

Abstract
Head and Neck Squamous Cell Carcinoma (HNSCC) is an aggressive epithelial cancer with poor overall response rates to checkpoint inhibitor therapy (CPI) despite CPI being the recommended treatment for recurrent or metastatic HNSCC. Mechanisms of resistance to CPI in HNSCC are poorly understood. To identify drivers of response and resistance to CPI in a unique patient who was believed to have developed three separate HNSCCs, we performed single-cell RNA-seq (scRNA-seq) profiling of two responding lesions and one progressive lesion that developed during CPI. Our results not only suggest interferon-induced APOBEC3-mediated acquired resistance as a mechanism of CPI resistance in the progressing lesion but further, that the lesion in question was actually a metastasis as opposed to a new primary tumor, highlighting the immense power of scRNA-seq as a clinical tool for inferring tumor origin and mechanisms of therapeutic resistance.
Subject terms: Molecular medicine, Translational research, Cancer genomics, Oral cancer
Introduction
Head and neck squamous cell carcinoma (HNSCC) will account for an estimated 54,000 new cases and 11,230 deaths in the United States in 20221. Despite advances in therapy, the prognosis of HNSCC remains poor with a 5-year survival of 67.4%, due to local and distant recurrences and the development of second primary cancers2–5. The development of multiple primary tumors is most commonly ascribed to field cancerization from tobacco exposure6 or from multisite infection from Human Papillomavirus (HPV) in HPV-associated HNSCC7–9. Differentiating a recurrence from a second primary cancer can be challenging and is based on clinical features such as anatomic location and time course, as opposed to genomic features. Importantly, treatment for a recurrent HNSCC differs considerably from that of a new primary tumor.
Anti-PD-1 checkpoint inhibitors (CPI) are now standard-of-care for recurrent or metastatic HNSCC10,11. However, most patients do not have a durable response to CPI12. Mechanisms of resistance to CPI in HNSCC remain poorly understood13. Most studies of CPI resistance focus on the immune microenvironment, particularly the deactivation and exhaustion of T and B cells. Biomarkers for understanding resistance to immunotherapy, including PD-L1, TIM3, LAG3, VISTA, GIFR, and TIGIT, have been identified in different cancers, including HNSCCs14–19. There has been less focus on tumor cell-intrinsic mechanisms of immunotherapy resistance in HNSCC. Recent studies in other tumor types, including work from our group, suggest that apolipoprotein B mRNA-editing enzyme, catalytic polypeptide-like 3 (APOBEC3) family members may be a mechanism of immune evasion20–24. Importantly, APOBEC is known to be active in HNSCC and is a significant source of DNA alterations25–27.
To identify drivers of response and resistance to CPI in a unique patient who was believed to have developed three separate HNSCCs, we performed single-cell RNA-seq (scRNA-seq) profiling of two responding (partial response, PR) lesions and one non-responding (progressive disease, PD) lesion that developed during CPI. Our results not only suggest interferon-induced APOBEC3-mediated acquired resistance as a mechanism of immunotherapy resistance in the progressing lesion but further, that the lesion in question was actually a metastasis as opposed to a new primary tumor, highlighting the immense power of scRNA-seq as a clinical tool for inferring tumor origin and mechanisms of resistance to therapy.
Case
The patient is a 50-year-old male smoker who was diagnosed with a squamous cell carcinoma (SCC) on the right tonsil in 2014 for which he received definitive chemoradiotherapy and subsequently, a salvage right cervical lymphadenectomy (Fig. 1). Four years later the patient developed a second cancer, this time, a left nasopharyngeal SCC (NPSCC; Tumor 1) with metastases to the left neck (Tumor 2) and the lung. Due to the lung metastasis, he was treated with the CPI nivolumab. The lung metastasis had a complete response and the primary NPSCC and neck lymph node metastases had partial responses which remained stable over time. The patient then presented with a new laryngeal mass which developed while on CPI, confirmed by biopsy to be SCC (Tumor 3). Because this cancer had a location in the aerodigestive tract remote from the existing cancer which had responded to CPI, and the classic appearance and anatomic positioning for a mucosally-based SCC arising from the vocal cord, it was presumed to represent a third primary malignancy. This cancer was therefore treated with a total laryngectomy and bilateral cervical lymphadenectomy.
Fig. 1. Timeline of cancer progression and treatment.
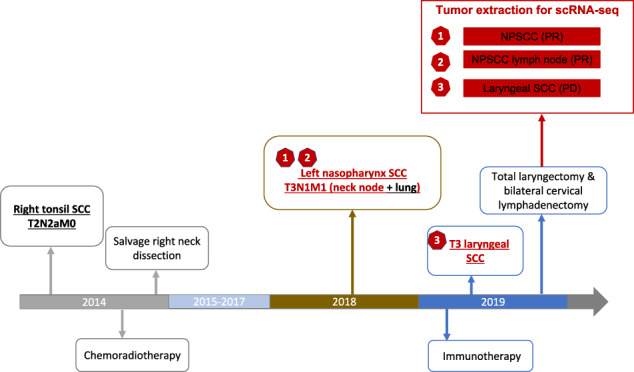
The tumors investigated are colored in red.
Results
To understand the heterogeneity in the patient response to CPI treatment, we performed scRNA-seq on the three isolated lesions (all extracted at the same time): Tumor 1—NPSCC (PR), Tumor 2—NPSCC lymph node (PR), and Tumor 3—laryngeal SCC (PD). After the removal of cells that did not pass QC (see “Methods”), a total of 11,470 cells remained for downstream analyses (NPSCC: n = 3818, NPSCC lymph node: n = 2870, laryngeal SCC: n = 4782), representing 18 different cell clusters (Fig. 2a, b, Supplemental Fig. 1A, B). The three specimens showed differing cell compositions (Fig. 2a, b, Supplemental Fig. 1C, D) with the NPSCC having the largest percentages of Treg and NK cells and the smallest percentage of epithelial cells, the laryngeal SCC having the largest number of B cells, and the NPSCC lymph node dominated by epithelial cells. Copy number alterations (CNAs), using T cells and B cells as reference “normal” cells, were used to confirm the malignant nature of epithelial cells (Fig. 2c). While copy number heterogeneity was observed in three tumors and different malignant cell clusters with distinct gene expression profiles (Fig. 2a, c), surprisingly, the NPSCC lymph node and the newly developed laryngeal SCC demonstrated nearly identical copy number alterations (Pearson’s correlation coefficient r = 0.92; Fig. 2d, e), including typical HNSCC altered regions such as gain of chromosome 3q and loss of 3p, as well as gain of 1q and loss of 11q, which were also observed in subclones of the primary NPSCC tumor (r = 0.75, 0.79 between NPSCC and NPSCC lymph node, laryngeal SCC, respectively). CNA analyses of 20 additional HNSCC’s scRNA-seq from Puram et al.28 showed that five matched pairs of primary tumors and lymph node metastases, have highly concordant CNA profiles, similar to the three lesions in this study, which is not observed in non-paired tumors (Supplemental Fig. 2, Fig. 2d). The probability of observing a CNA similarity of r >= 0.75 between any two random HNSCC was found to be extremely small (one-sided p-value = 0.023; Fig. 2e). Taken together, these analyses strongly suggest that the laryngeal SCC is a metastasis from the NPSCC as opposed to a new primary tumor.
Fig. 2. Copy number alteration profiles suggest the newly developed laryngeal SCC is a metastasis from the NPSCC, as opposed to a new primary tumor.

a UMAP plots and b bar graphs show different cell compositions among three tumors. Dysfunctional CD8+ T cells were split into three subtypes by their marker genes: CD8_dys_CXCL13 with marker gene CXCL13, CD8_dys_HSP with heat shock proteins, and CD8_dys_MTRNR with mitochondrially encoded RNA genes. NPSCC and NPSCC lymph node have distinct components of malignant cells especially Epithelail_1 and Epithelial_2 clusters, demonstrating the inter-tumor heterogeneity. c Heatmap of copy number alteration profiles inferred from two main malignant cell clusters of three tumors using T cell and B cell as “normal” references. Dark red indicates genomic amplifications and blue indicates deletions. The x-axis shows all autosomes in numerical order. The y-axis displays epithelial cells from different tumors (left: Epithelial_1; right: Epithelial_2). Nearly identical copy number alterations observed between the NPSCC lymph node and the laryngeal SCC are similar to those of subclones of the NPSCC tumor (red box). d Heatmap of Pearson’s correlation matrix of 23 HNSCC tumors’ CNA profiles. Correlation coefficients were calculated using mean CNA levels of tumors inferred from scRNA-seq using inferCNV. Five matched pairs of primary tumors (-P) and lymph node metastases (-LN) are in blue font. Matched pairs and three HNSCC tumors investigated in this study show higher similarity within the pairs (red squares), than between unpaired tumors. e Histograms of Pearson’s correlation coefficients of non-paired HNSCC tumors’ CNA profiles, excluding the five matched pairs and three tumors investigated in this study. Three tumors investigated here show higher similarity (r = 0.75, 0.79, and 0.92) than that would be expected randomly, with one-sided p-values of 0.023, 0.013, and 0.0017, respectively.
To identify mechanisms of resistance employed by the metastatic lesion (laryngeal SCC) which developed and progressed on CPI, the two regressing lesions were first combined to increase the statistical power (Fig. 2a) and then compared with the progressing lesion for differential gene expression analyses. Expression of immune-checkpoint factors in T-cell immunity, such as programmed death-1 (PD-1) and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) are critical in mediating response to CPI13. Of common T-cell exhaustion markers, LAG3 and PTPN6 were observed to be differentially expressed in the progressing laryngeal SCC, compared to the regressing lesions (Fig. 3a, b), suggesting the emergence of new inhibitory receptors in exhausted CD8+ T cells as one mechanism of CPI resistance.
Fig. 3. Increased expression of T cell exhaustion markers LAG3 and PTPN6, APOBEC3A, and interferon-inducible protein IFI6 in the progressing laryngeal SCC.

a Volcano plot of differentially expressed genes in dysfunctional CD8+ T cell (CD8_dys_CXCL13) in laryngeal SCC vs NPSCC and NPSCC lymph node shows increased expression of two T cell exhaustion markers LAG3 and PTPN6 (italicized pink font) in laryngeal SCC. Only genes with log2FoldChange larger than 0.25 were shown. Wilcoxon Rank Sum tests were performed and p values were adjusted by Bonferroni correction. Horizontal red line: −log10(0.05); vertical red lines: +/−log2(1.5). b Ridge plots of expression of LAG3 and PTPN6 in tumors with fold changes of 1.80, 1.79 and adjusted p values of 1.3e−5, 1.8e−5, respectively, comparing the laryngeal SCC (PD) to two regressing lesions (PR). c, d Volcano plots of differentially expressed genes in Epithelial_1(C) and Epithelial_2(D) in laryngeal SCC vs NPSCC and NPSCC lymph node. Commonly upregulated and down-regulated genes in laryngeal SCC were shown in italicized pink and dark green fonts, respectively, supporting increased APOBEC3A expression stimulated by interferon signaling. Horizontal red line: −log10(0.05); vertical red lines: +/−log2(1.8).
For malignant cells, we examined differentially expressed genes between the two largest malignant cell clusters in the progressing laryngeal SCC compared to the regressing nasopharyngeal tumors (Fig. 3c, d). Of the top 10 commonly upregulated genes in the laryngeal SCC, APOBEC3A was found to have fold changes of 2, 1.9 and adjusted p values of 9.4e−18, 1.9e−15, in the Epithelial 1 and Epithelial 2 clusters, respectively. An interferon-induced gene, interferon alpha inducible protein 6 (IFI6) also showed higher expression in the laryngeal SCC with fold changes of 2.04, 5.6, and adjusted p values of 5.2e−35, 7.8e−33 in the Epithelial 1 and Epithelial 2 clusters, respectively. Additional interferon-related genes such as IRF1, ISG15, and IFI27 were also upregulated in the Epithelial 2 cluster (Fig. 3d). Interferon signaling pathways were the most enriched signatures in gene set enrichment analysis of differentially expressed genes in the Epithelial 2 cluster (Fig. 4a). The co-upregulation of multiple interferon-related genes in the Epithelial 2 cluster (Figs. 3d and 4b) suggests increased APOBEC3A expression stimulated by interferon signaling. Increased APOBEC3A activity was further validated by the detection of DDOST558C>U RNA editing exclusively in the laryngeal SCC (Fig. 4c, Supplemental Fig. 3). Together, these findings support an interferon-induced upregulation of APOBEC3A as a mechanism of CPI resistance in the progressing Laryngeal SCC.
Fig. 4. Upregulated interferon-related genes and elevated APOBEC3A activity in Epithelial 2 cluster in the newly developed laryngeal tumor suggest an interferon-induced APOBEC3A upregulation driving acquired resistance to CPI.

a Gene set enrichment analysis of differentially expressed genes (absolute fold change >1.8 and adjusted P value <0.05) in Epithelial 2 cluster in laryngeal SCC vs two regressing lesions. b Dot plot of increased expression of APOBEC3A, ATF3, IFI6, IFI27, ISG15, and IRF1 in Epithelial 2 cluster in laryngeal SCC (PD) vs two regressing lesions (PR). c DDOST558C>U RNA editing is exclusively observed in laryngeal SCC, mainly in tumor cells. Epi: epithelial cell; non-Epi: non-epithelial cells.
Discussion
In this n-of-1 study, we demonstrate the potential clinical utility of scRNA-Seq for inferring tumor origin and mechanisms of therapeutic resistance. We performed single-cell transcriptomic profiling of three HNSCC lesions from the same patient, two of which responded to CPI and the third which developed de novo while the patient was receiving CPI. Clinically, the newly developed lesion was felt to be a new primary based on the clinical context, anatomic location, and appearance; however, in depth copy number alteration profiling suggests this was in fact a new metastasis from the existing NPSCC. Chromosomal abnormalities are widespread in human cancers and have been used previously to determine tumor origin. Worsham et al. reported common clonal origin of two synchronous primary HNSCC originating from separate sites based on shared aneuploidy patterns29. Recently, Puram et al. demonstrated concordant copy number alteration profiles between five matched primary HNSCC and lymph node metastases inferred from scRNA-seq, with validation of CNA profiles and their similarity using whole exome sequencing28. This finding holds important clinical implications as the patient was treated surgically for this presumed new cancer, while would have been treated non-surgically, if it was known to be a metastasis, suggesting that the resolution provided by scRNA-Seq in this setting, could have altered clinical care.
We further identified increased expression of T cell exhaustion markers LAG3 and PTPN6, as well as interferon-related genes and elevated APOBEC3A activity in this recurrence, suggesting a combination of immune-regulated and tumor cell-intrinsic APOBEC3A-mediated resistance to CPI in the progressing lesion. APOBEC-mediated mutagenesis is pervasive in multiple tumor types, such as bladder, cervical, breast, lung cancers, and particularly, HNSCC24–27,30–32. APOBEC mutagenesis has been reported to be involved in the acquisition of driver mutations24,33–35 and tumor subclone diversification35–37. Multiple studies have suggested the critical role of APOBEC3A upregulation in therapeutic resistance21,22. Law et al. showed that APOBEC-mediated therapeutic resistance develops while on therapy and suggest that the mechanism is likely C-to-U editing coupled with normal DNA repair process leading to DNA mutations (21). Our group demonstrated that certain therapeutics can induce APOBEC3A expression, leading to sustained mutagenesis in drug-tolerant cancer cells20. In addition, CRISPR-mediated deletion of APOBEC3A significantly reduced double-strand DNA breaks and delayed the emergence of drug resistance, indicating APOBEC3A as a potential target for preventing or delaying acquired resistance in lung cancer therapy20. Interestingly, others have found that APOBEC3A expression induces cellular PD-L1 and cell-surface PD-L1 expression23. APOBEC3A-induced PD-L1 upregulation requires its deaminase activity and suggests APOBEC3A as an immune evasion mechanism and a potential target for improving cancer therapeutic efficacy of PD-1/PD-L1 targeting therapy. While induction of PD-L1 by APOBEC3A would suggest a mechanism of immune evasion and resistance, such an effect should theoretically lead to increased sensitivity to CPI, which was not seen in this patient’s resistant tumor.
Interferons (IFNs) are considered the main modulators of the APOBEC3 family expression25,38. For instance, IFN-α can induce APOBEC3A expression through IFN-sensitive response elements in the putative promoter region of APOBEC3A39. While IFN-α itself was not differentially expressed in the PD laryngeal SCC, IFI6 and IFI27, two type I IFN-α stimulating genes, showed significantly higher expression in this new tumor. IFI6 and IFI27 have been reported to be highly expressed in HNSCC compared to normal tissues40 and have been shown to promote tumor development and metastatic potential of multiple cancers41,42. The detailed mechanisms of IFI6 and IFI27 regulating tumor growth however have not been well elucidated. Our finding of their co-upregulation with APOBEC3A in the newly developed tumor under CPI suggests they may promote tumor progression via upregulating APOBEC3A activity, further inducing APOBEC3A mutagenesis. Taken together, co-expression of high levels of APOBEC3A, IFI6, and IFI27 in the laryngeal SCC suggests that interferon-induced APOBEC3A upregulation could be a potential driver of therapeutic resistance to CPI in HNSCC. Increasing evidence combined with existing knowledge of the role APOBEC plays in mutagenesis in multiple cancer types strongly suggests APOBEC3A as a mediator of tumor response to therapy across multiple tumor types.
In summary, our findings support an interferon-induced APOBEC3A-mediated resistance to CPI in HNSCC, in addition to the deactivation and exhaustion of T cells. This study also demonstrates the power of scRNA sequencing in determining the origin of tumors, which has direct implications for treatment decisions. As this study lacks in vitro and in vivo studies to confirm the computational findings from scRNA-seq, future studies are needed to validate and further explore the role of APOBEC3A in HNSCC therapeutic resistance. Confirmation of the role of APOBEC-mediated resistance could help refine patient selection for enhanced therapeutic efficiency and approaches to overcoming CPI resistance.
Methods
Written informed consent to a protocol approved by the Dana-Farber/Harvard Cancer Center and Boston University Institutional Review Board was obtained prior to sample collection. The study was conducted in accordance with the U.S. Common Rule. Clinical Response was determined by RECIST Criteria Version 1.1.
We utilized single-cell RNA sequencing with the VDJ NextGEM v1.1 10x Genomics Chromium platform to profile three freshly isolated tumor samples collected at the time of total laryngectomy: (1) the nasopharyngeal tumor (PR), (2) the nasopharyngeal cancer lymph node metastasis (PR), and (3) the new laryngeal cancer (PD). Preprocessing of the single-cell RNA-seq data was done using the Cell Ranger pipeline provided by 10x Genomics. Quality control was performed with the following metrics: (1) doublet detection and removal by Scrublet43; (2) cell-level metric: number of genes >= 300 & <= 3000, unique molecular identifier (UMI) counts >= 500, mitochondrial ratio <0.2, and average expression of housekeeping genes >0.8, compiled based on Tirosh et al.44, where gene expression was normalized by log((raw_count_per_gene)/(total_count_per_cell)*10,000 + 1)); (3) gene-level metric: genes detected in <10 cells were excluded. Downstream analyses were performed using the Seurat package v4.0.345–48. Specifically, integrative analysis was applied to cluster together the same cell types across the three tumors, followed by the identification of marker genes for each cluster, and cell types were then manually annotated based on these marker genes (Supplemental Fig. 1A, B). Malignant cells were identified by copy number alterations inferred using inferCNV49, and Pearson’s correlation coefficients between tumors’ mean CNA levels were calculated to quantify their similarity. CNA analyses were also performed on 20 additional HNSCC’s scRNA-seq data from28, including 5 marched tumor-lymph node metastasis pairs, to get the distribution of similarity between paired and non-paired HNSCC’s CNAs as a reference for calculating the probabilities of observing similarity in the three tumors investigated here. Differential gene expression analyses were performed between the progressing lesion and regressing lesions using the Wilcoxon Rank Sum test and p values were adjusted by Bonferroni correction. Molecular Signatures Databse (MSigDB) hallmark gene sets50,51 provided in R package msigdbr (v7.4.1) were used for gene set enrichment analysis ran by clusterProfiler(v3.18.1)52.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary information
Acknowledgements
D.L.F. receives salary support from NIH/NIDCR 1K23DE029811. Funding for work contained in this manuscript was supported by Sundry funds from D.L.F. and L.W.
Author contributions
M.L.: Data analysis, manuscript writing, L.W.: conceptualization, funding MSF: data generation, analysis, manuscript writing, M.S.L.: data generation, conceptualization, manuscript writing. D.L.F.: conceptualization, manuscript writing, funding. All Authors gave final approval of the completed work and are accountable for accuracy and integrity.
Data availability
scRNA-Seq data files are available in GEO, accession number GSE213047.
Competing interests
Daniel L. Faden has received research funding from Bristol-Myers Squibb, holds equity in Illumina, receives in kind contributions from BostonGene and receives consulting fees from Merck, Noetic, Focus on Boston, and Chrystalis Biomedical Advisors. The other authors made no disclosures.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Michael S. Lawrence, Daniel L. Faden.
Supplementary information
The online version contains supplementary material available at 10.1038/s41698-022-00314-3.
References
- 1.Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA Cancer J. Clin. 2022;72:7–33. doi: 10.3322/caac.21708. [DOI] [PubMed] [Google Scholar]
- 2.Howlader, N. et al. SEER Cancer Statistics Review, 1975–2018 (National Cancer Institute, 2021).
- 3.Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2021. CA Cancer J. Clin. 2021;71:7–33. doi: 10.3322/caac.21654. [DOI] [PubMed] [Google Scholar]
- 4.Zhang WL, et al. Susceptibility of multiple primary cancers in patients with head and neck cancer: nature or nurture? Front Oncol. 2019;9:1275. doi: 10.3389/fonc.2019.01275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tabor MP, et al. Multiple head and neck tumors frequently originate from a single preneoplastic lesion. Am. J. Pathol. 2002;161:1051–1060. doi: 10.1016/S0002-9440(10)64266-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johnson DE, et al. Head and neck squamous cell carcinoma. Nat. Rev. Dis. Prim. 2020;6:92. doi: 10.1038/s41572-020-00224-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Faden DL, et al. HPV+ oropharyngeal squamous cell carcinomas from patients with two tumors display synchrony of viral genomes yet discordant mutational profiles and signatures. Carcinogenesis. 2021;42:14–20. doi: 10.1093/carcin/bgaa111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Faden DL, et al. Prospective assessment of multiple HPV-positive oropharyngeal squamous cell carcinomas. Oral. Oncol. 2021;117:105212. doi: 10.1016/j.oraloncology.2021.105212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Strober W, et al. Two for the price of one: prevalence, demographics and treatment implications of multiple HPV mediated Head and Neck Cancers. Oral. Oncol. 2020;100:104475. doi: 10.1016/j.oraloncology.2019.104475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ferris, R. L. et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N. Engl. J. Med.10.1056/NEJMoa1602252 (2016). [DOI] [PMC free article] [PubMed]
- 11.Burtness, B. et al. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): a randomised, open-label, phase 3 study. Lancet10.1016/s0140-6736(19)32591-7 (2019). [DOI] [PubMed]
- 12.Saada-Bouzid E, et al. Hyperprogression during anti-PD-1/PD-L1 therapy in patients with recurrent and/or metastatic head and neck squamous cell carcinoma. Ann. Oncol. 2017;28:1605–1611. doi: 10.1093/annonc/mdx178. [DOI] [PubMed] [Google Scholar]
- 13.Dos Santos LV, Abrahao CM, William WN., Jr. Overcoming resistance to immune checkpoint inhibitors in head and neck squamous cell carcinomas. Front. Oncol. 2021;11:596290. doi: 10.3389/fonc.2021.596290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deng WW, et al. LAG-3 confers poor prognosis and its blockade reshapes antitumor response in head and neck squamous cell carcinoma. Oncoimmunology. 2016;5:e1239005. doi: 10.1080/2162402X.2016.1239005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Herbst RS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563–567. doi: 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu JF, et al. T-cell immunoglobulin mucin 3 blockade drives an antitumor immune response in head and neck cancer. Mol. Oncol. 2017;11:235–247. doi: 10.1002/1878-0261.12029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schaer DA, et al. GITR pathway activation abrogates tumor immune suppression through loss of regulatory T cell lineage stability. Cancer Immunol. Res. 2013;1:320–331. doi: 10.1158/2326-6066.CIR-13-0086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu L, et al. Expression of VISTA correlated with immunosuppression and synergized with CD8 to predict survival in human oral squamous cell carcinoma. Cancer Immunol. Immunother. 2017;66:627–636. doi: 10.1007/s00262-017-1968-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu L, et al. Blockade of TIGIT/CD155 signaling reverses T-cell exhaustion and enhances antitumor capability in head and neck squamous cell carcinoma. Cancer Immunol. Res. 2019;7:1700–1713. doi: 10.1158/2326-6066.CIR-18-0725. [DOI] [PubMed] [Google Scholar]
- 20.Isozaki, H. et al. APOBEC3A drives acquired resistance to targeted therapies in non-small cell lung cancer. Preprint at bioRxiv10.1101/2021.01.20.426852 (2021).
- 21.Law EK, et al. The DNA cytosine deaminase APOBEC3B promotes tamoxifen resistance in ER-positive breast cancer. Sci. Adv. 2016;2:e1601737. doi: 10.1126/sciadv.1601737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sieuwerts AM, et al. Elevated APOBEC3B correlates with poor outcomes for estrogen-receptor-positive breast cancers. Horm. Cancer. 2014;5:405–413. doi: 10.1007/s12672-014-0196-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao, K. et al. Cytidine deaminase APOBEC3A regulates PD-L1 expression in cancer cells in a JNK/c-JUN-dependent manner. Mol. Cancer Res.10.1158/1541-7786.MCR-21-0219 (2021). [DOI] [PMC free article] [PubMed]
- 24.Faden DL, et al. Multi-modality analysis supports APOBEC as a major source of mutations in head and neck squamous cell carcinoma. Oral. Oncol. 2017;74:8–14. doi: 10.1016/j.oraloncology.2017.09.002. [DOI] [PubMed] [Google Scholar]
- 25.Faden DL, et al. APOBEC mutagenesis is tightly linked to the immune landscape and immunotherapy biomarkers in head and neck squamous cell carcinoma. Oral. Oncol. 2019;96:140–147. doi: 10.1016/j.oraloncology.2019.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Faden, D. L. et al. APOBEC mutagenesis is concordant between tumor and viral genomes in HPV-positive head and neck squamous cell carcinoma. Viruses13, 10.3390/v13081666 (2021). [DOI] [PMC free article] [PubMed]
- 27.Roberts SA, et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat. Genet. 2013;45:970–976. doi: 10.1038/ng.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Puram SV, et al. Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell. 2017;171:1611–1624.e1624. doi: 10.1016/j.cell.2017.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Worsham MJ, et al. Common clonal origin of synchronous primary head and neck squamous cell carcinomas: analysis by tumor karyotypes and fluorescence in situ hybridization. Hum. Pathol. 1995;26:251–261. doi: 10.1016/0046-8177(95)90054-3. [DOI] [PubMed] [Google Scholar]
- 30.Buisson, R. et al. Passenger hotspot mutations in cancer driven by APOBEC3A and mesoscale genomic features. Science364, 10.1126/science.aaw2872 (2019). [DOI] [PMC free article] [PubMed]
- 31.Buisson R, Lawrence MS, Benes CH, Zou L. APOBEC3A and APOBEC3B activities render cancer cells susceptible to ATR inhibition. Cancer Res. 2017;77:4567–4578. doi: 10.1158/0008-5472.CAN-16-3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Langenbucher A, et al. An extended APOBEC3A mutation signature in cancer. Nat. Commun. 2021;12:1602. doi: 10.1038/s41467-021-21891-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cancer Genome Atlas Research, N. et al. Integrated genomic and molecular characterization of cervical cancer. Nature. 2017;543:378–384. doi: 10.1038/nature21386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Henderson S, Chakravarthy A, Su X, Boshoff C, Fenton TR. APOBEC-mediated cytosine deamination links PIK3CA helical domain mutations to human papillomavirus-driven tumor development. Cell Rep. 2014;7:1833–1841. doi: 10.1016/j.celrep.2014.05.012. [DOI] [PubMed] [Google Scholar]
- 35.Jamal-Hanjani M, et al. Tracking the evolution of non-small-cell lung cancer. N. Engl. J. Med. 2017;376:2109–2121. doi: 10.1056/NEJMoa1616288. [DOI] [PubMed] [Google Scholar]
- 36.de Bruin EC, et al. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science. 2014;346:251–256. doi: 10.1126/science.1253462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McGranahan N, et al. Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci. Transl. Med. 2015;7:283ra254. doi: 10.1126/scitranslmed.aaa1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koning FA, et al. Defining APOBEC3 expression patterns in human tissues and hematopoietic cell subsets. J. Virol. 2009;83:9474–9485. doi: 10.1128/JVI.01089-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peng G, Lei KJ, Jin W, Greenwell-Wild T, Wahl SM. Induction of APOBEC3 family proteins, a defensive maneuver underlying interferon-induced anti-HIV-1 activity. J. Exp. Med. 2006;203:41–46. doi: 10.1084/jem.20051512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu L, et al. ATF3 downmodulates its new targets IFI6 and IFI27 to suppress the growth and migration of tongue squamous cell carcinoma cells. PLoS Genet. 2021;17:e1009283. doi: 10.1371/journal.pgen.1009283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cheriyath V, et al. G1P3 (IFI6), a mitochondrial localised antiapoptotic protein, promotes metastatic potential of breast cancer cells through mtROS. Br. J. Cancer. 2018;119:52–64. doi: 10.1038/s41416-018-0137-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang H, et al. Knockdown of IFI27 inhibits cell proliferation and invasion in oral squamous cell carcinoma. World J. Surg. Oncol. 2018;16:64. doi: 10.1186/s12957-018-1371-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wolock SL, Lopez R, Klein AM. Scrublet: computational identification of cell doublets in single-cell transcriptomic data. Cell Syst. 2019;8:281–291 e289. doi: 10.1016/j.cels.2018.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tirosh I, et al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature. 2016;539:309–313. doi: 10.1038/nature20123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hao Y, et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–3587.e3529. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Satija R, Farrell JA, Gennert D, Schier AF, Regev A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 2015;33:495–502. doi: 10.1038/nbt.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stuart T, et al. Comprehensive integration of single-cell data. Cell. 2019;177:1888–1902 e1821. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tickle, T., Tirosh, I., Georgescu, C., Brown, M. & Haas, B. InferCNV of the Trinity CTAT Project. https://github.com/broadinstitute/inferCNV (2019).
- 50.Liberzon A, et al. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
scRNA-Seq data files are available in GEO, accession number GSE213047.