

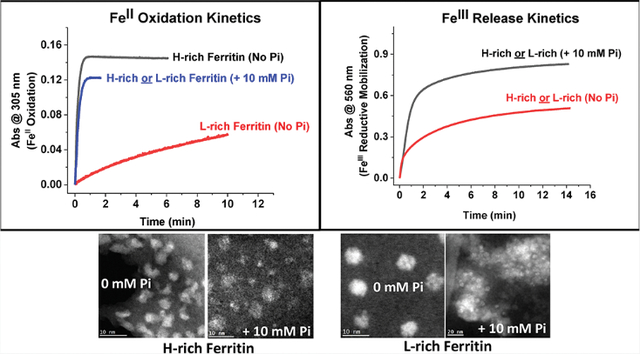
Abstract
Ferritins are highly conserved supramolecular protein nanostructures that play a key role in iron homeostasis. Thousands of iron atoms can be stored inside their hollow cavity as a hydrated ferric oxyhydroxide mineral. Although phosphate associates with the ferritin iron nanoparticles, the effect of physiological concentrations on the kinetics, structure, and reactivity of ferritin iron cores has not yet been explored. Here, the iron loading and mobilization kinetics were studied in the presence of 1–10 mM phosphate using homopolymer and heteropolymer ferritins having different H to L subunit ratios. In the absence of ferritin, phosphate enhances the rate of ferrous ion oxidation and forms large and soluble polymeric Fe(III)–phosphate species. In the presence of phosphate, Fe(II) oxidation and core formation in ferritin is significantly accelerated with oxidation rates several-fold higher than with phosphate alone. High-angle annular dark-field scanning transmission electron microscopy measurements revealed a strong phosphate effect on both the size and morphology of the iron mineral in H-rich (but not L-rich) ferritins. While iron nanoparticles in L-rich ferritins have spherical shape in the absence and presence of phosphate, iron nanoparticles in H-rich ferritins change from irregular shapes in the absence of phosphate to spherical particles in the presence of phosphate with larger size distribution and smaller particle size. In the presence of phosphate, the kinetics of iron-reductive mobilization from ferritin releases twice as much iron than in its absence. Altogether, our results demonstrate an important role for phosphate, and the ferritin H and L subunit composition toward the kinetics of iron oxidation and removal from ferritin, as well as the structure and reactivity of the iron mineral, and may have an important implication on ferritin iron management in vivo.
Graphical Abstract
INTRODUCTION
Iron is an essential element for almost all living organisms. It is a crucial cofactor for many enzymes and participates in numerous oxidation–reduction reactions. Proper iron transport and storage is pivotal to avoid the formation of reactive oxygen species and ultimately cellular damage. Among the storage mechanisms to protect against the potentially dangerous effects of free iron in the cell is ferritin, a ubiquitous iron storage and detoxification protein. Mammalian ferritin is 24-mer protein composed of two structurally similar but functionally different subunit types, named H and L with MW of ~21 and ~19 kDa, respectively.1 These subunits co-assemble in various H-to-L ratios with tissue-specific distribution (isoferritins) to form a 12 nm isoferritin spherical nanostructure, surrounding an 8 nm inner cavity that can accommodate thousands of iron atoms as an inorganic stable iron oxide mineral.1–3 The H subunit possesses a dinuclear ferroxidase center for the rapid oxidation of Fe(II) to Fe(III), whereas the L subunit lacks such center and consequently oxidizes iron at a much slower rate.1 While the mechanisms of iron mineralization in ferritin are relatively well understood, iron mobilization mechanisms have not been clearly elucidated.4 In vitro ferritin iron mobilization can be achieved using a variety of reducing agents, but in vivo iron retrieval may occur through a variety of processes, including proteolytic degradation of the ferritin shell, auxiliary iron mobilization mechanisms involving physiological reducing agents, and/or oxidoreductases.4 Recent studies have implicated a nuclear receptor coactivator-4 (NCOA4) protein in a specific mechanism called ferritinophagy, which involves ferritin degradation by lysosomal autophagy and consequent iron release.5,6
Historically, most characterization studies of human ferritins have mainly been performed with recombinant homopolymer H- or L-ferritins.1 However, ferritin exists largely as a heteropolymer cytoplasmic protein of different H-to-L subunit ratios, with homopolymer H-ferritin present in the mitochondria and in developing neurons, hepatocytes, corneal epithelial cells, and some cancer cells7 and homopolymer L-ferritin present in serum.1 Ferritins isolated from mammalian tissues consist of a mixture of H- and L-subunits with varying composition and iron contents. For instance, muscle, thymus, and red blood cell ferritins contain ~20 H and 4 L, liver and spleen ferritins ~2–3 H and 22–21 L, and brain and heart ferritins ~10–16 H and 14–8 L.1,8–12 Some hepatocellular carcinomas and fetal liver ferritins have an average subunit composition of ~17–21 H and ~7–3 L, whereas three types of neuroblastoma ferritins showed subunit compositions ~13 H and ~11 L, similarly to heart and brain ferritins.13 Generally speaking, L-rich ferritins are characteristic of organs that store relatively large amounts of iron (≥1,500 FeIII atoms/molecule), while H-rich ferritins are found in organs with a low average iron content (≤1000 FeIII atoms/molecule).8 While the mechanism through which ferritin H and L subunits co-assemble remains elusive, such assembly is likely a specific phenomenon since random interactions between H- and L-subunits would have led to the formation of isoferritin mixtures14 in each tissue or organ, ranging from homopolymer H- and H-rich ferritins to L-rich and homopolymer L- ferritins. In vitro heteropolymer ferritin reconstitution is a tedious process that yields very low amounts of functional proteins and may not represent naturally occurring ferritins; it occurs through denaturation and unfolding of recombinant homopolymers H- and L-subunits in high urea concentration and/or acidic pH, followed by “renaturation” of the two subunits at neutral pH.8,10,11,15–18 In vitro ferritin reconstitution shows a clear preference for the formation of heteropolymers over homopolymers with a remarkably narrow distribution, suggesting the presence of preferential interactions between H and L chains.8,10,11,18 This specific recognition is consistent with the fact that H and L homopolymers are poorly populated in mammalian tissues.
When isolated from various sources, native ferritin iron cores are associated with phosphate19–23 and are present in the form of inorganic iron-oxyhydroxide–phosphate nanoparticles.20 Phosphate-to-iron ratios between ~1:1 and ~1:3 P/Fe have been found in iron cores of bacterial and plant ferritins, much higher than those of vertebrate ferritins (i.e., ~1:10 and ~1:40 P/Fe in native ferritins extracted from horse spleen and rat liver, respectively).20,21 While the mechanism of phosphate incorporation remains elusive, it is proposed that after iron delivery, phosphate deposits on the mineral surface22 and that much of the phosphate associated with ferritin is loosely bound to the protein at readily accessible surface sites.23 Additionally, phosphate is known to react with Fe(II) to form both soluble and insoluble complexes. Precipitation reactions occur when Fe(II) and phosphate are present at nearly 1:1 ratios (Ksp ≈ 10−22 to 10−36),22 but soluble polymeric iron(III)–phosphate complexes (K > 108 M−1) form when phosphate is at least 10-fold excess over Fe(II)12,22 and have been shown to be poor substrates for iron loading into ferritin or transferrin.12
Despite being the most abundant intracellular anion, the concentration of cytosolic phosphate is unknown and debatable; its exact concentration in any given cell at any given time fluctuates and is dependent on the type of tissues and nutritional states. Noorwali et al.24 offered a range of values (1–14 mM) from different organisms and tissues. Other sources25 provided a range of phosphate concentration (1–5 mM) for the relatively idle human erythrocytes. Thus, it is reasonable to assume a range of 1–10 mM of free phosphate as physiologically relevant concentrations representative of human cells. Serum phosphate concentrations in healthy individuals are estimated at 1–2 mM and is about double in people with chronic kidney disease.26,27
The present work describes the effect of physiological concentrations of phosphate (i.e., 0–10 mM) on the iron oxidation and mobilization kinetics in recombinant human homopolymer and heteropolymer ferritins with different H and L subunit compositions and also on the morphology and reactivity of the iron mineral. Our results show that at physiological concentrations of phosphate (1 and 5 mM), ferritin H-homopolymer and H/L heteropolymer outcompete phosphate for Fe(II) binding and oxidation and exhibit oxidation rates that are up to 10-fold higher than those of phosphate alone. Phosphate was shown to accelerate Fe(II) oxidation in all types of ferritins tested in this study, with a significant effect (up to 50-fold higher rates) observed with L-rich ferritins. High-angle annular dark-field STEM measurements showed a strong phosphate effect on the shape of the iron core in H-rich but not in L-rich ferritins. Reductive iron mobilization kinetics revealed that in the presence of 5 or 10 mM phosphate, as much as twice the amount of iron is released from ferritin compared to that in the absence of phosphate. Altogether, our results demonstrate a role of phosphate in enhancing both iron uptake/oxidation and release kinetics, irrespective of the ferritin type (homopolymer or heteropolymer), and an effect from both phosphate and ferritin subunit composition on the morphology and structure of the iron core.
MATERIALS AND METHODS
Expression and Purification of Recombinant Heteropolymer Ferritins.
Recombinant human homopolymer and heteropolymer ferritins (with different H-to-L subunit ratios) were expressed in the Escherichia coli Rosetta-gami B strain using a recently engineered pWUR-FtH-TetO-FtL plasmid and different concentrations of inducers, as described in detail elsewhere.28 The pWUR-FTH-tetO-FTL plasmid-transformed cells (BL21(DE3) pLys or Rosetta gamie B strain of E. coli) were grown overnight, and 100 mL of this inoculum was used for a 1 L culture. The cells were grown at 37 °C using LB Lennox Broth (23.5 g of LB broth was dissolved into 1 L of DI water and autoclaved for 30 min at 120 °C) for 1–2 h until an optical density of 0.4–0.5 is reached. The LB Lennox formulation consisted of 1.0% tryptone, 0.5% yeast extract, 0.5% sodium chloride, 0.35% potassium phosphate dibasic, and pH 7.4. The culture was then induced with 10–1000 μM isopropyl β-D-1-thiogalactopyranoside (from Sigma-Aldrich) and 25–1600 ng/mL of anhydrotetracycline (IBA solutions) and incubated at 37 °C for 4 h. Protein purification and quantification were performed according to established protocols using size exclusion chromatography (Akta Go, GE Healthcare), native, and SDS-PAGE, and capillary gel electrophoresis (7100 model from Agilent Technologies).28 On average, ferritin yields were on the order of 10–15 mg per L of culture, irrespective of the H to L subunit composition of the ferritin samples.
Proteins and Chemicals.
Purified recombinant homopolymer and heteropolymer ferritins contained a small iron core (i.e., ~150 ± 30 Fe(III)/ferritin molecule for L- and L-rich ferritins and ~200 ± 50 Fe(III)/ferritin molecule for H- and H-rich ferritins), as determined by an iron-reductive mobilization assay.29,30 Therefore, to preserve the integrity of the ferritin shell and avoid oxidative damage due to harsh chemical treatments, we opted to use all purified ferritin samples in this study as expressed in E. coli and without the removal of this small iron core. Iron loading was performed as detailed in the text and reported in the figure captions.
Protein concentrations were determined using the Micro BCA Protein Assay Kit (Thermo Scientific). All chemicals were of reagent grade and used without further purification. Tris(hydroxymethyl)aminomethane buffer was purchased from G-Biosciences, dibasic anhydrous sodium phosphate from Fisher Chemicals, and FeSO4·7H2O from Fisher Chemicals. Fe(II) stock solutions were freshly prepared immediately before each experiment in a dilute HCl solution at pH 2.0. Stock solutions of 132 mM Tris buffer with varying concentrations of sodium phosphate (0, 1, 5, and 10 mM) were freshly prepared in deionized water and adjusted to pH 7.4 using 8 M HCl.
Light Absorption Spectroscopy.
UV–vis absorption spectroscopy was performed on a Varian Cary 50 Bio or Cary 60 spectrophotometers from Agilent Technologies. All experiments were conducted in a 1 mL quartz cuvette at 25.00 °C in 132 mM Tris in the presence of 0, 1, 5, or 10 mM sodium phosphate, pH 7.4.
Fe(II) Oxidation Kinetics.
The kinetics of iron oxidation in ferritin were followed at 305 nm where the Fe(III)oxo-(hydroxo) species absorbs.1 The instrument was zeroed using the iron-free ferritin solution, prepared in 132 mM Tris pH 7.4 buffer with varying concentrations of sodium phosphate (0, 1, 5, or 10 mM). Typically, 10 μL of a 4 mM (or 5 μL of an 8 mM) stock ferrous sulfate solution prepared in deionized H2O (pH ≈ 2) was injected with rapid stirring into a 1.0 mL aerobic protein solution (0.2 μM, to afford a concentration of 40 μM Fe(II) or 200 Fe(II)/protein). We note that freshly prepared phosphate or Tris-buffered solutions have slightly different Fe(II) oxidation rates than older solutions that were a few weeks old and stored at room temperature. Older phosphate solutions showed signs of cloudiness over time and were discarded. However, all our kinetics and their controls were performed using the same and relatively fresh buffer stocks. For all the iron oxidation and mineralization kinetics, the ferritin samples were prepared using the fresh buffer stock solutions described above (i.e., 132 mM Tris buffer in the presence of either 0, 1, 5, or 10 mM phosphate, pH 7.4).
Fe(II) Mobilization Kinetics.
The iron release kinetics were performed in a 1.0 mL UV–vis quartz cuvette (1 cm path length) hermetically sealed with a screw-on cap to prevent atmospheric oxygen diffusion inside the cell. In each iron release experiment, the iron-loaded ferritin samples prepared above were diluted three-fold using Tris buffer and 0, 1, 5, or 10 mM phosphate to afford a working solution of 0.067 μM ferritin. For the iron mobilization kinetics, these ferritin solutions were then mixed with FMN (5 mM), NADH (5 mM), and ferrozine (0.5 mM) and rapidly inverted several times for thorough mixing. The instrument was zeroed shortly after mixing all reagents (i.e., during the lag phase where very little reduction of the iron core occurs),30 and the kinetics of iron release were followed at 560 nm where the {Fe(II)-(ferrozine)3} species absorbs.31 Because all ferritin samples have been mineralized the same way (i.e., one injection of 200 Fe(II)/shell), and since they already contain a small iron core of ~200 Fe(III)/shell after expression and purification, the final concentration of iron present in all samples is ~26.8 μM (or a total of 400 Fe(III)/shell). Thus, our control experiment in these iron release kinetics consisted of soluble polymeric Fe(III)–phosphate species at 26.8 μM. To confirm the accuracy of the iron concentration in these samples, several control experiments were performed and consisted of freshly added 26.8 μM Fe(II) to an aqueous ferrozine solution, with and without phosphate. In all cases, absorbance values of (0.8 ± 0.1) were observed at 560 nm, as expected, due to the formation of {Fe(II)-(ferrozine)3} species.31 All iron oxidation and release experiments were performed two or three times to ensure reproducibility, and the kinetic traces were analyzed with OriginLab version 8.0 (OriginLab Corp).
Size Exclusion Chromatography.
Size exclusion chromatography was utilized to separate soluble polymeric Fe(III)–phosphate complexes (formed outside of the ferritin shell) from ferritin during iron loading. Ferritin samples (0.2 μM) in 50 mM MOPS and 100 mM NaCl pH 7.4 in the presence of 0, 1, 5, or 10 mM sodium phosphate were prepared in 1.0 mL reaction volumes, and either 10 or 20 μL of 4 mM or 2 mM ferrous sulfate, respectively, were added to the protein solution as described above for a total of 200 Fe(II)/shell or 40 μM Fe(II), respectively. For a control, 40 μM ferrous sulfate was added to the same buffer in the presence of 0, 1, 5, or 10 mM sodium phosphate (no ferritin). All samples were centrifuged to ensure that no precipitation was present (and there was none since soluble Fe(III)–phosphate polymers form under these conditions) and then loaded onto a GE Sephadex G-25 Superfine column (Cytiva Life Sciences) and eluted using the ÄKTA go protein purification chromatography system from Cytiva.
STEM.
Scanning transmission electron microscopy (STEM) images were obtained with a JEOL NEOARM instrument operating at 200 kV. Samples were deposited on lacey carbon supported on Cu grids, provided by Electron Microscopy Sciences. For imaging, the probe current was 150 pA with a camera length of 4 cm. Energy-dispersive X-ray spectroscopy (EDS) and electron energy-loss spectroscopy (EELS) measurements were performed with a camera length of 2 cm. The EDS detectors were provided by JEOL, and a Gatan K2 Summit camera was used to collect EELS data. A probe current of 500 pA was used for the EELS measurements.
Iron-loaded H-rich (H22:L2) and L-rich (or H4:L20) ferritin solutions (0.5 μM) were prepared by making 10 injections of Fe(II) ions, 3–5 min apart, from a ferrous sulfate solution to afford a ratio of 100 Fe(II)/shell per injection. Iron loading was monitored at 305 nm by UV–vis as explained above. The protein solutions were dissolved in 50 mM MOPS and 100 mM NaCl, with and without 10 mM phosphate, pH 7.40.
For phosphate quantification, we used a tool provided by Digital Micrograph and a software provided by Gatan to analyze EDS data and images. A background subtraction was performed using a top hat filter. A Casnati cross-sectional model was chosen without any correction mode.
RESULTS AND DISCUSSION
Iron Loading into Ferritin.
UV–vis spectrophotometric kinetic measurements were conducted to compare the ability of several recombinant human ferritins (homo- and heteropolymers) to oxidize Fe(II) and form mineral cores in the presence and absence of phosphate. Figure 1A shows an increase in absorbance at 305 nm (due to the formation of oxo/hydroxo Fe(III) species) following the addition of 40 μM Fe(II) (i.e., 200 Fe(II)/protein) to eight different ferritin samples with different H- to L-subunit ratios. For comparison, the iron oxidation rates in Tris buffer, in the absence or presence of 1, 5, and 10 mM phosphate, are displayed in Figure 1B. The average molar absorptivity value for the soluble Fe(III)–phosphate polymeric species (Fe(III)–Pi) is (2550 ± 250) M−1 cm−1, that of the oxidized iron core inside ferritin in the absence of phosphate is (3500 ± 500) M−1 cm−1 and that in the presence of phosphate is (3200 ± 300) M−1 cm−1, in accord with the published literature.1,22,28,32 The lower molar absorptivity value of the ferritin iron core in the presence of phosphate is likely an average value representing the two species present in solution (i.e., Fe(III)–Pi and ferritin iron core).
Figure 1.
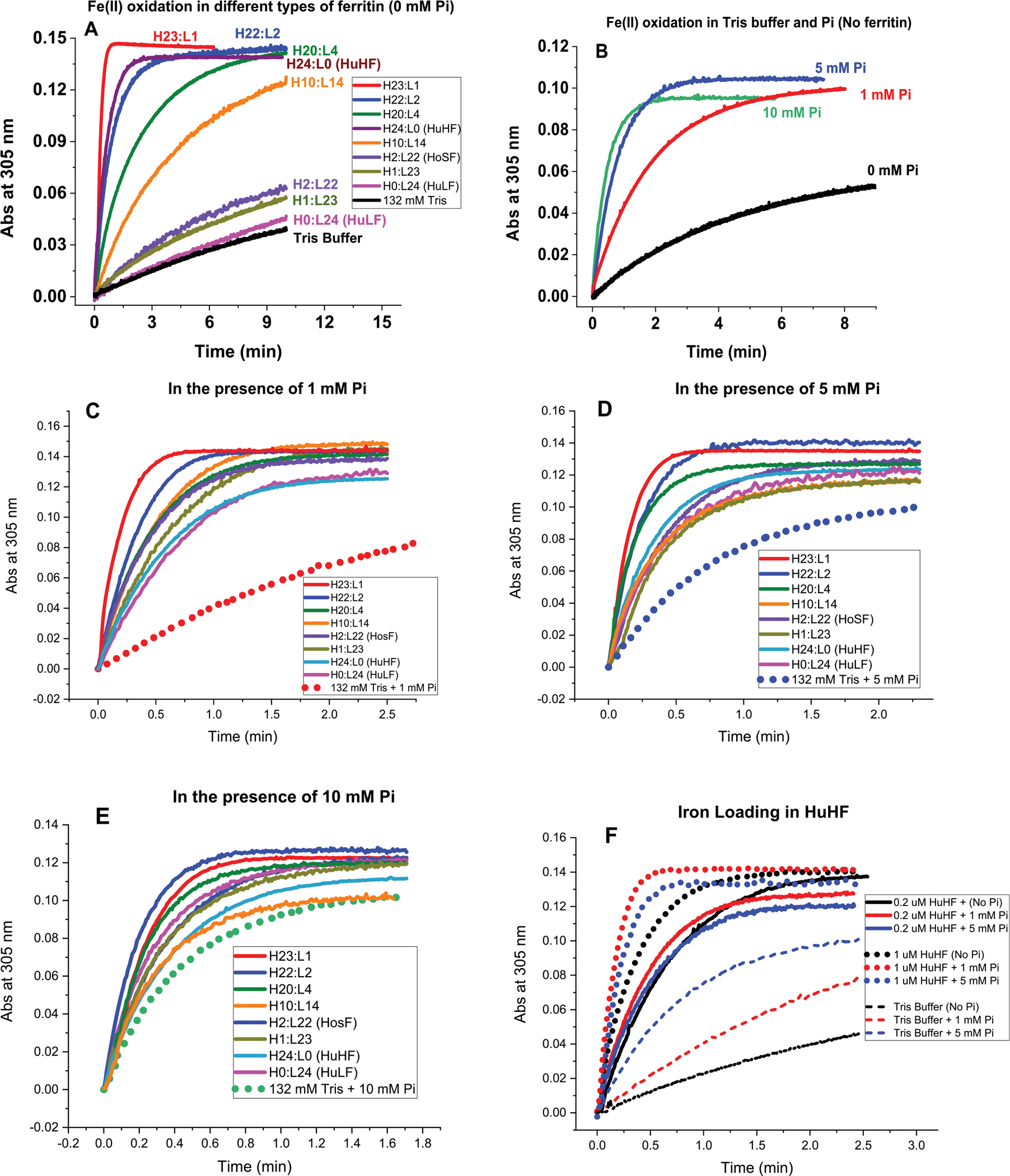
Iron oxidation kinetics at 305 nm in the presence and absence of phosphate using light absorption spectroscopy. Conditions (A–E): 132 mM Tris buffer with 0, 1, 5, or 10 mM sodium phosphate; 0.2 μM ferritin; 40 μM FeSO4; pH 7.4; and 25.0 °C. Panel F displays the iron oxidation kinetics of recombinant human H-ferritin at 0.2 and 1 μM ferritin in 1 and 5 mM phosphate compared to Tris buffer. Panel F: effect of ferritin concentration on iron loading into human H-ferritin. We note that the number of H and L subunits in any heteropolymer ferritin is likely an average value representing a heterogeneous mixture of heteropolymers whose H and L subunit composition could vary between 5 and 8%.
Effect of Phosphate on the Rates of Fe(II) Oxidation in Ferritin.
In the absence of ferritin, phosphate enhances the rapid oxidation of Fe(II) to Fe(III) to form polymeric Fe(III)–phosphate species (Figure 1B), with approximately 3–4 times higher rates in 5 and 10 mM phosphate compared to 1 mM (0.13 and 0.19 vs. 0.045 Abs/min, respectively). A much slower Fe(II) auto-oxidation kinetic is observed in Tris buffer in the absence of phosphate (0.0076 Abs/min or 6-, 17-, and 25-fold slower than the rates in 1, 5, and 10 mM phosphate). The absorbance at 305 nm and the lack of a visual precipitate upon sample centrifugation suggest that the polymeric iron(III)–phosphate complexes remained in solution, as confirmed in earlier studies at phosphate concentrations 10-fold excess over Fe(II).12
Figure 1C–E represents an overlay of the iron oxidation kinetics of eight ferritin samples in the presence of 1, 5, and 10 mM phosphate. Clearly, all ferritin samples (L-homopolymer, H-homopolymer, and different heteropolymer ferritins) outcompeted phosphate for Fe(II) binding and oxidation, particularly at the more physiologically relevant concentrations of 1 and 5 mM phosphate. For instance, in the presence of 1 or 5 mM phosphate, all ferritin samples tested here exhibited between 1.5- and 10-fold higher Fe(II) oxidation rates compared to control runs in the absence of ferritin, suggesting preferential oxidation of Fe(II) by ferritin and the formation of a mineral core. The fact that even L-ferritin (with no ferroxidase centers) outcompeted phosphate for Fe(II) oxidation suggests a role for L-subunits in facilitating the formation of a mineral core in the presence of phosphate. Alternatively, it is possible that phosphate itself is able to facilitate iron oxidation and deposition in ferritin, as shown in bacterial and archaeal ferritins where the presence of phosphate accelerated the rate of Fe(II) oxidation and its subsequent displacement to form an iron core.20,21,33 In the case of L-ferritin, high-density acidic residues on the inner surface of the protein cavity have been suggested to provide efficient sites for iron nucleation and mineralization and could serve as initial Fe(II) binding sites, whose oxidation is facilitated by the presence of phosphate.34 Even at the much higher phosphate concentration of 10 mM, all ferritin samples, including homopolymer L-ferritin, exhibited faster iron oxidation rates (Figure 1E), albeit only 1.5-fold higher in the case of L-ferritin. At the higher protein concentrations of 1 μM, homopolymer H-ferritin displayed Fe(II) oxidation rates of 0.5, 0.8, and 0.9 Abs/min at 0, 1, and 5 mM phosphate, respectively, compared to ~0.3 Abs/min at 0.2 μM and are about 20-, 15-, and 10-fold higher than those of the control kinetic runs in phosphate alone (Figure 1F). The iron oxidation rates displayed in Figure 2A show that variations in the H-to-L subunit ratios have a significant effect on the ability of ferritin to oxidize Fe(II) in the absence or presence of phosphate, with homopolymer H-ferritin (H24:L0) and heteropolymer H/L (up to ~4 L subunits per shell) displaying the least effect (i.e., 2–3-fold increase in rate between no phosphate and 10 mM phosphate), consistent with the presence of a larger number of ferroxidase centers responsible for rapid Fe(II) oxidation,1,28,32,35 and suggesting that H-rich heteropolymer ferritins containing a few L subunits (i.e., less than 4 L-subunits per 24-mer) are the most active and the least affected by the presence of phosphate. As the number of L-subunits increases, a gradual decrease in Fe(II) oxidation rates is observed (Figure 2B), reaching a plateau at >20 L subunits per shell, concomitant with a dramatic increase in rates (~15–50-fold higher) when phosphate is present.
Figure 2.
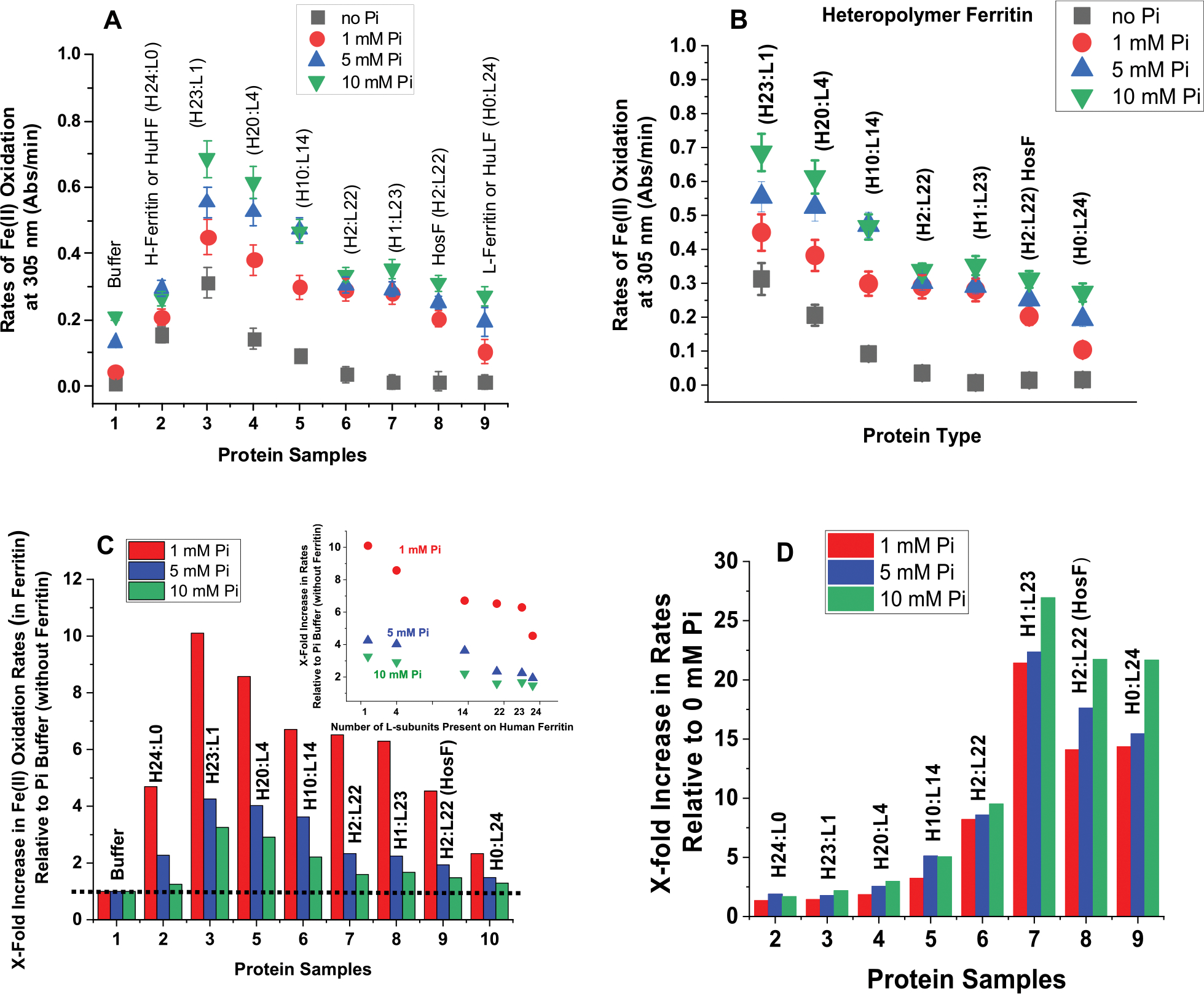
(A,B) Plots of Fe(II) oxidation rates as a function of ferritin type. (C) Normalized rates of Fe(II) oxidation in different types of ferritin in 1 mM (red), 5 mM (blue), and 10 mM (green) phosphate buffer relative to those in the absence of ferritin. (D) Normalized Fe(II) oxidation rates in ferritin in 1 mM (red), 5 mM (blue), and 10 mM (green) phosphate buffer relative to those in the absence of phosphate. The inset of (C) shows a linear decrease in Fe(II) oxidation rates as a function of the number of L-subunits present in human ferritin. The dotted line in (C) illustrates the Fe(II) oxidation rates in phosphate and represents our baseline. The experimental conditions are the same as those of Figure 1.
Further analysis of the kinetic data revealed up to 10-fold increase in the rates of Fe(II) oxidation in ferritin in the presence of 1, 5, and 10 mM of phosphate, relative to rates of Fe(II) oxidation in phosphate buffer alone (Figure 2C) and that phosphate increases the rates of Fe(II) oxidation in the different ferritin samples tested in this study by up to 25-fold compared to no-phosphate (Figure 2D). A negative linear dependence of the Fe(II) oxidation rates as a function of the number of L-subunits present in human ferritin is observed, consistent with the role of ferroxidase centers in the rapid oxidation of Fe(II) ions (Figure 2C, inset). Altogether, our results demonstrate a highly effective iron oxidation mechanism for ferritin, even in the presence of high concentrations of phosphate, where the latter plays a role in enhancing the rates of iron uptake and oxidation, irrespective of the ferritin types (homopolymer or heteropolymer).
Several mechanisms or proposals regarding the enhanced effect of phosphate on the rate of ferrous ion oxidation by ferritin have been discussed, suggesting that phosphate may play a role both at the ferroxidase site and on the surface of the mineral core during iron loading into ferritin. Some studies have suggested a shift to more negative values of the redox potential upon phosphate binding to ferrous ions at or near the ferroxidase centers of ferritins, thus facilitating iron oxidation.36–39 Alternatively, it has been proposed that phosphate may stimulate the movement of iron from the ferroxidase centers into the ferritin cavity or may bind on the surface of the mineral core and act as a ligand for incoming iron.36–40 Other studies have suggested that the enhanced rate of iron loading into ferritin in the presence of phosphate is based on solubility arguments, driven by the insolubility of the iron–phosphate complex; in other words, as a bioreactor, the ferritin shell guides the sequestration of the iron–phosphate complex inside the ferritin cavity.33,35 In these studies, it was suggested that the tetrahedral oxo-anions of phosphate and other analogues such as arsenate, vanadate, and molybdate stimulate the rate of iron loading into ferritin to produce ferritin mineral cores that contain up to 1:1 iron to oxo-anion ratios.
Effect of Phosphate on the Absorbance Spectra of Ferritin and on Iron Loading.
To determine the extent of iron loading into ferritin in the presence of phosphate, UV–vis absorbance scans of H-rich (H23:L1) and L-rich (H2:L22) ferritins loaded with iron were recorded (Figure 3A,B). In the absence of phosphate, the iron-loaded ferritins showed the typical and broad absorbance shoulder in the 300–350 nm range, characteristic of the oxidized ferritin iron core. In the presence of phosphate, a similar but lower absorbance feature with a less-enhanced shoulder is observed, suggesting the formation of a different iron core containing exogenously embedded phosphate. It is also possible that a potential competition between ferritin and phosphate for Fe(II), particularly in the case of L- and L-rich ferritins, lead to the formation of soluble polymeric Fe(III)–phosphate species, even though phosphate had a dramatic enhancement on the rates of iron oxidation in L and L-rich ferritin compared to Fe(II) oxidation by phosphate alone (Figure 2C,D). By contrast, the very rapid rate of Fe(II) oxidation in H- and H-rich ferritins in the absence of phosphate coupled with lower absorbance values in the presence of 5 and 10 mM phosphate strongly suggest that phosphate does not further enhance iron oxidation in these ferritins, but that its presence may have affected the structure of the iron core (more below, STEM results).
Figure 3.
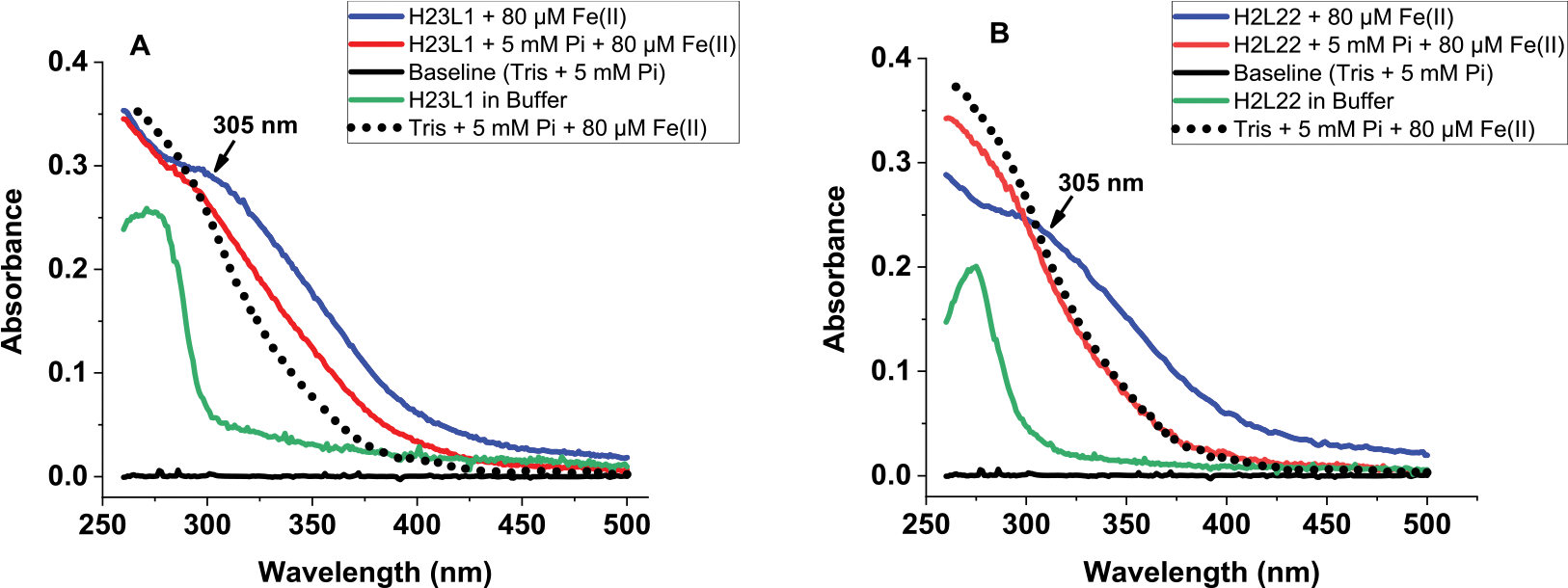
Absorbance scans of (A) H-rich (H23:L1) and (B) L-rich (H2:L22) ferritins in the presence (or absence) of iron and 5 mM phosphate. The spectrum of the Fe(III)–phosphate complex (as control) is shown as a dotted line. Conditions: 0.2 μM ferritin, 132 mM Tris buffer pH 7.4, and 80 μM Fe(II). Spectra were collected after 2 h of adding iron.
It is worth noting that, under our experimental conditions, the soluble Fe(III)–phosphate complex that forms outside of the ferritin cavity does not constitute a source of iron for ferritin, as reported earlier.22 Because the UV spectra of iron-loaded ferritin and of the Fe(III)–phosphate complex (used as a control) have significant absorbance around 300 nm, all iron oxidation kinetics reported here in the presence of phosphate reflect contributions from both species.
Size Exclusion Chromatography and High-Resolution STEM.
Attempts to fully and completely separate the Fe(III)-polymeric species from iron-loaded ferritin were unsuccessful. Elution peaks of different iron-loaded ferritin solutions passed over a GE Sephadex G-25 Superfine column (Cytiva Life Sciences) overlapped with those of control solutions containing the soluble polymeric phosphate species and eluted near the column’s void volume (~2–3 min), suggesting the presence of large molecular weight species with molecular mass close to that of ferritin (i.e., >450,000 Da, Figure 4). Other lower molecular mass species were also observed as judged by peaks eluting at a later time (~4–5 min). We were intrigued by an earlier study showing the ability of a BioRad P-10 Gel to bind very tightly Fe(III)–phosphate complexes, followed by their release from the resin once an anaerobic solution of sodium dithionite (1 mM) and bipyridyl (0.5 mM) were passed through.22 Repeating these earlier experiments using the same BioRad P-10 Gel showed very similar results to those obtained with our Sephadex G-25 column. Therefore, under our experimental conditions, we were unable to quantify the amount of Fe(II) bound to phosphate. Nonetheless, our detailed analysis of the kinetic experiments proved that all ferritin samples outcompeted phosphate for Fe(II), when 1 and 5 mM phosphate were present in solution, but to a lesser extent when a larger excess of phosphate (i.e., 10 mM) is used, particularly with L- and L-rich ferritins (Figure 2C, samples 7–9).
Figure 4.
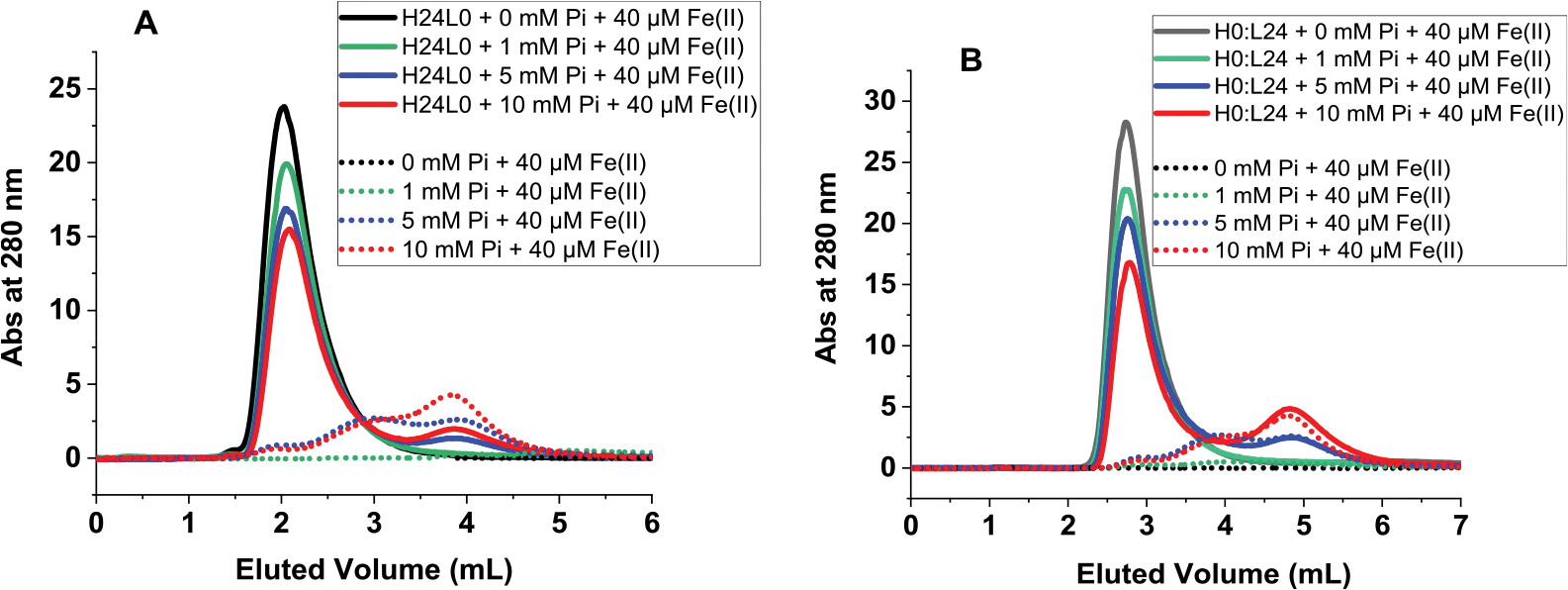
Size exclusion chromatography of iron-loaded ferritin in the presence of phosphate (straight lines) and of Fe(III)–phosphate complexes (dotted lines) used as controls. Conditions: (A) 0.2 μM H-ferritin and (B) 0.2 μM L-ferritin, 40 μM Fe(II) (or 200 Fe/shell), in 50 mM Mops, 100 mM NaCl, pH 7.4, and 0, 1, 5, or 10 mM phosphate.
To further investigate the effect of phosphate on the shape and Fe/P composition of the ferritin iron core, STEM images were obtained on a JEOL NEOARM instrument operating at 200 kV using two ferritin samples, an H-rich (H22:L2) and an L-rich (H4:L20) ferritin loaded with 1000 Fe/shell in the absence and presence of 10 mM phosphate.
Figure 5A shows high-angle annular dark-field STEM images of the two ferritin samples in the presence and absence of phosphate. The high-angle annular dark-field STEM images of Figures 5A/B show that the presence of phosphate did not alter the size or shape of the L-rich samples, which were spherical with a narrow size distribution. In contrast, the H-rich ferritin particles change from irregular shapes with rod- and crescent-like features in the absence of phosphate to more regular and spherical shapes in the presence of phosphate. Furthermore, the size of the iron core in the H-rich sample is also affected by the presence of phosphate and showed a larger size distribution with both large (~5 nm) and small (~2 nm) size nanoparticles.
Figure 5.
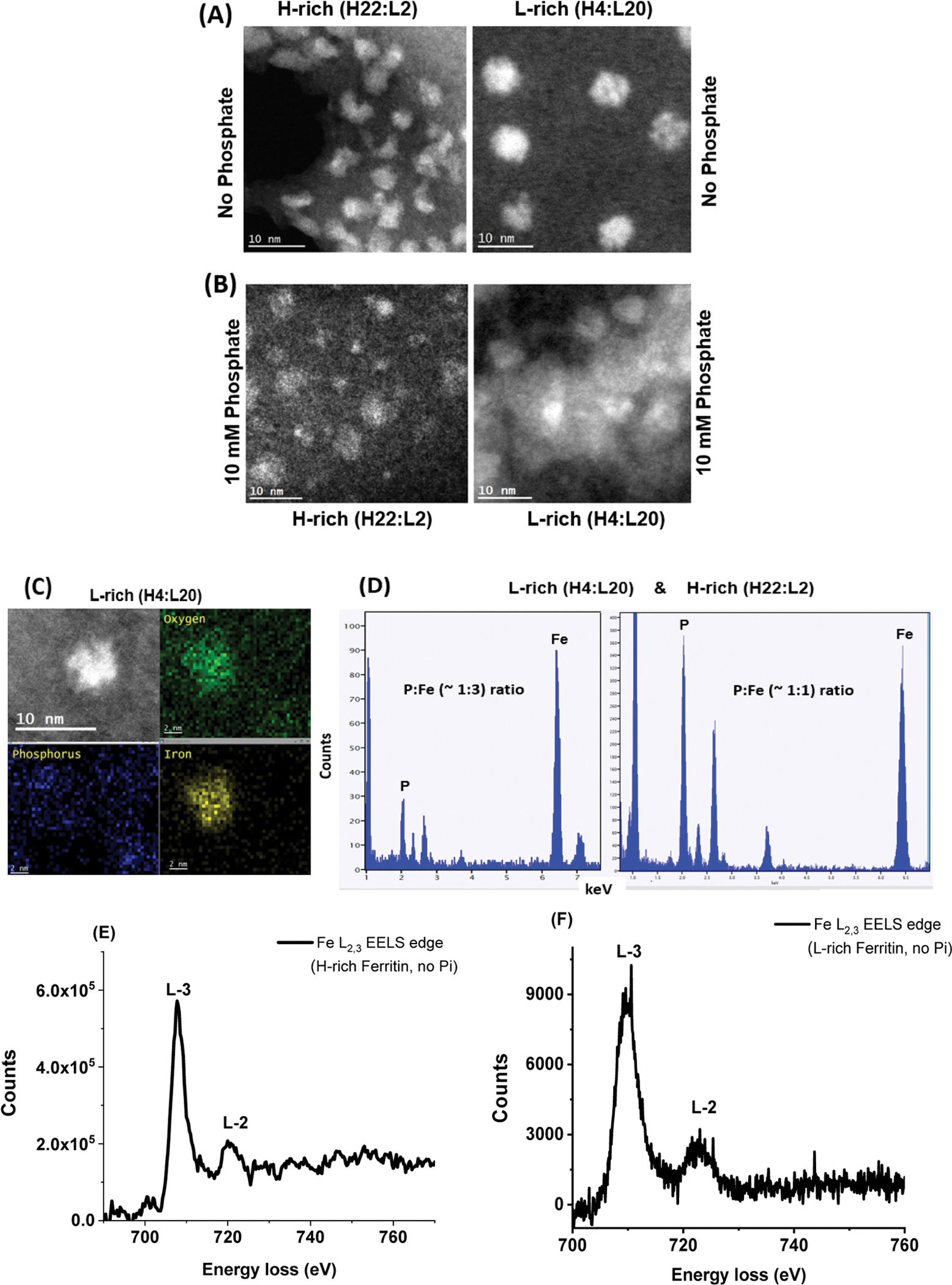
(A,B) STEM images of iron oxide nanoparticles formed within H-rich and L-rich ferritin in the absence (A) and presence (B) of 10 mM phosphate, at 10 nm magnification. (C,D) EDX spectra showing the estimated ratio of P/Fe in L-rich and H-rich ferritins. (E,F) STEM-EELS analysis shows the X-ray absorption spectra at the L3 and L2 edges of iron in H-rich and L-rich samples containing 1000 Fe atoms/core in the absence of phosphate.
Although the accumulation of Fe, P, and O inside the ferritin cavity is confirmed by EDX (Figure 5C,D), the Fe/P ratios are only relative and not quantitative and were estimated to be in the range of 1:1 and 3:1 Fe/P. EELS clearly indicates the presence of oxidized iron with the mineral core of H-rich and L-rich samples, as evidenced by the shape and position of the Fe L2,3 (Figure 5E,F) and O K EELS edge (not shown). Although our in vitro ferritin iron loading in the presence of 10 mM phosphate produced iron cores with a relativley low Fe/P ratios, it is possible that higher Fe/P ratios closer to those found in native ferritins (i.e., up to 10 Fe/P) might be observed with lower phosphate concentrations. Different iron/phosphate ratios might be produced in different tissues since the concentration of phosphate could vary significantly between tissues, as discussed earlier.24–27
Iron Mobilization from Ferritin.
To examine the influence of phosphate on the reductive mobilization of iron from ferritin, we followed the kinetics of iron reduction at 560 nm where the {Fe(II)-(ferrozine)3} complex absorbs (ε = 31,500 M−1 cm−1)31 using the FMN/NADH non-enzymatic redox system30,41,42 whereby NADH serves as the reducing agent, FMN as the electron-transfer mediator, and ferrozine as the Fe(II) chelator. Because the formation of FMNH2 from the oxidation of NADH is slow compared to its rapid reaction with O2, an initial lag phase (~2 min) is observed in our iron release kinetics, whose length depends on the concentration of reagents used.30,41–43 Once the level of dissolved O2 is almost completely depleted from solution, the iron release kinetics proceeded rapidly. Because an insignificant amount of iron (<5% of total iron) is released during the lag phase, and because it slightly varied in length from run to run and has no effect on our rate analysis, the lag phase was removed from all kinetic runs for simplicity. Furthermore, and because it is not feasible to separate the polymeric Fe(III)–phosphate species from ferritin, due to their similar elution time and MW (Figure 4), the iron release experiments presented here involve both species. Figure 6A–F shows the kinetic profiles of the iron release experiments from different ferritin samples, in the presence of phosphate, as indicated on each panel. A control experiment (Figure 6, panel J) using the soluble polymeric Fe(III)–phosphate species (in the absence of ferritin) showed similar iron release kinetics to those of the phosphate-containing ferritin samples (Figure 6). In contrast, in the absence of phosphate, the kinetics of iron release from all ferritin samples exhibit a 2-fold lower absorbance (i.e., 2-fold less the amount of iron released) in the presence of 5–10 mM phosphate, suggesting a role for phosphate in facilitating the removal of iron from ferritin. Surprisingly, the presence of phosphate strongly reduced the initial rates of iron release, particularly with L- and L-rich ferritins (Figure 6G,H,I). This finding suggests that the iron core structure/mobility in the L-rich ferritins differs from that of the H-rich ferritins, possibly because fewer ferroxidase centers reduce the rate of core formation and affect its crystallinity (more below).
Figure 6.
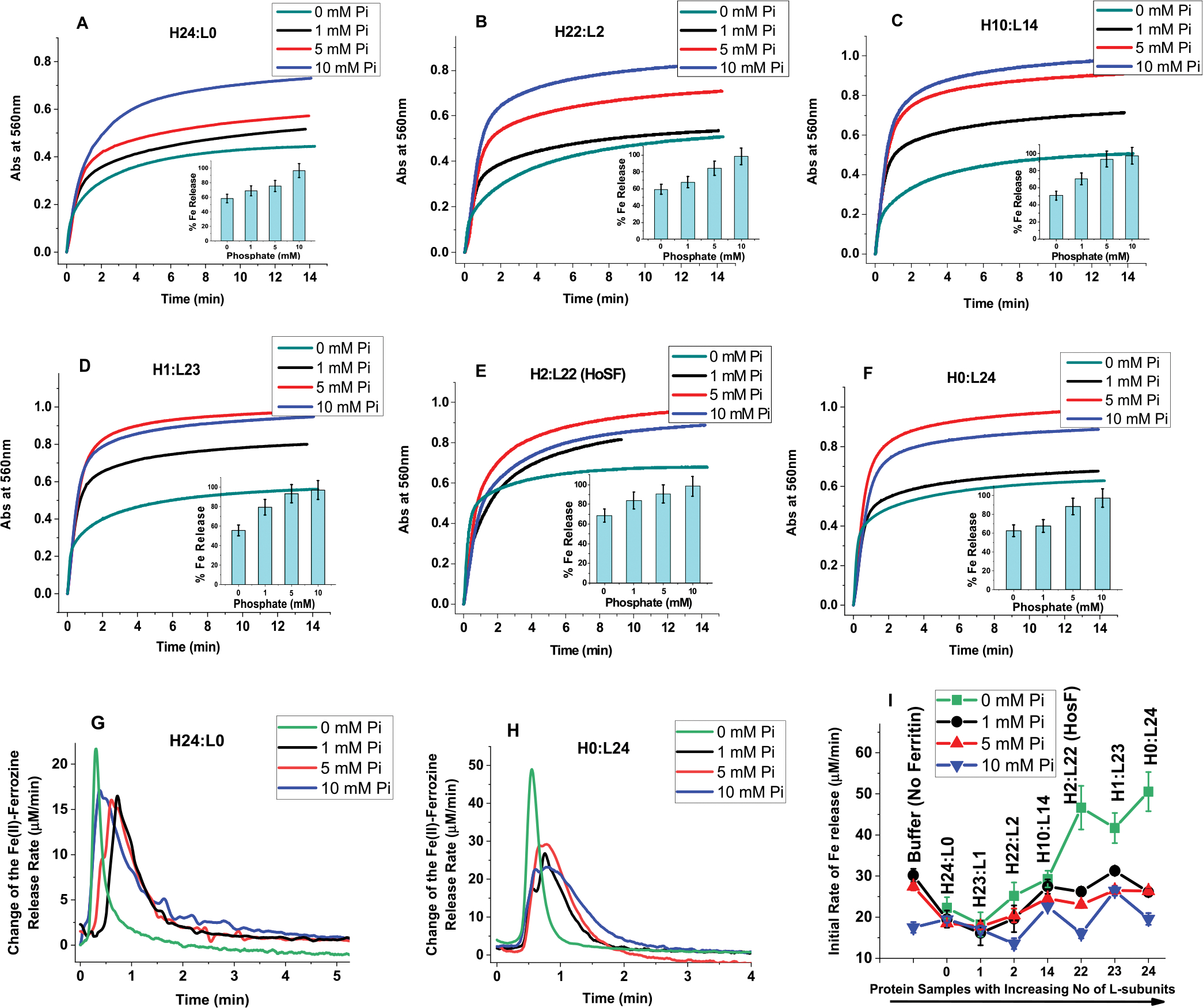
Effect of phosphate on the reductive mobilization of iron from human ferritin. (A–F) Kinetics of iron release from different ferritins with the insets representing percent of iron released as a function of phosphate concentration. (G,H) Change of the Fe(II)-ferrozine release rate vs time shown only for H- and L-homopolymers (H24:L0 and H0:L24) to illustrate the major differences between H and H-rich ferritins and L or L-rich ferritins. (I) Initial rate of iron release (calculated for the first 30 s of reaction) as a function of increasing ferritin L-subunit content. Conditions: 0.067 μM ferritin, 132 mM Tris pH 7.4 at 25.0 °C, 5 mM FMN, 0.5 mM ferrozine, 5 mM NADH, 13.4 μM Fe(II) freshly added (or 200 Fe/shell), and 0–10 mM phosphate as indicated on each panel. The ferritins samples used in these experiments are those of Figure 1, after 3-fold dilution, and thus, the total amount of iron present in each sample is 26.8 μM (i.e., freshly added 13.4 μM Fe(II) + an average of 13.4 μM Fe(III) already present in the purified ferritin; see M&M for more details). The control experiment was performed under the exact same conditions but in the absence of ferritin using 26.8 μM Fe(II) to account for the total amount of Fe(II) present in ferritin. In all cases, the iron mobilization kinetics were performed with freshly prepared samples, and no differences were observed when the kinetics are performed (or repeated) using 1 or 2 week-old sample storage in the fridge at 4 °C.
In theory, fewer ferritin ferroxidase centers decrease the rate of iron oxidation, and therefore, the rate of iron deposition and core formation and should lead to more crystalline iron cores. Differences in the rates of biomineral formation, biomineral order, degree of crystallinity, and iron turnover have been observed in natural ferritins and found to be partially associated with their subunit composition.44–46 Whereas the structure and core crystallinity of ferritin iron minerals across species can be quite variable with varying degrees of crystallinity, disordered mineral cores in animal ferritins are mostly observed in L-rich heteropolymers having a large number of catalytically inactive L-subunits, as those found in livers and spleens. In contrast, a more ordered ferritin core is typically reported in H-rich heteropolymers having more catalytically active H-subunits, as those found in hearts and brains.8,47 These differences between natural ferritins and recombinant homopolymer ferritins suggest that the morphology of the ferritin iron core depends on the ferritin subunits’ composition and is affected by the number of nucleation and ferroxidase sites present on the protein shell.48 Our results suggest that the ferritin’s iron core crystallinity is influenced by the rate of iron oxidation and deposition inside the protein, which is directly correlated with the composition of the ferritin shell (i.e., the ratio of H to L subunits) and the experimental conditions employed (i.e., low vs high iron loading and/or phosphate concentration). Furthermore, differences in ferritin iron core structures and/or crystallinity may also be responsible for differences in the rates of its demineralization (Figure 6I). For instance, in the absence of phosphate, a less-crystalline iron mineral in L- and L-rich ferritin releases its iron content faster than the more crystalline mineral of H- and H-rich ferritins. Notably, the conditions of iron uptake affect the core structure as shown by the evidence that natural ferritins with amorphous iron cores produced crystalline ferrihydrite cores upon in vitro reconstitution (i.e., when the iron core is reconstituted in vitro using native ferritin stripped of iron).8,49,50 It was also shown that the crystallinity of the mineral core is related to the phosphate content, varying from amorphous in plants and microbial ferritins (Fe/P ≈ 1:1) to nanocrystalline in animal ferritins (Fe/P ≈ 8:1),1,49–53 and the results of Figure 6 are consistent with these observations.
Recent studies using Mycobacterium tuberculosis (Mtb) ferritin showed that phosphate accelerates iron core formation but inhibited iron mobilization and was attributed to different stabilities of the core.21 Considering that bacterial ferritin has naturally higher proportions of phosphate compared to mammalian ferritins and is deeply embedded in the iron mineral, it is possible that phosphate has a stabilizing effect on the iron mineral in bacteria, rendering it less reactive. In contrast, our kinetic data show that the presence of phosphate in the reaction mixture enhanced the kinetics of iron mobilization (Figure 6A–F insets), possibly facilitating the iron exchange between FMN, NADH, and ferrozine. Collectively, the reactivity of the ferritin core toward reductive iron mobilization appears to depend on the morphology and structure of the iron core and on the concentration of phosphate present in the medium. While phosphate in mammalian ferritins appears to be largely surface adsorbed, it penetrates deeper into the iron core in bacterioferritins and plant ferritins,23,49,50 but in neither case, the mechanism by which iron mobilization is facilitated by phosphate is known.
CONCLUSIONS
In summary, this work demonstrates that physiological concentrations of phosphate (1–5 mM) significantly enhanced the kinetics of Fe(II) oxidation and Fe(III) reductive mobilization in human homopolymer and heteropolymer ferritins and altered the structure, stability, and reactivity of the iron core. The presence of higher phosphate concentrations (≥10 mM) may compete with ferritin iron loading, particularly the non-catalytically active L- and L-rich ferritins, which lack the catalytic diiron ferroxidase centers. Given the need to rapidly chelate free ferrous ions and prevent their participation in the generation of reactive oxygen species and subsequent cellular toxicity, cells that have predominantly L- or L-rich ferritin must employ an alternative mechanism for iron uptake that is different from that of H- or H-rich ferritins to minimize iron toxicity. One such mechanism could involve the recently identified iron chaperones, poly rC binding proteins 1 and 2 (PCBP1 and PCBP2), both of which binds tightly 3 Fe(II) ions and form redox-inactive Fe(II) complexes.54,55 Other, yet-to-be-identified, cytoplasmic iron chaperones, iron proteins, or small iron chelates might also be involved in transferring ferrous ions to these non-catalytically active ferritins. In addition to PCBP1 and PCBP2, the fast acquisition of iron by H- and H-rich ferritins suggests that rapid Fe(II) transfer or passive diffusion through the ferritin channels might be an alternative mechanism through which these catalytically active proteins acquire iron in the cytosol.
Funding
This work is supported by the National Institute of Health grant R15GM104879 (F.B.-A.), the National Science Foundation, Division of Molecular and Cellular Biosciences (MCB) award 1934666 (F.B.-A.), and a Cottrell Instrumentation Supplements Award from the Research Corporation for Science Advancement award #27452 (F.B.-A.). The STEM work was carried out in part at the Singh Center for Nanotechnology, which is supported by the NSF National Nanotechnology Coordinated Infrastructure Program under grant NNCI-2025608. Additional support to the Nanoscale Characterization Facility at the Singh Center has been provided by the Laboratory for Research on the Structure of Matter (MRSEC) supported by the National Science Foundation (DMR-1720530).
Footnotes
The authors declare no competing financial interest.
Contributor Information
Aliaksandra A. Reutovich, Department of Chemistry, State University of New York, Potsdam, New York 13676, United States
Ayush K. Srivastava, Department of Chemistry, State University of New York, Potsdam, New York 13676, United States
Gideon L. Smith, Department of Chemistry, State University of New York, Potsdam, New York 13676, United States
Alexandre Foucher, Department of Materials Science and Engineering, University of Pennsylvania, Philadelphia, Pennsylvania 19104, United States.
Douglas M. Yates, Department of Materials Science and Engineering, University of Pennsylvania, Philadelphia, Pennsylvania 19104, United States
Eric A. Stach, Department of Materials Science and Engineering, University of Pennsylvania, Philadelphia, Pennsylvania 19104, United States.
Georgia C. Papaefthymiou, Department of Physics, Villanova University, Villanova, Pennsylvania 19085, United States
Paolo Arosio, Department of Molecular and Translational Medicine, University of Brescia, 25121 Brescia, Italy.
Fadi Bou-Abdallah, Department of Chemistry, State University of New York, Potsdam, New York 13676, United States.
REFERENCES
- (1).Bou-Abdallah F The Iron Redox and Hydrolysis Chemistry of the Ferritins. Biochim. Biophys. Acta 2010, 1800, 719–731. [DOI] [PubMed] [Google Scholar]
- (2).Ford GC; Harrison PM; Rice DW; Smith JM; Treffry A; White JL; Yariv J Ferritin: Design and Formation of an Iron-Storage Molecule. Philos. Trans. R. Soc. London, Ser. B 1984, 304, 551–565. [DOI] [PubMed] [Google Scholar]
- (3).Harrison PM; Andrews SC; Artymiuk PJ; Ford GC; Guest JR; Hirzmann J; Lawson DM; Livingstone JC; Smith JMA; Treffry A; Yewdall SJ Probing Structure-Function Relations In Ferritin And Bacterioferritin. Adv. Inorg. Chem. 1991, 36, 449–486. [Google Scholar]
- (4).Melman A; Bou-Abdallah F Iron Mineralization and Core Dissociation in Mammalian Homopolymeric H-Ferritin: Current Understanding and Future Perspectives. Biochim. Biophys. Acta 2020, 1864, 129700. [DOI] [PubMed] [Google Scholar]
- (5).Mancias JD; Pontano Vaites L; Nissim S; Biancur DE; Kim AJ; Wang X; Liu Y; Goessling W; Kimmelman AC; Harper JW Ferritinophagy via NCOA4 Is Required for Erythropoiesis and Is Regulated by Iron Dependent HERC2-Mediated Proteolysis. ELife 2015, 4, No. e10308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Srivastava A; Flint N; Kreckel H; Gryzik H; Poli M; Arosio P; Bou-Abdallah F Thermodynamic and Kinetic Studies of the Interaction of Nuclear Receptor Coactivator-4 (NCOA4) with Human Ferritin. Biochemistry 2020, 59, 2707–2717. [DOI] [PubMed] [Google Scholar]
- (7).Alkhateeb AA; Connor JR Nuclear Ferritin: A New Role for Ferritin in Cell Biology. Biochim. Biophys. Acta 2010, 1800, 793–797. [DOI] [PubMed] [Google Scholar]
- (8).Harrison PM; Arosio P The Ferritins: Molecular Properties, Iron Storage Function and Cellular Regulation. Biochim. Biophys. Acta 1996, 1275, 161–203. [DOI] [PubMed] [Google Scholar]
- (9).Zhao G; Su M; Chasteen ND μ−1,2-Peroxo Diferric Complex Formation in Horse Spleen Ferritin. A Mixed H/L-Subunit Heteropolymer. J. Mol. Biol. 2005, 352, 467–477. [DOI] [PubMed] [Google Scholar]
- (10).Santambrogio P; Levi S; Cozzi A; Rovida E; Albertini A; Arosio P Production and Characterization of Recombinant Heteropolymers of Human Ferritin H and L Chains. J. Biol. Chem. 1993, 268, 12744–12748. [PubMed] [Google Scholar]
- (11).Levi S; Santambrogio P; Cozzi A; Rovida E; Corsi B; Tamborini E; Spada A; Albertini A; Arosio P The Role of the L-Chain in Ferritin Iron Incorporation. J. Mol. Biol. 1994, 238, 649–654. [DOI] [PubMed] [Google Scholar]
- (12).Hilton RJ; Seare MC; Andros ND; Kenealey Z; Orozco CM; Webb M; Watt RK Phosphate inhibits in vitro Fe3+ loading into transferrin by forming a soluble Fe(III)-phosphate complex: A potential non-transferrin bound iron species. J. Inorg. Biochem. 2012, 110, 1–7. [DOI] [PubMed] [Google Scholar]
- (13).Hann HW; Levy HM; Evans AE Serum Ferritin as a Guide to Therapy in Neuroblastoma. Cancer Res. 1980, 40, 1411–3. [PubMed] [Google Scholar]
- (14).Drysdale JW; Adelman TG; Arosio P; Casareale D; Fitzpatrick P; Harzard JT; Yokota M Human Isoferritins in Normal and Disease States. Semin. Hematol. 1977, 14, 71–88. [PubMed] [Google Scholar]
- (15).Lee J; Seo HY; Jeon ES; Park OS; Lee KM; Park CU; Kim KS Cooperative Activity of Subunits of Human Ferritin Heteropolymers in Escherichia coli. Biochem. Mol. Biol. 2001, 34, 365–370. [Google Scholar]
- (16).Kim HJ; Kim HM; Kim JH; Ryu KS; Park SM; Jahng KY; Yang MS; Kim DH Expression of Heteropolymeric Ferritin Improves Iron Storage in Saccharomyces cerevisiae. Appl. Environ. Microbiol. 2003, 69, 1999–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Rucker P; Torti FM; Torti SV Recombinant ferritin: modulation of subunit stoichiometry in bacterial expression systems. Protein Eng. 1997, 10, 967–973. [DOI] [PubMed] [Google Scholar]
- (18).Carmona F; Poli M; Bertuzzi M; Gianoncelli A; Gangemi F; Arosio P Study of Ferritin Self-assembly and Heteropolymer Formation by the use of Fluorescence Resonance Energy Transfer (FRET) Technology. Biochim. Biophys. Acta 2017, 1861, 522–532. [DOI] [PubMed] [Google Scholar]
- (19).Sevcenco A; Paravidino M; Vrouwenvelder JS; Wolterbeek HT; van Loosdrecht MM; Hagen WR Phosphate and Arsenate Removal Efficiency by Thermos Ferritin Enzyme from Pyrococcus Furiosus; Elsevier, 2015; Vol. 76, pp 181–186. [DOI] [PubMed] [Google Scholar]
- (20).Ebrahimi KH; Hagedoorn PL; Hagen WR Phosphate Accelerates Displacement of Fe(III) by Fe(II) in the Ferroxidase Center of Pyrococcus furiosus Ferritin. FEBS Lett. 2013, 587, 220–225. [DOI] [PubMed] [Google Scholar]
- (21).Parida A; Mohanty A; Kansara BT; Behera RK Impact of Phosphate on Iron Mineralization and Mobilization in Nonheme Bacterioferritin B from Mycobacterium tuberculosis. Inorg. Chem. 2020, 59, 629–641. [DOI] [PubMed] [Google Scholar]
- (22).Hilton RJ; David Andros ND; Watt RK The Ferroxidase Center is Essential for Ferritin Iron Loading in the Presence of Phosphate and Minimizes Side Reactions that Form Fe(III)-phosphate Colloids. BioMetals 2012, 25, 259–273. [DOI] [PubMed] [Google Scholar]
- (23).Trefry A; Harrison PM Incorporation and Release of Inorganic Phosphate in Horse Spleen Ferritin. Biochem. J. 1978, 171, 313–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Noorwali A; Preston CJ; Challa A; Russell RGG Measurement of Intracellular Inorganic Phosphate in Human Blood Red Cells, Leucocytes and Platelets. In Regulation of Phosphate and Mineral Metabolism; Massry SG, Letteri JM, Ritz E, Eds.; Advances in Experimental Medicine and Biology; Springer: Boston, MA, 1982; Vol 151. [DOI] [PubMed] [Google Scholar]
- (25).Ataullakhanov FI; Vitvitsky VM What determines the intracellular ATP concentration. Biosci. Rep. 2002, 22, 501–511. [DOI] [PubMed] [Google Scholar]
- (26).Tonelli M; Pannu N; Manns B Oral phosphate binders in patients with kidney failure. N. Engl. J. Med. 2010, 362, 1312–1324. [DOI] [PubMed] [Google Scholar]
- (27).Friedman EA An introduction to phosphate binders for the treatment of hyperphosphatemia in patients with chronic kidney disease. Kidney Int. 2005, 68, S2–S6. [DOI] [PubMed] [Google Scholar]
- (28).Srivastava AK; Arosio P; Poli M; Bou-Abdallah F A Novel Approach for the Synthesis of Human Heteropolymer Ferritins of Different H to L Subunit Ratios. J. Mol. Biol. 2021, 433, 167198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).McNally JR; Mehlenbacher MR; Luscieti S; Smith GL; Reutovich AA; Maura P; Arosio P; Bou-Abdallah F Mutant L-chain ferritins that cause neuroferritinopathy alter ferritin functionality and iron permeability. Metallomics 2019, 11, 1635–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Bou-Abdallah F; Paliakkara JJ; Melman G; Melman A Reductive Mobilization of Iron from Intact Ferritin: Mechanisms and Physiological Implication. Pharmaceuticals 2018, 11, 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Smith GL; Reutovich AA; Srivastava AK; Reichard RE; Welsh CH; Wilkinson T; Melman A; Bou-Abdallah F Complexation of Ferrous Ions by Ferrozine, 2,2’-Bipyridine and 1,10-Phenanthroline: Implication for the Quantification of Iron in Biological Systems. J. Inorg. Biochem. 2021, 220, 111460. [DOI] [PubMed] [Google Scholar]
- (32).Mehlenbacher M; Poli M; Arosio P; Santambrogio P; Levi S; Chasteen ND; Bou-Abdallah F Iron Oxidation and Core Formation in Recombinant Heteropolymeric Human Ferritins. Biochemistry 2017, 56, 3900–3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Cutler C; Bravo A; Ray AD; Watt RK Iron Loading into Ferritin can be Stimulated or Inhibited by the Presence of Cations and Anions: A Specific Role for Phosphate. J. Inorg. Biochem. 2005, 99, 2270–2275. [DOI] [PubMed] [Google Scholar]
- (34).Chasteen ND; Harrison PM Mineralization in Ferritin: An Efficient Means of Iron Storage. J. Struct. Biol. 1999, 126, 182–194. [DOI] [PubMed] [Google Scholar]
- (35).Polanams J; Ray AD; Watt RK Nanophase Iron Phosphate, Iron Arsenate, Iron Vanadate, and Iron Molybdate Minerals Synthesized within the Protein Cage of Ferritin. Inorg. Chem. 2005, 44, 3203–3209. [DOI] [PubMed] [Google Scholar]
- (36).Cheng YG; Chasteen ND Role of phosphate in initial iron deposition in apoferritin. Biochemistry 1991, 30, 2947–2953. [DOI] [PubMed] [Google Scholar]
- (37).Aitken-Rogers H; Singleton C; Lewin A; Taylor-Gee A; Moore GR; Le Brun NE Effect of phosphate on bacterioferritin-catalysed iron(II) oxidation. J. Biol. Inorg Chem. 2004, 9, 161–170. [DOI] [PubMed] [Google Scholar]
- (38).Huang HQ; Watt RK; Frankel RB; Watt GD Role of phosphate in iron(2+) binding to horse spleen holoferritin. Biochemistry 1993, 32, 1681–1687. [DOI] [PubMed] [Google Scholar]
- (39).Johnson JL; Cannon M; Watt RK; Frankel RB; Watt GD Forming the Phosphate Layer in Reconstituted Horse Spleen Ferritin and the Role of Phosphate in Promoting Core Surface Redox Reactions. Biochemistry 1999, 38, 6706–6713. [DOI] [PubMed] [Google Scholar]
- (40).Li C; Qi X; Li M; Zhao G; Hu X Phosphate facilitates Fe(II) oxidative deposition in pea seed (Pisum sativum) ferritin. Biochimie 2009, 91, 1475–1481. [DOI] [PubMed] [Google Scholar]
- (41).Melman G; Bou-Abdallah F; Vane E; Maura P; Arosio P; Melman A Iron Release from Ferritin by Flavin Nucleotides. Biochim. Biophys. Acta 2013, 1830, 4669–4674. [DOI] [PubMed] [Google Scholar]
- (42).Johnson LE; Wilkinson T; Arosio P; Melman A; Bou-Abdallah F Effect of Chaotropes on the Kinetics of Iron Release from Ferritin by Flavin Nucleotides. Biochim. Biophys. Acta 2017, 1861, 3257–3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Melman A; Bou-Abdallah F Iron Mineralization and Core Dissociation in Mammalian Homopolymeric H-Ferritin: Current Understanding and Future Perspectives. Biochim. Biophys. Acta 2020, 1864, 129700. [DOI] [PubMed] [Google Scholar]
- (44).Theil EC Ferritin: The Protein Nanocage and Iron Biomineral in Health and in Disease. Inorg. Chem. 2013, 52, 12223–12233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Michel FM; Ehm L; Antao SM; Lee PL; Chupas PJ; Liu G; Strongin DR; Schoonen MAA; Phillips BL; Parise JB The Structure of Ferrihydrite, A Nanocrystalline Material. Science 2007, 316, 1726–1729. [DOI] [PubMed] [Google Scholar]
- (46).Theil EC Ferritin protein nanocages use ion channels, catalytic sites, and nucleation channels to manage iron/oxygen chemistry. Curr. Opin. Chem. Biol. 2011, 15, 304–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).St. Pierre T; Tran KC; Webb J; Macey DJ; Heywood BR; Sparks NH; Wade VJ; Manna S; Pootrakul P Organ-specific crystalline structures of ferritin cores in?-thalassemia/hemoglobin E. Biol. Met. 1991, 4, 162–165. [DOI] [PubMed] [Google Scholar]
- (48).López-Castro JD; Delgado JJ; Perez-Omil JA; Gálvez N; Cuesta R; Watt RK; Domínguez-Vera M A new approach to the ferritin iron core growth: influence of the H/L ratio on the core shape. Dalton Trans. 2012, 41, 1320–1324. [DOI] [PubMed] [Google Scholar]
- (49).Mann S; Williams JM; Treffry A; Harrison PM Reconstituted and native iron-cores of bacterioferritin and ferritin. J. Mol. Biol. 1987, 198, 405–416. [DOI] [PubMed] [Google Scholar]
- (50).Wade VJ; Treffry A; Laulhère J-P; Bauminger ER; Cleton MI; Mann S; Briat J-F; Harrison PM Structure and composition of ferritin cores from pea seed (Pisum sativum). Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol 1993, 1161, 91–96. [DOI] [PubMed] [Google Scholar]
- (51).Michel FM; Barrón V; Torrent J; Morales MP; Serna J; Boily JF; Liu Q; Ambrosini A; Cismasu AC; Brown GE Ordered ferrimagnetic form of ferrihydrite reveals links among structure, composition, and magnetism. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 2787–2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Zhao G Phytoferritin and its implications for human health and nutrition. Biochim. Biophys. Acta 2010, 1800, 815–823. [DOI] [PubMed] [Google Scholar]
- (53).Rohrer JS; Islam QT; Watt GD; Sayers DE; Theil EC Iron environment in ferritin with large amounts of phosphate, from Azotobacter vinelandii and horse spleen, analyzed using extended X-ray absorption fine structure (EXAFS). Biochemistry 1990, 29, 259–264. [DOI] [PubMed] [Google Scholar]
- (54).Shi H; Bencze KZ; Stemmler TL; Philpott CC A Cytosolic Iron Chaperone that Delivers Iron to Ferritin. Science 2008, 320, 1207–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Leidgens S; Bullough KZ; Shi H; Li F; Shakoury-Elizeh M; Yabe T; Subramanian P; Hsu E; Natarajan N; Nandal A; Stemmler TL; Philpott CC Each Member of the Poly-r(C)-binding Protein 1 (PCBP) Family Exhibits Iron Chaperone Activity Toward Ferritin. J. Biol. Chem. 2013, 288, 17791–17802. [DOI] [PMC free article] [PubMed] [Google Scholar]