

Abstract
The interferon system protects mammals against virus infections. There are several types of interferons, which are characterized by their ability to inhibit virus replication and resultant pathogenesis by triggering both innate and cell-mediated immune responses. Virus infection is sensed by a variety of cellular pattern-recognition receptors and triggers the synthesis of interferons, which are secreted by the infected cells. In uninfected cells, cell surface receptors recognize the secreted interferons and activate intracellular signaling pathways that induce the expression of interferon-stimulated genes; the proteins encoded by these genes inhibit different stages of virus replication. To avoid extinction, almost all viruses have evolved mechanisms to defend themselves against the interferon system. Consequently, a dynamic equilibrium of survival is established between the virus and its host, an equilibrium that can be shifted to the host’s favor by the use of exogenous interferon as a therapeutic antiviral agent.
Keywords: virus infection, innate immunity, interferon-stimulated gene, pathogenesis, antiviral action, viral evasion, dsRNA, pattern-recognition receptor, interferon-λ
INTRODUCTION
Interferons (IFNs) are cytokines that act upon cells to impart resistance to virus replication. This property of the large family of IFN proteins was discovered by Isaacs and Lindenmann in 1957 (1). In general, virus infection triggers transient synthesis and secretion of IFNs to promote protection of as-yet-uninfected cells. IFNs induce the transcription of hundreds of IFN-stimulated genes (ISGs), whose protein products inhibit a variety of steps of virus replication (2). Specific ISG proteins are particularly effective against specific families of viruses, and optimal protection is achieved by the inhibition of more than one step of virus replication by different ISG proteins. Viruses have evolved many mechanisms to evade the antiviral actions of IFNs by blocking their synthesis and/or their actions (3). Thus, in vivo, the IFN system is the major mediator of the maintenance of host-virus homeostasis. In addition to their antiviral effects, IFNs affect other functional properties of cells, especially of the immune system; IFN activation of immune cells contributes to the inhibition of viral pathogenesis (4). This article focuses on the antiviral actions of IFNs, especially those mediated by the cell-intrinsic actions of ISG proteins.
INTERFERON CLASSIFICATION AND RECENT DISCOVERIES OF NOVEL INTERFERONS
The classification of IFNs is based on the cell surface receptors to which they bind, to activate intracellular signaling pathways. Type I, II, and III IFNs are distinguished by their distinct IFN receptor complexes (Figure 1) (5). Type I IFNs signal through the dimeric IFN-α/β receptor (IFNAR), consisting of IFNAR1 and IFNAR2; the type I IFNs comprise more than 10 subtypes of IFN-α, named IFN-α1, -α2, etc.; IFN-β, -κ, -ω, -ε; and limitin. Type II IFNs bind to the tetrameric IFN-γ receptor (IFNGR), consisting of two subunits each of IFNGR1 and IFNGR2; IFN-γ is the only known type II IFN. Type III IFNs, comprising IFN-λ1, -λ2, -λ3, and -λ4, trigger the IFN-λ receptor (IFNLR), a heterodimer of IFNLR1 and IL10RB (5–7).
Figure 1.
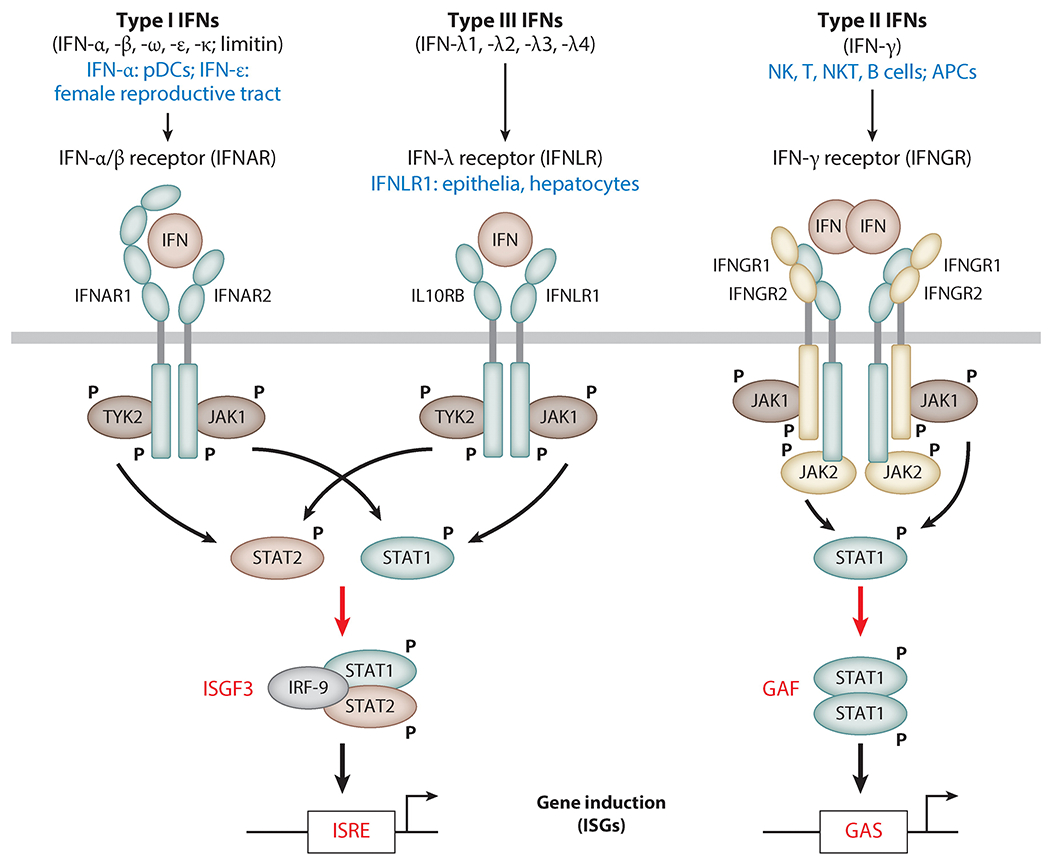
Major differences between the type I, II, and III IFN systems. Each type of IFN acts through its own cognate cell surface receptor. The Janus kinases JAK1, TYK2, and JAK2 phosphorylate STAT proteins and trigger their transcriptional activity. The three signaling systems are distinguished by their cell type–specific expression of IFNs or their receptor (blue), as well as by the different transcription factors they activate (red). Abbreviations: APC, antigen-presenting cell; GAS, gamma-activated site; IFN, interferon; ISG, interferon-stimulated gene; ISRE, IFN-stimulated response element; NK, natural killer; NKT, natural killer T; pDC, plasmacytoid dendritic cell. To download a PowerPoint slideshow illustrating further details about each IFN system, click the Interactive Figure button.
The biological responses to the three IFN types overlap in some aspects, such as their ability to inhibit virus replication in infected cells, but many effects of type I, II, and III IFN signaling are distinct, especially in vivo. These differences are largely based on two properties (Figure 1). First, the expression of an IFN or its cognate receptor is cell type or tissue specific; not all IFNs and all receptors are produced by all cells. Among type I IFNs, plasmacytoid dendritic cells (pDCs) are the dominant IFN-α producers in vivo after microbial infection, whereas IFN-ε is synthesized constitutively in the female reproductive tract (8–10). Whereas IFNAR is ubiquitously expressed, only cells of epithelial origin, as well as a few others, such as hepatocytes, have the ability to respond to type III IFNs (IFN-λ), because they express both IFNLR subunits (11–13). Type II IFNs (IFN-γ) are synthesized by cells of the immune system, mainly natural killer cells, natural killer T cells, B cells, and antigen-presenting cells (5). Such cell type–restricted expression allows for localized, targeted IFN effects and prevents uncontrolled systemic inflammation. Second, different IFN types activate different transcription factors to induce different groups of ISGs (14). These large sets of genes only partially overlap between IFNAR, IFNLR, and IFNGR. IFNAR and IFNLR, although unrelated, use the same intracellular signaling pathway to activate the transcription factor complex ISG factor 3 (ISGF3), and they therefore induce similar sets of genes (15). IFNGR, however, activates the transcription factor gamma-activated factor (GAF), which induces different genes (14, 16). Depending on the cell type, IFNAR signaling triggers activation of additional transcription factors, leading to further differentiation of IFN responses via differential induction of specific ISGs (17).
Type I and II IFNs and their receptors were cloned and sequenced in the late 1980s (18), and enormous research efforts have since elucidated the details of their signaling pathways. IFN-ε, a novel type I IFN, was discovered in 2004 (10); it had probably been overlooked because it is not inducible by microbial infection but is constitutively expressed with tissue restrictions. In 2003, two research teams independently discovered that a group of cytokines, formerly designated interleukin (IL)-29, IL-28A, and IL-28B, shared several biological properties of type I IFNs (19, 20). These cytokines used their own distinct receptor and hence were a new type of IFN, dubbed type III IFNs; they were later officially named IFN-λ1, -λ2, and -λ3. Because hepatocytes are one of the cell types that express their receptor, IFNLR, the IFN-λs have received a lot of attention in the context of chronic hepatitis C virus (HCV) infections in humans, in which polymorphisms in the IFN-λ gene locus have been found to correlate with treatment success. An additional member of the type III IFN family was discovered in 2013 (21). Surprisingly, not all humans are able to synthesize this novel IFN-λ4, whose gene is located in the same locus on chromosome 19 as those of the other IFN-λs. A two-nucleotide polymorphism (IFNL4 rs368234815) in its first exon determines whether or not the transcribed mRNA can be translated into the IFN-λ4 protein. The emerging allele, TT, contains a substitution and an extra nucleotide, leading to a codon frameshift and termination of translation (6, 21). The ancestral allele, −G, which allows for IFN-λ4 synthesis, is maintained especially in Africans as −G/−G or −G/TT but is almost lost in Asians; the inactive TT allele probably emerged in response to selection pressure by some pathogen other than HCV and is not found in other mammals (22).
INTERFERON SYNTHESIS TRIGGERED BY CELLULAR SENSORS OF VIRUS INFECTION
IFNs are highly effective in limiting virus replication and spread, but because they are normally not expressed, IFN synthesis needs to be triggered quickly and strongly upon host contact with the virus. Because all viruses replicate inside host cells, detecting the viral nucleic acids—e.g., RNA or DNA genomes or mRNAs—upon virus entry is an effective strategy for triggering innate immune responses. The viral nucleic acids are recognized by a specialized group of proteins known as pattern-recognition receptors (PRRs) (Figure 2) (23). Nucleic acid PRRs are either endosomal membrane proteins or cytosolic proteins. RNA viruses are recognized by the endosomal transmembrane Toll-like receptors TLR3, TLR7, and TLR8 (24) and by the cytoplasmic RNA helicases RIG-I, MDA5, and LGP2, also known as RIG-I-like receptors (RLRs) (25). DNA viruses are recognized by endosomal TLR9 or several cytoplasmic DNA sensors such as cyclic GMP-AMP synthase (cGAS), IFN-induced 16-kDa protein (IFI16), DDX41, and DAI (23, 26). Some of these receptors are expressed only in specific cell types and recognize specific families of viruses. TLRs are preferentially expressed in myeloid cells, such as plasmacytoid dendritic cells and macrophages, whereas RLRs and DNA sensors are expressed in many cell types, including epithelial cells and fibroblasts (27, 28). TLR3 is a dsRNA sensor involved in recognition of RNA viruses such as influenza A virus (FLUAV) and picornaviruses and DNA viruses such as herpes simplex virus 1 (HSV-1) (29–32). TLR7 and TLR8 are sensors of ssRNA from many viruses, including vesicular stomatitis virus (VSV) and FLUAV (33). RIG-I, MDA5, and LGP2, three closely related RLRs, are the key cytoplasmic sensors of viral RNA species (26, 34). RIG-I recognizes members of a variety of virus families, including paramyxoviruses, FLUAV, flaviviruses such as West Nile virus (WNV), rhabdoviruses such as VSV, and reoviruses. In contrast, MDA5 and LGP2 primarily recognize picornaviruses such as encephalomyocarditis virus (EMCV) (35). These specificities are based on the different RNA species that are recognized by different RLRs: RIG-I can bind short dsRNA and 5′-triphosphate or 5′-diphosphate ssRNA, generated during viral RNA genome replication as well as from incoming viral genomes (25, 36, 37), whereas MDA5 recognizes long dsRNA species and is also involved in sensing viral mRNAs lacking cap 2′-O-methylation (38, 39). The third RLR, LGP2, has a dual role during cytoplasmic RNA recognition: On the one hand, it binds dsRNA in tandem with MDA5 and thereby enhances MDA5 activation (40, 41); on the other hand, it can inhibit RIG-I activation by competing for dsRNA and by direct interaction with RIG-I (42). Viral CpG-containing DNA and genomic DNA from murine cytomegalovirus can be recognized in the endosomal lumen by TLR9, especially in plasmacytoid dendritic cells (43). AT-rich DNA from adenoviruses or herpesviruses is transcribed into RNA by RNA polymerase III, which generates ligands to activate RIG-I (44, 45). Several DNA sensors, such as cGAS, DDX41, and IFI16, use the adapter protein STING for downstream signaling (Figure 3) (26). It is unclear why so many cytoplasmic DNA receptors exist; they may act redundantly, or they may possess specificity for certain cell types or distinct features of DNA ligands (46).
Figure 2.
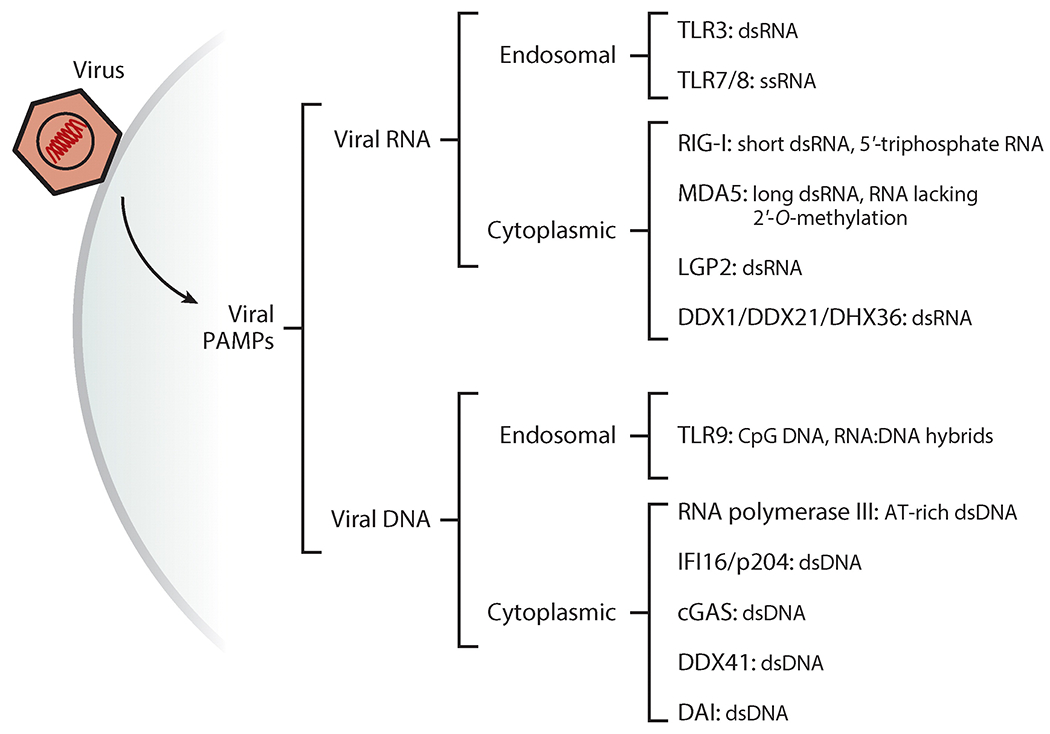
Cellular sensors detecting viral RNA and DNA. Viral nucleic acids are the key pathogen-associated molecular pattern (PAMP) for cells to recognize the presence of viruses. A large number of sensor proteins (pattern-recognition receptors) bind specific forms of nucleic acids in endosomes and in the cytoplasm and trigger signaling to elicit innate immune responses such as the induction of interferons.
Figure 3.
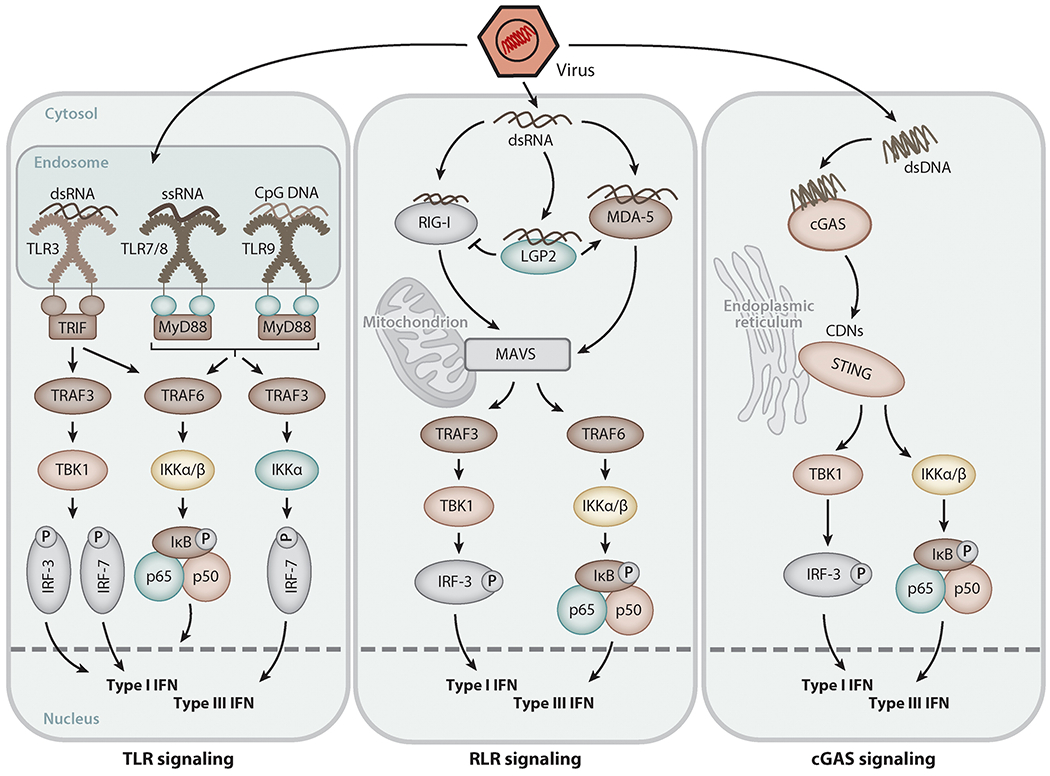
Signaling pathways for type I and III interferon (IFN) induction triggered by viral nucleic acids. Upon virus entry, viral RNA or DNA in endosomes or the cytoplasm is detected by receptor proteins [e.g., Toll-like receptors (TLRs), RIG-I-like receptors (RLRs), cyclic GMP-AMP synthase (cGAS)], which trigger signaling via adapter proteins such as TRIF, MyD88, MAVS, and STING to activate transcription factors of the IRF family as well as NF-κB, leading to induced transcription of the type I and type III IFN genes.
Activation of Pattern-Recognition Receptors
In the absence of virus infection, the PRRs remain in an inactive conformation to block any undesired activation of innate immune responses. Upon binding of viral molecular patterns, the PRRs become activated, leading to conformational changes, often dimerization or oligomerization, and interaction with downstream adapter proteins. We describe below some examples of PRR activation and how these steps are regulated.
The TLRs have multiple leucine-rich repeats in their ectodomain, in the endosomal lumen, where they can directly bind to nucleic acids (24). Activation of TLRs requires tyrosine phosphorylation of their cytoplasmic domains (47). For example, TLR3 is activated by tyrosine phosphorylation of two tyrosine residues, Tyr759 and Tyr858, in its cytoplasmic domain (48–50). In resting cells, the cytoplasmic domain of TLR3 remains in a closed conformation to avoid phosphorylation by kinases. Upon binding to dsRNA in the endosomal lumen, TLR3 dimerizes and changes its conformation, which allows the two critical tyrosine kinases, epidermal growth factor receptor (EGFR) and Src, to be recruited to the receptor (51, 52). The sequential recruitment of these tyrosine kinases triggers direct phosphorylation of the two tyrosine residues: EGFR phosphorylates Tyr858 and Src phosphorylates Tyr759. This activated TLR3 is able to recruit the adapter TRIF for downstream signaling (52).
RLRs are activated by dephosphorylation- and ubiquitination-dependent mechanisms (53). They possess a conserved RNA helicase domain for recognition of RNA, and RIG-I and MDA5 (but not LGP2) also possess two CARD domains, which interact with the critical adapter protein MAVS (26). In uninfected cells, phosphorylation of RIG-I CARDs by protein kinase C (PKC) maintains RIG-I’s closed conformation (54). Upon binding to viral RNA, RIG-I undergoes dephosphorylation by protein phosphatase 1 (PP1) (55). Then, TRIM25, a ubiquitin ligase, recognizes the dephosphorylated RIG-I and covalently adds ubiquitin moieties onto its CARD, inducing oligomerization and allowing for interaction with MAVS (56). RIG-I is also activated by binding to unanchored ubiquitin chains, produced by TRIM25 (57). One of several DNA sensors in the cytoplasm, cGAS, upon binding to dsDNA, produces cyclic dinucleotides from ATP and GTP; these cyclic dinucleotides directly bind and activate the endoplasmic reticulum-bound adapter STING, a common signaling platform utilized by DNA sensors (58).
Interferon Induction by Virus-Activated Pattern-Recognition Receptors
Virus detection by PRRs is critical for rapid activation of downstream signaling pathways that lead to the induction of type I and type III IFNs. Induction of IFN-β requires the synergistic promoter binding of three transcription factors: IRF-3, NF-κB, and AP1 (59, 60). IFN-α subtypes require IRF-7; IFN-λs utilize IRF-3, IRF-7, and NF-κB (61–64). These transcription factors are activated upon recognition of virus infection by PRRs. Both dsRNA and dsDNA, present in the endosome or cytoplasm, are capable of activating these transcription factors (Figure 3). Activated PRRs interact with their respective adapter protein: TLR3 uses TRIF, which is recruited to the intracellular domain of TLRs; TLR7, TLR8, and TLR9 use MyD88; RLRs use MAVS, located on the surface of mitochondria or peroxisomes; and several cytoplasmic DNA sensors use STING (65). In the following steps, TRIF and MAVS recruit signaling proteins of the TRAF family of ubiquitin ligases, as well as the IKKα/β and TBK1 kinases. These serine/threonine kinases phosphorylate and activate the downstream transcription factors. STING directly recruits the kinases without the need for TRAFs. IRF-3 remains in a closed conformation in the cytosol in uninfected cells, but upon PRR activation, it is phosphorylated by TBK1 on specific serine/threonine residues. This opens up its conformation, causing its dimerization and translocation to the nucleus, where it binds to specific sites, the IFN-stimulated response elements (ISREs), in the promoter regions of target genes such as IFN-β and IFN-λ (23). Another transcription factor, NF-κB, is activated by phosphorylation-dependent release from its inhibitor IκB to induce target genes, including genes encoding IFNs and proinflammatory cytokines such as interleukins and TNF-α. API is activated by all PRRs via mitogen-activated protein kinase (MAPK) pathways (23). Importantly, when IFNs are secreted by infected cells, they act on surrounding and remote cells to induce ISGs; the infected cell, however, benefits from a shortcut to induce ISGs: IRF-3 and IRF-7 can directly induce the transcription of ISGs, without the need for IFN, by binding to the ISREs in the ISG promoters (66, 67).
Interferon-Independent Antiviral Response to Pattern-Recognition Receptor Activation: The RIG-I-Induced IRF-3-Mediated Pathway of Apoptosis
The transcription factor IRF-3, apart from its role in inducing type I and III IFNs and ISGs in infected cells, has an independent additional function in the innate defense against viruses: Activation of IRF-3 by RLR signaling activates a direct proapoptotic effect, which we called the RIG-I-induced IRF-3-mediated pathway of apoptosis (RIPA) (68). RIPA does not require any of IRF-3’s transcriptional activities, and it is triggered by RIG-I activation, either directly by viral RNA or by RNA transcribed from viral DNA by RNA polymerase III. RIPA requires a unique set of signaling proteins, such as TRAF2 and TRAF6, to differentially activate IRF-3 for this pathway. In RIPA, IRF-3 directly activates the proapoptotic BH3-only protein BAX, an activator of the intrinsic apoptosis pathway. BAX is activated by its direct interaction with IRF-3 and the concomitant translocation of the IRF-3:BAX complex to the mitochondrial membrane. This causes mitochondrial dysfunction, release of cytochrome c into the cytosol, and subsequent activation of caspase 9 and caspase 3 to induce apoptosis (68). RIPA starts much later than IRF-3’s transcriptional activity; in the case of Sendai virus (SeV) infection, this is due to the PI3K/AKT pathway, which stabilizes the cellular pool of the apoptosis inhibitor XIAP. Once XIAP starts to be degraded, RIPA is initiated (69). RIPA provides a defense mechanism to the host: Absence of IRF-3 or caspase 3 causes viral persistence in SeV-infected cells, and RIPA-deficient, Bax-deficient cells and mice are highly susceptible to infection by SeV and EMCV (70, 71).
INTERFERON SIGNALING PATHWAYS
Following their synthesis and secretion, IFNs trigger biological effects by binding to their cognate cell surface receptor complex, resulting in conformational changes of the receptor’s intracellular domains and giving rise to a cascade of posttranslational modifications of intracellular signaling molecules. The JAK-STAT signaling pathway is the major effector; its main function is the activation of transcription factors that in turn induce transcription of ISGs (Figure 1) (17). The dimeric receptor complex IFNAR is engaged by binding of monomeric type I IFNs, stimulating the intracellular IFNAR-associated Janus kinases JAK1 and TYK2 to autophosphorylate some of their tyrosine residues, followed by tyrosine phosphorylation of the IFNAR subunits; these IFNAR phosphotyrosines create a docking platform for the STAT1 and STAT2 transcription factors. Due to proximity to the JAKs, the STATs are tyrosine phosphorylated (e.g., Tyr701 of STAT1) and dissociate from the receptor. The resulting STAT1-STAT2 heterodimer forms a complex with transcription factor IRF-9. This unique complex, ISGF3, translocates to the nucleus and binds to ISG promoters containing an ISRE to transcriptionally induce the respective genes (17, 72). Surprisingly, although it is unrelated to IFNAR, IFNLR utilizes the same JAK-STAT signaling pathway to induce a similar set of ISGs (15). In contrast, the IFN-γ receptor, IFNGR, cannot recruit TYK2 and therefore does not activate STAT2. In consequence, the STAT1 homodimer, GAF, is the dominant transcription factor activated by type II IFNs and activates the transcription of genes containing a gamma-activated site (GAS) in their promoters (73).
Besides the canonical activation of ISGF3 and GAF, IFNs activate additional signaling pathways that impact gene induction and provide negative feedback regulation of the receptor (17, 74). Figure 4 depicts examples from type I IFN–triggered signaling. Because IFNAR, like IFNGR, engages JAK1, IFNAR signaling activates not only ISGF3 but also GAF. This GAF activation, however, happens to a much lesser degree because the serine kinase IKKϵ skews the IFNAR pathway toward ISGF3 by serine phosphorylating STAT1, thereby inhibiting STAT1 homodimerization (75, 76). Further diversification of the sets of ISGs induced by type I IFNs occurs via heterodimerization of STAT1 with IRF-1 or subunits of NF-κB, as well as by activation of other STAT family members (STAT3–6), the availability of the latter being cell type dependent (77); all of those complexes can bind to promoters of additional genes that are otherwise not induced by ISGF3. Furthermore, various MAPK pathways are stimulated by type I IFNs, also contributing to the spectrum of induced genes (78). Apart from that, IFN-β can induce several genes that are not inducible by other type I IFNs, such as IFN-α2. This is based on the unique interaction of IFN-β (but not IFN-α2) with IFNAR1, even in the absence of IFNAR2, triggering gene induction via as-yet-unknown signaling pathways (79).
Figure 4.
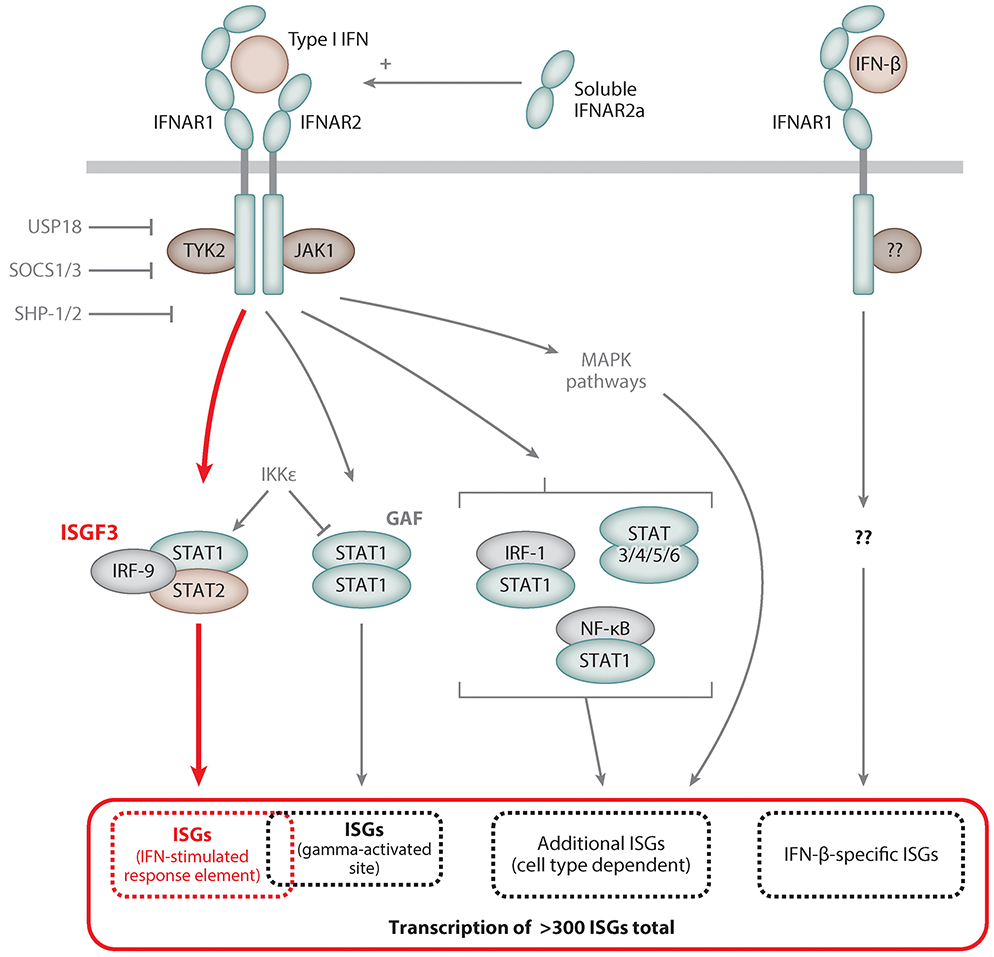
Type I interferon (IFN)-stimulated signaling pathways. Binding of type I IFNs to the dimeric type I IFN receptor, IFNAR, triggers activation of intracellular tyrosine kinases such as JAK1, TYK2, and MAPKs. These in turn activate transcription factor complexes containing STAT proteins, predominantly ISGF3, and others, depending on the cell type. MAPKs activate transcription factors of the ATF/JUN families. IKKϵ serine phosphorylates STAT1 and inhibits its dimerization in favor of ISGF3 formation. A unique interaction of IFN-β with IFNAR1 induces an additional set of genes not induced by other type I IFNs. The soluble splice variant IFNAR2a can enhance type I IFN signaling. Negative regulation of IFNAR signaling is provided by the IFN-stimulated gene (ISG) products USP18 and SOCS, which bind to IFNAR2 and TYK2, respectively, and the tyrosine phosphatases SHP-1 and SHP-2.
Type I and other IFN signaling pathways are fine-tuned in many ways, such as by posttranslational modification of their components through acetylation, methylation, SUMOylation, or palmitoylation (reviewed in 72) and by the generation of mRNA splice variants; for example, IFNAR2 also exists as a soluble IFNAR2a, which enhances type I IFN signaling, as well as IFNAR2b, a membrane-bound decoy receptor that dampens IFN responsiveness (80–82). Postactivation silencing of IFNAR signaling is achieved quickly after IFN exposure and is mediated by at least two proteins that are ISGs themselves and therefore accumulate in response to IFN: Suppressor of cytokine signaling 1 (SOCS1) binds and inhibits TYK2, whereas USP18 (UBP43) occupies IFNAR2 and hence blocks JAK1 activation (83, 84). Furthermore, IFN receptors are internalized into endosomes after IFN binding, thereby temporarily reducing IFN responsiveness of the cell (5). However, when STAT1, STAT2, and IRF-9 are abundant in a cell, as a consequence of their induction by IFN signaling, U-ISGF3, which is composed of unphosphorylated STATs and IRF-9, is capable of sustaining the induction of many ISGF3-inducible ISGs, even in the absence of IFN signaling (85).
IFNs have transcription-independent, direct effects on cellular metabolism as well (86). IFNAR engagement entails the activation of mammalian target of rapamycin (mTOR) complexes via the PI3K/AKT signaling pathway. mTOR kinase activity is a major facilitator of cellular translation via at least two routes: Translation initiation factor eIF4E is released from inhibition by 4E-BP1 when the latter is phosphorylated by mTOR, and mTOR-mediated p70S6K phosphorylation stimulates eIF4B. Both mechanisms lead to higher translation rates of capped mRNAs, such as ISG mRNAs synthesized after IFN exposure (Figure 5) (87, 88).
Figure 5.
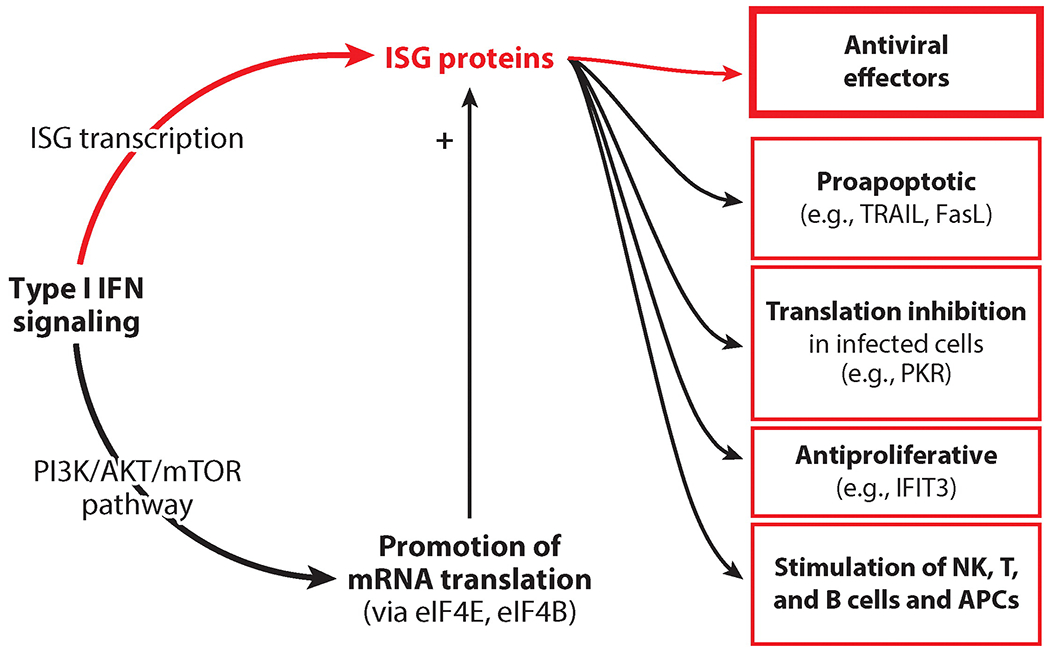
Interferon (IFN)-stimulated genes (ISGs): the mediators of the biological effects of IFNs. Type I IFN signaling transcriptionally induces ISGs, whose protein products are the effectors of IFN actions that inhibit virus life cycles. Other ISG proteins mediate proapoptotic effects or general inhibition of translation. Certain cell types show slowed proliferation after continuous IFN exposure. IFN action on immune cells, such as natural killer (NK) cells, T cells, B cells, and antigen-presenting cells (APCs), is important for shaping the innate and adaptive immune responses extrinsic to the infected cells. A transcription-independent effect of IFNs is the activation of mTOR kinase via the AKT pathway, a major promoter of translation.
ANTIVIRAL ACTIONS OF INTERFERON-INDUCED PROTEINS
Most IFN effects are mediated by the actions of the proteins encoded by the ISGs, which are transcriptionally induced by IFNs. The composition of specific sets of ISGs induced in a cell depends on the transcription factors activated by a specific type of IFN in that cell type and determines the range of biological effects (Figure 5). IFNs are powerful antiviral cytokines used therapeutically against viruses such as HCV (IFN-α2, IFN-λ1), hepatitis B virus (HBV) (IFN-α), and Kaposi’s sarcoma–associated herpesvirus (KSHV) (IFN-α). IFNs block viral pathogenesis by three different means. First, they induce cell-intrinsic innate immunity against viruses, which is the focus of this review. Second, they stimulate innate immune cells such as natural killer cells. Third, they help activate the adaptive immune system by activating B and T cells, as well as antigen-presenting cells, such as macrophages, in which they induce upregulation of major histocompatibility complexes in order to present microbial antigens more efficiently (4, 89). Many proteins that mediate or modulate IFN synthesis and IFN signaling are encoded by ISGs; these are constitutively expressed but induced further by IFN exposure. This family of ISGs includes the genes that encode the viral sensor proteins RIG-I, MDA5, TLRs, and cGAS and the transcription factors IRF-1, IRF-7, IRF-9, STAT1, and STAT2. SOCS1 and USP18, two negative feedback regulators of IFN signaling, are also encoded by ISGs (2). These proteins regulate the magnitude of action of the IFN system but do not affect virus replication per se.
Type I and III IFNs exert their innate antiviral effects through the actions of ISG proteins, which inhibit individual steps of virus life cycles—i.e., entry, genome release, transcription, translation, genome replication, assembly, and egress—within the infected cells (Figure 6) (2, 90). Several key concepts characterize the way these ISG proteins work against viruses. First, the antiviral activity of IFNs against a specific virus is mediated by a number of ISG proteins that act independently, but in concert, to inhibit the virus life cycle at multiple levels (91, 92). Second, different subsets of ISG proteins target different families of viruses (92, 93). Third, some ISG proteins show cell type–specific or tissue-specific antiviral activity; for example, murine Ifit2 inhibits VSV replication in brain neurons but not in lungs, despite being expressed in both organs (94). Fourth, some ISGs exist as small gene families of related members, such as the genes encoding MxA and MxB or IFIT1, IFIT2, IFIT3, and IFIT5. These, however, do not act redundantly; individual members show individual virus and tissue specificity of action. For example, MxA inhibits FLUAV and several other viruses, but only MxB inhibits HIV-1 (95, 96). Cell culture–based screens using overexpression or knockdown of individual ISGs can identify their virus specificity (92, 93, 97, 98), whereas cell type specificities and the biological relevance for disease outcome can be identified only by using individual ISG–knockout mice (94, 99, 100). Valuable insights have been gained from genetic analysis of patients, which demonstrates correlation between mutations in a specific ISG and susceptibility to infection by a specific virus (101). Only representative examples of ISG protein actions are highlighted here; more comprehensive reviews on this topic are available (2, 101).
Figure 6.
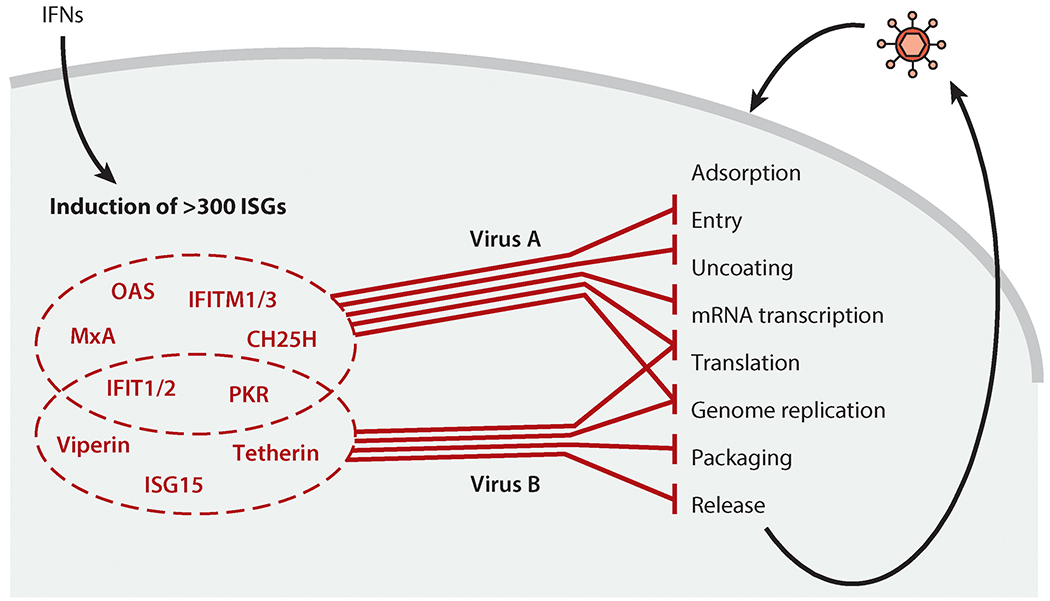
Inhibition of specific viruses by specific combinations of interferon (IFN)-stimulated genes (ISGs). IFNs induce the expression of hundreds of ISG proteins. Upon subsequent virus infection, a specific subset of ISG proteins inhibits multiple steps of the specific virus’s life cycle; each mechanism contributes to the overall inhibition. Additionally, some ISG proteins are inhibitory only in specific cell types. To download a PowerPoint slideshow illustrating further details about this process, click the Interactive Figure button.
Three long-known ISGs are the protein kinase PKR, the GTPase MxA, and the enzyme oligoadenylate synthetase (OAS). PKR is constitutively expressed but further induced by IFN; upon binding to (viral) dsRNA, it phosphorylates the translation initiation factor eIF2α, thereby inhibiting translation of cellular and viral mRNAs (102). Human cytoplasmic MxA and murine nuclear Mx1, on the other hand, have a specific spectrum of antagonized viruses and are both key antiviral effectors against influenza viruses; they bind to viral RNA-protein complexes, either trapping them and preventing their cytoplasmic trafficking (MxA) or inhibiting nuclear transcription of viral mRNA (Mx1) (96). OAS, upon binding to dsRNA, produces 2′,5′-oligoadenylates, which activate the latent ribonuclease, RNase L, which then degrades cellular and viral RNAs. Recent evidence suggests that the degradation products of RNase L can activate the RNA sensor RIG-I to enhance IFN induction within the infected cells (103, 104). Another family of ISGs is the IFITs; they are among the most strongly IFN-induced ISGs overall (14, 105, 106). Human IFIT1 and murine Ifit1 recognize viral mRNAs with either incompletely methylated caps (i.e., caps lacking ribose 2′-O-methylation) or with a triphosphate at their 5′ ends. This interaction inhibits mRNA translation or sequesters viral RNA, respectively, both of which inhibit the viral life cycle (107–110). Intriguingly, most viruses avoid IFIT1 action; murine Ifit2, however, is quite effective against viruses, strongly protecting against pathogenesis by a number of viruses, such as VSV, WNV, mouse hepatitis virus (MHV), and SeV (94, 99, 100, 111). Several ISG proteins inhibit the entry of enveloped viruses into cells by altering the cell membrane surface, thereby preventing fusion of viral and cellular membranes. This is achieved by cholesterol 25-hydroxylase (CH25H) generating 25-hydroxycholesterol, which presumably changes membrane properties, such as membrane fluidity, and thereby prevents the entry of a broad range of enveloped viruses (112–114). IFN-induced transmembrane protein 1 (IFITM1) and IFITM3 each inhibit the entry of a specific set of viruses, such as FLUAV, WNV, and Ebola virus, at different stages between receptor binding and entry (95). ISG15 is a small protein which, upon its IFN-induced synthesis, is conjugated to all newly synthesized cellular and viral proteins (a process termed ISGylation) by an enzyme complex similar to the ubiquitin ligases (115, 116). Antiviral effects of ISG15 were observed using Isg15-knockout mice infected with influenza or herpes viruses, but the requirement for protein ISGylation versus the action of unconjugated ISG15 was not uniform (117, 118). Inherited ISG15 deficiency in humans has recently shed new light onto the function of ISG15 during certain virus infections. The lack of ISG15 made patients more resistant to virus infections, because intracellular unconjugated ISG15 serves as a negative regulator of IFNAR signaling. ISG15 prevents degradation of IFNAR’s negative regulator USP18, and as a result, type I IFN signaling is strongly enhanced in the absence of ISG15 (119, 120). One example of an IFN-induced viral egress inhibitor is Tetherin (BST2); this transmembrane protein tethers HIV-1 virus particles to the cell surface, preventing their release, the last step of the virus life cycle (121–123).
Type I IFNs can also be detrimental to the host. High type I IFN levels are a major pathogenic factor in systemic lupus erythematosus and in septic shock caused by bacteria (124–126). Strikingly, in certain virus infections, type I IFN action is not protective. For example, it worsens pathogenesis during infections of mice with influenza virus or Sendai virus in the lungs, and it promotes persistent lymphocytic choriomeningitis virus (LCMV) infection (127–130). Furthermore, during influenza virus infection, high type I IFN expression in lungs has long been known to facilitate superinfection with bacteria such as Haemophilus influenzae and Streptococcus pneumoniae due to suppression of neutrophils and T helper 17 cell responses, among other mechanisms (131, 132).
ANTIVIRAL ACTIONS OF TYPE III INTERFERONS
Type III IFNs have major roles in specific viral pathogenesis in the intestine, lung, and liver (11). They prevent murine norovirus (MNV) infection in the gut, especially when the commensal bacterial flora has been previously removed by antibiotic treatment; furthermore, type III IFN injections are effective for curing persistent MNV infections(133, 134). The absence of commensal bacteria leads to abnormal intestinal morphology and lymphocyte function; strikingly, these are ameliorated by MNV-induced type I IFN (135). In humans, HCV replicates in hepatocytes. In over 70% of cases, it establishes a persistent infection; a third of these persistent infections progress to chronic liver diseases and carcinoma. Current therapy uses PEGylated IFN-α2 in combination with ribavirin and, more recently, with inhibitors of the viral protease (136). The major problem is a high percentage of IFN-α nonresponders—that is, patients who fail to eliminate the virus during therapy. As a novel alternative, IFN-λ1 has been successfully used in HCV therapeutic trials (137, 138). It may be more effective for two reasons: It has fewer side effects compared with IFN-α because of the cell type–restricted pattern of IFNLR expression (compared with ubiquitous IFNAR expression). Moreover, it has been suggested that, unlike IFNAR signaling, IFNLR signaling is not downregulated during prolonged treatment with IFN (139–141); the silencing of IFN-α2/IFNAR signaling, due to the continuous induction of ISG proteins such as USP18, is a major cause of nonresponsiveness (142). Another more subtle but equally important determinant of treatment outcome is the overall level of ISG expression in the infected liver before IFN-α therapy starts (Figure 7). Lower ISG expression and, concomitantly, relatively elevated HCV RNA levels in livers before therapy correlate with a higher likelihood of successful therapy and permanent eradication of HCV; similarly, spontaneous clearance of the virus without therapy is more likely with low liver ISG expression levels. On the contrary, high ISG expression levels predict failure of IFN therapy (143, 144). Possible causes for different ISG expression levels have emerged from many independent genetic studies correlating specific polymorphisms (one- or two-nucleotide substitutions) in the IFN-λ locus with therapy outcomes. The most conclusive example is the polymorphism rs368234815, described above, which determines whether or not the IFN-λ4 protein can be synthesized in infected cells (21, 145). Consequently, the production of IFN-λ4 by infected liver cells is a strong predictor of being an IFN-α therapy nonresponder (Figure 7) (12, 21, 143, 146).
Figure 7.
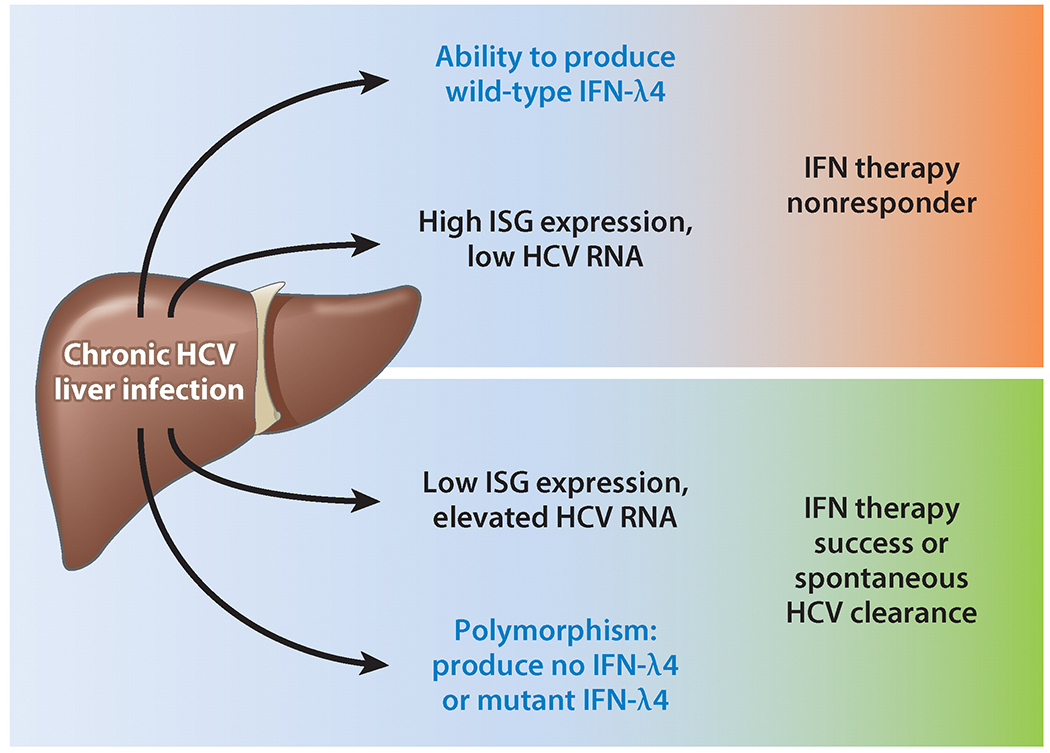
Interferon (IFN)-λ4 as a predictor of responsiveness to IFN-α therapy of chronic hepatitis C virus (HCV) infection. During chronic HCV infection, expression of IFN-λ4 or relatively high IFN-stimulated gene (ISG) expression levels in the liver before treatment correlates with a lack of response to IFN-α2 therapy (i.e., HCV cannot be cleared permanently from the liver). Inversely, being unable to produce IFN-λ4 or having low ISG expression levels makes spontaneous HCV clearance or successful therapy likely. Whether IFN-λ4 is responsible for elevated ISG expression levels is unknown.
VIRAL ANTAGONISM OF THE INTERFERON SYSTEM
To escape from the multipronged antiviral effects of the IFN system, viruses have evolved a variety of strategies. In fact, viral countermeasures target all levels of the IFN system, preventing IFN synthesis (Figure 8, steps ①–⑦), ablating IFN-stimulated JAK-STAT signaling (steps ⑧-–⑪), and inhibiting and/or evading ISG protein functions (step ⑫). Because several review articles have provided comprehensive accounts of these strategies (3, 147), only a few representative and novel examples are highlighted here.
Figure 8.
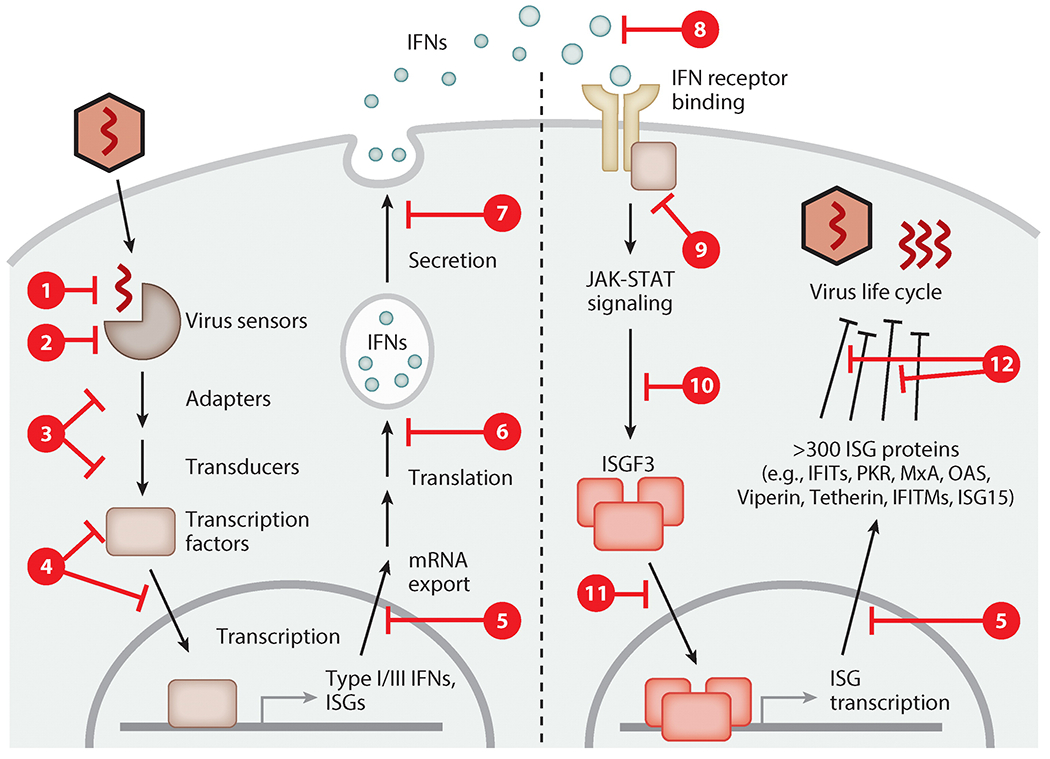
Principles of viral antagonism of the interferon (IFN) system. All mammalian viruses evolved to counteract one or multiple phases of the IFN system. The synthesis of IFNs is prevented by viruses by masking their nucleic acid (step ①), inactivation or cleavage of cellular recognition receptors or their adapter proteins (steps ② and ③), inhibition or degradation of key transcription factors such as IRF-3 (step ④), and generalized inhibition of host gene transcription, protein translation, or protein secretion (steps ⑤, ⑥, and ⑦). IFN signaling is blocked by viral expression of soluble decoy receptors (step ⑧), inhibition of Janus kinases (step ⑨), STAT protein sequestration or degradation (step ⑩), or prevention of transcription factor nuclear translocation (step ⑪). Lastly, the actions of antiviral interferon-stimulated gene (ISG) proteins are counteracted by direct antagonism or evasion (step ⑫). To download a PowerPoint slideshow illustrating further details about each step, click the Interactive Figure button.
The induction of IFN synthesis often starts with detection of viral RNA; several viral proteins, such as FLUAVNS1 and Ebola virus VP35, bind to viral RNA and hide it from detection by cellular sensors such as RIG-I (148, 149). RNA viruses in particular proficiently target RLR and TLR signaling pathways to prevent transcriptional induction of IFNs, very often by interfering with IRF-3 or IRF-7 phosphorylation, nuclear translocation, or promoter binding (3). Some examples include the relocation of the signaling components RIG-I, TBK1, and IRF-3 into viral inclusion bodies (bunyavirus NSs protein) (150) or degradation of these proteins within virally formed degradasomes (respiratory syncytial virus NS protein) (151). HCV, using its protease NS3/4A, specifically cleaves and thereby inactivates key signaling intermediates, such as the adapters MAVS and TRIF, blunting the RLR and TLR3 pathways (152). This discovery formed the basis for the development of new HCV protease inhibitors now used for treatment of HCV patients (136). Paramyxoviral V proteins show surprising sophistication in inhibiting virus detection by the sensor MDA5. Not only does V directly bind and inhibit both MDA5 and LGP2, but it also binds the cellular phosphatase PP1, keeping it from dephosphorylating MDA5 and hence maintaining MDA5 in an inactive conformation (153–156). The prime example of a multifunctional antihost protein is FLUAV NS1. It interferes with the ubiquitination of RIG-I by human TRIM25 or by murine Riplet ligases, preventing RIG-I activation (157–159). Also, NS1 functions in the nucleus by binding to CPSF and cellular RNAs to prevent poly(A) tailing and splicing of these RNAs, a generalized mechanism to prevent expression of host genes including IFN-β (149). VSV M protein achieves general host gene expression by blocking host mRNA transcription in the nucleus (160).
Signaling by IFNs can be targeted by soluble poxviral IFN decoy receptors, by virus-mediated degradation or relocation of STAT proteins, or by prevention of ISGF3 nuclear import in order to avoid the expression of ISGs in infected cells (3). A final means to antagonize the IFN system is to interfere with the functions of the ISG proteins themselves. An example is the antagonistic function of FLUAV NS1, which directly binds and inhibits PKR (161). Other viruses inhibit PKR by encoding small, partially double-stranded RNAs that stably bind PKR without activating it (Epstein-Barr virus EBER and adenovirus VA1) (162). MHV, a coronavirus, encodes a phosphodiesterase for the purpose of cleaving 2′,5′-oligoadenylates to efficiently prevent RNase L stimulation (163). Lastly, the anchoring of the egressing virus particles to the cell surface by Tetherin is antagonized by HIV, influenza viruses, and Ebola virus (123). The avoidance of ISG protein actions is another effective alternative to actively antagonizing them. The ISG protein IFIT1 inhibits translation of mRNAs lacking cap 2′-O-methylation at the first ribose (107–110). Viruses evade this inhibition in at least three different ways: (a) Viruses utilize the cellular mRNA capping machinery or transfer cellular 2′-O-methylated caps onto their own RNAs; (b) viruses encode a 2′-O-methyltransferase of their own (WNV, MHV, vaccinia virus, VSV, etc.); and (c) alphavirus mRNAs (Venezuelan equine encephalitis virus, Sindbis virus) form a secondary structure adjacent to the unmethylated cap to sterically prevent murine Ifit1 from binding (164). The evolutionary battle between the host and the virus can extend further; the host can, in turn, antagonize the viral antagonists, as seen with the multifunctional NS1 of FLUAV, which is conjugated to cellular ISG15 and thereby becomes inactivated (165). ISGs even coevolve with viral inhibitors such as the vaccinia virus K3L protein. K3L is a small eIF2α-mimicking PKR-binding protein that blocks PKR kinase function. In several primates, the interacting surface of PKR with eIF2α has changed over time to help overcome competitive binding by viral K3L (166).
FUTURE PERSPECTIVES
In spite of the wealth of knowledge acquired during the more than 50 years of research since the discovery of IFN, much remains to be learned. A major unanswered question is, Why so many type I IFNs? Are they redundant, or do they have different physiological roles? A second, more philosophical question is, If IFNs are antiviral, why do viruses induce their synthesis? In this context, a newly emerging concept postulates that IFNs, rather than protecting the host, may be partners in crime and augment viral pathogenesis (127, 129). A major underexplored research area addresses the nature of cross talk among IFNs and other cytokines that are also induced by virus infection (132). As our understanding of the actions of ISG proteins is increasing, it is intriguing to learn that a specific ISG protein can be antiviral only in a specific cell type (94). This observation suggests that an ISG protein, or its functional analogs, may have a more targeted and selective antiviral effect than an IFN, for therapeutic purposes. The biological implications of protein ISGylation are not yet fully understood. Moreover, it remains possible that not only ISG15 but other small IFN-induced proteins are also covalently linked to cellular and viral proteins. Another significant area of expansion of future investigation is on the role of IFNs in superinfections. There are intriguing reports on how IFNs produced by initial acute virus infection promote secondary bacterial infection (131, 167). In the same vein, it will be interesting to know how low levels of IFNs produced by chronic virus infection influence susceptibility to other infectious agents. In the recent past, important insights into the functions of specific components of the IFN system have been revealed by genetic analyses of familial cohorts that are susceptible to specific microbial infections (120); one expects that this kind of investigation will be a rich source of new information in the future, as systems analyses are more routinely performed to investigate human diseases.
Supplementary Material
SUMMARY POINTS.
The type I, II, and III IFNs and their cognate receptors form three biologically distinct systems. Their components are expressed with cell type restrictions and activate different transcription factors, inducing overlapping but distinct sets of ISGs.
Type I and III IFNs are transcriptionally induced upon detection of viral molecular patterns by cellular sensor proteins and are secreted. They act on the infected cell as well as surrounding and remote uninfected cells.
IFNs act through their cell surface receptors to engage the JAK-STAT pathway and other signaling pathways toward transcriptional induction of ISGs.
ISG proteins are the mediators of IFN actions.
IFNs are protective against most viruses but can be harmful during certain virus infections (e.g., influenza viruses, HCV), bacterial infections, or autoimmune diseases. High levels of type I and III IFN expression are observed in those scenarios.
Antiviral effector ISG proteins act in concert to inhibit multiple steps of virus life cycles; some ISG proteins inhibit only specific viruses, whereas some are active only in specific cell types.
Viruses antagonize the IFN system at multiple levels, including IFN synthesis, IFN signaling pathways, and the functions of individual ISG proteins.
ACKNOWLEDGMENTS
The authors’ research is supported by National Institutes of Health grants CA068782, AI073303, and CA062220.
Glossary
- IFN-stimulated gene (ISG)
a gene that is transcriptionally induced or upregulated upon IFN receptor signaling
- IFN-α/β receptor (IFNAR)
cell surface complex of IFNAR1 and IFNAR2; activated by type I IFNs
- IFN-γ receptor (IFNGR)
cell surface complex of IFNGR1 and IFNGR2; activated by type II IFNs
- IFN-λ receptor (IFNLR)
cell surface complex of IFNLR1 and IL10RB; activated by type III IFNs
- ISG factor 3 (ISGF3)
transcription factor consisting of STAT1, STAT2, and IRF-9; drives transcription of ISGs with an ISRE promoter
- Gamma-activated factor (GAF)
transcription factor consisting of a STAT1 homodimer; drives transcription of ISGs with a GAS promoter
- Pattern-recognition receptor (PRR)
cellular sensor protein in the cytoplasm, on the cell surface, or on endosomal membranes; recognizes microbial molecular patterns
- IFN-stimulated response element (ISRE)
promoter element responsive to ISGF3 and IRF transcription factors; drives transcription of many ISGs
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Isaacs A, Lindenmann J. 1957. Virus interference. I. The interferon. Proc. R. Soc. B 147:258–67 [DOI] [PubMed] [Google Scholar]
- 2.Schneider WM, Chevillotte MD, Rice CM. 2014. Interferon-stimulated genes: a complex web of host defenses. Annu. Rev. Immunol 32:513–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Versteeg GA, Garcia-Sastre A. 2010. Viral tricks to grid-lock the type I interferon system. Curr. Opin. Microbiol 13:508–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McNab F, Mayer-Barber K, Sher A, Wack A, O’Garra A. 2015. Type I interferons in infectious disease. Nat. Rev. Immunol 15:87–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Weerd NA, Nguyen T. 2012. The interferons and their receptors—distribution and regulation. Immunol. Cell Biol 90:483–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O’Brien TR, Prokunina-Olsson L, Donnelly RP. 2014. IFN-λ4: the paradoxical new member of the interferon lambda family. J. Interferon Cytokine Res 34:829–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pestka S, Krause CD, Walter MR. 2004. Interferons, interferon-like cytokines, and their receptors. Immunol. Rev 202:8–32 [DOI] [PubMed] [Google Scholar]
- 8.Fitzgerald-Bocarsly P, Dai J, Singh S. 2008. Plasmacytoid dendritic cells and type I IFN: 50 years of convergent history. Cytokine Growth Factor Rev. 19:3–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fung KY, Mangan NE, Cumming H, Horvat JC, Mayall JR, et al. 2013. Interferon-ε protects the female reproductive tract from viral and bacterial infection. Science 339:1088–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hardy MP, Owczarek CM, Jermiin LS, Ejdeback M, Hertzog PJ. 2004. Characterization of the type I interferon locus and identification of novel genes. Genomics 84:331–45 [DOI] [PubMed] [Google Scholar]
- 11.Mordstein M, Neugebauer E, Ditt V, Jessen B, Rieger T, et al. 2010. Lambda interferon renders epithelial cells of the respiratory and gastrointestinal tracts resistant to viral infections. J. Virol 84:5670–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sheahan T, Imanaka N, Marukian S, Dorner M, Liu P, et al. 2014. Interferon lambda alleles predict innate antiviral immune responses and hepatitis C virus permissiveness. Cell Host Microbe 15:190–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sommereyns C, Paul S, Staeheli P, Michiels T. 2008. IFN-lambda (IFN-λ) is expressed in a tissue-dependent fashion and primarily acts on epithelial cells in vivo. PLOS Pathog. 4:e1000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Der SD, Zhou A, Williams BR, Silverman RH. 1998. Identification of genes differentially regulated by interferon α, β, or γ using oligonucleotide arrays. PNAS 95:15623–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Doyle SE, Schreckhise H, Khuu-Duong K, Henderson K, Rosler R, et al. 2006. Interleukin-29 uses a type 1 interferon-like program to promote antiviral responses in human hepatocytes. Hepatology 44:896–906 [DOI] [PubMed] [Google Scholar]
- 16.Decker T, Kovarik P, Meinke A. 1997. GAS elements: a few nucleotides with a major impact on cytokine-induced gene expression. J. Interferon Cytokine Res 17:121–34 [DOI] [PubMed] [Google Scholar]
- 17.Platanias LC. 2005. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol 5:375–86 [DOI] [PubMed] [Google Scholar]
- 18.Pestka S. 2007. The interferons: 50 years after their discovery, there is much more to learn. J. Biol. Chem 282:20047–51 [DOI] [PubMed] [Google Scholar]
- 19.Kotenko SV, Gallagher G, Baurin VV, Lewis-Antes A, Shen M, et al. 2003. IFN-λs mediate antiviral protection through a distinct class II cytokine receptor complex. Nat. Immunol 4:69–77 [DOI] [PubMed] [Google Scholar]
- 20.Sheppard P, Kindsvogel W, Xu W, Henderson K, Schlutsmeyer S, et al. 2003. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat. Immunol 4:63–68 [DOI] [PubMed] [Google Scholar]
- 21.Prokunina-Olsson L, Muchmore B, Tang W, Pfeiffer RM, Park H, et al. 2013. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat. Genet 45:164–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Key FM, Peter B, Dennis MY, Huerta-Sanchez E, Tang W, et al. 2014. Selection on a variant associated with improved viral clearance drives local, adaptive pseudogenization of interferon lambda 4 (IFNL4). PLOS Genet. 10:e1004681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pandey S, Kawai T, Akira S. 2015. Microbial sensing by Toll-like receptors and intracellular nucleic acid sensors. Cold Spring Harb. Perspect. Biol 7:a016246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gay NJ, Symmons MF, Gangloff M, Bryant CE. 2014. Assembly and localization of Toll-like receptor signalling complexes. Nat. Rev. Immunol 14:546–58 [DOI] [PubMed] [Google Scholar]
- 25.Yoneyama M, Onomoto K, Jogi M, Akaboshi T, Fujita T. 2015. Viral RNA detection by RIG-I-like receptors. Curr. Opin. Immunol 32:48–53 [DOI] [PubMed] [Google Scholar]
- 26.Wu J, Chen ZJ. 2014. Innate immune sensing and signaling of cytosolic nucleic acids. Annu. Rev. Immunol 32:461–88 [DOI] [PubMed] [Google Scholar]
- 27.Kadowaki N, Ho S, Antonenko S, Malefyt RW, Kastelein RA, et al. 2001. Subsets of human dendritic cell precursors express different Toll-like receptors and respond to different microbial antigens. J. Exp. Med 194:863–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zarember KA, Godowski PJ. 2002. Tissue expression of human Toll-like receptors and differential regulation of Toll-like receptor mRNAs in leukocytes in response to microbes, their products, and cytokines. J. Immunol 168:554–61 [DOI] [PubMed] [Google Scholar]
- 29.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. 2001. Recognition of double-stranded RNA and activation of NF-κB by Toll-like receptor 3. Nature 413:732–38 [DOI] [PubMed] [Google Scholar]
- 30.Hardarson HS, Baker JS, Yang Z, Purevjav E, Huang CH, et al. 2007. Toll-like receptor 3 is an essential component of the innate stress response in virus-induced cardiac injury. Am. J. Physiol. Heart Circ. Physiol 292:H251–58 [DOI] [PubMed] [Google Scholar]
- 31.Le Goffic R, Balloy V, Lagranderie M, Alexopoulou L, Escriou N, et al. 2006. Detrimental contribution of the Toll-like receptor (TLR)3 to influenza A virus-induced acute pneumonia. PLOS Pathog. 2:e53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang SY, Jouanguy E, Ugolini S, Smahi A, Elain G, et al. 2007. TLR3 deficiency in patients with herpes simplex encephalitis. Science 317:1522–27 [DOI] [PubMed] [Google Scholar]
- 33.Lee HK, Lund JM, Ramanathan B, Mizushima N, Iwasaki A. 2007. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science 315:1398–401 [DOI] [PubMed] [Google Scholar]
- 34.Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, et al. 2006. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441:101–5 [DOI] [PubMed] [Google Scholar]
- 35.Gitlin L, Barchet W, Gilfillan S, Cella M, Beutler B, et al. 2006. Essential role of mda-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. PNAS 103:8459–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goubau D, Schlee M, Deddouche S, Pruijssers AJ, Zillinger T, et al. 2014. Antiviral immunity via RIG-I-mediated recognition of RNA bearing 5′-diphosphates. Nature 514:372–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weber M, Gawanbacht A, Habjan M, Rang A, Borner C, et al. 2013. Incoming RNA virus nucleocapsids containing a 5′-triphosphorylated genome activate RIG-I and antiviral signaling. Cell Host Microbe 13:336–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kato H, Takeuchi O, Mikamo-Satoh E, Hirai R, Kawai T, et al. 2008. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J. Exp. Med 205:1601–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zust R, Cervantes-Barragan L, Habjan M, Maier R, Neuman BW, et al. 2011. Ribose 2′-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nat. Immunol 12:137–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bruns AM, Leser GP, Lamb RA, Horvath CM. 2014. The innate immune sensor LGP2 activates antiviral signaling by regulating MDA5-RNA interaction and filament assembly. Mol. Cell 55:771–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Childs KS, Randall RE, Goodbourn S. 2013. LGP2 plays a critical role in sensitizing mda-5 to activation by double-stranded RNA. PLOS ONE 8:e64202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Saito T, Hirai R, Loo YM, Owen D, Johnson CL, et al. 2007. Regulation of innate antiviral defenses through a shared repressor domain in RIG-I and LGP2. PNAS 104:582–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Krug A, French AR, Barchet W, Fischer JA, Dzionek A, et al. 2004. TLR9-dependent recognition of MCMV by IPC and DC generates coordinated cytokine responses that activate antiviral NK cell function. Immunity 21:107–19 [DOI] [PubMed] [Google Scholar]
- 44.Ablasser A, Bauernfeind F, Hartmann G, Latz E, Fitzgerald KA, Hornung V. 2009. RIG-I-dependent sensing of poly (dA:dT) through the induction of an RNA polymerase III-transcribed RNA intermediate. Nat. Immunol 10:1065–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chiu YH, Macmillan JB, Chen ZJ. 2009. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 138:576–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Unterholzner L. 2013. The interferon response to intracellular DNA: Why so many receptors? Immunobiology 218:1312–21 [DOI] [PubMed] [Google Scholar]
- 47.Chattopadhyay S, Sen GC. 2014. Tyrosine phosphorylation in Toll-like receptor signaling. Cytokine Growth Factor Rev. 25:533–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sarkar SN, Elco CP, Peters KL, Chattopadhyay S, Sen GC. 2007. Two tyrosine residues of Toll-like receptor 3 trigger different steps of NF-κB activation. J. Biol. Chem 282:3423–27 [DOI] [PubMed] [Google Scholar]
- 49.Sarkar SN, Peters KL, Elco CP, Sakamoto S, Pal S, Sen GC. 2004. Novel roles of TLR3 tyrosine phosphorylation and PI3 kinase in double-stranded RNA signaling. Nat. Struct. Mol. Biol 11:1060–67 [DOI] [PubMed] [Google Scholar]
- 50.Sarkar SN, Smith HL, Rowe TM, Sen GC. 2003. Double-stranded RNA signaling by Toll-like receptor 3 requires specific tyrosine residues in its cytoplasmic domain. J. Biol. Chem 278:4393–96 [DOI] [PubMed] [Google Scholar]
- 51.Liu L, Botos I, Wang Y, Leonard JN, Shiloach J, et al. 2008. Structural basis of Toll-like receptor 3 signaling with double-stranded RNA. Science 320:379–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yamashita M, Chattopadhyay S, Fensterl V, Saikia P, Wetzel JL, Sen GC. 2012. Epidermal growth factor receptor is essential for Toll-like receptor 3 signaling. Sci. Signal 5:ra50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gack MU. 2014. Mechanisms of RIG-I-like receptor activation and manipulation by viral pathogens. J. Virol 88:5213–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Maharaj NP, Wies E, Stoll A, Gack MU. 2012. Conventional protein kinase C-α (PKC-α) and PKC-β negatively regulate RIG-I antiviral signal transduction. J. Virol 86:1358–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wies E, Wang MK, Maharaj NP, Chen K, Zhou S, et al. 2013. Dephosphorylation of the RNA sensors RIG-I and MDA5 by the phosphatase PP1 is essential for innate immune signaling. Immunity 38:437–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gack MU, Shin YC, Joo CH, Urano T, Liang C, et al. 2007. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 446:916–20 [DOI] [PubMed] [Google Scholar]
- 57.Zeng W, Sun L, Jiang X, Chen X, Hou F, et al. 2010. Reconstitution of the RIG-I pathway reveals a signaling role of unanchored polyubiquitin chains in innate immunity. Cell 141:315–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cai X, Chiu YH, Chen ZJ. 2014. The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol. Cell 54:289–96 [DOI] [PubMed] [Google Scholar]
- 59.Ford E, Thanos D. 2010. The transcriptional code of human IFN-β gene expression. Biochim. Biophys. Acta 1799:328–36 [DOI] [PubMed] [Google Scholar]
- 60.Panne D, Maniatis T, Harrison SC. 2007. An atomic model of the interferon-β enhanceosome. Cell 129:1111–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Genin P, Vaccaro A, Civas A. 2009. The role of differential expression of human interferon-A genes in antiviral immunity. Cytokine Growth Factor Rev. 20:283–95 [DOI] [PubMed] [Google Scholar]
- 62.Iversen MB, Paludan SR. 2010. Mechanisms of type III interferon expression. J. Interferon Cytokine Res 30:573–78 [DOI] [PubMed] [Google Scholar]
- 63.Onoguchi K, Yoneyama M, Takemura A, Akira S, Taniguchi T, et al. 2007. Viral infections activate types I and III interferon genes through a common mechanism. J. Biol. Chem 282:7576–81 [DOI] [PubMed] [Google Scholar]
- 64.Osterlund PI, Pietila TE, Veckman V, Kotenko SV, Julkunen I. 2007. IFN regulatory factor family members differentially regulate the expression of type III IFN (IFN-λ) genes. J. Immunol 179:3434–42 [DOI] [PubMed] [Google Scholar]
- 65.Broz P, Monack DM. 2013. Newly described pattern recognition receptors team up against intracellular pathogens. Nat. Rev. Immunol 13:551–65 [DOI] [PubMed] [Google Scholar]
- 66.Andersen J, VanScoy S, Cheng TF, Gomez D, Reich NC. 2008. IRF-3-dependent and augmented target genes during viral infection. Genes Immun. 9:168–75 [DOI] [PubMed] [Google Scholar]
- 67.Freaney JE, Kim R, Mandhana R, Horvath CM. 2013. Extensive cooperation of immune master regulators IRF3 and NFκB in RNA Pol II recruitment and pause release in human innate antiviral transcription. Cell Rep. 4:959–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chattopadhyay S, Marques JT, Yamashita M, Peters KL, Smith K, et al. 2010. Viral apoptosis is induced by IRF-3-mediated activation of Bax. EMBO J. 29:1762–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.White CL, Chattopadhyay S, Sen GC. 2011. Phosphatidylinositol 3-kinase signaling delays Sendai virus-induced apoptosis by preventing XIAP degradation. J. Virol 85:5224–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chattopadhyay S, Fensterl V, Zhang Y, Veleeparambil M, Yamashita M, Sen GC. 2013. Role of interferon regulatory factor 3-mediated apoptosis in the establishment and maintenance of persistent infection by Sendai virus. J. Virol 87:16–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chattopadhyay S, Yamashita M, Zhang Y, Sen GC. 2011. The IRF-3/Bax-mediated apoptotic pathway, activated by viral cytoplasmic RNA and DNA, inhibits virus replication. J. Virol 85:3708–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ivashkiv LB, Donlin LT. 2014. Regulation of type I interferon responses. Nat. Rev. Immunol 14:36–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schroder K, Hertzog PJ, Ravasi T, Hume DA. 2004. Interferon-γ: an overview of signals, mechanisms and functions. J. Leukoc. Biol 75:163–89 [DOI] [PubMed] [Google Scholar]
- 74.Durbin RK, Kotenko SV, Durbin JE. 2013. Interferon induction and function at the mucosal surface. Immunol. Rev 255:25–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ng SL, Friedman BA, Schmid S, Gertz J, Myers RM, et al. 2011. IκB kinase ε (IKKε) regulates the balance between type I and type II interferon responses. PNAS 108:21170–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rajsbaum R, Versteeg GA, Schmid S, Maestre AM, Belicha-Villanueva A, et al. 2014. Unanchored K48-linked polyubiquitin synthesized by the E3-ubiquitin ligase TRIM6 stimulates the interferon-IKε kinase-mediated antiviral response. Immunity 40:880–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Stark GR. 2007. How cells respond to interferons revisited: from early history to current complexity. Cytokine Growth Factor Rev. 18:419–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fish EN, Platanias LC. 2014. Interferon receptor signaling in malignancy: a network of cellular pathways defining biological outcomes. Mol. Cancer Res 12:1691–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.de Weerd NA, Vivian JP, Nguyen TK, Mangan NE, Gould JA, et al. 2013. Structural basis of a unique interferon-β signaling axis mediated via the receptor IFNAR1. Nat. Immunol 14:901–7 [DOI] [PubMed] [Google Scholar]
- 80.de Weerd NA, Samarajiwa SA, Hertzog PJ. 2007. Type I interferon receptors: biochemistry and biological functions. J. Biol. Chem 282:20053–57 [DOI] [PubMed] [Google Scholar]
- 81.Samarajiwa SA, Mangan NE, Hardy MP, Najdovska M, Dubach D, et al. 2014. Soluble IFN receptor potentiates in vivo type I IFN signaling and exacerbates TLR4-mediated septic shock. J. Immunol 192:4425–35 [DOI] [PubMed] [Google Scholar]
- 82.Gazziola C, Cordani N, Carta S, De Lorenzo E, Colombatti A, Perris R. 2005. The relative endogenous expression levels of the IFNAR2 isoforms influence the cytostatic and pro-apoptotic effect of IFNα on pleomorphic sarcoma cells. Int. J. Oncol 26:129–40 [PubMed] [Google Scholar]
- 83.Malakhova OA, Kim KI, Luo JK, Zou W, Kumar KG, et al. 2006. UBP43 is a novel regulator of interferon signaling independent of its ISG15 isopeptidase activity. EMBO J. 25:2358–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Piganis RA, de Weerd NA, Gould JA, Schindler CW, Mansell A, et al. 2011. Suppressor of cytokine signaling (SOCS) 1 inhibits type I interferon (IFN) signaling via the interferon α receptor (IFNAR1)-associated tyrosine kinase Tyk2. J. Biol. Chem 286:33811–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cheon H, Holvey-Bates EG, Schoggins JW, Forster S, Hertzog P, et al. 2013. IFNβ-dependent increases in STAT1, STAT2, and IRF9 mediate resistance to viruses and DNA damage. EMBO J. 32:2751–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kroczynska B, Mehrotra S, Arslan AD, Kaur S, Platanias LC. 2014. Regulation of interferon-dependent mRNA translation of target genes. J. Interferon Cytokine Res 34:289–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mohr I, Sonenberg N. 2012. Host translation at the nexus of infection and immunity. Cell Host Microbe 12:470–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kaur S, Lal L, Sassano A, Majchrzak-Kita B, Srikanth M, et al. 2007. Regulatory effects of mammalian target of rapamycin-activated pathways in type I and II interferon signaling. J. Biol. Chem 282:1757–68 [DOI] [PubMed] [Google Scholar]
- 89.Gonzalez-Navajas JM, Lee J, David M, Raz E. 2012. Immunomodulatory functions of type I interferons. Nat. Rev. Immunol 12:125–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Schoggins JW. 2014. Interferon-stimulated genes: roles in viral pathogenesis. Curr. Opin. Virol 6:40–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Karki S, Li MM, Schoggins JW, Tian S, Rice CM, MacDonald MR. 2012.Multiple interferon stimulated genes synergize with the zinc finger antiviral protein to mediate anti-alphavirus activity. PLOS ONE 7:e37398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, et al. 2011.A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472:481–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Schoggins JW, MacDuff DA, Imanaka N, Gainey MD, Shrestha B, et al. 2014. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature 505:691–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Fensterl V, Wetzel JL, Ramachandran S, Ogino T, Stohlman SA, et al. 2012. Interferon-induced Ifit2/ISG54 protects mice from lethal VSV neuropathogenesis. PLOS Pathog. 8:e1002712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bailey CC, Zhong G, Huang IC, Farzan M. 2014. IFITM-family proteins: the cell’s first line of antiviral defense. Annu. Rev. Virol 1:261–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Haller O, Staeheli P, Schwemmle M, Kochs G. 2015. Mx GTPases: dynamin-like antiviral machines of innate immunity. Trends Microbiol. 23:154–63 [DOI] [PubMed] [Google Scholar]
- 97.Li J, Ding SC, Cho H, Chung BC, Gale M Jr., et al. 2013. A short hairpin RNA screen of interferon-stimulated genes identifies a novel negative regulator of the cellular antiviral response. mBio 4:e00385–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Liu SY, Sanchez DJ, Aliyari R, Lu S, Cheng G. 2012. Systematic identification of type I and type II interferon-induced antiviral factors. PNAS 109:4239–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cho H, Shrestha B, Sen GC, Diamond MS. 2013. A role for Ifit2 in restricting West Nile virus infection in the brain. J. Virol 87:8363–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Fensterl V, Wetzel JL, Sen GC. 2014. Interferon-induced protein Ifit2 protects mice from infection of the peripheral nervous system by vesicular stomatitis virus. J. Virol 88:10303–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.MacMicking JD. 2012. Interferon-inducible effector mechanisms in cell-autonomous immunity. Nat. Rev. Immunol 12:367–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sadler AJ, Williams BR. 2008. Interferon-inducible antiviral effectors. Nat. Rev. Immunol 8:559–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chakrabarti A, Jha BK, Silverman RH. 2011. New insights into the role of RNase L in innate immunity. J. Interferon Cytokine Res 31:49–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Malathi K, Dong B, Gale M Jr., Silverman RH. 2007. Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature 448:816–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Fensterl V, Sen GC. 2011. The ISG56/IFIT1 gene family. J. Interferon Cytokine Res 31:71–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Fensterl V, Sen GC. 2015. Interferon-induced Ifit proteins: their role in viral pathogenesis. J. Virol 89:2462–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Daffis S, Szretter KJ, Schriewer J, Li J, Youn S, et al. 2010. 2′-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature 468:452–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Habjan M, Hubel P, Lacerda L, Benda C, Holze C, et al. 2013. Sequestration by IFIT1 impairs translation of 2′ O-unmethylated capped RNA. PLOS Pathog. 9:e1003663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kumar P, Sweeney TR, Skabkin MA, Skabkina OV, Hellen CU, Pestova TV. 2014. Inhibition of translation by IFIT family members is determined by their ability to interact selectively with the 5′-terminal regions of cap0-, cap1- and 5′ ppp-mRNAs. Nucleic Acids Res. 42:3228–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Pichlmair A, Lassnig C, Eberle CA, Gorna MW, Baumann CL, et al. 2011. IFIT1 is an antiviral protein that recognizes 5′-triphosphate RNA. Nat. Immunol 12:624–30 [DOI] [PubMed] [Google Scholar]
- 111.Butchi NB, Hinton DR, Stohlman SA, Kapil P, Fensterl V, et al. 2014. Ifit2 deficiency results in uncontrolled neurotropic coronavirus replication and enhanced encephalitis via impaired alpha/beta interferon induction in macrophages. J. Virol 88:1051–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Blanc M, Hsieh WY, Robertson KA, Kropp KA, Forster T, et al. 2013. The transcription factor STAT-1 couples macrophage synthesis of 25-hydroxycholesterol to the interferon antiviral response. Immunity 38:106–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Civra A, Cagno V, Donalisio M, Biasi F, Leonarduzzi G, et al. 2014. Inhibition of pathogenic non-enveloped viruses by 25-hydroxycholesterol and 27-hydroxycholesterol. Sci. Rep 4:7487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Liu SY, Aliyari R, Chikere K, Li G, Marsden MD, et al. 2013. Interferon-inducible cholesterol-25-hydroxylase broadly inhibits viral entry by production of 25-hydroxycholesterol. Immunity 38:92–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Durfee LA, Lyon N, Seo K, Huibregtse JM. 2010. The ISG15 conjugation system broadly targets newly synthesized proteins: implications for the antiviral function of ISG15. Mol. Cell 38:722–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Morales DJ, Lenschow DJ. 2013. The antiviral activities of ISG15. J. Mol. Biol 425:4995–5008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lenschow DJ, Lai C, Frias-Staheli N, Giannakopoulos NV, Lutz A, et al. 2007. IFN-stimulated gene 15 functions as a critical antiviral molecule against influenza, herpes, and Sindbis viruses. PNAS 104:1371–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Morales DJ, Monte K, Sun L, Struckhoff JJ, Agapov E, et al. 2015. Novel mode of ISG15-mediated protection against influenza A virus and Sendai virus in mice. J. Virol 89:337–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bogunovic D, Byun M, Durfee LA, Abhyankar A, Sanal O, et al. 2012.Mycobacterial disease and impaired IFN-γ immunity in humans with inherited ISG15 deficiency. Science 337:1684–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zhang X, Bogunovic D, Payelle-Brogard B, Francois-Newton V, Speer SD, et al. 2015. Human intracellular ISG15 prevents interferon-α/β over-amplification and auto-inflammation. Nature 517:89–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Liberatore RA, Bieniasz PD. 2011. Tetherin is a key effector of the antiretroviral activity of type I interferon in vitro and in vivo. PNAS 108:18097–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Neil SJ, Zang T, Bieniasz PD. 2008. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 451:425–30 [DOI] [PubMed] [Google Scholar]
- 123.Swiecki M, Omattage NS, Brett TJ. 2013. BST-2/tetherin: structural biology, viral antagonism, and immunobiology of a potent host antiviral factor. Mol. Immunol 54:132–39 [DOI] [PubMed] [Google Scholar]
- 124.Crow MK. 2014. Type I interferon in the pathogenesis of lupus. J. Immunol 192:5459–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Gough DJ, Messina NL, Clarke CJ, Johnstone RW, Levy DE. 2012. Constitutive type I interferon modulates homeostatic balance through tonic signaling. Immunity 36:166–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Karaghiosoff M, Steinborn R, Kovarik P, Kriegshauser G, Baccarini M, et al. 2003. Central role for type I interferons and Tyk2 in lipopolysaccharide-induced endotoxin shock. Nat. Immunol 4:471–77 [DOI] [PubMed] [Google Scholar]
- 127.Davidson S, Crotta S, McCabe TM, Wack A. 2014. Pathogenic potential of interferon αβ in acute influenza infection. Nat. Commun 5:3864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Teijaro JR, Ng C, Lee AM, Sullivan BM, Sheehan KC, et al. 2013. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science 340:207–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wetzel JL, Fensterl V, Sen GC. 2014. Sendai virus pathogenesis in mice is prevented by Ifit2 and exacerbated by interferon. J. Virol 88:13593–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wilson EB, Yamada DH, Elsaesser H, Herskovitz J, Deng J, et al. 2013. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science 340:202–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.McCullers JA. 2014. The co-pathogenesis of influenza viruses with bacteria in the lung. Nat. Rev. Microbiol 12:252–62 [DOI] [PubMed] [Google Scholar]
- 132.Davidson S, Maini MK, Wack A. 2015. Disease-promoting effects of type I interferons in viral, bacterial, and coinfections. J. Interferon Cytokine Res 35:252–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Baldridge MT, Nice TJ, McCune BT, Yokoyama CC, Kambal A, et al. 2015. Commensal microbes and interferon-λ determine persistence of enteric murine norovirus infection. Science 347:266–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Nice TJ, Baldridge MT, McCune BT, Norman JM, Lazear HM, et al. 2015. Interferon-λ cures persistent murine norovirus infection in the absence of adaptive immunity. Science 347:269–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kernbauer E, Ding Y, Cadwell K. 2014.An enteric virus can replace the beneficial function of commensal bacteria. Nature 516:94–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Welsch C, Jesudian A, Zeuzem S, Jacobson I. 2012. New direct-acting antiviral agents for the treatment of hepatitis C virus infection and perspectives. Gut 61(Suppl. 1):i36–46 [DOI] [PubMed] [Google Scholar]
- 137.Muir AJ, Shiffman ML, Zaman A, Yoffe B, de la Torre A, et al. 2010. Phase 1b study of pegylated interferon lambda 1 with or without ribavirin in patients with chronic genotype 1 hepatitis C virus infection. Hepatology 52:822–32 [DOI] [PubMed] [Google Scholar]
- 138.Ramos EL. 2010. Preclinical and clinical development of pegylated interferon-lambda 1 in chronic hepatitis C. J. Interferon Cytokine Res 30:591–95 [DOI] [PubMed] [Google Scholar]
- 139.Francois-Newton V, Magno de Freitas Almeida G, Payelle-Brogard B, Monneron D, Pichard-Garcia L, et al. 2011. USP18-based negative feedback control is induced by type I and type III interferons and specifically inactivates interferon α response. PLOS ONE 6:e22200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Makowska Z, Duong FH, Trincucci G, Tough DF, Heim MH. 2011. Interferon-β and interferon-λ signaling is not affected by interferon-induced refractoriness to interferon-α in vivo. Hepatology 53:1154–63 [DOI] [PubMed] [Google Scholar]
- 141.Sarasin-Filipowicz M, Wang X, Yan M, Duong FH, Poli V, et al. 2009. Alpha interferon induces long-lasting refractoriness of JAK-STAT signaling in the mouse liver through induction of USP18/UBP43. Mol. Cell. Biol 29:4841–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Sarasin-Filipowicz M, Oakeley EJ, Duong FH, Christen V, Terracciano L, et al. 2008. Interferon signaling and treatment outcome in chronic hepatitis C. PNAS 105:7034–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Terczynska-Dyla E, Bibert S, Duong FH, Krol I, Jorgensen S, et al. 2014. Reduced IFNλ4 activity is associated with improved HCV clearance and reduced expression of interferon-stimulated genes. Nat. Commun 5:5699. [DOI] [PubMed] [Google Scholar]
- 144.Urban TJ, Thompson AJ, Bradrick SS, Fellay J, Schuppan D, et al. 2010. IL28B genotype is associated with differential expression of intrahepatic interferon-stimulated genes in patients with chronic hepatitis C. Hepatology 52:1888–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Amanzada A, Kopp W, Spengler U, Ramadori G, Mihm S. 2013. Interferon-λ4 (IFNL4) transcript expression in human liver tissue samples. PLOS ONE 8:e84026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Aka PV, Kuniholm MH, Pfeiffer RM, Wang AS, Tang W, et al. 2014. Association of the IFNL4-ΔG allele with impaired spontaneous clearance of hepatitis C virus. J. Infect. Dis 209:350–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Randall RE, Goodbourn S. 2008. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J. Gen. Virol 89:1–47 [DOI] [PubMed] [Google Scholar]
- 148.Cardenas WB, Loo YM, Gale M Jr., Hartman AL, Kimberlin CR, et al. 2006. Ebola virus VP35 protein binds double-stranded RNA and inhibits alpha/beta interferon production induced by RIG-I signaling. J. Virol 80:5168–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Krug RM. 2015. Functions of the influenza A virus NS1 protein in antiviral defense. Curr. Opin. Virol 12:1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Santiago FW, Covaleda LM, Sanchez-Aparicio MT, Silvas JA, Diaz-Vizarreta AC, et al. 2014. Hijacking of RIG-I signaling proteins into virus-induced cytoplasmic structures correlates with the inhibition of type I interferon responses. J. Virol 88:4572–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Goswami R, Majumdar T, Dhar J, Chattopadhyay S, Bandyopadhyay SK, et al. 2013. Viral degradasome hijacks mitochondria to suppress innate immunity. Cell Res. 23:1025–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Horner SM, Gale M Jr. 2013. Regulation of hepatic innate immunity by hepatitis C virus. Nat. Med 19:879–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Childs K, Randall R, Goodbourn S. 2012. Paramyxovirus V proteins interact with the RNA helicase LGP2 to inhibit RIG-I-dependent interferon induction. J. Virol 86:3411–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Davis ME, Wang MK, Rennick LJ, Full F, Gableske S, et al. 2014. Antagonism of the phosphatase PP1 by the measles virus V protein is required for innate immune escape of MDA5. Cell Host Microbe 16:19–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Mesman AW, Zijlstra-Willems EM, Kaptein TM, de Swart RL, Davis ME, et al. 2014. Measles virus suppresses RIG-I-like receptor activation in dendritic cells via DC-SIGN-mediated inhibition of PP1 phosphatases. Cell Host Microbe 16:31–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Motz C, Schuhmann KM, Kirchhofer A, Moldt M, Witte G, et al. 2013. Paramyxovirus V proteins disrupt the fold of the RNA sensor MDA5 to inhibit antiviral signaling. Science 339:690–93 [DOI] [PubMed] [Google Scholar]
- 157.Gack MU, Albrecht RA, Urano T, Inn KS, Huang IC, et al. 2009. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe 5:439–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Mibayashi M, Martinez-Sobrido L, Loo YM, Cardenas WB, Gale M Jr., Garcia-Sastre A. 2007. Inhibition of retinoic acid-inducible gene I-mediated induction of beta interferon by the NS1 protein of influenza A virus. J. Virol 81:514–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Rajsbaum R, Albrecht RA, Wang MK, Maharaj NP, Versteeg GA, et al. 2012. Species-specific inhibition of RIG-I ubiquitination and IFN induction by the influenza A virus NS1 protein. PLOS Pathog. 8:e1003059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Gerlier D, Lyles DS. 2011. Interplay between innate immunity and negative-strand RNA viruses: towards a rational model. Microbiol. Mol. Biol. Rev 75:468–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Li S, Min JY, Krug RM, Sen GC. 2006. Binding of the influenza A virus NS1 protein to PKR mediates the inhibition of its activation by either PACT or double-stranded RNA. Virology 349:13–21 [DOI] [PubMed] [Google Scholar]
- 162.Sharp TV, Schwemmle M, Jeffrey I, Laing K, Mellor H, et al. 1993. Comparative analysis of the regulation of the interferon-inducible protein kinase PKR by Epstein-Barr virus RNAs EBER-1 and EBER-2 and adenovirus VAI RNA. Nucleic Acids Res. 21:4483–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Zhang R, Jha BK, Ogden KM, Dong B, Zhao L, et al. 2013. Homologous 2′,5′-phosphodiesterases from disparate RNA viruses antagonize antiviral innate immunity. PNAS 110:13114–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Diamond MS. 2014. IFIT1: a dual sensor and effector molecule that detects non-2′-O methylated viral RNA and inhibits its translation. Cytokine Growth Factor Rev. 25:543–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Tang Y, Zhong G, Zhu L, Liu X, Shan Y, et al. 2010. Herc5 attenuates influenza A virus by catalyzing ISGylation of viral NS1 protein. J. Immunol 184:5777–90 [DOI] [PubMed] [Google Scholar]
- 166.Elde NC, Child SJ, Geballe AP, Malik HS. 2009. Protein kinase R reveals an evolutionary model for defeating viral mimicry. Nature 457:485–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Navarini AA, Recher M, Lang KS, Georgiev P, Meury S, et al. 2006. Increased susceptibility to bacterial superinfection as a consequence of innate antiviral responses. PNAS 103:15535–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.