

Short abstract
Content available: Audio Recording
Listen to an audio presentation of this article.
INTRODUCTION
Primary sclerosing cholangitis (PSC) and autoimmune hepatitis (AIH) are the two most common autoimmune diseases to affect the pediatric liver. Both have recognized laboratory findings that can be helpful in clinical diagnosis along with typical autoantibody serologies of AIH and imaging patterns for PSC. Pathologic examination of liver biopsy is often a part of the diagnostic process. Both diseases PSC and AIH often demonstrate classic histologic features. However, the histologic differential diagnosis of PSC and AIH can be broad, including other infectious and inflammatory processes of the liver, the reciprocal diagnosis of PSC/AIH, PSC/AIH overlap syndromes, and in the post‐transplant setting rejection and de novo AIH of the liver. Given that both AIH and PSC are progressive conditions, with different mainstays of therapy, accurate diagnosis is important. Therefore, this review is focused on highlighting the common histologic presentations of AIH and PSC, as well as discussing the histology of some of the other less common autoimmune conditions of the pediatric liver that are often in the differential diagnosis.
AIH
AIH in children is typically classified clinically by the presence of serum autoantibodies antinuclear antibody (ANA) and/or anti‐smooth muscle antibody (AIH type 1) or anti‐liver kidney microsomal type 1 antibody and/or anti‐liver cytosol type 1 antibody (AIH type 2), 1 but antibody‐negative cases have also been reported. 2 Additionally, anti‐soluble liver antigen/liver‐pancreas antibodies, can be seen in either AIH type 1 or AIH type 2. 3 Regardless of serologic type, the classic histologic findings on liver biopsy are similar.These biopsies demonstrate portal and lobular inflammation. The inflammation is typically composed of lymphocytes and, most commonly, conspicuous plasma cells (Figure 1A); however, in some cases, plasma cells may be inconspicuous or even absent. The portal inflammation typically creates “interface hepatitis” where the limiting plate is disrupted and the inflammation involves the adjacent liver parenchyma; concomitant hepatocyte necrosis may or may not be present (Figure 1B). Lobular inflammation may also be present from mild to quite severe, with either scattered necrotic hepatocytes (acidophil bodies), or in more severe cases centrilobular and/or bridging necrosis (Figure 1C). Less common features include rosette formation of hepatocytes and/or emperipolesis of inflammatory cells by hepatocytes (Figure 1D); these features may help distinguish AIH from viral hepatitis. 4 More commonly reported in pediatric AIH than adult AIH, giant cell change of hepatocytes and hyaline droplets in Kupffer cells have also been described in a subset of cases. A significant portion of patients with AIH will have fibrosis at the time of initial diagnostic liver biopsy, and a subset with cirrhosis (more common in type 1 AIH). Fibrosis of the liver in AIH processes is thus staged/evaluated similarly to several other forms of chronic hepatitis, from portal fibrosis, to septate formation/periportal fibrosis, bridging fibrosis, and lastly cirrhosis.
FIGURE 1.
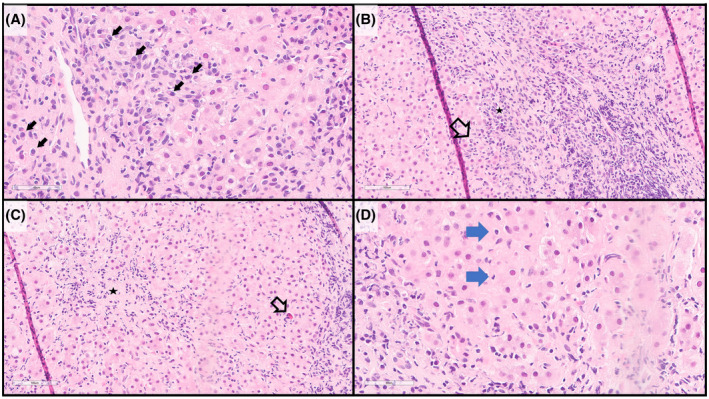
Histology of AIH (hematoxylin and eosin stains). (A) the inflammatory infiltrate often includes abundant and conspicuous plasma cells (arrows). (B) Interface hepatitis is characteristic, with the inflammatory infiltrate extending beyond the limiting plate into the lobule (arrow), including abundant and clustered plasma cells (star). (C) Lobular inflammation (star) with scattered acidophil bodies (necrotic hepatocytes) may also be present (arrow). (D) Emperipolesis of inflammatory cells by hepatocytes may be observed (arrows)
The differential diagnosis of AIH includes chronic viral hepatitis, Wilson's disease, drug‐induced liver injury, or PSC‐AIH overlap syndrome. Many of the aforementioned may be excluded with laboratory testing (viral hepatitis, Wilson's disease) and/or clinical correlation (drug‐induced liver injury). It is important to be aware that plasma cells may not always be abundant or present in the biopsy, and biopsies from patients receiving immunosuppressive agents may show decreased amounts of inflammation overall. Additionally, mild bile duct inflammation and proliferation may occur in AIH, and these features do not necessitate a concurrent diagnosis of PSC; however, duct destruction is generally not seen in AIH alone (Table 1).
TABLE 1.
Comparison of histologic, clinical, and laboratory findings in pediatric autoimmune liver disease
| Histologic features | Clinical findings | Laboratory studies | |
|---|---|---|---|
| AIH |
|
|
|
| PSC |
|
|
|
| ASC |
|
Younger at presentation than classic PSC; associated with IBD |
|
PSC
PSC is primarily seen as a rare, male‐predominant pauci‐immune disease with an association with inflammatory bowel disease (a majority of pediatric PSC patients have IBD). 5 Other causes of sclerosing cholangitis in children include manifestations of cystic fibrosis, mutations in ABCB4 (MDR3), and immunodeficiencies. 6 The majority of PSC cases demonstrate involvement of large hepatic and extrahepatic bile ducts. This large duct involvement then shows characteristic imaging findings on endoscopic retrograde cholangiopancreatography (ERCP) and/or magnetic resonance cholangiopancreatography (MRCP) (the latter favored in the pediatric population), with “beaded appearance” secondary to alternating stricture and dilatation of bile ducts. However, small duct PSC can also occur and is more prevalent in children than in adults, with approximately 15%–35% of pediatric PSC presenting with small duct disease. 7 Small duct only involvement may not demonstrate characteristic imaging findings, and diagnosis relies more heavily on tissue biopsy. Regardless of underling etiologies and/or small or large duct involvement, similar histopathology is observed.
The histopathologic features of PSC are that of a cholestatic pattern of injury. The characteristic “fibro‐obliterative duct lesion” typically involves medium sized ducts, or small sized ducts (in small duct PSC), in a discontinuous fashion and, thus, may not be seen in every biopsy. When present, the fibro‐obliterative duct lesions appear as a round fibrotic scar in place of the bile duct. Alternatively, a spectrum of duct atrophy with concentric periductal fibrosis (A.K.A. “onion‐skin fibrosis”) may also be seen (Figure 2A, B). Other features include duct injury, ductular proliferation (Figure 2C), duct loss, cholestasis, and extravasated bile (Figure 2D), which may lead to xanthomatous reaction. Inflammation is typically mild (in contrast to AIH), and often when present it is concentrated around the biliary epithelium. Similar to AIH, fibrosis is progressive from portal, periportal, bridging, and nodule‐forming (cirrhosis) (Figure 2E).
FIGURE 2.
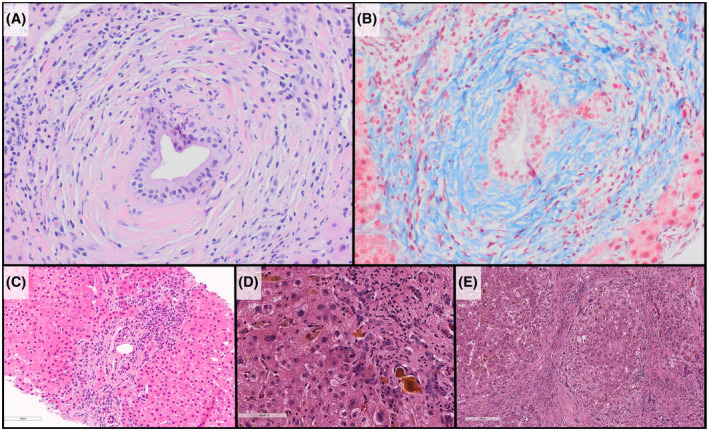
Histology of PSC. (A) Characteristic periductal fibrosis is seen in portal tracts (hematoxylin and eosin stains). (B) Masson trichrome stain highlights the dense collagen fibrosis surrounding the residual ductal epithelium. (C) Ductular reaction/proliferation is seen, likely as a result of downstream biliary obstruction. (D) Longstanding PSC leads to a cholestatic pattern of cirrhosis with inspissated bile, extravasated bile, and canalicular bile all be seen in this image. (E) Cirrhotic nodules, in an explanted liver of a child with history of PSC
The histopathologic differential diagnosis of PSC is namely to exclude a secondary cause of sclerosing cholangitis, such as a duct obstructive process (e.g., Caroli disease, stones), immune deficiency, or cystic fibrosis. If the biopsy shows significant inflammation, features of hepatitis, and/or positive serologies are present, consideration of autoimmune sclerosing cholangitis should be made (Table 1).
AUTOIMMUNE SCLEROSING CHOLANGITIS (AIH AND PSC OVERLAP SYNDROME)
The term “overlap syndrome” has been used to describe autoimmune hepatopathies with overlapping clinical, serologic, or histologic characteristics. In pediatrics, this typically involves overlap of AIH with PSC, which is also known as autoimmune sclerosing cholangitis. Histologically, features of both cholestatic and hepatitic patterns of injury may be seen. However, identification of both histologic components may be challenging, and a high index of suspicion based on clinical and/or laboratory findings is often required for appropriate diagnosis. Of particular challenge is often the identification of biliary injury and/or loss when the characteristic concentric periductal fibrosis of PSC is not seen/sampled on liver biopsy; as such features of AIH may predominate in the liver biopsy from such cases (Table 1). Discussion between the pediatric pathologist and gastroenterologist is often key for this diagnosis.
POST‐LIVER TRANSPLANT DE NOVO AIH (AIH‐LIKE DISEASE IN THE TRANSPLANTED LIVER)
De novo AIH occurs in the setting of a post‐transplant patient as a form of liver allograft dysfunction of unclear etiology, where the original reason for transplantation was for something other than AIH. Histologically, de novo AIH shows similar features to AIH to include interface hepatitis with lymphoplasmacytic infiltration, with varying degrees of fibrosis. In patients with a history of AIH, it is best to prescribe these histologic findings of recurrent AIH. However, some have observed a wider range of histologic patterns in cases of de novo hepatitis, which alone may have limited specificity, and as such, correlation with serologic findings is critical. 8 Of note, while de novo AIH was first described in pediatric patients, a similar histologic pattern is seen in adult allograft livers. However, in adults this is hypothesized to have a different pathobiology and is now classified by some as form of atypical rejection, “plasma cell‐rich rejection.” 9
SUMMARY
In addition to clinical history, laboratory studies, and imaging findings, histopathologic examination of liver biopsy continues to play a critical role in the diagnosis and classification of autoimmune liver diseases in the pediatric setting. While some pediatric liver biopsies may show classic or characteristic pathologic features of autoimmune liver disease, many cases may have a degree of ambiguity necessitating a partnership between the hepatology and pathology teams, and all cases ultimately require clinicopathologic correlation for definitive patient diagnosis of underlying disease process.
CONFLICT OF INTEREST
All authors declare no conflicts of interest and/or disclosures with relationship to preparation of this manuscript.
Mau B, Hakar M, Lin HC, Davis JL. A review of histopathologic features of pediatric autoimmune liver disease. Clinical Liver Disease. 2022;20:116–119. 10.1002/cld.1228
REFERENCES
- 1. Homberg JC, Abuaf N, Bernard O, Islam S, Alvarez F, Khalil SH, et al. Chronic active hepatitis associated with antiliver/kidney microsome antibody type 1: a second type of “autoimmune” hepatitis. Hepatology. 1987;7(6):1333–9. [DOI] [PubMed] [Google Scholar]
- 2. Sokollik C, McLin VA, Vergani D, Terziroli Beretta‐Piccoli B, Mieli‐Vergani G. Juvenile autoimmune hepatitis: a comprehensive review. J Autoimmun. 2018;95:69–76. [DOI] [PubMed] [Google Scholar]
- 3. Vitozzi S, Djilali‐Saiah I, Lapierre P, Alvarez F. Anti‐soluble liver antigen/liver‐pancreas (SLA/LP) antibodies in pediatric patients with autoimmune hepatitis. Autoimmunity. 2002;35(8):485–92. [DOI] [PubMed] [Google Scholar]
- 4. de Boer YS, van Nieuwkerk CM, Witte BI, Mulder CJ, Bouma G, Bloemena E. Assessment of the histopathological key features in autoimmune hepatitis. Histopathology. 2015;66(3):351–62. [DOI] [PubMed] [Google Scholar]
- 5. Miloh T, Arnon R, Shneider B, Suchy F, Kerkar N. A retrospective single‐center review of primary sclerosing cholangitis in children. Clin Gastroenterol Hepatol. 2009;7(2):239–45. [DOI] [PubMed] [Google Scholar]
- 6. Mieli‐Vergani G, Vergani D, Baumann U, Czubkowski P, Debray D, Dezsofi A, et al. Diagnosis and Management of Pediatric Autoimmune Liver Disease: ESPGHAN hepatology committee position statement. J Pediatr Gastroenterol Nutr. 2018;66(2):345–60. [DOI] [PubMed] [Google Scholar]
- 7. Valentino PL, Wiggins S, Harney S, Raza R, Lee CK, Jonas MM. The natural history of primary Sclerosing cholangitis in children: a large single‐center longitudinal cohort study. J Pediatr Gastroenterol Nutr. 2016;63(6):603–9. [DOI] [PubMed] [Google Scholar]
- 8. Pongpaibul A, Venick RS, McDiarmid SV, Lassman CR. Histopathology of de novo autoimmune hepatitis. Liver Transpl. 2012;18(7):811–8. [DOI] [PubMed] [Google Scholar]
- 9. Kerkar N, Vergani D. De novo autoimmune hepatitis ‐is this different in adults compared to children? J Autoimmun. 2018;95:26–33. [DOI] [PubMed] [Google Scholar]