

Abstract
Viruses rely on an array of cellular metabolites to replicate and form progeny virions. One set of these molecules, polyamines, are small aliphatic molecules, which are abundant in most cells, that support virus infection; however, the precise roles of polyamines in virus infection remain incompletely understood. Recent work demonstrated that polyamine metabolism supports cellular cholesterol synthesis through translation of the key transcription factor SREBP2. Here, we show that the bunyavirus Rift Valley fever virus (RVFV) relies on both cholesterol and polyamines for virus infection. Depletion of cellular cholesterol or interruption of cholesterol trafficking negatively impacts RVFV infection. Cholesterol is incorporated into RVFV virions and mediates their infectivity in a polyamine-dependent manner; we find that the virus derived from polyamine-depleted cells lacks cholesterol within the virion membrane. Conversely, we find that virion-associated cholesterol is linked to the incorporation of spermidine within the virion. Our prior work demonstrated that polyamines facilitate pH-mediated fusion and genome release, which may be a consequence of cholesterol depletion within virions. Thus, our work highlights the metabolic connection between polyamines and cholesterol synthesis to impact bunyavirus infection. These data demonstrate the connectedness between cellular metabolic pathways and reveal potential avenues of therapeutic intervention.
Keywords: polyamines, bunyaviruses, Rift Valley fever virus, cholesterol, metabolism
Bunyaviruses are common pathogens and are considered the largest family of viruses.1 The bunyavirus Rift Valley fever virus (RVFV) is a significant human pathogen causing regular outbreaks in Africa and the Middle East. The mosquito-transmitted virus is associated with significant pathologies in humans and ruminants, including spontaneous abortion and encephalitis. The virus has exhibited significant spreading, especially as climate change accelerates and mosquito habitats change, though it has yet to spread beyond Africa and the Arabian peninsula.2 Fortunately, vaccine development for RVFV has shown promise3,4 with significant drawbacks, specifically to the lack of approval for human use. Thus, the ability to effectively combat RVFV as well as other bunyaviruses requires a better understanding of their fundamental biology leading to a greater understanding of their pathogenesis.
The ability of RVFV to infect and cause pathology relies on its ability to remain infectious during transmission. RVFV virions are composed of three genome segments sheathed by a nucleoprotein (N). These genomes are encapsulated in a lipid membrane studded with Gn and Gc, the viral glycoproteins that mediate attachment and entry of virions to susceptible cells.5 RVFV, like other bunyaviruses, enters cells by engagement of cellular attachment factors and receptors6 followed by endocytosis.7 Upon acidification of the endosome, the viral lipid envelope fuses with the endosome, releasing viral genomes and initiating infection.
We recently demonstrated that, during the process of viral entry, polyamines play an important role in mediating infectivity.8 Polyamines are small, positively charged biomolecules within cells, comprising carbon chains and amino groups. These molecules are involved in cell cycling, cellular transcription and translation, and modulating signaling pathways.9 Additionally, polyamines are critical for multiple functions in RNA viruses,10,11 including enhancing polymerase12 and protease13,14 activity and mediating cellular attachment.15 In bunyaviruses, polyamines associate with virions and mediate entry,8 maintaining infectivity16 and promoting infection upon endosomal acidification.
Fusion of the virion membrane with the cellular membrane upon exposure to reduced pH mediates release of the bunyavirus genome and infection.17 The composition of the RVFV membrane remains incompletely described, though we know that certain host cell-derived molecules are incorporated into the virion membrane and affect its function. One of the critical components of cellular (and frequently viral) membranes is cholesterol,18–22 which mediates membrane flexibility and fluidity. These two properties are crucial to cellular and virion membrane function, and thus, bunyaviruses rely on cellular cholesterol for the production of progeny virions as well as their infectivity following release from infected cells.18,19,21 How cholesterol is incorporated and how it functions in bunyavirus infection remains incompletely understood.
We recently found that polyamines facilitate cholesterol synthesis to impact enterovirus infection.23 Here, we investigated the connections between polyamines and cholesterol synthesis in the context of bunyavirus infection using RVFV as our model system. We found that RVFV is sensitive to manipulations of cellular cholesterol, specifically at early stages in infection. We further find that polyamines facilitate cholesterol incorporation into RVFV virions and virions derived from polyamine-depleted conditions are insensitive to cholesterol inhibitors. Additionally, we find that polyamine incorporation into RVFV virions relies on virion-associated cholesterol. Thus, we find that cholesterol and polyamines (specifically spermidine) are intricately linked in RVFV virions and mediate viral attachment and entry. These data have important implications for the composition of RVFV virions as well as the functions of small molecules like polyamines and cholesterol in the infectivity of bunyavirus particles.
RESULTS
Cellular Cholesterol Facilitates Rift Valley Fever Virus Infection.
To confirm the role for cholesterol in RVFV infection, we tested the inhibitor U18666A,24 which targets NPC1-mediated cholesterol transport from the lysosome to the cell’s plasma membrane and impacts cellular cholesterol distribution, for antiviral activity. Prior work demonstrated antiviral activity of U18666A in other viral families.25–29 To test for antiviral activity against RVFV, we treated Vero cells with increasing doses of U18666A for 24 h prior to infection at a multiplicity of infection (MOI) of 0.1 plaque-forming units (PFUs) per cell. As previously shown with other viruses, we observed a dose-dependent decrease in RVFV titers at 48 h post infection (hpi) with an IC50 value of 9.23 μM (Figure 1A). We observed no significant toxicity of U18666A at the doses used (Figure 1B), but we did observe significant toxicity over 40 μM with a CC50 value of 85.6 μM. To determine if U18666A was antiviral over several rounds of infection, we treated Vero cells with 10 μM U18666A and measured viral titers over several days of infection (Figure 1C). We observed that U18666A-treated cells produced reduced viral titers over the entire time course. The half-life of U18666A (in serum) is approximately 22.4 h,30 suggesting that some of the loss of antiviral activity later in infection could be due to degradation of the molecule.
Figure 1.
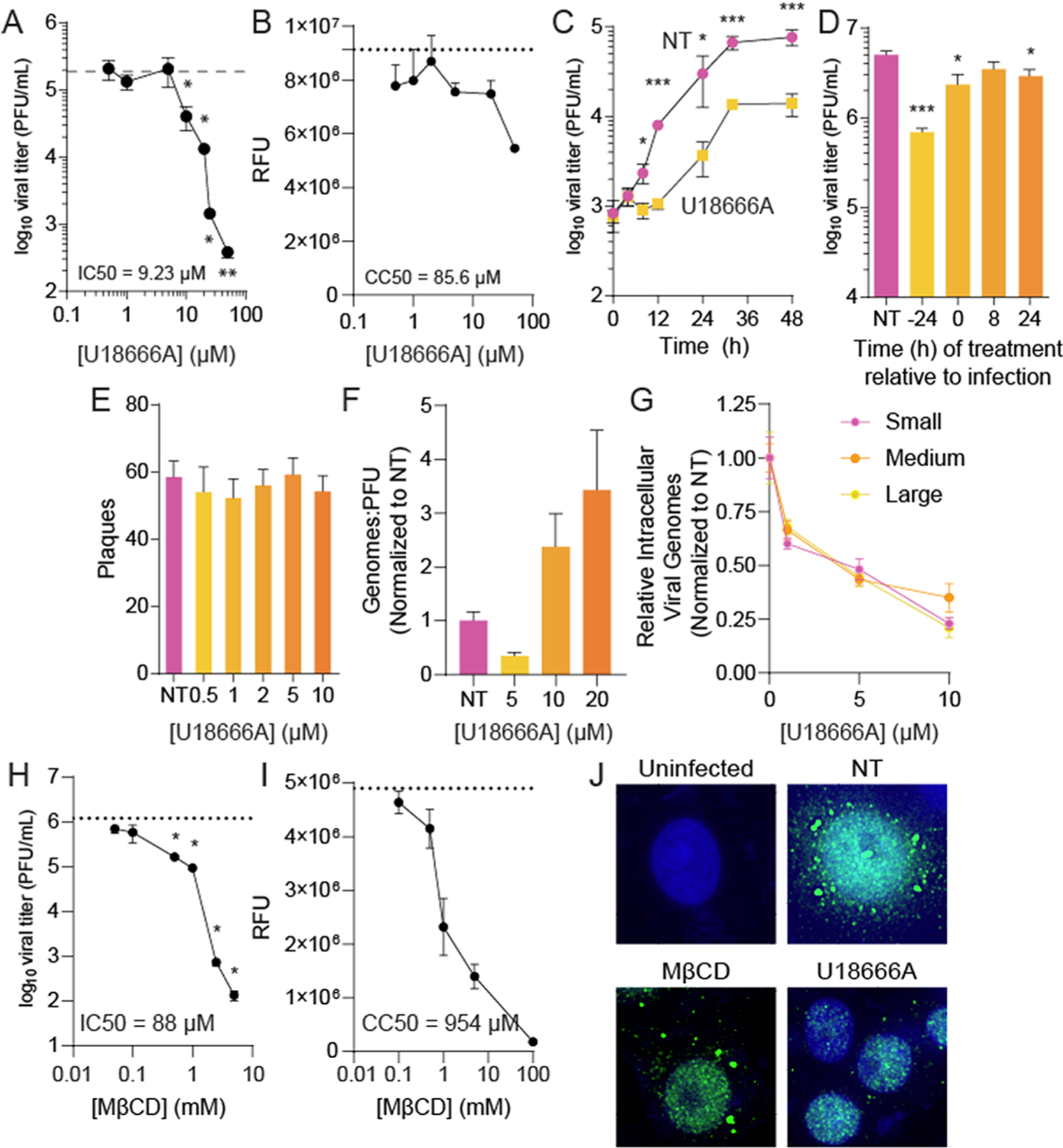
Manipulating cellular cholesterol inhibits RVFV infection. (A) Vero cells were treated with increasing doses of U18666A for 24 h prior to infection with RVFV at MOI 0.1. Viral titers were measured at 48 hpi by the plaque assay. (B) Cells were treated as in A, and cellular viability was measured 64 h after treatment. (C) Cells were treated with 10 μM U18666A prior to infection with RVFV at MOI 0.01. Viral titers were measured at the times indicated by the plaque assay. (D) Cells were treated with 10 μM U18666A at the times indicated relative to infection with RVFV at MOI 5. Viral titers were determined at 24 hpi. (E) Cells were treated with increasing doses of U18666A, and RVFV attachment was measured by a plaque-formation binding assay. (F) The ratio of viral genomes measured by qRT-PCR to infectious virus as measured by the plaque assay was measured from samples produced as in A. (G) Cells were treated with U18666A, and cell-associated viral genomes were measured by qRT-PCR at 24 hpi for the small, medium, and large genome segments. (H) Cells were treated for 4 h with increasing doses of MβCD prior to infection with RVFV at MOI 0.1. Viral titers were measured at 48 hpi. (I) Cells were treated as in H, and cellular viability was measured. (J) Cells were treated with 20 μM U18666A or 1 mM MβCD and infected at MOI 5. Cells were stained for viral polymerase (L, green) at 16 hpi. *p < 0.05, **p < 0.01, ***p < 0.001 by Student’s T-test (N ≥ 3).
To determine the step(s) of virus infection affected by U18666A and the subsequent defective cholesterol transport, we added U18666A to Vero cells at distinct times post infection and measured viral titers at 24 hpi. We observed that treating with U18666A was potently antiviral when it is added to the cell culture before infection and the antiviral effect was diminished when added to the cells at later points in infection (Figure 1D). These data suggest a role for cholesterol in early stages of infection, and thus, we tested whether U18666A might inhibit viral attachment to susceptible cells, as observed for other viruses.28,29 To measure attachment, we treated cells with increasing doses of U18666A and performed a binding assay in which we applied a viral inoculum of 1000 pfu for 5 min. Afterward, we washed the cells with PBS and overlaid with an agarose-containing medium, lacking U18666A, thus limiting cellular exposure to U18666A to immediately before and during the attachment of virus to the cells. After four days, the agarose-containing medium was removed, the cells were fixed and stained, and plaques were revealed, indicating attachment and infection. We found that U18666A had no discernible effect on the number of plaques formed (Figure 1E), suggesting that the inhibitor did not impact viral attachment to susceptible cells.
Since U18666A changes cellular cholesterol trafficking, we hypothesized that this would result in changes to RVFV infectivity, resulting in altered particle-to-PFU ratios. To test whether the virus derived from U18666A-treated cells had changes in specific infectivity, we measured the number of viral genomes compared to the viral titer (genomes:PFU ratio) from infected, treated cells. Despite viral titers dropping 100-fold (Figure 1A) with U18666A treatment, we observed only a modest change in specific infectivity with the genome-to-PFU ratio increasing only threefold (Figure 1F), suggesting that U18666A’s activity did not directly impact virion infectivity. When we measured cell-associated viral genomes for all three viral segments (small, medium, and large), we observed a significant reduction in abundance with U18666A treatment, correlating with the reduction in viral titers (Figure 1G).
To confirm our results with U18666A, which works by preventing cholesterol trafficking to the cellular plasma membrane,24 we used methyl-β-cyclodextran (MβCD), which functions to remove cholesterol from membranes. We treated cells with increasing doses of MβCD for 4 h prior to infection with RVFV, at which time the MβCD was removed from cells, and we found that MβCD treatment reduced viral titers in a dose-dependent manner with an IC50 value of 88 μM (Figure 1H). To confirm that MβCD was not toxic at these concentrations, we measured cellular viability and found a CC50 value of 954 μM (Figure 1I).
We next considered if U18666A might contribute to the structure of viral replication compartments upon the establishment of productive infection. We infected Vero cells, treated with 20 μM U18666A or left untreated, with RVFV at MOI 5 for 16 h, and subsequently fixed and stained for viral polymerase (L). We find that staining for L was sparser in U18666A-treated cells compared to untreated cells and the signal was primarily nuclear, fitting with the reduced titers observed in Figure 1A. This suggests that U18666A and its effects on cellular cholesterol may disturb viral replication compartments, as measured by L staining, perhaps when establishing these replication compartments. With MβCD treatment, we observed fewer infected cells, though the polymerase stains were indistinguishable from untreated (NT) conditions.
Finally, we tested these inhibitors in primary cells: murine embryonic fibroblasts (MEFs; Figure 2A,B) and human foreskin fibroblasts (HFFs; Figure 2C,D). We find that both U18666A and MβCD treatments significantly reduced RVFV replication in these cell types, similar to that in Vero cells. Thus, plasma membrane-associated cholesterol is important for RVFV infection and replication, as demonstrated by the two molecules that affect cholesterol levels via distinct mechanisms, including those in primary cells.
Figure 2.
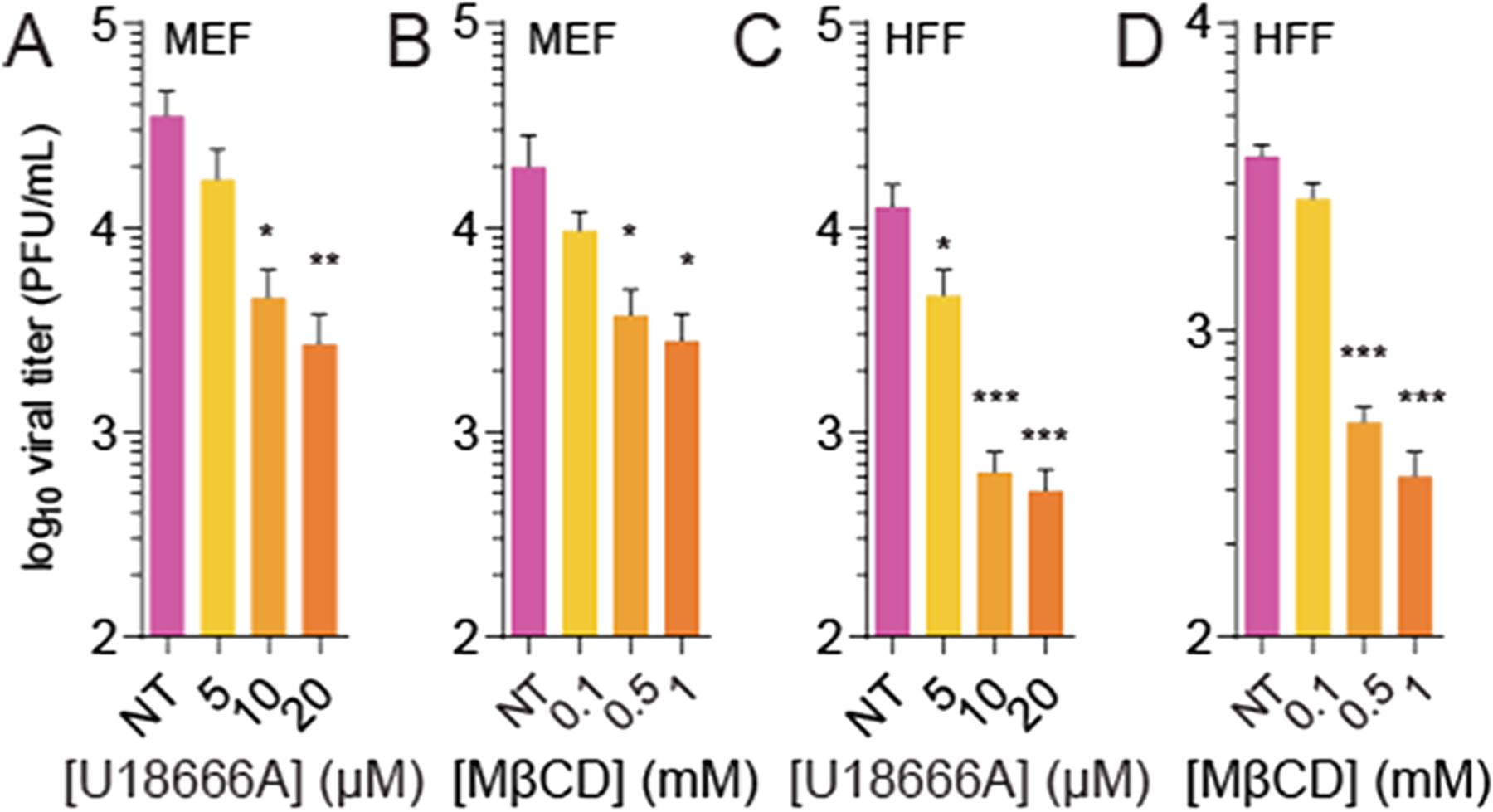
U18666A and MβCD inhibit RVFV infection in primary cells. Murine embryonic fibroblasts (MEF) were treated with (A) U18666A or (B) MβCD and subsequently infected with RVFV at MOI 0.1. Viral titers were measured at 48 hpi. Human foreskin fibroblasts (HFF) were similarly treated, infected, and assayed with (C) U18666A or (D) MβCD. Error bars represent one standard error of the mean. *p < 0.05, **p < 0.01, ***p < 0.001 by the Student’s T-test (N ≥ 3).
Cholesterol Depletion Reduces Rift Valley Fever Virus Infectivity.
Many viruses incorporate cholesterol in progeny virions, and others have shown that this virion-associated cholesterol is important for infectivity.18,22 To determine if virion-associated cholesterol mediated infectivity, we directly incubated MβCD with RVFV virions for 4 h prior to titering virus. In accordance with previous work on a related bunyavirus, bunyamweravirus,21 we observed a dose-dependent decrease in viral titers (Figure 3A), suggesting that MβCD reduces virion infectivity. Importantly, in these assays, the MβCD used to incubate with the virus was diluted out of samples upon titration (at least 1:100 and up to 100,000-fold), so residual MβCD would have minimal effects on cell-membrane cholesterol levels. To confirm that MβCD was not directly virucidal, we incubated RVFV with MβCD and titrated cholesterol into the incubation. MβCD functions by removing or adding cholesterol to membranes, so we hypothesized that we could maintain an infectious virus by incorporating cholesterol in the membranes via MβCD. We observed that 100 μM cholesterol fully rescued RVFV titers when incubated concurrently with MβCD (Figure 3B), though mevalonate (“mev”), a cholesterol precursor molecule, had no effect, suggesting that MβCD reduced viral titers by reducing virion-associated cholesterol levels.
Figure 3.
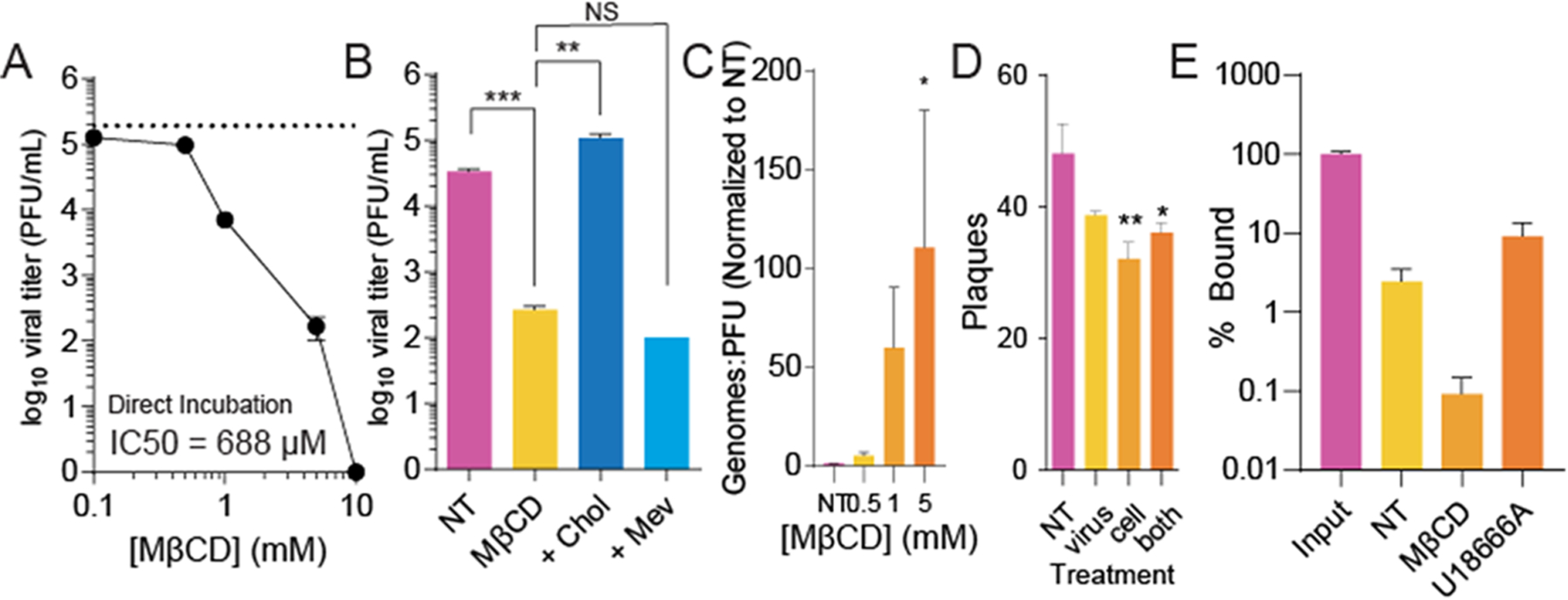
Cholesterol depletion within the virion reduces RVFV infectivity. (A) Purified RVFV was treated for 4 h with increasing doses of MβCD and viral titers measured by the plaque assay. (B) RVFV was treated with 1 mM MβCD and supplemented with 100 μM cholesterol (“chol”) or mevalonate (“mev”). Viral titers were measured by the plaque assay after 4 h of incubation. (C) Viral genomes were quantified and compared to viral titers in RVFV samples treated with increasing doses of MβCD. (D) Viral attachment was measured by incubating the virus, cells, or both with 1 mM MβCD for 4 h prior to the plaque-mediated attachment assay. (E) Viral attachment was similarly measured by qRT-PCR, measuring cell-associated viral genomes after treatment with 1 mM MβCD (p = 0.09 vs NT) and 10 μM U18666A (p = 0.08 vs NT). *p < 0.05, **p < 0.01, ***p < 0.001 by Student’s T-test (N ≥ 3).
In addition to measuring virion titers after MβCD treatment, we measured viral genomes, anticipating that they would not decrease with incubation, as suggested previously for bunyamweravirus.21 To this end, we treated RVFV with increasing doses of MβCD and then extracted viral RNA, generated cDNA, and measured the quantity of viral genomes by qPCR with primers specific for the small genome segment. We found that MβCD treatment resulted in modest changes in the viral genome content, and when we compared this to the titer of the viruses (genomes/titer), we observed a significant increase in this ratio (Figure 3C). Together, these data suggest that cholesterol depletion within virions does not disrupt the virions but affects their specific infectivity. These data are also in line with prior work21 demonstrating that MβCD does not destroy bunyavirus particles, as measured by electron microscopy of treated virions. To determine whether this change in specific infectivity was due to the virions’ inability to bind and enter susceptible cells, we treated both cells and virus with 1 mM MβCD for 4 h and then performed an attachment assay as with U18666A. We found that incubation of the cells or the virus with MβCD reduced viral attachment, as measured by the number of plaques formed after attachment (Figure 3D). To confirm these phenotypes, we performed an additional virus attachment assay, but instead of counting plaques as a surrogate for viral attachment, we measured the number of cell-associated viral genomes, immediately after washing away the unbound virus via qRT-PCR (Figure 3E). We find that approximately 2.5% viral genomes attached after a 5 min incubation period, compared to the number of viral genomes in the input sample. This value decreases with MβCD treatment but is unchanged with U18666A treatment, similar to our results in Figures 1E and 3D. These data suggest that cholesterol present within the virion membrane and cholesterol within the cytoplasmic membranes contribute to RVFV attachment.
Cholesterol Supplementation Rescues Virus Replication in Polyamine-Depleted Cells.
Polyamine depletion reduces the replication of many RNA viruses,10,11 including RVFV.16 We hypothesized that supplementing cholesterol to polyamine-depleted cells might rescue virus replication within these cells. To this end, we treated cells with the small-molecule inhibitor difluoromethylornithine (DFMO) to deplete polyamines and then supplemented increasing doses of cholesterol into the cellular media prior to infection with RVFV. DFMO reduces cellular polyamines by inhibiting the rate-limiting enzyme ornithine decarboxylase (ODC1), which subsequently reduces viral titers.10,16,31 When we measured viral titers at 48 hpi, we observed that DFMO significantly reduced viral titers and that cholesterol modestly but significantly rescued RVFV replication in both Vero (Figure 4A) and BHK-21 cells (Figure 4B), suggesting that cholesterol supplementation can overcome polyamine depletion to facilitate RVFV replication.
Figure 4.
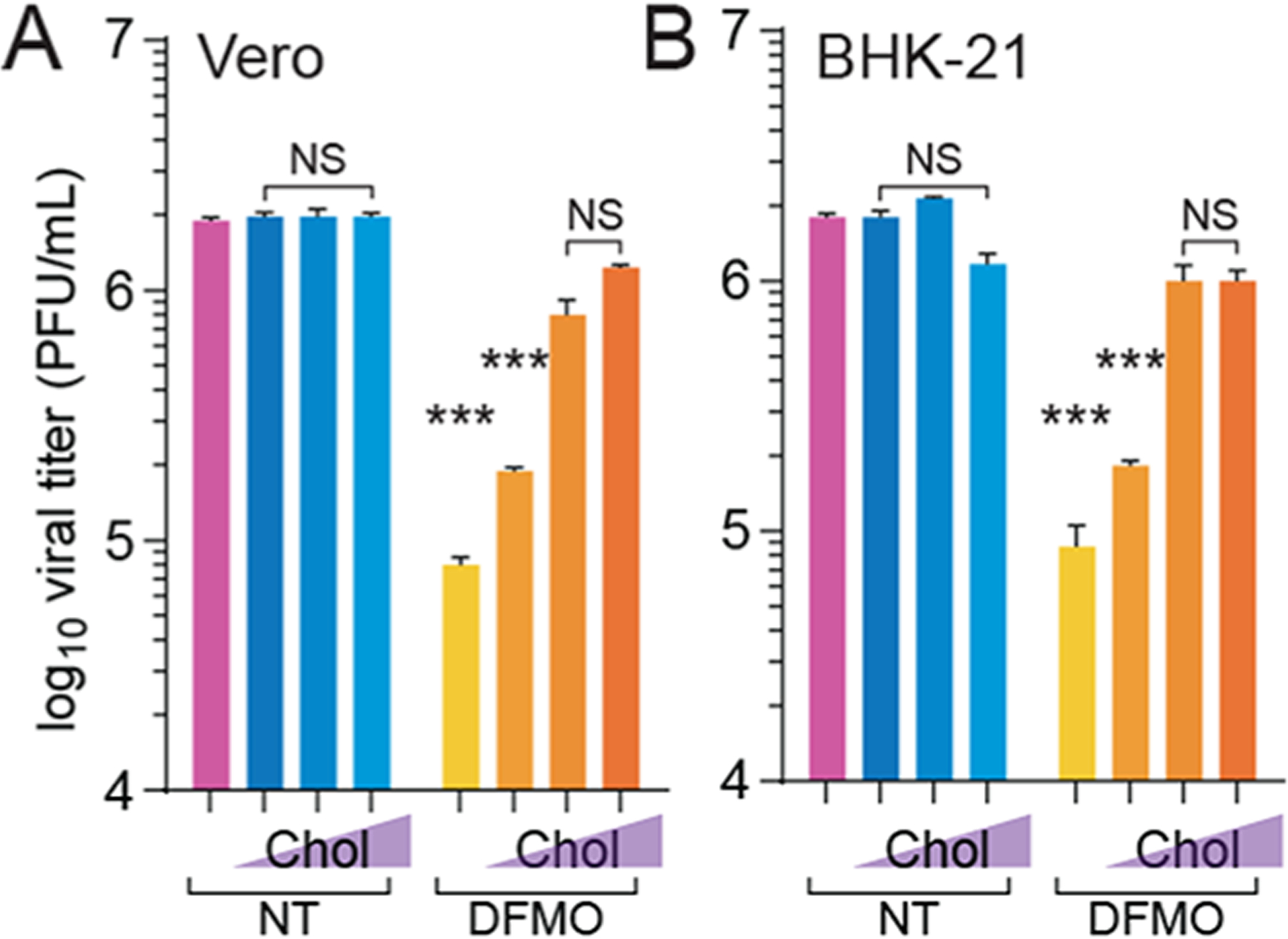
Cholesterol supplementation rescues bunyavirus replication in polyamine-depleted cells. (A) Vero cells were treated with 1 mM DFMO for four days and subsequently supplemented with cholesterol for 8 h prior to infection with RVFV for 48 h. Viral titers were measured by the plaque assay. (B) BHK-21 cells were treated, infected, and analyzed as in A. NS not significant, *p < 0.05, **p < 0.01, ***p < 0.001 by Student’s T-test comparing treatment to untreated conditions (N ≥ 3).
Polyamine Depletion Limits Cholesterol Incorporation into Rift Valley Fever Virus Particles.
We recently demonstrated that cellular polyamines promote cholesterol synthesis through polyamine-mediated SREBP2 translation and activity.23 We thus hypothesized that polyamine depletion via DFMO and subsequent cholesterol depletion would result in reduced RVFV replication. If DFMO reduces cellular cholesterol, we considered that virions derived from these cells would have reduced cholesterol as well. To test this, we isolated virions from untreated and DFMO-treated conditions and then incubated these viruses with MβCD, which reduces virion infectivity by removing cholesterol. We found that virions from untreated conditions were sensitive to MβCD (normalizing untreated conditions to 100%) with titers dropping more than 10-fold; however, virions from DFMO conditions were insensitive to MβCD treatment (Figure 5A). To confirm the cholesterol content of these virions, we purified virions from untreated or DFMO-treated polyamine-depleted cells, as previously reported,8 and we measured the total cholesterol within the samples. We previously found that purification of virions from untreated and DFMO-treated conditions yields similar numbers of viral particles,8 and here, we normalized by measuring viral genomes (as a surrogate for viral particles) prior to measuring the cholesterol content. We found that DFMO treatment of the cells led to a significant reduction in virion-associated cholesterol (Figure 5B), suggesting that DFMO-mediated cellular cholesterol depletion affects cholesterol within the virion membrane. To confirm the fact that the cells where these virions were derived from exhibited reduced cholesterol, we also measured the bulk cellular cholesterol with DFMO treatment, observing a modest dose-dependent decrease in cholesterol abundance with increasing DFMO concentration (Figure 5C). Finally, we treated cells with a combination of U18666A and DFMO. We subsequently infected the cells with RVFV and measured viral titers at 48 hpi. We observed that U18666A reduced viral titers in a dose-dependent manner, but when combined with DFMO-mediated polyamine depletion, U18666A did not have any additive effect on viral titers (Figure 5D). These data may suggest that polyamine depletion limits RVFV at least partially through the reduction in cellular cholesterol synthesis.
Figure 5.
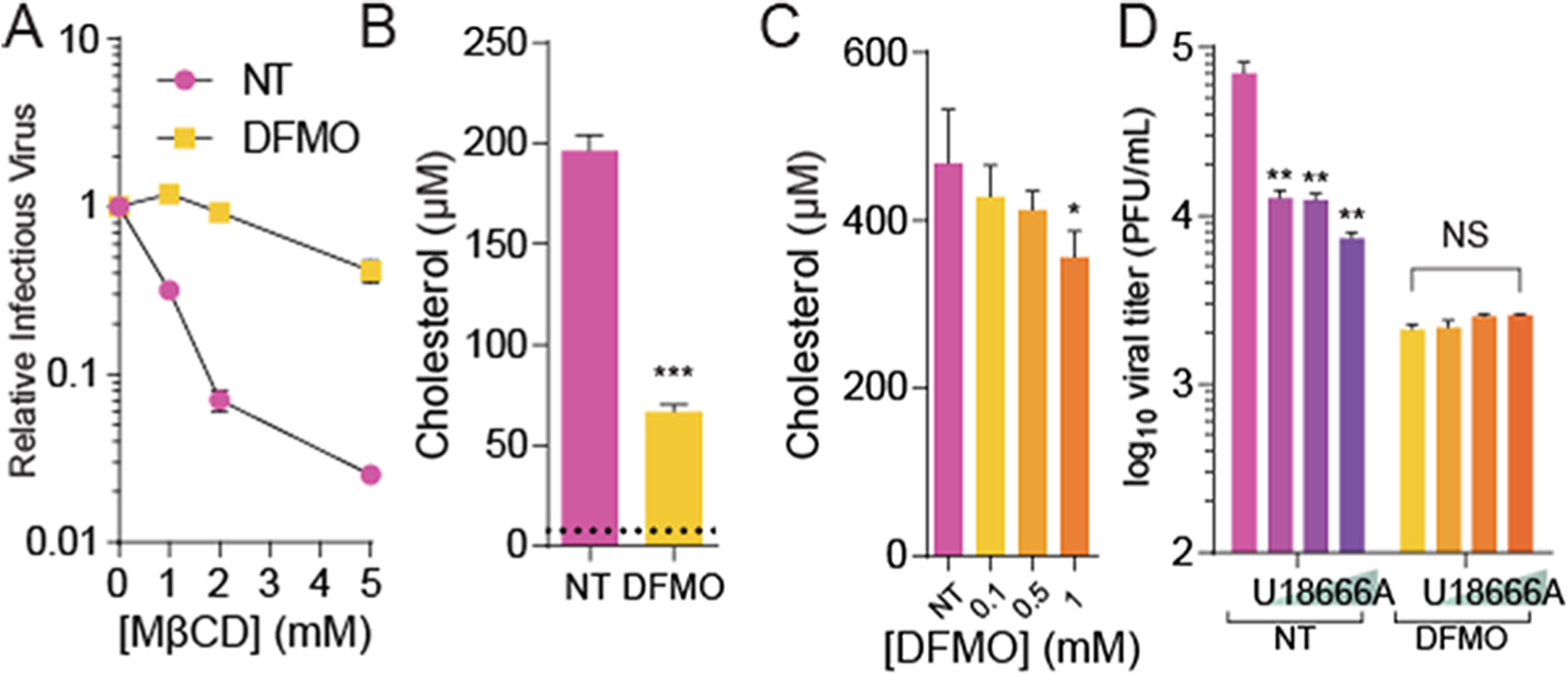
Polyamine depletion limits cholesterol incorporation into RVFV particles. (A) RVFV derived from untreated and polyamine-depleted (DFMO) conditions was treated for 4 h with increasing doses of MβCD and infectious virus measured by the plaque assay. (B) RVFV from untreated and DFMO treatment conditions was purified, and the associated cholesterol was measured. The limit of detection is noted by the dashed line. (C) Vero cells were treated with increasing doses of DFMO for four days and cellular cholesterol was measured. (D) Vero cells were treated with U18666A in conjunction with 1 mM DFMO prior to infection with RVFV at MOI 0.1 Viral titers were measured by the plaque assay after 48 h of infection. *p < 0.05, **p < 0.01, ***p < 0.001 by Student’s T-test (N ≥ 3).
MβCD Treatment Reduces Virion-Associated Polyamines.
We previously demonstrated that RVFV incorporates the polyamine spermidine into virions and this polyamine supports viral entry.8 Because we see that RVFV from DFMO-treated cells has reduced cholesterol levels (Figure 5B), we hypothesized that cholesterol and spermidine incorporation into virions may be linked. As an initial test, we treated RVFV with MβCD to remove cholesterol, purified as previously reported,8 and then detected spermidine within these virions by thin-layer chromatography. We observed that RVFV had detectable levels of spermidine (Figure 6A; see arrow); however, treatment with MβCD depleted this signal. Incubation of RVFV with cholesterol or MβCD in combination with cholesterol had no effect on virion-associated spermidine, and disruption of RVFV with SDS (prior to purification) removed spermidine. Others have reported that bunyamweravirus particles remain intact with MβCD treatment,21 suggesting that MβCD treatment of RVFV did not reduce the spermidine signal due to virion disruption. We confirmed that virion proteins were present in our samples by running a western blot for Gn after treatment with MβCD, and we observed significant amounts of Gn, comparable to untreated RVFV (Figure 6B). As expected, disruption with SDS removed Gn from the samples. Together, these data suggest that virion-associated spermidine relies on cholesterol within the membrane and spermidine within the virion may be associated with this cholesterol or the virion membrane to affect viral entry.
Figure 6.
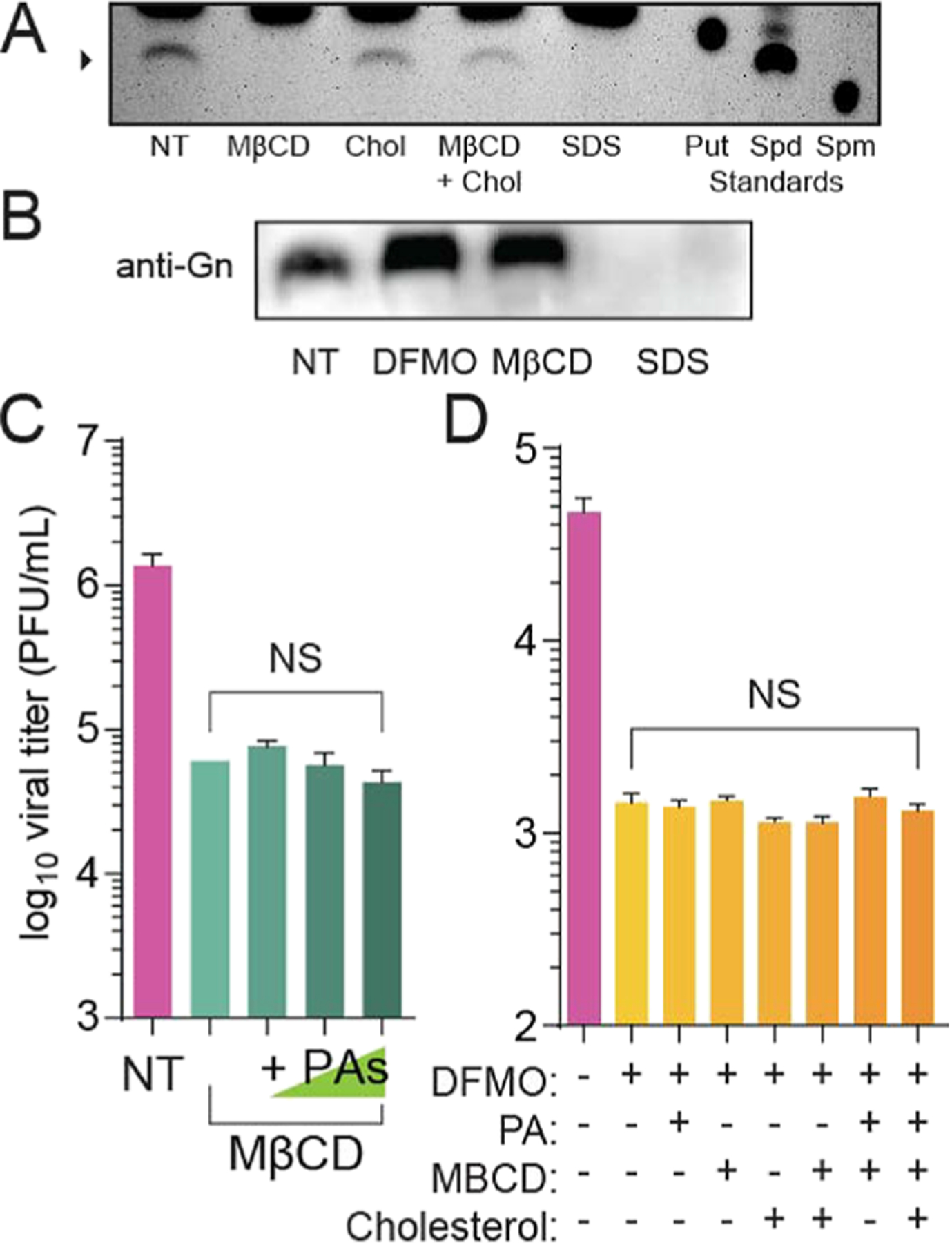
MβCD treatment reduces virion-associated polyamines. (A) RVFV was either not treated (NT) or treated with MβCD, cholesterol, or a combination and purified, and associated polyamines were measured by thin-layer chromatography (putrescine - Put, spermidine - Spd, and spermine - Spm). Virions were fully disrupted with 10% SDS. (B) RVFV Gn protein level measured by western blot from viruses from untreated conditions or DFMO, MβCD, or SDS treatment. (C) RVFV was treated with MβCD for 4 h in combination with increasing doses of polyamines. Viral titers were measured by the plaque assay. (D) Virions lacking polyamines and cholesterol (+DFMO) were treated with 10 μM polyamines, 1 mM MβCD, 50 μg/mL cholesterol, or a combination, and the infectious virus was measured by the plaque assay after 4 h of incubation. NS = not significant by Student’s T-test (N ≥ 3). The chromatogram and western blot are representative of at least three independent experiments.
Because we see this relationship between virion cholesterol and polyamine levels, we considered that MβCD might reduce RVFV titers by affecting polyamine levels. To test this, we treated RVFV with MβCD for 4 h and supplemented with increasing doses of polyamines. We found that, despite incubating with polyamines and MβCD, RVFV remained sensitive to MβCD (Figure 6C). Conversely, we considered that polyamine depletion through DFMO limits cholesterol in virions and this mediates infectivity. To test this, we derived RVFV from untreated and DFMO-treated conditions and then treated these virions with MβCD and an excess of cholesterol to favor incorporation of cholesterol back into these virions. We found that RVFV from DFMO-treated cells exhibited significantly reduced titers that were not rescued when incubated with MβCD and cholesterol (Figure 6D). These data suggest that cellular cholesterol and spermidine are necessary for virion infectivity and manipulation of either polyamine levels or cholesterol impacts these two molecules and their incorporation into virions.
DISCUSSION
Polyamines function at distinct stages in virus replication depending on the viral family,10 suggesting that diverse viruses have evolved unique ways of coopting cellular polyamines for their replication. We previously demonstrated that RVFV relies on polyamines for the production of infectious virions16 and further demonstrated that virion-associated polyamines are critical for viral entry.8 Other viruses, especially DNA viruses, incorporate polyamines into the virion,32,33 though their precise role(s) within the virion are incompletely understood. A potential explanation is that polyamines serve as a counter charge to the negatively charged viral genome, providing a charge balance within the virion. However, whether these polyamines affect virus entry in DNA viruses akin to RVFV is unknown. Here, we show that, in addition to spermidine’s role in RVFV as a virion component,8 polyamines also facilitate the synthesis and incorporation of cholesterol in RVFV, a key sterol that mediates viral attachment and entry21,34 as well as fusion.35 The relative contribution of spermidine and cholesterol to our observed phenotypes is unresolved, but our data suggest that these two molecules are linked in their incorporation into virions (Figure 7). Without cholesterol, spermidine is not incorporated into virions, and without polyamines, cholesterol is not incorporated. Understanding whether these two molecules interact directly within the virions may provide additional insight, though such experiments are technically challenging.
Figure 7.
Working model. RVFV relies on polyamines for cholesterol synthesis for replication, including incorporation of cholesterol in virions. In cells with polyamines, virions contain both polyamines and cholesterol. In polyamine-depleted cells (e.g., when treated with DFMO), cholesterol and polyamine levels are depleted, leading to reduced infectivity.
Cellular cholesterol plays multiple roles in RNA virus infection, including for bunyaviruses like RVFV.21,36 The recently discovered receptor for RVFV Lrp16 is an LDL receptor family protein, which interacts directly with Gn to facilitate viral entry. Additionally, cholesterol is important for membrane fusion following receptor engagement and prior to genome release into the cytoplasm to initiate infection.21 It is unclear whether cholesterol levels within the cell change during infection, though type I interferon signaling interfaces with cholesterol synthesis, including the synthesis of 25-hydroxycholesterol, an antiviral molecule.37 Our prior work found that RVFV derived from polyamine-depleted cells required a lower pH for membrane fusion and release of viral genomes.8 At the time, we were unaware of the effect of polyamines on cholesterol synthesis and the subsequent depletion of cholesterol within these virions. The current work may support a role for virion-associated cholesterol in mediating viral attachment and entry, possibly at the stage of membrane fusion.35,38,39 It is currently unclear if the inhibitors used in this work could impact Lrp1 expression or function or whether other virus attachment factors, such as heparan sulfates, might be affected under polyamine-depleted conditions. We previously reported that the virion morphology does not grossly change upon polyamine depletion;16 thus, the reduced infectivity is likely due to a combination of host- and virus-specific effects of polyamine depletion. Further work, including biochemical assays to measure fusion, will clarify these phenotypes and measure the contribution of virion-associated spermidine and cholesterol to membrane fusion.
As an arbovirus, RVFV is transmitted by mosquitos, specifically Aedes and Culex species, and the cycling of RVFV through arthropod and mammalian hosts is essential to maintaining fitness.40 Intriguingly, mosquitos require dietary cholesterol and encode several sterol transporting proteins.41 Thus, cholesterol levels within the mosquito vary, and this can translate to disparate levels of cholesterol within virions.42 In the case of the Mayaro virus, an alphavirus related to the chikungunya virus, reduced cholesterol incorporation into virion membranes due to reduced mosquito cell-associated cholesterol does not affect stability or infectivity of the particles. However, virions derived from mammalian cells require this cholesterol for stability and infectivity.43 The authors posit that depleted cholesterol affects lipid membrane organization, which may explain differences in virions derived from arthropod versus mammalian hosts. Our results, using mammalian cell culture systems, support these findings as depleting cholesterol, either through MβCD or polyamine depletion, significantly reduced RVFV titers.
The connection between polyamines and cholesterol synthesis has important implications for the effects of connected metabolic pathways on virus infection. Prior work showed that serum sterol levels were reduced in rats and mice treated with DFMO.44,45 Our work, both here and in a previous one,23 demonstrates that polyamines support cholesterol synthesis through translation of SREBP2, a critical transcription factor for cholesterol biosynthetic genes. We previously showed that Coxsackievirus B3 attachment relied on both polyamines and the connected cholesterol synthetic pathway for infection. Here, we expand these findings to the enveloped virus RVFV, further demonstrating that virus-associated cholesterol within the envelope is also depleted when RVFV is derived from polyamine-depleted cells. Further, we show that cholesterol incorporation into virions is related to polyamine levels and vice versa: depletion of cholesterol from RVFV also depletes virion-associated spermidine. While the full mechanism remains to be elucidated, these data support a role for polyamines in cholesterol synthesis and the collaboration between these molecules to support infection.
METHODS
Cell Culture.
Cells were maintained at 37°C in 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM; Life Technologies) with bovine serum and penicillin–streptomycin. Vero cells (BEI Resources) were supplemented with 10% newborn calf serum (GeminiBio), and Huh7, BHK-21, MEF, and HFF cells were supplemented with 10% fetal bovine serum (GeminiBio).
Drug Treatment.
Difluoromethylornithine (DFMO; TargetMol) was diluted to 100 mM in water. Methyl-β-cyclodextran (MβCD, Cayman Chemical) was diluted to 100 mM in dimethyl sulfoxide (DMSO, Sigma-Aldrich). U18666A (Cayman Chemical) was diluted to 20 mM in DMSO (Sigma-Aldrich). U18666A treatment was maintained throughout infection (unless otherwise stated), and MβCD was removed from cells after 4 h of treatment. Cells were seeded with a fresh medium with 2% serum and attached overnight. For DFMO, cells were treated and incubated for 96 h. During infection, media was cleared and saved from the cells. The same medium was used to replenish cells following infection. For MβCD treatment on cells, the drug was added and incubated for 4 h. For MβCD treatment on the virus, the drug was directly incubated with the virus for 4 h at 25°C. For U18666A, cells were treated and incubated for 24 h. Polyamines (Sigma-Aldrich) were added to the cells at the time of infection.
Infection, Enumeration of Viral Titers, and Virion Purification.
RVFV MP-1246 was derived from Huh7 cells. For infection, the virus was diluted in serum-free DMEM for a multiplicity of infection (MOI) of 0.1 on Vero cells, unless otherwise indicated. The viral inoculum was overlain on cells for 10 to 30 min, and the cells were washed with PBS before replenishment of media. Viral titers were determined as previously described.16 Viral purification, modified from a protocol for alphavirus purification,47 was performed by a clarifying supernatant with low-speed centrifugation. The supernatant was collected and centrifuged at 4000 × g for 5 min at 4°C to remove cellular debris. The supernatant was collected and further centrifuged at 5300 × g for 16 h at 15 °C. The medium was aspirated with a serological pipette, and the purified virus was resuspended in serum-free DMEM.
Thin-Layer Chromatography Determination of Polyamines.
Polyamines were separated by thin-layer chromatography as previously described.8,48 For all samples, the virus was collected, purified, and centrifuged. The pellets were washed with PBS and then resuspended in 100 μL of 2% perchloric acid. Samples were then incubated overnight at 4°C. A volume of 100 μL of the supernatant was combined with 200 μL of 5 mg/mL dansyl chloride (Sigma-Aldrich) in acetone and 100 μL of saturated sodium bicarbonate. Samples were incubated in the dark overnight at room temperature. Excess dansyl chloride was cleared by incubating the reaction with 100 μL of 150 mg/mL proline (Sigma-Aldrich). Dansylated polyamines were extracted with 50 μL of toluene (Sigma-Aldrich) and centrifuged. A volume of 5 μL of the sample was added in small spots to the TLC plate (silica gel matrix; Sigma-Aldrich) and exposed to ascending chromatography with 1:1 cyclohexane:ethylacetate. The plate was dried and visualized via exposure to UV.
RNA Purification, cDNA Synthesis, and Viral Genome Quantification.
The medium was cleared from cells, and a TRIzol reagent (Zymo Research) was directly added. The lysate was then collected, and RNA was purified according to the manufacturer’s protocol utilizing the Direct-zol RNA Miniprep Plus kit (Zymo Research). The purified RNA was subsequently used for cDNA synthesis using high-capacity cDNA reverse transcription kits (Thermo Fisher), according to the manufacturer’s protocol, with 10–100 ng of RNA and random hexamer primers. Following cDNA synthesis, qRT-PCR was performed using the QuantStudio3 (Applied Biosystems by Thermo Fisher) and SYBR green master mix (DotScientific). Primers against the RVFV small genome were 5′-CAG-CAG-CAA-CTC-GTG-ATA-GA-3′ (forward) and 5′-CCC-GGA-GGA-TGA-TGA-TGA-AA-3′ (reverse). Primers against the RVFV medium genome were 5′-GGA-ACT-AGG-GAA-GAC-TGA-GAG-A-3′ (forward) and 5′-CTG-CTG-AAG-GGT-GGA-AACA-3′ (reverse). Primers against the RVFV large genome were 5′-CTC-CAC-TAA-CCC-AGA-GAT-GAT-TG-3′ (forward) and 5′-CTC-CTG-GCT-TGA-GGT-CTT-AAC-3′ (reverse). Primers were verified for linearity using eightfold serially diluted cDNA and checked for specificity via melt curve analysis. The number of viral genomes was divided by the viral titer, as determined by the plaque assay, to measure the genome-to-PFU ratio. Values obtained were normalized to untreated conditions to obtain the relative genome-to-PFU ratio.
Intracellular and Viral Cholesterol Abundance Assay.
To measure intracellular cholesterol abundance, Vero cells were plated at a density of 5000 cells/well in a 96-well plate in DMEM with 2% NBCS. Cells were treated with DFMO for 96 h. The medium was removed from cells followed by a PBS wash. To measure viral cholesterol levels, Vero cells were plated and either treated with 1 mM DFMO or untreated. After 4 days, cells were infected with RVFV MP-12 for 48 h at MOI 0.1. Viruses in the supernatant were collected, concentrated, and purified as described above. To measure the total intracellular or viral cholesterol abundance, we used the Cholesterol/Cholesterol Ester-Glo Assay (Promega) in accordance to the manufacturer’s protocol. Luciferase levels were measured with Renilla luciferase. Values were normalized to a cholesterol standard curve to obtain the relative cholesterol content.
Western Blot.
Samples were collected with the Bolt LDS buffer and Bolt reducing agent (Invitrogen) and run on polyacrylamide gels. Gels were transferred using the iBlot 2 gel transfer device (Invitrogen). Membranes were probed with primary antibodies for the Rift Valley fever glycoprotein, Gn, (1:1000, BEI Resources). Membranes were treated with a SuperSignal West Pico PLUS chemiluminescent substrate (ThermoFisher Scientific) and visualized on a ProteinSimple FluorChem E imager.
Binding Assay.
Vero cells were seeded in six-well plates and grown to confluence in DMEM with 2% NBCS. The cells were treated with U18666A for 24 h at 0.5, 1, 2, 5, and 10 μM and with MβCD for 4 h at 1 mM. After treatment, the cells were placed on ice, and the medium was aspirated from the cells and replaced with 0.5 mL of the serum-free medium containing 1000 PFU of RVFV MP-12. The infected cells were incubated on ice for 5 min. After the specified time, the cells were washed three times with PBS and then overlaid with 0.8% agarose containing DMEM with 2% NBCS. The plates were incubated at 37°C for 4 days for MP-12 plaques to develop. The cells were fixed with 4% formalin, and the plaques were visualized with crystal violet staining.
qPCR-Based Binding Assay.
Viral attachment was measured via qPCR as previously described.15 Vero cells were treated with 10 μM U18666A for 24 h and 1 mM MβCD for 4 h. After treatment, the cells were placed on ice, and the medium was aspirated and replaced with 0.5 mL of the serum-free medium containing 1000 PFU of RVFV MP-12. The infected cells were incubated on ice for 5 min. The cells were then washed three times with PBS, and the TRIzol Reagent (Zymo Research) was added. The lysate was collected, and the RNA was extracted and converted to cDNA, as described above. Viral genomes were quantified as previously described.16 Relative genomes were calculated using the ΔCT method normalized to β-actin qRT-PCR control. Primers for β-actin were 5′-CAC-TCT-TCC-AGC-CTT-CCT-TC-3′ (forward) and 5′-GTA-CAG-GTC-TTT-GCG-GAT-GT-3′ (reverse).
Immunofluorescence.
Vero cells were seeded on cover-slips and treated with 20 μM U18666A for 24 h or with 1 mM MβCD for 4 h. Cells were subsequently infected with RVFV MP-12 at MOI 5 for 16 h and fixed with 4% formalin at room temperature. Cells were blocked with 0.2% Triton X-100 and 2% BSA in PBS at room temperature. Following blocking, cells were incubated with an anti-L antibody (1:500; SinoBiological) for 1 h at room temperature. Cells were washed three times with PBS and incubated with Cy3-conjugated affinipure goat anti-rabbit IgG(H + L) (1:500; Proteintech) for 1 h at room temperature. Cells were washed three times with PBS prior to mounting the coverslips on microscope slides using mounting media containing DAPI (Biotium) for staining of cell nuclei. Images were collected with a DeltaVision wide-field fluorescence microscope (Applied Precision) equipped with a CoolSNAP HQ digital camera (Photometrics) and a 40× objective lens. Excitation light of the required wavelength was generated with an Insight SSI solid-state illumination module (Applied Precision). All acquired images were analyzed using Imaris v. 8.4.1 (Bitplane).
Cell Viability Assay.
Vero cells were seeded in a 96-well plate and drug-treated with U18666A and MβCD as described above. A CellTiter-Glo reagent (Promega) was added in equal volume to the cells, and they were placed on a shaker for 2 min to induce lysis. Contents were further incubated at room temperature for 10 min to allow for the luminescence to stabilize. The luminescent signal was measured with the Veritas Microplate Luminometer (Turner Biosystems) using the Promega CellTiter-Glo protocol. Viable cells were counted and compared to untreated conditions.
Statistical Analysis.
Prism 6 (GraphPad) was used to generate graphs and perform statistical analysis. For all analyses, one-tailed Student’s T-test was used to compare groups, unless otherwise noted, with a = 0.05. ANOVA was used to compare groups of data, as indicated, with α = 0.05. For tests of sample proportions, p values were derived from calculated Z scores with two tails and α = 0.05.
ACKNOWLEDGMENTS
This work was supported by no. R35GM138199 from NIGMS (BCM). We thank Dr. Susan Uprichard for the Huh7 cells. Dr. Mitch Denning provided the HFFs. Dr. Liliana Radoshevich provided the MEFs.
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/acsinfecdis.2c00071
The authors declare no competing financial interest.
Contributor Information
Vincent Mastrodomenico, Department of Microbiology and Immunology, Loyola University Chicago Stritch School of Medicine, Maywood, Illinois 60153, United States.
Natalie J. LoMascolo, Department of Microbiology and Immunology, Loyola University Chicago Stritch School of Medicine, Maywood, Illinois 60153, United States
Yazmin E. Cruz-Pulido, Department of Microbiology and Immunology and Infectious Diseases and Immunology Research Institute, Loyola University Chicago Stritch School of Medicine, Maywood, Illinois 60153, United States
Christina R. Cunha, Department of Microbiology and Immunology, Loyola University Chicago Stritch School of Medicine, Maywood, Illinois 60153, United States
Bryan C. Mounce, Department of Microbiology and Immunology and Infectious Diseases and Immunology Research Institute, Loyola University Chicago Stritch School of Medicine, Maywood, Illinois 60153, United States.
REFERENCES
- (1).Elliott RM Emerging Viruses: The Bunyaviridae. Mol. Med 1997, 3, 572–577. [PMC free article] [PubMed] [Google Scholar]
- (2).Rolin AI; Berrang-Ford L; Kulkarni MA The Risk of Rift Valley Fever Virus Introduction and Establishment in the United States and European Union. Emerging Microbes Infect. 2013, 2, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Ikegami T; Makino S Rift Valley Fever Vaccines. Vaccine 2009, 27, D69–D72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Faburay B; LaBeaud AD; McVey DS; Wilson WC; Richt JA Current Status of Rift Valley Fever Vaccine Development. Vaccines 2017, 5, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Wright D; Kortekaas J; Bowden TA; Warimwe GM Rift Valley Fever: Biology and Epidemiology. J. Gen. Virol 2019, 100, 1187–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Ganaie SS; Schwarz MM; McMillen CM; Price DA; Feng AX; Albe JR; Wang W; Miersch S; Orvedahl A; Cole AR; Sentmanat MF; Mishra N; Boyles DA; Koenig ZT; Kujawa MR; Demers MA; Hoehl RM; Moyle AB; Wagner ND; Stubbs SH; Cardarelli L; Teyra J; McElroy A; Gross ML; Whelan SPJ; Doench J; Cui X; Brett TJ; Sidhu SS; Virgin HW; Egawa T; Leung DW; Amarasinghe GK; Hartman AL Lrp1 Is a Host Entry Factor for Rift Valley Fever Virus. Cell 2021, 184, 5163–5178.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Harmon B; Schudel BR; Maar D; Kozina C; Ikegami T; Tseng C-TK; Negrete OA Rift Valley Fever Virus Strain MP-12 Enters Mammalian Host Cells via Caveola-Mediated Endocytosis. J. Virol 2012, 86, 12954–12970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Mastrodomenico V; Esin JJ; Qazi S; Khomutov MA; Ivanov AV; Mukhopadhyay S; Mounce BC Virion-Associated Polyamines Transmit with Bunyaviruses to Maintain Infectivity and Promote Entry. ACS Infect. Dis 2020, 6, 2490–2501. [DOI] [PubMed] [Google Scholar]
- (9).Igarashi K; Kashiwagi K Polyamines: Mysterious Modulators of Cellular Functions. Biochem. Biophys. Res. Commun 2000, 271, 559–564. [DOI] [PubMed] [Google Scholar]
- (10).Firpo MR; Mounce BC Diverse Functions of Polyamines in Virus Infection. Biomolecules 2020, 10, 628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Mounce BC; Olsen ME; Vignuzzi M; Connor JH Polyamines and Their Role in Virus Infection. Microbiol. Mol. Biol. Rev 2017, 81, No. e00029–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Mounce BC; Poirier EZ; Passoni G; Simon-Loriere E; Cesaro T; Prot M; Stapleford KA; Moratorio G; Sakuntabhai A; Levraud J-P; Vignuzzi M Interferon-Induced Spermidine-Spermine Acetyltransferase and Polyamine Depletion Restrict Zika and Chikungunya Viruses. Cell Host Microbe 2016, 20, 167–177. [DOI] [PubMed] [Google Scholar]
- (13).Dial CN; Tate PM; Kicmal TM; Mounce BC Coxsackievirus B3 Responds to Polyamine Depletion via Enhancement of 2A and 3C Protease Activity. Viruses 2019, 11, 403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Hulsebosch BM; Mounce BC Polyamine Analog Diethylnorspermidine Restricts Coxsackievirus B3 and Is Overcome by 2A Protease Mutation In Vitro. Viruses 2021, 13, 310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Kicmal TM; Tate PM; Dial CN; Esin JJ; Mounce BC Polyamine Depletion Abrogates Enterovirus Cellular Attachment. J. Virol 2019, No. e01054–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Mastrodomenico V; Esin JJ; Graham ML; Tate PM; Hawkins GM; Sandler ZJ; Rademacher DJ; Kicmal TM; Dial CN; Mounce BC Polyamine Depletion Inhibits Bunyavirus Infection via Generation of Noninfectious Interfering Virions. J. Virol 2019, No. e00530–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Gonzalez-Scarano F; Pobjecky N; Nathanson N La Crosse Bunyavirus Can Mediate PH-Dependent Fusion from Without. Virology 1984, 132, 222–225. [DOI] [PubMed] [Google Scholar]
- (18).Aizaki H; Morikawa K; Fukasawa M; Hara H; Inoue Y; Tani H; Saito K; Nishijima M; Hanada K; Matsuura Y; Lai MMC; Miyamura T; Wakita T; Suzuki T Critical Role of Virion-Associated Cholesterol and Sphingolipid in Hepatitis C Virus Infection. J. Virol 2008, 82, 5715–5724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Carro AC; Damonte EB Requirement of Cholesterol in the Viral Envelope for Dengue Virus Infection. Virus Res. 2013, 174, 78–87. [DOI] [PubMed] [Google Scholar]
- (20).Albulescu L; Wubbolts R; van Kuppeveld FJM; Strating JR P. M. Cholesterol Shuttling Is Important for RNA Replication of Coxsackievirus B3 and Encephalomyocarditis Virus. Cell. Microbiol 2015, 17, 1144–1156. [DOI] [PubMed] [Google Scholar]
- (21).Charlton FW; Hover S; Fuller J; Hewson R; Fontana J; Barr JN; Mankouri J Cellular Cholesterol Abundance Regulates Potassium Accumulation within Endosomes and Is an Important Determinant in Bunyavirus Entry. J. Biol. Chem 2019, 294, 7335–7347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Tang Q; Liu P; Chen M; Qin Y Virion-Associated Cholesterol Regulates the Infection of Human Parainfluenza Virus Type 3. Viruses 2019, 11, 438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Firpo MR; Petite MJ; LoMascolo NJ; Shah PS; Mounce BC Polyamines and EIF5A Hypusination Facilitate SREBP2 Translation and Cholesterol Synthesis to Enhance Enterovirus Attachment and Infection. bioRxiv November 1, 2021, p 2021.11.01.465941, DOI: 10.1101/2021.11.01.465941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Lu F; Liang Q; Abi-Mosleh L; Das A; De Brabander JK; Goldstein JL; Brown MS Identification of NPC1 as the Target of U18666A, an Inhibitor of Lysosomal Cholesterol Export and Ebola Infection. eLife 2015, 4, No. e12177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Doki T; Tarusawa T; Hohdatsu T; Takano T In Vivo Antiviral Effects of U18666A Against Type I Feline Infectious Peritonitis Virus. Pathogens 2020, 9, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Elgner F; Ren H; Medvedev R; Ploen D; Himmelsbach K; Boller K; Hildt E The Intracellular Cholesterol Transport Inhibitor U18666A Inhibits the Exosome-Dependent Release of Mature Hepatitis C Virus. J. Virol 2016, 90, 11181–11196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Liang X-D; Zhang Y-N; Liu C-C; Chen J; Chen X-N; Sattar Baloch A; Zhou B U18666A Inhibits Classical Swine Fever Virus Replication through Interference with Intracellular Cholesterol Trafficking. Vet. Microbiol 2019, 238, 108436. [DOI] [PubMed] [Google Scholar]
- (28).Poh MK; Shui G; Xie X; Shi P-Y; Wenk MR; Gu F U18666A, an Intra-Cellular Cholesterol Transport Inhibitor, Inhibits Dengue Virus Entry and Replication. Antiviral Res. 2012, 93, 191–198. [DOI] [PubMed] [Google Scholar]
- (29).Sabino C; Basic M; Bender D; Elgner F; Himmelsbach K; Hildt E Bafilomycin A1 and U18666A Efficiently Impair ZIKV Infection. Viruses 2019, 11, 524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Cenedella RJ; Sarkar CP; Towns L Studies on the Mechanism of the Epileptiform Activity Induced by U18666A. II. Concentration, Half-Life and Distribution of Radiolabeled U18666A in the Brain. Epilepsia 1982, 23, 257–268. [DOI] [PubMed] [Google Scholar]
- (31).Mounce BC; Cesaro T; Moratorio G; Hooikaas PJ; Yakovleva A; Werneke SW; Smith EC; Poirier EZ; Simon-Loriere E; Prot M; Tamietti C; Vitry S; Volle R; Khou C; Frenkiel M-P; Sakuntabhai A; Delpeyroux F; Pardigon N; Flamand M; Barba-Spaeth G; Lafon M; Denison MR; Albert ML; Vignuzzi M Inhibition of Polyamine Biosynthesis Is a Broad-Spectrum Strategy against RNA Viruses. J. Virol 2016, 90, 9683–9692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Gibson W; Roizman B Compartmentalization of Spermine and Spermidine in the Herpes Simplex Virion. Proc. Natl. Acad. Sci. U. S. A 1971, 68, 2818–2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Lanzer W; Holowczak JA Polyamines in Vaccinia Virions and Polypeptides Released from Viral Cores by Acid Extraction. J. Virol 1975, 16, 1254–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Ripa I; Andreu S; López-Guerrero JA; Bello-Morales R Membrane Rafts: Portals for Viral Entry. Front. Microbiol 2021, 12, 631274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Yang S-T; Kreutzberger AJB; Lee J; Kiessling V; Tamm LK The Role of Cholesterol in Membrane Fusion. Chem. Phys. Lipids 2016, 199, 136–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Simon M; Johansson C; Mirazimi A Crimean-Congo Hemorrhagic Fever Virus Entry and Replication Is Clathrin-, PH- and Cholesterol-Dependent. J. Gen. Virol 2009, 90, 210–215. [DOI] [PubMed] [Google Scholar]
- (37).Robertson KA; Ghazal P Interferon Control of the Sterol Metabolic Network: Bidirectional Molecular Circuitry-Mediating Host Protection. Front. Immunol 2016, 7, 634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Meng Y; Heybrock S; Neculai D; Saftig P Cholesterol Handling in Lysosomes and Beyond. Trends Cell Biol. 2020, 30, 452–466. [DOI] [PubMed] [Google Scholar]
- (39).Sousa IP Jr.; Carvalho CAM; Gomes AM O. Current Understanding of the Role of Cholesterol in the Life Cycle of Alphaviruses. Viruses 2021, 13, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Moutailler S; Roche B; Thiberge J-M; Caro V; Rougeon F; Failloux A-B Host Alternation Is Necessary to Maintain the Genome Stability of Rift Valley Fever Virus. PLoS Neglected Trop. Dis 2011, 5, No. e1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Talyuli OAC; Bottino-Rojas V; Taracena ML; Soares ALM; Oliveira JHM; Oliveira PL The Use of a Chemically Defined Artificial Diet as a Tool to Study Aedes Aegypti Physiology. J. Insect Physiol 2015, 83, 1–7. [DOI] [PubMed] [Google Scholar]
- (42).Hafer A; Whittlesey R; Brown DT; Hernandez R Differential Incorporation of Cholesterol by Sindbis Virus Grown in Mammalian or Insect Cells. J. Virol 2009, 83, 9113–9121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Sousa IP Jr.; Carvalho CAM; Ferreira DF; Weissmüller G; Rocha GM; Silva JL; Gomes AMO Envelope Lipid-Packing as a Critical Factor for the Biological Activity and Stability of Alphavirus Particles Isolated from Mammalian and Mosquito Cells *. J. Biol. Chem 2011, 286, 1730–1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Pirinen E; Gylling H; Itkonen P; Yaluri N; Heikkinen S; Pietilä M; Kuulasmaa T; Tusa M; Cerrada-Gimenez M; Pihlajamäki J; Alhonen L; Jänne J; Miettinen TA; Laakso M Activated Polyamine Catabolism Leads to Low Cholesterol Levels by Enhancing Bile Acid Synthesis. Amino Acids 2010, 38, 549–560. [DOI] [PubMed] [Google Scholar]
- (45).Brown AP; Morrissey RL; Crowell JA; Levine BS Difluoromethylornithine in Combination with Tamoxifen in Female Rats: 13-Week Oral Toxicity Study. Cancer Chemother. Pharmacol 1999, 44, 475–483. [DOI] [PubMed] [Google Scholar]
- (46).Ikegami T; Won S; Peters CJ; Makino S Rescue of Infectious Rift Valley Fever Virus Entirely from CDNA, Analysis of Virus Lacking the NSs Gene, and Expression of a Foreign Gene. J. Virol 2006, 80, 2933–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Rayaprolu V; Ramsey J; Wang JC-Y; Mukhopadhyay S Alphavirus Purification Using Low-Speed Spin Centrifugation. Bio-Protoc. 2018, 8, No. e2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Madhubala R Thin-Layer Chromatographic Method for Assaying Polyamines. Polyamine Protoc. 1997, 79, 131–136. [DOI] [PubMed] [Google Scholar]