

Summary
Long-range gene editing by homology-directed repair (HDR) in hematopoietic stem/progenitor cells (HSPCs) often relies on viral transduction with recombinant adeno-associated viral vector (AAV) for template delivery. Here, we uncover unexpected load and prolonged persistence of AAV genomes and their fragments, which trigger sustained p53-mediated DNA damage response (DDR) upon recruiting the MRE11-RAD50-NBS1 (MRN) complex on the AAV inverted terminal repeats (ITRs). Accrual of viral DNA in cell-cycle-arrested HSPCs led to its frequent integration, predominantly in the form of transcriptionally competent ITRs, at nuclease on- and off-target sites. Optimized delivery of integrase-defective lentiviral vector (IDLV) induced lower DNA load and less persistent DDR, improving clonogenic capacity and editing efficiency in long-term repopulating HSPCs. Because insertions of viral DNA fragments are less frequent with IDLV, its choice for template delivery mitigates the adverse impact and genotoxic burden of HDR editing and should facilitate its clinical translation in HSPC gene therapy.
Keywords: gene editing, CRISPR-Cas9, hematopoietic stem/progenitor cells, DNA double-strand breaks, DNA damage response, adeno-associated vector, integrase-defective lentiviral vector, genotoxicity, p53 pathway, inverted terminal repeats
Graphical abstract
Highlights
-
•
AAV DNA activates p53, thereby reducing HSPC engraftment
-
•
AAV inverted terminal repeats (ITRs) can get trapped at DNA breaks in edited HSPCs
-
•
AAV ITRs are transcriptionally competent in human HSPCs
-
•
Integrase-defective lentiviral vector outperforms AAV as DNA template to edit HSPCs
Naldini and colleagues describe p53-mediated cellular sensing and frequent integration of transcriptionally competent AAV ITRs at induced DNA breaks as inadvertent consequences of templated gene editing of human HSPCs. To alleviate such potentially adverse effects, they propose an optimized gene editing protocol using integrase-defective lentiviral vectors for template delivery.
Introduction
Programmable nucleases enable editing by inducing site-specific DNA double-strand break (DSB) (Doudna, 2020). DSB repair occurs by non-homologous or microhomology-mediated end joining (NHEJ or MMEJ) or by high-fidelity HDR, a mechanism active in S/G2 cell cycle phases, which can exploit an exogenous template to introduce new sequences of interest. Although induction of DSBs triggers a detrimental p53-mediated DNA damage response (DDR) and impacts genome integrity, HDR-based editing remains the best-performing strategy to achieve long-range editing (Naldini, 2019). HDR, however, is constrained in long-term repopulating (LT)-HSPCs by the requirement for cell cycle progression (Filippo et al., 2008) and efficient template delivery. So far, recombinant AAV2/6 has been the preferred template vector in human HSPCs (Dever et al., 2016; Kuo et al., 2018; Rai et al., 2020; Schiroli et al., 2017; Wang et al., 2015). However, we reported that concomitant exposure of HSPCs to DSB and AAV2/6 leads to robust DDR, reducing repopulation potential and graft clonal diversity in transplanted hematochimeric mice (Ferrari et al., 2020; Pattabhi et al., 2019; Pavani et al., 2020; Romero et al., 2019; Schiroli et al., 2019). Transient expression of a dominant-negative p53 mutant protein (GSE56) dampens DDR and rescues in part its adverse impact (Ferrari et al., 2020). Whether an alternative delivery platform or further engineering of AAV can mitigate the vector-induced DDR is unknown.
Until now, investigation of the potential source of genotoxicity during gene editing mainly focused on off-target nuclease activity and the occurrence of genomic rearrangements at the target site (Adikusuma et al., 2018; Boutin et al., 2021; Kosicki et al., 2018; Lattanzi et al., 2021; Leibowitz et al., 2021; Nahmad et al., 2022; Turchiano et al., 2021). The genotoxic risk ascribed to DNA template delivery, its persistence, and its integration in edited cells (Colella et al., 2018; Penaud-Budloo et al., 2018; Schnödt and Büning, 2017) remains poorly investigated, especially for HSPCs. Most studies of AAV-based gene therapy have shown good safety profile (Kuzmin et al., 2021) except for severe hepatotoxicity in patients treated with high systemic vector doses (Mullard, 2021) and neurotoxicity in some non-human primates (NHPs) (Keiser et al., 2021; Sondhi et al., 2020), although the pathogenesis of these adverse effects remains to be clarified. Several studies also reported integration of fragmented or full-length AAV DNA in the genome of transduced cells (Dalwadi et al., 2021a; Gil-Farina et al., 2016; Inagaki et al., 2008; Kaeppel et al., 2013; McCarty et al., 2004; Miller et al., 2004; Nakai et al., 1999, Nakai et al., 2003, Nakai et al., 2005; Schultz and Chamberlain, 2008). Insertions near cancer genes were associated to development of hepatocellular carcinoma in some mouse models and clonal expansion of hepatocytes in dogs (Dalwadi et al., 2021b; Ferla et al., 2021; Nguyen et al., 2021; Rosas et al., 2012; Walia et al., 2015). Nuclease expression by AAV was shown to promote efficient integration of AAV DNA at the nuclease on-/off-target sites (Hanlon et al., 2019; Nelson et al., 2019).
Here, we assessed the impact of single-strand (ss) AAV2/6 and other AAV genome forms on the fitness and genome integrity of edited human HSPCs and explored the use of IDLV as alternative platform for template delivery.
Results
The AAV genome induces p53-dependent DDR constraining the hematopoietic graft
We performed HDR-based gene editing in human HSPCs by targeting the AAVS1 safe harbor (Lombardo et al., 2011) with Cas9 ribonucleoprotein (RNP) and a highly specific (HS) guide RNA (gRNA) (Schiroli et al., 2019). Cells were then transduced with CsCl purified ssAAV2/6 carrying homologies for AAVS1 and the green fluorescent protein (GFP) expressed by the human phosphoglycerate kinase (PGK) promoter (Figure 1A). Each component of the procedure contributed to p53 activation, as measured by CDKN1A (p21) induction, which delayed cell growth and decreased HSPC clonogenic potential (Figures S1A–S1D). ssAAV2/6 was the main culprit of toxicity. Incremental ssAAV2/6 doses progressively increased editing in bulk and the most primitive HSPCs (CD34+CD133+CD90+) from cord blood (CB) or mobilized peripheral blood (mPB), at the expense of exacerbated p53 activation (Figures 1B, 1C, S1E, and S1F), which reduced clonogenic potential (Figure 1D) and repopulation in transplanted mice (Figures 1E, 1F, S1G, and S1H). The proportion of edited cells within the graft was higher early after reconstitution, likely reflecting higher permissiveness to HDR editing of short-term repopulating cells. Purification of ssAAV2/6 by immune-affinity chromatography did not improve the editing protocol (Figures S1I–S1O; Table S1). The AAV DNA was responsible for p53 triggering, as shown by comparing HSPCs edited in the presence of “empty” or “full” AAV particles (Figures 1G–1K, S1I, S1J, and S1P–S1S). Editing multiple sites with different AAV constructs sharing the same AAV2 ITRs but different cargos (Figure S1T) showed similar p21 induction, cumulative with that induced by RNP (Figure 1L), pointing to the ITRs as responsible for DDR activation.
Figure 1.

AAV DNA is the culprit of p53 activation
(A) Experimental workflow.
(B) Percentage of GFP+ human cord-blood (CB) HSPCs transduced with incremental ssAAV2/6 doses (viral genomes (vg)/cell) (n = 3). Median.
(C) Fold change expression of CDKN1A relative to untreated cells (UT) at 1 day (D) after editing (n = 4). Median.
(D) Number of colonies grown from treated HSPCs as indicated (n = 1, 3, 3, and 3). Median.
(E and F) Percentage of circulating hCD45+ (E) and GFP+ cells within the human graft (F) in mice transplanted with the outgrown progeny of starting-matched limiting cell doses of CB (n = 9 and 10) or mPB (n = 5) HSPCs edited as indicated. Mean ± SEM, linear mixed effect model (LME) followed by post-hoc analysis; results are shown for the last time point.
(G–I) Percentage of GFP+ cells within CB HSPC subpopulations (G), fold change expression of CDKN1A over time relative to UT (H), number of colonies grown from HSPCs (I) after editing in the presence of “full” (5×105 viral capsids/cell, equivalent to 2 × 104 vg/cell) or “empty” (5 × 107 viral capsids/cell) ssAAV2/6 particles (n = 3). Median.
(J and K) Percentage of circulating hCD45+ (J) and GFP+ cells within the human graft (K) in mice transplanted with the outgrown progeny of starting-matched limiting cell doses of CB HSPCs edited as indicated (n = 9 and 10). Mean ± SEM, LME followed by post-hoc analysis for (J); results are shown for the last time point. “RNP + Full” corresponds to “RNP + ssAAV2/6 (2 × 104)” in (E) and (F).
(L) Fold change expression of CDKN1A relative to UT at 1 day after AAVS1, IL2RG, or CD40L editing with the indicated treatments (n = 3). Median.
See also Figure S1.
AAV ITRs are the main culprit of p53 activation via MRN complex in edited HSPCs
Editing with ssAAV5/6, whose ITR sequences evolutionary diverged along the AAV phylogenic tree (Gao et al., 2004) and contain fewer putative p53 binding sites (Figure S2A), resulted in similar p21 induction and clonogenic potential, at comparable editing efficiency, as with ssAAV2/6 (Figure S2B–S2F), suggesting that common structural features of AAV ITRs were responsible for DDR.
We then compared ss- with self-complementary (sc)-AAV2/6, which is encapsidated as double-strand genome folded at an intervening ITR sequence (Figure S2G) and found equal editing efficiencies (Figures 2A, S2H, and S2I), as also previously reported (Bak et al., 2018), but exacerbated p21 induction, drastically impairing cell growth and clonogenicity (Figures 2B, 2C, and S2J). Quantification of AAV DNA, using different set of probes along the genome (Figure 2D) showed substantial and persistent amounts of AAV genomes in treated HSPCs, consistent with robust induction of DDR which, in turn, by halting cell proliferation, may prevent effective dilution of the episomal DNA (Figure 2E). We also found DNA sequences from the AAV plasmid E. coli replication origin (Figure S2K), although at 10- to 30-fold lower abundance than the AAV genome, likely due to reverse packaging of plasmid backbone in the viral particle (Chadeuf et al., 2005; Tran et al., 2022; Wright, 2008). Intracellular AAV copies per human genome (CG) were on average 10-fold higher and more persistent for sc- than ss-AAV2/6, again in line with increased toxicity and the near abrogation of engraftment of edited cells (Figures 2E–2G). Targeted deep sequencing of AAVS1 in hematopoietic organs of transplanted mice showed that full ssAAV2/6 particles decreased indels diversity, and thus clonal complexity, in a dose-dependent manner, which was exacerbated when using scAAV2/6 (Figures 2H and S2L).
Figure 2.

AAV ITRs trigger p53 activation via MRN sensing
(A–C) Percentage of GFP+ cells within HSPC subpopulations (A, n = 7, Mann-Whitney test), fold change expression of CDKN1A over time relative to UT (B, n = 7, LME followed by post-hoc analysis; results are shown for the last time point), number of colonies grown from HSPCs (C, n = 9, Kruskal-Wallis test with Dunn’s multiple comparisons) after AAVS1 editing with the indicated AAVs. Median with 95% confidence interval (CI).
(D) Panel of digital droplet (dd) PCR probes tiling ss- and sc-AAV2/6 genomes (“ITR,” “ITR+cargo,” and “GFP”) and 3′ AAVS1 vector-genome junction (“3′ TI” or “3′ HDR”).
(E) Intracellular CG of the indicated AAV features retrieved over time from HSPCs edited with two doses of ss- or sc-AAV2/6 (n = 3). See STAR Methods for details. Median.
(F and G) Percentage of circulating hCD45+ (F) and GFP+ cells within the human graft (G) in mice transplanted with the outgrown progeny of starting-matched limiting cell doses of CB HSPCs edited as indicated (n = 5). Mean ± SEM, LME followed by post-hoc analysis; results are shown for the last time point.
(H) Number of unique indels retrieved by deep sequencing AAVS1 in human splenocytes from mice in Figures 1E and 1J and in (F) (n = 9, 9, 10, and 5). Median with quartiles. Kruskal-Wallis test with Dunn’s multiple comparisons.
(I) Quantification of NBS1+ cells and distribution of the percentage of foci bearing cells over time from HSPCs edited with the indicated treatments in the presence or absence of E1B55K and E4orf6 (Ad5; n = 11, 7, 1, 6, 6, 2, 1, and 3).
(J) Percentage of HDR-edited alleles and GFP+ cells in CB HSPCs edited in the presence or absence of E1B55K and/or E4orf6 (n = 3 for HDR and n = 4 for GFP). Median.
(K) Representative fluorescence microscopy images from (J).
(L) GFP relative fluorescence intensity (RFI) over the “RNP + ssAAV2/6” condition in bulk and primitive HSPCs from (J) (n = 3). Median.
(M) Fold change expression of APOBEC3H relative to UT 1 day after editing in experiments from (J) (n = 4). Median.
(N) Percentage of HDR-edited alleles and GFP+ cells in mPB HSPCs edited with different AAV variants as indicated (n = 3). Median.
(O) Intracellular CG of the indicated AAV features retrieved 1 day after treatment in HSPCs edited using different AAV variants (n = 3). See STAR Methods. Median.
(P) Fold change expression of CDKN1A over time relative to UT from experiments in (O) (n = 3). Median.
See also Figure S2.
The MRN complex, which recognizes free DNA ends at DSB and engages the repair machinery (Reginato and Cejka, 2020; Wienert et al., 2019), binds the AAV ITR and lowers transgene expression in transduced cells (Cervelli et al., 2008; Lentz and Samulski, 2015; Zhou et al., 2017). Upon staining for the MRN subunit NBS1, we found that the number of cells bearing NBS1+ nuclear foci (di Micco et al., 2021) increased upon RNP or ss-/sc-AAV2/6 treatments and was exacerbated when combining both treatments (Figures 2I and S2M). We then induced transient expression of Adenovirus serotype 5 (Ad5) proteins E1B55K and E4orf6 during editing, previously shown to inhibit MRN activity by promoting MRE11 degradation (Blanchette et al., 2008). Although HDR efficiency was similar among treatments (Figure 2J), the fraction and fluorescence intensity of GFP+ cells were higher upon editing in the presence of both Ad5 proteins (Figures 2J–2L), in agreement with previous reports (Chu et al., 2015). Ad5 proteins decreased the percentage of NBS1+ cells in samples treated with RNP and AAV (Figure 2I), which correlates with the nearly abrogated induction of the p53 downstream effectors APOBEC3H (Figure 2M) and p21 (Ferrari et al., 2020), highlighting a central role of MRN in triggering the AAV-dependent DDR. However, MRN inhibition was highly detrimental for cell proliferation, likely due to interference with DNA repair (Figures S2N and S2O).
Nuclear entry of the viral genome was required for both HDR and DDR sensing, as shown by the absence of AAV-dependent increase in p21 response when editing with ssAAV2/6 lacking VP1, which successfully enters cells but fails nuclear translocation of the viral genome (François et al., 2018) (Figures 2N–2P and S2P). AAV genome engineering by either modified 3′ and 5′ AAV ITRs (deleted of the B-B’ and/or C-C’ reactive sequences) or mutated 3′ ITR D-sequence (resulting in encapsidation of single polarity [sp] genomes into the viral capsid [Zhou et al., 2008]) did not mitigate p21 induction despite showing similar ITR CG content (Figures 2N–2P, S2Q, and S2R). Moreover, ITR-deleted AAVs were less efficient at inducing HDR, suggesting lower proficiency at second strand synthesis and making hardly possible to dissociate template function from innate sensing.
Trapping of transcriptionally active AAV ITRs at nuclease target site is an inadvertent consequence of HDR editing
Deep sequencing of the edited AAVS1 highlighted the presence of alleles carrying insertions of AAV sequences with variable lengths (Figures 3A and S3A) in two thirds of mice receiving HSPCs edited with full ssAAV2/6 particles (Figure 3B), accounting for up to 3% of the total allele diversity. Higher abundances were found in mice from the scAAV group which, having the lowest graft clonality, may show relative overrepresentation of any contributing clone. More than 55% of inserted AAV sequences aligned to ITRs, with prevalence of the 3′ ITR (Figures 3A and 3C). Interestingly, we often observed indels at one or both genomic junctions, suggesting NHEJ- or MMEJ-mediated trapping (Figure S3B). A minor subset of inserts mapped inside the transgene cassette adjacent to the proximal portion of the homology arms, possibly reflecting aborted HDR events (Figures 3A, 3C, and S3B). Trapping of AAV fragments at AAVS1 was not detected in xenografts derived from HSPCs only transduced with AAV (Figure S3C).
Figure 3.

Integration of AAV ITR fragments at the target site
(A) Heatmap showing alignment and normalized abundance (Log(counts per million, CPM)) of reads bearing integrated DNA fragments (length ≥ 20 bp) across the AAV genome for each mouse from Figure 2H (n = 9, 9, 10, and 5). Untreated (UT) samples sequenced and analyzed in parallel are shown (n = 2). HA, homology arms; PolyA, bovine growth hormone polyadenylation signal. The low signal in UT samples accounts for background noise of the analysis.
(B) Percentage of AAVS1 alleles in human splenocytes from (A) carrying integrated DNA fragments. The proportion of mice within each group carrying at least one event of DNA fragment integration is shown above (n = 9, 9, 10, and 5). Median with quartiles, Kruskal-Wallis test with Dunn’s multiple comparisons.
(C) Pie chart showing the proportion of fragments aligning to each region of the AAV or human genome within the total number of retrieved DNA fragments from experiments in (A) (n = 28 events in 23 mice).
(D) Percentage of alleles as in (B) from experiments in Figure S3F (n = 23, 11, 15, and 16). Median with quartiles, Kruskal-Wallis test with Dunn’s multiple comparisons.
(E) Pie chart as in (C) for experiments in Figure S3F (n = 53 events in 65 mice).
(F and G) RFI over time of GFP+ CD4+ T cells (F) and mPB HSPCs (G) transduced with the promoter-less ssAAV2/6 shown in Figure S3L (n = 4, 4, 4, and 3 for T cells, n = 3 for HSPCs). Cells transduced with the PGK-GFP ssAAV2/6 are shown as reference. Median.
(H) Capillary electrophoretic analysis showing transcript amplification at the expected molecular weight in three donors (D) from Figure 3F.
See also Figure S3.
These findings were reproduced when analyzing 65 samples from the long-term xenograft of mice belonging to four previously published experiments (Figures S3D and S3E), further showing that inclusion of HDR editing enhancers (GSE56 and the Adenovirus 5 E4orf6/7 protein) (Ferrari et al., 2020) did not influence the frequency or pattern of AAV fragments integrations at the target site (Figures 3D, 3E, and S3F). Because the AAV donor used in these experiments carried a barcoded sequence to allow clonal tracking of edited cells, we proved the occurrence of cells bearing aborted HDR events in the mouse graft by two independent analyses (Figures S3F and S3G). We also found three trapping events carrying sequences aligning to the human cellular genome, two of which mapped to the q-arm of chromosome 19 (the same of AAVS1) (Figure 3E). Integration of DNA fragments of AAV origin was confirmed in xenotransplanted human HSPCs (Figures S3H and S3I) or T cells edited at CD40LG (Figures S3J and S3K) from two previously published experiments (Vavassori et al., 2021).
Putative transcriptional activity of AAV ITRs (Earley et al., 2020; Haberman et al., 2000) might be of concern upon integration in the human genome. To assess whether AAV2 ITRs have promoter activity in human hematopoietic cells, we designed ssAAV2/6 devoid of promoter and carrying GFP downstream of either the 5′ or the 3′ ITR (Figure S3L). Transduced human primary hematopoietic cells showed detectable GFP expression peaking 48 h after transduction and progressively decreasing over time (Figures 3F, 3G, S3M, and S3N). Successful amplification of the spliced GFP transcript confirmed the presence of transcriptional start site(s) upstream of the splicing donor site and therefore within the ITR sequence (Figure 3H). When aligning sequencing reads from transcriptomic analyses on edited CD4+ T cells, we found low but consistent signal covering the non-coding region within the AAV genome nearby the ITRs, supporting ITR-driven transcription upon their integration (Figure S3O).
Overall, these findings reveal unanticipated editing outcomes that may aggravate the genotoxic burden of AAV-based HDR editing in primary hematopoietic cells, including LT-HSPCs.
Unbiased genome-wide analysis reveals AAV DNA inserted at nuclease on- and off-target sites in LT-HSPCs
We then developed a methodology for unbiased genome-wide retrieval of AAV integration sites (IS), using several primers to amplify junctions involving different portions of the AAV genome (Figure 4A). Sequenced amplicons were analyzed by an ad-hoc bioinformatics pipeline (Recombinant Adeno-Associated Vector Integration analysis, RAAVIoli; STAR Methods and Figure S4A).
Figure 4.

Unbiased genome-wide retrieval of AAV IS reveals frequent integration events at nuclease on- and off-target sites in edited LT-HSPCs
(A) Schematic representation of the PCR primer sets for retrieval of AAV IS. Inner PCR primers for nested amplification have blue tails.
(B) Number of AAV IS from hematopoietic organs of mice transplanted with HSPCs edited as indicated at AAVS1 (n = 5, 3, 7, and 8) or RAG1 (n = 6). Median.
(C) Genome-wide distribution of AAV integrations retrieved from mice transplanted with HSPCs edited at AAVS1 (shown in black) and RAG1 (shown in red).
(D) Genomic views of AAV IS within the AAVS1 (top) and RAG1 (bottom) target sites. Genomic coordinates and scale are indicated. Black rectangles indicate the position of AAV IS, horizontal bars indicate homology arms, and the protospacer sequence targeted by each gRNA.
(E) Pie charts indicating the frequency of AAV features found at the nuclease on-/off-target sites upon AAVS1 (left) and RAG1 (right) editing.
(F) Heatmap of the 3′ AAV ITR secondary structure with red scale indicating the frequency of AAV insertions at the indicated nucleotide position for editing AAVS1 (top) or RAG1 (bottom).
(G) Percentage of IS represented by the indicated number of genomes in IS datasets derived from AAVS1- and RAG1-edited cells. Mean ± SEM.
(H–K) CG measured by the “ITR+cargo” probe system within: human splenocytes of mice from Figures 1E, 1J, and 2F. (H) n = 9, 9, 10, and 5; median with quartiles, Kruskal-Wallis with Dunn’s multiple comparisons, circulating hCD45+ cells in transplanted mice from previously published experiments (Ferrari et al., 2020). (I) n = 3, 4, 5, and 4; median, human hematopoietic lineages in transplanted mice from previously published experiments (Ferrari et al., 2020). (J) n = 3, 5, 5, and 4; median), and human splenocyte of serially transplanted mice from previously published experiments (Ferrari et al., 2020). (K) n = 6, 6, 4, and 4; median.
(L) CG measured by the “ITR+cargo” or “ITR” ddPCR probe systems within human splenocytes of mice from Figures 1E, 1J, and 2F, and UT samples. Paired values are linked by a black line (n = 20, 5, 9, and 3). The dashed red line indicates the background noise threshold determined by UT samples. The ITR signal in the “empty” group was comparable with the threshold except for one mouse, possibly in agreement with the finding that fractionated empty AAV particles may carry ITR fragments at low abundance (Tai et al., 2018).
(M) “ITR” CG within human splenocyte of mice transplanted with HSPCs edited at different loci (n = 20, 23, and 41). Median with quartiles.
See also Figure S4.
We analyzed genomic DNA samples from bone marrow (BM) and spleen of 33 mice transplanted with CB or mPB HSPCs edited at AAVS1 with RNP carrying a high (HS, from Figure 1J) or low (LS) specificity gRNA and empty or full ssAAV template (Figures S4B and S4C for CB cells) or treated with ssAAV alone (Figures S4D and S4E for mPB). In the CB dataset, we identified 130 non-redundant IS, ranging from 1 to 11 unique IS per mouse, whereas no IS were retrieved from the empty AAV group (Figure 4B). Similar number of IS were identified in all other treatment groups, whether differing for the AAV dose or guide, and among the different PCR systems used for the amplification step (Figures 4B and S4F). The AAVS1 editing site was the preferential target for integration accounting for 21% IS (Figures 4C and S4G; Table 1; and Data Table S1). These IS tightly clustered into the protospacer sequences targeted by the two different gRNAs used, proving that AAV integration was promoted by the nuclease-induced DSB (Figure 4D). Interestingly, among the other IS identified at lower frequency, two of them, LAMC3 and LRR1, were also captured by in silico (Haeussler et al., 2016) and GUIDE-Seq specificity analyses for the LS gRNA (Data Table S2). AAV integrations at these sites occurred within genomic regions homologous to the gRNA, proving their origin from RNP off-target activity (Figure S4H). In addition, 3 independent IS mapped to the PGK1 gene (Figures S4G and S4I), suggesting that homology between the vector-contained and cellular PGK1 promoter sequence might favor recombination at this transcriptionally active locus. Corresponding findings were obtained in the smaller mPB dataset, where we retrieved AAVS1, PGK1, and BCL11A integrations in the RNP-treated group. Remaining IS in both datasets, and those found in the AAV-only treated group, were unique and may thus represent insertions at random spontaneous DNA breaks.
Table 1.
Most frequent genes targeted by AAV and IDLV integration
| Chr | GeneID | N IS | N mice | % | PCR | Off-target | |
|---|---|---|---|---|---|---|---|
| AAVS1 (AAV) | 19 | PPP1R12C | 27 | 12 | 20.8 | B/E/F | |
| 2 | LINC00486 | 4 | 3 | 3.1 | E/F | ||
| 9 | LAMC3 | 3 | 2 | 2.3 | E/F | LS | |
| X | PGK | 3 | 3 | 2.3 | B | ||
| X | CD40LG | 2 | 2 | 1.5 | A | ||
| 7 | SLC4A2 | 2 | 2 | 1.5 | B | ||
| 3 | ERC2 | 2 | 1 | 1.5 | F | ||
| 9 | MOB3B | 2 | 1 | 1.5 | F | ||
| 15 | ISG20 | 2 | 1 | 1.5 | E | ||
| 20 | PANK2 | 2 | 1 | 1.5 | F | ||
| 14 | LRR1 | 1 | 1 | 0.8 | F | LS | |
| RAG1 (AAV) | 11 | RAG1 | 18 | 5 | 56.3 | I/H | |
| 2 | SCG2 | 2 | 1 | 6.3 | I | ||
| 12 | PPFIA2 | 2 | 1 | 6.3 | I | ||
| 17 | TIMM22 | 2 | 1 | 6.3 | H | ||
| AAVS1 (IDLV) | 9 | LAMC3 | 6 | 3 | 11.3 | B/L | LS |
| 19 | PPP1R12C | 5 | 3 | 9.4 | B/L | ||
| X | PGK1 | 3 | 3 | 5.7 | B | ||
| 1 | DISP3 | 2 | 2 | 3.8 | B | ||
| 11 | OR4C46 | 4 | 1 | 7.5 | L | ||
| 7 | LINC00972 | 2 | 1 | 3.8 | L | ||
| 22 | SYN3 | 2 | 1 | 3.8 | L | ||
| 22 | ZDHHC8 | 1 | 1 | 1.9 | L | LS |
Chr, chromosome; Gene ID, RefSeq Gene Symbol of the gene targeted by IS; N IS, number of vector integrations; N mice, number of mice harboring vector integrations targeting the indicated gene; %, percentage of IS targeting the indicated gene; PCR, PCR sets from which the IS were identified; Off-target, gene containing a sequence recognized as putative gRNA off-target. 12 of 18 RAG1 IS were within the gRNA target site.
See also Data Table S1.
Nearly all integrations at AAVS1 and two predicted off-target sites involved AAV ITRs, with prevalence of the 3′ ITR (Figure 4E). A more granular inspection identified some preferred nucleotide breakpoints within the A–A’ and C–C’ loop regions of 3′ ITR (Figure 4F), as previously reported (Hanlon et al., 2019; Nguyen et al., 2021). More than half of AAV-containing clones were represented by >5 to nearly 100 genomes, implying their proliferation and relevant contribution to the graft (Figure 4G).
A similar analysis was performed on 6 mice repopulated with BM HSPCs edited at the intron 1 of RAG1 (Figures 4A, S4B, and S4J), using a promoter-less codon-optimized RAG1 sequence as template delivered by ssAAV, modeling correction of mutant alleles causing primary immunodeficiency. The RAAVIoli platform identified 32 unique AAV IS, 12 of which were at the RAG1 editing site (Figures 4B–4D and S4F). As for AAVS1 editing, ITR fragments were the most frequent portion of AAV inserted with similar preferences for the nucleotide breakpoint (Figures 4E–4G).
Since short-read sequencing of the edited locus and genome-wide IS identification may underestimate the overall frequency of trapping events, we performed ddPCR analyses to get an independent genome-wide quantification of AAV genome trapping events. Long-term human xenografts from Figure 1 showed detectable signal when probing for the AAV sequence spanning from the ITR trs to the homology arm (“ITR+cargo”) in nearly all mice, with median 0.05 or 0.1 CG depending on the group. These are higher frequencies than those estimated from the target site sequencing and were similar for the ssAAV2/6 and scAAV2/6 groups and null in the empty AAV (Figures 4H and S4K). We then extended the analysis on long-term AAVS1-edited HSPC xenografts from previously published experiments (Ferrari et al., 2020) and found more copies in the presence of GSE56 and/or E4orf6/7 (Figures S4L and S4M). Of note, ITR+cargo CG were stable over time and across lineages, including BM-derived CD34+ HSPCs, and upon serial transplantation (Figures 4I–4K and S4N). Altogether, these results rule out that the ITR+cargo signal may come from carryover of episomal AAV genomes. When performing ddPCR with the ITR probe on the same samples, we found equal or higher signal in most mice (Figure 4L). Long-term xenografts of mice transplanted with mPB HSPCs edited at other genomic loci, CD40L and IL2RG, showed 1.5-fold higher and 1.5-fold lower median ITR CG, respectively, compared with AAVS1-edited grafts (Figure 4M), highlighting consistent occurrence but variable extent of AAV integration at different target sites. Whether integration of AAV DNA occurs mostly as individual elements in a sizable fraction of cells treated for editing or as concatemers in only a small fraction of them cannot be determined by this analysis.
Overall, these analyses identified reproducible and significant occurrence of integration of AAV DNA at DSB induced by editing at nuclease on- and off-target sites, mainly involving a specific region of the AAV ITR, which might trigger or be prone to capture during DSB repair.
IDLV editing shows low frequency of fragments trapping at the nuclease target sites in LT-HSPCs
We then performed AAVS1 editing in CB HSPCs using IDLV as alternative template delivery and cyclosporin-H (CsH) as transduction enhancer (Petrillo et al., 2018). Despite editing efficiencies were in the same range of AAV-based editing experiments, only one mouse of 26 analyzed by targeted deep sequencing showed a single event of IDLV fragment integration, likely due to aborted HDR (Figures 5A, 5B, and S5A–S5D).
Figure 5.

Unbiased genome-wide retrieval of IDLV IS in gene-edited HSPCs
(A) Number of unique indels retrieved by deep sequencing AAVS1 in human BM cells of mice from S5C (n = 10, 6, and 10). Median with quartiles. Kruskal-Wallis test with Dunn’s multiple comparisons.
(B) Percentage of alleles in human BM cells from Figure S5C carrying integration of DNA fragments as in Figure 3B (n = 10, 6, and 10). Median with 95% CI.
(C) Schematic representation of the PCR primer sets for retrieval of IDLV IS as in Figure 4A.
(D) Number of IDLV IS retrieved from transplanted mice (n = 4). Median.
(E) Genomic view of IDLV IS within AAVS1 as in Figure 4D.
(F) Pie charts indicating the frequency of IDLV features found at the IS breakpoints either at (left) or outside (right) nuclease target sites.
See also Figure S5.
We then screened for genome-wide IDLV integration in 4 long-term engrafted mice transplanted with CB HSPCs edited at AAVS1 with the LS RNP described above (Figures 5C, S5E, and S5F). We identified 53 unique IS, ranging from 6 to 25 unique IS per mouse, all represented by multiple genomes (Figures 5D and S5G; Table 1; and Data Table S1). Preferential IS correspond to the LS gRNA protospacer sequence within AAVS1 (Figures 5E, S5H, and S5I) and the off-target LAMC3 also captured by the AAV template in Figure S4H. Other IS mapped to another predicted off-target site, ZDHH8 (Table 1; Figure S5I) and PGK1 (Figure S5J). These findings confirm that IDLV can be captured at nuclease on- and off-target sites, similarly to what we described above for AAV, with preferential occurrence of the IDLV SIN LTR at the vector-genome junction (Figure 5F). Such trapping, however, should mostly involve the whole genome or large fragments thereof, as our targeted deep sequencing analysis found only low occurrence of short IDLV derived sequences at the editing sites.
Optimized IDLV editing shows a more favorable toxicity and safety profile than AAV and reaches higher editing efficiencies in LT-HSPCs
We further tailored IDLV editing for the clinically relevant mPB HSPCs, combining CsH, GSE56, and E4orf6/7. By testing different doses, timings, and rounds of transduction, we reached up to 12% editing in the most primitive HSPCs by two hits of IDLV at the highest dose in presence of all enhancers (Figures 6A and S6A). When comparing this optimized IDLV protocol with the optimized AAV-based one, we found that the latter was 3-times more efficient within committed progenitors, whereas the difference flattened in the most primitive compartment (Figures 6A, 6B, and S6B). Notably, IDLV transduction better preserved HSPC clonogenic capacity, yielding 2-fold more colonies than the AAV-based protocol (Figure 6C). This finding was consistent with a shorter wave of p21 induction and NBS1 foci after editing (Figures 6D, 6E, and S6C), and the substantially lower content and faster decay over time of intracellular IDLV DNA, compared with AAV (Figures 6F and S6D; compared with Figure 2E).
Figure 6.

Optimized IDLV-based editing protocol results in higher editing efficiency than the AAV-based one in the long-term graft
(A) Percentage of GFP+ cells within mPB HSPC subpopulations after AAVS1 editing using IDLV in the presence or absence of CsH and GSE56/E4orf6/7. Transduction was performed once 24 or 12 h before electroporation (1 hit) or twice with the second round of transduction after electroporation (2 hits), using multiplicity of infection (MOI) of 150 transducing units (TU)/cell per hit. The optimized ssAAV2/6 protocol (2 × 104 vg/cell, with GSE56/E4orf6/7) (Ferrari et al., 2020) was performed in parallel (n = 1, 2, 2, 5, 4, and 5). Median. Opt: with GSE56/E4orf6/7 mRNA.
(B–D) Percentage of HDR-edited alleles (B) n = 2; median, number of grown colonies (C) n = 9; median with 95% CI, Friedman test with Dunn’s multiple comparisons, fold change expression over time of CDKN1A relative to UT (D) n = 15 at 1 day, 13 at 4 days, 11 at 7 days; median with 95% CI, LME followed by post-hoc analysis; results are shown for the last time point after editing mPB HSPCs as indicated.
(E) Quantification of NBS1+ cells and distribution of the percentage of foci bearing cells over time from HSPCs treated as indicated (n = 11, 7, 8, 3, and 9). Mean.
(F) Intracellular CG of the indicated IDLV features retrieved over time from HSPCs edited with the optimized 1-hit or 2-hits protocols (n = 3) performed in parallel to those reported in Figure 2E. See STAR Methods. Median.
(G) Heatmaps representing CG measured by the indicated probe systems in single colonies plated 4 days after editing as indicated (n = 26, 16, 40, 20, 39, and 19). To account for the residual presence of episomal DNA, a lower threshold was set at 0.5 CG (white boxes).
(H) Heatmaps as in (G) showing CG in colonies plated 4 days after B2M editing in the presence of the unrelated AAVS1 repair templates (n = 9, 30, 10, 32, 10, and 23).
(I) CG of an AAVS1 sequence telomeric to the left HA (n = 19, 15, 26, 14, 16, 39, 15, 19, 39, and 17). Mean. Red lines indicate cut-off values for colonies carrying long-range deletions or duplications (red dots).
(J and K) Percentage of circulating hCD45+ (J) and GFP+ cells within the human graft (K) in mice transplanted with the outgrown progeny of starting-matched saturating cell doses of mPB HSPCs edited as indicated (n = 5). Mean ± SEM, LME followed by post-hoc analysis; results are shown for the last time point.
(L) CG measured by HIV and ITR probes within human BM cells from mice in (K) (n = 5). Median with quartiles. Kruskal-Wallis test with Dunn’s multiple comparisons.
(M) Percentage of AAVS1 alleles within human splenocytes from Figure S6J carrying ITR or LTR DNA fragments (length ≥ 20 bp) (n = 5). Median with quartiles. Kruskal-Wallis test with Dunn’s multiple comparisons.
(N) Pie charts as in Figure 3C from experiment in (J).
(O) Heatmap as in (G) for colonies plated from FACS-sorted CD34+ HSPCs harvested at the end of the experiment from BM of xenotransplanted mice (n = 12 and 13).
See also Figure S6.
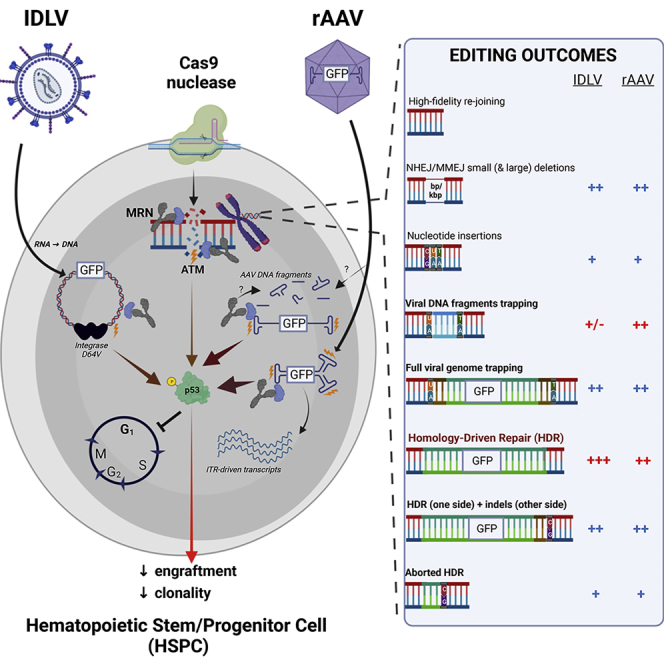
We then screened with panels of probes tiling the vectors genome, >200 randomly picked colonies outgrown from sorted GFP+ or GFP− HSPCs edited at AAVS1 with AAV or IDLV protocols. For both templates, nearly all GFP+ colonies scored positive for the payload and on-target integration (Figure 6G). Among them, from 45% to 60%, according to the treatment, carried putatively precise mono- or bi-allelic HDR-mediated integration, given the absence of any signal for the assays probing for viral features. The remaining colonies tested positive also for one or more viral features, suggesting the occurrence of HDR on one junction and NHEJ/MMEJ on the other of the same edited allele, or, less likely, the occurrence of full HDR on one allele and vector trapping on the other, or targeted integration of concatemers (with GFP probe >2; an outcome predominantly observed for AAV), or possibly concomitant off-target ITR/vector trapping. Analysis of colonies from sorted GFP− cells uncovered some GFP and on-target integration clones, which were more abundant for the two-hit IDLV protocol, likely due to delayed GFP expression after sorting (Figure 6G). Notably, integration of viral DNA fragments in the absence of GFP payload and on-target integration signal was reported exclusively for the AAV-based editing protocol. Analysis of colonies plated from vector transduced-only HSPCs showed none of them carrying vector integration (Figure S6E).
To further evaluate trapping of viral vector independently from HDR, we transduced HSPCs with the same AAVS1 ssAAV2/6 or IDLV templates and edited them in the unrelated B2M locus. Colonies derived from the low proportion of sorted GFP+ B2M-edited HSPCs mostly contained full-length vector trapping events (positive for payload and viral features) and confirmed the tendency of AAV to integrate more frequently as longer concatemers (Figure 6H). Even more strikingly, we found no integration event out of 55 colonies for IDLV and 7 of 30 (23%) for AAV among the GFP− colonies, confirming the higher tendency of AAV to integrate as DNA fragments, particularly ITR sequences.
We also probed colonies edited at AAVS1 for bearing long-range deletions at the target locus (Figure S6F). When probing for a sequence about 800 bp telomeric to the nuclease target site, 7% to 15% of AAVS1-edited colonies harbored only one copy of the allele, regardless of the viral vector (Figure 6I). Neither transduction with AAV nor the use of HDR enhancers aggravated the burden of large deletions at the target site (Figure S6G). However, none of the colonies lacked copies of AURKC, a gene 2.1 Mbp telomeric to the target site (Figures S6F and S6H).
We then edited human mPB HSPCs pooled from 3 donors with the different enhanced protocols and HDR templates (Figure S6I) and transplanted them into mice to evaluate repopulation capacity. Although the best-performing IDLV and AAV protocols showed a similar rate of GFP+ cells with medians of 18% in the graft early post-transplant (Figures 6J and 6K), there was a progressive decrease with time in the AAV group, consistent with previous findings (Ferrari et al., 2020; Schiroli et al., 2017, Schiroli et al., 2019). On the contrary, IDLV editing showed stable marking throughout the follow-up with a median of 15% at the end of the study (Figures 6J, 6K, S6J, and S6K), thus outperforming AAV at editing LT-HSPCs. This outcome was confirmed throughout hematopoietic lineages (Figures S6L and S6M), with no differences in composition across treatments (Figure S6N). We then measured the copies of IDLV and AAV within the human graft and found detectable signal for both platforms in conditions yielding highly edited grafts (Figures 6L and S6O). However, deep sequencing of the edited locus from mice splenocytes showed a higher number and proportion of alleles harboring integrated DNA fragments for the AAV-based protocol than the IDLV ones (Figures S6P and S6Q), which became more evident when computing the fraction of alleles carrying ITR or LTR sequences (Figures 6M and 6N). Aborted HDR events were retrieved for both viral templates. Two alleles in the AAV group showed integration of fragments derived from reverse packaged AAV transfer plasmid backbone (Figure 6N). Colonies generated from CD34+ harvested from the BM of human hematochimeric mice 14 weeks post-transplant confirmed engraftment and persistence of some clones carrying integrated AAV features (Figure 6O).
Overall, these results support the choice of IDLV as repair template for HDR editing because of the lower cytotoxic and genotoxic potential and the higher rates of editing in LT-HSPCs.
Discussion
Our findings uncover an unexpected genotoxic burden of editing HSPCs, which is mainly due to the high content of template DNA required for efficient HDR and its persistence in the treated cells together with a variable amount of fragments carried over or generated from the viral platform chosen for delivery. This DNA can be incorporated during the repair process at the target as well as off-target and spontaneous DNA DSBs. Moreover, it triggers a sustained p53-dependent DDR exacerbated by some intrinsic features of viral DNA, which together with the nuclease-induced DNA DSBs, impact cell viability and clonogenic capacity. Altogether, these factors concur to generate a highly heterogeneous genetic configuration at the genomic sites vulnerable to the editing machinery in a clonally shrunken hematopoietic graft derived from the treated HSPCs. These include precise insertion of the templated sequence by HDR, integration of the whole viral DNA, its fragments, or concatemers, at one or both sides of the break, mediated by NHEJ or MMEJ, as well as deletions encompassing the targeted locus (Figure S6R). It appears that all these genetic outcomes occur independently, including at each side of the same DSB, as we could not find any correlation with the investigated parameters. Although the consequences of most unintended genetic outcomes are likely to be context-specific and often not deleterious, our findings highlight the limited precision of HDR editing and call for strategies to improve it. Here, we show that the choice of template delivery platform as well as its dose and configuration strongly affect the type and frequency of most investigated adverse effects. Compared with the commonly used AAV platform, optimized IDLV delivery can significantly mitigate both the genotoxic risk and adverse cellular impact of editing, although allowing a higher rate of HDR editing in LT-HSPCs.
From a mechanistical standpoint, the DDR induced by AAV was ascribed to the amount of DNA translocated into the nucleus rather than the capsid of the viral particle, similar to what has been previously described for LV (Piras et al., 2017). The high burden and persistence of DDR were likely due to the high intracellular viral DNA load in infected cells, the detectable transfer of plasmid DNA from the producer cells by reverse packaging, and the carryover and generation of genomic fragments enriched for the ITR (Dalwadi et al., 2021a; Lecomte et al., 2015; Tai et al., 2018), which preferentially trigger DDR and become trapped at DNA DSBs. Indeed, the stronger and more sustained DDR triggered by scAAV than ssAAV agrees with the presence of three cis ITR motifs in the former and its higher stability when translocated into the nucleus (McCarty et al., 2003; Wang et al., 2003). Although a role of the MRN complex in sensing AAV ITRs was previously reported in cell lines (Cervelli et al., 2008; Lentz and Samulski, 2015; Zhou et al., 2017), we document massive formation in AAV-treated HSPCs of NBS1 nuclear foci that are prerequisite for p53 induction and might also contribute to the generation of ITR-comprising viral genomic fragments. These findings may have broader implications in the context of AAV gene therapy, in which ITR-driven p53 activation should be investigated as a potential source of treatment-emergent toxicity upon the delivery of high vector doses (Food and Drug Administration, 2021). Although HSPCs were expected to clear non-integrated vector DNA by their rapid proliferation, activation of p53 strongly limits this escape mechanism by preventing cell cycle progression. Although we identified AAV ITRs as the main cause of DDR induction and source of DNA fragments integrated in HSPCs, we could not dissociate the cellular sensing triggered by ITR from their crucial role in genome replication and packaging (Hirsch, 2014; Wilmott et al., 2019). These results emphasize the challenge in engineering stealth AAV vectors as sequences essential for function (e.g., RBE) and structural conformations that are obligate replication intermediates (e.g., hairpins and DNA free ends) are also inherently linked to DDR triggering. Up to date, transient p53 inhibition remains the most valid option to abrogate this response and improve HSPC editing outcomes for clinical translation.
A relevant safety concern raising from the frequent occurrence of AAV ITR trapping in the genome is their previously described transcription-promoting activity (Earley et al., 2020; Haberman et al., 2000), which we show to be present also in HSPCs and for commonly used AAV ITRs lacking a hepatocyte-specific enhancer/promoter. Possible adverse impacts of ITR-originated transcription might be aberrant expression of genes flanking their insertion and/or cross-packaged contaminating genetic material and cytotoxicity, as reported by a recent NHP study (Keiser et al., 2021). On the contrary, the full deletion of all enhancer and promoter sequences from the SIN LTR of commonly used LV (Bukovsky et al., 1999; Zufferey et al., 1998) safeguards against residual transcription originating from these sequences upon inadvertent IDLV integration.
Although AAV has long been considered a non-integrating platform, the recent development of sensitive technologies for unbiased retrieval of genomic insertions from a background of excess episomal DNA (Breton et al., 2020; Nguyen et al., 2021) has uncovered a significant rate of vector integration in tissues following in vivo administration (Hanlon et al., 2019; Nelson et al., 2019), which may build-up with time as spontaneous DSBs occur in the cellular genome. Gene editing, however, creates an abundance of genomic baits when AAV DNA is at its peak concentration, thus providing ample opportunity for capture of both intact and fragmented DNA at on- and off-target sites of the nuclease, albeit occurring in competition with HDR at the former ones. This behavior was reproducibly found at all editing target sites investigated in the study, ruling out a confounding effect of AAVS1, which was originally reported as preferential integration site for wild-type AAV, but not for its derived recombinant vectors, which lack the Rep protein (Ponnazhagan et al., 1997). Trapping at nuclease-induced DSBs also occurs with other DNA delivery platform such as IDLV, and indeed, some of us exploited it for the assessment of in vivo specificity of editing nucleases (Gabriel et al., 2011), providing a foundation for commonly used technologies such as GUIDE-Seq. The actual extent of trapping and the preferential occurrence of some vector genomic features at the insertion site may however vary with each platform. The higher intracellular content of AAV DNA and the abundance of its ITR-containing fragments, which are prone to recruit DNA repair components, may explain the more frequent trapping of AAV fragments at the nuclease target sites observed here in comparison with IDLV. Integration of the latter may rather occur by aborted HDR, comprising most of the vector genome.
Unbiased genome scanning by specialized PCR protocols that ensure the coverage of different vector portions and the use of the RAAVIoli bioinformatics pipelines described in this study confirmed occurrence of AAV and IDLV integration events at predicted editing off-target sites. Further application of this technology may help to better characterize the specificity of a designer nuclease complex and uncover unpredicted biases for off-target insertion originating from specific configuration of the template, such as shown here for the propensity of vector harboring sequences derived from the PGK promoter to integrate at the endogenous constitutively transcribed locus. The observed integration of vector DNA at predicted off-target sites recall the importance of gRNA selection based on its specificity to avoid the occurrence of such undesired events.
In rare cases, we found cellular genomic sequences trapped at the editing site of HSPCs treated with either AAV or IDLV. Intriguingly, 3 of 5 events mapped to chromosome 19 where the editing target site is found. Such preference may be suggestive of chromothripsis, which entails chromosomal fragmentation and rearrangement triggered by DSB and represents a rare genotoxic outcome of editing (Leibowitz et al., 2021).
Because the life cycle and the structural features of AAV and IDLV, as well as the experimental methods to quantitate them, are different, a proper comparison at matched infectious doses remains difficult. We thus compared the maximal effective dose of each vector, which fell in the same range of editing efficiency, and found a lower cytotoxicity profile of IDLV, which we ascribe to reduced intracellular DNA load and, possibly, more efficient genome disposal. These same features might also explain the better proficiency of IDLV at editing LT-HSPCs, which are less permissive to transduction and more sensitive to DDR and its adverse impact. Further advantages of IDLV for HDR template delivery in HSPC editing are as follows: (1) the excellent safety and efficacy track record of LV-mediated HSPC gene therapy trials, which show robust polyclonal repopulation by vector treated HSPCs (Ferrari et al., 2021a), (2) the larger cargo capacity, which enables more complex design of the therapeutic cassette (e.g., by including selector markers and purging unintended genetic addition or deletion at the target site), and (3) the lower concern for immune rejection of ex-vivo-edited HSPCs upon presentation of residual viral antigens. Nevertheless, our clonal analysis and genome-wide quantification uncovered a similar extent of unintended integration events occurring at the target site for IDLV and AAV. These include HDR occurring at only one side of the DNA DSB, full trapping of the vector genome and concatemer insertion. Cells bearing these imprecise repair outcomes may fail to rescue gene function when editing aims to in-frame restoration of the endogenous coding sequence, such as “one-size-fits-all” approaches targeting a corrective cDNA into an exon (Dever et al., 2016; Genovese et al., 2014; Hubbard et al., 2016). In most other cases, however, such insertions would still be compatible with the expected corrective outcome.
Despite the imprecision of genetic outcome, HDR editing remains the most feasible and often unique approach to long-range gene correction when disease-causing mutations are several and scattered over the gene sequence or to target integration of a transgene cassette to safe harbors. The strategies demonstrated here to uncover and alleviate the inadvertent consequences of HDR editing and improve its efficiency in LT-HSCs should facilitate safer and more effective clinical translation.
Limitations of the study
The experimental model of human HSPC repopulation used in this study is constrained by the xenogeneic host and only represents a limited surrogate of human hematopoiesis, albeit being routinely adopted for preclinical studies. AAV integration at nuclease on- and off-target sites, although reproducible, was observed for a limited number of investigated sites and may thus not have captured site-specific influences or represent universal features. Moreover, neither ITR-driven transcription nor DDR were evaluated in engrafting hematopoietic cells in this study. Whether these unintended events raise concern of genotoxicity, presently this remains only hypothetical because of the lack of suitable models to assess the consequence of rare genotoxic events. Our findings should not raise a barrier but rather inform a more comprehensive risk-benefit assessment for future clinical translation of HDR gene editing.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD133/2-PE, anti-human | Miltenyi Biotec | RRID:AB_2654900 |
| CD34-VioBlue, anti-human | Miltenyi Biotec | RRID:AB_2726008 |
| CD34-PE-Vio770, anti-human | BD Biosciences | RRID:AB_2868855 |
| CD90-APC, anti-human | BD Biosciences | RRID:AB_398677 |
| CD45-VioBlue, anti-human | BioLegend | RRID:AB_2174123 |
| CD45-APC, anti-human | BD Biosciences | RRID:AB_2868745 |
| CD45-APC-Vio770, anti-human | BD Biosciences | RRID:AB_2868859 |
| CD19-PE, anti-human | BD Biosciences | RRID:AB_2868815 |
| CD3-PE-Vio770, anti-human | BioLegend | RRID:AB_314052 |
| CD13-APC, anti-human | BD Biosciences | RRID:AB_398624 |
| CD33-PE-Vio770, anti-human | Miltenyi Biotec | RRID:AB_2726125 |
| CD38-PerCP/Cyanine5.5, anti-human | BioLegend | RRID:AB_2562183 |
| CD4-VioBlue, anti-human | BioLegend | RRID:AB_2174123 |
| CD8-APC-Vio770, anti-human | BD Biosciences | RRID:AB_1645736 |
| Rabbit anti-NBS1 Antibody | Novus Biologicals | RRID:AB_10078050 |
| Anti-Digoxigenin-AP antibody | Sigma-Aldrich | RRID:AB_514494 |
| DAPI | Sigma-Aldrich | D9542; CAS: 28718-90-3 |
| Aqua-Poly/Mount solution | TebuBio | Cat# 18606-20 |
| Live/Dead Fixable Dead Cell Stain Kit | Thermo Fisher | Cat# L34957 |
| 7-aminoactinomycin D | Sigma Aldrich | SML1633; CAS: 7240-37-1 |
| Biological samples | ||
| Mobilized Leukopak | AllCells | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| 16,16-dymethylprostaglandin E2 | Cayman Cat | Cat# 14750 |
| StemSpan SFEM | VODEN | Cat# 09650 |
| Recombinant human stem cell factor | Peprotech | Cat# 300-07 |
| Recombinant human thrombopoietin | Peprotech | Cat# 300-18 |
| Recombinant human Flt3 ligand | Peprotech | Cat# 300-19 |
| Recombinant human IL6 | Peprotech | Cat# 200-06 |
| StemRegenin 1 (SR1) | BioVision | Cat# 1967 |
| UM171 | STEMCell Technologies | Cat# 72912 |
| MethoCult H4434 | STEMCell Technologies | Cat# 04434 |
| CD34 MicroBead Kit, human | Miltenyi Biotec | Cat# 130-046-702 |
| Iscove’s DMEM, 1X | Corning | Cat# 15-016-CVR |
| Fetal Bovine Serum South America Origin | EuroClone | Cat# FA30WS1810500 |
| Penicillin-Streptomycin (Pen-Strep) (Pen 10.000 U/mL; Strep 10.000 μg/mL) | Lonza | Cat# 17-602E |
| L-Glutamine 200mM | EuroClone | Cat# ECB3000D |
| Alt-R® Cas9 Electroporation Enhancer | Integrated DNA Technologies | Cat# 1075916 |
| Cyclosporin H | Sigma | SML1575; CAS: 83602-39-5 |
| Poly-L-lysine solution | Sigma-Aldrich | P8920; CAS: 25988-63-0 |
| PFA | Santa Cruz Biotechnology | sc-281692 |
| AAVS1 gRNA (high specificity, HS) | Schiroli et al. (2019) | N/A |
| AAVS1 gRNA (low specificity, LS) | Schiroli et al. (2019) | N/A |
| CD40L gRNA | Vavassori et al. (2021) | N/A |
| IL2RG gRNA | Schiroli et al (2017) | N/A |
| B2M gRNA | Gaudelli et al. (2020) | N/A |
| Critical commercial assays | ||
| AmpPure XP Reagent | Beckman Coulter | Cat #A63881 |
| QIAamp DNA Micro Kit | QIAGEN | Cat# 56304 |
| RNeasy Plus Micro Kit | QIAGEN | Cat# 74034 |
| QIAquick PCR Purification Kit | QIAGEN | Cat# 28104 |
| SuperScript VILO IV cDNA Synthesis Kit | Thermo Fisher | Cat# 11754050 |
| P3 Primary Cell 4D-Nucleofector X Kit | Lonza | Cat# V4XP-3032 |
| T7 Endonuclease I | New England Biolabs | Cat# M0302L |
| RNase-Free DNase Set | QIAGEN | Cat# 79254 |
| QuickExtract | Epicentre | Cat# FS99060 |
| CliniMACS CD34 Reagent System | Miltenyi Biotec | Cat# 200-074-012 |
| MEGAscript T7 Transcription Kit | Thermo Fisher | Cat# AM1333 |
| GoTaq G2 DNA Polymerase | Promega | Cat# M7401 |
| Deposited data | ||
| Targeted deep sequencing data | This paper | GEO: GSE197386 |
| RAAVIoli sequencing data | This paper | GEO: GSE197388 |
| Raw data from main figures | This paper | Mendeley: https://doi.org/10.17632/5kjcyzbkd9.1 |
| Experimental models: Cell lines | ||
| HEK293T cells | ATCC | RRID:CVCL_0063 |
| K-562 cells | ATCC | RRID:CVCL_0004 |
| HeLa cells | ATCC | RRID:CVCL_0030 |
| CB-CD34 | Lonza | Cat# 2C-101 |
| G-CSF or G-CSF + Plerixafor mPB CD34+ HSPCs | In house purification | N/A |
| Experimental models: Organisms/strains | ||
| NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJArc Mus musculus |
Jackson laboratory (IACUC 749 and 1206) | Cat# ARC:NSG, RRID:IMSR_ARC:NSG |
| Oligonucleotides | ||
| See Data Table S2 | This paper | N/A |
| Recombinant DNA | ||
| AAVS1 PGK-GFP ssAAV2/6 | Schiroli et al. (2019) | N/A |
| AAVS1 PGK-GFP ssAAV5/6 | This paper | N/A |
| AAVS1 PGK-GFP dVP1 | This paper | N/A |
| AAVS1 PGK-GFP scAAV2/6 | This paper | N/A |
| AAVS1 PGK-GFP spAAV2/6 | This paper | N/A |
| AAVS1 PGK-GFP dBC-AAV2/6 | This paper | N/A |
| AAVS1 PGK-GFP dC-AAV2/6 | This paper | N/A |
| CD40L partial cDNA ssAAV2/6 | Vavassori et al. (2021) | N/A |
| CD40L partial cDNA.IRES-NGFR ssAAV2/6 | Vavassori et al. (2021) | N/A |
| IL2RG PGK-GFP ssAAV2/6 | Schiroli et al. (2017) | N/A |
| pCCLsin.cPPT.AAVS1.PGK.GFP.pA.WPRE | Schiroli et al. (2017) | N/A |
| pVax.GSE56-Ad5-E4orf6/7.WPRE.pA | Ferrari et al. (2020) | N/A |
| pVax.GSE56.WPRE | Schiroli et al. (2019) | N/A |
| pVax.Ad5-E4orf6.WPRE.pA | Ferrari et al. (2020) | N/A |
| pVax.Ad5-E1B55K.WPRE | Ferrari et al (2020) | N/A |
| Software and algorithms | ||
| QuantStudio Real-Time PCR software v.1.1 | Applied Biosystem | https://www.thermofisher.com/us/en/home.html |
| BDFACS Diva software | BD Biosciences | N/A |
| FCS Express Flow | De Novo Software | https://www.denovosoftware.com/ |
| GraphPad Prism v.9.3.1 | GraphPad Software | https://www.graphpad.com/ |
| SnapGene v.6.1 | Dotmatics | https://www.snapgene.com |
| TapeStation software | Agilent | https://www.agilent.com/en/product/automated-electrophoresis/tapestation-systems/tapestation-software/tapestation-software-379381 |
| QuantaSoft | Biorad | https://www.bio-rad.com/ |
| R statistical software | The R Foundation | https://www.r-project.org |
| LAS X Leica Software | Leica Mycrosystems | https://www.leica-microsystems.com/it/prodotti/software-per-microscopi/dettagli/product/leica-las-x-ls/ |
| CRISPResso2 | Clement et al. (2019) | http://crispresso.pinellolab.org/submission |
| RAAVIoli computer code | This paper | https://github.com/calabrialab/Code_AAV_integrations |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Luigi Naldini (naldini.luigi@hsr.it).
Materials availability
The reagents described in this manuscript are available under material transfer agreement with IRCCS Ospedale San Raffaele and Fondazione Telethon; requests for materials should be addressed to the lead contact.
Experimental model and subject details
Mice
All experiments and procedures involving animals were performed with the approval of the Animal Care and Use Committee of the San Raffaele Hospital (IACUC no. 749 and 1206) and authorized by the Italian Ministry of Health and local authorities accordingly to Italian law. NOD-SCID-IL2Rg−/− (NSG) female mice (The Jackson Laboratory) were held in specific pathogen-free conditions.
Cell lines and primary cell culture
HEK293T and K-562 cells were cultured in Iscove’s modified Dulbecco’s medium (IMDM, Corning) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Euroclone), 100 IU ml−1 penicillin, 100 μg ml−1 streptomycin and 2% glutamine. For AAV production, HEK293 cells were cultured in DMEM supplemented with 2% heat-inactivated FBS, 100 IU ml−1 penicillin, 100 μg ml−1 streptomycin. For IDLV production, HEK293T cells were cultured in IMDM without phenol red supplemented with 10% heat-inactivated FBS, 100 IU ml−1 penicillin and 100 μg ml−1 streptomycin. For the Infectious Center Assay (ICA), Hela cells were cultured in DMEM 4.5 g/l glucose, supplemented with 10% heat-inactivated FBS and 1% penicillin and streptomycin.
CB CD34+ HSPCs were purchased frozen from Lonza according to the TIGET-HPCT protocol approved by OSR Ethical Committee and seeded at the concentration of 5x105 cells per ml in serum-free StemSpan SFEM (STEMCELL Technologies) supplemented with 100 IU ml−1 penicillin, 100 μg ml−1 streptomycin, 2% glutamine, 100 ng ml−1 hSCF (PeproTech), 100 ng ml−1 hFlt3-L (PeproTech), 20 ng ml−1 hTPO (PeproTech) and 20 ng ml−1 hIL-6 (PeproTech), 10 μM 16,16-Dimethyl Prostaglandin E2 (at the beginning of the culture, Cayman), 1 μM SR1 (Biovision) and 50nM UM171 (STEMCELL Technologies).
G-CSF or G-CSF + Plerixafor mPB CD34+ HSPCs were purified in house with the CliniMACS CD34 Reagent System (Miltenyi Biotec) from Mobilized Leukopak (AllCells) according to the TIGET-HPCT protocol approved by OSR Ethical Committee and following the manufacturer’s instructions. HSPCs were seeded at the concentration of 5x105 cells per ml in serum-free StemSpan SFEM supplemented with 100 IU ml−1 penicillin, 100 μg ml−1 streptomycin, 2% glutamine, 300 ng ml−1 hSCF, 300 ng ml−1 hFlt3-L, 100 ng ml−1 hTPO and 10 μM 16,16-Dimethyl Prostaglandin E2 (at the beginning of the culture), 1 μM SR1 and 35 nM UM171.
All cells were cultured in a 5% CO2 humidified atmosphere at 37 °C.
Method details
CRISPR/Cas9 nucleases
Genomic target sequences of gRNAs were previously reported (AAVS1-HS, IL2RG, CD40LG, B2M) (Gaudelli et al., 2020; Schiroli et al., 2019; Vavassori et al., 2021) or will be reported in detail elsewhere (RAG1). The genomic target sequence of AAVS1-LS gRNA is the following: 5’-GTCCCCTCCACCCCACAGTG GGG-3’. RNP complexes to be delivered by electroporation were assembled by incubating at 1:1.5 (AAVS1, IL2RG, B2M, RAG1) or 1:2 (CD40LG) molar ratio Streptococcus pyogenes (Sp)Cas9 protein (Aldevron) with pre-annealed synthetic Alt-R crRNA:tracrRNA (Integrated DNA Technologies) for 10 min at 25 °C. For some AAVS1 experiments, synthetic single gRNA from Synthego was used and assembled with Cas9 protein following the same procedure.
HDR viral donor templates
The design of ssAAV2/6 transfer vector constructs for AAVS1, CD40L and IL2RG editing were previously reported (Ferrari et al., 2020; Gaudelli et al., 2020; Schiroli et al., 2019; Vavassori et al., 2021) and are schematized in Figure S1T. For some AAVS1 experiments the barcoded ssAAV2/6 cassette described in (Ferrari et al., 2020) was used interchangeably with the non-barcoded one. The design of ssAAV2/6 construct for RAG1 editing will be described elsewhere. All the cargo cassettes are flanked by AAV serotype-2 derived ITRs, unless otherwise specified. The 5’ AAV ITR sequence bears a 11-bp deletion (5’-AAAGCCCGGGC-3’) that can be rescued during viral genome replication by exploiting the wild-type 3’ AAV ITR sequence (Samulski et al., 1983). This conventional ssAAV vector contains either positive [+] or negative [-] polarity genome, equally separated in mature virions during vector production.
The ssAAV5/6 transfer vector construct for AAVS1 editing contained the same payload of the ssAAV2/6. 5’ and 3’ AAV ITRs originating from the wild-type genome of AAV5 (GenBank ID NC_006152.1, 167 nucleotides on both sides) were cloned in place of ITR-2 (Figure S2A).
The scAAV2/6 transfer vector construct contained the enhanced GFP transgene under the control of the human PGK promoter and a bovine growth hormone (BGH) polyadenylation signal (Figure S2G). This cassette was flanked by homology arms for AAVS1. The left homology arm was shortened to fit within the scAAV size limit of encapsidation (about 2.8 kb). The 5’ AAV ITR sequence contains the trs deletion (5’-CCAACTCCATCACTAGG-3’) to avoid strand cleavage during viral genome replication, thus enabling AAV packaging in the self-complementary isoform (McCarty et al., 2003).
The engineered ssAAV2/6 transfer vector constructs contained the same payload as the AAVS1 ssAAV2/6 but with i) replacement of the 5’ ITR D sequence by a randomly designed 20 nucleotides lacking a RBE site, thus allowing exclusive encapsidation of the positive DNA strand. (Zhou et al., 2008) for the spAAV2/6 construct, or ii) deletions of B-B’ and/or C-C’ sequences at both 3’ and 5’ AAV ITRs for the ssAAV2/6-ITRdBC and ssAAV2/6-ITRdC constructs (Figure S2P).
The IDLV transfer vector construct for AAVS1 editing was previously reported (Ferrari et al., 2020) and is schematized in Figures 5C and S6D.
Vector maps were designed with SnapGene software v.6.0.2 (from GSL Biotech, available at snapgene.com).
AAV production and quality controls
Most recombinant AAV2/6 and the ssAAV5/6 were produced at the Vector Core of the UMR1089 (CPV, INSERM, University of Nantes). For ssAAV2/6 and spAAV2/6, the pDP6 helper plasmid (Grimm et al., 2003) was used. For ssAAV5/6, a construct (named Rep5/Cap6-Ad) was generated to encompass: the full wild-type AAV5 rep (GenBank ID NC_006152.1, cloned in place of the AAV2 rep in the pDP6), the AAV6 cap gene and the adenoviral helper functions (i.e., E2A, VA RNA, and E4 Ad). For ssAAV2/6-dVP1, a construct (named Rep2/Cap6-dVP1) was generated by mutating the ATG VP1 start codon in TGA to avoid VP1 translation initiation(François et al., 2018). ITR-deleted AAVs were produced at lower yields.
HEK293 cells were seeded in Cell-Stacks 5 and transfected with the two plasmids by calcium phosphate precipitation method. Cell pellets were harvested after 3 d; then, a freeze/thaw action and benzonase treatment were performed to lyse the cells and release AAV particles, which were precipitated by polyethylene glycol and purified by double CsCl density gradient ultracentrifugation, unless otherwise specified. Instead, for experiments in Figures S1K–S1O, AAV purification was performed by affinity chromatography (AVB). AAV were formulated in 1X DPBS (Thermo Scientific, Illkirch, France), sterile filtered (0.22 μm), aliquoted and frozen at -80°C. Vector genome titers (vg ml-1) were determined using a qPCR assay specific for ITR (Aurnhammer et al., 2012; D’Costa et al., 2016) or the payload.
Empty and full ssAAV2/6 particles fractionated by CsCl gradient, and ssAAV2/6 purified by AVB affinity chromatography, were analyzed by analytical ultracentrifugation (AUC) (Figure S1I; Table S1).
For SDS-PAGE analysis of AAV preparations, vectors were denatured for 5 min at 95°C in Laemmli buffer and loaded on 10% Tris-Glycine polyacrylamide gels (Life Technologies). Following electrophoresis, gels were transferred on nitrocellulose membranes for western blot analysis. Membranes were probed with monoclonal antibody B1 (Kleinschmidt), which recognizes VP1, VP2 and VP3 capsid proteins. Goat anti-mouse-HRP secondary antibody was used (Dako, P0447).
Dot blot to assess the polarity aspect of both ssAAV2/6 and spAAV2/6 were performed. Briefly, DNA was extracted by phenol-chloroform method from 3μl of purified AAV and dosed. 1 μg and 0.1 μg of DNA were loaded on a Zeta-probe membrane (Bio-Rad) using a Bio-Dot apparatus (Bio-Rad). After UV cross-linking, membranes were blocked and hybridized using DIG Easy Hyb, DIG Wash and Block buffer (Roche). Probes targeting the GFP sequence were used at a final concentration of 100 ng/μl to detect the AAV negative (5’-/5DigN/AGGTGAACTTCAAGATCCGCCACAACATCG/3Dig_N/-3’) or positive strand (5’-/5DigN/CGATGTTGTGGCGGATCTTGAAGTTCACCT/3Dig_N/-3). Membranes were washed according to manufacturer’s protocol. Anti- Digoxigenin (DIG)-AP antibodies were used at a 1:5,000 dilution (Figure S2Q).
Infectious Center Assays (ICA) were performed for AAV2/6 preparations, as previously described (Zolotukhin et al., 1999). Briefly, HeLa RC32 cells were seeded in 48-well plates at 6x104 cells/well and transduced the day after in duplicate by adding 10-fold dilutions of the AAV preparations, in presence of wild-type Ad5 at MOI of 500 transducing units (TU)/cell. Cells were harvested 24-26 h post-infection and filtered through Zeta-Probe nylon membranes (Bio-Rad) using a vacuum device. Membranes were hybridized overnight with vector-specific probes generated with the PCR Fluorescein Labeling Mix (Sigma-Aldrich), and detection was performed using the CDP-Star labeling kit (Sigma-Aldrich). Titers were determined by counting dots (i.e., AAV-infected cells) on membrane autoradiography.
IDLV production and quality controls
IDLVs were manufactured by transient quadri-transfection of HEK293T cells, followed by DNase treatment, anion exchange chromatography, concentration, gel filtration and final sterilizing filtration; the purification workflow was previously optimized in order to remove >99% of DNA and protein impurities while preserving vector biological activity (Soldi et al., 2020). HEK293T cells were seeded in 6 ten-tray cell factories (Corning) and transiently transfected in the presence of calcium phosphate with the following plasmids: the transfer vector construct described above and in(Ferrari et al., 2020), the envelope plasmid encoding for VSV.G, the third-generation packaging plasmids pRSV.REV and pGag-Pol pMDLg/pRRE.D64VInt encoding for a catalytically inactive integrase (Lombardo et al., 2007). In addition, the pAdvantage plasmid was used (Nature Technologies). After 14 h, the transfection mixture was removed and replaced by fresh medium supplemented with 1 mM Sodium Butyrate (Sigma). After 30 h, 6 l of culture supernatant were harvested, clarified through 0.8-0.45 μm filtration (Sartorius) and treated with benzonase (Merck) at 16 U ml-1 final concentration for 4 h at 4 °C. Anion exchange chromatography was performed using the AKTA Avant 150 system (Cytiva): IDLV particles were loaded on a column containing Toyopearl DEAE-650C resin (Tosoh) and eluted by DPBS/NaCl linear gradient. The eluted vector was diluted in DPBS, further treated with benzonase at 50 U mL-1 for 2 h at 4 °C and concentrated through a MWCO 100 kDa VivaFlow cassette (Sartorius). Gel filtration was performed using the AKTA Avant 150 system and a column filled with Sepharose 6FF resin (Cytiva); the IDLV was eluted in DPBS, sterilized by filtration using 0.2 μm polyethersulphone filter (Sartorius), concentrated by MWCO 100 kDa Vivaspin (Sartorius) obtaining approximately a final volume of 2.3 ml, aliquoted and stored at -80°C.
The infectious titer was determined as previously described (Soldi et al., 2020) with minor modifications. HEK293T cells were transduced with serial dilutions of the purified IDLV in the presence of polybrene; after 3 d, the cells were collected, the DNA extracted and the CG determined by ddPCR, using primers described previously (Mátrai et al., 2011) and human TELO as normalizer. The infectious titer was expressed as TU ml-1 and calculated as: CG x number of cells x (1/dilution factor). As positive control a CEM cell line stably carrying four vector integrants was used. The physical titer was measured by HIV-1 Gag 24 antigen immunocapture assay (Perkin Elmer) following manufacturer’s instructions. IDLV specific infectivity was calculated as ratio between the infectious titer and physical titer. The total particles concentration and aggregation were measured by multi-angle dynamic light scattering (MADLS) technology using Zetasizer Ultra (Malvern Panalytical) following manufacturer’s instructions. The endotoxin level was determined by LAL kinetic chromogenic method, using the Endosafe® PTS™ system and a single use cartridge with sensitivity of 0.005-0.5 EU ml-1 (Charles River).
The results of analytical tests conducted on the IDLV preparation used throughout the present study were the following: infectious titer = 3.1x109 TU ml-1; physical titer = 47.0 p24 mg ml-1; infectivity = 6.7x104 TU/p24 ng; total particles concentration = 4.75x1011 pp ml-1; aggregates = 1.3 %; endotoxin = 11.7 EU/108 TU.
mRNA in vitro transcription
All constructs for mRNA in vitro transcription and the methods for their preparation, quantification and quality assessment were previously described (Ferrari et al., 2020; Schiroli et al., 2019).
Gene editing of human HSPCs and analyses
Gene editing protocols for human HSPCs have been previously described in detail (Ferrari et al., 2021b) and is shown in Figures 1A and S6I. Briefly, for AAV-based gene editing, after 3 d of stimulation 1x105–5x105 cells were washed with ten volumes of DPBS and electroporated using P3 Primary Cell 4D-Nucleofector X Kit and Nucleofector 4D device (program EO-100) (Lonza). Cells were electroporated according to the manufacturer’s instructions with RNPs at a final concentration of 1.25–2.5 μM together with 0.1 nmol of Alt-R Cas9 Electroporation Enhancer (Integrated DNA Technologies) only for two-parts gRNAs. AAV transduction was performed 15 min after electroporation at a dose of 2x104 vg/cell, unless otherwise specified.
For one-hit IDLV-based gene editing, after 2 or 2.5 d of stimulation 1x105–5x105 cells were treated with 8 μM cyclosporin H (CsH, Sigma) and then transduced with purified IDLV at MOI of 150, unless otherwise specified. After 24 or 12 h, cells were washed with DPBS and electroporated as described above. For two-hits IDLV-based gene editing, another round of transduction in presence of 8 μM CsH was performed immediately after electroporation with purified IDLV at MOI of 150, unless otherwise specified.
When indicated, in vitro transcribed mRNAs were added to the electroporation mixture at the following final concentrations: 150 μg/μl GSE56; 250 μg/μl GSE56/E4orf6/7; 100 μg/μl E4orf6; 150 μg/μl E1B55K. Four days after the editing procedure, cells were collected to analyze by flow cytometry the percentage of cells expressing the GFP marker within HSPC subpopulations and to extract genomic (g)DNA for molecular analyses, unless otherwise indicated.
Flow cytometry
Immunophenotypic analyses were performed on the fluorescence activated cell sorting (FACS) Canto II (BD Pharmingen). From 0.5x105 to 2x105 cells (either from culture or mouse samples) were analyzed by flow cytometry. Cells were stained for 15 min at 4°C with antibodies listed in the key resources table in a final volume of 100 μl and then washed with DPBS + 2% heat-inactivated FBS. Single stained and fluorescence-minus-one-stained cells were used as controls. The Live/Dead Fixable Dead Cell Stain Kit (Thermo Fisher) or 7-aminoactinomycin D (Sigma Aldrich or Biolegend) were included during sample preparation according to the manufacturer’s instructions to identify dead cells. Gating strategies for flow cytometry analyses are provided in Figure S7.
Cell sorting was performed on a BD FACSAria Fusion (BD Biosciences) using BDFACS Diva software and equipped with four lasers: blue (488 nm), yellow/green (561 nm), red (640 nm) and violet (405 nm). Cells were sorted with an 85 mm nozzle. Sheath fluid pressure was set at 45 psi. A highly pure sorting modality (four-way purity sorting) was chosen. Sorted cells were collected in 1.5 ml Eppendorf tubes containing 500 μl of DPBS or HSPC medium. Data were analyzed with FCS Express 6 Flow or 7 Flow.
Quantification of NHEJ and HDR editing efficiency
gDNA was isolated with QIAamp DNA Micro Kit (QIAGEN) according to the manufacturer’s instructions. Unless otherwise specified, nuclease activity was measured using a mismatch-sensitive endonuclease T7 assay (New England Biolabs) on PCR-based amplification products of the targeted locus, as previously described (Ferrari et al., 2021b; Schiroli et al., 2017). Digested DNA fragments were resolved and quantified by capillary electrophoresis on 4200 TapeStation System (Agilent) according to the manufacturer’s instructions. In the other cases, NHEJ efficiency was quantified as the percentage of alleles containing indels from deep-sequencing data (see below).
For HDR ddPCR analysis, 5–50 ng of gDNA were analyzed using the QX200 Droplet Digital PCR System (Bio-Rad) according to the manufacturer’s instructions. HDR ddPCR primers and probes were designed on the junction between the vector sequence and the targeted locus, as shown in Figures 2D and S6D and previously described (Ferrari et al., 2020, Ferrari et al., 2021b; Vavassori et al., 2021). Human TTC5 (Bio-Rad) was used as normalizer.
Primers and probes for NHEJ and HDR editing quantification are listed in Data Table S3.
Gene expression analyses
Total RNA was extracted using RNeasy Plus Micro Kit (QIAGEN), according to the manufacturer’s instructions and DNAse treatment was performed using RNase-free DNAse Set (QIAGEN). Complementary DNA was synthesized with SuperScript VILO IV cDNA Synthesis Kit (Thermo Fisher) with EzDNAse treatment. cDNA was then used for quantitative PCR (qPCR) in a Viia7 Real-time PCR thermal cycler using TaqMan Gene Expression Assays (Applied Biosystems) mapping to genes listed in Data Table S3. Data were analyzed with QuantStudio Real-Time PCR software v.1.1 (Applied Biosystem). The Ct value considered for each sample was calculated as mean of the two/three technical replicates performed. Relative expression of each target gene (Ct) was first normalized to HPRT1 and then represented as fold changes of the ΔΔCt relative to the untreated sample.
Clonogenic assay
Colony-forming-unit cell assay was performed 1 d after editing, unless otherwise specified, by plating in three technical replicate 600 HSPCs/each in methylcellulose-based medium (MethoCult H4434, STEMCELL Technologies) supplemented with 100 IU ml−1 penicillin and 100 μg ml−1 streptomycin. For the experiment in Figure 6O, CD34+GFP+ cells were FACS-sorted from BM cells of xenotransplanted mice, and 1200 cells were plated as described above. Two weeks after plating, colonies were counted and classified according to morphological criteria as erythroid or myeloid. The mean of the three technical replicates was calculated and considered as an individual biological replicate. For experiments in Figures 6G–6I, 6O, S6E, S6G, and S6H, single colonies were manually picked and analyzed for CG quantification as described below.
Immunofluorescence analysis
Multitest slides (MP Biomedicals, 096041505) were coated with Poly-L-lysine solution (Sigma-Aldrich, P8920-500ML). After three washes with PBS solution, 0.3-0.5x105 cells were seeded on covers for 20 min and fixed with 4% PFA (Santa Cruz Biotechnology, sc-281692) for 20 min. Cells were then permeabilized with 0.1% Triton X-100. After blocking with 0.5% BSA and 0.2% fish gelatin in DPBS, cells were stained with the indicated primary antibodies (53BP1 Antibody, Bethyl Laboratories; Anti-phospho Histone H2A.X (Ser139) Antibody, clone JBW301, Merck; NBS1 Antibody, Novus Biologicals). Cells were than washed with DPBS and incubated with Alexa Fluor 568- and 647-labeled secondary antibodies (Invitrogen/Thermo Scientific). Nuclear DNA was stained with DAPI at 0.2 μg ml-1 concentration (Sigma-Aldrich, D9542) and covers were mounted with Aqua-Poly/Mount solution (TebuBio, 18606-20) on glass slides (Bio-Optica). Fluorescent images were acquired using Leica SP5 Confocal microscopes. Quantification of DDR foci in immunofluorescence images was conducted using Cell Profiler.
CD34+ HSPC xenotransplantation experiments in NSG mice
For transplantation of CB and mPB CD34+ HSPCs, the outgrowths of 1.5x105 and 1x106 culture-initiating HSPCs, respectively, were injected intravenously 24 h after editing into sub-lethally irradiated NSG mice (150–180 cGy). The lower, limiting doses were used for CB-derived cells when aiming to better report difference in HSPC engraftment potential between protocols. Sample size for each experiment was determined by the total number of available treated cells. Mice were randomly distributed to each experimental group. Human CD45+ cell engraftment and the presence of GFP+ edited cells were monitored by serial collection of blood from the mouse tail or the retro-orbital plexus. At the end of the experiment (>18 weeks after transplantation for CB HSPCs and ≥14 weeks for mPB HSPCs after transplantation), mice were euthanized and blood, BM and spleen were collected for end-point analyses.
Quantification of viral vectors copies per human genome
gDNA was isolated with QIAamp DNA Micro Kit from DNAse-treated in vitro culture cells pellet and in vivo samples, or with QuickExtract (Epicentre) from single colonies according to the manufacturer’s instructions. For ddPCR analyses, 5–50 ng of gDNA for in vitro/vivo samples and ≥2 μl of gDNA for each assay for single colonies were analyzed using the QX200 Droplet Digital PCR System according to the manufacturer’s instructions. Primers and probes were designed as shown in Figures 2D and S6D and are listed in Data Table S3. Human TTC5 (Bio-Rad) was used for normalization, except for experiments in Figures 2E and 6F in which GAPDH (Bio-Rad) was used. To increase robustness and reduce variability, single colonies not reaching 300 normalizer-positive droplets were excluded from the analyses. For intracellular CG quantification in Figures 2E and 6F, cells edited in presence of heat-inactivated AAV or IDLV, respectively, were used as controls for each condition and their CG values subtracted to the paired treatment samples to account for the low but detectable signal coming from extracellular cell-associated viral DNA. For the same reason, cell pellets were pre-treated with DNase (QIAGEN) before extraction.
Retrieval of AAV and IDLV integration sites: library preparation and bioinformatic analyses by RAAVIoli pipeline
To assess AAV or IDLV integration, we adopted a Sonication-based Linker-Mediated PCR method (SLiM), as previously described (Cesana et al., 2021; Gentner et al., 2021). Briefly, genomic DNA was sheared using a Covaris E220 Ultrasonicator (Covaris Inc.), generating fragments with a target size of 1,000 bp. The fragmented DNA was subjected to end repair, 3’ adenylation and ligation (NEBNext® Ultra™ DNA Library Prep Kit for Illumina®, New England Biolabs) to custom linker cassettes (LC) (Integrated DNA Technologies). LC sequences contain an 8-nucleotide barcode for sample identification. Ligation products were subjected to 35 cycles of exponential PCR with primers (available upon request) complementary to different regions of the AAV or IDLV genomes (Figures 4A and 5C) and to the LC. For each set of AAV or IDLV specific primers, the procedure was performed in technical replicates (n = 2-3) using 80-100 ng of sheared DNA each. Next, ten additional PCR cycles were done to include sequences required for sequencing and a second 8-nucleotide DNA barcode. PCR products were quantified by qPCR using the Kapa Biosystems Library Quantification Kit for Illumina, following the manufacturer’s instructions. qPCR was performed in triplicate on each PCR product diluted 10:3, and the concentrations were calculated by plotting the average Ct values against the provided standard curve. Finally, the amplification products were sequenced by Illumina Next/Novaseq platforms (Illumina).
After sequencing, a dedicated bioinformatics pipeline was developed to analyze the amplified sequences for IS identification. Briefly, the approach of the pipeline is to identify all the different sub-sequences belonging to the vector or the target reference genome (such as human hg19) from each input raw read (cleaned by barcodes and amplicon sequences) and to precisely recognize the vector-target genome junction and the integration locus. Once the pipeline aligned each paired-end read with Burrows-Wheeler aligner - Maximal Exact Match (BWA-MEM), all the resulting alignments are parsed to identify the vector junction and the integration point even in the presence of insertions or deletions between them. To parse the alignments, we used the information embedded within the CIGAR (Concise Idiosyncratic Gapped Alignment Report) string (the compressed report of the alignment result). This step returns the list of all identified chimeric reads. Specific details of the pipelines will be reported in a follow-up methodological paper. Here are reported the main steps of the pipeline: i) quality checks of input sequences were run using FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) while adapters and PhiX reads were removed with Flexbar (Dodt et al., 2012), and the initial 12 random nucleotides with Trimmomatic (Bolger et al., 2014); ii) sequences were then aligned to the AAV genome to identify the AAV or IDLV portions using BWA-MEM (Li and Durbin, 2009); iii) among AAV and IDLV containing sequences, we selected only those starting with the sequence of the last nucleotides adopted for PCR amplification plus 10 vector-specific nucleotides; iv) we then aligned the selected reads on a hybrid genome composed of both AAV and human genome (release hg19/GRCh37 downloaded from UCSC Genome Browser website). The alignments were then processed with a custom Python software to identify integration loci and vector rearrangements using the CIGAR string (Figure S4A). Three different types of sequencing reads containing the IS are identified by the pipeline: i) reads characterized by a precise homology breakpoint (± 2 nucleotides) between the integrated vector and the host chromosomal sequences; ii) reads with a microhomology (MHoR) sequence between the vector and the host chromosomal sequences; and iii) reads containing an insertion of nucleotides between the vector and the host chromosomal sequences. These criteria agree with previous works showing that short stretches of nucleotides that unambiguously mapped to the provirus and the host genome or had a random origin can be present at AAV vector-host genome junctions (examples are shown in Figure S4G) (Miller et al., 2004; Nakai et al., 2003, Nakai et al., 2005). Unique IS were identified considering the AAV/human genome breakpoint and the number and type of AAV rearrangements, such that two reads of the same PCR were assigned to the same IS if both alignments on human genome and the AAV junctions are aligned within a window of 10 bases. Moreover, potential indels in between AAV junction and genomic locus were included in the identification window to distinguish one or two IS. To remove potential PCR artefacts, all analyses were performed considering integration events identified by at least 3 independent fragments of DNA. We applied this approach to source-aligned reads and the resulting table with all independent IS from AAV and IDLV reads are available in GEO (GSE197388). The abundance of each IS was performed by counting for the same IS the number of different DNA fragments containing a genomic segment variable in size depending on the shear site position and that will be unique for each different cell genome contained in the starting cell population. Therefore, the number of different shear sites assigned to an IS will be proportional to the initial number of contributing cells in the population, thus the clonal abundance of each IS in the starting sample does not consider the biases introduced by PCR amplification (Cesana et al., 2021).
Deep sequencing of the target locus and bioinformatic analyses
PCR amplicons for individual samples were generated by nested PCR using primers listed in Data Table S3 and starting from >50-100 ng of purified gDNA. The first PCR step was performed with GoTaq G2 DNA Polymerase (Promega) according to manufacturer instruction using the following amplification protocol: 95°C x 5’min, (95°C x 0.5 min, 60°C x 0.5 min, 72°C x 0.2 5min) x 20 cycles, 72°C x 5 min. The second PCR step was performed with GoTaq G2 DNA Polymerase (Promega) according to manufacturer instruction using 5μl of the first-step PCR product and the following amplification protocol: 95°C x 5min, (95°C x 0.5 min, 60°C x 0.5 min, 72°C x 0.3 min) x 20 cycles, 72°C x 5 min. Second-step PCR primers were endowed with tails containing P5/P7 sequences, i5/i7 Illumina tags to allow multiplexed sequencing and R1/R2 primer binding sites (Data Table S3). PCR amplicons were separately purified performing double-side selection with AmpPure XP beads (Beckman Coulter) or QIAquick PCR Purification Kit (QIAGEN). Concentration and quality of amplicons were assessed by QuantiFluor ONE dsDNA system and 4200 Tapestation System (Agilent). Amplicons from up to 36 differently tagged samples were multiplexed at equimolar ratios and run by the Center for Omic Sciences (COSR) at San Raffaele or by Genewiz (Azenta Life Sciences) on MiSeq 2x300bp paired end sequencing (Illumina).
Sequencing data were analyzed with CRISPResso2 (Clement et al., 2019), which enables detection and quantification of insertions, mutations, and deletions in reads from gene editing experiments. In details, for each sample, input NGS reads were trimmed using the Trimmomatic software (http://www.usadellab.org/cms/?page=trimmomatic) based on the phred33 score to get rid of low-quality positions (score < 30) and to remove Illumina adapters, keeping only trimmed sequences longer than 100 bp (CRISPResso2 options: --trim_sequences --trimmomatic_command trimmomatic --trimmomatic_options_string 'ILLUMINACLIP:TruSeq3-PE-2.fa:2:30:10 MINLEN:100'). Then, each couple of paired-end reads was merged using the FLASh software to produce a single contig, which was mapped to the input amplicon reference (AAV or IDLV, depending on the experiment). The gRNA sequence was provided to focus the analysis on the target region, and the quantification window was set to 1 bp per side around the cut site. Identified alleles were quantified by measuring the number of reads and their relative abundance based on total read counts. Finally, we post-processed the CRISPResso2 allele outputs by correcting all the mismatch positions outside the quantification window, which are likely to be the result of amplification/sequencing errors, and we re-quantified the total read counts and the corresponding relative abundances. Alleles showing a relative abundance lower than the false positive threshold (set at 0.2% based on UT samples) were filtered out.
For quantification and characterization of trapped fragments into CRISPResso2 alleles, we extracted insertions longer than 20 bp and with relative abundance >0.2% and we locally aligned these fragments against the viral vector genome (AAV or IDLV, depending on the experiment) with the Bowtie2 software (options: --local -L 10). Coverage of vector genome was assessed with the genomeCoverageBed utility from the bedtools suite, and the corresponding normalized abundance (LogCPM) was computed. Since deep sequencing of the edited locus drops out the alleles carrying targeted integration of the cassette, we calculate the percentage of alleles with trapping events in the total human graft of each mouse using the following formula: % alleles with trapping events measured by targeted deep sequencing x 100 / (100 - % HDR (measured by ddPCR)).
GUIDE-Seq
For GUIDE-Seq analysis 3x105 K562 cells were electroporated with 25 pmol CRISPR-Cas9 delivered as RNP and 200 pmol dsODN (Tsai et al., 2015) using SF Cell Line 4D-Nucleofector X Kit and Nucleofector 4D device (program FF-120), according to the manufacturer’s instructions. Successful dsODN integration at the on-target sites was confirmed by restriction fragment length polymorphism assay using NdeI enzyme (NEB). Library preparation was performed as in (Tsai et al., 2015). GUIDE-Seq computational analysis was performed as previously described (Schiroli et al., 2019).
Quantification and statistical analysis
The number of biologically independent samples, animals or experiments is indicated by “n”. For some experiments, different HSPC donors were pooled to account for donor-related variability and reach the number of cells needed for the analyses. Data were summarized as median with 95% CI or mean ± s.e.m. depending on data distribution. Inferential techniques were applied in presence of adequate sample sizes (n ≥ 5), otherwise only descriptive statistics are reported. Two-tail tests were performed throughout the study. The Mann-Whitney test was performed to compare two independent groups, while in presence of more than two independent groups the Kruskal-Wallis test followed by post hoc analysis using Dunn’s test was used. In presence of dependent observations, the Friedman test with Dunn’s multiple comparisons or linear mixed-effects models (LME) (Bates et al., 2015; Pinheiro and Bates, 2011) were performed. The last procedures were applied to properly account for the dependence structure among observations, by including additional (nested) random-effect terms, thus considering in the model unobservable sources of heterogeneity among experimental units. When analyzing time courses, treatment group indicator and time variables, along with their interaction, were included as covariates in the model to identify potential differences in growth dynamics of treatment groups. A random intercept model was estimated and, when necessary, nested random effects were considered (for example, to account for repeated measures of cells per mouse within experiments). Quadratic polynomial models were also specified to better capture nonlinear trends. Standard transformations (logarithm, square/cubic root, ordered quantile normalization) were applied to outcome variables before entering in the LME model in order to satisfy model regression assumptions.
Post-hoc analysis after LME was performed, considering all the pairwise comparisons of treatment groups at a fixed timepoint. P values were adjusted using Bonferroni’s correction. In all the analyses, the significance threshold was set at 0.05, while “NS” means not significant. Analyses were performed using GraphPad Prism v.9.3.1 (GraphPad) and R statistical software. Detailed results of statistical analyses are shown in Data Table S4.
Acknowledgments
We thank the San Raffaele Flow Cytometry facility and the Center for Omics Sciences; A. Auricchio and M. Doria (TIGEM and InnovaVector, Naples), V. Blouin and F. Balter (INSERM UMR1089, Nantes) for AAV; Integrated Structural Biology (Grenoble) for analytical ultracentrifugation analysis; T. Plati, C. Asperti, G. Desantis, T. Di Tomaso, M. Biffi, I. Cuccovillo, M. Radrizzani, L. Sergi, M. Soldi, R. De Marco, and L. Rudilosso for technical help; P. Genovese (DFCI, Boston) for initial input on the study; and C. Di Serio for coordinating CUSSB (Vita-Salute San Raffaele University). Some schematics were created using www.BioRender.com. This work was supported by grants to: L.N. from Telethon (TIGET grant E4), Italian Ministry of Health (PE-2016-02363691 and E-Rare-3 JTC 2017), Italian Ministry of University and Research (PRIN 2017 Prot. 20175XHBPN), EU Horizon 2020 Program (UPGRADE), and the Louis-Jeantet Foundation through the 2019 Jeantet-Collen Prize for Translational Medicine; M.P.B. from EU Horizon 2020 Program (UPGRADE); A.K.R. from ERC (ImmunoStem – 819815); E.M. from Telethon (TIGET grant B1); R.D.M. from Telethon (TIGET grant E5) and The New York Stem Cell Foundation; and A. Conti was supported by Lady Tata Memorial Trust International Award for Research in Leukemia 2020-2021. R.D.M. is a New York Stem Cell Foundation – Robertson investigator. A. Conti is an EHA and ASH-TRTH early career hematological scientist. M.L. and D. Canarutto conducted this study as partial fulfillment of their PhD.
Author contributions
S.F. and A.J. performed most research, interpreted data, and designed the study. D. Cesana, C.C., and A. Calabria performed and developed genome-wide integration studies coordinated by E.M.; M.L. generated AAV variants supervised by E.A., O.A., and M.P.-B.; S.B. performed bioinformatic analyses supervised by I.M.; A.Varesi, D. Canarutto, and V.V. performed research. A. Conti performed DDR foci analyses supervised by R.D.M.; M.C.C. performed RAG1 experiments supervised by A.Villa. G.U. performed a first set of IDLV experiments supervised by A.K.-R.; L.A. and S.E. provided technical help. C.B. and F.C. performed statistical analyses. L.N. supervised research and coordinated the work. S.F., A.J., D. Cesana., and L.N. wrote the manuscript.
Declaration of interests
L.N., A.K.-R., A.V., M.C.C., S.F., and A.J. are inventors of patents on gene editing owned and managed by the San Raffaele Scientific Institute and Telethon Foundation. L.N. is founder, quota holder, and consultant of GeneSpire, a startup company aiming to develop ex vivo gene editing and exploiting IDLV.
Published: October 6, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.stem.2022.09.001.
Supplemental information
Data and code availability
Targeted deep-sequencing and RAAVIoli data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
Raw data from main figures have been deposited at Mendeley and are publicly available as of the date of publication. DOIs are listed in the key resources table.
All original code has been deposited at GitHub and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Adikusuma F., Piltz S., Corbett M.A., Turvey M., McColl S.R., Helbig K.J., Beard M.R., Hughes J., Pomerantz R.T., Thomas P.Q. Large deletions induced by Cas9 cleavage. Nature. 2018;560:E8–E9. doi: 10.1038/s41586-018-0380-z. [DOI] [PubMed] [Google Scholar]
- Aurnhammer C., Haase M., Muether N., Hausl M., Rauschhuber C., Huber I., Nitschko H., Busch U., Sing A., Ehrhardt A., Baiker A. Universal real-time PCR for the detection and quantification of adeno-associated virus serotype 2-derived inverted terminal repeat sequences. Hum. Gene Ther. Methods. 2012;23:18–28. doi: 10.1089/hgtb.2011.034. [DOI] [PubMed] [Google Scholar]
- Bak R.O., Dever D.P., Porteus M.H. CRISPR/Cas9 genome editing in human hematopoietic stem cells. Nat. Protoc. 2018;13:358–376. doi: 10.1038/nprot.2017.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates D., Mächler M., Bolker B., Walker S. Fitting linear mixed-effects models using lme4. J. Stat. Softw. 2015;67:1–48. doi: 10.18637/jss.v067.i01. [DOI] [Google Scholar]
- Blanchette P., Kindsmüller K., Groitl P., Dallaire F., Speiseder T., Branton P.E., Dobner T. Control of mRNA export by adenovirus E4orf6 and E1B55K proteins during productive infection requires E4orf6 ubiquitin ligase activity. J. Virol. 2008;82:2642–2651. doi: 10.1128/JVI.02309-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger A.M., Lohse M., Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutin J., Rosier J., Cappellen D., Prat F., Toutain J., Pennamen P., Bouron J., Rooryck C., Merlio J.P., Lamrissi-Garcia I., et al. CRISPR-Cas9 globin editing can induce megabase-scale copy-neutral losses of heterozygosity in hematopoietic cells. Nat. Commun. 2021;12:4922. doi: 10.1038/s41467-021-25190-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breton C., Clark P.M., Wang L., Greig J.A., Wilson J.M. ITR-Seq, a next-generation sequencing assay, identifies genome-wide DNA editing sites in vivo following adeno-associated viral vector-mediated genome editing. BMC Genomics. 2020;21:239. doi: 10.1186/s12864-020-6655-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukovsky A.A., Song J.P., Naldini L. Interaction of human immunodeficiency virus-derived vectors with wild-type virus in transduced cells. J. Virol. 1999;73:7087–7092. doi: 10.1128/JVI.73.8.7087-7092.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervelli T., Palacios J.A., Zentilin L., Mano M., Schwartz R.A., Weitzman M.D., Giacca M. Processing of recombinant AAV genomes occurs in specific nuclear structures that overlap with foci of DNA-damage-response proteins. J. Cell Sci. 2008;121:349–357. doi: 10.1242/jcs.003632. [DOI] [PubMed] [Google Scholar]
- Cesana D., Calabria A., Rudilosso L., Gallina P., Benedicenti F., Spinozzi G., Schiroli G., Magnani A., Acquati S., Fumagalli F., et al. Retrieval of vector integration sites from cell-free DNA. Nat. Med. 2021;27:1458–1470. doi: 10.1038/s41591-021-01389-4. [DOI] [PubMed] [Google Scholar]
- Chadeuf G., Ciron C., Moullier P., Salvetti A. Evidence for encapsidation of prokaryotic sequences during recombinant adeno-associated virus production and their in vivo persistence after vector delivery. Mol. Ther. 2005;12:744–753. doi: 10.1016/j.ymthe.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Chu V.T., Weber T., Wefers B., Wurst W., Sander S., Rajewsky K., Kühn R. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat. Biotechnol. 2015;33:543–548. doi: 10.1038/nbt.3198. [DOI] [PubMed] [Google Scholar]
- Clement K., Rees H., Canver M.C., Gehrke J.M., Farouni R., Hsu J.Y., Cole M.A., Liu D.R., Joung J.K., Bauer D.E., Pinello L. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol. 2019;37:224–226. doi: 10.1038/s41587-019-0032-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colella P., Ronzitti G., Mingozzi F. Emerging issues in AAV-mediated in vivo gene therapy. Mol. Ther. Methods Clin. Dev. 2018;8:87–104. doi: 10.1016/j.omtm.2017.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Costa S., Blouin V., Broucque F., Penaud-Budloo M., François A., Perez I.C., le Bec C., Moullier P., Snyder R.O., Ayuso E. Practical utilization of recombinant AAV vector reference standards: focus on vector genomes titration by free ITR qPCR. Mol. Ther. Methods Clin. Dev. 2016;5:16019. doi: 10.1038/mtm.2016.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalwadi D.A., Calabria A., Tiyaboonchai A., Posey J., Naugler W.E., Montini E., Grompe M. AAV integration in human hepatocytes. Mol. Ther. 2021;29:2898–2909. doi: 10.1016/j.ymthe.2021.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalwadi D.A., Torrens L., Abril-Fornaguera J., Pinyol R., Willoughby C., Posey J., Llovet J.M., Lanciault C., Russell D.W., Grompe M., Naugler W.E. Liver injury increases the incidence of HCC following AAV gene therapy in mice. Mol. Ther. 2021;29:680–690. doi: 10.1016/j.ymthe.2020.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dever D.P., Bak R.O., Reinisch A., Camarena J., Washington G., Nicolas C.E., Pavel-Dinu M., Saxena N., Wilkens A.B., Mantri S., et al. CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nature. 2016;539:384–389. doi: 10.1038/nature20134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- di Micco R., Krizhanovsky V., Baker D., d’Adda di Fagagna F. Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 2021;22:75–95. doi: 10.1038/s41580-020-00314-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodt M., Roehr J.T., Ahmed R., Dieterich C. FLEXBAR-flexible barcode and adapter processing for next-generation sequencing platforms. Biology. 2012;1:895–905. doi: 10.3390/biology1030895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doudna J.A. The promise and challenge of therapeutic genome editing. Nature. 2020;578:229–236. doi: 10.1038/s41586-020-1978-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earley L.F., Conatser L.M., Lue V.M., Dobbins A.L., Li C., Hirsch M.L., Samulski R.J. Adeno-associated virus serotype-specific inverted terminal repeat sequence role in vector transgene expression. Hum. Gene Ther. 2020;31:151–162. doi: 10.1089/hum.2019.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferla R., Alliegro M., Dell’Anno M., Nusco E., Cullen J.M., Smith S.N., Wolfsberg T.G., O’Donnell P., Wang P., Nguyen A.-D., et al. Low incidence of hepatocellular carcinoma in mice and cats treated with systemic adeno-associated viral vectors. Mol. Ther. Methods Clin. Dev. 2021;20:247–257. doi: 10.1016/j.omtm.2020.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari G., Thrasher A.J., Aiuti A. Gene therapy using haematopoietic stem and progenitor cells. Nat. Rev. Genet. 2021;22:216–234. doi: 10.1038/s41576-020-00298-5. [DOI] [PubMed] [Google Scholar]
- Ferrari S., Beretta S., Jacob A., Cittaro D., Albano L., Merelli I., Naldini L., Genovese P. BAR-Seq clonal tracking of gene-edited cells. Nat. Protoc. 2021;16:2991–3025. doi: 10.1038/s41596-021-00529-x. [DOI] [PubMed] [Google Scholar]
- Ferrari S., Jacob A., Beretta S., Unali G., Albano L., Vavassori V., Cittaro D., Lazarevic D., Brombin C., Cugnata F., et al. Efficient gene editing of human long-term hematopoietic stem cells validated by clonal tracking. Nat. Biotechnol. 2020;38:1298–1308. doi: 10.1038/s41587-020-0551-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Food and Drug Administration . Toxicity risks of adeno-associated virus (AAV) vectors for gene therapy (GT) https://www.fda.gov/media/151599/download; 2021. Cellular, T. and G.T.A.C. (CTGTAC) M. #70. [Google Scholar]
- François A., Bouzelha M., Lecomte E., Broucque F., Penaud-Budloo M., Adjali O., Moullier P., Blouin V., Ayuso E. Accurate titration of infectious AAV particles requires measurement of biologically active vector genomes and suitable controls. Mol. Ther. Methods Clin. Dev. 2018;10:223–236. doi: 10.1016/j.omtm.2018.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel R., Lombardo A., Arens A., Miller J.C., Genovese P., Kaeppel C., Nowrouzi A., Bartholomae C.C., Wang J., Friedman G., et al. An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nat. Biotechnol. 2011;29:816–823. doi: 10.1038/nbt.1948. [DOI] [PubMed] [Google Scholar]
- Gao G., Vandenberghe L.H., Alvira M.R., Lu Y., Calcedo R., Zhou X., Wilson J.M. Clades of adeno-associated viruses are widely disseminated in human tissues. J. Virol. 2004;78:6381–6388. doi: 10.1128/JVI.78.12.6381-6388.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudelli N.M., Lam D.K., Rees H.A., Solá-Esteves N.M., Barrera L.A., Born D.A., Edwards A., Gehrke J.M., Lee S.-J., Liquori A.J., et al. Directed evolution of adenine base editors with increased activity and therapeutic application. Nat. Biotechnol. 2020;38:892–900. doi: 10.1038/s41587-020-0491-6. [DOI] [PubMed] [Google Scholar]
- Genovese P., Schiroli G., Escobar G., di Tomaso T.D., Firrito C., Calabria A., Moi D., Mazzieri R., Bonini C., Holmes M.C., et al. Targeted genome editing in human repopulating haematopoietic stem cells. Nature. 2014;510:235–240. doi: 10.1038/nature13420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentner B., Tucci F., Galimberti S., Fumagalli F., de Pellegrin M., Silvani P., Camesasca C., Pontesilli S., Darin S., Ciotti F., et al. Hematopoietic stem- and progenitor-cell gene therapy for Hurler syndrome. N. Engl. J. Med. 2021;385:1929–1940. doi: 10.1056/NEJMoa2106596. [DOI] [PubMed] [Google Scholar]
- Gil-Farina I., Fronza R., Kaeppel C., Lopez-Franco E., Ferreira V., D’Avola D., Benito A., Prieto J., Petry H., Gonzalez-Aseguinolaza G., Schmidt M. Recombinant AAV integration is not associated With hepatic genotoxicity in nonhuman Primates and patients. Mol. Ther. 2016;24:1100–1105. doi: 10.1038/mt.2016.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm D., Kay M.A., Kleinschmidt J.A. Helper virus-free, optically controllable, and two-plasmid-based production of adeno-associated virus vectors of serotypes 1 to 6. Mol. Ther. 2003;7:839–850. doi: 10.1016/S1525-0016(03)00095-9. [DOI] [PubMed] [Google Scholar]
- Haberman R.P., McCown T.J., Samulski R.J. Novel transcriptional regulatory signals in the adeno-associated virus terminal repeat A/D junction element. J. Virol. 2000;74:8732–8739. doi: 10.1128/jvi.74.18.8732-8739.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeussler M., Schönig K., Eckert H., Eschstruth A., Mianné J., Renaud J.B., Schneider-Maunoury S., Shkumatava A., Teboul L., Kent J., Concordet J.-P. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 2016;17:148. doi: 10.1186/s13059-016-1012-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanlon K.S., Kleinstiver B.P., Garcia S.P., Zaborowski M.P., Volak A., Spirig S.E., Muller A., Sousa A.A., Tsai S.Q., Bengtsson N.E., et al. High levels of AAV vector integration into CRISPR-induced DNA breaks. Nat. Commun. 2019;10:4439. doi: 10.1038/s41467-019-12449-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch M.L. Adeno-associated virus inverted terminal repeats stimulate gene editing. Gene Ther. 2015;22:190–195. doi: 10.1038/gt.2014.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard N., Hagin D., Sommer K., Song Y., Khan I., Clough C., Ochs H.D., Rawlings D.J., Scharenberg A.M., Torgerson T.R. Targeted gene editing restores regulated CD40L function in X-linked hyper-IgM syndrome. Blood. 2016;127:2513–2522. doi: 10.1182/blood-2015-11-683235. [DOI] [PubMed] [Google Scholar]
- Inagaki K., Piao C., Kotchey N.M., Wu X., Nakai H. Frequency and spectrum of genomic integration of recombinant adeno-associated virus serotype 8 vector in neonatal mouse liver. J. Virol. 2008;82:9513–9524. doi: 10.1128/JVI.01001-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeppel C., Beattie S.G., Fronza R., van Logtenstein R., Salmon F., Schmidt S., Wolf S., Nowrouzi A., Glimm H., von Kalle C., et al. A largely random AAV integration profile after LPLD gene therapy. Nat. Med. 2013;19:889–891. doi: 10.1038/nm.3230. [DOI] [PubMed] [Google Scholar]
- Keiser M.S., Ranum P.T., Yrigollen C.M., Carrell E.M., Smith G.R., Muehlmatt A.L., Chen Y.H., Stein J.M., Wolf R.L., Radaelli E., et al. Toxicity after AAV delivery of RNAi expression constructs into nonhuman primate brain. Nat. Med. 2021;27:1982–1989. doi: 10.1038/s41591-021-01522-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosicki M., Tomberg K., Bradley A. Repair of double-strand breaks induced by CRISPR–Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018;36:765–771. doi: 10.1038/nbt.4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo C.Y., Long J.D., Campo-Fernandez B., de Oliveira S., Cooper A.R., Romero Z., Hoban M.D., Joglekar A.V.v., Lill G.R., Kaufman M.L., et al. Site-specific gene editing of human hematopoietic stem cells for X-linked hyper-IgM syndrome. Cell Rep. 2018;23:2606–2616. doi: 10.1016/j.celrep.2018.04.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmin D.A., Shutova M.V., Johnston N.R., Smith O.P., Fedorin V.V., Kukushkin Y.S., van der Loo J.C.M., Johnstone E.C. The clinical landscape for AAV gene therapies. Nat. Rev. Drug Discov. 2021;20:173–174. doi: 10.1038/D41573-021-00017-7. [DOI] [PubMed] [Google Scholar]
- Lattanzi A., Camarena J., Lahiri P., Segal H., Srifa W., Vakulskas C.A., Frock R.L., Kenrick J., Lee C., Talbott N., et al. Development of β-globin gene correction in human hematopoietic stem cells as a potential durable treatment for sickle cell disease. Sci. Transl. Med. 2021;13:eabf2444. doi: 10.1126/scitranslmed.abf2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecomte E., Tournaire B., Cogné B., Dupont J.-B., Lindenbaum P., Martin-Fontaine M., Broucque F., Robin C., Hebben M., Merten O.-W., et al. Advanced characterization of DNA molecules in rAAV vector preparations by single-stranded virus next-generation sequencing. Mol. Ther. Nucleic Acids. 2015;4:e260. doi: 10.1038/mtna.2015.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leibowitz M.L., Papathanasiou S., Doerfler P.A., Blaine L.J., Sun L., Yao Y., Zhang C.Z., Weiss M.J., Pellman D. Chromothripsis as an on-target consequence of CRISPR–Cas9 genome editing. Nat. Genet. 2021;53:895–905. doi: 10.1038/s41588-021-00838-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lentz T.B., Samulski R.J. Insight into the mechanism of inhibition of adeno-associated virus by the Mre11/Rad50/Nbs1 complex. J. Virol. 2015;89:181–194. doi: 10.1128/JVI.01990-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardo A., Cesana D., Genovese P., di Stefano B., Provasi E., Colombo D.F., Neri M., Magnani Z., Cantore A., lo Riso P., et al. Site-specific integration and tailoring of cassette design for sustainable gene transfer. Nat. Methods. 2011;8:861–869. doi: 10.1038/nmeth.1674. [DOI] [PubMed] [Google Scholar]
- Lombardo A., Genovese P., Beausejour C.M., Colleoni S., Lee Y.L., Kim K.A., Ando D., Urnov F.D., Galli C., Gregory P.D., et al. Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nat. Biotechnol. 2007;25:1298–1306. doi: 10.1038/nbt1353. [DOI] [PubMed] [Google Scholar]
- Mátrai J., Cantore A., Bartholomae C.C., Annoni A., Wang W., Acosta-Sanchez A., Samara-Kuko E., de Waele L., Ma L., Genovese P., et al. Hepatocyte-targeted expression by integrase-defective lentiviral vectors induces antigen-specific tolerance in mice with low genotoxic risk. Hepatology. 2011;53:1696–1707. doi: 10.1002/hep.24230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarty D.M., Fu H., Monahan P.E., Toulson C.E., Naik P., Samulski R.J. Adeno-associated virus terminal repeat (TR) mutant generates self-complementary vectors to overcome the rate-limiting step to transduction in vivo. Gene Ther. 2003;10:2112–2118. doi: 10.1038/sj.gt.3302134. [DOI] [PubMed] [Google Scholar]
- McCarty D.M., Young S.M., Samulski R.J. Integration of adeno-associated virus (AAV) and recombinant AAV vectors. Annu. Rev. Genet. 2004;38:819–845. doi: 10.1146/annurev.genet.37.110801.143717. [DOI] [PubMed] [Google Scholar]
- Miller D.G., Petek L.M., Russell D.W. Adeno-associated virus vectors integrate at chromosome breakage sites. Nat. Genet. 2004;36:767–773. doi: 10.1038/ng1380. [DOI] [PubMed] [Google Scholar]
- Mullard A. Gene therapy community grapples with toxicity issues, as pipeline matures. Nat. Rev. Drug Discov. 2021;20:804–805. doi: 10.1038/d41573-021-00164-x. [DOI] [PubMed] [Google Scholar]
- Nahmad A.D., Reuveni E., Goldschmidt E., Tenne T., Liberman M., Horovitz-Fried M., Khosravi R., Kobo H., Reinstein E., Madi A., et al. Frequent aneuploidy in primary human T cells after CRISPR–Cas9 cleavage. Nat. Biotechnol. 2022:1–7. doi: 10.1038/s41587-022-01377-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai H., Iwaki Y., Kay M.A., Couto L.B. Isolation of recombinant adeno-associated virus vector-cellular DNA junctions from mouse liver. J. Virol. 1999;73:5438–5447. doi: 10.1128/JVI.73.7.5438-5447.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai H., Montini E., Fuess S., Storm T.A., Grompe M., Kay M.A. AAV serotype 2 vectors preferentially integrate into active genes in mice. Nat. Genet. 2003;34:297–302. doi: 10.1038/ng1179. [DOI] [PubMed] [Google Scholar]
- Nakai H., Wu X., Fuess S., Storm T.A., Munroe D., Montini E., Burgess S.M., Grompe M., Kay M.A. Large-scale molecular characterization of adeno-associated virus vector integration in mouse liver. J. Virol. 2005;79:3606–3614. doi: 10.1128/JVI.79.6.3606-3614.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naldini L. Genetic engineering of hematopoiesis: current stage of clinical translation and future perspectives. EMBO Mol. Med. 2019;11 doi: 10.15252/emmm.201809958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson C.E., Wu Y., Gemberling M.P., Oliver M.L., Waller M.A., Bohning J.D., Robinson-Hamm J.N., Bulaklak K., Castellanos Rivera R.M., Collier J.H., et al. Long-term evaluation of AAV-CRISPR genome editing for Duchenne muscular dystrophy. Nat. Med. 2019;25:427–432. doi: 10.1038/s41591-019-0344-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen G.N., Everett J.K., Kafle S., Roche A.M., Raymond H.E., Leiby J., Wood C., Assenmacher C.A., Merricks E.P., Long C.T., et al. A long-term study of AAV gene therapy in dogs with hemophilia A identifies clonal expansions of transduced liver cells. Nat. Biotechnol. 2021;39:47–55. doi: 10.1038/s41587-020-0741-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattabhi S., Lotti S.N., Berger M.P., Singh S., Lux C.T., Jacoby K., Lee C., Negre O., Scharenberg A.M., Rawlings D.J. In vivo outcome of homology-directed repair at the HBB gene in HSC Using Alternative donor template delivery methods. Mol. Ther. Nucleic Acids. 2019;17:277–288. doi: 10.1016/j.omtn.2019.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavani G., Laurent M., Fabiano A., Cantelli E., Sakkal A., Corre G., Lenting P.J., Concordet J.-P., Toueille M., Miccio A., Amendola M. Ex vivo editing of human hematopoietic stem cells for erythroid expression of therapeutic proteins. Nat. Commun. 2020;11:3778. doi: 10.1038/s41467-020-17552-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penaud-Budloo M., François A., Clément N., Ayuso E. Pharmacology of recombinant adeno-associated virus production. Mol. Ther. Methods Clin. Dev. 2018;8:166–180. doi: 10.1016/j.omtm.2018.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrillo C., Thorne L.G., Unali G., Schiroli G., Giordano A.M.S., Piras F., Cuccovillo I., Petit S.J., Ahsan F., Noursadeghi M., et al. Cyclosporine H overcomes innate immune restrictions to improve lentiviral transduction and gene editing in human hematopoietic stem cells. Cell Stem Cell. 2018;23:820–832.e9. doi: 10.1016/j.stem.2018.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinheiro J., Bates D. Linear and nonlinear mixed effects models (nlme) Package R Software Stat. Comput. CRAN Repos. 2011:0–21. [Google Scholar]
- Piras F., Riba M., Petrillo C., Lazarevic D., Cuccovillo I., Bartolaccini S., Stupka E., Gentner B., Cittaro D., Naldini L., Rudnitski A.K. Lentiviral vectors escape innate sensing but trigger p53 in human hematopoietic stem and progenitor cells. EMBO Mol. Med. 2017;9:1198–1211. doi: 10.15252/emmm.201707922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponnazhagan S., Erikson D., Kearns W.G., Zhou S.Z., Nahreini P., Wang X.S., Srivastava A. Lack of site-specific integration of the recombinant adeno-associated virus 2 genomes in human cells. Hum. Gene Ther. 1997;8:275–284. doi: 10.1089/hum.1997.8.3-275. [DOI] [PubMed] [Google Scholar]
- Rai R., Romito M., Rivers E., Turchiano G., Blattner G., Vetharoy W., Ladon D., Andrieux G., Zhang F., Zinicola M., et al. Targeted gene correction of human hematopoietic stem cells for the treatment of Wiskott - Aldrich syndrome. Nat. Commun. 2020;11:4034. doi: 10.1038/s41467-020-17626-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reginato G., Cejka P. 91–92. Amst; 2020. The MRE11 complex: A versatile toolkit for the repair of broken DNA; p. 102869. DNA Repair. [DOI] [PubMed] [Google Scholar]
- Romero Z., Lomova A., Said S., Miggelbrink A., Kuo C.Y., Campo-Fernandez B., Hoban M.D., Masiuk K.E., Clark D.N., Long J., et al. Editing the sickle cell disease mutation in human hematopoietic stem cells: comparison of endonucleases and homologous donor templates. Mol. Ther. 2019;27:1389–1406. doi: 10.1016/j.ymthe.2019.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosas L.E., Grieves J.L., Zaraspe K., la Perle K.M., Fu H., McCarty D.M. Patterns of scAAV vector insertion associated With oncogenic events in a mouse model for genotoxicity. Mol. Ther. 2012;20:2098–2110. doi: 10.1038/mt.2012.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samulski R.J., Srivastava A., Berns K.I., Muzyczka N. Rescue of adeno-associated virus from recombinant plasmids: gene correction within the terminal repeats of AAV. Cell. 1983;33:135–143. doi: 10.1016/0092-8674(83)90342-2. [DOI] [PubMed] [Google Scholar]
- San Filippo J., Sung P., Klein H. Mechanism of eukaryotic homologous recombination. Annu. Rev. Biochem. 2008;77:229–257. doi: 10.1146/annurev.biochem.77.061306.125255. [DOI] [PubMed] [Google Scholar]
- Schiroli G., Conti A., Ferrari S., della Volpe L., Jacob A., Albano L., Beretta S., Calabria A., Vavassori V., Gasparini P., et al. Precise gene editing preserves hematopoietic stem cell function following transient p53-mediated DNA damage response. Cell Stem Cell. 2019;24:551–565.e8. doi: 10.1016/j.stem.2019.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiroli G., Ferrari S., Conway A., Jacob A., Capo V., Albano L., Plati T., Castiello M.C., Sanvito F., Gennery A.R., et al. Preclinical modeling highlights the therapeutic potential of hematopoietic stem cell gene editing for correction of SCID-X1. Sci. Transl. Med. 2017;9:eaan0820. doi: 10.1126/scitranslmed.aan0820. [DOI] [PubMed] [Google Scholar]
- Schnödt M., Büning H. Improving the quality of adeno-associated viral vector preparations: the challenge of product-related impurities. Hum. Gene Ther. Methods. 2017;28:101–108. doi: 10.1089/hgtb.2016.188. [DOI] [PubMed] [Google Scholar]
- Schultz B.R., Chamberlain J.S. Recombinant adeno-associated virus transduction and integration. Mol. Ther. 2008;16:1189–1199. doi: 10.1038/MT.2008.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldi M., Sergi Sergi L., Unali G., Kerzel T., Cuccovillo I., Capasso P., Annoni A., Biffi M., Rancoita P.M.V., Cantore A., et al. Laboratory-scale lentiviral vector production and purification for enhanced ex vivo and in vivo genetic engineering. Mol. Ther. Methods Clin. Dev. 2020;19:411–425. doi: 10.1016/j.omtm.2020.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sondhi D., Kaminsky S.M., Hackett N.R., Pagovich O.E., Rosenberg J.B., De B.P., Chen A., van de Graaf B., Mezey J.G., Mammen G.W., et al. Slowing late infantile Batten disease by direct brain parenchymal administration of a rh.10 adeno-associated virus expressing CLN2. Sci. Transl. Med. 2020;12:eabb5413. doi: 10.1126/scitranslmed.abb5413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai P.W.L., Xie J., Fong K., Seetin M., Heiner C., Su Q., Weiand M., Wilmot D., Zapp M.L., Gao G. Adeno-associated virus genome population sequencing achieves full vector genome resolution and reveals human-vector chimeras. Mol. Ther. Methods Clin. Dev. 2018;9:130–141. doi: 10.1016/j.omtm.2018.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran N.T., Lecomte E., Saleun S., Namkung S., Robin C., Weber K., Devine E., Blouin V., Adjali O., Ayuso E., et al. Human and insect cell-produced recombinant adeno-associated viruses show differences in genome heterogeneity. Hum. Gene Ther. 2022;33:371–388. doi: 10.1089/hum.2022.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai S.Q., Zheng Z., Nguyen N.T., Liebers M., Topkar V.V.v., Thapar V., Wyvekens N., Khayter C., Iafrate A.J., Le L.P., et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 2015;33:187–197. doi: 10.1038/nbt.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turchiano G., Andrieux G., Klermund J., Blattner G., Pennucci V., el Gaz M., Monaco G., Poddar S., Mussolino C., Cornu T.I., et al. Quantitative evaluation of chromosomal rearrangements in gene-edited human stem cells by CAST-Seq. Cell Stem Cell. 2021;28:1136–1147.e5. doi: 10.1016/j.stem.2021.02.002. [DOI] [PubMed] [Google Scholar]
- Vavassori V., Mercuri E., Marcovecchio G.E., Castiello M.C., Schiroli G., Albano L., Margulies C., Buquicchio F., Fontana E., Beretta S., et al. Modeling, optimization, and comparable efficacy of T cell and hematopoietic stem cell gene editing for treating hyper-IgM syndrome. EMBO Mol. Med. 2021;13:e13545. doi: 10.15252/emmm.202013545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walia J.S., Altaleb N., Bello A., Kruck C., LaFave M.C., Varshney G.K., Burgess S.M., Chowdhury B., Hurlbut D., Hemming R., et al. Long-term correction of Sandhoff disease following intravenous delivery of rAAV9 to mouse neonates. Mol. Ther. 2015;23:414–422. doi: 10.1038/mt.2014.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Exline C.M., Declercq J.J., Llewellyn G.N., Hayward S.B., Li P.W.L., Shivak D.A., Surosky R.T., Gregory P.D., Holmes M.C., Cannon P.M. Homology-driven genome editing in hematopoietic stem and progenitor cells using ZFN mRNA and AAV6 donors. Nat. Biotechnol. 2015;33:1256–1263. doi: 10.1038/nbt.3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Ma H.-I., Li J., Sun L., Zhang J., Xiao X. Rapid and highly efficient transduction by double-stranded adeno-associated virus vectors in vitro and in vivo. Gene Ther. 2003;10:2105–2111. doi: 10.1038/sj.gt.3302133. [DOI] [PubMed] [Google Scholar]
- Wienert B., Wyman S.K., Richardson C.D., Yeh C.D., Akcakaya P., Porritt M.J., Morlock M., Vu J.T., Kazane K.R., Watry H.L., et al. Unbiased detection of CRISPR off-targets in vivo using DISCover-Seq. Science. 2019;364:286–289. doi: 10.1126/science.aav9023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilmott P., Lisowski L., Alexander I.E., Logan G.J. A User’s guide to the inverted terminal repeats of adeno-associated virus. Hum. Gene Ther. Methods. 2019;30:206–213. doi: 10.1089/hgtb.2019.276. [DOI] [PubMed] [Google Scholar]
- Wright J.F. Manufacturing and characterizing AAV-based vectors for use in clinical studies. Gene Ther. 2008;15:840–848. doi: 10.1038/gt.2008.65. [DOI] [PubMed] [Google Scholar]
- Zhou Q., Tian W., Liu C., Lian Z., Dong X., Wu X. Deletion of the B-B’ and C-C’ regions of inverted terminal repeats reduces rAAV productivity but increases transgene expression. Sci. Rep. 2017;7:5432. doi: 10.1038/s41598-017-04054-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X., Zeng X., Fan Z., Li C., McCown T., Samulski R.J., Xiao X. Adeno-associated virus of a single-polarity DNA genome is capable of transduction in vivo. Mol. Ther. 2008;16:494–499. doi: 10.1038/sj.mt.6300397. [DOI] [PubMed] [Google Scholar]
- Zolotukhin S., Byrne B.J., Mason E., Zolotukhin I., Potter M., Chesnut K., Summerford C., Samulski R.J., Muzyczka N. Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Ther. 1999;6:973–985. doi: 10.1038/sj.gt.3300938. [DOI] [PubMed] [Google Scholar]
- Zufferey R., Dull T., Mandel R.J., Bukovsky A., Quiroz D., Naldini L., Trono D. Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery. J. Virol. 1998;72:9873–9880. doi: 10.1128/JVI.72.12.9873-9880.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Targeted deep-sequencing and RAAVIoli data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
Raw data from main figures have been deposited at Mendeley and are publicly available as of the date of publication. DOIs are listed in the key resources table.
All original code has been deposited at GitHub and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.