D (ReoT3D) host factors in U87MG cells. Significance of enrichment was determined via MAGeCK analysis (y-axis). All genes are plotted as bubbles on the x-axis, each representing a specific gene. A subset is color-coded according to function, labels show gene names. (B) Crystal violet staining after infection with ReoT3D representative of n=3 biologically independent replicates. “mock”: non-infected controls. (C) RT-qPCR quantification of ReoT3D RNA in infected U87MG and 293FT cells at MOI 1 at 72 hpi (mean ± SEM, n= 3) (D) U87MG or 293FT cells were infected with MOI of 1 ReoT3D virus. At 72 hpi, cells were lysed and viral titers determined via plaque assay (mean ± SEM, n = 3, ***p<0.001, ****p<0.0001; significance determined via one-way ANOVA with post-hoc Dunnett’s multiple comparisons test for (C) and (D)). (E) Time course of VSV-EBOV-GP infection of 293FT cells wild-type and LYSET KO (mean ± SEM, n = 3). (F) Bar-graph depicting independent infections of A549-ACE2 cells ± LYSET KO with SARS-CoV-2-mNeon using flow cytometry (mean ± SD, n = 3, ****p<0.0001; significance determined via unpaired, two-tailed student’s t test). (G) Infection of SARS-CoV-2 Delta and Omicron Spike reporter virus particles in Huh7.5.1 cell lines with or without LYSET KO. Cells were engineered to stably express ACE2 or ACE2 in combination with TMPRSS2, as indicated. Luciferase activity was measured at 72 hpi and normalized to wild-type cells (mean ± SEM, n = 6, ***p<0.001, ****p<0.0001; significance determined via significance was determined by unpaired, two-tailed student’s t-test with Welch’s correction).
LYSET is critical for M6P-mediated lysosomal enzyme transport
The identification of a previously uncharacterized transmembrane protein potentially involved in cathepsin sorting was unexpected, because the key proteins involved in lysosomal targeting are well-established (13). To investigate if LYSET impacted lysosomal trafficking of cathepsins, we analyzed cathepsin B (CTSB) protein levels in 293FT and U87MG wild-type and KO cells (Fig. 2A, and fig. S4A). In wild-type cells, the majority of CTSB was present in its mature form following auto-catalytic cleavage in lysosomes. Only low levels were found in the extracellular medium, indicating efficient lysosomal sorting and trafficking. In contrast, LYSET deficiency resulted in aberrant secretion of CTSB precursor forms into the medium concomitant with a near complete loss of mature CTSB in the cell lysate (Fig. 2A, and fig. S4A). The dysregulation of cathepsin transport was as pronounced as observed after disruption of core M6P formation components (GNPTAB and S1P). We extended the analysis to a larger panel of cathepsins in 293FT and HAP1 cells (fig. S4B, C) and measured cathepsin activity using a quenched cell-permeable probe (BMV-109) that covalently links to active cysteine cathepsins and gains fluorescence (18). Compared to wild-type cells, LYSET KO cells displayed a strongly decreased fluorescence after live cell labeling suggesting a global loss of cysteine cathepsin protease activity (Fig. 2B, and fig. S4D, E). This indicated a severe defect in lysosomal protein targeting because activity requires auto-catalytic cleavage in lysosomes. To investigate whether the missorting was specific to cathepsins or pointed to a more general defect, we analyzed the consequences of LYSET deficiency for additional lysosomal proteins and in different cellular contexts. We consistently found large increases in secretion of typical soluble lysosomal proteins into the extracellular medium, while their intracellular protein levels and proteolytic maturation were strongly decreased in KO cells generated in primary human fibroblasts, U87MG, 293FT, HAP1 and SK-MEL-30 cells (fig. S5). In addition, we observed an increase in LC3B type II levels associated with autophagic membranes in cell lysates (fig. S5C). Autophagy markers or the accumulation of an autophagosome-specific dye were markedly elevated in LYSET and GNPTAB KO cells compared to wild-type cells under basal conditions and not further elevated by blocking lysosomal hydrolase activities with chloroquine (fig. S6A) or a combination of chloroquine and rapamycin (fig. S6B). This was expected because the content of autophagic vesicles is degraded by lysosomal hydrolases upon fusion with lysosomes (19). To more comprehensively assess the effect of LYSET knockout on a primary cell type, we used quantitative proteomic approaches in mouse embryonic fibroblasts (MEFs). We first generated C57BL/6J mice with deletions in Lyset by gene editing. We designed sgRNAs to excise a region in the second exon of Lyset resulting in two mouse lines with distinct out of frame deletion variants (of Δ184 and Δ199 nucleotides) (fig. S7, A–C and E). To generate mice with Lyset gene deletions (referred to as Lyset KO), mice were bred as compound heterozygotes (Δ184/Δ199) or homozygotes. We verified loss of Lyset protein expression in different tissues (fig. S7D). To investigate the extracellular proteome, we collected and concentrated serum-free conditioned medium from wild-type and Lyset KO MEFs. In parallel, we prepared cell lysates to determine the intracellular proteome. Using data independent acquisition (DIA) mass spectrometry (fig. S8A (20)), we detected and quantified more than 4,000 proteins in the intracellular proteome and more than 1,000 proteins in the secretome, showing a high correlation between replicates (fig. S8, B–E, and table S3). Only a small subset of proteins were differentially expressed in Lyset KO versus wild-type cells. The majority of proteins found in higher abundance in the secretome of Lyset KO MEFs were well-characterized luminal lysosomal proteins (indicated in red), while these proteins were depleted from the intracellular proteome (Fig. 2C, and fig. S9A). Protein levels of two lysosomal enzymes known to traffic independently from M6P (Gba and Acp2) were unaffected (Fig. 2C), which we confirmed using specific activity assays (fig. S10). Moreover, membrane lysosomal proteins (indicated in blue) were unaffected. Similar results were obtained using parallel reaction monitoring (PRM), a targeted method that provides more sensitive quantification of a smaller subset of lysosomal proteins (fig. S9B (20)). Thus, LYSET deficiency results in a severe defect in lysosomal trafficking that is specific for M6P cargoes.
Fig. 2. LYSET is required for global lysosomal enzyme transport via M6P-tagging.
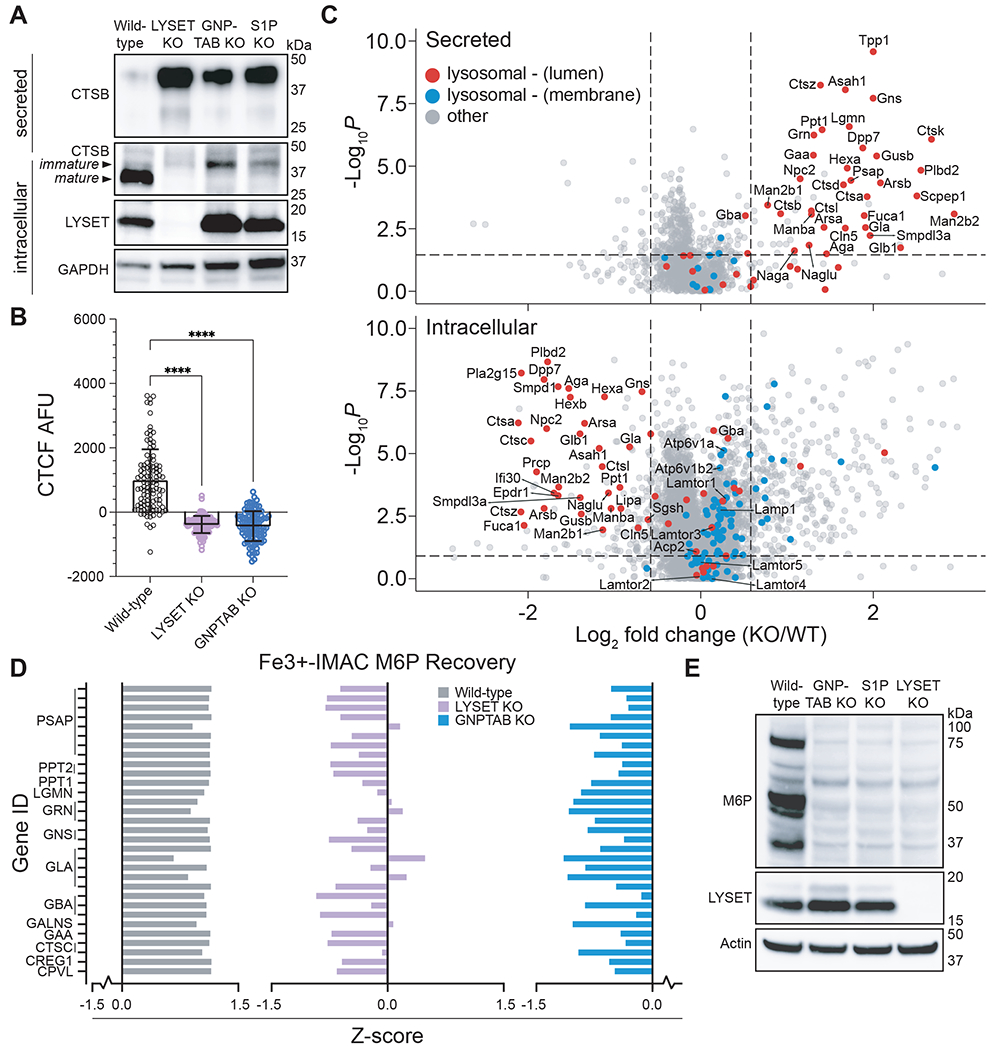
(A) Immunoblot analysis of cathepsin B (CTSB) in lysates (intracellular) and in extracellular medium (secreted) from wild-type cells and clonal 293FT cell lines containing knockout (KO) mutations in indicated genes. (B) Cathepsin activity in cells was determined by quantification of BMV-109 fluorescence signal in live 293FT wild-type and KO cells as the raw corrected total cellular fluorescence (CTCF) in arbitrary fluorescence units (AFU) (mean ± SD, n = 100 cells, **** p<0.0001; significance determined via one-way ANOVA with a post-hoc Dunnett’s multiple comparisons test). (C) Proteomic analysis of wild-type and LYSET KO MEFs by unbiased data independent acquisition (DIA). DIA was used for intracellular and secreted proteins. (D) Z-score analysis of individual peptides that contain the M6P moiety as determined using glycoproteomics. Peptides were derived from indicated lysosomal proteins in wild-type, LYSET KO and GNPTAB KO 293FT cells; n = 3 replicates for each cell line. (E) Immunoblot analysis of M6P-tagged proteins from 293FT wild-type, GNPTAB KO, S1P KO, and LYSET KO cells using an M6P-specific single-chain antibody fragment (M6P).
Key steps in lysosomal enzyme sorting are the modification with M6P by GlcNAc-1-phosphotransferase in the Golgi apparatus and the subsequent binding to M6P-specific receptors that mediate the transport to the lysosome (13). To distinguish between an early defect in tagging or a later defect in M6P recognition, we used an unbiased proteomic approach to detect M6P modifications directly on glycoproteins isolated from wild-type cells and cells deficient in LYSET. This method, based on immobilized metal ion affinity chromatography (Fe3+-IMAC), allows for enrichment of glycopeptides containing the negatively charged M6P modification followed by glycopeptide identification using mass spectrometry (fig. S11A) (21)). In wild-type 293FT cells we readily identified glycopeptides containing M6P from canonical lysosomal enzymes (Fig. 2D; fig. S11B; and table S4). As expected, these M6P glycopeptides were absent in cells lacking GNPTAB. The LYSET KO cells displayed a similar absence of M6P modified glycopeptides. This was confirmed in an orthologous assay using a single-chain M6P-specific antibody fragment (22). Lysates of 293FT and HAP1 cells devoid of LYSET showed a large decrease in M6P immunoreactive glycoproteins, comparable to decreases observed with GNPTAB KO and S1P KO cells (Fig. 2E, and fig. S11C). Thus, LYSET is essential for an early step in lysosomal enzyme transport --the generation of the M6P modification.
LYSET binds to GNPTAB and regulates its function by mediating proper Golgi localization
LYSET is a small, poorly characterized protein containing two transmembrane domains. It co-localizes with GNPTAB in the Golgi apparatus (Fig. 3A–C and fig. S12A–C). We performed immunoprecipitation (IP) followed by immunoblotting experiments to test if LYSET interacts with GNPTAB. After co-expression of epitope-tagged LYSET and GNPTAB we detected GNPTAB-MYC in the LYSET-FLAG IP and LYSET-FLAG in the GNPTAB-MYC IP (Fig. 3D). These reciprocal co-IPs appeared to be specific because the endogenous Golgi protein Giantin was not detected, nor were LYSET or GNPTAB found in control IPs with epitope-tagged mNeonGreen. This interaction suggests that LYSET plays a role in modulating the function of the GlcNAc-1-phosphotransferase complex. Because pathogenic mutations in GNPTAB have been reported that lead to mislocalization of the GlcNAc-1-phosphotransferase complex (23, 24), we hypothesized that LYSET might function by retaining GNPTAB in the Golgi apparatus. We examined this by immunofluorescence analysis for endogenous GNPTAB and co-staining with antibodies for the Golgi apparatus (GM130) and lysosomes (LAMP2). The staining pattern in LYSET KO HAP1 cells was clearly distinct from wild-type cells. While in wild-type cells GNPTAB was localized predominantly in the Golgi apparatus, LYSET KO resulted in a loss of Golgi co-localization and a gain in localization to lysosomal structures (Fig 4A). To investigate this further, we characterized the expression of the endogenous GlcNAc-1-phosphotransferase complex in subcellular fractions. We used sequential centrifugation to isolate a 20K fraction enriched in lysosomes and a 100K fraction enriched in ER/Golgi membranes (25). LYSET deficiency resulted in a near complete loss of endogenous GlcNAc-1-phosphotransferase α-subunit protein levels in the 100K fraction (Fig. 4B). This loss was post-transcriptional because GNPTAB mRNA levels did not substantially differ between wild-type and LYSET KO cells (fig. S13). Inhibition of proteasomal degradation with epoxomicin did not rescue expression of GNPTAB in LYSET KO cells (fig. S14A, B). In contrast, preventing lysosomal degradation by blocking organellar acidification (bafilomycin A1 or by protease inhibition (PI; E64d/leupeptin/pepstatin A) increased GNPTAB protein levels specifically in the lysosome-enriched 20K fraction (Fig. 4C, fig. S14C–E). Moreover, upon bafilomycin treatment we observed immunoreactive bands of higher electrophoretic mobility likely corresponding to partial cleavage products (Fig. 4C), which was also observed in magnetite-purified lysosomes (fig. S14F). These results suggest that GNPTAB is degraded in lysosomes. Immunofluorescence microscopy in LYSET KO cells revealed that the majority of endogenous GNPTAB remained co-localized with a lysosomal marker upon treatment with both inhibitors (fig. S14G, H). However, in bafilomycin- but not PI-treated cells some GNPTAB co-localized with the Golgi apparatus (fig. S14G, H). The latter could be because of a disturbance of the pH within the Golgi apparatus by bafilomycin that could lead to aberrant post Golgi trafficking (26).These data support a model in which LYSET interacts with GNPTAB and is important for proper localization of GNPTAB in Golgi stacks. In the absence of LYSET, GNPTAB is mislocalized to the lysosomes where it is degraded.
Fig. 3. LYSET binds to GNPTAB and co-localizes with GNPTAB/GNPTG in Golgi apparatus cisternae.
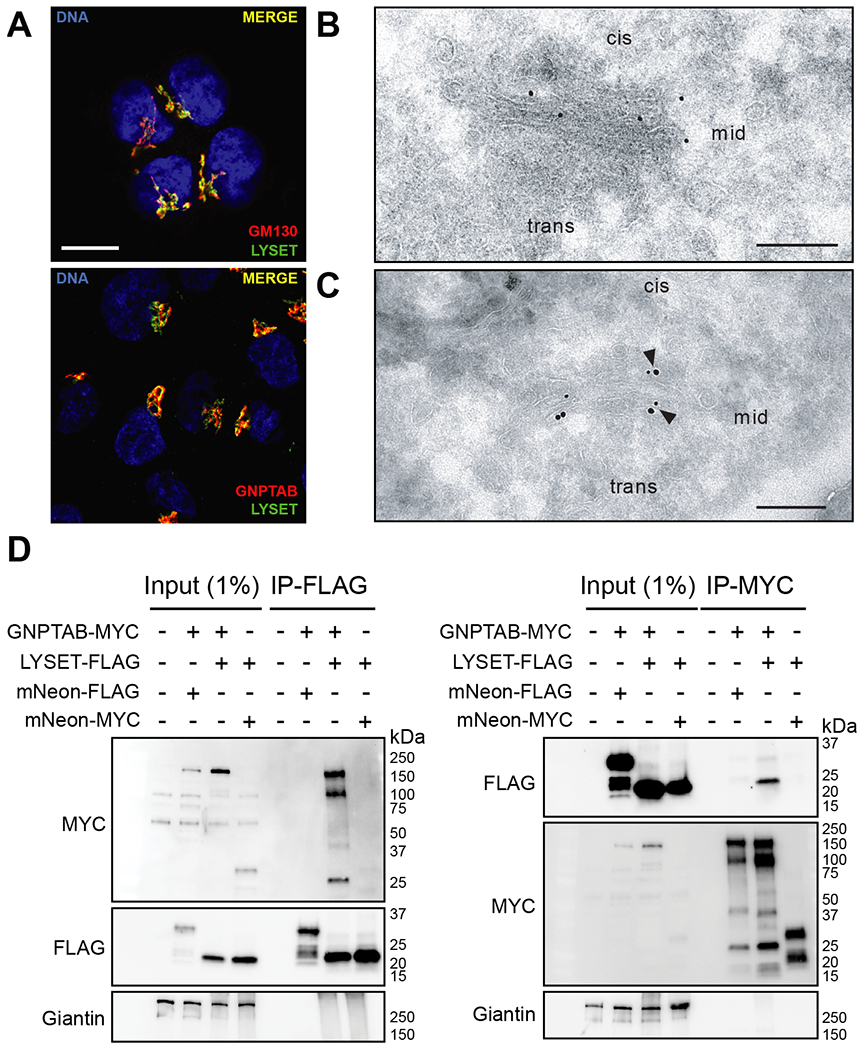
(A) Immunofluorescence microscopy in HAP1 cells showing co-localization of LYSET with the cis-Golgi marker GM130, and GNPTAB. Scale bar, 10 μm (B) Transmission electron microscopy (TEM) immunogold staining (15 nm gold) shows LYSET in Golgi cisternae of SK-MEL-30 wild-type cells using ultrathin sections. (C) TEM double immunogold staining on ultrathin sections for LYSET (15 nm gold) and GNPTG (10 nm gold) in the Golgi apparatus of SK-MEL-30 wild-type cells. Arrowheads indicate co-localization. Scale bar, 200 nm. (D) Immunoprecipitation (IP) on lysates of cells expressing epitope-tagged proteins using anti-FLAG (left panel) or anti-MYC (right panel) magnetic beads, followed by immunoblot analysis with indicated antibodies. Input lysates are also analyzed.
Fig. 4. LYSET KO triggers mislocalization of GNPTAB to the lysosome where it is degraded.
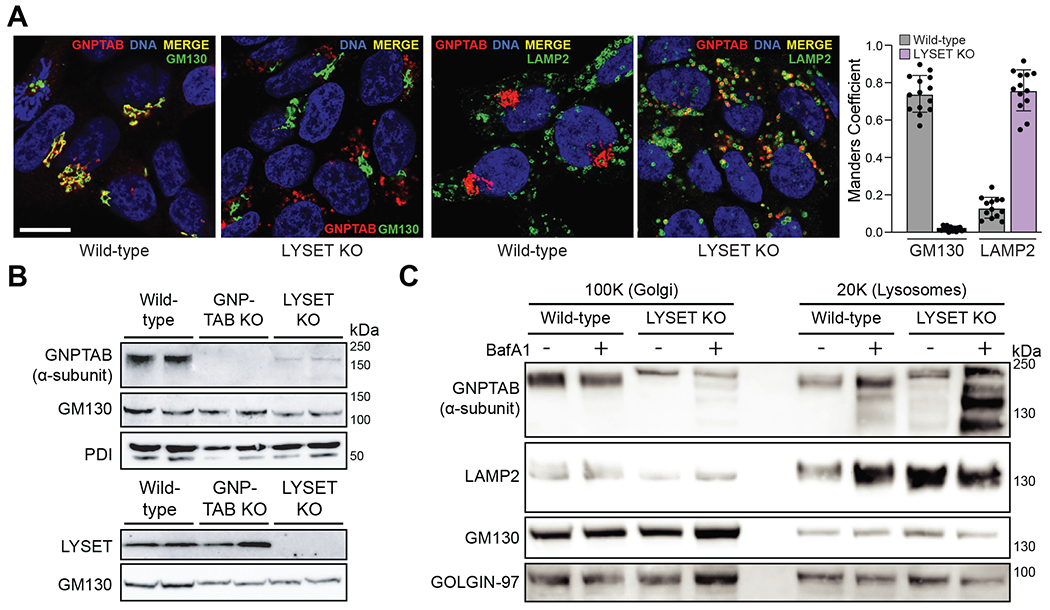
(A) Colocalization of GNPTAB with GM130 and LAMP2 in wild-type and LYSET KO cells and Manders colocalization quantification. Colocalization analysis was performed on at least n = 12 separate micrograph images. Scale bar, 10 μm (B) Immunoblot analysis of GNPTAB in 100K enriched ER/Golgi-fractions from wild-type, GNPTAB KO, and LYSET KO HAP1 cells. The antibody is specific for the α-subunit domain of GNPTAB. Golgi-marker (GM130) and ER-marker (PDI) proteins were used as loading controls. (C) Immunoblot analysis of 20K and 100K fractions from wild-type and LYSET KO HAP1 cells with or without bafilomycin A1 (BafA1) treatment. LAMP2, GM130 and GOLGIN-97 proteins were used as controls for preparation and loading.
LYSET’s role in lysosomal transport provides a disease mechanism for a previously described genetic disorder
It has long been recognized that mutations in genes encoding the core components of lysosomal enzyme trafficking cause specific inherited syndromes including mucolipidosis II (MLII) in which defective GNPTAB mutations are etiological (27, 28). Recently, biallelic LYSET mutations have been described in individuals from two families with a recessive genetic disorder with characteristics of mucopolysaccharidoses/mucolipidosis (29). However, the mechanistic basis to link LYSET with disease was tentative. Our results suggest that defects in lysosomal protein sorting caused by LYSET deficiency might underlie this genetic disorder. Elevated serum level of lysosomal enzymes is a defining feature of MLII. The measurement of these enzymes in serum is used diagnostically to distinguish MLII from other metabolic diseases that cause similar clinical features (28). Compared to wild-type mice, serum from Lyset KO mice showed markedly higher enzyme activities of all tested lysosomal enzymes including α-mannosidase, β-hexosaminidase, α-L-fucosidase and β-galactosidase (Fig. 5A). Furthermore, electron microscopy (EM) analysis of Lyset KO mouse embryonic fibroblasts (MEFs) showed obvious morphological changes of lysosomes (Fig. 5B). Lysosomes with electron dense material were accumulated in the cytoplasm, another characteristic feature of lysosomal storage disorders (28). Quantitative analysis of EM images revealed that lysosomes were significantly more numerous and larger in Lyset KO MEFs while the cell area was not significantly different compared to wild-type cells (Fig. 5C, and fig. S15A, B). In line with this, aberrant lysosomes were also observed in human cells lacking LYSET (fig. S16). In addition, Lyset KO MEFs were resistant to infection by VSV-EBOV-GP, reinforcing our results in human cell lines (fig. 5D, and movie S9 and S10). Despite showing characteristic MLII phenotypes including increased lysosomal enzyme serum levels and storage materials in lysosomes, Lyset KO mice did not display obvious clinical symptoms observed in human patients with LYSET mutations. Similarly, the Gnptab KO mouse is not as deleterious as in MLII disease (30).
Fig. 5. Lyset KO mice display characteristics used to diagnose ML-II disease including elevated blood serum levels of lysosomal enzymes and aberrant lysosomes in isolated cells.
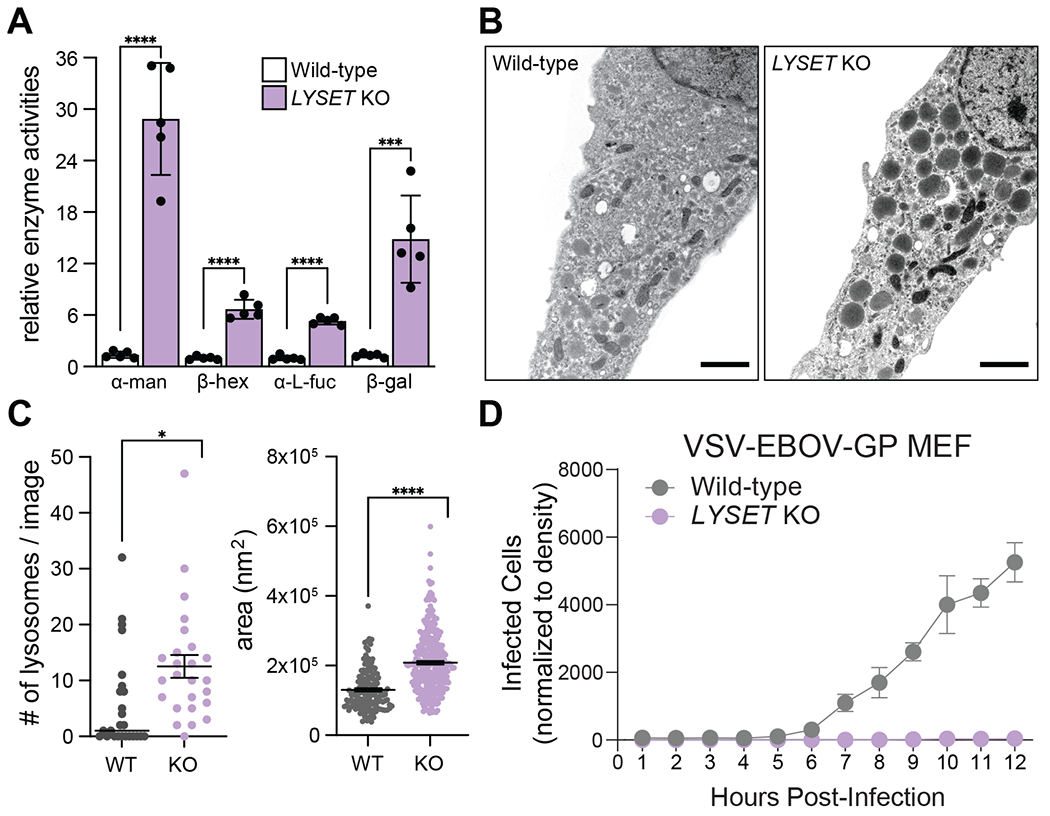
(A) Relative enzyme activities of α-mannosidase (α-man), β-hexosaminidase (β-hex), α-L-fucosidase (α-L-fuc) and β-galactosidase (β-gal) from blood sera of adult wild-type (WT) and Lyset KO mice; WT activities set to 1; mean ± SD, n = 5 mice, ***p<0.001, ****p<0.0001, unpaired student’s t test (two-tailed). (B) Electron micrographs of WT and Lyset KO MEFs. Scale bar, 1 μm. (C) Numbers of lysosomes and areas counted for 25 independent images for WT and Lyset KO (KO) MEFs; mean ± SEM; unpaired, two-tailed student’s t test with Welch’s correction; *P<0.05, ****P< 0.0001. (D) Time course of VSV-EBOV-GP infection of WT and KO MEFs (mean ± SD, n = 3)
To link the human patient mutations in LYSET (29) directly to lysosomal protein sorting we first established that lentiviral complementation of wild-type LYSET (isoform 1 and isoform 2) could restore CTSB missorting and maturation in 293FT LYSET KO cells (fig. S17). Subsequently, we tested complementation by LYSET containing the Y72Ter or R45W pathogenic mutations (29). As controls we used wild-type LYSET as well as three nonsynonymous LYSET variants not associated with disease, which are frequent in the population. R45W displayed slightly lower protein expression levels. As expected, Y72Ter was not detected because this frameshift mutation leads to a premature stop codon. While all controls corrected CTSB missorting, Y72Ter and R45W failed to do so (Fig. 6A). Similarly, R45W expression did not rescue the lysosomal storage defects observed using electron microscopy in LYSET KO cells (fig. S18). Moreover, the pathogenic allele R45W did not rescue the loss of endogenous GNPTAB in 100K fractions (Fig. 6B). Co-immunoprecipitation experiments showed a severe defect of R45W in binding with GNPTAB (Fig 6C and fig. S19). Because R45W fails to rescue the lysosomal trafficking defect, these results suggest that the interaction of LYSET with GNPTAB is critical for GNPTAB function and that mutations in LYSET that affect this interaction can contribute to disease development. Thus, LYSET deficiency causes defining features of mucolipidosis II and patient mutant alleles are compromised in the role of LYSET in M6P-dependent lysosomal protein sorting.
Fig. 6. Pathogenic LYSET mutations fail to restore lysosomal transport defects in LYSET KO cells.
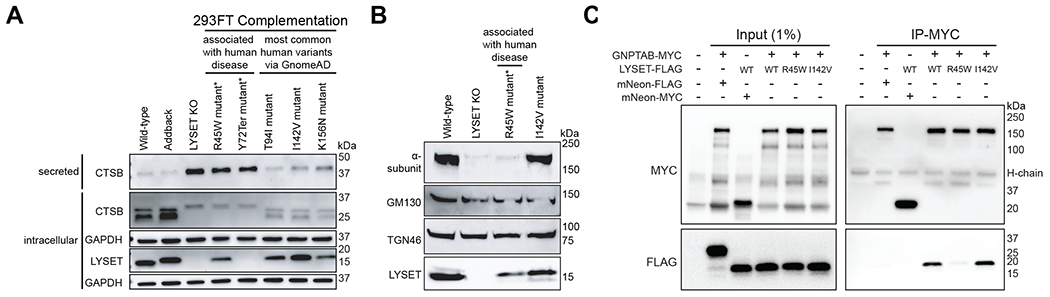
(A) Immunoblot of 293FT lysates or supernatants from LYSET KO cells complemented with wild-type LYSET (Addback) or human variants. R45W and Y72Ter are patient-mutations (22); T94I, I142V and K156N variants are not disease-associated. (B) Immunoblots of 100K enriched-Golgi/ER fractions from 293FT KO cells complemented with indicated LYSET alleles. (C) Immunoprecipitation (IP) on lysates of cells expressing epitope-tagged proteins using anti-MYC magnetic beads followed by immunoblot analysis with indicated antibodies.
Conclusion
Here, we establish LYSET as a Golgi-resident protein essential for M6P-mediated lysosomal protein trafficking. Our results support a model in which LYSET is essential for the activity of GlcNAc-1-phosphotransferase by binding to it and retaining it in the Golgi apparatus. LYSET is relevant to human disease because patients with biallelic LYSET mutations suffer a genetic inherited disorder (29). Based on our elucidation of the role of LYSET in cell physiology, we propose that this disorder is similar to MLII. Because the clinical symptoms of mucolipidosis and mucopolysaccharidosis overlap and not all cases can be explained by mutations in known disease genes, LYSET sequencing may help to identify disease-causing mutations in remaining patients. Furthermore, as an important component of lysosomal function, LYSET is essential for infection by diverse highly pathogenic viruses that rely on endo-lysosomal activation by cathepsins.
Materials and methods summary
A detailed version of the materials and methods is provided in the supplementary materials. In brief, genome-scale CRISPR/Cas9 knockout libraries were generated in eHAP and U87MG cells. Libraries were infected with reovirus type 3D to select for a cell population containing mutations that confer resistance to viral infection. After genomic DNA isolation, PCR was used to amplify sequences encoding guide RNAs and their abundance was quantified using next generation sequencing. Statistical analysis was performed to identify and rank genes that were enriched in the selected population compared to the uninfected population.
LYSET KO mutations were introduced in different human cell lines using CRISPR/Cas9. Cells were infected with reovirus type 3D and viral infection was assessed using crystal violet assay, RT-qPCR and plaque forming assay. For infections with VSV pseudotyped with Ebolavirus GP or SARS-CoV-2 S, viral entry was assayed using live cell imaging with an Incucyte system to monitor VSV-encoded GFP expression.
The endogenous expression of proteins involved in M6P lysosomal protein transport was determined in extracellular medium, intracellular lysates and subcellular organelle fractions in wild-type and KO cells using immunoblotting, lysosomal enzyme activity assays and mass-spectrometry. Binding between LYSET and GNPTAB was determined using co-immunoprecipitations from lysates of cells transfected with plasmids encoding epitope-tagged proteins.
All experiments involving mice were approved by Stanford’s Institutional Animal Care and Use Committee. C57BL/6J zygotes were pronuclear injected with Cas9 RNPs targeting Lyset (Tmem251) with two synthetic gRNAs to introduce a frameshift mutation.
For electron microscopy, cells were fixed, osmicated, and Epon polymerized. Ultrathin sections (60 nm) were prepared and examined in an EM902 microscope. For post-embedding immunogold labeling, ultrathin sections (60 nm) were prepared from cryoprotected cell pellets (2.3 M sucrose), collected on Carbon-Formvar-coated nickel grids, and incubated with one or two antibodies followed by protein A-coupled to colloidal gold particles. Images were acquired with a JEM- 2100Plus Transmission Electron Microscope.
For immunofluorescence, cells treated or not for 24 h with lysosomal protease inhibitors (Leupeptin, Pepstatin A and E64d) or bafilomycin A1 were fixed with 4% PFA, blocked and permeabilized in BSA/saponin buffer followed by incubations with primary antibodies, Alexa Fluor-coupled secondary antibodies and Hoechst 33342. Images were acquired using a LSM880 confocal microscope equipped with an Airyscan detector. For in situ labeling of glucocerebrosidase (GBA) GBA inhibody MDW933 was used prior to fixation. BMV-109 probes were applied for live cell imaging of cysteine cathepsin activities on an Axiovert 200 M confocal microscope. Autophagic vacuole accumulation was visualized using the CYTO-ID Autophagy detection kit. All images were processed using ImageJ (https://imagej.nih.gov/ij/notes.html). The Manders colocalization coefficient was determined by means of the JACoP plug-in.
For mass-spectrometry, proteins were extracted from cell lysates and conditioned media by chloroform/methanol, digested by trypsin in the presence of RapiGest, and the concentration of desalted samples was determined using a peptide assay. For each sample, 1 μg of peptide was analyzed by UHPLC-MSMS using self-packed columns in combination with a Dionex Ultimate 3000 nano-UHPLC system coupled to an Orbitrap Fusion Lumos mass spectrometer. Data acquisition was performed using parallel reaction monitoring (PRM), with a previously established assay for the analysis of lysosomal proteins, and data independent acquisition (DIA) for both sample types. Raw files were analyzed by Skyline (PRM) and Spectronaut (DIA) with determination of significantly regulated proteins based on fold-change and p/q-values. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD029609.
For glycoproteomics, cells were lysed and protein digestion was performed using an S-trap mini column protocol. Peptides were then enriched with immobilized metal affinity chromatography with iron-functionalized magnetic beads. Subsequently, peptide mixtures were separated over an EasySpray reversed phase LC column. The mobile phases were driven and controlled by a Dionex Ultimate 3000 RPLC nano system. An integrated loading pump was used to load peptides onto a trap column, which was put in line with the analytical column. Eluted peptides were analyzed on an Orbitrap Fusion Tribrid MS system. All data were searched with Byonic using the entire human proteome downloaded and an N-glycan database of 183 N-glycans that included M6P glycans. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD029746.
Supplementary Material
Acknowledgments:
The authors thank Allison Dupzyk, Benjamin Waldman, Alex Johnson and Christopher LaPointe for insightful comments. We thank Mike Bassik and Denise Monack for access to their respective Incucyte microscopes, Emanuela Szpotowicz, Chudamani Raithore and Jane Rehberg for technical assistance, and Robert Hardt for support with DIA and PRM data analysis and visualization. We thank Fabienne Seyfried, Gudrun Dubberke, and Friedrich Koch-Nolte, Institute of Immunology, UKE, Hamburg, for assistance with the production of mAb RG95-A96. We thank Dr. Wilhelm Palm for sharing unpublished data. Some images were created with BioRender.com.
Funding:
This work was funded in part by the National Institutes of Health (NIH) T32 AI007502 (C.M.R.), NIH T32 GM007276 (L.D.V.), NIH T32 AI007502 (A.R.), NIH R01 AI140186 (J.E.C.), NIH R01 AI141970 (J.E.C.), NIH R01 AI130123 (J.E.C.), NIH R01 GM058867 (C.R.B.), NIH R01 AI134907 (P.-Y.S), NIH K00CA21245 (N.M.R.), Deutsche Forschungsgemeinschaft (DFG) CRC877 (T.B., M.S.), DFG FOR2625 TP7 (P.R.M., D.W., T.B.), DFG 240245660-SFB1129 (R.B.), DFG 272983813-TRR 179 (R.B.), Helmholtz Association’s Initiative and Networking Fund KA1-Co-02 “COVIPA” (R.B.), Burroughs Wellcome Fund Investigator Investigators in the Pathogenesis of Infectious Disease (J.E.C.), Stanford ChEM-H Innovative Medicines Accelerator (J.E.C), Sealy Smith Foundation (P.-Y.S), Kleberg Foundation (P.-Y.S), John S. Dunn Foundation (P.-Y.S), Amon G. Carter Foundation (P.-Y.S), Gillson Longenbaugh Foundation (P.-Y.S), and Summerfield Robert Foundation (P.-Y.S).
Footnotes
Competing interests: J.E.C. consulted for Janssen BioPharma on topics unrelated to this study. C.R.B. is a co-founder and Scientific Advisory Board member of Lycia Therapeutics, Palleon Pharmaceuticals, Enable Bioscience, Redwood Biosciences (a subsidiary of Catalent) and InterVenn Biosciences. C.A.B. reports compensation for consulting and/or SAB membership from Catamaran Bio, DeepCell Inc., Immunebridge, Sangamo Therapeutics, Bicycle Tx, and Revelation Biosciences on topics unrelated to this study. The remaining authors declare no competing interests.
Data and materials availability:
All data presented are available in the main text or the supplementary materials. FASTQ files for CRISPR screens are available at ArrayExpress (Accession# E-MTAB-11158). Acquired glycoproteomics data are available at ProteomeXchange with identifier PXD029746 and DIA and PRM data with identifier PXD029609. Cell lines and LYSET knockout mice generated in the laboratory of J.E.C. are available upon request to J.E.C. under a material transfer agreement from Stanford University. Huh7.5.1 derivatives require an additional material transfer agreement from Apath.
References and Notes
- 1.Mercer J, Schelhaas M, Helenius A, Virus entry by endocytosis. Annual review of biochemistry 79, 803–833 (2010). [DOI] [PubMed] [Google Scholar]
- 2.Mainou BA, Dermody TS, In search of cathepsins: how reovirus enters host cells. DNA and cell biology 31, 1646–1649 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chandran K, Sullivan NJ, Felbor U, Whelan SP, Cunningham JM, Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science (New York, N.Y.) 308, 1643–1645 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shirato K, Kawase M, Matsuyama S, Middle East respiratory syndrome coronavirus infection mediated by the transmembrane serine protease TMPRSS2. Journal of virology 87, 12552–12561 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hoffmann M et al. , SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 181, 271–280.e278 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Simmons G et al. , Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proceedings of the National Academy of Sciences of the United States of America 102, 11876–11881 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Doench JG et al. , Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nature biotechnology 34, 184–191 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ebert DH, Deussing J, Peters C, Dermody TS, Cathepsin L and cathepsin B mediate reovirus disassembly in murine fibroblast cells. The Journal of biological chemistry 277, 24609–24617 (2002). [DOI] [PubMed] [Google Scholar]
- 9.Mach L, Biosynthesis of lysosomal proteinases in health and disease. Biological chemistry 383, 751–756 (2002). [DOI] [PubMed] [Google Scholar]
- 10.Tiede S et al. , Mucolipidosis II is caused by mutations in GNPTA encoding the alpha/beta GlcNAc-1-phosphotransferase. Nature medicine 11, 1109–1112 (2005). [DOI] [PubMed] [Google Scholar]
- 11.Raas-Rothschild A et al. , Molecular basis of variant pseudo-hurler polydystrophy (mucolipidosis IIIC). The Journal of clinical investigation 105, 673–681 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marschner K, Kollmann K, Schweizer M, Braulke T, Pohl S, A key enzyme in the biogenesis of lysosomes is a protease that regulates cholesterol metabolism. Science (New York, N.Y.) 333, 87–90 (2011). [DOI] [PubMed] [Google Scholar]
- 13.Braulke T, Bonifacino JS, Sorting of lysosomal proteins. Biochimica et biophysica acta 1793, 605–614 (2009). [DOI] [PubMed] [Google Scholar]
- 14.Takada A et al. , A system for functional analysis of Ebola virus glycoprotein. Proceedings of the National Academy of Sciences of the United States of America 94, 14764–14769 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xie X et al. , An Infectious cDNA Clone of SARS-CoV-2. Cell host & microbe 27, 841–848.e843 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hui KPY et al. , SARS-CoV-2 Omicron variant replication in human bronchus and lung ex vivo. Nature 603, 715–720 (2022). [DOI] [PubMed] [Google Scholar]
- 17.Meng B et al. , Altered TMPRSS2 usage by SARS-CoV-2 Omicron impacts infectivity and fusogenicity. Nature 603, 706–714 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Verdoes M et al. , Improved quenched fluorescent probe for imaging of cysteine cathepsin activity. Journal of the American Chemical Society 135, 14726–14730 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boonen M, van Meel E, Oorschot V, Klumperman J, Kornfeld S, Vacuolization of mucolipidosis type II mouse exocrine gland cells represents accumulation of autolysosomes. Molecular biology of the cell 22, 1135–1147 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mosen P, Sanner A, Singh J, Winter D, Targeted Quantification of the Lysosomal Proteome in Complex Samples. Proteomes 9, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Čaval T et al. , Targeted Analysis of Lysosomal Directed Proteins and Their Sites of Mannose-6-phosphate Modification. Molecular & cellular proteomics : MCP 18, 16–27 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Müller-Loennies S, Galliciotti G, Kollmann K, Glatzel M, Braulke T, A novel single-chain antibody fragment for detection of mannose 6-phosphate-containing proteins: application in mucolipidosis type II patients and mice. The American journal of pathology 177, 240–247 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Meel E, Qian Y, Kornfeld SA, Mislocalization of phosphotransferase as a cause of mucolipidosis III αβ. Proceedings of the National Academy of Sciences of the United States of America 111, 3532–3537 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee WS, Jennings BC, Doray B, Kornfeld S, Disease-causing missense mutations within the N-terminal transmembrane domain of GlcNAc-1-phosphotransferase impair endoplasmic reticulum translocation or Golgi retention. Human mutation 41, 1321–1328 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Graham JM, Fractionation of Subcellular Organelles. Curr Protoc Cell Biol 69, 3.1.1–3.1.22 (2015). [DOI] [PubMed] [Google Scholar]
- 26.Axelsson MA et al. , Neutralization of pH in the Golgi apparatus causes redistribution of glycosyltransferases and changes in the O-glycosylation of mucins. Glycobiology 11, 633–644 (2001). [DOI] [PubMed] [Google Scholar]
- 27.Leroy JG, Demars RI, Mutant enzymatic and cytological phenotypes in cultured human fibroblasts. Science (New York, N.Y.) 157, 804–806 (1967). [DOI] [PubMed] [Google Scholar]
- 28.Khan SA, Tomatsu SC, Mucolipidoses Overview: Past, Present, and Future. International journal of molecular sciences 21, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ain NU et al. , Biallelic TMEM251 variants in patients with severe skeletal dysplasia and extreme short stature. Human mutation 42, 89–101 (2021). [DOI] [PubMed] [Google Scholar]
- 30.Gelfman CM et al. , Mice lacking alpha/beta subunits of GlcNAc-1-phosphotransferase exhibit growth retardation, retinal degeneration, and secretory cell lesions. Investigative ophthalmology & visual science 48, 5221–5228 (2007). [DOI] [PubMed] [Google Scholar]
- 31.Essletzbichler P et al. , Megabase-scale deletion using CRISPR/Cas9 to generate a fully haploid human cell line. Genome research 24, 2059–2065 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carette JE et al. , Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature 477, 340–343 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klein S et al. , SARS-CoV-2 structure and replication characterized by in situ cryo-electron tomography. Nature communications 11, 5885 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Franke M, Braulke T, Storch S, Transport of the GlcNAc-1-phosphotransferase α/β-subunit precursor protein to the Golgi apparatus requires a combinatorial sorting motif. The Journal of biological chemistry 288, 1238–1249 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Karczewski KJ et al. , The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li W et al. , Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426, 450–454 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Campeau E et al. , A versatile viral system for expression and depletion of proteins in mammalian cells. PloS one 4, e6529 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang R et al. , Genetic Screens Identify Host Factors for SARS-CoV-2 and Common Cold Coronaviruses. Cell 184, 106–119.e114 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dieterle ME et al. , A Replication-Competent Vesicular Stomatitis Virus for Studies of SARS-CoV-2 Spike-Mediated Cell Entry and Its Inhibition. Cell host & microbe 28, 486–496.e486 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wong AC, Sandesara RG, Mulherkar N, Whelan SP, Chandran K, A forward genetic strategy reveals destabilizing mutations in the Ebolavirus glycoprotein that alter its protease dependence during cell entry. Journal of virology 84, 163–175 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sanjana NE, Shalem O, Zhang F, Improved vectors and genome-wide libraries for CRISPR screening. Nature methods 11, 783–784 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li W et al. , MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol 15, 554 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ran FA et al. , Genome engineering using the CRISPR-Cas9 system. Nature protocols 8, 2281–2308 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Slot JW, Geuze HJ, Cryosectioning and immunolabeling. Nature protocols 2, 2480–2491 (2007). [DOI] [PubMed] [Google Scholar]
- 45.De Pace R et al. , Mucolipidosis II-related mutations inhibit the exit from the endoplasmic reticulum and proteolytic cleavage of GlcNAc-1-phosphotransferase precursor protein (GNPTAB). Human mutation 35, 368–376 (2014). [DOI] [PubMed] [Google Scholar]
- 46.Radons J et al. , Stimulation of the biosynthesis of lactosamine repeats in glycoproteins in differentiating U937 cells and its suppression in the presence of NH4Cl. Eur J Cell Biol 57, 184–192 (1992). [PubMed] [Google Scholar]
- 47.Bolte S, Cordelières FP, A guided tour into subcellular colocalization analysis in light microscopy. J Microsc 224, 213–232 (2006). [DOI] [PubMed] [Google Scholar]
- 48.Kuo CL et al. , Activity-Based Probes for Glycosidases: Profiling and Other Applications. Methods Enzymol 598, 217–235 (2018). [DOI] [PubMed] [Google Scholar]
- 49.Aerts JM et al. , A procedure for the rapid purification in high yield of human glucocerebrosidase using immunoaffinity chromatography with monoclonal antibodies. Anal Biochem 154, 655–663 (1986). [DOI] [PubMed] [Google Scholar]
- 50.Markmann S et al. , Lrp1/LDL Receptor Play Critical Roles in Mannose 6-Phosphate-Independent Lysosomal Enzyme Targeting. Traffic 16, 743–759 (2015). [DOI] [PubMed] [Google Scholar]
- 51.HaileMariam M et al. , S-Trap, an Ultrafast Sample-Preparation Approach for Shotgun Proteomics. Journal of proteome research 17, 2917–2924 (2018). [DOI] [PubMed] [Google Scholar]
- 52.Marx H et al. , A proteomic atlas of the legume Medicago truncatula and its nitrogen-fixing endosymbiont Sinorhizobium meliloti. Nature biotechnology 34, 1198–1205 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Riley NM, Malaker SA, Driessen MD, Bertozzi CR, Optimal Dissociation Methods Differ for N- and O-Glycopeptides. Journal of proteome research 19, 3286–3301 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Saba J, Dutta S, Hemenway E, Viner R, Increasing the productivity of glycopeptides analysis by using higher-energy collision dissociation-accurate mass-product-dependent electron transfer dissociation. International journal of proteomics 2012, 560391 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu SW, Pu TH, Viner R, Khoo KH, Novel LC-MS2 product dependent parallel data acquisition function and data analysis workflow for sequencing and identification of intact glycopeptides. Analytical chemistry 86, 5478–5486 (2014). [DOI] [PubMed] [Google Scholar]
- 56.Singh C, Zampronio CG, Creese AJ, Cooper HJ, Higher energy collision dissociation (HCD) product ion-triggered electron transfer dissociation (ETD) mass spectrometry for the analysis of N-linked glycoproteins. Journal of proteome research 11, 4517–4525 (2012). [DOI] [PubMed] [Google Scholar]
- 57.Bern M, Kil YJ, Becker C, Byonic: advanced peptide and protein identification software. Current protocols in bioinformatics Chapter 13, Unit13.20 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.UniProt-Consortium, UniProt: a worldwide hub of protein knowledge. Nucleic acids research 47, D506–d515 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Čaval T, Zhu J, Heck AJR, Simply Extending the Mass Range in Electron Transfer Higher Energy Collisional Dissociation Increases Confidence in N-Glycopeptide Identification. Analytical chemistry 91, 10401–10406 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pino LK et al. , The Skyline ecosystem: Informatics for quantitative mass spectrometry proteomics. Mass spectrometry reviews 39, 229–244 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Müller T, Winter D, Systematic Evaluation of Protein Reduction and Alkylation Reveals Massive Unspecific Side Effects by Iodine-containing Reagents. Molecular & cellular proteomics : MCP 16, 1173–1187 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ponnaiyan S, Akter F, Singh J, Winter D, Comprehensive draft of the mouse embryonic fibroblast lysosomal proteome by mass spectrometry based proteomics. Sci Data 7, 68 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cox J, Mann M, MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nature biotechnology 26, 1367–1372 (2008). [DOI] [PubMed] [Google Scholar]
- 64.Mi H, Muruganujan A, Ebert D, Huang X, Thomas PD, PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic acids research 47, D419–d426 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.MacLean B et al. , Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics (Oxford, England) 26, 966–968 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tyanova S et al. , The Perseus computational platform for comprehensive analysis of (prote)omics data. Nature methods 13, 731–740 (2016). [DOI] [PubMed] [Google Scholar]
- 67.N. R. Council, Guide for the Care and Use of Laboratory Animals 8th edn. (National Academies Press, 2010). [Google Scholar]
- 68.U. S. D. o. H. a. H. S. N. I. o. Health, Public Health Service Policy on Humane Care and Use of Laboratory Animals. (Office of Laboratory Animal Welfare, 2015). [Google Scholar]
- 69.Diep J et al. , Enterovirus pathogenesis requires the host methyltransferase SETD3. Nature microbiology 4, 2523–2537 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Suter A et al. , Overlapping functions of lysosomal acid phosphatase (LAP) and tartrate-resistant acid phosphatase (Acp5) revealed by doubly deficient mice. Development 128, 4899–4910 (2001). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data presented are available in the main text or the supplementary materials. FASTQ files for CRISPR screens are available at ArrayExpress (Accession# E-MTAB-11158). Acquired glycoproteomics data are available at ProteomeXchange with identifier PXD029746 and DIA and PRM data with identifier PXD029609. Cell lines and LYSET knockout mice generated in the laboratory of J.E.C. are available upon request to J.E.C. under a material transfer agreement from Stanford University. Huh7.5.1 derivatives require an additional material transfer agreement from Apath.