

Abstract
Macrophages use an array of innate immune sensors to detect intracellular pathogens and to tailor effective antimicrobial responses. In addition, extrinsic activation with the cytokine interferon gamma (IFNγ) is often required as well to tip the scales of the host-pathogen balance toward pathogen restriction. However, little is known about how host pathogen sensing impacts the anti-microbial IFNγ-activated state. We observed that in the absence of IRF3, a key downstream component of pathogen sensing pathways, IFNγ-primed macrophages more efficiently restricted the intracellular bacterium Legionella pneumophila and the intracellular protozoan parasite Trypanosoma cruzi. This effect did not require IFNAR, the receptor for Type I interferons known to be induced by IRF3, nor the sensing adaptors MyD88/TRIF, MAVS, or STING. This effect also did not involve differential activation of STAT1, the major signaling protein downstream of both Type 1 and Type 2 interferon receptors. IRF3-deficient macrophages displayed a significantly altered IFNγ-induced gene expression program, with upregulation of microbial restriction factors such as Nos2. Finally, we found that IFNγ-primed but not unprimed macrophages largely excluded the activated form of IRF3 from the nucleus following bacterial infection. These data are consistent with a relationship of mutual inhibition between IRF3 and IFNγ-activated programs, possibly as a component of a partially reversible mechanism for modulating the activity of potent innate immune effectors (such as Nos2) in the context of intracellular infection.
Keywords: host-pathogen interactions, innate immunity, immunomodulation
Summary sentence:
IRF3 modulates multiple responses of IFNγ-primed macrophages to intracellular bacteria, including production of nitric oxide; meanwhile, IFNγ reciprocally inhibits IRF3, contributing to immune homeostasis.
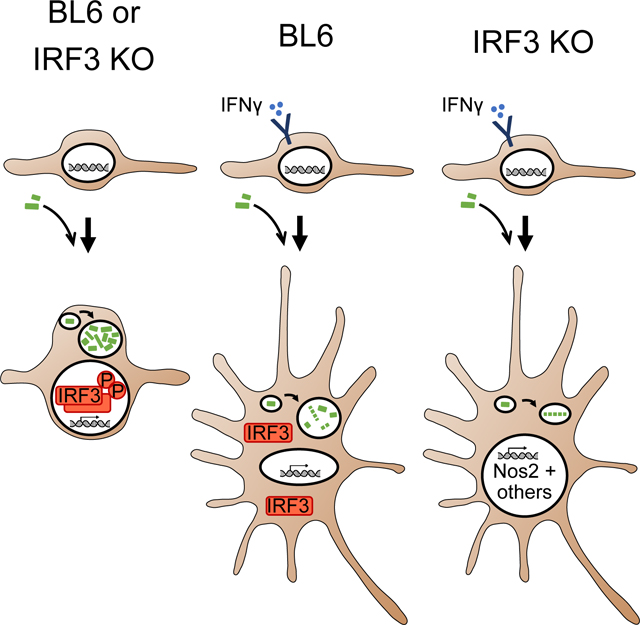
Graphical Abstract
INTRODUCTION
Interferon gamma (IFNγ) is a potent activator of macrophage defense mechanisms that restrict intracellular pathogens, such as the bacterial agents of Legionnaire’s disease, salmonellosis, tularemia, pneumonic plague, and tuberculosis, and the parasitic agents of Chagas disease, toxoplasmosis, and leishmaniasis 1,2. While macrophages depend mostly on NK, NKT, and T cells for the production of IFNγ, macrophages also collect information about infection using their own pathogen sensors that regulate cell-intrinsic defenses. Previous studies suggest a synergy between several pathogen sensing pathways and IFNγ activation in macrophages at the level of signaling and transcription 3–7. For example, IFNγ-mediated restriction of the cytosolic bacterial pathogen Shigella flexneri in fibroblasts was found to be dependent on the activity of the RIG-I/MAVS RNA sensing pathway and on the upregulation of this pathway by IRF1 8. Despite our knowledge of microbial ligands and the pathways induced when they are sensed, we know little about whether and how these innate sensing pathways affect restriction of bacteria in macrophages activated by IFNγ.
Legionella pneumophila is a Gram-negative vacuolar bacterial pathogen of unicellular amoeba that can opportunistically colonize mammalian macrophages and, in humans, lead to Legionnaire’s disease or Pontiac fever. Several innate sensing pathways are known to mediate macrophage responses to L. pneumophila 9, including inflammasome 10–12, Toll-like receptor (TLR) 13,14, and cytosolic DNA 15–17 and RNA 18 sensing pathways. Despite the vacuolar localization of L. pneumophila, there is evidence that bacterial nucleic acids and other pathogen ligands reach sensors throughout the macrophage via the specialized secretion system Dot/icm 19,20. Infected macrophages with an intact NAIP5-NLRC4 inflammasome pathway undergo rapid pyroptosis upon exposure to bacterial flagellin 21. Resting macrophages that lack elements of the NAIP5 pathway or are infected with flagellin-deficient L. pneumophila are permissive for bacterial growth 20–22. However, macrophages in this scenario become restrictive upon addition of IFNγ 23.
To test the hypothesis that innate sensing pathways interact with the IFNγ-induced anti-microbial state, we quantified IFNγ-induced restriction of intracellular bacteria in bone-marrow derived macrophages (BMMs) from mice deficient in key components of pathogen sensing pathways, including the TLR, DNA- and RNA-sensing pathways. We found a strong increase in restriction of L. pneumophila in IFNγ-primed BMMs deficient in the key sensing pathway protein IRF3. This restriction is independent of Type I IFN signaling through IFNAR, as well as of signaling through MAVS, STING, and MyD88/TRIF. We then sought to determine how IRF3 modifies the anti-bacterial state, and in turn, to determine whether its activity is subject to reciprocal control by IFNγ.
MATERIALS AND METHODS
Ethics Statement
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. Mice were handled according to all applicable institutional, state, and federal animal care guidelines, under animal care protocols approved by the Massachusetts General Hospital Animal Care and Use Committee (animal welfare assurance # A3596–01, protocol # 2003N000284).
Mice
IRF3-deficient mice (intrinsically deficient in expression of the critically overlapping gene Bcl2L12, and therefore designated Irf3−/−/Bcl2L12−/− mice24) were provided by Dr. Meixiong Wu with permission from Dr. Tadatsugu Taniguchi. Irf7−/− mice were provided by Dr. Evelyn Kurt-Jones. Irf3−/−/Bcl2L12−/−/Irf7−/− and Ifnar−/− mice were provided by Dr. Kate Fitzgerald. Sting−/− mice were provided by Dr. Glen Barber. Bone marrow derived from Mavs−/− mice was provided by Dr. Akiko Iwasaki. Myd88−/−/Trif−/− mice were provided by Dr. Ruslan Medzhitov. Age and sex-matched C57BL/6J mice were obtained from Jackson laboratories.
BMM isolation and culture
Bone marrow was collected from femurs and tibiae of 2–6 month old mice. Red blood cells were lysed using TAC RBC lysis buffer (Sigma). Cells were passed through a 70 μm cell strainer and plated on non-tissue culture treated petri dishes in RPMI-1640 medium, supplemented with 10% FBS, L-glutamine, penicillin/streptomycin, MEM nonessential amino acids, HEPES, sodium pyruvate, β-mercaptoethanol, heat-inactivated FBS, and human MCSF (10ng/ml, R&D Systems). BMMs were collected by pipetting and reseeded into assay plates in supplemented media without penicillin/streptomycin after 5 days of differentiation, and stimulated or infected after 7–12 days of differentiation. BMMs were seeded in 96-well tissue culture-treated assay plates at 4×104-5×104 cells/well, unless otherwise noted. BMMs were primed overnight for 16–24 hours, unless otherwise noted. Cytokines used to stimulate BMMs were from Millipore (IFNγ) or Peprotech (TNFα). Nos2 inhibitors (L-NOARG, L-NIL, 1400W) were from Sigma.
Bacterial strains and parasites
The bioluminescent L. pneumophila strain LP02 ahpC::lux ΔflaA was previously described 25. T. cruzi strain CL-Brener trypomastigotes were provided by Dr. Ricardo Gazzinelli.
L. pneumophila and T. cruzi infections and quantification
L. pneumophila strains were maintained on N-(2-acetamido)-2-aminoethanesulfonic acid (ACES) buffered charcoal-yeast extract agar supplemented with FeNO3, cysteine, and thymidine. For experimental assays, L. pneumophila was grown in ACES-buffered yeast extract broth at 37°C to a density greater than 3 OD600. Bacteria were washed with PBS twice before infection, then resuspended in fresh BMM media. BMMs were infected at a moiety of infection (MOI) of 4 by replacing cell culture media with bacterial suspension as above. Infected BMMs were centrifuged for 10 minutes at 1400rpm in an Allegra X15-R centrifuge (Beckman Coulter), then incubated for two hours in a cell culture incubator. Media was then removed and replaced with fresh supplemented BMM media containing thymidine. Bioluminescence was measured over two days after infection using the Envision Multilabel Reader (Perkin Elmer).
Trypomastigote forms of T. cruzi CL-Brener were maintained in LLC-MK2 cells as described 26 and harvested from supernatants. For T. cruzi infection, IFNAR-blocking antibody (Leinco) was included in cell media starting at one day prior to infection. BMMs were infected with purified trypomastigotes at an MOI of 5 by replacing media with a suspension of parasites in media, and incubated for two hours in a cell culture incubator. BMMs were washed three times with warm PBS, and media was removed and replaced with fresh BMM media containing cytokines and IFNAR-blocking antibody at 2hpi as well as daily from 3dpi onward. For extracellular parasite quantification, well contents were agitated briefly to resuspend trypomastigotes, 20ul of supernatant was applied to a Neubauer hemocytometer, and motile trypomastigotes were counted manually. For intracellular parasite quantification, infected macrophages were washed three times with PBS, fixed with 4% paraformaldehyde for 10 minutes, permeabilized with 0.25% Triton-X, stained with 1ug/ml 4’,6-diamidino-2-phenylindole (DAPI) for 5 minutes, and imaged using a confocal microscope (Olympus) at 20X. BMMs and parasites were quantified on the basis of brightfield images (BMM traces) and DAPI staining (BMM nuclei and parasite nuclei/kinetoplastids).
For quantification of nitrite/nitrate byproducts of NO synthesis in infected or stimulated BMMs, the Griess assay (Promega) was used according to the kit instructions. Absorbance was quantified with the Envision Multilabel Reader (Perkin Elmer).
Measurement of type I interferon activity in BMM supernatants
Activity of type I interferons was measured using a p53−/− mouse embryonic fibroblast (MEF) reporter cell line with a stably integrated Cignal lentiviral ISRE-luciferase construct (Qiagen). ISRE-Luc reporter MEFs were seeded in 96-well plates at a density of 6×103 cells/well. BMMs were cultured in 96-well plates and infected with L. pneumophila as described above, or with Sendai virus (Charles River Labs) at an MOI of 2. Plates were centrifuged at each timepoint to separate supernatant from cells and bacterial debris, and supernatants were collected and frozen. Thawed supernatants were transferred to ISRE-Luc reporter MEFs and incubated for 24 hours. Luciferase reporter activity was assayed by addition of firefly luciferase substrate (Promega), and luminescence was quantified with the Envision Multilabel Reader (Perkin Elmer).
RNA-seq
Total RNA was prepared using RNeasy columns (Qiagen). RNA-seq libraries were prepared as described previously 27. Briefly, poly-A mRNA was captured using selection beads (Oligo-dT Dynabeads, Life Technologies). mRNA was fragmented using zinc chloride (Ambion), and 3’ ends were dephosphorylated prior to ligation of RNA adapters (Illumina) using T4 RNA ligase (New England Biosciences). Reverse transcription was performed using reverse transcriptase (Agilent), ssDNA was removed using ExoSap-IT (Affymetrix), and RNA was removed using acetic acid and sodium hydroxide. DNA adapters (Illumina) were ligated using T4 RNA ligase (New England Biosciences). PCR was performed with barcoded primers (Illumina) and Phusion DNA polymerase (New England Biosciences) to barcode and amplify libraries. Sequencing was performed using a HiSeq 2500 (Illumina). Quantification of read counts per gene were obtained using the RSEM package in R 28, followed by TMM normalization across samples 29. Data were filtered to include only significantly expressed transcripts, defined as those with for which transcript-wise maximum expression value (across conditions) fell above the 50th percentile across all normalized counts (TMM ≥5.74), using a custom script in R. Differentially expressed genes were identified using DESeq2, and p-values were adjusted using the Benjamini-Hochberg correction. Heatmaps were generated by plotting the z-score across each row (gene) using the heatmap.2 function in R.
Western blotting
Bone marrow cells from B6 mice were seeded into 6-well tissue culture-treated plates at 1.5×106 cells/well, then cultured for 7 days in the presence of MCSF. BMMs were stimulated with 10U/ml IFNγ for 24h, and infected with L. pneumophila at an MOI of 4. Media was changed completely before infection and at 2hpi. Cells were washed three times in PBS and lysed in RIPA buffer (Boston Bioproducts) with protease inhibitor (Roche).
Nuclear and cytoplasmic protein extracts were collected at the indicated timepoints using the Thermo NE-PER Nuclear Protein Extraction Kit (Pierce). Protein from 3×106 macrophages was loaded into each well of a Bis-Tris 4–12% polyacrylamide gel (Novex), separated by electrophoresis, transferred to a polyvinylidene fluoride membrane, blocked with 5% milk in Tris-buffered saline with Tween (TBST), and incubated overnight at 4°C with primary antibodies in 5% nonfat dry milk in TBST or, for phospho-protein antibodies, in 5% bovine serum albumin (BSA) in TBST. Membranes were washed, incubated with secondary antibody in 5% BSA in TBST for one hour at room temperature, washed again, and incubated with for 5min with detection reagent (Amersham). Antibodies were from Abcam (β-actin), Cell Signaling Technologies (IRF3, IRF3pSer396 [murine IRF3pSer388], IKKεpSer172, TBK1, TBK1pSer172), Jackson Immunoresearch (HRP-conjugated secondary antibodies), and Santa Cruz (TBP).
Immunofluorescence microscopy
BMMs were seeded onto 8-well glass chamber slides (Nunc) at 2×104 cells/well. At each timepoint, BMMs were washed three times with PBS, then fixed with 4% paraformaldehyde (Electron Microscopy Sciences) in PBS for 10 minutes. BMMs were permeabilized with 0.25% Triton-X and blocked with 5% goat or donkey serum (Jackson Immunoresearch) and 5% BSA (Cell Signaling Technologies). Primary antibody incubation was done overnight at 4°C, followed by washing three times with PBS and incubation with Alexa-fluorophore conjugated secondary antibody (Life Technologies) for one hour in the dark. BMMs were then washed, counterstained with DAPI, mounted on coverslips, and imaged using a confocal microscope (Olympus) at 63X.
shRNA knockdown
High titer lentivirus encoding shRNA targeting TBK1 or control pLKO.1 lentivirus was obtained from The RNAi Consortium at the Broad Institute 30. Bone marrow cells were seeded at 1.5×106 cells/well in non-tissue culture treated 6-well plates and cultured in the presence of MCSF. At day 3, cells were spin-infected with shRNA-encoding lentivirus supplemented with 8 μg/mL polybrene (Sigma). Fresh MCSF-supplemented media was exchanged at day 4. At day 5, infected cells were selected by adding puromycin (Life Technologies) to a final concentration of 4.5 μg/mL. At day 7, surviving cells were collected and reseeded in 96-well plates for L. pneumophila infection assays as described above.
Statistical Analyses
Growth curve analysis (Mirman, 2014) was used to compare time courses of bacterial bioluminescence. Mixed-effect models were constructed using the package lme4 in R version 4.0.2. In each comparison, time, the key dependent variables (genotype, IFNβ treatment, or anti-IFNAR antibody treatment) as well as the interaction between these terms were treated as fixed effects, while biological (mouse) replicates, with nested technical (well) replicates, were treated as random effects. For growth curves with predominantly linear trends after the first timepoint, a linear model was used with the first data point omitted. For growth curves with predominantly concave or convex trends, a second-order (quadratic) orthogonal polynomial was fit to the timepoint values and the linear and quadratic terms (as well as their interaction with the key dependent variable) were used as fixed effects. Statistical significance (p-values) of the fixed-effect terms were determined with an F-test using Satterthwaite degrees of freedom and type III sum of squares implemented with the lmerTest package in R.
Statistical significance for the Griess nitrite assay was calculated using the unpaired Student’s t test. For T. cruzi, the unpaired Wilcoxon rank-sum test was used where indicated. Significance was defined as (*** P≤0.0005; ** P≤0.005; * P≤0.05).
RESULTS
IRF3 inhibits the maintenance but not the establishment of the IFNγ-activated state
Because pathogen sensors are often highly redundant for bacterial sensing, we focused on the role of transcription factors that are downstream of multiple sensors. We analyzed the impact of two transcription factors, IRF3 and IRF7, which are essential elements downstream of many innate immune sensors, on the IFNγ-activated antibacterial state in macrophages. We designed experiments to test whether pathogen sensing alters (strengthens or attenuates) the maintenance of a pre-existing IFNγ-primed state, as well as whether pathogen sensing alters the establishment of such a state during initial exposure to IFNγ (Fig. 1A). To study the effects of IRF3 and IRF7 on maintenance of a pre-existing IFNγ-primed state, we primed bone marrow-derived macrophages (BMMs) with IFNγ, replacing media with IFNγ-free media at the time of L. pneumophila infection (Fig 1A, top). Conversely, to test the effects of IRF3 and IRF7 on establishment of the IFNγ-activated state, unprimed BMMs were first infected with bacteria and then stimulated with IFNγ (Fig. 1A, bottom).
Fig. 1: Contribution of IRF3 and IRF7 to the maintenance and establishment of the IFNγ-activated state in BMMs.

(A) Design of the L. pneumophila infection assay to assess the effects of genetic perturbations on the maintenance (top) or establishment (bottom) of the IFNγ-activated state.
(B-D) BMMs from B6 and (B) Irf3−/−/Bcl2L12−/−/Irf7−/− (labeled IRF3/IRF7 DKO), (C) Irf3−/−/Bcl2L12−/− (labeled IRF3 KO), and (D) Irf7−/− (labeled IRF7 KO) mice were stimulated with IFNγ at concentrations of 0, 3, 10, or 100U/ml either before infection (i.e., ‘primed’) (B, C-D top panels) or at 2hpi (C-D bottom panels), then infected with L. pneumophila. Bacterial bioluminescence was measured over two days of infection. In (B, C, and D), data points show represent biological replicates (mice) (one, two, and three, respectively) and error bars represent the standard error of the mean across technical replicates (wells) (four, five, and five, respectively). p-values indicate the significance of the effect of genotype on bacterial growth over time under the conditions in each sub-panel, based on a linear mixed-effect model.
BMMs were infected with LP02 ΔflaA lux, a flagellin-deficient strain that does not induce pyroptosis through NAIP5/NLRC4, and is constitutively bioluminescent in a manner that has been shown to correlate closely with bacterial growth by CFU 25. As expected, the bacteria exhibited logarithmic growth in unprimed B6 BMMs, but were highly restricted in B6 BMMs primed with IFNγ in a dose-dependent manner (Fig. 1B), as expected for flagellin-deficient L. pneumophila 23. We found that unprimed IRF3/IRF7-deficient BMMs were slightly more permissive to L. pneumophila compared to B6 BMMs (Fig. 1B, top), but this was not statistically significant (p=0.28). Surprisingly, lack of IRF3 and IRF7 significantly enhanced IFNγ-mediated restriction of bacteria in IFNγ-primed BMMs (Fig. 1B, top; 3 U/ml IFNγ: p=3.0e-7, 10U/ml IFNγ: p=1.1e-11), in contrast to our original hypothesis of synergy between innate sensing and IFNγ-mediated host defense.
Further experiments with macrophages deficient in either IRF3 or IRF7 showed that lack of IRF3 is wholly responsible for the enhanced bacterial restriction observed in IFNγ-primed IRF3/IRF7-deficient BMMs (Fig. 1C; 3 U/ml IFNγ: p=0.012, 10U/ml IFNγ: p=3.7e-3, 100U/ml IFNγ: p=4.7e-3), while IRF7-deficient BMMs are phenotypically identical to B6 BMMs in our model of IFNγ-mediated bacterial restriction (Fig. 1D). Together, our results suggest that IRF3 inhibits the maintenance of the IFNγ-primed state in L. pneumophila infected BMMs.
We next asked whether IRF3/7 also affect the establishment of the IFNγ-activated state by stimulating BMMs with IFNγ at two hours post-infection (hpi). Under these conditions, IFNγ-mediated bacterial restriction was similar in B6 and IRF3-deficient as well as IRF7-deficient BMMs (Fig. 1C–D, bottom), suggesting that IRF3-mediated pathways involved in sensing of L. pneumophila affect the maintenance, but not the establishment of the IFNγ-activated state.
The adaptors MYD88/TRIF, MAVS, and STING do not account for the inhibitory effects of IRF3 on restriction of bacteria by IFNγ-primed BMMs
To determine the effects of other innate pathogen-sensing pathways on IFNγ-dependent bacterial restriction, we first considered the roles of the Toll-like receptors (TLRs), membrane-associated sensors of bacterial components and nucleic acids. We used mice lacking both MyD88 and TRIF to eliminate signaling downstream of TLR ligand sensing. We found that MyD88/TRIF deficiency did not increase normal permissiveness to L. pneumophila growth in unprimed BMMs, but led to slightly increased growth in BMMs primed with IFNγ (Fig. 2, top; 3 U/ml IFNγ: p=4.3e-3, 10U/ml IFNγ: p=5.9e-4), consistent with a model in which macrophage-intrinsic TLR signaling enhances maintenance of the anti-bacterial state established by priming with IFNγ.
Fig. 2: Contribution of MyD88 and TRIF to the maintenance and establishment of the IFNγ-activated state in BMMs.

Myd88−/−Trif−/− BMMs were compared to B6 BMMs using the L. pneumophila growth/restriction assay described in Fig. 1. Data points represent three biological replicates (mice) per genotype, and error bars represent the standard error of the mean across five technical replicates (wells). p-values indicate the significance of the effect of genotype on bacterial growth over time under the conditions in each sub-panel, based on a linear mixed-effect model.
To further investigate whether the TLR and IFNγ signaling pathways synergize when activated at the same time, we stimulated macrophages with IFNγ at 2hpi. In this scenario, Myd88−/−/Trif−/− BMMs exhibited a significant reduction in IFNγ-dependent bacterial restriction compared to B6 BMMs across a wide range of concentrations of IFNγ (Fig. 2, bottom; 3 U/ml IFNγ: p=1.4e-6, 10U/ml IFNγ: p=2.1e-9, 100U/ml IFNγ: p=2.1e-7). In contrast to our results with IRF3, we conclude that TLR pathways not only significantly synergize with simultaneous IFNγ signaling to establish the anti-bacterial state in macrophages, but also, given sufficiently strong IFNγ priming, facilitate the maintenance and robustness of this state.
We next asked whether intracellular nucleic acid sensing affects IFNγ-dependent bacterial restriction. Previous work in macrophages had identified key roles for MAVS 15,18 and STING 16 in sensing L. pneumophila RNA and DNA, respectively. Activation of these sensing pathways was associated with IRF3 activation by phosphorylation and nuclear translocation, and with transcription of type I interferons, key transcriptional targets of IRF3. We therefore hypothesized that deficiency in STING or MAVS might phenocopy deficiency in IRF3. In contrast to these expectations, and similar to the results in Myd88−/−/Trif−/− BMMs, we found evidence of a positive, rather than negative, role for MAVS and/or STING-dependent sensing pathways in both the establishment (Fig. S1, top; Sting−/−: 3 U/ml IFNγ: p=9.1e-3, 100U/ml IFNγ: p=0.02; Mavs−/−: 100U/ml IFNγ: p=0.031), and maintenance (Fig. S1, bottom; Sting−/−: 3 U/ml IFNγ: p=2.1e-3, 10U/ml IFNγ: p=5.5e-5) of the IFNγ-activated state.
IFNAR signaling does not inhibit the IFNγ-activated state during L. pneumophila infection of macrophages
We next considered how the downstream effects of IRF3 activation repress the IFNγ-induced anti-bacterial state. Since Type I IFNs are potent immune regulators induced by IRF3 activation 19, we tested a role for signaling through IFNAR, the Type I interferon receptor. The targets of Type I and II interferons partially overlap 31–33, and prior studies have shown that Type I and II IFN-stimulated pathways can reinforce 34,33,35–41 or antagonize 33,42–44 each other in a context-dependent manner. In the setting of bacterial and mycobacterial infection, however, prior work has shown antagonizing effects of Type I IFN on IFNγ-dependent macrophage activation through a variety of mechanisms 45–47, including induction of antagonistic effectors 43 and downregulation of the IFNγ receptor 44. Based on these prior data, we expected IFNAR deficiency to phenocopy lack of IRF3 and enhance IFNγ-dependent bacterial restriction.
Contrary to our expectations of antagonistic effects between Type I and Type II IFN pathways in the context of bacterial infection, we found that IFNAR deficiency slightly decreased restriction of L. pneumophila growth in IFNγ-primed BMMs (Fig. 3A, top right; 10U/ml IFNγ: p=1.4e-3). We also noted a small, but statistically insignificant increase in bacterial growth in unprimed Ifnar−/− BMMs, as shown before 25 (Fig. 3A, left), as well as in BMMs stimulated with IFNγ during infection (Fig. 3A, bottom right). To assess signaling via IFNAR a different way, IFNAR-blocking antibodies were used during infection and subsequent incubation. Inhibition of IFNAR signaling in this way did not consistently affect growth or restriction of L. pneumophila in resting or IFNγ-primed B6 or IRF3/IRF7-deficient BMMs (Fig. S2A, first row) except for a slight enhancement in bacterial restriction in B6 BMMs at 10U/ml IFNγ (p=0.014). Even at this condition, however, the enhancing effect of IRF3/IRF7 deficiency on bacterial restriction was not abrogated by anti-IFNAR antibody (Fig. S2A, first vs second row). In BMMs stimulated with IFNγ during infection, a small increase in IFNγ-mediated restriction of L. pneumophila was also seen in IFNAR-blocked BMMs compared to BMMs treated with isotype control antibody in B6 BMMs (Fig. S2A, third row; 10U/ml IFNγ: p=0.015) as well as in IRF3/IRF7-deficient BMMs (Fig. S2A, fourth row; 3U/ml IFNγ: p=1.7e-3, 10U/ml IFNγ: p=2.5e-3).
Fig. 3: Lack of contribution of Type I interferons to IRF3-mediated suppression of the IFNγ-activated state in primed BMMs.

(A) Ifnar−/− BMMs were compared to B6 BMMs using the L. pneumophila growth/restriction assay as described in Fig. 1. Data represent three biological replicates (mice) per genotype, and error bars represent the standard error of the mean across five technical replicates (wells). p-values indicate the significance of the effect of genotype on bacterial growth over time under the conditions in each sub-panel, based on a linear mixed-effect model.
(B) Irf3−/−/Bcl2L12−/−/Irf7−/− (labeled IRF3/IRF7 DKO) and B6 BMMs were used in the L. pneumophila growth/restriction assay as described in Fig. 1, and were additionally treated with 50U IFNβ or mock-treated (vehicle) starting at 2hpi. Error bars represent the standard error of the mean across four to five technical replicates (wells). p-values indicate the significance of the effect of IFNβ treatment on bacterial growth over time under the conditions in each sub-panel, based on a linear mixed-effect model.
These results suggest that IRF3-mediated repression of the IFNγ-primed state proceeds through a novel, IFNAR-independent mechanism. In fact, we found that the level of endogenous production of Type I IFNs by B6 BMMs during L. pneumophila infection is low. Supernatants from L. pneumophila-infected B6 BMMs were unable to activate ISRE-driven transcription in a reporter cell line, in contrast to the robust ISRE activation observed using supernatants from BMMs infected with Sendai virus (Fig. S2B). In addition, IFNβ transcript levels in L. pneumophila infected BMMs were less than 0.1% of those in Sendai virus-infected BMMs, and were not increased by IFNγ priming.
Even when added exogenously at high levels, Type I IFNs do not suppress the IFNγ-primed state. In fact, in both B6 and IRF3/IRF7-deficient IFNγ-primed BMMs infected with L. pneumophila, stimulation with IFNβ at 2hpi appeared to potentiate IFNγ-dependent effectors, significantly enhancing bacterial restriction throughout the course of infection (Fig. 3B, top; B6: 3U/ml IFNγ: p=0.012, 10U/ml IFNγ: p=2.9e-9; IRF3/IRF7 DKO: 3U/ml IFNγ: p=1.8e-8, 10U/ml IFNγ: p=4.3e-13). In unprimed BMMs, exogenous IFNβ also modestly contributed to bacterial restriction, but the magnitude of the effect was smaller than in IFNγ-primed BMMs (Fig. 3B, left; p=2.3e-4). Notably, the relative timing of IFNβ and IFNγ stimuli was critical to its qualitative effect on the IFNγ-activated state. In contrast to our results with IFNγ-primed BMMs, in both B6 and IRF3-deficient unprimed BMMs stimulated with IFNγ 2h after L. pneumophila infection, simultaneous stimulation with exogenous IFNβ significantly attenuated IFNγ-dependent bacterial restriction (Fig. 3B, bottom; B6: 3U/ml IFNγ: p=5.6e-4, 10U/ml IFNγ: p=4.9e-10, 100U/ml IFNγ: p=3.2e-8; IRF3/IRF7 DKO: 3U/ml IFNγ: p=1.7e-4, 10U/ml IFNγ: p=2.0e-5, 10U/ml IFNγ: p=3.1e-10), likely by direct inhibition of IFNγ signaling 43,44,46,47. In summary, while Type I and II interferons clearly interact to modulate anti-bacterial activities, Type I IFN signaling is not required for the observed inhibitory effect of IRF3 on the maintenance of the IFNγ-primed state in BMMs.
STAT1 phosphorylation downstream of IFNγ signaling is similar between B6 and IRF3-deficient BMMs
After finding that type I IFN signaling through IFNAR is unlikely to contribute to the inhibitory effects of IRF3 on IFNγ-primed BMMs, we asked whether type II IFN signaling by IFNγ itself is affected by the presence of absence of IRF3. Engagement of the IFNγ receptor has been shown to result in crosslinking of the receptor, followed by activation of at least two distinct kinase classes: Janus kinases 1 and 2 and phosphatidylinositol 3-kinase; these pathways converge at the transcription factor STAT1 which is subsequently phosphorylated at Y701 and S72748. In our prior work, we have shown that knockdown of Stat1 transcripts in BMMs using shRNA completely eliminates IFNγ-mediated restriction of L. pneumophila in these cells (not shown), supporting the model that STAT1 is essential to IFNγ priming. We therefore hypothesized that increased STAT1pY01 and/or STAT1pS727 may contribute to enhanced IFNγ-induced antibacterial activity in IRF3-deficient BMMs relative to B6 BMMs. Contrary to this hypothesis, however, we observed similar or slightly decreased levels of STAT1pY01, and similar levels of STAT1pS727 in IRF3-deficient IFNγ-primed BMMs compared to similarly treated B6 BMMs (Fig. 4). Therefore, our data do not support a role for differential activation of STAT1 downstream of IFNγ signaling in the observed inhibition of IFNγ-mediated antibacterial activity by IRF3.
Fig. 4: Stat1 phosphorylation is not enhanced in IRF3-deficient BMMs after IFNγ priming.

Irf3−/−/Bcl2L12−/− (labeled KO) and B6 BMMs were unprimed or were primed with 10U/ml IFNγ, and infected with L. pneumophila. Cytoplasmic protein extracts were collected at the indicated timepoints after infection, and analyzed by Western blotting for STAT1pY701, STAT1pS727, total STAT1, and β-actin. Results are representative of three independent experiments. The slight increase in STAT1pY701 seen in IFNγ-primed Irf3−/−/Bcl2L12−/− BMMs relative to B6 BMMs at 0hpi was not seen in the other two experiments, and is considered to be insignificant.
IFNγ inhibits L. pneumophila-induced phosphorylation of IRF3
We hypothesized that IFNγ-primed BMMs might partially overcome IRF3-mediated repression in order to mount an effective antimicrobial response. We asked whether IRF3 activation is affected by IFNγ priming in infected BMMs in the L. pneumophila bacterial infection model. We started by monitoring the activity of nuclear IRF3 based on detection of phosphorylation status at mouse Ser388 (human Ser396) 49–51 in BMM nuclear lysates by Western blot. We found that IRF3pSer388 is absent from the nuclei of uninfected BMMs, but is strongly detected in the nucleus of unprimed BMMs within one hour of infection by L. pneumophila, where it remains through at least 8hpi (Fig. 5A). In IFNγ-primed BMMs, however, IRF3pSer388 is only weakly detected in nuclear extracts at all timepoints after infection (Fig. 5A). These data suggest that IFNγ priming disrupts canonical IRF3 activation downstream of L. pneumophila infection.
Fig. 5: Effect of L. pneumophila infection on IRF3 phosphorylation and distribution in IFNγ-primed vs. unprimed BMMs.

(A) B6 BMMs were primed with 10U/ml IFNγ and infected with L. pneumophila. Nuclear protein extracts were collected at the indicated timepoints after infection, and analyzed by Western blotting for murine IRF3pSer388 and TBP. Data are representative of three independent experiments.
(B) Immunofluorescence microscopy analysis of IRF3 localization in B6 BMMs primed with 0, 10, or 100 U/ml IFNγ, then fixed 2 hours after infection with L. pneumophila or stimulation with LPS.
(C) B6 BMMs were primed with 10U/ml IFNγ and infected with L. pneumophila. Cytoplasmic protein extracts were collected at the indicated timepoints after infection, and analyzed by Western blotting for IKKεpSer172, TBK1pSer172, TBK1, and β-actin.
(D) B6 BMMs were stimulated with 10U/ml IFNγ and infected with L. pneumophila. Cytoplasmic protein extracts were collected at the indicated timepoints after infection, and analyzed by Western blotting for murine IRF3pSer388, IRF3, and β-actin.
To further investigate the differential localization of IRF3 in innate immune responses based on IFNγ priming status, we performed immunofluorescence microscopy of unprimed or IFNγ-primed B6 BMMs before or after L. pneumophila infection or after LPS stimulation, which has also been shown to activate IRF3 phosphorylation and nuclear localization 52. Consistent with the results of Western blot analysis in L. pneumophila-infected BMMs (Fig. 5A), we observed IRF3 in the nuclei of unprimed BMMs two hours after either infection with L. pneumophila or stimulation with LPS. In IFNγ-primed BMMs, however, IRF3 was found in an unexpected cytosolic speckling pattern, which persisted after either infection or LPS stimulation (Fig. 5B).
IRF3 phosphorylation at Ser388 requires the activity of IKKε and TBK1 49,53, kinases which are themselves regulated by phosphorylation 54. We next monitored IKKε and TBK1 phosphorylation to determine whether the significant decrease in IRF3pSer388 we observed in IFNγ-primed, infected BMMs is due to decreased activity of these kinases. We found that IKKε and TBK1 are indeed phosphorylated in BMMs following L. pneumophila infection, and that this response remains intact in macrophages primed with IFNγ (Fig. 5C). This suggests that IRF3 is inhibited directly in IFNγ-primed and infected BMMs, rather than by regulation of its kinases.
Further supporting the lack of connection between IRF3 phosphorylation and the activation of its activating kinases, we found that genetic deficiency in IKKε or shRNA-mediated knockdown of TBK1 did not phenocopy IRF3 deficiency (Fig. S3). We note that we could not assess the ability of BMMs deficient in both Ikkε and TBK1 to phenocopy IRF3 deficiency, because of the confounding requirement for at least one of these kinases for bacterial growth in unprimed BMMs (Fig. S3, left). Finally, we found that phosphorylated IRF3 does not accumulate in the cytoplasm of IFNγ-primed BMMs (not shown), suggesting that IRF3 is not stably phosphorylated in these cells. Together, these data suggest that despite the presence of activated IKKε and TBK1, IRF3 is not stably phosphorylated at Ser388 in response to L. pneumophila infection in IFNγ-primed BMMs. Therefore, while L. pneumophila infection leads to early phosphorylation and nuclear localization of IRF3 in unprimed BMMs, IFNγ priming appears to significantly inhibit these events.
IRF3/7 deficiency affects the transcriptional programs of unprimed, IFNγ-primed, and L. pneumophila-infected BMMs
To identify genes that may account for the observed inhibition of the IFNγ-induced anti-bacterial state, we used RNA sequencing to quantify gene expression in B6 or mutant IRF3/IRF7-deficient BMMs in the settings of rest, IFNγ priming, and/or L. pneumophila infection. We observed 93 significantly expressed genes that were up- or down-regulated by ≥3-fold in IFNγ-primed IRF3/IRF7-deficient BMMs vs. B6 (WT) BMMs following infection with L. pneumophila (Fig. S4A–H). Among these, 43 genes were differentially expressed based on IRF3/IRF7 genotype only in BMMs that were IFNγ-primed prior to infection (Fig. S4A), including genes encoding intracellular iron homeostasis regulators (Lcn2, Ftl2), transcription factors (Lyl1, Egr3, Epas1, Batf3, Krc), viral response and cytokine genes (Oasl2, Ifit3, Mx1, Il23a), a macrophage-derived inflammatory neuropeptide (Npy)55 a GPCR ligand (Niacr), a p47 GTPase (Gm12250), and genes involved in apoptotic (Chek2, Nptx1, Serpinb2, Fah) or non-apoptotic cell death (Cd00if). Some of the 93 genes (in Fig. S4A–H) were IRF3/IRF7-regulated in IFNγ-primed BMMs, but not unprimed BMMs, regardless of L. pneumophila infection status (Fig. S4D). These included inducible nitric oxide synthase (Nos2) as well as chemokines (Gdf15, Cxcl11), scavenger receptors (Scarf1), and a cation channel implicated in macrophage activation (Trpv4). In contrast, some of the 93 genes were IRF3/IRF7-regulated in infected BMMs regardless of IFNγ-priming status, but not in uninfected BMMs (Fig. S4B). This subset included genes encoding the transcription factor Plac8 and the IFNγ-inducible GTPase Gm4951/Ifgga2. In addition, expression of 28 of the 93 genes was also IRF3/IRF7-regulated at baseline in unprimed BMMs as well as in IFNγ-primed infected BMMs (Fig. S4E), including the viral response gene Oasl1, a homolog of iron homeostasis protein mitoferrin (AK08749), as well as genes encoding secreted factors including G-CSF (Csf3) and the acute-phase reactant serum amyloid A3 (Saa3). A non-overlapping set of 139 genes were differentially regulated based on genotype in BMMs that were either primed with IFNγ or infected with L. pneumophila, but not both (Fig. S4I–S4M); the relevance of these genes to our infection model is unclear. We conclude that several IFNγ-induced effectors are dysregulated in infected cells in the absence of IRF3/IRF7, and thus may contribute to the enhanced microbial restriction observed in IRF3/IRF7-deficient BMMs.
Nos2 is upregulated, but is not required for superior IFNγ-mediated restriction of L. pneumophila in IRF3-deficient BMMs
Inducible nitric oxide (iNOS, Nos2) is one of the canonical mediators of the IFNγ-mediated response to intracellular bacteria and parasites 56–59. Nos2 transcription was synergistically induced in macrophages by priming with 10U/ml IFNγ and by L. pneumophila infection. Interestingly, transcript levels in IFNγ-primed macrophages were approximately fivefold higher in IRF3-deficient BMMs relative to wild type BMMs before or after infection (Fig. S4D), suggesting that increased Nos2 may play a role in the enhanced capacity of IRF3-deficient BMMs to restrict intracellular bacteria.
Consistent with the trend in Nos2 transcript levels, IRF3-deficient BMMs primed with 100U/ml of IFNγ produced significantly more nitrite metabolites (a marker of Nos2 activity) in response to bacterial infection than did identically primed B6 BMMs (Fig. 6A, right). However, this relative increase in nitrite production was not seen in IRF3-deficient BMMs primed with 10U/ml IFNγ (Fig. 6A, middle), which nevertheless exhibit enhanced bacterial restriction relative to B6 BMMs (Fig. 1C). To further test the role of Nos2, we used selective or nonselective Nos2 inhibitors to treat macrophages during IFNγ stimulation and infection. Treatment with the inhibitors L-NIL, 1400W and L-NOARG suppressed nitrite production by both B6 and IRF3-deficient BMMs (Fig. 6A), but did not affect restriction of L. pneumophila in either B6 or IRF3-deficient IFNγ-primed BMMs (Fig. 6B). Together, these results suggest that enhanced Nos2 activity is dispensable for the enhanced IFNγ-dependent restriction of L. pneumophila in IRF3-deficient BMMs, the latter being dependent on redundant mechanisms.
Fig. 6: Increased activity of IFNγ-induced Nos2 is observed in IRF3-deficient BMMs, but is dispensable for increased restriction of L. pneumophila.

(A) Irf3−/−/Bcl2L12−/− (labeled IRF3 KO) and B6 BMMs were unprimed or primed with 3 (not shown), 10, or 100 U/ml IFNγ, then infected with L. pneumophila. The Nos2 inhibitors L-NOARG, L-NIL or 1400W were used to treat BMMs before and after infection at concentrations 5-fold (low), 25-fold (medium), or 125-fold (high) times the IC50 of the compound. Nitrite levels in supernatants two days after infection were measured using the Griess assay to assess Nos2 activity. Data represent two technical replicates per sample. Asterisks indicate the significance of the difference in nitrite production between mutant and B6 genotypes, calculated using the unpaired Student’s t test.
(B) Irf3−/−/Bcl2L12−/− BMMs were compared to B6 BMMs using the L. pneumophila growth/restriction assay described in Fig. 1. BMMs were mock-treated or treated with the Nos2 inhibitors L-NOARG, L-NIL, or 1400W at concentrations 5-fold (low, not shown), 25-fold (medium, not shown), or 125-fold (high) times the IC50 of each inhibitor for two hours before infection and thereafter. Data for conditions not shown are not significantly different from the data shown. Error bars represent the standard error of the mean across four technical replicates (wells). p-values indicate the significance of the effect of genotype on bacterial growth over time under the conditions in each sub-panel, based on a linear mixed-effect model.
IRF3 suppresses IFNγ-activated defense mechanisms against the intracellular parasite T. cruzi
The role of Nos2 in pathogen restriction varies with pathogen species. Unlike IFNγ, Nos2 has in fact been shown to be dispensable for restriction of L. pneumophila in mouse macrophages and in murine models in vivo 60,61. However, the role of Nos2 in intracellular pathogen restriction in macrophages is well known to vary among microbe species 56,62. For example, the intracellular protozoan parasite T. cruzi, the etiologic agent of Chagas disease, has been shown to be restricted in a Nos2-dependent manner in IFNγ-activated macrophages 63. In order to assess the relevance of IFNγ-dependent enhanced Nos2 induction that we observed in IRF3-deficient BMMs to a range of intracellular pathogens, we applied the T. cruzi infection model to compare IRF3-deficient and B6 BMMs. This infection model differs from the L. pneumophila model in several notable ways, including co-stimulation of BMMs with TNFα to potentiate Nos2-dependent restriction of T. cruzi in response to IFNγ 64,65, and a known impact of Type I IFN on T. cruzi infection 66, which we addressed by using IFNAR-blocking antibody throughout the infections described.
In macrophages co-stimulated with 100U/ml IFNγ and TNFα, IRF3-deficient BMMs produced significantly more Nos2 activity-byproduct nitrites than B6 BMMs both before (top) and after (bottom) infection with T. cruzi (Fig. S5A). IRF3-deficient BMMs primed with 100U/ml IFNγ and co-stimulated with TNFα released significantly fewer parasites into cell supernatants at 6 and 7 days post-infection (dpi) than similarly treated B6 BMMs (Fig. S5B, top). This IRF3-dependent effect was Nos2-dependent, since it was abolished in BMMs treated with the Nos2 inhibitor L-NIL (Fig. S5B, bottom). Microscopic examination of intracellular T. cruzi parasites revealed a significantly lower proportion of infected BMMs at 5dpi among IRF3-deficient BMMs primed with 100U/ml IFNγ and co-stimulated with TNFα, compared to similarly treated B6 BMMs (Fig. S4C). Under these conditions, IRF3-deficient BMMs also exhibited a lower median burden of parasites per infected BMM compared to B6 BMMs (Fig. S5D, top), though this was not statistically significant. Treatment with L-NIL to inhibit Nos2 activity largely eliminated the difference in T. cruzi infection rates between IRF3-deficient and B6 BMMs (Fig. S5C, bottom right). These results suggest that restriction of intracellular T cruzi in IFNγ-primed BMMs requires Nos2, both of which are negatively affected by IRF3.
DISCUSSION
Our studies of the role of innate sensing pathways in regulating IFNγ-mediated innate immunity in macrophages have led us to identify a novel inhibitory role for IRF3 in the maintenance of the IFNγ-activated state. This is in contrast to prior work that has identified positive synergies between IFNγ and pathogen sensing pathways 3–7, as well as our observations on the positive effect of the sensing/signaling proteins MyD88, TRIF, and IFNAR on the establishment and the maintenance of the IFNγ-induced anti-bacterial state. We also noted a lack of effect of the sensing proteins STING and MAVS on the maintenance of the IFNγ-primed state. The inhibitory action of IRF3 is therefore not likely to depend on MyD88/TRIF, STING, MAVS or IFNAR for its effects. In addition, our data show that IRF3 deficiency does not enhance IFNγ signaling itself, based on assessments of phosphorylation of the IFNGR signaling mediator STAT1 at either Y701 or S727. One limitation of our work, as with all work using Irf3−/−/Bcl2L12−/− mice, is that these mice also lack expression of BCL2L12, due to overlap of a critical 5’ segment of Bcl2L12 with the targeted portion of Irf3 with on the opposite strand 24. While Bcl2L12 deficiency could theoretically contribute to any phenotype observed in cells from these mice, our RNAseq data indicate that Bcl2L12 is not transcribed at a notable level under the conditions of our experiments in B6 BMMs.
Many genes are dysregulated in IRF3/IRF7-deficient BMMs when compared to B6 BMMs that are primed with IFNγ and/or infected with L. pneumophila, suggesting that IRF3 is a critical regulator of gene expression during establishment and maintenance of the anti-bacterial state. A canonical activity of IRF3 is the induction of Type I interferons and downstream signaling via IFNAR, and Type I IFNs can in turn antagonize the effects of IFNγ 42–45,67. However, the effects of IRF3 that we observe on IFNγ-primed BMMs appear to be independent of IFNAR. In our model, expression of IFNα and IFNβ in IFNγ-primed and L. pneumophila-infected BMMs was low, as measured both by RNA-seq and by quantification of ISRE activation by supernatants. IFNAR deficiency did not enhance IFNγ-mediated activity of these BMMs, in contrast to what prior data would suggest; meanwhile, the addition of exogenous Type I IFN in our model system synergized with rather than antagonized IFNγ-mediated bacterial restriction. Therefore, we propose that the IFNγ-inhibiting activity of IRF3 at the transcriptional level lies beyond Type I interferons and their targets. Several candidate mechanisms are discussed below.
Increased transcription and activity of Nos2 is observed in IFNγ-primed BMMs lacking IRF3, and is required for enhanced IFNγ-mediated restriction of T. cruzi in these cells relative to B6 BMMs. Nos2 is therefore a critical IRF3-modulated element affecting antimicrobial activity in IFNγ-primed BMMs. In the L. pneumophila infection model, however, our experiments with Nos2 inhibitors showed that it is dispensable for IFNγ-mediated bacterial restriction in either IRF3-deficient or B6 BMMs. Our results are consistent with prior observations regarding the redundancy of Nos2 in restriction of L. pneumophila in a human monocyte cell line 68, as well as with recent results in mouse BMMs69.
The mechanism of enhanced effector function of Nos2 on T. cruzi in IRF3-deficient BMMs has not yet been addressed in our work, but at least two broad possibilities exist. First, elevated relative levels of nitric oxide may directly decrease parasite burdens by direct antimicrobial effect of reactive nitrogen intermediates. Alternatively, it may decrease parasite burdens indirectly, through its signaling properties that modulate host cell function 70. These possibilities may be addressed in further studies.
Another set of pathways that may be affected by IRF3 deficiency involve iron trafficking in the infected macrophage. While not tested further here, another potential effector targeted by the inhibitory activity of IRF3 is lipocalin 2 (Lcn2), which was synergistically induced by IFNγ and L. pneumophila in IRF3/IRF7-deficient but not B6 BMMs. LCN2 restricts iron availability in the vacuoles of pathogenic bacteria including S. typhimurium, M. avium, C. pneumoniae, and B. abortus, in some cases in an IFNγ-dependent manner 71,72. Further work may assay iron content within IRF3-deficient and wildtype macrophages directly, though assessing iron located in the relevant cellular compartments (eg, Legionella-containing vacuoles) in bioavailable forms can be challenging. Interestingly, LCN2 has been shown to play an immunomodulatory role in pulmonary, intestinal, and hepatic infection and inflammation models 73,74, suggesting that iron content alone may not fully reflect the potential role of LCN2 in potentiating IFNγ-mediated immune responses in IRF3-deficient BMMs. The connection between iron metabolism and nitric oxide signaling in this setting is another area for possible further study, as NO has been shown to induce expression of the iron transporter ferroportin (Slc40a1) 75, which has in turn has been shown to affect cytokine levels in mouse macrophages 76. We have not noted a significant difference in transcription of ferroportin in our model, but further studies of protein-level expression of this and other regulators of iron metabolism are warranted.
How might IRF3 alter the transcriptional program of wildtype IFNγ-primed BMMs to inhibit the production of Nos2, Lcn2, and other antimicrobial effectors? We had originally hypothesized that IRF3 is activated downstream of microbial pattern recognition receptors or other sensors after infection. However, our RNA-seq analysis revealed profound transcriptional differences between B6 and IRF3/IRF7-deficient BMMs even prior to infection, in BMMs that were either completely unstimulated or primed with IFNγ only. These results are consistent with a tonic baseline activity of IRF3, a function separate from and in addition to its known role as an acute immune signaling mediator. One potential model by which tonic activity of IRF3 might take place is by altering chromatin conformation at the promoters or enhancers of IFNγ-activated genes. Mechanistically, IRF3 may induce a repressor that destabilizes the IFNγ-primed state, as seen in other contexts 77, or it may directly repress a promoter element. Further studies with ChIP-Seq and ATAC-Seq using wildtype or IRF3-deficient BMMs could address these possibilities.
Since we have observed that IRF3 translocation to the nucleus is drastically reduced in IFNγ-primed BMMs, it is also possible that cytosolic rather than nuclear IRF3 affects gene expression indirectly, through a noncanonical mechanism. For example, IRF3 might bind and sequester a transcription factor needed to maintain the IFNγ-primed state – a model of action previously demonstrated for cytosolic IRF3 in T cells 78. In certain cellular contexts, mounting evidence implicates IRF3 as a cytoplasmic effector in non-transcriptional processes, including induction of apoptosis in virally infected cells79 and inhibition of NFκB signaling80 by direct binding to cytosolic Bax or IKKβ, respectively. Future studies involving overexpression of constitutively active IRF3 have the potential to elucidate these mechanisms, though studies would likely require using a macrophage cell line model due to the low efficiency of transduction of overexpression constructs in primary BMMs.
The speckled cytosolic distribution of IRF3 we have observed in IFNγ-primed BMMs both before and after L. pneumophila infection (or LPS stimulation), in lieu of phosphorylation and nuclear localization in infected non-primed BMMs, shows that the function of IRF3 itself is differentially regulated in the context of the IFNγ-primed state. Recruitment of IRF3 to cytosolic puncta has been observed in other studies, suggesting that this localization may play a role in its activation 81,82 or degradation 83. Along this direction, preliminary studies in our laboratory shown selective expression of an IRF3 phosphatase in IFNγ-primed BMMs; further work will be required to determine whether this antagonist of IRF3 is functionally active in this setting, and whether it is recruited to IRF3-containing speckles. In addition, it is possible that recruitment of IRF3 to cytosolic foci could prevent its phosphorylation by active IKKε/TBK1 kinases, or could prevent the translocation of phosphorylated IRF3 to the nucleus. Studies using overexpression of IRF3 mutants that cannot translocate to the nucleus 79 may be valuable in future studies to elucidate the contribution of cytoplasmic IRF3 to inhibition of the IFNγ-activated state.
Our work with T. cruzi-infected BMMs confirmed that IRF3 can play a Type I IFN-independent role in suppressing IFNγ-mediated responses to intracellular parasites as well as bacteria, despite significant differences in pathogen detection, restriction, virulence and life cycle. Key differences in the experimental system used with T. cruzi infection should be considered in interpreting these results; in particular, since TNFα is required to co-stimulate BMMs together with IFNγ, and since macrophages were stimulated with IFNγ and TNFα both before (priming) and after T. cruzi infection. For instance, the decrease in proportion of infected macrophages observed in IRF3-deficient vs. B6 BMMs over the course of T. cruzi infection across all conditions, including in macrophages activated by TNFα alone, suggests an iNOS-independent and possibly TNFα–induced mechanism that preferentially restricts the spread of T. cruzi in populations of IRF-deficient macrophages. Interestingly, recent work with the intracellular parasite Toxoplasma gondii showed that IRF3 is critical to the intracellular growth of this parasite in MEFs in a manner independent of Type I IFNs 84. This study noted a genotype-dependent decrease in per-cell infectious burden of T. gondii in unprimed macrophages, a result we did not observe in our work with T. cruzi. This difference may be accounted for by differences in the biology or lifecycle of the two parasitic species, or may indicate a distinct set of IFNγ-independent mechanisms restricting T. cruzi in IRF3-deficient- BMMs.
When considered together with our observations regarding the effects of IRF3 on the IFNγ-primed state, these results are consistent with an antagonistic relationship between exogenous macrophage activation via IFNγ and bacterial sensing via IRF3 in the modulation of the anti-bacterial state. Macrophages carry out important roles in modulating their environment, including maintaining innate immunity, priming adaptive immunity, and repairing tissue damage85. The presence of an intrinsic IRF3-dependent suppressor mechanism for IFNγ-mediated effectors in macrophages is likely part of a homeostatic process that controls potentially harmful immune activation by powerful antimicrobial effectors such as nitric oxide. Since IRF3 is known to be activated during viral infection, its actions may be essential to preclude the triggering of unneeded antibacterial or antiparasitic defense mechanisms by IFNγ, which is produced by activated T cells during viral infection. Further work using co-infection models will be useful to address this possibility. In addition, future experiments in animals will test the role of IRF3 in balancing the need for host protection against intracellular bacteria and parasites with the risk of tissue damage caused by activated macrophages during infection with these pathogens in vivo 86.
Supplementary Material
Fig. S1: Contribution of MAVS and STING to the maintenance and establishment of the IFNγ-activated state in BMMs
Fig. S2: Low expression of Type I IFN in IFNγ-primed BMMs infected by L. pneumophila
Fig. S3: Variation in gene expression due to IRF3/7 deficiency in BMMs primed with IFNγ and/or infected with L. pneumophila
Fig. S4: Effect of IRF3 on nitrite production and intracellular pathogen restriction in IFNγ and TNFα-primed BMMs infected by T. cruzi
Fig. S5: Contribution of IRF3 kinases to intracellular bacterial growth and the maintenance and establishment of the IFNγ-activated state in BMMs
ACKNOWLEDGMENTS
The authors would like to acknowledge the excellent technical assistance provided by Weibo Li, Tom Eisenhaure, Matthew Roy, David Lieb (Hacohen Lab); Rafael B. Polidoro (Gazzinelli Lab); and Donna Neuberg (Dana Farber Cancer Institute). We thank Deb Hung (Broad Institute) and Barbara Burleigh (Harvard School of Public Health) for generously sharing lab space for infections, and we are grateful to Jonathan Kagan (Boston Children’s Hospital) as well as Ricardo Gazzinelli and Kate Fitzgerald (University of Massachusetts – Worcester) for critical discussions. This work was supported by the NIH Director’s New Innovator award DP2OD002230 to NH and the NIH MSTP fellowship T32GM007753 to KM.
ABBREVIATIONS
- B6
C57BL/6J
- BMM
bone marrow-derived macrophage
- DAPI
4’,6-diamidino-2-phenylindole
- Dot/Icm
defect in organelle trafficking/intracellular multiplication
- FBS
fetal bovine serum
- GTPase
guanosine triphosphate hydrolase
- HPI
hours post infection
- IFN
interferon
- IFNAR
type I interferon receptor
- IKK
inhibitor of NFκB (IκB) kinase
- IL
interleukin
- iNOS
inducible nitric oxide synthase
- IRF
interferon response factor
- ISRE
interferon-sensitive response element
- LPS
lipopolysaccharide
- MAVS
mitochondrial antiviral-signaling protein
- MCSF
macrophage colony-stimulating factor
- MEF
mouse embryonic fibroblast
- MOI
multiplicity of infection
- mRNA
messenger RNA
- MYD88
myeloid differentiation primary response gene 88
- NAIP
NLR family, apoptosis inhibitory protein
- NFκB
nuclear factor κ-light-chain-enhancer of activated B cells
- NK
natural killer
- NKT
natural killer T
- NLRC
NOD-like receptor (NLR) with N-terminal caspase activating and recruitment domain
- NO
nitric oxide
- NOD
nucleotide-binding oligomerization domain
- RIG-I
retinoic acid-inducible gene I
- RNAi
RNA interference
- shRNA
short hairpin RNA
- STING
stimulator of type I IFN gene
- TBK
tank-binding kinase
- TLR
toll-like receptor
- TNF
tumor necrosis factor
- TRIF
toll-interleukin 1 receptor (TIR)-domain-containing adaptor inducing IFNβ
Footnotes
AUTHORSHIP
Designed experiments: KM, RR, MRM, NH. Conducted experiments: KM, RR, KS, MRM. Analyzed data: KM, AMS, SS. Interpreted results: all authors. Wrote paper: KM. Revised paper: KM, RR, JC, NH.
DISCLOSURES
The authors declare no conflicts of interest.
REFERENCES
- 1.Murray HW. Current and future clinical applications of interferon-gamma in host antimicrobial defense. Intensive Care Med. 1996;22:S456–S461. [DOI] [PubMed] [Google Scholar]
- 2.Bustamante J, Boisson-Dupuis S, Abel L, et al. Mendelian susceptibility to mycobacterial disease: Genetic, immunological, and clinical features of inborn errors of IFN-γ immunity. Semin Immunol. 2014;26:454–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sweet MJ, Stacey KJ, Kakuda DK, et al. IFN-gamma primes macrophage responses to bacterial DNA. J Interferon Cytokine Res Off J Int Soc Interferon Cytokine Res. 1998;18:263–271. [DOI] [PubMed] [Google Scholar]
- 4.Schroder K, Sweet M, Hume D. Signal integration between IFNgamma and TLR signalling pathways in macrophages. Immunobiology. 2006;211:511–524. [DOI] [PubMed] [Google Scholar]
- 5.Gough D, Levy D, Johnstone R, et al. IFNgamma signaling-does it mean JAK-STAT? Cytokine Growth Factor Rev. 2008;19:383–394. [DOI] [PubMed] [Google Scholar]
- 6.Hu X, Chakravarty SD, Ivashkiv LB. Regulation of interferon and Toll-like receptor signaling during macrophage activation by opposing feedforward and feedback inhibition mechanisms. Immunol Rev. 2008;226:41–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qiao Y, Giannopoulou EG, Chan CH, et al. Synergistic Activation of Inflammatory Cytokine Genes by Interferon-γ-Induced Chromatin Remodeling and Toll-like Receptor Signaling. Immunity. 2013;39:454–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jehl S, Nogueira C, Zhang X, et al. IFNγ inhibits the cytosolic replication of Shigella flexneri via the cytoplasmic RNA sensor RIG-I. PLoS Pathog;8. Epub ahead of print 2012. DOI: 10.1371/journal.ppat.1002809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Massis LM, Zamboni DS. Innate immunity to legionella pneumophila. Front Microbiol;2. Epub ahead of print 2010. DOI: 10.3389/fmicb.2011.00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Casson CN, Shin S. Inflammasome-mediated cell death in response to bacterial pathogens that access the host cell cytosol: lessons from legionella pneumophila. Front Cell Infect Microbiol;3. Epub ahead of print 2012. DOI: 10.3389/fcimb.2013.00111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pilla DM, Hagar JA, Haldar AK, et al. Guanylate binding proteins promote caspase-11-dependent pyroptosis in response to cytoplasmic LPS. Proc Natl Acad Sci U S A. 2014;111:6046–6051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cerqueira DM, Pereira MSF, Silva ALN, et al. Caspase-1 but Not Caspase-11 Is Required for NLRC4-Mediated Pyroptosis and Restriction of Infection by Flagellated Legionella Species in Mouse Macrophages and In Vivo. J Immunol Baltim Md 1950. 2015;195:2303–2311. [DOI] [PubMed] [Google Scholar]
- 13.Akamine M, Higa F, Arakaki N, et al. Differential roles of Toll-like receptors 2 and 4 in in vitro responses of macrophages to Legionella pneumophila. Infect Immun. 2004;73:352–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Archer KA, Alexopoulou L, Flavell RA, et al. Multiple MyD88-dependent responses contribute to pulmonary clearance of Legionella pneumophila. Cell Microbiol. 2008;11:21–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chiu Y-H, Macmillan J, Chen Z. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell. 2009;138:576–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lippmann J, Müller HC, Naujoks J, et al. Dissection of a type I interferon pathway in controlling bacterial intracellular infection in mice. Cell Microbiol. 2011;13:1668–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ge J, Gong Y-N, Xu Y, et al. Preventing bacterial DNA release and absent in melanoma 2 inflammasome activation by a Legionella effector functioning in membrane trafficking. Proc Natl Acad Sci U S A. 2012;109:6193–6198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Monroe K, McWhirter S, Vance R. Identification of host cytosolic sensors and bacterial factors regulating the type I interferon response to Legionella pneumophila. PLoS Pathog;5. Epub ahead of print 2009. DOI: 10.1371/journal.ppat.1000665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stetson D, Medzhitov R. Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity. 2006;24:93–9103. [DOI] [PubMed] [Google Scholar]
- 20.Ren T, Zamboni D, Roy C, et al. Flagellin-deficient Legionella mutants evade caspase-1- and Naip5-mediated macrophage immunity. PLoS Pathog;2. Epub ahead of print 2006. DOI: 10.1371/journal.ppat.0020018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Molofsky A, Byrne B, Whitfield N, et al. Cytosolic recognition of flagellin by mouse macrophages restricts Legionella pneumophila infection. J Exp Med. 2006;203:1093–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Derré I, Isberg RR. Macrophages from mice with the restrictive Lgn1 allele exhibit multifactorial resistance to Legionella pneumophila. Infect Immun. 2004;72:6221–6229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Akamine M, Higa F, Haranaga S, et al. Interferon-gamma reverses the evasion of Birc1e/Naip5 gene mediated murine macrophage immunity by Legionella pneumophila mutant lacking flagellin. Microbiol Immunol. 2007;51:279–287. [DOI] [PubMed] [Google Scholar]
- 24.Nakajima A, Nishimura K, Nakaima Y, et al. Cell type-dependent proapoptotic role of Bcl2L12 revealed by a mutation concomitant with the disruption of the juxtaposed Irf3 gene. Proc Natl Acad Sci. 2009;106:12448–12452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coers J, Vance RE, Fontana MF, et al. Restriction of Legionella pneumophila growth in macrophages requires the concerted action of cytokine and Naip5/Ipaf signalling pathways. Cell Microbiol. 2007;9:2344–2357. [DOI] [PubMed] [Google Scholar]
- 26.Zingales B, Andrews NW, Kuwajima VY, et al. Cell surface antigens of Trypanosoma cruzi: possible correlation with the interiorization process in mammalian cells. Mol Biochem Parasitol. 1982;6:111–124. [DOI] [PubMed] [Google Scholar]
- 27.Schwartz S, Agarwala SD, Mumbach MR, et al. High-resolution mapping reveals a conserved, widespread, dynamic mRNA methylation program in yeast meiosis. Cell. 2013;155:1409–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moffat J, Grueneberg DA, Yang X, et al. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell. 2006;124:1283–1298. [DOI] [PubMed] [Google Scholar]
- 31.Decker T, Kovarik P, Meinke A. GAS elements: a few nucleotides with a major impact on cytokine-induced gene expression. J Interferon Cytokine Res Off J Int Soc Interferon Cytokine Res. 1997;17:121–134. [DOI] [PubMed] [Google Scholar]
- 32.Sanda C, Weitzel P, Tsukahara T, et al. Differential gene induction by type I and type II interferons and their combination. J Interferon Cytokine Res Off J Int Soc Interferon Cytokine Res. 2006;26:462–472. [DOI] [PubMed] [Google Scholar]
- 33.Ng S-L, Friedman B, Schmid S, et al. IκB kinase epsilon (IKK(epsilon)) regulates the balance between type I and type II interferon responses. Proc Natl Acad Sci U S A. 2011;108:21170–21175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Decker T, Müller M, Stockinger S. The yin and yang of type I interferon activity in bacterial infection. Nat Rev Immunol. 2005;5:675–687. [DOI] [PubMed] [Google Scholar]
- 35.Abadie A, Besançon F, Wietzerbin J. Type I interferon and TNFalpha cooperate with type II interferon for TRAIL induction and triggering of apoptosis in SK-N-MC EWING tumor cells. Oncogene. 2004;23:4911–4920. [DOI] [PubMed] [Google Scholar]
- 36.Tan H, Derrick J, Hong J, et al. Global transcriptional profiling demonstrates the combination of type I and type II interferon enhances antiviral and immune responses at clinically relevant doses. J Interferon Cytokine Res Off J Int Soc Interferon Cytokine Res. 2005;25:632–649. [DOI] [PubMed] [Google Scholar]
- 37.Zhang X-NN, Liu J-XX, Hu Y-WW, et al. Hyper-activated IRF-1 and STAT1 contribute to enhanced interferon stimulated gene (ISG) expression by interferon alpha and gamma co-treatment in human hepatoma cells. Biochim Biophys Acta. 2005;1759:417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peng T, Zhu J, Hwangbo Y, et al. Independent and cooperative antiviral actions of beta interferon and gamma interferon against herpes simplex virus replication in primary human fibroblasts. J Virol. 2008;82:1934–1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Changotra H, Jia Y, Moore TN, et al. Type I and type II interferons inhibit the translation of murine norovirus proteins. J Virol. 2009;83:5683–5692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gough D, Messina N, Hii L, et al. Functional crosstalk between type I and II interferon through the regulated expression of STAT1. PLoS Biol;8. Epub ahead of print 2010. DOI: 10.1371/journal.pbio.1000361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kropp K, Robertson K, Sing G, et al. Reversible inhibition of murine cytomegalovirus replication by gamma interferon (IFN-γ) in primary macrophages involves a primed type I IFN-signaling subnetwork for full establishment of an immediate-early antiviral state. J Virol. 2011;85:10286–10299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yoshida R, Murray H, Nathan C. Agonist and antagonist effects of interferon alpha and beta on activation of human macrophages. Two classes of interferon gamma receptors and blockade of the high-affinity sites by interferon alpha or beta. J Exp Med. 1988;167:1171–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Teles R, Graeber T, Krutzik S, et al. Type I interferon suppresses type II interferon-triggered human anti-mycobacterial responses. Science. 2013;339:1448–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rayamajhi M, Humann J, Penheiter K, et al. Induction of IFN-alphabeta enables Listeria monocytogenes to suppress macrophage activation by IFN-gamma. J Exp Med. 2010;207:327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fertsch D, Schoenberg DR, Germain RN, et al. Induction of macrophage Ia antigen expression by rIFN-gamma and down-regulation by IFN-alpha/beta and dexamethasone are mediated by changes in steady-state levels of Ia mRNA. J Immunol Baltim Md 1950. 1987;139:244–249. [PubMed] [Google Scholar]
- 46.Kearney S, Delgado C, Lenz LL. Differential effects of type I and II interferons on myeloid cells and resistance to intracellular bacterial infections. Immunol Res. 2013;55:187–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McNab FW, Ewbank J, Howes A, et al. Type I IFN induces IL-10 production in an IL-27-independent manner and blocks responsiveness to IFN-γ for production of IL-12 and bacterial killing in Mycobacterium tuberculosis-infected macrophages. J Immunol Baltim Md 1950. 2014;193:3600–3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nguyen H, Ramana CV, Bayes J, et al. Roles of phosphatidylinositol 3-kinase in interferon-gamma-dependent phosphorylation of STAT1 on serine 727 and activation of gene expression. J Biol Chem. 2001;276:33361–33368. [DOI] [PubMed] [Google Scholar]
- 49.Fitzgerald K, McWhirter S, Faia K, et al. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol. 2003;4:491–496. [DOI] [PubMed] [Google Scholar]
- 50.Servant M, Grandvaux N, tenOever B, et al. Identification of the minimal phosphoacceptor site required for in vivo activation of interferon regulatory factor 3 in response to virus and double-stranded RNA. J Biol Chem. 2003;278:9441–9447. [DOI] [PubMed] [Google Scholar]
- 51.Chen W, Srinath H, Lam SS, et al. Contribution of Ser386 and Ser396 to activation of interferon regulatory factor 3. J Mol Biol. 2008;379:251–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kawai T, Takeuchi O, Fujita T, et al. Lipopolysaccharide Stimulates the MyD88-Independent Pathway and Results in Activation of IFN-Regulatory Factor 3 and the Expression of a Subset of Lipopolysaccharide-Inducible Genes. J Immunol. 2001;167:5887–5894. [DOI] [PubMed] [Google Scholar]
- 53.McWhirter SM, Fitzgerald KA, Rosains J, et al. IFN-regulatory factor 3-dependent gene expression is defective in Tbk1-deficient mouse embryonic fibroblasts. Proc Natl Acad Sci U S A. 2004;101:233–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kishore N, Huynh QK, Mathialagan S, et al. IKK-i and TBK-1 are enzymatically distinct from the homologous enzyme IKK-2: comparative analysis of recombinant human IKK-i, TBK-1, and IKK-2. J Biol Chem. 2002;277:13840–13847. [DOI] [PubMed] [Google Scholar]
- 55.Fujiwara S, Hoshizaki M, Ichida Y, et al. Pulmonary phagocyte-derived NPY controls the pathology of severe influenza virus infection. Nat Microbiol. Epub ahead of print November 19, 2018. DOI: 10.1038/s41564-018-0289-1. [DOI] [PubMed] [Google Scholar]
- 56.MacMicking J, Xie QW, Nathan C. Nitric oxide and macrophage function. Annu Rev Immunol. 1997;15:323–350. [DOI] [PubMed] [Google Scholar]
- 57.Fang FC. Antimicrobial reactive oxygen and nitrogen species: concepts and controversies. Nat Rev Microbiol. 2004;2:820–832. [DOI] [PubMed] [Google Scholar]
- 58.Nahrevanian H. Involvement of nitric oxide and its up/down stream molecules in the immunity against parasitic infections. Braz J Infect Dis Off Publ Braz Soc Infect Dis. 2009;13:440–448. [DOI] [PubMed] [Google Scholar]
- 59.Weiss G, Schaible UE. Macrophage defense mechanisms against intracellular bacteria. Immunol Rev. 2015;264:182–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gebran S, Yamamoto Y, Newton C, et al. Inhibition of Legionella pneumophila growth by gamma interferon in permissive A/J mouse macrophages: role of reactive oxygen species, nitric oxide, tryptophan, and iron(III). Infect Immun. 1994;62:3197–3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Heath L, Chrisp C, Huffnagle G, et al. Effector mechanisms responsible for gamma interferon-mediated host resistance to Legionella pneumophila lung infection: the role of endogenous nitric oxide differs in susceptible and resistant murine hosts. Infect Immun. 1996;64:5151–5160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chakravortty D, Hensel M. Inducible nitric oxide synthase and control of intracellular bacterial pathogens. Microbes Infect Inst Pasteur. 2003;5:621–627. [DOI] [PubMed] [Google Scholar]
- 63.Gazzinelli R, Oswald I, Hieny S, et al. The microbicidal activity of interferon-gamma-treated macrophages against Trypanosoma cruzi involves an L-arginine-dependent, nitrogen oxide-mediated mechanism inhibitable by interleukin-10 and transforming growth factor-beta. Eur J Immunol. 1992;22:2501–2506. [DOI] [PubMed] [Google Scholar]
- 64.Muñoz-Fernández M, Fernández M, Fresno M. Synergism between tumor necrosis factor-alpha and interferon-gamma on macrophage activation for the killing of intracellular Trypanosoma cruzi through a nitric oxide-dependent mechanism. Eur J Immunol. 1992;22:301–307. [DOI] [PubMed] [Google Scholar]
- 65.Silva J, Vespa G, Cardoso M, et al. Tumor necrosis factor alpha mediates resistance to Trypanosoma cruzi infection in mice by inducing nitric oxide production in infected gamma interferon-activated macrophages. Infect Immun. 1995;63:4862–4867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chessler A-DC, Caradonna K, Da’dara A, et al. Type I interferons increase host susceptibility to Trypanosoma cruzi infection. Infect Immun. 2011;79:2112–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Takaoka A, Mitani Y, Suemori H, et al. Cross talk between interferon-gamma and -alpha/beta signaling components in caveolar membrane domains. Science. 2000;288:2357–2360. [DOI] [PubMed] [Google Scholar]
- 68.Neumeister B, Bach V, Faigle M, et al. Induction of iNOS in human monocytes infected with different Legionella species. FEMS Microbiol Lett. 2001;202:31–38. [DOI] [PubMed] [Google Scholar]
- 69.Price JV, Russo D, Ji DX, et al. IRG1 and Inducible Nitric Oxide Synthase Act Redundantly with Other Interferon-Gamma-Induced Factors To Restrict Intracellular Replication of Legionella pneumophila. mBio;10. Epub ahead of print 12 2019. DOI: 10.1128/mBio.02629-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bogdan C. Nitric oxide synthase in innate and adaptive immunity: an update. Trends Immunol. 2015;36:161–178. [DOI] [PubMed] [Google Scholar]
- 71.Nairz M, Haschka D, Demetz E, et al. Iron at the interface of immunity and infection. Front Pharmacol. 2014;5:152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hop HT, Arayan LT, Huy TXN, et al. Lipocalin 2 (Lcn2) interferes with iron uptake by Brucella abortus and dampens immunoregulation during infection of RAW 264.7 macrophages. Cell Microbiol. Epub ahead of print November 22, 2017. DOI: 10.1111/cmi.12813. [DOI] [PubMed] [Google Scholar]
- 73.Guglani L, Gopal R, Javier R-M, et al. Lipocalin 2 regulates inflammation during pulmonary mycobacterial infections. PloS One;7. Epub ahead of print 2011. DOI: 10.1371/journal.pone.0050052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Moschen AR, Adolph TE, Gerner RR, et al. Lipocalin-2: A Master Mediator of Intestinal and Metabolic Inflammation. Trends Endocrinol Metab TEM. 2017;28:388–397. [DOI] [PubMed] [Google Scholar]
- 75.Nairz M, Schleicher U, Schroll A, et al. Nitric oxide-mediated regulation of ferroportin-1 controls macrophage iron homeostasis and immune function in Salmonella infection. J Exp Med. 2013;210:855–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang L, Harrington L, Trebicka E, et al. Selective modulation of TLR4-activated inflammatory responses by altered iron homeostasis in mice. J Clin Invest. 2009;119:3322–3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chow E, Castrillo A, Shahangian A, et al. A role for IRF3-dependent RXRalpha repression in hepatotoxicity associated with viral infections. J Exp Med. 2006;203:2589–2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ysebrant de Lendonck L, Tonon S, Nguyen M, et al. Interferon regulatory factor 3 controls interleukin-17 expression in CD8 T lymphocytes. Proc Natl Acad Sci U S A. 2013;110:E3189–E3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chattopadhyay S, Sen GC. RIG-I-like receptor-induced IRF3 mediated pathway of apoptosis (RIPA): a new antiviral pathway. Protein Cell. 2017;8:165–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang X-A, Zhang R, She Z-G, et al. Interferon regulatory factor 3 constrains IKKβ/NF-κB signaling to alleviate hepatic steatosis and insulin resistance. Hepatol Baltim Md. 2014;59:870–885. [DOI] [PubMed] [Google Scholar]
- 81.Kaiser WJ, Upton JW, Mocarski ES. Receptor-interacting protein homotypic interaction motif-dependent control of NF-kappa B activation via the DNA-dependent activator of IFN regulatory factors. J Immunol Baltim Md 1950. 2008;181:6427–6434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Funami K, Sasai M, Ohba Y, et al. Spatiotemporal mobilization of Toll/IL-1 receptor domain-containing adaptor molecule-1 in response to dsRNA. J Immunol Baltim Md 1950. 2007;179:6867–6872. [DOI] [PubMed] [Google Scholar]
- 83.Jefferson M, Whelband M, Mohorianu I, et al. The pestivirus N terminal protease N(pro) redistributes to mitochondria and peroxisomes suggesting new sites for regulation of IRF3 by N(pro.). PloS One;9. Epub ahead of print 2014. DOI: 10.1371/journal.pone.0088838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Majumdar T, Chattopadhyay S, Ozhegov E, et al. Induction of interferon-stimulated genes by IRF3 promotes replication of Toxoplasma gondii. PLoS Pathog. 2015;11:e1004779. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 85.Gordon S, Plüddemann A. Tissue macrophages: heterogeneity and functions. BMC Biol;15. Epub ahead of print December 2017. DOI: 10.1186/s12915-017-0392-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kanwar JR, Kanwar RK, Burrow H, et al. Recent advances on the roles of NO in cancer and chronic inflammatory disorders. Curr Med Chem. 2009;16:2373–2394. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1: Contribution of MAVS and STING to the maintenance and establishment of the IFNγ-activated state in BMMs
Fig. S2: Low expression of Type I IFN in IFNγ-primed BMMs infected by L. pneumophila
Fig. S3: Variation in gene expression due to IRF3/7 deficiency in BMMs primed with IFNγ and/or infected with L. pneumophila
Fig. S4: Effect of IRF3 on nitrite production and intracellular pathogen restriction in IFNγ and TNFα-primed BMMs infected by T. cruzi
Fig. S5: Contribution of IRF3 kinases to intracellular bacterial growth and the maintenance and establishment of the IFNγ-activated state in BMMs