

Abstract
Extracellular vesicles (EV) are important mediators of cell communication and physiology. EVs are frequently investigated by transiently transfecting cells with plasmid DNA to produce EVs modified with protein(s) or nucleic acid(s) of interest. DNA‐transfection reagent complexes (DTC) are approximately the same size as EVs, raising the possibility that some purification procedures may fail to separate these two species and activity arising from carryover DTC may be improperly attributed to EVs. We find that differential ultracentrifugation, a commonly employed EV isolation procedure, does not separate EVs from DTC present in the cell culture supernatant of transiently transfected cells. We demonstrate that the biological activity of an EV‐directed Cre recombinase is due to contaminating plasmid DNA and not EV‐mediated delivery of Cre protein. Moreover, steps commonly taken to remove plasmid DNA from EV samples, such as media exchanges and treatment with nucleases, are ineffective at avoiding this artefact. Due to the pernicious nature of plasmid DNA in these cellular assays, some reports of EV function are likely artefacts produced by contaminating DTC. EVs and DTC can be separated by density gradient ultracentrifugation, highlighting the importance of validating elimination of DTC when using transient transfection of EV‐producing cells to interrogate EV function.
Keywords: artefact, Cre, delivery, exosome, extracellular vesicle, purification, transfection
1. INTRODUCTION
Extracellular vesicles (EVs) are released by all cells examined to date (An et al., 2007; Brown et al., 2015; Colombo et al., 2014; Schwechheimer & Kuehn, 2015; Stanly et al., 2016) and have been implicated in a variety of physiological and pathological processes [reviewed in (Stoorvogel et al., 2002; Tkach & Théry, 2016; Van Niel et al., 2018)]. EVs can mediate their effects on recipient cells through the interaction of molecules on the EV surface (proteins, glycans, or lipids) with receptors on recipient cells or by delivering cargo to recipient cells (Pegtel et al., 2010; Prada & Meldolesi, 2016). While the exact mechanisms responsible for EV cargo delivery are unclear, harnessing the natural communication network employed by EVs is of particular interest for developing EV‐based therapeutics (Madhusoodanan, 2020; Sabanovic et al., 2021; Witwer & Wolfram, 2021).
Previous reports have demonstrated the ability of EVs to deliver a range of cargo types, including DNA, RNA and protein (Didiot et al., 2016; Hung & Leonard, 2016; Pan et al., 2012; Pegtel et al., 2010; Sterzenbach et al., 2017; Segel et al., 2021; Ye et al., 2020; Yim et al., 2016). However, the degree of functional delivery varies significantly across reports and may be due to the variety of EV sources, production methods, isolation procedures, and reporter systems used. Reports of the ability of EVs to deliver nucleotides vary widely, with some reporting robust EV‐mediated RNA knockdown (Didiot et al., 2016; Pan et al., 2012; Pegtel et al., 2010), while others report no discernible functional delivery (Hung & Leonard, 2016). Similarly, reported efficiencies for protein delivery range from high levels for Cre (Yim et al., 2016) to low amounts for Cre, Cas9, and curcumin (Sterzenbach et al., 2017; Ye et al., 2020; Yim et al., 2016). In many cases, reported EV effects are modest and require sensitive reporter systems to detect; however, the sensitivity of these reporter systems can make them prone to artefacts and it is important to ensure that proper controls are included to rule out non‐EV sources of activity (Kooijmans et al., 2013).
It was recently reported that EVs loaded with Cre recombinase edited a reporter cell line and that this activity was significantly increased in the presence of drugs traditionally used to enhance endosomal escape of DNA‐transfection reagent complexes, such as chloroquine (Heath et al., 2019). We sought to replicate this finding by following the same procedures: transfecting EV‐producing cells with an EV‐targeted Cre, purifying EVs by differential ultracentrifugation, and treating reporter cells with the resuspended ultracentrifuge pellet (UCP). While we were able to detect Cre activity in EV‐containing UCP samples, we observed that the level of Cre protein did not correlate with the level of Cre activity, similar to what was observed by Heath et al. (2019). Here we report that when using transient transfection and standard EV purification methods, that EVs are dispensable for editing reporter cells; instead, Cre activity is due to DNA‐transfection reagent complexes (DTC) that co‐purify with EVs in the UCP. Although DTC are biophysically distinct from EVs in terms of charge and density, they are similar in size (Almofti et al., 2003; Ma et al., 2007), making it unlikely that some purification methods, such as differential ultracentrifugation, are able to separate these two species. Here, our data suggest that reported activity of some EV studies may be due to a transfection reagent artefact, rather than EV‐mediated cargo delivery and highlights the need for rigorous purification and characterization of EV samples.
2. MATERIALS AND METHODS
2.1. Cell culture and transfection conditions
Expi293F cells (ThermoFisher Scientific) were maintained in Expi293 Expression Medium (ThermoFisher Scientific) at 37°C in 8% CO2, 80% humidity, at 150 RPM in a Multitron incubator (Infors HT); all components required for cell growth are present in the media without the addition of serum. On the day of transfection, cells were counted on a Vi‐CELL XR cell counter (Beckman Coulter), seeded at 2.5 million cells/mL, and transfected with DNA complexed with transfection reagents according to manufacturers’ protocols (see Table S1) or as indicated in specific figures. Expifectamine (ThermoFisher Scientific) was used for all experiments, unless noted otherwise. To generate the stable Cre reporter cell line, media was changed 24 h after transfection into media with HygromycinB (200 μg/mL) for selection; cells typically required 3 weeks of selection, at which point viability recovered to > 95%. Transiently transfected cells were cultured for 4 days post transfection before harvesting cell culture supernatant. Cells were above 95% viability at the time of seeding and transfection, and above 90% viability at the time of cell culture harvest.
2.2. EV purification by differential and density gradient ultracentrifugation
Upon culture termination, cells were removed by centrifugation at 6000 × g for 20 min at room temperature, the supernatant was collected and then centrifuged at 16,000 × g for 30 min to remove large cell debris. Supernatant was collected and 1 mM MgCl2 and benzonase (20 U/mL, Millipore) were added and incubated at 37°C for 2–3 h. Media was transferred to 100 mL Quick‐Seal Ultra‐Clear tubes (Beckman Coulter) and centrifuged for 60 min at 133,900 × g at 4°C in a 45 Ti fixed‐angle rotor (Beckman Coulter, k‐factor 133). Supernatant was discarded, and the ultracentrifuge pellet (UCP) was resuspended in PBS.
For fractionation experiments, UCP was diluted to 1 mL with PBS and mixed with 3 mL of 60% iodixanol solution (OptiPrep, Sigma), and pipetted into the bottom of a 13‐mL Ultra‐Clear tube (Beckman Coulter). Lower‐density solutions were prepared by diluting 60% iodixanol with homogenization buffer (250 mM sucrose, 10 mM TrisHCl, 1 mM EDTA, pH 7.4) and successive layers of 30% (3 mL), 23% (2 mL), and 18% (2 mL) iodixanol were layered on top of samples, with a final layer of 1 mL PBS added to the top. Gradients were spun at 150,000 × g for 16 h at 4°C in a SW 41 Ti swinging‐bucket rotor (Beckman Coulter, k‐factor 124). The interfaces between each layer were collected, transferred to a 13‐mL Ultra‐Clear tube (Beckman Coulter), mixed with PBS to a final volume of 13 mL, and spun for 3 h at 133,900 × g at 4°C in an SW 41 Ti rotor. Resulting pellets were resuspended in 100 μL of PBS and stored at 4°C for less than 1 week before analysis.
2.3. Western blot analysis
Samples were diluted with an equal volume of 2× Laemmli buffer (Bio‐Rad) and heated at 95°C for 10 min prior to loading into 4–20% TGX stain‐free precast gels (Bio‐Rad). Gels were imaged on a ChemiDoc imaging system (Bio‐Rad) prior to transfer to a PVDF membrane using a Trans‐Blot Turbo semi‐dry transfer system (Bio‐Rad). Blots were blocked in 1% casein for 1 h, incubated with primary antibodies (diluted 1:1000 in blocking buffer) for 1–3 h, washed three times with PBST (0.05% Tween20), incubated with HRP‐conjugated secondary antibodies (diluted 1:5000 in blocking buffer) for 1 h, washed three times in PBST, and imaged following incubation with chemiluminescent substrate (see Table S1 for additional information). Densitometric quantitation of band intensity was performed using FIJI software (Schindelin et al., 2012).
2.4. Reporter cell activity assay
Reporter cells were plated at 100,000 cells per well in a 24‐well plate or 20,000 cells per well in a 96‐well plate. Samples were added to cells with or without 50 μM chloroquine (Heath et al., 2019), as indicated; significant reduction in cell viability was observed for chloroquine concentrations above 50 μΜ (Figure S2), so this dose was used for all subsequent experiments. Cells were incubated at 37°C and 5% CO2 in a static cell culture incubator for 24 h, washed with PBS, and fresh media was added. 48 h after media exchange, cells were resuspended by gentle pipetting, pelleted (500 × g for 3 min), washed once with PBS, and resuspended in 100 μL PBS. Flow cytometry was performed using a CytoFlex LX (Beckman Coulter) and gating and data analysis performed using FCS Express 7 (De Novo Software).
2.5. Nanoparticle tracking analysis
Size distributions and particle concentrations were measured by nanoparticle tracking analysis using a NanoSight NS300 (Malvern). Samples were analysed immediately after diluting 1000−10,000× in 0.22‐μm filtered PBS. Three 30‐s acquisitions were made for each sample under continuous flow and analysed with NanoSight NTA software (v.3.4, Malvern).
2.6. Quantification and statistical analysis
Experimental replicates are defined in the figure legends for each experiment. Statistical analyses were performed in GraphPad Prism 9 using the statistical tests indicated in figure legends. Values are expressed as mean ± standard deviation (SD); differences with a P value < 0.05 were considered significant.
2.7. PCR analysis
Prior to PCR analysis, samples were either untreated or extracted with phenol‐chloroform. Extracted samples were mixed with one volume of phenol:chloroform:isoamyl alcohol (25:24:1), vortexed for 20 s, and then centrifuged in a benchtop centrifuge (Beckman 5424) at 16,000 × g for 5 min. The upper aqueous phase was removed, transferred to a fresh 1.5 mL tube, to which was added (in order): sodium acetate (0.1 volume of 3 M sodium acetate pH 5.2), ethanol (4 volumes of 100%), after which samples were placed at −20°C overnight. Samples were then centrifuged at 4°C for 10 min. at 16,000 × g (Beckman 5424) to pellet DNA, supernatant was removed, and the pellet washed with ice cold 70% ethanol. The DNA pellet was air dried for 10 min before dissolving in 30 μL of sterile water. Standard PCR conditions were used employing Cre‐specific primers: Forward (5′‐ gagctcggatcgatatctg −3′) and Reverse (5′‐ ctggatcagttatctatgcg −3′). PCR was performed using an Eppendorf Master Cycler Pro (Eppendorf). PCR samples were analysed on 2% agarose gels with SYBR Safe DNA Gel Stain, (E‐Gel 48 Agarose, Thermo Fisher) and visualized using a ChemiDoc imaging system (Bio‐Rad).
2.8. EVtrack
Experimental details have been submitted to the EV‐TRACK knowledgebase (evtrack.org; EV‐TRACK ID: EV210301) (Van Deun et al., 2017).
3. RESULTS
3.1. Cre reporter cells and Cre‐loaded EVs
We sought to determine if adding EVs loaded with Cre recombinase to cells stably expressing a Cre reporter would result in functional protein delivery, as has recently been reported (Heath et al., 2019). To this end, we generated an Expi293F cell line stably expressing a “stoplight” reporter cassette consisting of mCherry in‐frame with a 3′ Stop codon flanked by LoxP sites, upstream of a GFP transgene (Figure 1a). This construct results in stable expression of mCherry, but not GFP, in the absence of Cre (Figure 1a); upon Cre‐mediated recombination the region encoding mCherry and the stop codon are excised, resulting in GFP expression (Figure 1a). Both mCherry and GFP are fused with N‐terminal nuclear localization sequences to concentrate fluorescent signal in the nucleus (Figure 1a). To load EVs with Cre protein, we utilized an EV‐localizing peptide derived from BASP‐1 (MGGKLSKKKK) (Dooley et al., 2021) fused to the amino‐terminus of Cre (Figure 1b). Transient transfection with BASP1‐Cre plasmid resulted in expression of GFP in > 40% of reporter cells, demonstrating that Cre was functional with an N‐terminal fusion (Figure 1a). To generate EVs carrying Cre protein, we transiently transfected Expi293F cells with BASP1‐Cre plasmid, and cell culture supernatant was processed by differential ultracentrifugation to generate an EV‐enriched UCP (Figure 1c). Adding the UCP from BASP1‐Cre transfected cells to reporter cells resulted in GFP expression in a subset of cells, indicating Cre‐mediated recombination (Figure 1d‐e). Consistent with previous reports (Heath et al., 2019),the levels of functional Cre delivery was low (∼1%) in the absence of endosomal escape enhancing compounds, and was significantly enhanced (∼5 fold) by 50 μM chloroquine treatment (Figure 1e).
FIGURE 1.
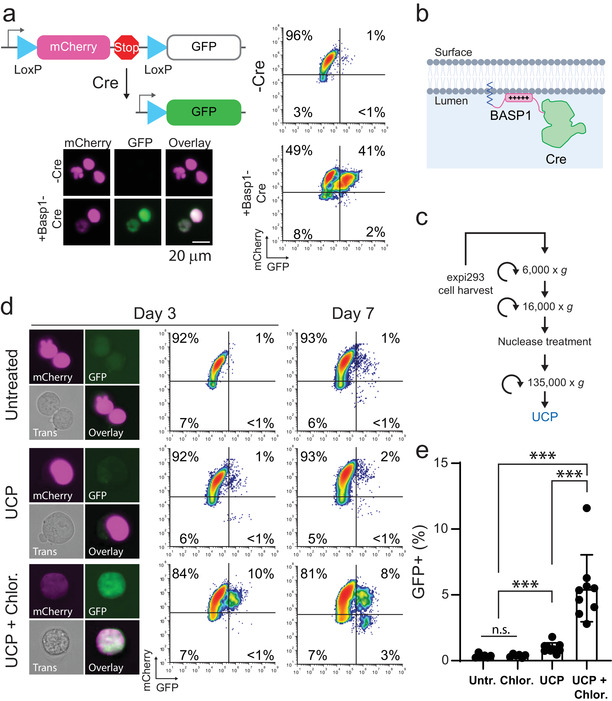
Ultracentrifuge pellets contain Cre activity that is enhanced by an endosome escaping agent. (A) Schematic of Cre “stoplight” reporter system. GFP is expressed only after Cre‐mediated excision of the mCherry and Stop codon. Flow cytometry and microscopy of stable reporter cells either untreated or transiently transfected with BASP1‐Cre plasmid. Scale Bar = 20 μm. (B) Schematic of Cre protein fused to Basp1 N‐terminal peptide (amino acids 1–10) for localization to the EV lumen. (C) Schematic of EV purification procedure. (E) Representative images and flow cytometry plots of reporter cells 3 and 7 days after addition of UCP isolated from cells transfected with Expifectamine +/‐ 50 μM chloroquine. Plot axes are the same as in (A). (E) Bar graph showing the percentage of GFP+ reporter cells on day 3 from nine independent experiments (N = 9, ***P < 0.001, n.s. not significant: P > 0.05, Mann‐Whitney U‐test)
3.2. Cre activity is not associated with EVs
While our results demonstrate that the UCP from Cre‐transfected cells can edit a reporter cell line (Figure 1d‐e), the UCP contains both EVs and many types of non‐EV material (Dooley et al., 2021; Jeppesen et al., 2019), opening up the possibility that the observed Cre activity may come from non‐EV sources. To determine if Cre protein had been successfully loaded into EVs using the BASP1 fusion, we used an iodixanol density gradient (Figure 2a) to separate the UCP into EV‐enriched and EV‐depleted fractions (Dooley et al., 2021). Cre and the EV markers CD9 and CD81 were detected in the UCP and after density fractionation were primarily found in the low‐density fraction F1 (Figure 2b, Figure S1c). As the majority of Cre protein co‐purifies with CD9 and CD81 in F1, we conclude that the Cre protein in the UCP is associated with bona fide EVs (Figure 2b).
FIGURE 2.
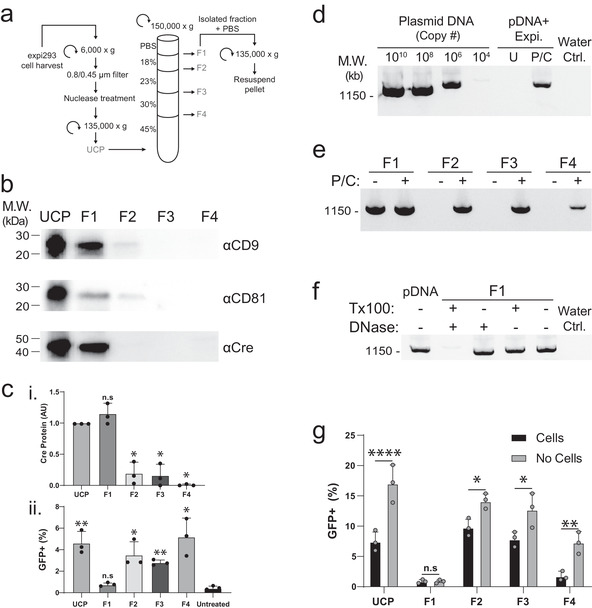
Cre activity present in UCP is biochemically separable from Cre protein and EVs. (A) Schematic of differential centrifugation and iodixanol density gradient used to separate UCP isolated from cells transfected with Expifectamine into four fractions (F1‐4). (B) Western blot analysis of UCP and density gradient fractions with antibodies against Cre and the EV markers CD9 and CD81. Equal particle numbers (1e10 particles) were loaded in each lane. (C) Quantification of Cre protein by Western blot densitometry (i); statistical comparison of each density gradient fraction relative to UCP (*, P < 0.0005, n.s., not significant, Mann‐Whitney U‐test). (ii) Quantification of GFP+ reporter cells 3 days after treatment with 50 μM chloroquine and either UCP or F1‐4 analysed by flow cytometry; statistical comparison of each sample relative to untreated controls. (*, P < 0.05; **, P < 0.005; n.s., not significant, P > 0.05; unpaired Mann‐Whitney U test). (D‐F) Agarose gels stained with SYBR safe DNA dye showing PCR amplification from the indicated samples. (D) Control plasmid (lanes 1–4); lanes 5–6: plasmid complexed with Expifectamine reagent, treated with nuclease, followed by phenol chloroform extraction (P/C) or column purification (U) prior to PCR amplification. Water Ctrl. is a no‐plasmid control containing all other PCR components. (E) Density gradient fractions F1‐4 were processed with (+) or without (‐) phenol chloroform extraction (P/C) prior to PCR amplification. (F) PCR amplification of control plasmid (pDNA) or F1 material pre‐treated with the indicated compounds. Water Ctrl. is a no‐plasmid control containing all other PCR components. (G) Quantification of GFP+ reporter cells treated with 50 μM chloroquine and either UCP or F1‐4. For the No Cells condition, a mock transfection was performed where no cells were included in the transient transfection with Cre plasmid; samples were otherwise processed identically. Cells analysed 4 days post treatment, with no media exchange for either condition (*, P < 0.05; **, P < 0.005; ****, P < 0.0001; n.s., not significant: P > 0.05; two‐way ANOVA)
To our surprise, reporter cells showed evidence of Cre activity when treated with density gradient fractions F2 and F3 but not F1, the EV‐enriched fraction containing most of the Cre protein (Figure 2b‐c). The observation that Cre activity was not correlated with Cre protein levels in the iodixanol density gradient fractions led us to question if Cre plasmid DNA from the transient transfection may carry over through our purification process, despite the inclusion of a nuclease treatment step in our purification protocol (Figure 1c, Figure 2a). To test this possibility, we sought to determine if we could detect DNA in the gradient fractions using PCR. We first validated that our Cre‐specific primers were functional and that we could detect DNA complexed with transfection reagent. We found that our primers amplified a single DNA band of the expected size (∼1300 b.p.) when used on BASP1‐Cre plasmid DNA (Figure 2d). To mimic our transfection and UCP purification conditions, plasmid DNA was complexed with Expifectamine transfection reagent and then treated with benzonase (see Methods) to digest non‐complexed DNA. Without further processing we were unable to detect plasmid DNA by PCR; however, phenol‐chloroform extraction of this sample enabled detection of the plasmid DNA (Figure 2d). When we examined fractions F1‐4 with PCR, we were surprised to observe the presence of DNA in all density gradient fractions (Figure 2e).
We noted that DNA in F1 was detectable without phenol‐chloroform extraction, while this step was required to detect DNA in fractions F2‐4 (Figure 2e). This suggests that DNA in F1 is biochemically distinct from that found in F2‐4 and from purified DNA complexed with transfection reagent, which require phenol‐chloroform extraction to be detected by PCR (Figure 2d‐e). DNA detected in F1 is unlikely to be free plasmid DNA because cell culture harvest is treated with nuclease prior to ultracentrifugation (Figure 2a). To determine if F1 DNA was still resistant to nuclease digestion, we incubated F1 with DNase I and observed similar DNA levels as untreated F1 (Figure 2f). We reasoned that the factor protecting F1 DNA from nuclease digestion was unlikely to be transfection reagent, as this prevents detection by PCR without phenol‐chloroform extraction (Figure 2d) which is not required to detect F1 DNA (Figure 2e‐f). Another possibility is that DNA could be contained within EVs found in F1 (Figure 2b); consistent with this hypothesis, F1 treated with detergent (1% TritonX‐100) is no longer resistant to nuclease digestion (Figure 2f).
The observation that DNA was present in density gradient fractions lead us to examine if DTC were sufficient to explain the observed Cre activity, or if a cellular contribution (EVs or other factor) were required. To test this, we performed a matched transfection and mock transfection where DTC were added to cell culture media in either the presence or absence of cells. Cultures with and without cells were incubated under the same conditions for 3 days, and then processed identically by differential and density gradient ultracentrifugation (Figure 2a). Once added to reporter cells, the UCP from the no‐cell condition contained twice the Cre activity of the UCP from the cell supernatant (Figure 2g); moreover, both samples showed the highest Cre activity in F2/3, with little to no activity present in F1 (Figure 2g). The observation that Cre activity is observed in the UCP and density gradient fractions F2‐4 in the absence of cells, and therefore in the absence of EVs, suggests that plasmid DNA polyplexes survive the cell culture conditions and differential ultracentrifugation EV purification protocol and are sufficient to edit Cre reporter cells.
3.3. Transfection reagents co‐sediment with EVs
Our experiments up to this point had been conducted using only one transfection reagent (Expifectamine) so it was possible that the artefact we had observed was specific to this reagent. We therefore sought to determine if other common transfection reagents including cationic lipids (Lipofectamine 2000 and Lipofectamine 3000) and polymers (polyethyleneimine and PEI Max) also co‐purified with EVs in the UCP. An equal number of cells were transfected with equivalent amounts of DNA complexed with transfection reagent (Table S1), and UCP material was harvested after 4 days using differential ultracentrifugation (Figure 1c). When UCPs were added to reporter cells we observed a range of Cre activity with all reagents across six independent experiments (Figure 3a). While Expifectamine gave the most reproducibly high level of editing, all tested transfection reagents yielded UCPs that produced significant numbers of GFP+ reporter cells when compared to untreated controls (Figure 3a). Of note, there existed significant variability in the amount of Cre activity present within each group across experiments: some experiments showed levels of activity barely above background whereas other experiments yielded responses in the 5–15% range (Figure 3a). The underlying cause of this variability is not immediately clear but underscores the need for proper negative controls to be run with every experiment to rule out potentially confounding background activity derived from non‐EV material.
FIGURE 3.
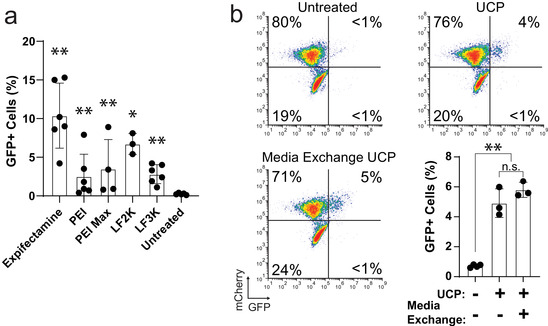
Transfection reagent‐DNA complexes are resistant to removal techniques. (A) Quantification of GFP+ reporter cells treated with 50 μM chloroquine and UCP from cells transfected using one of five different transfection reagents. Cells analysed 3 days post addition and compared to untreated controls (N = 6, *P < 0.05, **P < 0.01, Mann‐Whitney U‐test). (B) Flow cytometry data of reporter cells treated with 50 μM chloroquine and UCP isolated from cells following a media exchange 24 h post‐transfection with Expifectamine. Bar graph shows percentage of GFP+ reporter cells (N = 3; **, P < 0.01; n.s., not significant: P > 0.5; Welch's unequal variances t‐test). Cells analysed 3 days post addition by flow cytometry
3.4. Transfection reagent complexes persist through media exchange
As carryover DTC appeared to be responsible for the Cre activity observed in UCP samples (Figures 2 and 3), we sought methods to remediate this artefact. Two commonly used approaches are nuclease treatment to digest extracellular nucleic acids and media exchange to remove free transfection reagent complexes. Our EV purification strategy includes a step in which clarified cell culture supernatant is treated with the DNA/RNA endonuclease benzonase (Figure 1a), which was not effective in removing plasmid DNA complexed with transfection reagents (Figure 2d), as has been reported by others (Tros De Ilarduya et al., 2002). We therefore sought to determine if exchanging the cell culture media post‐transfection would remove enough transfection‐reagent‐DNA complexes to eliminate or reduce levels of Cre activity in the UCP. Following transient transfection, cells were either left in media containing DNA complexes or moved to fresh media 1 day after transfection. Four days post‐transfection, cell culture supernatant was harvested and processed by differential centrifugation (Figure 1c). Surprisingly, the UCP from cells that had undergone a media exchange after transfection contained approximately the same levels of Cre activity (Figure 3b), indicating that a complete media exchange did not eliminate DTC present in the UCP.
4. DISCUSSION
The lack of correlation between the abundance of Cre protein and Cre activity in iodixanol density gradient fractions first alerted us to a potential issue with a non‐EV source of Cre activity in our experiments. We subsequently found that Cre activity was not associated with EVs loaded with Cre protein, but in fact was associated with DTC that co‐purified with EVs by differential ultracentrifugation. We confirmed these results by demonstrating that even in the absence of cells (and therefore EVs), DTC can survive in media for extended periods of time and can be isolated using standard EV purification methods. Moreover, all transfection reagents examined showed detectable levels of this artefact, and steps commonly taken to remediate potential plasmid DNA contamination, such as nuclease treatment or media exchanges following transfection, were ineffective. Taken together, our data show that differential ultracentrifugation, the most commonly employed EV purification method (Théry et al., 2018), co‐purifies a mix of DTC and EVs which can lead to incorrectly attributing activity to EVs. In this particular case, an iodixanol density gradient was able to separate EVs from contaminating DTC, but appropriate tests should be run to rule out this potential artefact when testing EV activity from transiently transfected cells.
We were initially surprised that exchanging media the day after transfection was not sufficient to eliminate Cre activity from the cell culture supernatant (Figure 3). A possible explanation is that DTC are released from transfected cells after media exchange, as has been reported for lipid nanoparticles (LNPs) (Sahay et al., 2013). Another possibility is that DTC could be released in a complex with EVs, as has been suggested for LNPs (Maugeri et al., 2019). Here we show that plasmid DNA is detected in an EV‐enriched iodixanol density gradient fraction by PCR (F1) following transient transfection of EV‐producing cells (Figure 2e). Although not dispositive, DNA in F1 is rendered sensitive to nuclease digestion following detergent treatment, consistent with the DNA being packaged inside of vesicles (Figure 2f). We do not believe that DNA in F1 is complexed with transfection reagent for two reasons: (1) DNA in F1 can be amplified by PCR without phenol‐chloroform extraction (Figure 2e‐f) and (2) Cre activity is absent in F1 (Figure 2c, g). Higher‐density iodixanol gradient fractions that contain significant Cre activity (F2‐4, Figure 2c, g) all require phenol‐chloroform extraction for detection of DNA by PCR (Figure 2e), similar to what is seen with purified plasmid DNA complexed with transfection reagent (Figure 2d). The density gradient fractions that exhibit Cre activity when added to reporter cells lack significant levels of Cre protein (Figure 2b) but contain Cre DNA that biochemically resembles DNA complexed with transfection reagent (Figure 2d‐f). While the EV‐enriched fraction F1 also contains DNA, it appears to be localized inside vesicles and not associated with transfection reagent (Figure 2e‐f).
Why then would chloroquine enhance delivery of Cre DNA contained in DTC (Figure 1e) but not Cre protein in EVs (Figure 2c)? A possible explanation is that chloroquine is thought to enhance delivery of nucleic acids through the “proton sponge effect”, essentially causing rupture of endolysosomal membranes and release of their contents into the cytosol (Varkouhi et al., 2011). This type of release would present a topological problem for EV‐mediated delivery as the cargo would still be located within the intact vesicle, whereas DTC would be able to transfect the cell; this would explain why DNA located inside EVs was unable to transduce recipient cells while the same plasmid in DTC was effective (Figure 2). A productive method of EV cargo delivery would entail either disruption of both the endosomal and EV membranes, or fusion of the EV membrane with the endosomal or plasma membrane of the recipient cell. Evidence for the latter mechanism has been recently demonstrated in the context of engineered EVs by the functional delivery of mRNA only when the EVs carry a fusogenic protein such as VSVg or Syncytin (Segel et al., 2021).
Several artefacts that can confound EV studies have been reported, and in most cases arise from contaminating factors that are not adequately separated from EVs. Examples include the co‐purification of soluble proteins (Whittaker et al., 2020), lipoproteins (Karimi et al., 2018; Coumans et al., 2017), or nucleic acid precipitates (Kooijmans et al., 2013). Although the artefact described here was resistant to techniques commonly employed to reduce plasmid DNA contamination (nuclease treatment and media exchanges), other approaches can mitigate this issue. One way to avoid this problem entirely is to generate cell lines stably expressing constructs of interest so that no transfection reagent can carry over to purified EVs. In cases where stable cell line generation is not feasible, methods that isolate EVs from cell culture supernatants based primarily on size (differential centrifugation, filtration, size exclusion chromatography) may be particularly prone to the artefact described here. Ideally these approaches would be coupled with orthogonal techniques that select for EVs based on a different property such as density (gradient centrifugation), charge (ion exchange chromatography), or molecular markers (immunoaffinity) to eliminate unwanted DTC. Once a purification strategy has been employed, proper controls to rule out activity arising from this artefact should be included, such as employing a construct that should be functional but not enriched in EVs (e.g., molecule of interest without an EV‐localizing fusion).
CONFLICTS OF INTEREST
All authors are current or former employees of Codiak BioSciences, which is developing EV‐based therapeutics. Russell E. McConnell, Madeleine Youniss, Bhargavee Gnanasambandam, Palak Shah, Wei Zhang and Jonathan D. Finn are inventors of patents relating to EV‐based therapeutics.
Supporting information
Supplemental information
ACKNOWLEDGEMENTS
The authors would like to thank Eric Lander, Sriram Sathyanarayanan, and Doug Williams for helpful discussions.
McConnell, R. E. , Youniss, M. , Gnanasambandam, B. , Shah, P. , Zhang, W. , & Finn, J. D. (2022). Transfection reagent artefact likely accounts for some reports of extracellular vesicle function. Journal of Extracellular Vesicles, 11, e12253. 10.1002/jev2.12253
Russell E. McConnell and Madeleine Youniss contributed equally to this work.
REFERENCES
- Almofti, M. R. , Harashima, H. , Shinohara, Y. , Almofti, A. , Li, W. , & Kiwada, H. (2003). Lipoplex size determines lipofection efficiency with or without serum. Molecular Membrane Biology, 20(1), 35–43. 10.1080/09687680210035104 [DOI] [PubMed] [Google Scholar]
- An, Q. , Van Bel, A. J. E. , & Hückelhoven, R. (2007). Do plant cells secrete exosomes derived from multivesicular bodies? Plant Signaling & Behavior, 2(1), 4–7. 10.4161/psb.2.1.3596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, L. , Wolf, J. M. , Prados‐Rosales, R. , & Casadevall, A. (2015). Through the wall: Extracellular vesicles in gram‐positive bacteria, mycobacteria and fungi. Nature Reviews Microbiology, 13, 620–630. 10.1038/nrmicro3480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo, M. , Raposo, G. , & Théry, C. (2014). Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annual Review of Cell and Developmental Biology, 255–289. 10.1146/annurev-cellbio-101512-122326 [DOI] [PubMed] [Google Scholar]
- Coumans, F. A. W. , Brisson, A. R. , Buzas, E. I. , Dignat‐George, F. , Drees, E. E. E. , El‐Andaloussi, S. , Emanueli, C. , Gasecka, A. , Hendrix, A. , Hill, A. F. , Lacroix, R. , Lee, Y. , van Leeuwen, T. G. , Mackman, N. , Mäger, I. , Nolan, J. P. , van der Pol, E. , Pegtel, D. M. , Sahoo, S. , … Nieuwland, R. (2017). Methodological guidelines to study extracellular vesicles. Circulation Research, 1632–1648. 10.1161/CIRCRESAHA.117.309417 [DOI] [PubMed] [Google Scholar]
- Didiot, M. C. , Hall, L. M. , Coles, A. H. , Haraszti, R. A. , Godinho, B. M. D. C. , Chase, K. , Sapp, E. , Ly, S. , Alterman, J. F. , Hassler, M. R. , Echeverria, D. , Raj, L. , Morrissey, D. V. , DiFiglia, M. , Aronin, N. , & Khvorova, A. (2016). Exosome‐mediated delivery of hydrophobically modified SiRNA for huntingtin MRNA silencing. Molecular Therapy, 24(10), 1836–1847. 10.1038/mt.2016.126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dooley, K. , McConnell, R. E. , Xu, K. , Lewis, N. D. , Haupt, S. , Youniss, M. R. , Martin, S. , Sia, C. L. , McCoy, C. , Moniz, R. J. , Burenkova, O. , Sanchez‐Salazar, J. , Jang, S. C. , Choi, B. , Harrison, R. A. , Houde, D. , Burzyn, D. , Leng, C. , Kirwin, K. , … Williams, D. E. (2021). A versatile platform for generating engineered extracellular vesicles with defined therapeutic properties. Molecular Therapy, 29(5), 1729–1743. 10.1016/j.ymthe.2021.01.020. Epub 2021 Jan 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath, N. , Osteikoetxea, X. , De Oliveria, T. M. , Lázaro‐Ibáñez, E. , Shatnyeva, O. , Schindler, C. , Tigue, N. , Mayr, L. M. , Dekker, N. , Overman, R. , & Davies, R. (2019). Endosomal escape enhancing compounds facilitate functional delivery of extracellular vesicle cargo. Nanomedicine, 14(21), 2799–2814. 10.2217/nnm-2019-0061 [DOI] [PubMed] [Google Scholar]
- Heath, N. , Osteikoetxea, X. , Oliveria, T. M. D. , Lázaro‐Ibáñez, E. , Shatnyeva, O. , Schindler, C. , Tigue, N. , Mayr, L. M. , Dekker, N. , Overman, R. , & Davies, R. (2019). Endosomal escape enhancing compounds facilitate functional delivery of extracellular vesicle cargo. Nanomedicine, 14(21), 2799–2814. 10.2217/nnm-2019-0061 [DOI] [PubMed] [Google Scholar]
- Hung, M. E. , & Leonard, J. N. (2016). A platform for actively loading cargo RNA to elucidate limiting steps in EV‐mediated delivery. Journal of Extracellular Vesicles, 5(1), 10.3402/jev.v5.31027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeppesen, D. K. , Fenix, A. M. , Franklin, J. L. , Higginbotham, J. N. , Zhang, Q. , Zimmerman, L. J. , Liebler, D. C. , Ping, J. , Liu, Q. , Evans, R. , Fissell, W. H. , Patton, J. G. , Rome, L. H. , Burnette, D. T. , & Coffey, R. J. (2019). Reassessment of exosome composition. Cell, 177(2), 428–445.e18. 10.1016/j.cell.2019.02.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimi, N. , Cvjetkovic, A. , Jang, S. C. , Crescitelli, R. , Hosseinpour Feizi, M. A. , Nieuwland, R. , Lötvall, J. , & Lässer, C. (2018). Detailed analysis of the plasma extracellular vesicle proteome after separation from lipoproteins. Cellular and Molecular Life Sciences, 75(15), 2873–2886. 10.1007/s00018-018-2773-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooijmans, S. A. A. , Stremersch, S. , Braeckmans, K. , De Smedt, S. C. , Hendrix, A. , Wood, M. J. A. , Schiffelers, R. M. , Raemdonck, K. , & Vader, P. (2013). Electroporation‐induced SiRNA precipitation obscures the efficiency of SiRNA loading into extracellular vesicles. Journal of Controlled Release, 172(1), 229–238. 10.1016/j.jconrel.2013.08.014 [DOI] [PubMed] [Google Scholar]
- Kooijmans, S. A. A. , Stremersch, S. , Braeckmans, K. , De Smedt, S. C. , Hendrix, A. , Wood, M. J. A. , Schiffelers, R. M. , Raemdonck, K. , & Vader, P. (2013). Electroporation‐induced SiRNA precipitation obscures the efficiency of SiRNA loading into extracellular vesicles. Journal of Controlled Release, 172(1), 229–238. 10.1016/j.jconrel.2013.08.014 [DOI] [PubMed] [Google Scholar]
- Ma, B. , Zhang, S. , Jiang, H. , Zhao, B. , & Lv, H. (2007). Lipoplex morphologies and their influences on transfection efficiency in gene delivery. Journal of Controlled Release, 184–194. 10.1016/j.jconrel.2007.08.022 [DOI] [PubMed] [Google Scholar]
- Madhusoodanan, J. (2020). The therapeutic potential of exosomes. Nature, S10–S11. 10.1038/d41586-020-01375-9 [DOI] [Google Scholar]
- Maugeri, M. , Nawaz, M. , Papadimitriou, A. , Angerfors, A. , Camponeschi, A. , Na, M. , Hölttä, M. , Skantze, P. , Johansson, S. , Sundqvist, M. , Lindquist, J. , Kjellman, T. , Mårtensson, I. L. , Jin, T. , Sunnerhagen, P. , Östman, S. , Lindfors, L. , & Valadi, H. (2019). Linkage between endosomal escape of LNP‐MRNA and loading into EVs for transport to other cells. Nature Communication, 10(1), 10.1038/s41467-019-12275-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan, Q. , Ramakrishnaiah, V. , Henry, S. , Fouraschen, S. , Ruiter, P. E. D. , Kwekkeboom, J. , Tilanus, H. W. , Janssen, H. L. A. , & Laan, L. J. W. V. D. (2012). Hepatic cell‐to‐cell transmission of small silencing RNA can extend the therapeutic reach of RNA interference (RNAi). Gut, 61(9), 1330–1339. 10.1136/gutjnl-2011-300449 [DOI] [PubMed] [Google Scholar]
- Pegtel, D. M. , Cosmopoulos, K. , Thorley‐Lawson, D. A. , Van Eijndhoven, M. A. J. , Hopmans, E. S. , Lindenberg, J. L. , De Gruijl, T. D. , Würdinger, T. , & Middeldorp, J. M. (2010). Functional delivery of viral MiRNAs via exosomes. PNAS, 107(14), 6328–6333. 10.1073/pnas.0914843107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prada, I. , & Meldolesi, J. (2016). Binding and fusion of extracellular vesicles to the plasma membrane of their cell targets. International Journal of Molecular Sciences, 17(8), 1296. 10.3390/ijms17081296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabanovic, B. , Piva, F. , Cecati, M. , & Giulietti, M. (2021). Promising extracellular vesicle‐based vaccines against viruses, including SARS‐CoV‐2. Biology, 1–14. 10.3390/biology10020094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahay, G. , Querbes, W. , Alabi, C. , Eltoukhy, A. , Sarkar, S. , Zurenko, C. , Karagiannis, E. , Love, K. , Chen, D. , Zoncu, R. , Buganim, Y. , Schroeder, A. , Langer, R. , & Anderson, D. G. (2013). Efficiency of SiRNA delivery by lipid nanoparticles is limited by endocytic recycling. Nature Biotechnology, 31(7), 653–658. 10.1038/nbt.2614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin, J. , Arganda‐Carreras, I. , Frise, E. , Kaynig, V. , Longair, M. , Pietzsch, T. , Preibisch, S. , Rueden, C. , Saalfeld, S. , Schmid, B. , Tinevez, J. Y. , White, D. J. , Hartenstein, V. , Eliceiri, K. , Tomancak, P. , & Cardona, A. (2012). Fiji: An open‐source platform for biological‐image analysis. Nature Methods, 676–682. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwechheimer, C. , & Kuehn, M. J. (2015). Outer‐membrane vesicles from gram‐negative bacteria: Biogenesis and functions. Nature Reviews Microbiology, 16, 605–619. 10.1038/nrmicro3525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segel, M. , Lash, B. , Song, J. , Ladha, A. , Liu, C. C. , Jin, X. , Mekhedov, S. L. , Macrae, R. K. , Koonin, E. V. , & Zhang, F. (2021). Mammalian retrovirus‐like protein PEG10 packages its own MRNA and can be pseudotyped for MRNA delivery. Science (80‐.), 373(6557), 882–889. 10.1126/science.abg6155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanly, C. , Fiume, I. , Capasso, G. , & Pocsfalvi, G. (2016). Isolation of exosome‐like vesicles from plants by ultracentrifugation on sucrose/deuterium oxide (D2O) density cushions. In Methods in Molecular Biology. Humana Press Inc., (vol. 1459, pp 259–269). 10.1007/978-1-4939-3804-9_18 [DOI] [PubMed] [Google Scholar]
- Sterzenbach, U. , Putz, U. , Low, L. H. , Silke, J. , Tan, S. S. , & Howitt, J. (2017). Engineered exosomes as vehicles for biologically active proteins. Molecular Therapy, 25(6), 1269–1278. 10.1016/j.ymthe.2017.03.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoorvogel, W. , Kleijmeer, M. J. , Geuze, H. J. , & Raposo, G. (2002). The biogenesis and functions of exosomes. Traffic, 3, 321–330. [DOI] [PubMed] [Google Scholar]
- Théry, C. , Witwer, K. W. , Aikawa, E. , Alcaraz, M. J. , Anderson, J. D. , Andriantsitohaina, R. , Antoniou, A. , Arab, T. , Archer, F. , Atkin‐Smith, G. K. , Ayre, D. C. , Bach, J. M. , Bachurski, D. , Baharvand, H. , Balaj, L. , Baldacchino, S. , Bauer, N. N. , Baxter, A. A. , Bebawy, M. , … Beckham, C. (2018). Minimal Information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and Update of the MISEV2014 Guidelines. Journal of Extracellular Vesicles, 7(1), 10.1080/20013078.2018.1535750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tkach, M. , & Théry, C. (2016). Communication by extracellular vesicles: Where we are and where we need to go. Cell, 164(6), 1226–1232. 10.1016/j.cell.2016.01.043 [DOI] [PubMed] [Google Scholar]
- Tros De Ilarduya, C. , Arangoa, M. A. , Moreno‐Aliaga, M. J. , & Düzgüneş, N. (2002). Enhanced gene delivery in vitro and in vivo by improved transferrin‐lipoplexes. Biochimica et Biophysica Acta ‐ Biomembranes, 1561(2), 209–221. 10.1016/S0005-2736(02)00348-6 [DOI] [PubMed] [Google Scholar]
- Van Deun, J. , Mestdagh, P. , Agostinis, P. , Akay, Ö. , Anand, S. , Anckaert, J. , Martinez, Z. A. , Baetens, T. , Beghein, E. , Bertier, L. , Berx, G. , Boere, J. , Boukouris, S. , Bremer, M. , Buschmann, D. , Byrd, J. B. , Casert, C. , Cheng, L. , Cmoch, A. , … Daveloose, D. (2017). EV‐TRACK: Transparent reporting and centralizing knowledge in extracellular vesicle research. Nature Methods, 14(3), 228–232. 10.1038/NMETH.4185 [DOI] [PubMed] [Google Scholar]
- Van Niel, G. , D'Angelo, G. , & Raposo, G. (2018). Shedding light on the cell biology of extracellular vesicles. Nature Reviews Molecular Cell Biology, 19(4), 213–228. 10.1038/nrm.2017.125 [DOI] [PubMed] [Google Scholar]
- Varkouhi, A. K. , Scholte, M. , Storm, G. , & Haisma, H. J. (2011). Endosomal escape pathways for delivery of biologicals. Journal of Controlled Release, 151(3), 220–228. 10.1016/J.JCONREL.2010.11.004 [DOI] [PubMed] [Google Scholar]
- Whittaker, T. E. , Nagelkerke, A. , Nele, V. , Kauscher, U. , & Stevens, M. M. (2020). Experimental artefacts can lead to misattribution of bioactivity from soluble mesenchymal stem cell paracrine factors to extracellular vesicles. Journal of Extracellular Vesicles, 9(1), 10.1080/20013078.2020.1807674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witwer, K. W. , & Wolfram, J. (2021). Extracellular vesicles versus synthetic nanoparticles for drug delivery. Nature Reviews Materials, 103–106. 10.1038/s41578-020-00277-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye, Y. , Zhang, X. , Xie, F. , , Xu, B. , Xie, P. , Yang, T. , Shi, Q. , Zhang, C. Y. , Zhang, Y. , Chen, J. , Jiang, X. , & Li, J. (2020). An engineered exosome for delivering SgRNA:Cas9 ribonucleoprotein complex and genome editing in recipient cells. Biomaterials Science, 8(10), 2966–2976. 10.1039/d0bm00427h [DOI] [PubMed] [Google Scholar]
- Yim, N. , Ryu, S. W. , Choi, K. , Lee, K. R. , Lee, S. , Choi, H. , Kim, J. , Shaker, M. R. , Sun, W. , Park, J. H. , Kim, D. , Heo, W. D. , & Choi, C. (2016). Exosome engineering for efficient intracellular delivery of soluble proteins using optically reversible protein‐protein interaction module. Nature Communication, 7(1), 1–9. 10.1038/ncomms12277 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental information