

Abstract

N-heterospirocycles are interesting structural units found in both natural products and medicinal compounds but have relatively few reliable methods for their synthesis. Here, we enlist the photocatalytic generation of N-centered radicals to construct β-spirocyclic pyrrolidines from N-allylsulfonamides and alkenes. A variety of β-spirocyclic pyrrolidines have been constructed, including drug derivatives, in moderate to very good yields. Further derivatization of the products has also been demonstrated as has a viable scale-up procedure, making use of flow chemistry techniques.
Introduction
Developing new and versatile routes to desirable organic architectures constitutes an important aspect of the molecular assembly of pharmaceutically and agrochemically relevant molecules. One such novel class is spirocyclic derivatives, which feature in natural products and are increasingly both modeled as and observed in medicinal chemistry compounds.1 Heterospirocycles are particularly interesting for several reasons such as being isosteric alternatives to hydrophobic aromatic motifs,2 and they are also amenable to wide-ranging derivatization and heteroatom incorporation to adjust biological profiles.3
Of the methods developed to prepare azaheterospirocycles in recent years, most require multistep procedures;3a,3c,3d,4,5 however, the latest applications of photocatalysis have made this possible in few synthetic steps.6 Indeed, the development of highly efficient light sources, especially LEDs, has triggered a renaissance of interest in photochemical reactions.7 As such, photocatalysis has arisen as a valuable tool that enables the construction of complex molecular structures in a modular fashion under mild conditions via the intermediacy of highly reactive species.8 Of the methods developed to prepare spirocyclic pyrrolidines, most have enabled the preparation of α-spirocyclic pyrrolidines, overcoming some of the difficulties associated with preparing α-tertiary amines (Figure 1).3c,4b−4d,4g,4i,6a β-spirocyclic pyrrolidine scaffolds, despite their increased uptake as medicinal targets, have by comparison received less attention and thus often rely on multistep syntheses.3a−3c,4f,4h
Figure 1.

Strategies Developed to Access Spirocyclic Pyrrolidines.
The generation of nitrogen-centered radicals (NCRs) has been heavily influenced by the emergence of photocatalysis, facilitating several modes of activation of free N–H and N–X bonds.9 The utility of NCRs is enhanced by the ability to attach different groups to the nitrogen atom, conveniently modulating their reactivity.9b,10 As such, many NCR strategies have been shown to successfully incorporate nitrogen into aliphatic substrates and also to construct nitrogen-containing heterocycles, scaffolds, and readily decorated fragments.9 Of these modes, homolytic cleavage of N–X bonds has proved useful and can be executed, among other methods, by direct photoexcitation with UV light or by photosensitization.9 Our group has taken interest in this area and has previously disclosed a one-pot, two-step pyrrolidine ring-forming reaction using electron-rich or activated alkenes, a chlorinating agent, N-allylsulfonamides, and a photosensitizer (either organic or inorganic).11 Here, we report development on this strategy that makes use of the matched electrophilicity of arylsulfonamides and exocyclic olefins that enables access to a variety of β-spirocyclic pyrrolidines in moderate to excellent yields. Further derivatization and scale-up by the use of flow chemistry have also been explored.
Results and Discussion
Adopting N-allyltoluenesulfonamide (1a), 1,3-dichloro-5,5′-dimethylhydantoin, and methylenecyclohexane (2a) as model substrates and (Ir[dF(CF3)ppy]2(dtbpy))PF6 (PC1) as the photocatalyst in the CH2Cl2 solvent, we set about finding viable reaction conditions (Table 1). We were pleased to find that spirocycle 3a was formed in 80% yield by NMR and was isolated in 76% yield (entry 1). N-Boc, N-methyl, and N-acetamide-protected allylamine were all unsuccessful when investigated as alternatives to the sulfonamide-protected allylamine. Likewise, N-chlorosuccinimide and trichloroisocyanuric acid alternative chlorinating reagents gave inferior yields of 1a. As expected, removal of the photocatalyst from the reaction mixture decreased the yield of the product substantially with a large quantity of the N–Cl intermediate remaining unconverted, evidencing its function as a photocatalyst (entry 2). Removal of the light source almost completely prevented any conversion of the intermediate (entry 3). Interestingly, performing the reaction under air only mildly decreased the yield of 3a (entry 4). Switching the solvent to acetonitrile failed to deliver any identifiable spirocyclic product 3a, instead producing 1a as the sole product presumably by hydrogen atom transfer (HAT) of the nitrogen-centered radical from the solvent (entry 5). Using 1,2-dichloroethane (DCE) as the reaction solvent did however give 3a in 62% yield (entry 6). Both increasing and decreasing the reaction concentration for the photochemical step made little impact on the conversion to the product (entries 7 and 8). Reducing the stoichiometry of 2a did decrease the yield (entry 9) and increased the proportion of 1a recovered from the reaction mixture, likely by increasing the rate of HAT from the solvent relative to olefin addition. Increasing the quantity of 2a only had a marginal improvement on the yield (entry 10) and so was not adopted for the remainder of the experiments so as not to unnecessarily expend other potentially valuable olefins. Adopting only a 5 min prestir for N-chlorination in step 1 still gave spirocycle 3a in 76% isolated yield (entry 11). Adopting an organic photocatalyst (4CzIPN) instead of (Ir[dF(CF3)ppy]2(dtbpy))PF6 was also able to deliver 3a in 72% yield, demonstrating the option of switching out expensive inorganic photocatalysts for easily prepared organic alternatives (entry 12).
Table 1. Reaction Optimization.

| entry | deviation from standard conditions | yield (%)a |
|---|---|---|
| 1 | none | 80 (76)b |
| 2 | no photocatalyst | 14c |
| 3 | no light | <5c |
| 4 | under air | 72 |
| 5 | MeCN solvent | 0d |
| 6 | DCE solvent | 62 |
| 7 | 0.1 M (step 2) | 80 |
| 8 | 0.025 M (step 2) | 78 |
| 9 | 2 equiv 2a | 64 |
| 10 | 5 equiv 2a | 82 |
| 11 | 5 min (step 1) | 76b |
| 12 | 4CzIPN used as PC | 72b |
Reaction conditions: 0.2 mmol, 1.0 equiv of N-allyltoluenesulfonamide, 1.5 equiv of 1,3-dichloro-5,5′-dimethylhydantoin, 3.0 equiv of methylenecyclohexane, 0.5 mol % (Ir[dF(CF3)ppy]2(dtbpy))PF6 (PC1), 4 mL total (step 2) of dichloromethane (DCM). 1H NMR conversion to the product.
Isolated yield.
Unreacted N–Cl intermediate observed as the major component by 1H NMR.
N-allyltoluenesulfonamide alone observed by 1H NMR.
With the new reaction conditions established, we set about evaluating the suitability of a range of N-allylsulfonamides (Table 2). The common amine-protecting group (N-tosyl)allylamine underwent spirocyclization in 76% isolated yield to 3a. N-(4-Bromobenzenesulfonyl)allylamine (1b) was less well-suited to the conditions, providing 3b in modest yield. Electron-withdrawn aromatic sulfonamides were well tolerated under the conditions, producing 3c and 3d in very good and good yields, respectively. Heterocyclic sulfonamide 3e was tolerated, albeit modestly, as was 4-(trifluoromethoxy)benzenesulfonamide 3f. Sulfone-containing spirocycle 3g was produced in very good yield. N-allylsulfonamides 1h and 1i produced spirocycles 3h and 3i, respectively, in good and moderate yields, demonstrating the opportunity to have variants of the methyl substituent on the pyrrolidine ring. Alternative halogenating agents were able to yield the desired spirocyclic scaffolds but with alternative halogen atom substituents on the pyrrolidine methyl substituent, providing alkyl bromide 3j and alkyl iodide 3k in very good yields. N-methanesulfonylallylamine (1l) gave no conversion to the product, instead giving full return of the starting material.
Table 2. N-Allylsulfonamide Scopea.


Reaction conditions: 0.2 mmol, 1.0 equiv of N-allyltoluenesulfonamide, 1.5 equiv of 1,3-dichloro-5,5′-dimethylhydantoin, 3.0 equiv of methylenecyclohexane, 0.5 mol % (Ir[dF(CF3)ppy]2(dtbpy))PF6, 4 mL total (step 2) of DCM. a1.5 equiv of 1,3-dibromo-5,5′-dimethylhydantoin was used instead of 1,3-dichloro-5,5′-dimethylhydantoin.
1.5 equiv of 1,3-diiodo-5,5′-dimethylhydantoin was used instead of 1,3-dichloro-5,5′-dimethylhydantoin.
Starting material 1l was the sole observed product in the crude 1H NMR spectrum.
We subsequently turned our attention to the olefin component (Table 3). Aliphatic and aromatic substituted olefins were tolerated well under the reaction conditions, producing separable mixtures of N-heterospirocycle diastereomers (3m and 3n, respectively). The presence of a large number of benzylic protons in the olefin component (2o) however led to a diminished yield of 3o and a large recovery of the HAT product 1a. Spirocyclization of 2-methyleneadamantane (2p) was successful, showing tolerance for α-hindered olefins to produce 3p in a decent yield. Other medicinally and synthetically useful moieties such as gem-difluoro- and acetal-protected ketones were well tolerated, producing 3q and 3r in good yields. Aliphatic rings of varying sizes were also tolerated from small rings (3s), through medium-sized rings (3a and 3t), to macrocyclic rings (3u and 3v), all of which were obtained in good to very good yields. Most noteworthy of these are the macrocyclic rings 3u and 3v (spirocyclic 12- and 15-membered aliphatic rings respectively), which would pose synthetic challenges by other means. Spirocycles constituting two heterocycles were then our main point of focus for novel structures and it indeed proved fruitful. Oxo-heterocycles oxetane-containing bis-spirocycle 3w and tetrahydropyran-containing spirocycle 3x were obtained in 55 and 46% yields, respectively, from their corresponding alkenes. The unusual bis-spirocyclic oxazoline 3y was accessed in an acceptable 39% yield and sulfone-containing heterocycle 3z was also well-suited. Observing recent interest in bis-azaheterocycles for applications such as bioavailable strained-type linkers,3 we next turned our focus to preparing a selection of bis-azaheterospirocycles with orthogonal protecting groups and with both chloromethyl and bromomethyl moieties. Azetidine 3aa was prepared in reasonable yield as was bis-pyrrolidine spirocycle 3ab, though the latter was an inseparable 1:1 mixture of diastereomers. Piperidine ring-containing spirocycles could be successfully prepared in good to very good yields with or without orthogonal protecting groups and with either chloromethyl or bromomethyl substituents (3ac–3af) and also demonstrated the potential to switch out the expensive [Ir] photocatalyst for an easily prepared organic photocatalyst, with no diminishment in yield (3af). Finally, bis-azaheterospirocycles 3ag and 3ah were prepared in moderate and good yields, respectively.
Table 3. Exocyclic Olefin Scopea.


Reaction conditions: 0.2 mmol, 1.0 equiv of N-allyltoluenesulfonamide, 1.5 equiv of 1,3-dihalo-5,5′-dimethylhydantoin, 3.0 equiv of olefin, 0.5 mol % (Ir[dF(CF3)ppy]2(dtbpy))PF6, 4 mL total (step 2) of DCM. aDiastereomeric ratios not able to be determined due to inseparability and substantial overlapping of peaks in the 1H NMR spectra of the isolated diastereomers.
2 equiv of olefin component used.
0.5 mol % 4CzIPN used as photocatalyst instead.
The inevitable formation of the pendant halomethyl group on the pyrrolidine ring opens up opportunities for further diversification of the products. We have previously shown how a pyrrolidine chloromethyl substituent can be altered by nucleophilic substitution and elimination; however, to open up other modes of activation such as cross-coupling, an sp2-Cl moiety was required.11 The realization that other halogens can be installed instead of just chlorine (as found previously) serves to facilitate other derivatization options for the products. As such, we set about to demonstrate a few of these options (Table 4). First, in situ hydrodehalogenation of the alkyl halide using (tristrimethylsilyl)silane (TTMSS) produced derivatives 4a–c in good yields (Table 4a). The hydrodehalogenation reaction to 4a from alkyl bromide 3j was notably slower than from alkyl iodide 3k, requiring 24 h of 470 nm LED irradiation to achieve full conversion (though producing 4a in superior yield). This poses an attractive one-pot (double-photochemical), three-step telescoped method to the hydrodehalogenated β-spirocyclic pyrrolidine products. Alkyl bromide 3ad was further derivatized by an unoptimized Kornblum oxidation to aldehyde 5 in 40% yield and was also amenable to a previously reported photoredox-catalyzed cross-electrophile coupling reaction12 with 3-bromopyridine, forming tris-azaheterocycle 6 in 32% yield (Table 4b).
Table 4. Halomethyl Derivatizations.

Recognizing the ability to easily construct interesting N-heterospirocycles using this method, we became interested in its possible application toward drug derivatization. Sulfonamides are frequently found among APIs, agrochemicals, and natural products and are, therefore, highly desirable targets for selective modification.13 The opportunity to install spirocyclized pyrrolidine rings on sulfonamide-containing drugs using this method is particularly attractive due to the tethered halogen substituent. This can be removed, cross-coupled, or transformed to a plethora of other desirable functional groups, thus opening up a wider range of chemical space to be explored for biological activity. We were therefore pleased to find that a selection of drug molecules could be derivatized in moderate to excellent yields (Table 5). Celecoxib derivatives 3ai and 3aj were prepared in moderate and very good yields, respectively, showing that a variety of spirocyclic compounds from sulfonamide-containing drugs are possible. Valdecoxib, methazolamide, and ethoxzolamide derivatives 3ak, 3aj, and 3am were also successfully prepared. Using the telescoped one-pot hydrodehalogenation procedure (Table 4a), probenecid analogue 4d was obtained in excellent yield. Examples 3ak, 3aj, 3am, and 4d are all compliant with Lipinski’s rule of 5, relating to the favorable physicochemical properties of drug molecules.14
Table 5. Drug Derivativesa.


Reaction conditions: 0.2 mmol, 1.0 equiv of N-allyltoluenesulfonamide, 1.5 equiv of 1,3-dihalo-5,5′-dimethylhydantoin, 3.0 equiv of olefin, 0.5 mol % Ir[dF(CF3)ppy]2(dtbpy)PF6, 4 mL total (step 2) of DCM. a1,3-diiodo-5,5′-dimethylhydantoin used and the resulting alkyl iodide subjected to hydrodehalogenation conditions with 4.0 equiv TTMSS in situ as in Table 4a.
With the application of this method to make a variety of N-heterospirocyclic structures and derivatives illustrated, we turned our attention to addressing the scalability of the reaction. Despite the attraction of photochemistry for facilitating the construction of novel compounds, photochemical reactions are notorious for their poor efficiency, reproducibility, and scalability.15 Many groups, including ours, have found that adopting continuous flow platforms can have several advantages over their batch counterparts by mitigating some of the issues faced with photochemistry such as superior heat distribution, mixing, and photon flux as a result of the small cross section of tubing used in flow setups.7c,16
Perhaps the most attractive aspect of continuous processing, however, particularly in industrial settings, is safety and hazard containment.17 Continuous flow setups have been employed to avoid the build-up of large quantities of hazardous intermediates by generating them in situ, performing the reaction and then quenching any left unreacted, all within a short period of time and in a relatively small volume of tubing.18 This reduces chemical hazards associated with mechanical failure of the reactor and/or leakage, while also providing greater control over thermal runaway pathways due to the high surface-area/volume ratio of flow tubing compared to batch reactors.18,19N-Haloamines, despite their utility in synthesis,20 are often overlooked for large-scale applications due to concerns around toxicity and instability. Indeed, continuous flow platforms have been applied to handling N-chloramines in the synthesis successfully to mitigate some of their associated challenges.21 As such, we saw the potential to tackle both the photochemical scale-up and the N-haloamine instability using a one-flow reaction to execute a decagram scale-up of one particular spirocycle as a demonstration of the process.
Previously, we have made efforts to reduce the overall cost of multigram-scale photocatalytic reactions by employing a photocatalyst recycling method.16g To further such efforts, we reasoned here that the use of a high-power UV lamp for the scaled procedure would make the presence of a photocatalyst unnecessary by facilitating direct photocleavage of the N–X bond. Transferring the batch procedure into flow with the use of a 61 W 365 nm LED, we found the reaction to be equally successful both with and without a photocatalyst. By employing 1.0 equiv of 1a, 2.0 equiv of 1,3-dibromo-5,5′-dimethylhydantoin, 3.0 equiv of olefin 2ad, and a residence time of just 2.5 min in the photoreactor, we were able to prepare bis-azaheterospirocycle 3ad in 71% isolated yield on a 0.2 mmol scale. We then set about performing the scaled-up one-flow two-step reaction so as to generate the N–Br intermediate in situ and consume it directly from the output feed (Scheme 1). With a residence time of 3.33 min at room temperature, the output of reactor R1 was then mixed at a Y-piece with another inlet delivering a solution of olefin 2ad. The resulting solution was then flowed into a UV-150 photoreactor at 34 °C with a residence time of 2.50 min and the output collected, dried, and purified by chromatography to give 13.20 g of 3ad (70% yield) in 3 h. Using this reactor setup, a throughput of 105.6 g day–1 can be achieved for this particular spirocycle.
Scheme 1. Multigram Continuous Flow Construction of β-Spirocyclic Pyrrolidine 3ad.

In summary, we have described a new method to prepare a range of β-spirocyclic pyrrolidines from N-allylsulfonamides, halogenating agents, and exocyclic olefins. Furthermore, we have shown how these products can be further derivatized via the pendant halomethyl group present in the product and also the formation of spirocyclic drug derivatives using this method. Finally, we have demonstrated that this transformation can be executed efficiently in a continuous flow platform on a decagram scale using high-power LEDs, while also mitigating potential hazards surrounding the handling of N-halo intermediates.
Experimental Section
General Methods
All procedures below were conducted under inert nitrogen atmosphere unless stated otherwise. Reagents were supplied by Sigma-Aldrich, Alfa Aesar, Acros, TCI, and Fluorochem and were used as received. Dichloromethane (extra) dry was purchased from Acros Organics and used for all spirocyclization reactions. All substrates were synthesized using the batch photochemical setup (as shown in the Supporting Information (SI)), consisting of a coiled LED strip (8–9 W total power output) on the inside of the tubing (4 cm in height, 8 cm in diameter). Each reaction used commercially available 6 mL microwave vials, cleaned and oven-dried before use. The vials were placed individually in the center of the photoreactor tube and were fan-cooled from above (under these conditions, the reaction temperature did not exceed 35 °C). Flow reactions and scale-up experiment were performed using a Vapourtec E-series comprising three peristaltic pumps and a photoreactor, with an 8 bar BPR at the output. The light source used was a commercially available 61 W radiant power 365 nm (peak intensity) LED from Vapourtec with an irradiation band ranging 350–400 nm. The reactor coil constituted 10 mL total volume of FEP tubing (ID = 1.0 mm). Work-up solvents were obtained from commercial sources and distilled prior to use. Petroleum ether (Pet ether) refers to the fractions of petrol collected between 40 and 60 °C b.p. Flash column chromatography was conducted either using a Biotage SPX system with single-use disposable silica columns of appropriate size (SiliaSep Flash Cartridges 12 or 25 g 40–60 μm ISO04/012) or manually with silica gel 60 (230–400 mesh particle size, 40–63 μm particle size). Thin-layer chromatography analysis was carried out using silica gel 60 F254 precoated glass-backed plates and visualized under UV light (254 nm) or with permanganate or vanillin stains. 1H NMR, 13C{1H} NMR, and 19F NMR were obtained using a 500 MHz Bruker Avance III (Smart Probe 500 MHz) Spectrometer or a Bruker AV400 (Avance 400 MHz) Spectrometer. Chemical shifts (d) are referenced to residual CDCl3 in parts per million (ppm). Coupling constants J are quoted in hertz (Hz). Proton and carbon multiplicity is recorded as singlet (s), doublet (d), double of doublets (dd), triplet (t), quartet (q), pentet/quintet (p), heptet (hept.) multiplet (m), and broad (br), or combinations thereof. All compounds examined were dried in vacuo to remove the residual solvents. 1H NMR signals are reported to two decimal places and 13C{1H} signals to one decimal place. Assignments are made according to the labeling of structures in the experimental section and derived from two-dimensional (2D) spectra, all of which are accessible by request from the authors. The operating frequency used for all nuclei in the included 2D spectra (see the SI) are those reported in the corresponding 1H and 13C NMR spectra captions immediately above the 2D spectra. High-resolution mass spectra (HRMS) were obtained on a Waters Vion IMS QTOF spectrometer. Infrared spectra were recorded neat on a PerkinElmer Spectrum One Fourier transform infrared (FTIR) spectrometer with a universal attenuated total reflection (ATR) sampling accessory, and selected peaks are reported.
General Procedure A: Preparation of N-Allylsulfonamides 1a, 1b, 1c, 1d, 1e, 1f, 1g, 1l, 1aa, and 1ak (Schotten–Baumann-Like Conditions)
N-Allylamine (1.1 equiv) and
dry Et3N (1.1 equiv) were dissolved in DCM (0.25 M) under
N2. The mixture was then stirred and cooled to 0 °C.
The corresponding sulfonyl chloride (1.0 equiv) was added dropwise
if liquid, or in small portions if solid. The resulting reaction mixture
was then stirred at 0 °C for 1 h and then allowed to warm to
room temperature and continue to react for a further 15 h. The crude
solution was then added to sat. NaHCO3, the layers were
separated, and the aqueous layer was extracted with DCM (×3).
The combined organic phases were then washed with brine, dried (MgSO4), filtered, and then concentrated in vacuo to yield the desired N-allylsulfonamide. The literature
data for compounds 1a,111b,221d,221e,111f,231h,241i,251l,22 and 1aa(22) matched the data obtained using this
procedure.
Methyl 4-(N-(But-3-en-1-yl)sulfamoyl)benzoate (1c)
N-Allylamine (165 μL,
2.2 mmol), dry Et3N (307 μL, 2.2 mmol), DCM (8 mL),
and methyl 4-(chlorosulfonyl)benzoate (469 mg, 2.0 mmol) were subjected
to General Procedure A to give compound 1c as a white
solid (444 mg, 87%). 1H NMR (400 MHz, CDCl3)
δ 8.17 (app d, J = 8.6 Hz, 2H, H4), 7.94 (app d, J = 8.6 Hz, 2H, H5),
5.76–5.60 (m, 1H, H8), 5.15 (dd, 1H, J = 17.1, 1.2 Hz, H9), 5.09 (dd, 1H, J = 10.2, 1.2 Hz, H9′), 4.80 (t, J = 6.1 Hz, 1H, N–H), 3.96 (s, 3H, H1), 3.63 (tt, J = 6.1, 1.4 Hz, 2H, H7). 13C{1H} NMR (101 MHz, CDCl3) δ 165.8 (C2), 144.2 (C3), 134.0 (C6), 132.8 (C8), 130.5 (C4), 127.2 (C5), 118.2 (C9), 52.8 (C1), 45.9 (C7). Mp: 80.0–81.9
°C. IR: νmax 3231, 2920, 1701, 1434, 1333, 1287,
1156 cm–1. HRMS: m/z calculated for C11H14NO4S+, 256.0638 [M + H]+. Found m/z 256.0632, Δ = −2.3 ppm.
N-Allyl-4-(methylsulfonyl)benzenesulfonamide (1g)
N-Allylamine (330 μL,
4.4 mmol), dry Et3N (614 μL, 4.4 mmol), DCM (16 mL),
and 4-(methylsulfonyl)benzenesulfonyl chloride (1.02 g, 4.0 mmol)
were subjected to General Procedure A to give compound 1c as a white solid (809 mg, 74%). 1H NMR (400 MHz, CDCl3) δ 8.09 (m, J = 8.5 Hz, 4H, H3 + H4), 5.72 (ddt, J = 16.5, 11.2,
5.9 Hz, 1H, H7), 5.19 (d, J = 17.2 Hz,
1H, H8), 5.14 (d, J = 10.2 Hz, 1H, H8′), 4.64 (t, J = 5.9 Hz, 1H, N–H),
3.68 (t, J = 6.0 Hz, 2H, H6), 3.10 (s,
3H, H1).13C{1H} NMR (101 MHz, CDCl3) δ 145.7 (C5), 144.5 (C2), 132.6
(C7), 128.6 (C3 or C4), 128.3 (C3 or C4), 118.5 (C8), 46.0 (C6), 44.5 (C1). Mp: 147.0–148.6 °C. IR: νmax 3268, 2922, 1435, 1386, 1326, 1308, 1282, 1147 cm–1. HRMS: m/z calculated for C10H14NO4S2+, 276.0359
[M + H]+. Found m/z 276.0354,
Δ = −1.7 ppm. Rf (30% EtOAc in Pet ether) = 0.21.
N-Allyl-4-(5-methyl-3-phenylisoxazol-4-yl)benzenesulfonamide (1ak)
N-Allylamine (124 μL, 1.65 mmol), dry Et3N (229 μL, 1.65 mmol), DCM (6 mL), and 4-(methylsulfonyl)benzenesulfonyl chloride (500 mg, 1.5 mmol) were subjected to General Procedure A to give compound 1c as a white solid (531 mg, 99%). 1H NMR (400 MHz, CDCl3) δ 7.86 (app d, J = 7.7 Hz, 2H, H11), 7.36 (m, 7H, H1 + H2 + H3 + H10), 5.73 (ddt, J = 16.4, 11.0, 5.7 Hz, 1H, H14), 5.17 (d, J = 17.2 Hz, 1H, H15), 5.11 (d, J = 10.2 Hz, 1H, H15′), 4.69 (bs, 1H, N–H), 3.67 (bs, 2H, H13), 2.49 (s, 3H, H8). 13C{1H} NMR (101 MHz, CDCl3) δ 167.4 (C7), 161.2 (C5), 139.4 (C9), 135.4 (C12), 133.0 (C14), 130.5 (C10), 129.9 (C1), 128.8 (C2 or C3), 128.6 (C2 or C3 + C4), 127.6 (C11), 117.9 (C15), 114.6 (C6), 45.9 (C13), 11.9 (C8). Mp: 91.0–93.3 °C. IR: νmax 3293, 1623, 1412, 1394, 1329, 1164 cm–1. HRMS: m/z calculated for C19H19N2O3S+, 355.1111 [M + H]+. Found m/z 355.1109, Δ = −0.5 ppm. Rf (30% EtOAc in Pet ether) = 0.21.
General Procedure B: Preparation of Sulfonamides 1h, 1i, 1ai, and 1al (SN2 Conditions)

Alkyl bromide (1.0 equiv), K2CO3 (2.0 equiv), KI (0.10 equiv), and the corresponding sulfonamide (2.0 equiv) were combined in a round-bottom flask and the mixture subjected to a nitrogen atmosphere. MeCN (0.33 M) was then added and the resultant mixture was heated to 60 °C for 12 h and then cooled to room temperature. The mixture was then filtered and the filtrate concentrated in vacuo. The residue was then subjected to flash chromatography to yield the desired sulfonamide. The literature data for compounds 1h,261i,27 and 1ai(22) matched the data obtained using this procedure.
(E)-N-(5-(N-Allylsulfamoyl)-3-methyl-1,3,4-thiadiazol-2(3H)-ylidene)acetamide (1al)
Allyl bromide (347 μL, 4.0 mmol),
K2CO3 (1.107 g, 8.0 mmol), KI (67 mg, 0.4 mmol),
MeCN (12 mL), and methazolamide (1.89 g, 8.0 mmol) were subjected
to General Procedure B (chromatography eluent: 20–40% EtOAc
in Pet ether) to give compound 1al as a white solid (668
mg, 60%). 1H NMR (400 MHz, CDCl3) δ 5.93–5.73
(m, 2H, H7 + N–H), 5.28 (d, J =
17.1 Hz, 1H, H8), 5.20 (d, J = 10.2 Hz,
1H, H8′), 4.00 (s, 3H, H5), 3.85 (t, J = 5.8 Hz, 2H, H6), 2.36 (s, 3H, H1). 13C{1H} NMR (101 MHz, CDCl3)
δ 181.5 (C2), 165.4 (C3), 156.2 (C4), 132.4 (C7), 118.7 (C8), 46.4 (C6), 38.6 (C5), 26.7 (C1). Mp: 143.6–146.1
°C. IR: νmax 3081, 2881, 1588, 1489, 1457, 1385,
1353, 1312, 1147 cm–1. HRMS: m/z calculated for C8H13N4O3S2+, 277.0424 [M + H]+. Found m/z 277.0418, Δ =
−2.0 ppm. Rf (30% EtOAc in Pet ether) = 0.16.
N-Allyl-6-ethoxybenzo[d]thiazole-2-sulfonamide (1am)
Allyl bromide (241 μL, 2.0 mmol), K2CO3 (276 mg, 2.0 mmol), KI (33.2 mg, 0.2 mmol), MeCN (8 mL), and ethoxzolamide (516.6 mg, 2.0 mmol) were subjected to General Procedure B (chromatography eluent: 20–40% EtOAc in Pet ether) to give compound 1am as a white solid (178 mg, 30%). 13C{1H} NMR (400 MHz, CDCl3) δ 8.02 (d, J = 9.1 Hz, 1H, H7), 7.33 (d, J = 2.5 Hz, 1H, H4), 7.18 (dd, J = 9.1, 2.5 Hz, 1H, H8), 5.80 (ddt, J = 17.0, 10.3, 5.8, 1H, H11), 5.34 (t, J = 6.0 Hz, 1H, N–H), 5.24 (dq, J = 17.1, 1.5 Hz, 1H, H12), 5.12 (dq, J = 10.3, 1.2 Hz, 1H, H12′), 4.12 (q, J = 7.0 Hz, 2H, H2), 3.86 (tt, J = 6.1, 1.4 Hz, 2H, H10), 1.48 (t, J = 7.0 Hz, 3H, H1). 13C{1H} NMR (101 MHz, CDCl3) δ 162.7 (C9), 159.0 (C3), 146.8 (C5), 138.4 (C6), 132.7 (C11), 125.8 (C7), 118.4 (C12), 118.3 (C8), 104.2 (C4), 64.4 (C2), 46.6 (C10), 14.8 (C1). Mp: 115.0–116.1 °C. IR: νmax 3111, 2978, 2935, 1599, 1486, 1471, 1432, 1395, 1330, 1254, 1224 cm–1. HRMS: m/z calculated for C12H15N2O3S2+, 299.0519 [M + H]+. Found m/z 299.0524, Δ = 1.7 ppm. Rf (3% EtOAc in Pet ether) = 0.49.
General Procedure C: Preparation of Exocyclic Olefins 2m, 2n, 2o, 2p, 2q, 2r, 2t, 2u, 2v, 2w, 2x, and 2z (Wittig Conditions)
Following a typical Wittig reaction
procedure, methyltriphenylphosphonium bromide (1.5 equiv) was suspended
in dry tetrahydrofuran (THF) or Et2O (0.2 M) under N2 and then cooled to 0 °C. Fresh tBuOK (1.5 equiv) was added in one portion and the mixture was
stirred for 30 min at 0 °C. Ketone was then added dropwise if
liquid or in small portions if solid. After 1 h, the reaction mixture
was warmed to room temperature and stirred for a further 16 h. The
crude product mixture was then filtered and the filtrate then poured
into aqueous saturated NH4Cl solution. The two phases were
separated and the aqueous phase was extracted with diethyl ether (×3).
The combined organic phases were washed with brine, dried (MgSO4), filtered, and the solvent was removed in vacuo. The crude residue was then purified by silica flash column chromatography
to yield the desired alkene precursor. The literature data for compounds 2m,282n,292o,302p,312q,322r,332t,342u,352x,34 and 2z(36) matched the data
obtained using this procedure.
1-Methyl-3-methylenecyclopentadecane (2v)
Methyltriphenylphosphonium bromide (1.607 g, 4.5 mmol), KOtBu (505 mg, 4.5 mmol), dry THF (15 mL), and muscone (715.2 mg, 776
μL, 3.0 mmol) were subjected to General Procedure C (chromatography
eluent: 0–3% Et2O in Pet ether) to give 2v as a colorless oil (609.3 mg, 86%). 1H NMR (400 MHz,
CDCl3) δ 4.74 (s, 1H, H16), 4.69 (s, 1H,
H16′), 2.10 (dd, J = 13.5, 6.0
Hz, 1H, H2), 2.06–1.89 (m, 2H, H15),
1.76 (dd, J = 13.4, 7.9 Hz, 1H, H2′), 1.68–1.56 (m, 1H, H3), 1.53–1.04 (m,
22H, H4–14), 0.84 (d, J = 6.6 Hz,
3H, H17). 13C{1H} NMR (101 MHz, CDCl3) δ 149.4 (C1), 110.9 (C16), 44.3
(C2), 35.6 (C4), 35.2 (C15), 29.6
(C3), 27.7 (C5–14), 27.2 (C5–14), 27.0 (C5–14), 27.0 (C5–14),
26.9 (C5–14), 26.8 (2C, C5–14),
26.8 (C5–14), 26.6 (C5–14), 25.4
(C5–14), 20.4 (C17). IR: νmax 2923, 2855, 1642, 1458, 1375 cm–1. HRMS: m/z calculated for C17H32Na+, 259.2396 [M + Na]+. Found m/z 259.2395, Δ = −0.5 ppm.
Rf (5% Et2O in Pet ether) = 0.78.
7-Methylene-2-oxaspiro[3.5]nonane (2w)
Methyltriphenylphosphonium bromide (0.4432 g, 1.21 mmol), KOtBu (136 mg, 1.21 mmol), dry Et2O (4 mL), and 2-oxaspiro[3.5]nonan-7-one (113.0 mg, 0.81 mmol) were subjected to General Procedure C to give 2w as a colorless oil (105.0 mg, 94%). 1H NMR (400 MHz, CDCl3) δ 4.64 (s, 2H, H1), 4.42 (s, 4H, H6), 2.15–2.03 (m, 4H, H3), 1.90–1.77 (m, 4H, H4). 13C{1H} NMR (126 MHz, CDCl3) δ 147.5 (C2), 108.0 (C1), 82.0 (C6), 40.2 (C5), 36.6 (C4), 31.4 (C3). HRMS: m/z calculated for C9H14ONH4+, 156.1383 [M + NH4]+. Found m/z 156.1376, Δ = −4.5 ppm. Rf (10% EtOAc in Pet ether) = 0.33.
General Procedure D: Preparation of Heterocyclic Exocyclic Olefins 2aa, 2ab, 2ac, and 2ag
To the N-Boc-protected olefin*, 4.0 N HCl in 1,4-dioxane was added dropwise (4 equiv) over a period of 10 min to keep the reaction temperature below 30 °C and then stirred at room temperature for 3 h. The solvent was then removed in vacuo to yield crude aminium chloride. The crude was then taken up in DCM (0.67 M). To this solution was added water (3:2 DCM:H2O solvent), K2CO3 (2.4 equiv), and the corresponding sulfonyl chloride (1.0 equiv) with vigorous stirring. After stirring for 16 h, the layers were separated and the organic layer extracted with DCM (×2). The combined organic layers were then washed with brine, dried (MgSO4), and the solvent then removed in vacuo. The crude residue was then purified by flash chromatography to yield the desired heterocyclic olefin. The literature data for compounds 2aa(37) and 2ac(37) matched the data obtained using this procedure.
*Where the N-Boc-protected
olefin was not commercially available, it was instead prepared from
the corresponding ketone using the Wittig reaction conditions of General
Procedure C.
3-Methylene-1-((4-nitrophenyl)sulfonyl)pyrrolidine (2ab)
tert-Butyl 3-methylenepyrrolidine-1-carboxylate
(1.833 g, 10.0 mmol), 4.0 N HCl in 1,4-dioxane (20 mL), K2CO3 (3.33 g, 24.0 mmol), and nosyl chloride (2.22 g, 10.0
mmol) were subjected to General Procedure D (chromatography eluent
20% EtOAc in Pet ether) to give 2ab as a white solid
(1.655 g, 62%). 1H NMR (400 MHz, CDCl3) δ
8.39 (app d, J = 8.8 Hz, 2H, H2), 8.01
(app d, J = 8.8 Hz, 2H, H3), 5.01–4.96
(m, 1H, H9), 4.96–4.91 (m, 1H, H9′) 3.85 (s, 2H, H5), 3.37 (t, J = 7.1
Hz, 2H, H8), 2.53 (t, J = 7.1 Hz, 2H,
H7). 13C{1H} NMR (101 MHz, CDCl3) δ 150.4 (C1), 143.1 (C6), 142.3
(C4), 128.9 (C3), 124.5 (C2), 108.4
(C9), 51.9 (C5), 48.3 (C8), 31.9
(C7). Mp: 109.0–111.6 °C. IR: νmax 3110, 1535, 1346, 1314, 1161 cm–1. HRMS: m/z calculated for C11H13N2O4S+, 269.0590 [M + H]+. Found m/z 269.0579, Δ
= −4.1 ppm.
2-Methylene-7-tosyl-7-azaspiro[3.5]nonane (2ag)
tert-Butyl 2-methylene-7-azaspiro[3.5]nonane-7-carboxylate (2.37 g, 10.0 mmol), 4.0 N HCl in 1,4-dioxane (20 mL), K2CO3 (3.32 g, 24.0 mmol), and tosyl chloride (1.90 g, 10.0 mmol) were subjected to General Procedure D (chromatography eluent 30% Et2O in Pet ether) to give 2ab as a white solid (2.27 g, 78%). 1H NMR (400 MHz, CDCl3) δ 7.65 (app d, J = 8.0 Hz, 2H, H4), 7.34 (app d, J = 8.0 Hz, 2H, H3), 4.80 (s, 2H, H11), 3.04–2.85 (m, 4H, H6), 2.46 (s, 3H, H1), 2.33 (s, 4H, H9), 1.78–1.64 (m, 4H, H7). 13C{1H} NMR (101 MHz, CDCl3) δ 143.9 (C10), 143.5 (C5), 133.5 (C2), 129.7 (C3), 127.8 (C4), 108.0 (C11), 43.7 (C6), 41.8 (C9), 36.1 (C7), 33.0 (C8), 21.7 (C1). Mp: 129.6–132.9 °C. IR: νmax 2920, 2846, 1462, 1348, 1328, 1158 cm–1. HRMS: m/z calculated for C16H22NO2S+, 292.1366 [M + H]+. Found m/z 292.1362, Δ = −1.3 ppm. Rf (30% Et2O in Pet ether) = 0.40.
General Procedure E: Preparation of β-Halomethylated γ-Spirocyclic Pyrrolidines
Conditions E1: In a 6 mL microwave vial was placed N-allylsulfonamide (0.2 mmol, 1.0 equiv), 1,3-dichloro-5,5′-dimethylhydantoin (0.3 mmol, 1.5 equiv), and [Ir(dF(CF3)ppy)2(dtbpy)]PF6 (0.001 mmol, 0.5 mol %). The vial was then capped, degassed, and backfilled with N2 (×3), and then dry DCM (1 mL, 0.2 M) was added. The resulting solution was then stirred at room temperature for 10 min. The reaction mixture was checked by TLC (20% EtOAc in P.E.) and then dry DCM (3 mL, 0.05 M total) and the corresponding olefin (0.6 mmol, 3.0 equiv)* were added. The reaction mixture was then stirred under 470 nm LED irradiation with fan-cooling (see the SI for the setup) for 3 h. The crude reaction mixture was then concentrated in vacuo and then purified by silica flash column chromatography to yield the desired spirocyclic pyrrolidine product. Conditions E2: Same conditions, but using 4CzIPN (0.001 mol, 0.5 mol %) as the photosensitizer. Conditions E3: Same conditions, but using 1,3-dibromo-5,5′-dimethylhydantoin (0.3 mmol, 1.5 equiv) as the halogenating reagent. Conditions E4: Same conditions, but using 1,3-diiodo-5,5′-dimethylhydantoin (0.3 mmol, 1.5 equiv) as the halogenating reagent.
*If the olefin was a solid, it was predissolved in the aforementioned 3 mL of dry DCM added to the reaction mixture after the 10 min prestir, ensuring that the solution was flushed with N2.
General Procedure F: Preparation of β-Methylated γ-Spirocyclic Pyrrolidines
Conditions F1: In a 6 mL microwave vial was placed N-allylsulfonamide (0.2 mmol, 1.0 equiv), diiodo-5,5′-dimethylhydantoin (0.3 mmol, 1.5 equiv), and [Ir(dF(CF3)ppy)2(dtbpy)]PF6 (0.001 mmol, 0.5 mol %). The vial was then capped, degassed, and backfilled with N2 (×3), and then dry DCM (1 mL, 0.2 M) was added. The resulting solution was then stirred at room temperature for 10 min. The reaction mixture was checked by TLC (20% EtOAc in P.E.), and then dry DCM (3 mL, 0.05 M total) and the corresponding olefin (0.6 mmol, 3.0 equiv)* were added. The reaction mixture was then stirred under 470 nm LED irradiation with fan-cooling (see the SI for the setup) for 3 h. Tris(trimethylsilyl)silane (247 μL, 0.8 mmol, 4.0 equiv) was then added by syringe to the reaction mixture and the resulting solution was immediately resubjected to 470 nm LED irradiation with fan-cooling for 14 h. The crude reaction mixture was then concentrated in vacuo and then purified by silica flash column chromatography to yield the desired spirocyclic pyrrolidine product. Conditions F2: Same conditions, but using 1,3-dibromo-5,5′-dimethylhydantoin (0.3 mmol, 1.5 equiv) as the halogenating reagent.
*If the olefin was a solid, it was predissolved in the aforementioned 3 mL of dry DCM added to the reaction mixture after the 10 min prestir, ensuring that the solution was flushed with N2.
Scale-Up Procedure
Scale-up procedure was executed using a Vapourtec E-series fitted with a UV-150 photoreactor with 61 W 365 nm LED and a 10 mL heated jacket coil reactor (see the SI for the detailed setup). Independent solutions of N-allyltoluenesulfonamide 1a (7.40 g, 35.0 mmol, 0.2 M), 1,3-dibromo-5,5′-dimethylhydantoin (20.01 g, 70 mmol, 0.2 M), and 4-methylene-1-tosylpiperidine 2ad (26.39 g, 105 mmol, 0.6 M) in dry DCM under nitrogen were prepared (total volume of reagent solutions was 700 mL) and stirred throughout the experiment at room temperature. By use of the embedded peristaltic pumps on the Vapourtec E-series apparatus, the solutions of 1a and 1,3-dibromo-5,5′-dimethylhydantoin were mixed (1.00 and 2.00 mL min–1 flow rates, respectively) at a Y-piece and then passed through a coil reactor (10 mL reactor volume, 28 °C, tR = 3.33 min). The output of this reactor was then mixed at a Y-piece with an inlet stream of the 2ad solution (1.00 mL min–1) and the output fed into the UV-150 photoreactor with 61 W 365 nm LED attachment (LED was operated at 100% power). The output was collected via an 8 bar BPR in a 1 L RBF. Upon completion of the reaction (3 h), the solvent of the product solution was removed in vacuo and the residue subjected to chromatography (15–25% EtOAc in Pet ether) to produce bis-azaheterospirocycle 3ad as a white crystalline solid (13.20 g, 70%).
Characterization of Products 3a-al, 4a–d, 5, and 6
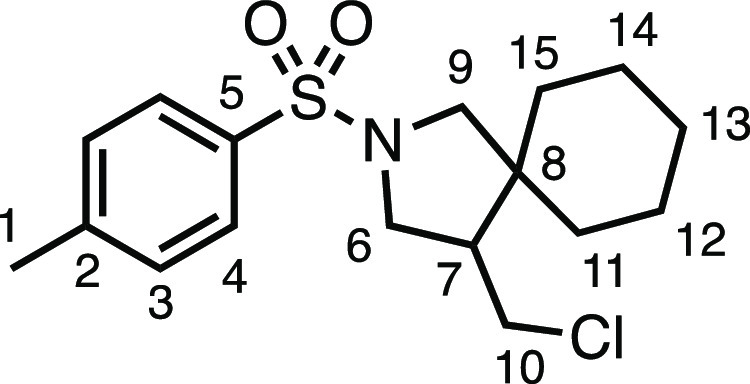
4-(Chloromethyl)-2-tosyl-2-azaspiro[4.5]decane (3a)
The compound was prepared according to General Procedure
E1. Flash column chromatography (5–30% Et2O in Pet
ether) afforded the product as a colorless oil (52.0 mg, 76%). 1H NMR (400 MHz, CDCl3) δ 7.72 (app d, J = 8.1 Hz, 2H, H4), 7.32 (app d, J = 8.1 Hz, 2H, H3), 3.56 (dd, J = 10.2,
7.4 Hz, 1H, H6), 3.51 (dd, J = 10.9, 4.1
Hz, 1H, H10), 3.32 (d, J = 10.1 Hz, 1H,
H9), 3.22 (dd, J = 10.2, 6.9 Hz, 1H, H6′), 3.09 (app t, J = 10.9 Hz, 1H,
H10′), 3.04 (d, J = 10.1 Hz, 1H,
H9′), 2.42 (s, 3H, H1), 2.06 (dtd, J = 10.9, 7.1, 4.2 Hz, 1H, H7), 1.60–1.43
(m, 3H, H11–H15), 1.37–1.00 (m,
7H, H11–H15). 13C{1H} NMR (101 MHz, CDCl3) δ 143.6 (C5),
133.8 (C2), 129.8 (C3), 127.5 (C4), 56.4 (C9), 50.5 (C6), 50.2 (C7), 44.9 (C8), 43.4 (C10), 35.7 (C11–15), 29.1 (C11–15), 25.8 (C11–15), 23.3 (C11–15), 22.8 (C11–15), 21.6 (C1). IR: νmax 2928, 2854, 1340,
1158 cm–1. HRMS: m/z calculated for C17H25ClNO2S+, 342.1289 [M + H]+. Found m/z 342.1295, Δ = 1.6 ppm. Rf (30% Et2O in
Pet ether) = 0.35.
2-((4-Bromophenyl)sulfonyl)-4-(chloromethyl)-2-azaspiro[4.5]decane (3b)
The compound was prepared according to
General Procedure E1. Flash column chromatography (5–10% EtOAc
in Pet ether) afforded the product as a white solid (22.7 mg, 28%). 1H NMR (400 MHz, CDCl3) δ 7.77–7.60
(m, 4H, H2 + H3), 3.58 (dd, J = 10.7, 7.8 Hz, 1H, H5), 3.54 (dd, J = 11.1, 3.8 Hz, 1H, H9), 3.34 (d, J =
10.1 Hz, 1H, H8), 3.25 (dd, J = 10.3,
6.9 Hz, 1H, H5′), 3.14 (t, J =
10.8 Hz, 1H, H9′), 3.05 (d, J =
10.1 Hz, 1H, H8′), 2.11 (dtd, J = 11.0, 7.1, 4.2 Hz, 1H, H6), 1.61–1.46 (m, 3H,
H10–14), 1.40–1.32 (m, 1H, H10–14), 1.27–1.06 (m, 6H, H10–14). 13C{1H} NMR (101 MHz, CDCl3) δ 136.1 (C4), 132.5 (C2), 129.0 (C3), 127.9 (C1), 56.5 (C8), 50.6 (C5), 50.2 (C6), 45.0 (C7), 43.4 (C9), 35.7 (C10–14), 29.1 (C10–14), 25.8 (C10–14), 23.4 (C10–14), 22.8 (C10–14). Mp: 127.9–130.9 °C. IR: νmax 2935, 2852, 1333, 1160, 1142 cm–1. HRMS: m/z calculated for C16H22BrClNO2S+, 406.0238 [M + H]+. Found m/z 406.0241, Δ =
0.7 ppm. Rf (10% EtOAc in Pet ether) = 0.12.
Methyl 4-((4-(Chloromethyl)-2-azaspiro[4.5]decan-2-yl)sulfonyl)benzoate (3c)
The compound was prepared according to
General Procedure E1. Flash column chromatography (10% EtOAc in Pet
ether) afforded the product as a white solid (57.2 mg, 74%). 1H NMR (400 MHz, CDCl3) δ 8.18 (app d, J = 8.4 Hz, 2H, H4), 7.90 (app d, J = 8.4 Hz, 2H, H5), 3.95 (s, 3H, H1), 3.59
(dd, J = 10.3, 7.4 Hz, 1H, H7), 3.51 (dd, J = 10.9, 4.0 Hz, 1H, H11), 3.34 (d, J = 10.1 Hz, 1H, H10), 3.25 (dd, J = 10.3, 6.9 Hz, 1H, H7′), 3.10 (app t, J = 10.8 Hz, 1H, H11′), 3.07 (d, J = 10.1 Hz, 1H, H10′), 2.08 (dtd, J = 10.9, 7.0, 4.1 Hz, 1H, H8), 1.60–0.99
(m, 10H, H12–16). 13C{1H}
NMR (101 MHz, CDCl3) δ 165.8 (C2), 140.8
(C3), 134.0 (C6), 130.4 (C4), 127.4
(C5), 56.4 (C10), 52.8 (C1), 50.5
(C7), 50.1 (C8), 44.9 (C9), 43.3
(C11), 35.6 (C12–16), 29.0 (C12–16), 25.7 (C12–16), 23.3 (C12–16), 22.7 (C12–16). Mp: 94.0–96.0 °C.
IR: νmax 2932, 2849, 1724, 1346, 1277, 1163 cm–1. HRMS: m/z calculated
for C18H25ClNO4S+, 386.1191
[M + H]+. Found m/z 386.1189,
Δ = 0.5 ppm. Rf (30% EtOAc in Pet ether) = 0.35.
2-((3,5-Bis(trifluoromethyl)phenyl)sulfonyl)-4-(chloromethyl)-2-azaspiro[4.5]decane (3d)
The compound was prepared according to
General Procedure E1. Flash column chromatography (5% EtOAc in Pet
ether) afforded the product as a white solid (61.5 mg, 66%). 1H NMR (400 MHz, CDCl3) δ 8.28 (s, 2H, H4), 8.09 (s, 1H, H3), 3.62 (dd, J = 10.3, 7.4 Hz, 1H, H6), 3.56 (dd, J = 11.0, 4.1 Hz, 1H, H10), 3.41 (d, J = 10.2 Hz, 1H, H9), 3.29 (dd, J = 10.3,
6.6 Hz, 1H, H6′), 3.24–3.11 (app t, 1H, J = 10.8 Hz, H10′), 3.24–3.11 (d, J = 10.2 Hz, 1H, H9′), 2.18 (dtd, J = 10.8, 7.0, 4.1 Hz, 1H, H7), 1.63–1.48
(m, 3H, H11–15), 1.43–1.34 (m, 1H, H11–15), 1.32–1.10 (m, 6H, H11–15). 13C{1H} NMR (101 MHz, CDCl3)
δ 140.5 (C5), 133.1 (q, J = 34.5
Hz, C2), 127.5 (q, 3.9 Hz, C4), 126.3 (hept., J = 3.4 Hz, C3) 122.6 (q, J =
273.4 Hz, C1), 56.7 (C9), 50.5 (C6), 50.0 (C7), 45.1 (C8), 43.1 (C10), 35.6 (C11–15), 29.1 (C11–15), 25.8 (C11–15), 23.2 (C11–15), 22.7 (C11–15). Mp: 111.0–114.0 °C.
IR: νmax 3084, 2940, 2867, 1353, 1283, 1269, 1166,
1127 cm–1. HRMS: m/z calculated for C18H21ClF6NO2S+, 464.0880 [M + H]+. Found m/z 464.0881, Δ = 0.2 ppm. Rf (10%
EtOAc in Pet ether) = 0.45.
3-(5-((4-(Chloromethyl)-2-azaspiro[4.5]decan-2-yl)sulfonyl)thiophen-2-yl)-5-(trifluoromethyl)isoxazole (3e)
The compound was prepared according to
General Procedure E1. Flash column chromatography (5–10% EtOAc
in Pet ether) afforded the product as a colorless oil (32.0 mg, 34%). 1H NMR (400 MHz, CDCl3) δ 7.61 (d, J = 3.9 Hz, 1H, H7), 7.52 (d, J = 3.9 Hz, 1H, H6), 6.99 (s, 1H, H3), 3.67
(dd, J = 10.5, 7.5 Hz, 1H, H9), 3.57 (dd, J = 10.9, 4.0 Hz, 1H, H13), 3.44 (d, J = 10.2 Hz, 1H, H12), 3.35 (dd, J = 10.5, 6.9 Hz, 1H, H9′), 3.20 (app t, J = 10.9 Hz, 1H, H13′), 3.18 (d, J = 10.2 Hz, 1H, H12′), 2.16 (dtd, J = 11.0, 7.1, 4.2 Hz, 1H, H10), 1.60–1.11
(m, 10H, H14–18). 13C{1H}
NMR (101 MHz, CDCl3) δ 160.1 (q, J = 43.2 Hz, C2), 156.9 (C4), 140.0 (C8), 134.4 (C5), 132.1 (C7), 128.5 (C6), 117.7 (q, J = 270.9 Hz, C1), 103.7
(app d, J = 2.1 Hz, C3), 56.7 (C12), 50.8 (C9), 50.2 (C10), 45.2 (C11), 43.3 (C13), 35.7 (C14–18), 29.2 (C14–18), 25.8 (C14–18), 23.4 (C14–18), 22.8 (C14–18). IR: νmax 2932, 1553, 1350, 1314, 1153 cm–1. HRMS: m/z calculated for C18H21ClF3N2O3S2+, 469.0629 [M + H]+. Found m/z 469.0628, Δ = −0.2 ppm. Rf (10% EtOAc in
Pet ether) = 0.12.
4-(Chloromethyl)-2-((4-(trifluoromethoxy)phenyl)sulfonyl)-2-azaspiro[4.5]decane (3f)
The compound was prepared according to
General Procedure E1. Flash column chromatography (5–10% EtOAc
in Pet ether) afforded the product as a colorless oil (22.1 mg, 27%). 1H NMR (400 MHz, CDCl3) δ 7.90 (app d, J = 8.6 Hz, 2H, H3), 7.37 (app d, J = 8.6 Hz, 2H, H4), 3.60 (dd, J = 10.3,
7.4 Hz, 1H, H6), 3.54 (dd, J = 10.9, 4.0
Hz, 1H, H10), 3.35 (d, J = 10.1 Hz, 1H,
H9), 3.26 (dd, J = 10.3, 6.9 Hz, 1H, H6′), 3.14 (t, J = 10.8 Hz, 1H, H10′), 3.08 (d, J = 10.1 Hz, 1H, H9′), 2.12 (dtd, J = 11.0, 7.1, 4.2
Hz, 1H, H7), 1.52 (m, 3H, H11–15), 1.39–1.31
(m, 1H, H11–15), 1.26–1.05 (m, 6H, H11–15). 13C{1H} NMR (101 MHz,
CDCl3) δ 152.4 (q, J = 1.9 Hz, C2), 135.5 (C5), 129.5 (C4), 121.1 (q, J = 1.1 Hz, C3), 120.4 (q, J = 259.3 Hz, C1), 56.4 (C9), 50.6 (C6), 50.2 (C7), 45.0 (C8), 43.3 (C10), 35.6 (C11–15), 29.1 (C11–15), 25.8 (C11–15), 23.3 (C11–15), 22.8 (C11–15). IR: νmax 2931,
2858, 1347, 1251, 1208, 1157 cm–1. HRMS: m/z calculated for C17H22ClF3NO3S+, 412.0955 [M +
H]+. Found m/z 412.0958,
Δ = 0.7 ppm. Rf (10% EtOAc in Pet ether) = 0.23.
4-(Chloromethyl)-2-((4-(methylsulfonyl)phenyl)sulfonyl)-2-azaspiro[4.5]decane (3g)
The compound was prepared according to
General Procedure E1. Flash column chromatography (30–40% EtOAc
in Pet ether) followed by recrystallization by vapor diffusion (DCM
with Pet ether antisolvent) afforded the product as a white solid
(76.2 mg, 83%). 1H NMR (400 MHz, CDCl3) δ
8.12 (app d, J = 8.3 Hz, 2H, H4), 8.03
(app d, J = 8.3 Hz, 2H, H3), 3.60 (dd, J = 10.2, 7.5 Hz, 1H, H6), 3.54 (dd, J = 10.9, 4.0 Hz, 1H, H10), 3.37 (d, J = 10.1 Hz, 1H, H9), 3.27 (dd, J = 10.2, 6.9 Hz, 1H, H6′), 3.14 (t, J = 10.9 Hz, 1H, H10′), 3.10 (s, 3H, H1), 3.08 (d, J = 10.1 Hz, 1H, H9′), 2.13 (dtd, J = 10.9, 7.5, 4.0 Hz, 1H, H7), 1.73–1.43 (m, 3H, H11–H15),
1.41–1.04 (m, 7H, H11–H15). 13C{1H} NMR (101 MHz, CDCl3) δ
144.4 (C2), 142.3 (C5), 128.5 (C3), 128.3 (C4), 56.5 (C9), 50.6 (C6), 50.0 (C7), 45.0 (C8), 44.4 (C1), 43.3 (C10), 35.6 (C11–15), 29.0 (C11–15), 25.7 (C11–15), 23.3 (C11–15), 22.8 (C11–15). Mp: 170.0–174.1
°C. IR: νmax 2923, 2851, 1336, 1307, 1285, 1164,
1145 cm–1. HRMS: m/z calculated for C17H25ClNO4S2+, 406.0908 [M + H]+. Found m/z 406.0915, Δ = 1.8 ppm. Rf (40%
EtOAc in Pet ether) = 0.28.
4-(Chloro(phenyl)methyl)-2-tosyl-2-azaspiro[4.5]decane (3h)
The compound was prepared according to General
Procedure E1. Flash column chromatography (5–10% EtOAc in Pet
ether) afforded a single diastereomer of the product as a white solid
(53.0 mg, 63%). 1H NMR (400 MHz, CDCl3) δ
7.76 (app d, J = 8.2 Hz, 2H, H4), 7.46–7.20
(m, 7H, H3 + H17 + H18 + H19), 4.82 (d, J = 9.4 Hz, 1H, H10), 3.79
(dd, J = 10.3, 8.1 Hz, 1H, H6), 3.61 (d, J = 10.1 Hz, 1H, H9), 3.34 (t, J = 10.0 Hz, 1H, H6′), 2.93 (d, J = 10.1 Hz, 1H, H9′), 2.55–2.35 (m, 4H,
H1 + H7), 1.45 (s, 1H, H11–15), 1.30–0.68 (m, 8H, H11–15), 0.43 (td, J = 13.2, 3.8 Hz, 1H, H11–15). 13C{1H} NMR (126 MHz, CDCl3) δ 143.6 (C5), 140.6 (C16), 134.0 (C2), 129.8 (C3), 128.9 (C19), 128.8 (C4 or C17 or C18), 127.6 (C4 or C17 or C18), 127.6 (C4 or C17 or C18), 63.1 (C10), 56.9 (C9), 54.9 (C7), 51.1 (C6), 45.2 (C8), 35.8 (C11–15), 27.9 (C11–15), 25.7 (C11–15), 23.6 (C11–15), 22.5 (C11–15), 21.7 (C1). Mp: 139.4–144.8 °C. IR: νmax 2943, 1342, 1168 cm–1. HRMS: m/z calculated for C23H29ClNO2S+, 418.1602 [M + H]+. Found m/z 418.1586, Δ =
−3.8 ppm. Rf (20% EtOAc in Pet ether) = 0.39.
4-(1-Chloroethyl)-2-tosyl-2-azaspiro[4.5]decane (3i)
The compound was prepared according to General Procedure
E1. Flash column chromatography (5–10% EtOAc in Pet ether)
afforded two separated diastereoisomers (3:2 d.r.) (total 26.8 mg,
38%). Major diastereoisomer (colorless oil, 16.0
mg): 1H NMR (400 MHz, CDCl3) δ 7.73 (app
d, J = 8.1 Hz, 2H, H4), 7.31 (app d, J = 8.1 Hz, 2H, H3), 4.12–4.03 (m, 1H,
H10), 3.61 (dd, J = 10.3, 8.4 Hz, 1H,
H6), 3.36 (d, J = 9.7 Hz, 1H, H9), 3.28 (dd, J = 10.3, 6.9 Hz, 1H, H6′), 3.14 (d, J = 9.7 Hz, 1H, H9′), 2.42 (s, 3H, H1), 1.97–1.89 (m, 1H, H7), 1.62–1.48 (m, 3H, H11–15), 1.45 (d, J = 6.7 Hz, 3H, H16), 1.41–1.10 (m, 7H,
H11–15). 13C{1H} NMR (101
MHz, CDCl3) δ 143.4 (C5), 134.0 (C2), 129.7 (C3), 127.7 (C4), 57.0 (C10), 56.4 (C9), 54.7 (C7), 48.9 (C6), 45.0 (C8), 36.9 (C11–15),
29.0 (C11–15), 25.9 (C11–15),
25.8 (C16), 23.4 (C11–15), 23.0 (C11–15), 21.7 (C1). HRMS: m/z calculated for C18H27ClNO2S+, 356.1445. [M + H]+. Found m/z 356.1447, Δ = 0.5 ppm. Rf (20%
EtOAc in Pet ether) = 0.36. Minor diastereoisomer (colorless oil, 10.8 mg): 1H NMR (400 MHz, CDCl3) δ 7.72 (d, J = 8.1 Hz, 2H, H4), 7.33 (d, J = 8.1 Hz, 2H, H3), 4.01
(p, J = 6.6 Hz, 1H, H10), 3.47–3.35
(m, 2H, H6 + H9), 3.05 (t, J = 10.0 Hz, 2H, H6′ + H9′), 2.44
(s, 3H, H1), 2.08 (q, J = 8.1 Hz, 1H,
H7), 1.84–1.74 (m, 1H, H11–16),
1.56–1.06 (m, 12H, H11–16). 13C{1H} NMR (101 MHz, CDCl3) δ 143.7 (C5), 133.7 (C2), 129.8 (C3), 127.6 (C4), 56.7 (C9), 56.0 (C10), 55.2 (C7), 49.3 (C6), 45.1 (C8), 36.8 (C11–15), 28.5 (C11–15), 25.8 (C11–15), 24.1 (C16), 23.8 (C11–15), 22.9 (C11–15), 21.7 (C1). HRMS: m/z calculated for C18H27ClNO2S+, 356.1445 [M + H]+. Found m/z 356.1447, Δ =
0.5 ppm. Rf (20% EtOAc in Pet ether) = 0.42.
4-(Bromomethyl)-2-tosyl-2-azaspiro[4.5]decane (3j)
The compound was prepared according to General Procedure
E3. Flash column chromatography (5–10% EtOAc in Pet ether)
afforded the product as a colorless oil (54.3 mg, 71%). 1H NMR (400 MHz, CDCl3) δ 7.72 (app d, J = 8.1 Hz, 2H, H4), 7.32 (app d, J =
8.1 Hz, 2H, H3), 3.61 (dd, J = 10.3, 7.4
Hz, 1H, H6), 3.38 (dd, J = 9.9, 3.8 Hz,
1H, H10), 3.37 (d, J = 10.1 Hz, 1H, H9), 3.19 (dd, J = 10.3, 7.3 Hz, 1H, H6′), 3.03 (d, J = 10.1 Hz, 1H, H9′), 2.94 (dd, J = 11.4, 10.1 Hz, 1H,
H10′), 2.42 (s, 3H, H1), 2.12 (dtd, J = 11.2, 7.3, 3.7 Hz, 1H, H7), 1.60–1.43
(m, 3H, H11–15), 1.37–1.00 (m, 7H, H11–15). 13C{1H} NMR (101 MHz,
CDCl3) δ 143.5 (C5), 133.7 (C2), 129.7 (C3), 127.4 (C4), 56.3 (C9), 51.5 (C6), 50.5 (C7), 45.4 (C8), 35.5 (C11–15), 31.5 (C11–15), 28.8 (C11–15), 25.7 (C11–15), 22.6 (C11–15), 21.6 (C1). IR: νmax 2924, 2854, 1340, 1157 cm–1. HRMS: m/z calculated for C17H25BrNO2S+, 386.0784 [M + H]+. Found m/z 346.0789, Δ =
1.2 ppm. Rf (10% EtOAc in Pet ether) = 0.29.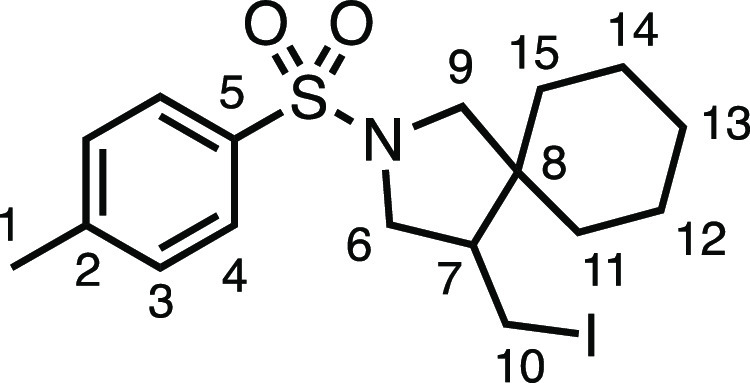
4-(Iodomethyl)-2-tosyl-2-azaspiro[4.5]decane (3k)
The compound was prepared according to General Procedure
E4. Flash column chromatography (5–10% EtOAc in Pet ether)
afforded the product as an off-white crystalline solid (70.4 mg, 81%). 1H NMR (400 MHz, CDCl3) δ 7.73 (app d, J = 8.1 Hz, 2H, H4), 7.33 (app d, J = 8.1 Hz, 2H, H3), 3.67 (dd, J = 10.2,
7.4 Hz, 1H, H6), 3.45 (d, J = 10.1 Hz,
1H, H9), 3.18 (dd, J = 9.7, 3.4 Hz, 1H,
H10), 3.10 (dd, J = 10.2, 7.9 Hz, 1H,
H6′), 3.01 (d, J = 10.1 Hz, 1H,
H9′), 2.70 (dd, J = 12.1, 9.7 Hz,
1H, H10′), 2.43 (s, 3H, H1), 2.11 (app
dtd, J = 11.2, 7.6, 3.4 Hz, 1H, H7), 1.60–1.28
(m, 5H, H11–15), 1.23–1.04 (m, 5H, H11–15). 13C{1H} NMR (101 MHz,
CDCl3) δ 143.6 (C5), 133.9 (C2), 129.8 (C3), 127.5 (C4), 56.6 (C9), 53.2 (C6), 51.4 (C7), 45.8 (C8), 35.5 (C11–15), 28.4 (C11–15), 25.9 (C11–15), 23.5 (C11–15), 22.6 (C11–15), 21.7 (C1), 3.4 (C10). Mp: 99.5–102.5 °C. IR: νmax 2925, 2859, 1337, 1160 cm–1. HRMS: m/z calculated for C17H25INO2S+, 434.0646 [M + H]+. Found m/z 434.0656, Δ = 2.4 ppm. Rf (20%
EtOAc in Pet ether) = 0.59. Spectroscopic data consistent with those
previously reported.38
8-(tert-Butyl)-4-(chloromethyl)-2-tosyl-2-azaspiro[4.5]decane (3m)
The compound was prepared according to
General Procedure E1. Flash column chromatography (5–25% Et2O in Pet ether) afforded two separate diastereoisomers (4:3
d.r.) (total 57.0 mg, 72%). Major diastereoisomer (white solid, 29.0 mg): 1H NMR (400 MHz, CDCl3) δ 7.72 (app d, J = 8.0 Hz, 2H, H4), 7.32 (app d, J = 8.0 Hz, 2H, H3),
3.62 (d, J = 11.0 Hz, 1H, H6), 3.45 (m,
2H, H6′ + H10), 3.01 (d, J = 9.8 Hz, 1H, H9), 2.91 (d, J = 9.8
Hz, 1H, H9′), 2.73 (t, J = 11.2
Hz, 1H, H10′), 2.43 (s, 3H, H1), 2.27
(dt, J = 10.3, 5.2 Hz, 1H, H7), 1.68–1.43
(m, 4H, H11–15), 1.26–1.14 (m, 1H, H11–15), 1.06–0.86 (m, 4H, H11–15), 0.81 (s, 9H, H17). 13C{1H} NMR
(101 MHz, CDCl3) δ 143.6 (C5), 133.7 (C2), 129.8 (C3), 127.6 (C4), 59.2 (C9), 50.0 (C6), 47.7 (C13), 44.8 (C8), 44.2 (C10), 44.0 (C7), 36.8 (C11–15), 32.5 (C16), 30.5 (C11–15), 27.6 (C17), 24.1 (C11–15), 23.4 (C11–15), 21.7 (C1). Mp: 148.8–150.8
°C. IR: νmax 2944, 1335, 1155 cm–1. HRMS: m/z calculated for C21H33ClNO2S+, 398.1915 [M
+ H]+. Found m/z 398.1921,
Δ = 1.4 ppm. Rf (30% Et2O in Pet ether) = 0.88. Minor diastereoisomer (white solid, 28.0 mg): 1H NMR (400 MHz, CDCl3) δ 7.73 (app d, J = 8.0 Hz, 2H, H4), 7.33 (app d, J =
8.0 Hz, 2H, H3), 3.59 (dd, J = 10.2, 7.7
Hz, 1H, H6), 3.51 (dd, J = 10.8, 4.1 Hz,
1H, H10), 3.34 (d, J = 10.0 Hz, 1H, H9), 3.17 (dd, J = 10.5, 5.8 Hz, 1H, H6′), 3.14 (app t, J = 8.4 Hz 1H, H10′), 3.03 (d, J = 10.0 Hz, 1H, H9′), 2.44 (s, 3H, H1), 2.02 (dtd, J = 11.6, 7.7, 4.1 Hz, 1H, H7), 1.62 (s, 1H,
H11 or H12 or H14 or H15), 1.41–1.22 (m, 3H, H11–15), 1.09–0.85
(m, 5H, H11–15), 0.82 (s, 9H, H17). 13C{1H} NMR (101 MHz, CDCl3) δ
143.7 (C5), 133.8 (C2), 129.8 (C3), 127.6 (C4), 55.9 (C9), 51.0 (C7), 50.8 (C6), 47.9 (C13), 44.7 (C8), 43.4 (C10), 36.0 (C11–15), 32.5 (C16), 29.4 (C11–15), 27.6 (C17),
24.4 (C11–15), 23.6 (C11–15),
21.7 (C1). Mp: 128.0–131.0 °C. IR: νmax 2944, 2859, 1345, 1155 cm–1. HRMS: m/z calculated for C21H33ClNO2S+, 398.1915 [M + H]+. Found m/z 398.1925, Δ =
2.4 ppm. Rf (30% Et2O in Pet ether) = 0.72. Spectroscopic
data consistent with those in the literature.11
4-(Chloromethyl)-8-phenyl-2-tosyl-2-azaspiro[4.5]decane (3n)
The compound was prepared according to General
Procedure E1. Flash column chromatography (5–30% Et2O in Pet ether) afforded two separate diastereoisomers (3:2 d.r.)
(total 60.0 mg, 72%). Major diastereoisomer (white
solid, 35.0 mg): 1H NMR (400 MHz, CDCl3) δ
7.75 (app d, J = 8.1 Hz, 2H, H4), 7.35
(app d, J = 8.1 Hz, 2H, H3), 7.29 (t, J = 7.5 Hz, 2H, H17), 7.22–7.13 (m, 3H,
H18 + H19), 3.65 (d, J = 11.0
Hz, 1H, H6), 3.55 (d, J = 10.9 Hz, 1H,
H6′), 3.57–3.51 (m, 1H, H10),
3.12 (d, J = 9.8 Hz, 1H, H9), 3.01 (d, J = 9.8 Hz, 1H, H9′), 2.79 (t, J = 11.0 Hz, 1H, H10′), 2.55–2.37
(m, 5H, H7 + H1 + H13), 1.86–1.73
(m, 2H, H11 or H12 or H14 or H15), 1.69–1.19 (m, 6H, H11 or H12 or H14 or H15). 13C{1H} NMR (101 MHz, CDCl3) δ 146.0 (C16),
143.7 (C5), 133.7 (C2), 129.8 (C3), 128.6 (C17), 127.6 (C4), 126.8 (C18), 126.4 (C19), 59.1 (C9), 50.1 (C6), 44.6 (C8), 44.5 (C7), 44.0 (C10), 43.3 (C13), 36.3 (C11 or C12 or
C14 or C15), 30.6 (C11 or C12 or C14 or C15), 30.1 (C11 or C12 or C14 or C15), 29.9 (C11 or C12 or C14 or C15), 21.7 (C1). Mp: 133.5–136.0 °C. IR: νmax 2925, 2850, 1342, 1163 cm–1. HRMS: m/z calculated for C23H29ClNO2S+, 418.1602 [M + H]+. Found m/z 418.1608, Δ = 1.5 ppm. Minor diastereoisomer (white solid, 25.0 mg): 1H NMR (400 MHz, CDCl3) δ 7.76 (app d, J = 8.1 Hz, 2H, H4), 7.34 (app d, J =
8.1 Hz, 2H, H3), 7.30 (d, J = 7.5 Hz,
2H, H17), 7.21 (t, J = 7.5 Hz, 1H, H19), 7.17 (d, J = 7.5 Hz, 2H, H18), 3.64 (dd, J = 10.2, 7.7 Hz, 1H, H6), 3.58 (dd, J = 10.8, 4.2 Hz, 1H, H10), 3.50 (d, J = 10.1 Hz, 1H, H9), 3.26–3.15
(m, 3H, H6′ + H9′ + H10′), 2.43 (m, 4H, H1 + H13), 2.12 (dtd, J = 11.7, 7.8, 4.3 Hz, 1H, H7), 1.77 (d, J = 13.4 Hz, 2H, H11 or H12 or H14 or H15), 1.60–1.20 (m, 7H, H11 or H12 or H14 or H15). 13C{1H} NMR (101 MHz, CDCl3) δ 146.3 (C16), 143.7 (C5), 133.7 (C2), 129.9 (C3), 128.6 (C17), 127.6 (C4), 126.8 (C18), 126.4 (C19), 55.9 (C9), 51.0 (C7), 50.7 (C6), 44.4 (C8), 44.0 (C13), 43.2 (C10), 35.7 (C11 or C12 or C14 or C15), 31.0 (C11 or C12 or C14 or C15), 30.4 (C11 or C12 or C14 or C15), 29.2 (C11 or C12 or C14 or C15),
21.7 (C1). Mp: 146.0–149.0 °C. IR: νmax 2929, 1340, 1163 cm–1. HRMS: m/z calculated for C23H29ClNO2S+, 418.1602 [M + H]+. Found m/z 418.1610, Δ =
1.9 ppm.
4′-(Chloromethyl)-1′-tosyl-1,3-dihydrospiro[indene-2,3′-pyrrolidine] (3o)
The compound was prepared according to
General Procedure E1. Flash column chromatography (10–20% EtOAc
in Pet ether) afforded the product as a yellow oil (16.0 mg, 21%). 1H NMR (400 MHz, CDCl3) δ 7.73 (app d, J = 8.1 Hz, 2H, H4), 7.36 (app d, J = 8.1 Hz, 2H, H3), 7.17–7.05 (m, 4H, H13–16), 3.65 (dd, J = 10.5, 7.2 Hz, 1H, H6), 3.45–3.35 (m, 2H, H6′ + H10), 3.26 (d, J = 9.8 Hz, 1H, H9), 3.21
(d, J = 9.8 Hz, 1H, H9′), 3.08
(t, J = 10.9 Hz, 1H, H10′), 2.88
(d, J = 15.9 Hz, 1H, H11), 2.81 (d, J = 15.8 Hz, 2H, H18), 2.73 (d, J = 15.8 Hz, 1H, H18′), 2.56 (d, J = 15.9 Hz, 1H, H11′), 2.47 (s, 3H, H1), 2.36 (dtd, J = 10.8, 6.8, 4.3 Hz, 1H, H7). 13C{1H} NMR (101 MHz, CDCl3)
δ 143.8 (C5), 141.0 (C12 or C17), 140.8 (C12 or C17), 133.9 (C2) 129.9 (C3), 127.6 (C4), 127.1 (C13–16), 127.1 (C13–16), 124.7 (C13–16), 124.6 (C13–16), 59.1 (C9), 53.0 (C8), 51.3 (C6), 49.4 (C7), 43.6 (C10), 43.0 (C18), 38.2 (C11), 21.7 (C1). IR: νmax 1340, 1157 cm–1. HRMS: m/z calculated for C20H23ClNO2S+, 376.1133 [M
+ H]+. Found m/z 376.1137,
Δ = 1.1 ppm. Rf (30% EtOAc in Pet ether) = 0.49.
(1R,3S,5r,7r)-4′-(Chloromethyl)-1′-tosylspiro[adamantane-2,3′-pyrrolidine] (3p)
The compound was prepared according to
General Procedure E1. Flash column chromatography (5–10% EtOAc
in Pet ether) afforded the product as an off-white solid (45.5 mg,
58%). 1H NMR (400 MHz, CDCl3) δ 7.74 (app
d, J = 8.0 Hz, 2H, H4), 7.32 (app d, J = 8.0 Hz, 2H, H3), 3.76 (d, J = 10.6 Hz, 1H, H9), 3.68 (d, J = 10.8
Hz, 1H, H6), 3.56–3.44 (m, 1H, H10),
3.30 (dd, J = 10.7, 5.5 Hz, 1H, H6′), 2.78 (d, H = 10.6 Hz, 1H, H9′), 2.75 (app t, J = 11.2 Hz,1H, H10′), 2.55 (dt, J = 11.7, 4.2 Hz, 1H, H7), 2.43 (s, 3H, H1), 1.93–1.42 (m, 14H, H11–19). 13C{1H} NMR (101 MHz, CDCl3)
δ 143.6 (C5), 133.9 (C2), 129.7 (C3), 127.5 (C4), 54.2 (C9), 50.8 (C8), 49.0 (C6), 45.9 (C7), 43.7 (C10), 38.0 (C11–19), 34.5 (C11–19), 34.0 (C11–19), 33.6 (C11–19), 33.5 (C11–19), 32.0 (C11–19), 31.8 (C11–19), 27.1 (C11–19), 27.0 (C11–19), 21.7 (C1). Mp: 118.3–120.2
°C. IR: νmax 2897, 1344, 1163 cm–1. HRMS: m/z calculated for C21H29ClNO2S+, 394.1602 [M
+ H]+. Found m/z 394.1608,
Δ = 1.5 ppm. Rf (10% EtOAc in Pet ether) = 0.33.
4-(Chloromethyl)-8,8-difluoro-2-tosyl-2-azaspiro[4.5]decane (3q)
The compound was prepared according to
General Procedure E1. Flash column chromatography (10% EtOAc in Pet
ether) afforded the product as a white solid (48.9 mg, 65%). 1H NMR (400 MHz, CDCl3) δ 7.73 (app d, J = 8.0 Hz, 2H, H4), 7.35 (app d, J = 8.0 Hz, 2H, H3), 3.61 (dd, J = 10.4,
7.7 Hz, 1H, H6), 3.49 (dd, J = 10.9, 4.2
Hz, 1H, H10), 3.35 (d, J = 10.1 Hz, 1H,
H9), 3.26 (dd, J = 10.4, 7.0 Hz, 1H, H6′), 3.15 (t, J = 10.6 Hz, 1H, H10′), 3.09 (d, J = 10.1 Hz, 1H, H9), 2.44 (s, 3H, H1), 2.22–2.12 (m, 1H, H7), 1.95 (dd, J = 15.2, 6.2 Hz, 2H, H12 + H14), 1.61–1.78 (m, 3H, H12′ + H14′ + H11 or H15), 1.48–1.30
(m, 3H, H11 + H15). 13C{1H} NMR (101 MHz, CDCl3) δ 144.0 (C5),
133.6 (C2), 130.0 (C3), 127.5 (C4), 122.4 (dd, J = 239.7, 240.1 Hz, C13), 55.4 (d, J = 1.7 Hz, C9), 50.5 (C6), 49.2 (d, J = 1.8 Hz, C7), 43.6
(d, J = 1.1 Hz, C8), 42.9 (C10), 31.6 (dd, J = 8.6, 1.2 Hz, C11 or
C15), 31.18 (dd, J = 25.5, 23.7 Hz, C12 or C14), 30.67 (dd, J = 25.5,
24.0 Hz, C12 or C14), 25.2 (d, J = 9.9 Hz, C11 or C15), 21.7 (C1). 19F NMR (376 MHz, CDCl3) δ −94.1
(d, J = 237.5 Hz), −103.5 (d, J = 236.6 Hz). Mp: 101.0–103.0 °C. IR: νmax 2963, 2941, 2920, 2861, 1338, 1163 cm–1. HRMS: m/z calculated for C17H23ClF2NO2S+, 378.1101 [M +
H]+. Found m/z 378.1101,
Δ = 0.1 ppm. Rf (20% EtOAc in Pet ether) = 0.31.
12-(Chloromethyl)-10-tosyl-1,4-dioxa-10-azadispiro[4.2.48.25]tetradecane (3r)
The
compound was prepared according to General Procedure E1. Flash column
chromatography (10–20% EtOAc in Pet ether, then 5% MeCN in
toluene) afforded the product as a white solid (54.2 mg, 68%). 1H NMR (400 MHz, CDCl3) δ 7.72 (app d, J = 8.1 Hz, 2H, H4), 7.33 (app d, J = 8.1 Hz, 2H, H3), 3.89 (m, 4H, H16 + H17), 3.60 (dd, J = 10.3, 7.4 Hz, 1H, H6), 3.52 (dd, J = 10.8, 4.0 Hz, 1H, H10), 3.35 (d, J = 10.2 Hz, 1H, H9), 3.24 (dd, J = 10.3, 6.9 Hz, 1H, H6′), 3.14 (d, J = 10.8 Hz, 1H, H9′), 3.10 (app t, J = 10.3 Hz, 1H, H10′), 2.43 (s, 3H, H1), 2.14 (dtd, J = 11.0,
7.1, 4.1 Hz, 1H, H7), 1.69–1.27 (m, 8H, H11 + H12 + H14 + H15). 13C{1H} NMR (126 MHz, CDCl3) δ 143.8 (C5), 133.7 (C2), 129.9 (C3), 127.5 (C4), 107.9 (C13), 64.5 (C16 or C17), 64.4 (C16 or C17), 55.8 (C9),
50.7 (C6), 49.5 (C7), 44.0 (C8),
43.3 (C10), 32.8 (C11 or C12 or C14 or C15), 32.1 (C11 or C12 or C14 or C15), 31.6 (C11 or C12 or C14 or C15), 26.2 (C11 or C12 or C14 or C15), 21.7 (C1). Mp: 110.7–113.0 °C. IR: νmax 2926, 1338, 1161 cm–1. HRMS: m/z calculated for C19H27ClNO4S+, 400.1344 [M + H]+. Found m/z 400.1352, Δ = 2.0 ppm. Rf (20%
EtOAc in Pet ether) = 0.08.
8-(Chloromethyl)-6-tosyl-6-azaspiro[3.4]octane (3s)
The compound was prepared according to General Procedure
E1. Flash column chromatography (5–30% Et2O in Pet
ether) afforded the product as a colorless oil (48.0 mg, 77%). 1H NMR (400 MHz, CDCl3) δ 7.71 (app d, J = 8.0 Hz, 2H, H4), 7.32 (app d, J = 8.0 Hz, 2H, H3), 3.48 (dd, J = 10.9,
4.2 Hz, 1H, H10), 3.40 (dd, J = 10.5,
6.6 Hz, 1H, H6), 3.34 (d, J = 9.8 Hz,
1H, H9), 3.28 (dd, J = 10.4, 4.4 Hz, 1H,
H6′), 3.20 (d, J = 9.8 Hz, 1H,
H9), 2.93 (t, J = 10.7 Hz, 1H, H10′), 2.42 (s, 3H, H1), 2.18 (dq, J = 10.4,
4.4 Hz, 1H, H7), 2.05–1.94 (m, 1H, H11 or H13), 1.91–1.59 (m, 5H, H11 + H12 + H13). 13C{1H} NMR (101
MHz, CDCl3) δ 143.7 (C5), 133.7 (C2), 129.8 (C3), 127.6 (C4), 58.2 (C9), 50.1 (C6), 49.4 (C7), 47.5 (C8), 43.3 (C10), 32.6 (C11 or C13), 26.0 (C11 or C13), 21.6 (C1),
16.3 (C12). IR: νmax 2929, 1341, 1155
cm–1. HRMS: m/z calculated for C15H21ClNO2S+, 314.0976 [M + H]+. Found m/z 314.0975, Δ = −0.3 ppm. Rf (30% Et2O in Pet ether) = 0.25.
4-(Chloromethyl)-2-tosyl-2-azaspiro[4.6]undecane (3t)
The compound was prepared according to General Procedure
E1. Flash column chromatography (10% EtOAc in Pet ether) afforded
the product as a colorless oil (40.7 mg, 57%). 1H NMR (400
MHz, CDCl3) δ 7.71 (app d, J = 8.1
Hz, 2H, H4), 7.32 (app d, J = 8.1 Hz,
2H, H3), 3.57 (app t, J = 10.1 Hz, 1H,
H6), 3.55 (dd, J = 11.6, 5.1 Hz, 1H, H10), 3.25 (d, J = 9.7 Hz, 1H, H9), 3.20 (dd, J = 10.4, 7.6 Hz, 1H, H6′), 3.13 (t, J = 10.9 Hz, 1H, H10′), 2.97 (d, J = 9.7 Hz, 1H, H9′), 2.43 (s, 3H, H1), 2.08 (dtd, J = 11.5,
7.6, 4.2 Hz, 1H, H7), 1.59–1.17 (m, 12H, H11–16). 13C{1H} NMR (101 MHz, CDCl3)
δ 143.6 (C5), 134.0 (C2), 130.0 (C3), 127.5 (C4), 59.3 (C9), 51.7 (C7), 51.1 (C6), 47.8 (C8), 43.6 (C10), 38.6 (C11–16), 31.8 (C11–16), 29.6 (C11–16), 29.5 (C11–16), 23.9 (C11–16), 23.6 (C11–16), 21.7 (C1). IR: νmax 2922, 2854, 1341,
1158 cm–1. HRMS: m/z calculated for C18H27ClNO2S+, 356.1445 [M + H]+. Found m/z 356.1445, Δ = −0.1 ppm. Rf (20% EtOAc in
Pet ether) = 0.43.
4-(Chloromethyl)-2-tosyl-2-azaspiro[4.11]hexadecane (3u)
The compound was prepared according to General Procedure
E1. Flash column chromatography (5–10% EtOAc in Pet ether)
afforded inseparable diastereomers of the product as a colorless oil
(44.9 mg, 53%). 1H NMR (400 MHz, CDCl3) δ
7.72 (app d, J = 8.1 Hz, 2H, H4), 7.32
(app d, J = 8.1 Hz, 2H, H3), 3.57 (dd, J = 10.7, 6.0 Hz, 1H, H6), 3.55 (app t, J = 10.1 Hz, 1H, H10), 3.36 (dd, J = 10.7, 4.8 Hz, 1H, H6′), 3.07–2.92 (m,
3H, H9 + H9′ + H10′), 2.43 (s, 3H, H1), 2.16 (ddt, J = 11.3,
8.3, 4.4 Hz, 1H, H7), 1.38–1.10 (m, 22H, H11–21). 13C{1H} NMR (101 MHz, CDCl3)
δ 143.6 (C2), 133.7 (C5), 129.8 (C3), 127.6 (C4), 57.4 (C9), 51.2 (C6), 48.9 (C7), 47.5 (C8), 44.7 (C10), 32.3 (C11–21), 27.1 (C11–21), 26.8 (C11–21), 26.6 (C11–21), 26.1 (C11–21), 22.8 (C11–21), 22.7 (C11–21), 22.3 (C11–21), 22.2 (C11–21), 21.7 (C1), 19.6 (C11–21). IR: νmax 2930, 2859, 1345,
1158 cm–1. HRMS: m/z calculated for C23H37ClNO2S+, 426.2228 [M + H]+. Found m/z 426.2239, Δ = 2.6 ppm. Rf (20% EtOAc in Pet ether)
= 0.56.
4-(Chloromethyl)-7-methyl-2-tosyl-2-azaspiro[4.14]nonadecane (3v)
The compound was prepared according to
General Procedure E1. Flash column chromatography (5–10% EtOAc
in Pet ether) afforded an inseparable mixture of diastereomers as
a colorless oil (52.2 mg, 54%). 1H NMR (400 MHz, CDCl3) δ 7.72 (app d, J = 7.7 Hz, 2H, H4), 7.33 (app d, J = 7.7 Hz, 2H, H3), 3.66–3.41 (m, 2H, H6 + H10), 3.33
(app dq, J = 11.0, 6.3, 5.8 Hz, 1H, H6′), 3.25–2.71 (m, 3H, H9 + H9′ + H10′), 2.43 (s, 3H, H1), 2.26–1.95
(m, 1H, H7), 1.45–0.91 (m, 27H, H11–24), 0.83 (app t, J = 6.9 Hz, 3H, H25). 13C{1H} NMR (101 MHz, CDCl3) δ
143.7, 143.6, 143.6, 134.0, 133.7, 133.6, 129.8, 129.8, 127.7, 127.6,
127.6, 59.1, 57.8, 56.9, 51.4, 51.2, 50.5, 50.2, 48.6, 48.2, 47.8,
47.5, 44.7, 44.4, 44.3, 43.8, 39.7, 37.9, 37.4, 37.3, 37.0, 36.0,
31.9, 28.5, 28.2, 28.0, 28.0, 27.8, 27.7, 27.6, 27.2, 27.1, 27.00,
27.0, 26.9, 26.9, 26.8, 26.8, 26.8, 26.3, 26.1, 25.9, 25.9, 25.7,
25.6, 25.6, 25.6, 25.5, 25.5, 25.1, 24.8, 24.7, 23.2, 23.0, 22.8,
22.6, 22.5, 22.1, 21.7, 21.5, 21.2. IR: νmax 2925,
2855, 1345, 1160 cm–1. HRMS: m/z calculated for C27H45ClNO2S+, 482.2866 [M + H]+. Found m/z 482.2860, Δ = 1.2 ppm. Rf (20% EtOAc in
Pet ether) = 0.56.
11-(Chloromethyl)-9-tosyl-2-oxa-9-azadispiro[3.2.47.24]tridecane (3w)
The compound
was prepared according to General Procedure E1. Flash column chromatography
(20–40% EtOAc in Pet ether) afforded the product as a colorless
oil (41.9 mg, 55%). 1H NMR (500 MHz, CDCl3)
δ 7.71 (app d, J = 8.1 Hz, 2H, H4), 7.33 (app d, J = 8.1 Hz, 2H, H3),
4.38–4.21 (m, 4H, H16 + H17), 3.55 (dd, J = 10.4, 7.5 Hz, 1H, H6), 3.44 (dd, J = 10.9, 4.4 Hz, 1H, H10), 3.26 (d, J = 10.2 Hz, 1H, H9), 3.23 (dd, J = 10.5, 6.8 Hz, 1H, H6′), 3.0899 (app t, J = 10.7 Hz, 1H, H10′), 3.04 (d, J = 10.2 Hz, 1H, H9′), 2.44 (s, 3H, H1), 2.10–2.03 (m, 1H, H7), 1.91 (t, J = 11.9 Hz, 2H, H12 or H14), 1.51–1.39
(m, 2H, H11–15), 1.33 (td, J =
12.8, 12.2, 3.6 Hz, 1H, H11–15), 1.24–1.05
(m, 3H, H11–15). 13C{1H} NMR
(126 MHz, CDCl3) δ 143.8 (C2), 133.6 (C5), 129.8 (C3), 127.5 (C4), 82.0 (C16 or C17), 81.4 (C16 or C17), 55.6 (C9), 50.4 (C6), 49.5 (C7), 43.9 (C8), 43.1 (C10), 39.7 (C13), 32.0 (C11 or C12 or C14 or C15), 32.0 (C11 or C12 or C14 or C15), 31.6 (C11 or C12 or C14 or C15), 25.5 (C11 or C12 or C14 or C15), 21.7 (C1). IR:
νmax 2922, 2858, 1339, 1157 cm–1. HRMS: m/z calculated for C19H27ClNO3S+, 384.1395 [M
+ H]+. Found m/z 384.1399,
Δ = 1.1 ppm. Rf (40% EtOAc in Pet ether) = 0.13.
4-(Chloromethyl)-2-tosyl-8-oxa-2-azaspiro[4.5]decane (3x)
The compound was prepared according to General
Procedure E1. Flash column chromatography (20–40% EtOAc in
Pet ether) afforded the product as a colorless oil (31.5 mg, 46%). 1H NMR (400 MHz, CDCl3) δ 7.72 (app d, J = 8.1 Hz, 2H, H4), 7.33 (app d, J = 8.1 Hz, 2H, H3), 3.79–3.68 (m, 2H, H12 + H13), 3.57 (dd, J = 10.5, 7.4 Hz,
1H, H6), 3.52 (dd, J = 10.9, 4.2 Hz, 1H,
H10), 3.43–3.32 (m, 3H, H9 + H12′ + H13′), 3.26 (dd, J = 10.5,
6.6 Hz, 1H, H6′), 3.19 (d, J =
10.2 Hz, 1H, H9′), 3.12 (t, J =
10.7 Hz, 1H, H10′), 2.43 (s, 3H, H1),
2.18–2.04 (m, 1H, H7), 1.66 (ddd, J = 13.5, 11.5, 4.6 Hz, 1H, H11–14), 1.48 (ddd, J = 13.1, 11.6, 4.5 Hz, 1H, H11–14), 1.19
(dq, J = 13.5, 2.4 Hz, 1H, H11–14), 1.15–1.07 (m, 1H, H11–14). 13C{1H} NMR (101 MHz, CDCl3) δ 143.9, (C2), 133.6 (C5), 129.9 (C3), 127.5 (C4), 65.0 (C12 or C13), 64.6 (C12 or C13), 55.4 (C9), 50.2 (C6),
50.1 (C7), 42.9 (C8), 42.7 (C10),
35.3 (C11 or C14), 29.6 (C11 or C14), 21.7 (C1). IR: νmax 2923,
2851, 1339, 1156 cm–1. HRMS: m/z calculated for C16H23ClNO3S+, 344.1082 [M + H]+. Found m/z 344.1076, Δ = −1.7 ppm. Rf (40%
EtOAc in Pet ether) = 0.22.
(5r,7r)-4″-(Chloromethyl)-2′-phenyl-1″-tosyldispiro[adamantane-2,4′-oxazole-5′,3″-pyrrolidine] (3y)
The compound was prepared according to
General Procedure E1. Flash column chromatography (10–30% Et2O in Pet ether) afforded the product as an off-white solid
(36.8 mg, 39%). 1H NMR (400 MHz, CDCl3) δ
7.78 (d, J = 7.7 Hz, 2H, H4), 7.52 (d, J = 7.7 Hz, 2H, H14), 7.46 (t, J = 7.4 Hz, 1H, H16), 7.32 (app q, J =
7.6 Hz, 4H, H3 + H15), 4.13 (d, J = 11.8 Hz, 1H, H9), 3.98 (dd, J = 9.5,
8.0 Hz, 1H, H6), 3.77 (d, J = 11.8 Hz,
1H, H9′), 3.74 (dd, J = 11.1, 3.4
Hz, 1H, H10), 3.20 (t, J = 10.0 Hz, 1H,
H6), 3.13 (t, J = 11.1 Hz, 1H, H10), 2.81 (tdd, J = 11.0, 8.0, 3.3, 1H, H7), 2.68 (d, J = 12.2 Hz, 1H, H17 or H23), 2.55 (d, J = 12.6 Hz, 1H, H17 or H23), 2.46 (s, 3H, H1), 1.96–1.46
(m, 12H, H18–22 + H24–25). 13C{1H} NMR (126 MHz, CDCl3) δ
158.2 (C11), 143.7 (C5), 134.1 (C2), 131.7 (C16), 129.9 (C3), 128.4 (C15), 128.2 (C14), 127.9 (C4), 127.1 (C13), 95.4 (C8), 75.4 (C12), 56.5 (C9), 51.4 (C6), 47.4 (C7), 42.6 (C10), 38.4 (C18–22 or C24–25), 36.2
(C18–22 or C24–25), 34.7 (C17 or C23), 34.6 (C18–25), 34.6
(C18–25), 33.7 (C18–22 or C24–25), 33.6 (C18–22 or C24–25), 30.5 (C18–22 or C24–25), 27.6
(C18–22 or C24–25), 26.7 (C18–22 or C24–25), 21.8 (C1). Mp: 86.7–90.0 °C. IR: νmax 2908,
1653, 1335, 1162 cm–1. HRMS: m/z calculated for C29H34ClN2O3S+, 525.1973 [M + H]+. Found m/z 525.1978, Δ = 1.0 ppm.
4-(Chloromethyl)-2-tosyl-8-thia-2-azaspiro[4.5]decane 8,8-Dioxide (3z)
The compound was prepared according to
General Procedure E1. Flash column chromatography (20–30% EtOAc
in Pet ether) followed by vapor diffusion recrystallization from DCM
(Pet ether antisolvent) afforded the product as off-white crystals
(43.6 mg, 56%). 1H NMR (400 MHz, CDCl3) δ
7.73 (app d, J = 8.1 Hz, 2H, H4), 7.36
(app d, J = 8.1 Hz, 2H, H3), 3.64 (dd, J = 10.6, 8.0 Hz, 1H, H6), 3.52 (dd, J = 11.2, 4.7 Hz, 1H, H10), 3.49 (d, J = 10.1 Hz, 1H, H9), 3.30–3.17 (m, 2H,
H6′ + H10′), 3.12 (d, J = 10.3 Hz, 1H, H9′), 3.00–2.85
(m, 4H, H12 + H13), 2.45 (s, 3H, H1), 2.26 (dtt, J = 12.7, 8.2, 4.4 Hz, 2H, H7 + H11 or H14), 1.99 (td, J = 12.8, 10.8, 5.1 Hz, 1H, H11 or H14), 1.81–1.67
(m, 2H, H11 or H14). 13C{1H} NMR (101 MHz, CDCl3) δ 144.4 (C5),
133.4 (C2), 130.1 (C3), 127.5 (C4), 54.8 (C9), 50.4 (C6), 49.2 (C7), 48.7 (C12 or C13), 47.9 (C12 or
C13), 43.0 (C8), 42.2 (C10), 33.0
(C11 or C14), 26.6 (C11 or C14), 21.7 (C1). Mp: 176.0–178.0 °C.
IR: νmax 2916, 2854, 1340, 1161 cm–1. HRMS: m/z calculated for C16H23ClNO4S2+,
392.0751 [M + H]+. Found m/z 392.0748, Δ = −0.8 ppm.
8-(Bromomethyl)-6-((4-nitrophenyl)sulfonyl)-2-tosyl-2,6-diazaspiro[3.4]octane (3aa)
The compound was prepared according to
General Procedure E3. Flash column chromatography (DCM) followed by
recrystallization by vapor diffusion from DCM (Pet ether as the antisolvent)
afforded the product as a white crystalline solid (57.8 mg, 53%). 1H NMR (400 MHz, CDCl3) δ 8.39 (app d, J = 8.8 Hz, 2H, H2), 7.97 (app d, J = 8.8 Hz, 2H, H3), 7.69 (app d, J =
8.2 Hz, 2H, H13), 7.40 (app d, J = 8.1
Hz, 2H, H14), 3.74 (d, J = 8.8 Hz, 1H,
H8), 3.60 (d, J = 8.3 Hz, 1H, H10), 3.60 (d, J = 8.4 Hz, 1H, H10′), 3.53 (dd, J = 10.7, 7.4 Hz, 1H, H5), 3.45 (d, J = 8.8 Hz, 1H, H8′), 3.34 (s, 2H, H11), 3.18 (dd, J = 10.6,
8.1 Hz, 1H, H9), 3.17 (t, J = 10.5 Hz,
1H, H5′), 2.87 (t, J = 10.1 Hz,
1H, H9′), 2.48 (s, 3H, H16), 2.37 (qt, J = 6.9, 4.2 Hz, 1H, H6). 13C{1H} NMR (101 MHz, CDCl3) δ 150.5 (C4), 145.1 (C12), 142.3 (C1), 131.0 (C15), 130.2 (C14), 128.7 (C3), 128.5 (C13), 124.7 (C2), 59.1 (C10 or C11),
57.0 (C10 or C11), 54.3 (C8), 51.3
(C5), 46.7 (C6), 42.4 (C7), 29.5
(C9), 21.8 (C16). Mp: 189.8–192.4 °C.
IR: νmax 2887, 1527, 1346, 1333, 1164, 1150 cm–1. HRMS: m/z calculated
for C20H23BrN3O6S2+, 544.0206 [M + H]+. Found m/z 544.0215, Δ = 1.7 ppm. Rf (DCM)
= 0.10.
4-(Chloromethyl)-7-((4-nitrophenyl)sulfonyl)-2-tosyl-2,7-diazaspiro[4.4]nonane (3ab)
The compound was prepared according to
General Procedure E1. Flash column chromatography (30% EtOAc in Pet
ether) afforded the product as an inseparable 1:1 mixture of diastereomers
(yellow oil, 50.2 mg, 49%). 1H NMR (400 MHz, CDCl3) δ 8.38 (app d, J = 8.6 Hz, 2H, H16), 7.97 (app d, J = 8.6 Hz, 2H, H15),
7.72–7.57 (m, 2H, H4), 7.34 (app d, J = 7.9 Hz, 2H, H3), 3.55–3.32 (m, 3H, H6–13), 3.30–2.84 (m, 7H, H6–13), 2.44 (s, 3H,
H1), 2.25 (tt, J = 8.8, 4.4 Hz, 1H, H7), 1.87–1.58 (m, 2H, H11). 13C{1H} NMR (101 MHz, CDCl3) δ 150.4, 150.4,
144.4, 144.3, 142.4, 142.2, 133.0, 132.9, 130.1, 130.1, 128.7, 128.6,
127.6, 124.7, 124.7, 57.0 (C12), 56.5 (C13),
56.3 (C13″), 51.9 (C12″), 51.5
(C8), 51.2 (C8″), 50.9 (C6), 50.8 (C6″), 47.6 (C7), 46.9 (C6), 46.1 (C6″), 45.9 (C7″), 43.1 (C10), 42.8 (C10″), 35.0 (C11), 29.8 (C11″), 21.7 (C1 + C1″). IR: νmax 2922, 1528, 1346, 1159
cm–1. HRMS: m/z calculated for C21H25ClN3O6S2+, 514.0868 [M + H]+. Found m/z 514.0842, Δ = −5.0 ppm.
Rf (30% EtOAc in Pet ether) = 0.29.
4-(Chloromethyl)-2,8-ditosyl-2,8-diazaspiro[4.5]decane (3ac)
The compound was prepared according to General
Procedure E1. Flash column chromatography (20–30% EtOAc in
Pet ether) afforded the product as a white solid (75.5 mg, 76%). 1H NMR (400 MHz, CDCl3) δ 7.64 (app d, J = 8.1 Hz, 2H, H4 or H16), 7.60 (app
d, J = 8.2 Hz, 2H, H4 or H16), 7.35 (app d, J = 8.1 Hz, 2H, H3 or
H17), 7.30 (app d, J = 8.1 Hz, 2H, H3 or H17), 3.53 (app dt, J = 10.4,
5.3 Hz, 3H, H6 + H12 + H13), 3.44
(dd, J = 10.8, 4.2 Hz, 1H, H10), 3.18
(dd, J = 10.6, 7.1 Hz, 1H, H6′),
3.14 (d, J = 10.0 Hz, 1H, H9), 3.08 (t, J = 10.8 Hz, 1H, H10′), 2.89 (d, J = 10.1 Hz, 1H, H9′), 2.47 (s, 3H, H1 or H19), 2.43 (s, 3H, H1 or H19), 2.25 (qd, J = 12.1, 2.3 Hz, 2H, H12′ + H13′), 2.05 (dtd, J = 11.3,
7.3, 4.5 Hz, 1H, H7), 1.72 (td, J = 12.8,
4.3 Hz, 1H, H11 or H14), 1.52 (td, J = 12.7, 4.2 Hz, 1H, H11 or H14), 1.39–1.26
(m, 2H, H11′ + H14′). 13C{1H} NMR (101 MHz, CDCl3) δ 144.2 (C5 or C15), 144.1 (C5 or C15), 133.3 (C2 or C18), 132.8 (C2 or
C18), 130.0 (C3 or C17), 129.9 (C3 or C17), 127.7 (C4 or C16), 127.5 (C4 or C16), 54.9 (C9),
50.2 (C6), 49.6 (C7), 43.6 (C12 or
C13), 43.1 (C12 or C13), 42.7 (C8), 42.6 (C10), 34.1 (C11 or C14), 28.2 (C11 or C14), 21.7 (C1 +
C19). Mp: 102.8–104.0 °C. IR: νmax 2843, 1341, 1328, 1157; cm–1. HRMS: m/z calculated for C23H30ClN2O4S2+, 497.1330 [M + H]+. Found m/z 497.1335, Δ
= 0.9 ppm. Rf (30% EtOAc in Pet ether) = 0.23.
4-(Bromomethyl)-2,8-ditosyl-2,8-diazaspiro[4.5]decane (3ad)
The compound was prepared according to General
Procedure E3. Flash column chromatography (20% EtOAc in Pet ether)
afforded the product as a white solid (84.6 mg, 78%). 1H NMR (400 MHz, CDCl3) δ 7.64 (app d, J = 8.3 Hz, 2H, H4 or H16), 7.59 (app d, J = 8.3 Hz, 2H, H4 or H16), 7.35 (app
d, J = 8.0 Hz, 2H, H3 or H17), 7.30 (app d, J = 8.0 Hz, 2H, H3 or
H17), 3.57 (dd, J = 10.5, 7.7 Hz, 3H,
H6 + H12 + H13), 3.29 (dd, J = 10.1, 3.9 Hz, 1H, H10), 3.20 (d, J = 10.2 Hz, 1H, H9), 3.16 (dd, J = 10.6, 7.5 Hz, 1H, H6′), 2.90 (app t, J = 10.6 Hz, 1H, H10′), 2.87 (d, J = 10.1 Hz, 2H, H9′), 2.47 (s, 3H, H1 or H19), 2.43 (s, 3H, H1 or H19), 2.30–2.16 (m, 2H, H12′ + H13′), 2.11 (dtd, J = 11.3, 7.6, 3.9 Hz, 1H, H7), 1.73 (td, J = 12.9, 12.4, 4.5 Hz, 1H, H11 or H14), 1.51 (td, J = 12.7, 4.3 Hz,
1H, H11 or H14), 1.36–1.26 (m, 2H, H11′ + H14′). 13C{1H} NMR (101 MHz, CDCl3) δ 144.2 (C5 or
C15), 144.1 (C5 or C15), 133.4 (C2 or C18), 132.8 (C2 or C18), 130.1 (C3 or C17), 129.9 (C3 or
C17), 127.7 (C4 or C16), 127.5 (C4 or C16), 54.9 (C9), 51.3 (C6), 50.0 (C7), 43.6 (C12 or C13),
43.2 (C8), 43.0 (C12 or C13), 34.0
(C11 or C14), 30.2 (C10), 28.0 (C11 or C14), 21.7 (C1 or C19), 21.7 (C1 or C19). Mp: Decomposed above 160
°C. IR: νmax 2923, 2845, 1340, 1328, 1157; cm–1. HRMS: m/z calculated
for C23H30BrN2O4S2+, 541.0825 [M + H]+. Found m/z 541.0830, Δ = 0.9 ppm. Rf (30%
EtOAc in Pet ether) = 0.24.
4-(Chloromethyl)-2-((4-nitrophenyl)sulfonyl)-8-tosyl-2,8-diazaspiro[4.5]decane (3ae)
The compound was prepared according to
General Procedure E1. Flash column chromatography (DCM) afforded the
product as a white solid (55.7 mg, 53%). 1H NMR (400 MHz,
CDCl3) δ 8.37 (app d, J = 8.8 Hz,
2H, H2), 7.96 (app d, J = 8.8 Hz, 2H,
H3), 7.62 (app d, J = 8.2 Hz, 2H, H15), 7.36 (app d, J = 8.1 Hz, 2H, H16), 3.64–3.54 (m, 3H, H5 + H11 + H12), 3.47 (dd, J = 11.2, 4.1 Hz, 1H, H9), 3.27 (dd, J = 10.6, 6.8 Hz, 1H, H5′), 3.22 (d, J = 9.9 Hz, 1H, H8), 3.18–3.09 (m, 1H, H9′), 2.97 (d, J = 9.9 Hz, 1H, H8′), 2.48 (s, 3H, H1), 2.39–2.26 (m, 2H, H11′ + H12′), 2.13 (dtd, J = 11.3, 7.5, 4.4
Hz, 1H, H6), 1.79 (td, J = 13.4, 4.3 Hz,
1H, H10 or H13), 1.67–1.55 (m, 1H, H10 or H13), 1.47 (d, J = 13.6 Hz,
2H, H10′ + H13′). 13C{1H} NMR (101 MHz, CDCl3) δ 150.3 (C1), 144.1 (C17), 142.5 (C4), 132.8 (C14), 130.0 (C16), 128.5 (C3), 127.6 (C15), 124.5 (C2), 55.0 (C8), 50.1 (C5), 49.3 (C6), 43.4 (C11 or C12), 43.0 (C11 or C12), 42.9 (C7),
42.5 (C9), 34.1 (C10 or C13), 28.2
(C10 or C13), 21.6 (C18). Mp: Decomposed
above 200 °C. IR: νmax 2853, 1529, 1347, 1327,
1161 cm–1. HRMS: m/z calculated for C22H27ClN3O6S2+, 528.1024 [M + H]+. Found m/z 528.1020, Δ = −0.7 ppm.
Rf (DCM) = 0.13.
4-(Bromomethyl)-2-((4-nitrophenyl)sulfonyl)-8-tosyl-2,8-diazaspiro[4.5]decane (3af)
The compound was prepared according to
General Procedure E2. Flash column chromatography (DCM) followed by
recrystallization by vapor diffusion from DCM (Pet ether as the antisolvent)
afforded the product as a white crystalline solid (68.9 mg, 60%). 1H NMR (400 MHz, CDCl3) δ 8.38 (app d, J = 8.8 Hz, 2H, H2), 7.97 (app d, J = 8.8 Hz, 2H, H3), 7.62 (app d, J =
8.1 Hz, 2H, H18), 7.36 (app d, J = 8.1
Hz, 2H, H19), 3.63 (dd, J = 10.5, 7.8
Hz, 3H, H5 + H11 + H12), 3.37–3.20
(m, 3H, H5′ + H8 + H9), 2.95
(app t, J = 10.5 Hz, 1H, H9′),
2.92 (d, J = 9.9 Hz, 1H, H8′),
2.48 (s, 3H, H21), 2.38–2.23 (m, 2H, H11′ + H12′), 2.18 (dtd, J = 11.1,
7.5, 3.8 Hz, 1H, H6), 1.80 (td, J = 13.4,
4.3 Hz, 1H, H10 or H13), 1.65–1.56 (m,
1H, H10 or H13), 1.43 (d, J = 13.5 Hz, 2H, H10′ + H13′). 13C{1H} NMR (101 MHz, CDCl3) δ
150.4 (C1), 144.3 (C17), 142.7 (C4), 133.0 (C14), 130.1 (C16), 128.6 (C3), 127.7 (C15), 124.7 (C2), 55.1 (C8), 51.3 (C5), 49.9 (C6), 43.6 (C11 or C12), 43.5 (C7), 43.5 (C11 or
C12), 43.0 (C11 or C12), 34.1 (C10 or C13), 30.0 (C9), 28.0 (C10 or C13), 21.8 (C18). Mp: 206.8–209.9
°C. IR: νmax 2853, 1529, 1347, 1327, 1161 cm–1. HRMS: m/z calculated
for C22H27BrN3O6S2+, 572.0519 [M + H]+. Found m/z 572.0517, Δ = −0.4 ppm.
Rf (DCM) = 0.16.
4-(Chloromethyl)-2-((4-nitrophenyl)sulfonyl)-10-tosyl-2,10-diazadispiro[4.1.57.15]tridecane (3ag)
The compound
was prepared according to General Procedure E1. Flash column chromatography
(20–30% EtOAc in Pet ether) afforded the product as a white
solid (56.8 mg, 50%). 1H NMR (400 MHz, CDCl3) δ 8.36 (app d, J = 8.7 Hz, 2H, H2), 7.98 (app d, J = 8.7 Hz, 2H, H3),
7.58 (app d, J = 8.0 Hz, 2H, H18), 7.30
(app d, J = 8.0 Hz, 2H, H19), 3.40 (dt, J = 10.7, 5.5 Hz, 2H, H5 + H9), 3.30
(td, J = 7.8, 3.6 Hz, 2H, H5 + H8), 3.18 (d, J = 9.7 Hz, 1H, H8′), 2.94–2.75 (m, 5H, H9′ + H14 + H15), 2.42 (s, 3H, H21), 2.17 (app dq, J = 10.2, 5.4, 4.5 Hz, 1H, H6), 1.78 (d, J = 12.9 Hz, 1H, H10 or H12), 1.70–1.49
(m, 7H, H10 + H12 + H13 + H16). 13C{1H} NMR (101 MHz, CDCl3)
δ 150.3 (C4), 143.8 (C17), 142.8 (C1), 133.0 (C20), 129.8 (C19), 128.5 (C3), 127.7 (C18), 124.5 (C2), 59.7 (C8), 50.3 (C6), 49.7 (C5), 43.1 (C10 or C12), 42.8 (C9 or C14 or C15), 42.8 (C9 or C14 or C15), 42.8 (C9 or C14 or C15), 40.8 (C7), 38.0 (C13 or C16),
37.2 (C13 or C16), 35.4 (C10 or C12), 31.0 (C11), 21.6 (C21). Mp: 188.0–189.0
°C. IR: νmax 2921, 1528, 1347, 1328, 1161 cm–1. HRMS: m/z calculated
for C25H31ClN3O6S2+, 568.1338 [M + H]+. Found m/z 568.1336, Δ = −0.3 ppm.
Rf (40% EtOAc in Pet ether) = 0.56.
4-(Bromomethyl)-2-((4-nitrophenyl)sulfonyl)-10-tosyl-2,10-diazadispiro[4.1.57.15]tridecane (3ah)
The compound
was prepared according to General Procedure E3. Flash column chromatography
(20–30% EtOAc in Pet ether) afforded the product as a white
solid (77.4 mg, 63%). 1H NMR (400 MHz, CDCl3) δ 8.35 (app d, J = 8.8 Hz, 2H, H2), 7.98 (app d, J = 8.8 Hz, 2H, H3),
7.57 (app d, J = 8.1 Hz, 2H, H18), 7.30
(app d, J = 8.1 Hz, 2H, H19), 3.42–3.36
(m, 1H, H5), 3.34–3.23 (m, 3H H5′ + H8 + H9), 3.20 (d, J =
9.8 Hz, 1H, H8′), 2.92–2.75 (m, 4H, H14 + H15), 2.63 (t, J = 10.6 Hz,
1H, H9′), 2.42 (s, 3H, H21), 2.22 (app
tt, J = 6.4, 3.9 Hz, 1H, H6), 1.76 (d, J = 12.9 Hz, 1H, H10 or H12), 1.66–1.47
(m, 7H, H10 + H12 + H13 + H16). 13C{1H} NMR (101 MHz, CDCl3)
δ 150.2 (C4), 143.8 (C17), 142.7 (C1), 133.0 (C20), 129.8 (C19), 128.5 (C3), 127.6 (C18), 124.5 (C2), 59.5 (C8), 50.6 (C5), 50.5 (C6), 42.8 (C10 or C12 or C14 or C15),
42.8 (C10 or C12 or C14 or C15), 42.8 (C10 or C12 or C14 or C15), 41.3 (C7), 37.9 (C13 or
C16), 37.2 (C13 or C16), 35.3 (C10 or C12), 30.9 (C11), 30.7 (C9), 21.6 (C21). Mp: Decomposed above 200 °C. IR: νmax 2921, 2841, 1535, 1350, 1330, 1161 cm–1. HRMS: m/z calculated for C25H31BrN3O6S2+, 612.0832 [M + H]+. Found m/z 612.0840, Δ = 1.3 ppm. Rf (30% EtOAc in Pet ether)
= 0.25.
4-(Chloromethyl)-2-((4-(5-(p-tolyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl)phenyl)sulfonyl)-10-tosyl-2,10-diazadispiro[4.1.57.15]tridecane (3ai)
The compound
was prepared according to General Procedure E1. Flash column chromatography
(15–30% EtOAc in Pet ether, then 5% MeCN in toluene) afforded
the product as an off-white crystalline solid (74.2 mg, 50%). 1H NMR (400 MHz, CDCl3) δ 7.79 (app d, J = 8.7 Hz, 2H, H27), 7.61 (app d, J = 8.2 Hz, 2H, H12), 7.49 (app d, J =
8.7 Hz, 2H, H28), 7.31 (app d, J = 8.0
Hz, 2H, H11), 7.16 (app d, J = 8.1 Hz,
2H, H3), 7.08 (app d, J = 8.1 Hz, 2H,
H4), 6.74 (s, 1H, H7), 3.43 (dd, J = 11.1, 4.1 Hz, 1H, H18), 3.39 (dd, J = 10.7, 6.6 Hz, 1H, H14), 3.29 (d, J = 9.5 Hz, 1H, H17), 3.28 (dd, J = 10.6,
4.2 Hz, 2H, H14′), 3.12 (d, J =
9.6 Hz, 1H, H17′), 2.98–2.77 (m, 5H, H18′ + H23 + H24), 2.43 (s, 3H,
H30), 2.37 (s, 3H, H1), 2.14 (ddt, J = 10.4, 6.6, 4.0 Hz, 1H, H15), 1.80 (d, J = 12.9 Hz, 1H, H19), 1.66–1.51 (m, 7H, H19′ + H21 + H22 + H25). 13C{1H} NMR (101 MHz, CDCl3) δ 145.4 (C6), 144.3 (d, J = 38.6 Hz, C8),
143.8 (C26), 142.9 (C13), 140.0 (C2), 136.3 (C10), 133.1 (C29), 129.9 (C3), 129.8 (C28), 128.9 (C4), 128.5 (C12), 127.8 (C27), 125.8 (C5), 125.7 (C11), 121.2 (q, J = 269.2 Hz, C9), 106.5
(C7), 59.8 (C17), 50.5 (C15), 49.8
(C14), 43.3 (C21), 43.0 (C14 or C23 or C24), 42.9 (C18 or C23 or C24), 42.9 (C14 or C23 or C24), 40.8 (C16), 38.2 (C22 or C25), 37.3 (C22 or C25), 35.6 (C19),
31.1 (C20), 21.7 (C30), 21.5 (C1). 19F NMR (376 MHz, CDCl3) δ −62.4 (CF3). Mp: 199.0–201.5 °C. IR: νmax 2923, 1346, 1235, 1161 cm–1. HRMS: m/z calculated for C36H38ClF3N4 O4S2+, 747.2048
[M + H]+. Found m/z 747.2049,
Δ = 0.2 ppm. Rf (30% EtOAc in Pet ether) = 0.40.
4-(Chloromethyl)-2-((4-(5-(p-tolyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl)phenyl)sulfonyl)-2-azaspiro[4.5]decane (3aj)
The compound was prepared according to General
Procedure E1. Flash column chromatography (10–30% Et2O in Pet ether) afforded the product as a white solid (89.0 mg, 81%). 1H NMR (400 MHz, CDCl3) δ 7.83 (app d, J = 8.5 Hz, 2H, H12), 7.50 (app d, J = 8.5 Hz, 2H, H11), 7.16 (app d, J =
8.0 Hz, 2H, H3), 7.09 (app d, J = 8.0
Hz, 2H, H4), 6.74 (s, 1H, H7), 3.56 (td, J = 10.5, 5.9 Hz, 2H, H14 + H18),
3.56 (td, J = 10.5, 5.9 Hz, 2H, H14 +
H18), 3.33 (d, J = 10.0 Hz, 1H, H17), 3.24 (dd, J = 10.2, 7.0 Hz, 1H, H14′), 3.14 (t, J = 10.8 Hz, 1H, H18′), 3.05 (d, J = 10.0 Hz, 1H, H17′), 2.37 (s, 3H, H1), 2.15–2.04
(m, 1H, H15), 1.61–1.08 (m, 10H, H19–20). 13C{1H} NMR (101 MHz, CDCl3)
δ 145.4 (C6), 144.2 (q, J = 38.5
Hz, C8), 142.7 (C13), 139.9 (C5),
136.5 (C10), 129.9 (C3), 128.8 (C4), 128.4 (C12), 125.8 (C2), 125.7 (C11), 121.2 (q, J = 269.2 Hz, C9), 106.4
(d, J = 1.8 Hz, C7), 56.5 (C17), 50.6 (C14), 50.1 (C15), 45.0 (C16), 43.3 (C18), 35.7 (C19–23), 29.2 (C19–23), 25.8 (C19–23), 23.3 (C19–23), 22.8 (C19–23), 21.4 (C1). Mp: 82.0–86.0 °C. IR: νmax 2925, 1471, 1347, 1235, 1159, 1129 cm–1. HRMS: m/z calculated for C27H30ClF3N3O2S+, 552.1694
[M + H]+. Found m/z 552.1696,
Δ = 0.4 ppm. Rf (30% Et2O in Pet ether) = 0.34.
4-(4-((4-(Chloromethyl)-2-azaspiro[4.5]decan-2-yl)sulfonyl)phenyl)-5-methyl-3-phenylisoxazole (3ak)
The compound was prepared according to
General Procedure E1. Flash column chromatography (10–30% Et2O in Pet ether) afforded the product as a colorless oil (70.0
mg, 72%). 1H NMR (400 MHz, CDCl3) δ 7.84
(app d, J = 8.2 Hz, 2H, H11), 7.42–7.27
(m, 7H, H1 + H2 + H3 + H10), 3.61 (dd, J = 10.3, 7.3 Hz, 1H, H13), 3.54 (dd, J = 10.9, 3.9 Hz, 1H, H17), 3.32 (d, J = 9.9 Hz, 1H, H16), 3.28
(dd, J = 10.4, 6.6 Hz, 1H, H13′), 3.14 (d, J = 10.2 Hz, 1H, H16′), 3.09 (app t, J = 10.8 Hz, 1H, H17′), 2.47 (s, 3H, H8), 2.17–2.06 (m, 1H, H14), 1.57–1.12 (m, 10H, H18–22). 13C{1H} NMR (101 MHz, CDCl3) δ 167.3 (C7), 161.1 (C5), 136.0 (C9), 135.4 (C12), 130.4 (C10), 129.8 (C1), 128.8 (C2 or C3), 128.6 (C4), 128.5 (C2 or C3), 127.8 (C11), 114.6 (C6),
56.3 (C16), 50.4 (C13), 49.9 (C14), 45.0 (C15), 43.3 (C17), 35.6 (C18–22), 29.2 (C18–22), 25.8 (C18–22), 23.2 (C18–22), 22.8 (C18–22), 11.8 (C8). IR: νmax 2928, 2855, 1343,
1159 cm–1. HRMS: m/z calculated for C26H30ClN2 O3S+, 485.1660 [M + H]+. Found m/z 485.1667, Δ = 1.3 ppm. Rf (30%
EtOAc in Pet ether) = 0.69.
N-(5-((4-(Chloromethyl)-2-azaspiro[4.5]decan-2-yl)sulfonyl)-3-methyl-1,3,4-thiadiazol-2(3H)-ylidene)acetamide (3al)
The compound
was prepared according to General Procedure E1. Flash column chromatography
(10–30% EtOAc in Pet ether, then 3–5% MeCN in toluene)
afforded the product as a white solid (36.7 mg, 45%). 1H NMR (400 MHz, CDCl3) δ 4.00 (s, 3H, H5), 3.80 (dd, J = 10.6, 7.4 Hz, 1H, H6), 3.61 (dd, J = 11.0, 4.1 Hz, 1H, H10), 3.57 (d, J = 10.2 Hz, 1H, H9), 3.48
(dd, J = 10.6, 6.8 Hz, 1H, H6′),
3.30 (t, J = 10.7 Hz, 1H, H10′),
3.29 (d, J = 10.1 Hz, 1H, H9′),
2.36 (s, 3H, H1), 2.25 (dtd, J = 10.9,
7.1, 4.1 Hz, 1H, H7), 1.65–1.16 (m, 10H, H11–15). 13C{1H} NMR (101 MHz, CDCl3)
δ 181.1 (C2), 164.9 (C3), 153.9 (C4), 56.8 (C9), 50.9 (C6), 50.1 (C7), 45.3 (C8), 43.1 (C10), 38.6 (C5), 35.6 (C11–15), 29.1 (C11–15), 26.7 (C1), 25.8 (C11–15), 23.3 (C11–15), 22.7 (C11–15). Mp: 157.0–158.4
°C. IR: νmax 2929, 2848, 1618, 1486, 1366, 1301,
1242, 1163 cm–1. HRMS: m/z calculated for C15H24ClN4 O3S2+, 407.0979 [M + H]+. Found m/z 407.0976, Δ =
0.8 ppm. Rf (30% EtOAc in Pet ether) = 0.18.
2-((8-(Chloromethyl)-6-azaspiro[3.4]octan-6-yl)sulfonyl)-6-ethoxybenzo[d]thiazole (3am)
The compound was
prepared according to General Procedure E1. Flash column chromatography
(7–10% EtOAc) afforded the product as an off-white solid (29.5
mg, 37%). 1H NMR (500 MHz, CDCl3) δ 8.03
(d, J = 9.1 Hz, 1H, H7), 7.34 (d, J = 2.5 Hz, 1H, H4), 7.17 (dd, J = 9.1, 2.5 Hz, 1H, H8), 4.11 (q, J =
7.0 Hz, 2H, H2), 3.71 (dd, J = 10.6, 6.5
Hz, 1H, H10), 3.68 (d, J = 10.0 Hz, 1H,
H13), 3.61 (dd, J = 10.6, 4.4 Hz, 1H,
H10′), 3.56 (d, J = 10.3 Hz, 1H,
H13′), 3.56 (dd, J = 10.7, 3.9
Hz, 1H, H14), 3.19 (t, J = 10.8 Hz, 1H,
H14′), 2.37–2.28 (m, 1H, H11),
2.05 (td, J = 10.6, 9.9, 6.3 Hz, 1H, H15), 1.94–1.71 (m, 5H, H15′-17), 1.47
(t, J = 7.0 Hz, 3H, H1). 13C{1H} NMR (126 MHz, CDCl3) δ 160.5 (C9), 159.0 (C3), 147.0 (C5), 138.1 (C6), 125.9 (C7), 118.3 (C8), 104.1 (C4), 64.4 (C2), 58.7 (C13), 50.7 (C10), 49.6 (C11), 47.7 (C12), 43.1 (C14), 32.6 (C17), 26.0 (C15), 16.3 (C16), 14.8 (C1). Mp: 100.0–102.1 °C.
IR: νmax 2980, 2927, 2874, 1599, 1485, 1470, 1365,
1255, 1226, 1156 cm–1. HRMS: m/z calculated for C17H22ClN2O3S2+, 407.0755 [M + H]+. Found m/z 401.0761, Δ =
1.4 ppm. Rf (20% EtOAc in Pet ether) = 0.28.
4-Methyl-2-tosyl-2-azaspiro[4.5]decane (4a)
The compound was prepared according to General Procedure F1. Flash
column chromatography (5% EtOAc in Pet ether) afforded the product
as a white solid (46.5 mg, 76%). 1H NMR (400 MHz, CDCl3) δ 7.71 (app d, J = 8.1 Hz, 2H, H4), 7.30 (app d, J = 8.1 Hz, 2H, H3), 3.44 (dd, J = 9.5, 7.4 Hz, 1H, H6),
3.38 (d, J = 10.1 Hz, 1H, H9), 2.94 (d, J = 10.1 Hz, 1H, H9′), 2.85 (dd, J = 9.5, 8.5 Hz, 1H, H6′), 2.41 (s, 3H,
H1), 1.73 (h, J = 7.2 Hz, 1H, H7), 1.58–1.39 (m, 3H, H11–15), 1.28–0.85
(m, 7H, H11–15), 0.76 (d, J = 7.0
Hz, 3H, H10). 13C{1H} NMR (101 MHz,
CDCl3) δ 143.3 (C5), 134.3 (C2), 129.6 (C3), 127.5 (C4), 56.2 (C9), 53.3 (C6), 44.2 (C8), 42.2 (C7), 35.1 (C11–15), 28.0 (C11–15), 26.1 (C11–15), 23.7 (C11–15), 22.8 (C11–15), 21.6 (C1), 12.0 (C10). Mp: 92.2–95.4 °C. IR: νmax 2925, 2857, 1335, 1153 cm–1. HRMS: m/z calculated for C17H26NO2S+, 308.1679 [M + H]+. Found m/z 308.1674, Δ = −1.6 ppm.
Rf (10% EtOAc in Pet ether) = 0.35.
4-Methyl-2,8-ditosyl-2,8-diazaspiro[4.5]decane (4b)
The compound was prepared according to General Procedure
F1. Flash column chromatography (10–30% EtOAc in Pet ether)
afforded the product as a white solid (59.0 mg, 64%). 1H NMR (400 MHz, CDCl3) δ 7.62 (app d, J = 8.6 Hz, 2H, H4 or H16), 7.60 (app d, J = 8.6 Hz, 2H, H4 or H16), 7.35 (app
d, J = 8.1 Hz, 2H, H3 or H17), 7.28 (app d, J = 8.0 Hz, 2H, H3 or
H17), 3.52 (app t, J = 11.8 Hz, 2H, H12 + H13), 3.40 (dd, J = 9.9, 7.6
Hz, 1H, H6), 3.17 (d, J = 10.1 Hz, 1H,
H9), 2.83 (dd, J = 9.8, 8.5 Hz, 1H, H6′), 2.78 (d, J = 10.2 Hz, 1H, H9′), 2.47 (s, 3H, H1 or H19),
2.42 (s, 3H, H1 or H19), 2.27–2.12 (m,
2H, H12′ + H13′), 1.72 (dt, J = 14.8, 7.3 Hz, 1H, H7), 1.63 (td, J = 13.1, 4.5 Hz, 1H, H11 or H14),
1.47 (td, J = 12.7, 4.1 Hz, 1H, H11 or
H14), 1.22–1.14 (m, 1H, H11′ or
H14′), 1.10–1.01 (m, 1H, H11′ or H14′), 0.74 (d, J = 6.9 Hz,
3H, H10). 13C{1H} NMR (101 MHz, CDCl3) δ 144.0 (C2 or C18), 143.7 (C2 or C18), 133.8 (C5 or C15), 132.9 (C5 or C15), 130.0 (C3 or
C17), 129.8 (C3 or C17), 127.7 (C4 or C16), 127.4 (C4 or C16), 54.7 (C9), 52.9 (C6), 43.9 (C12 or C13), 43.1 (C12 or C13), 42.1
(C8), 41.7 (C7), 33.4 (C11 or C14), 27.2 (C11 or C14), 21.7 (C1 or C19), 21.7 (C1 or C19), 11.9
(C10). Mp: 61.1–64.2 °C. IR: νmax 2923, 1339, 1155 cm–1. HRMS: m/z calculated for C23H31N2O4S2+, 463.1720 [M + H]+. Found m/z 463.1727, Δ
= 1.5 ppm. Rf (30% EtOAc in Pet ether) = 0.22.
8-Methyl-6-((4-(methylsulfonyl)phenyl)sulfonyl)-6-azaspiro[3.4]octane (4c)
The compound was prepared according to
General Procedure F1. Flash column chromatography (20–40% EtOAc
in Pet ether) afforded the product as an off-white crystalline solid
(46.4 mg, 68%). 1H NMR (400 MHz, CDCl3) δ
8.09 (app dd, J = 6.7, 1.7 Hz, 2H, H3),
8.01 (app dd, J = 6.7, 1.7 Hz, 2H, H4),
3.37 (dd, J = 9.6, 6.5 Hz, 1H, H6), 3.36
(d, J = 9.7 Hz, 1H, H9), 3.24 (d, J = 9.7 Hz, 1H, H9′), 3.09 (s, 3H, H1), 2.89 (dd, J = 9.7, 6.2 Hz, 1H, H6′), 1.90 (app ddt, J = 13.5, 9.6, 6.5 Hz, 2H, H7 + H11), 1.81–1.64 (m, 4H, H12 + H13), 1.56–1.46 (m, 1H, H11′), 0.81 (d, J = 6.9 Hz, 3H, H10). 13C{1H} NMR (101 MHz, CDCl3) δ
144.1 (C2), 142.8 (C5), 128.3 (C3 or C4), 128.3 (C3 or C4), 58.4
(C9), 53.3 (C6), 48.0 (C8), 44.4
(C1), 40.8 (C7), 30.1 (C12), 25.7
(C11), 15.7 (C13), 12.8 (C10). Mp:
148.0–151.0 °C. IR: νmax 3096, 2925,
1335, 1308, 1283, 1157 cm–1. HRMS: m/z calculated for C15H22NO4S2+, 344.0984 [M + H]+. Found m/z 344.0980, Δ = −1.3 ppm.
Rf (40% EtOAc in Pet ether) = 0.28.
Methyl 4-((8-Methyl-6-azaspiro[3.4]octan-6-yl)sulfonyl)benzoate (4d)
The compound was prepared according to General Procedure F1. Flash column chromatography (5–10% EtOAc in Pet ether, then 0–2% MeCN in toluene) afforded the product as a white solid (58.1 mg, 90%). 1H NMR (400 MHz, CDCl3) δ 8.17 (app d, J = 8.5 Hz, 2H, H4), 7.89 (app d, J = 8.5 Hz, 2H, H5), 3.94 (s, 3H, H1), 3.36 (dd, J = 9.7, 6.6 Hz, 1H, H7), 3.34 (d, J = 9.8 Hz, 1H, H10), 3.24 (d, J = 9.8 Hz, 1H, H10′), 2.89 (dd, J = 9.7, 6.0 Hz, 1H, H7′), 1.87 (dt, J = 12.5, 6.5 Hz, 2H, H8 + H12), 1.81–1.58 (m, 4H, H13 + H14), 1.54–1.42 (m, 1H, H12′), 0.78 (d, J = 6.9 Hz, 3H, H11). 13C{1H} NMR (101 MHz, CDCl3) δ 165.8 (C2), 141.3 (C3), 133.8 (C6), 130.3 (C4), 127.4 (C5), 58.3 (C10), 53.3 (C7), 52.7 (C1), 48.0 (C9), 40.9 (C8), 30.3 (C14), 25.7 (C12), 15.7 (C13), 12.8 (C11). Mp: 83.0–84.3 °C. IR: νmax 2954, 2870, 1726, 1336, 1275, 1154 cm–1. HRMS: m/z calculated for C16H22NO4S+, 324.1264 [M + H]+. Found m/z 324.1264, Δ = 0.1 ppm. Rf (20% EtOAc in Pet ether) = 0.29.
Derivatization Procedures to 5 and 6

2,8-Ditosyl-2,8-diazaspiro[4.5]decane-4-carbaldehyde (5)
The compound was prepared following an adapted
literature procedure.39 A 20 mL microwave
vial was charged with spirocycle 3ad (199.1 mg, 0.368
mmol, 1.0 equiv) and AgBF4 (214.7 mg, 1.162 mmol, 3.0 equiv),
placed under nitrogen, and then dry DMSO (7 mL) was added. The mixture
was then heated to 100 °C for 16 h before being allowed to cool
to room temperature. Et3N (61 μL, 0.44 mmol, 1.2
equiv) was then added and the solution stirred at room temperature
for 1 h. The mixture was then poured into Et2O (100 mL)
and washed with H2O (100 mL). The aqueous layer was then
extracted with Et2O (2 × 50 mL) and the organic layers
combined. The solvent was removed in vacuo and the
crude product subjected to flash column chromatography (20–30%
EtOAc in Pet ether), which afforded the product as a white crystalline
solid (70.7 mg, 40%). 1H NMR (400 MHz, CDCl3) δ 9.52 (d, J = 2.1 Hz, 1H, H10), 7.66 (app d, J = 8.2 Hz, 4H, H4 or
H16), 7.61 (app d, J = 8.2 Hz, 4H, H4 or H16), 7.36 (app d, J = 8.0
Hz, 4H, H3 or H17), 7.32 (app d, J = 8.0 Hz, 4H, H3 or H17), 3.61–3.34
(m, 4H, H6 + H6′ + H12 + H13), 3.09 (d, J = 10.1 Hz, 1H, H9), 3.04 (d, J = 10.1 Hz, 1H, H9′), 2.63 (ddd, J = 7.9, 6.2, 2.0 Hz, 1H, H7), 2.51–2.30 (m, 8H, H1 + H12′ + H13′ + H19), 1.88 (ddd, J = 13.6, 11.3, 4.3 Hz, 1H, H11), 1.71–1.56 (m,
2H, H11′ + H14), 1.40 (d, J = 13.5 Hz, 1H, H14′). 13C{1H} NMR (101 MHz, CDCl3) δ 199.3 (C10),
144.3 (C5 or C15), 144.3 (C5 or C15), 133.2 (C2 or C18), 132.9 (C2 or C18), 130.1 (C3 or C17), 130.0 (C3 or C17), 127.7 (C4 or
C16), 127.6 (C4 or C16), 57.8 (C7), 55.0 (C9), 45.5 (C6), 44.1 (C8), 43.5 (C12 or C13), 43.2 (C12 or C13), 34.5 (C11), 29.8 (C14),
21.7 (C1 + C19). Mp: 70.0–73.1 °C.
IR: νmax 2923, 2847, 1720, 1597, 1340, 1327, 1157;
cm–1. HRMS: m/z calculated for C23H29N2O5S2+, 477.1512 [M + H]+. Found m/z 477.1520, Δ = 1.7 ppm. Rf (30%
EtOAc in Pet ether) = 0.16.
4-(Pyridin-3-ylmethyl)-2,8-ditosyl-2,8-diazaspiro[4.5]decane (6)
The compound was prepared following the general procedure as disclosed by MacMillan.12 To an 8 mL microwave vial equipped with a stir bar was added photocatalyst (Ir[dF(CF3)ppy]2(dtbbpy))PF6 (5.6 mg, 0.01 equiv), 4-(bromomethyl)-2,8-ditosyl-2,8-diazaspiro[4.5]decane (3ad) (406.1 mg, 0.75 mmol, 1.5 equiv), 3-bromopyridine (48 μL, 0.50 mmol, 1.0 equiv), tris(trimethylsilyl)silane (154 μL, 0.50 mmol, 1.0 equiv), and anhydrous sodium carbonate (106 mg, 1.0 mmol, 2.0 equiv). The vial was sealed and placed under nitrogen before 4 mL of 1,2-dimethoxyethane (DME) was added. To a separate vial were added NiCl2·glyme (1 mg, 5 μmol, 0.01 equiv) and 4,4′-di-tert-butyl-2,2′-bipyridine (1.3 mg, 5 μmol, 0.01 equiv). The catalyst vial was sealed, purged with nitrogen, and then to it was added 2 mL of DME. The precatalyst solution was sonicated for 5 min and 1 mL (0.5 mol % catalyst, 2.5 μmol, 0.005 equiv) was syringed into the reaction vessel. The solution was degassed by sparging with nitrogen while stirring for 10 min before sealing with Parafilm. The reaction was stirred and irradiated with a 470 nm LED with fan-cooling (see the SI for setup) for 20 h. The reaction was then concentrated in vacuo and purified by chromatography (50–60% EtOAc in Pet ether) to give tris-azaheterocycle 6 (86.4 mg, 32%). 1H NMR (400 MHz, CDCl3) δ 8.51–8.43 (m, 1H, H23), 8.29–8.23 (m, 1H, H24), 7.63 (app dd, J = 6.6, 1.6 Hz, 2H, H4 or H16), 7.58 (app dd, J = 6.6, 1.7 Hz, 2H, H4 or H16), 7.37 (d, J = 8.0 Hz, 2H, H3 or H17), 7.34 (dt, J = 7.8, 1.9 Hz, 1H, H21), 7.29 (d, J = 7.9 Hz, 2H, H3 or H17), 7.21 (dd, J = 7.7, 4.8 Hz, 1H, H22), 3.65 (d, J = 11.7 Hz, 2H, H12 + H13), 3.31 (d, J = 10.2 Hz, 1H, H9), 3.17 (dd, J = 10.1, 7.6 Hz, 1H, H6), 2.92 (dd, J = 10.1, 8.6 Hz, 1H, H6′), 2.83 (d, J = 10.2 Hz, 1H, H9′), 2.70 (dd, J = 13.7, 3.4 Hz, 1H, H10), 2.49 (s, 3H, H1 or H19), 2.44 (s, 3H, H1 or H19), 2.27 (td, J = 12.1, 2.4 Hz, 1H, H12′), 2.19 (dtd, J = 14.8, 7.5, 7.0, 4.1 Hz, 2H, H13′ + H10′), 1.89 (dtd, J = 11.6, 8.2, 3.5 Hz, 1H, H7), 1.81 (td, J = 13.0, 4.5 Hz, 1H, H14), 1.65 (td, J = 12.6, 4.1 Hz, 1H, H11), 1.35–1.24 (m, 2H, H11′ + H14′). 13C{1H} NMR (126 MHz, CDCl3) δ 149.8 (C24), 148.2 (C23), 144.2 (C5 or C15), 144.0 (C5 or C15), 136.2 (C21), 134.8 (C20), 133.7 (d, J = 0.6 Hz, C2 or C18), 132.8 (d, J = 0.7 Hz, C2 or C18), 130.1 (C3 or C17), 129.9 (C3 or C17), 127.7 (C4 or C16), 127.4 (C4 or C16), 123.8 (C22), 54.9 (C9), 50.6 (C6), 49.3 (C7), 43.9 (C13), 43.1 (C12), 42.6 (C8), 33.7 (C14), 31.0 (C10), 27.8 (C11), 21.8 (C1 or C19), 21.7 (C1 or C19). Mp: 174.6–177.7 °C. IR: νmax 2844, 1338, 1326, 1157 cm–1. HRMS: m/z calculated for C28H34N3O4S2+, 539.1985 [M + H]+. Found m/z 539.1997, Δ = 2.2 ppm. Rf (50% EtOAc in Pet ether) = 0.09.
Acknowledgments
O.M.G. acknowledges support from the EPSRC (EP/S024220/1) SynTech Automated Centre for Chemical Synthesis, Centre for Doctoral Training in Cambridge, UK. S.V.L. thanks the American Chemical Society for support through receipt of the Arthur C. Cope Award.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.2c01684.
Details of experimental setup, NMR spectra, and X-ray crystallographic data (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Zheng Y.-J.; Tice C. M.; Singh S. B. The use of spirocyclic scaffolds in drug discovery. Bioorg. Med. Chem. Lett. 2014, 24, 3673–3682. 10.1016/j.bmcl.2014.06.081. [DOI] [PubMed] [Google Scholar]; b Johansson L.; Anders M.. Azetidine derivatives for treatment of melanin related disorders. Int. Patent WO2013/011285A1, 2013.; c Smith L. K.; Baxendale I. R. Total syntheses of natural products containing spirocarbocycles. Org. Biomol. Chem. 2015, 13, 9907–9933. 10.1039/C5OB01524C. [DOI] [PubMed] [Google Scholar]; d Chen H.; Colletti S. L.; Demong D.; Guo Y.; Miller M.; Nair A.; Plummer C.; Xiao D.; Yang D.-Y.. Antidiabetic bicyclic compounds. Int. Patent WO2016/022742A1, 2016.; e Zheng Y.-J.; Tice C. M. The utilization of spirocyclic scaffolds in novel drug discovery. Expert Opin. Drug Discovery 2016, 11, 831–834. 10.1080/17460441.2016.1195367. [DOI] [PubMed] [Google Scholar]; f Luttens A.; Gullberg H.; Abdurakhmanov E.; Vo D. D.; Akaberi D.; Talibov V. O.; Nekhotiaeva N.; Vangeel L.; Jonghe S. D.; Jochmans D.; Krambrich J.; Tas A.; Lundgren B.; Gravenfors Y.; Craig A. J.; Atilaw Y.; Sandström A.; Moodie L. W. K.; Lundkvist Å.; van Hemert M. J.; Neyts J.; Lennerstrand J.; Kihlberg J.; Sandberg K.; Danielson U. H.; Carlsson J. Ultralarge Virtual Screening Identifies SARS-CoV-2 Main Protease Inhibitors with Broad-Spectrum Activity against Coronaviruses. J. Am. Chem. Soc. 2022, 144, 2905–2920. 10.1021/jacs.1c08402. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Bora D.; Kaushal A.; Shankaraiah N. Anticancer potential of spirocompounds in medicinal chemistry: A pentennial expedition. Eur. J. Med. Chem. 2021, 215, 113263 10.1016/j.ejmech.2021.113263. [DOI] [PubMed] [Google Scholar]
- Ritchie T. J.; MacDonald S. J. F. The impact of aromatic ring count on compound developability: Are too many aromatic rings a liability in drug design?. Drug Discovery Today 2009, 14, 1011–1020. 10.1016/j.drudis.2009.07.014. [DOI] [PubMed] [Google Scholar]
- For some recent applications of bis-azaheterospirocycles, see:; a Brown D. G.; Bernstein P. R.; Griffin A.; Wesolowski S.; Labrecque D.; Tremblay M. C.; Sylvester M.; Mauger R.; Edwards P. D.; Throner S. R.; Folmer J. J.; Cacciola J.; Scott C.; Lazor L. A.; Pourashraf M.; Santhakumar V.; Potts W. M.; Sydserff S.; Giguere P.; Levesque C.; Dasser M.; Groblewski T. Discovery of spirofused piperazine and diazepane amides as selective histamine-3 antagonists with in vivo efficacy in a mouse model of cognition. J. Med. Chem. 2014, 57, 733–758. 10.1021/jm4014828. [DOI] [PubMed] [Google Scholar]; b Ikeda S.; Sugiyama H.; Tokuhara H.; Murakami M.; Nakamura M.; Oguro Y.; Aida J.; Morishita N.; Sogabe S.; Dougan D. R.; Gay S. C.; Qin L.; Arimura N.; Takahashi Y.; Sasaki M.; Kamada Y.; Aoyama K.; Kimoto K.; Kamata M. Design and Synthesis of Novel Spiro Derivatives as Potent and Reversible Monoacylglycerol Lipase (MAGL) Inhibitors: Bioisosteric Transformation from 3-Oxo-3,4-dihydro-2H-benzo[b][1,4]oxazin-6-yl Moiety. J. Med. Chem. 2021, 64, 11014–11044. 10.1021/acs.jmedchem.1c00432. [DOI] [PubMed] [Google Scholar]; c Kargbo R. B. Discovery of Spirocyclic Androgen Receptor Protein Degraders for the Treatment of Prostate Cancer. ACS Med. Chem. Lett. 2021, 12, 1635–1636. 10.1021/acsmedchemlett.1c00498. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Chan B. K.; Seward E.; Lainchbury M.; Brewer T. F.; An L.; Blench T.; Cartwright M. W.; Chan G. K. Y.; Choo E. F.; Drummond J.; Elliott R. L.; Gancia E.; Gazzard L.; Hu B.; Jones G. E.; Luo X.; Madin A.; Malhotra S.; Moffat J. G.; Pang J.; Salphati L.; Sneeringer C. J.; Stivala C. E.; Wei B.; Wang W.; Wu P.; Heffron T. P. Discovery of Spiro-azaindoline Inhibitors of Hematopoietic Progenitor Kinase 1 (HPK1). ACS Med. Chem. Lett. 2022, 13, 84–91. 10.1021/acsmedchemlett.1c00473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For selected recent examples, see:; a Wang Y.-Y.; Bode J. W. Olefin Amine (OLA) Reagents for the Synthesis of Bridged Bicyclic and Spirocyclic Saturated N-Heterocycles by Catalytic Hydrogen Atom Transfer (HAT) Reactions. J. Am. Chem. Soc. 2019, 141, 9739–9745. 10.1021/jacs.9b05074. [DOI] [PubMed] [Google Scholar]; b Espinosa M.; Noda H.; Shibasaki M. Synthesis of Unprotected Spirocyclic β-Prolines and β-Homoprolines by Rh-Catalysed C-H Insertion. Org. Lett. 2019, 21, 9296–9299. 10.1021/acs.orglett.9b03198. [DOI] [PubMed] [Google Scholar]; c Sveiczer A.; North A. J. P.; Mateu N.; Kidd S. L.; Sore H. F.; Spring D. R. Spirocycles as Rigified sp3-Rich Scaffolds for a Fragment Collection. Org. Lett. 2019, 21, 4600–4604. 10.1021/acs.orglett.9b01499. [DOI] [PubMed] [Google Scholar]; d Shennan B. D. A.; Smith P. W.; Ogura Y.; Dixon D. J. A modular and divergent approach to spirocyclic pyrrolidines. Chem. Sci. 2020, 11, 10354–10360. 10.1039/D0SC03676E. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Safrygin A.; Dar’in D.; Bakulina O.; Krasavin M. Synthesis of spirocyclic tetrahydroisoquinolines (spiroTHIQs) via the Castagnoli-Cushman reaction. Tetrahedron Lett. 2020, 61, 152408 10.1016/j.tetlet.2020.152408. [DOI] [Google Scholar]; f Lin S.; Chen Y.; Li F.; Shi C.; Shi L. Visible-light-driven spirocyclization of epoxides via dual titanocene and photoredox catalysis. Chem. Sci. 2020, 11, 839–844. 10.1039/C9SC05601G. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Peshkov A. A.; Makhmet A.; Bakulina O.; Kanov E.; Gainetdinov R.; Peshkov V. A.; Dar’in D.; Krasavin M. A General Approach to Spirocyclic Piperidines via Castagnoli-Cushman Chemistry. Synthesis 2022, 54, 2604–2615. 10.1055/s-0040-1719878. [DOI] [Google Scholar]; h Chalyk B. A.; Butko M. V.; Yanshyna O. O.; Gavrilenko K. S.; Druzhenko T. V.; Mykhailiuk P. K. Synthesis of Spirocyclic Pyrrolidines: Advanced Building Blocks for Drug Discovery. Chem. – Eur. J. 2017, 23, 16782–16786. 10.1002/chem.201702362. [DOI] [PubMed] [Google Scholar]; i Melnykov K. P.; Artemenko A. N.; Ivanenko B. O.; Sokolenko Y. M.; Nosik P. S.; Ostapchuk E. N.; Grygorenko O. O.; Volochnyuk D. M.; Ryabukhin S. V. Scalable Synthesis of Biologically Relevant Spirocyclic Pyrrolidines. ACS Omega 2019, 4, 7498–7515. 10.1021/acsomega.9b00896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For reviews, see:; a Fang X.; Wang C.-J. Catalytic asymmetric construction of spiropyrrolidines via 1,3-dipolar cycloaddition of azomethine ylides. Org. Biomol. Chem. 2018, 16, 2591–2601. 10.1039/C7OB02686B. [DOI] [PubMed] [Google Scholar]; b Gataullin R. R. Advances in the Synthesis of Benzo-Fused Spiro Nitrogen Heterocycles: New Approaches and Modification of Old Strategies. Helv. Chim. Acta 2020, 103, e2000137 10.1002/hlca.202000137. [DOI] [Google Scholar]; c Yang W.-C.; Zhang M.-M.; Feng J.-G. Recent Advances in the Construction of Spiro Compounds via Radical Dearomatization. Adv. Synth. Catal. 2020, 362, 4446–4461. 10.1002/adsc.202000636. [DOI] [Google Scholar]; d Alves N. G.; Alves A. J. S.; Soares M. I. L.; Pinho e Melo T. M. V. D. Recent Advances in the Synthesis of Spiro-β-Lactams and Spiro-δ-Lactams. Adv. Synth. Catal. 2021, 363, 2464–2501. 10.1002/adsc.202100013. [DOI] [Google Scholar]; e Westphal R.; Filho E. V.; Medici F.; Benaglia M.; Greco S. J. Stereoselective Domino Reactions in the Synthesis of Spiro Compounds. Synthesis 2022, 54, 2927–2975. 10.1055/a-1771-0641. [DOI] [Google Scholar]; f Ganesh M.; Suraj S. Expeditious entry into carbocyclic and heterocyclic spirooxindoles. Org. Biomol. Chem. 2022, 20, 5651–5693. 10.1039/D2OB00767C. [DOI] [PubMed] [Google Scholar]
- For recent examples, see:; a Flodén N. J.; Trowbridge A.; Willcox D.; Walton S. M.; Kim Y.; Gaunt M. J. Streamlined Synthesis of C(sp3)-Rich N-Heterospirocycles Enabled by Visible-Light-Mediated Photocatalysis. J. Am. Chem. Soc. 2019, 141, 8426–8430. 10.1021/jacs.9b03372. [DOI] [PubMed] [Google Scholar]; b Zhu M.; Zheng C.; Zhang X.; You S.-L. Synthesis of Cyclobutane-Fused Angular Tetracyclic Spiroindolines via Visible-Light-Promoted Intramolecular Dearomatization of Indole Derivatives. J. Am. Chem. Soc. 2019, 141, 2636–2644. 10.1021/jacs.8b12965. [DOI] [PubMed] [Google Scholar]; c Oderinde M. S.; Mao E.; Ramirez A.; Pawluczyk J.; Jorge C.; Cornelius L. A. M.; Kempson J.; Vetrichelvan M.; Pitchai M.; Gupta A.; Gupta A. K.; Meanwell N. A.; Mathur A.; Dhar T. G. M. Synthesis of Cyclobutane-Fused Tetracyclic Scaffolds via Visible-Light Photocatalysis for Building Molecular Complexity. J. Am. Chem. Soc. 2020, 142, 3094–3103. 10.1021/jacs.9b12129. [DOI] [PubMed] [Google Scholar]; d Becker M. R.; Wearing E. R.; Schindler C. S. Synthesis of azetidines via visible-light-mediated intermolecular [2+2] photocycloadditions. Nat. Chem. 2020, 12, 898–905. 10.1038/s41557-020-0541-1. [DOI] [PubMed] [Google Scholar]; e Xiong Y.; Großkopf J.; Jandl C.; Bach T. Visible Light-Mediated Dearomative Hydrogen Atom Abstraction/Cyclisation Cascade of Indoles. Angew. Chem., Int. Ed. 2022, 61, e202200555 10.1002/anie.202200555. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Dai Y.; Liang S.; Zeng G.; Huang H.; Zhao X.; Cao S.; Jiang Z. Asymmetric [3+2] photocycloadditions of cyclopropylamines with electron-rich and electron-neutral olefins. Chem. Sci. 2022, 13, 3787–3795. 10.1039/D1SC07044D. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Yang M.; Hua J.; Wang H.; Ma T.; Liu C.; He W.; Zhu N.; Hu Y.; Fang Z.; Guo K. Photomediated Spirocyclization of N-Benzyl Propiolamide with N-Iodosuccinimide for Access to Azaspiro[4.5]deca-6,9-diene-3,8-dione. J. Org. Chem. 2022, 87, 8445–8457. 10.1021/acs.joc.2c00579. [DOI] [PubMed] [Google Scholar]; h Kalaitzakis D.; Kampouropoulos I.; Sofiadis M.; Montagnon T.; Vassilikogiannakis G. Access to high value sp3-rich frameworks using photocatalyzed [2+2]-cycloadditions of γ-alkylidene-γ-lactams. Chem. Commun. 2022, 58, 8085–8088. 10.1039/D2CC03009H. [DOI] [PubMed] [Google Scholar]
- a Tsao J. Y.; Han J.; Haitz R. H.; Pattison M. The Blue LED Nobel Prize: Historical Context and Human Benefit. Ann. Phys. 2015, 527, A53–A61. 10.1002/andp.201570058. [DOI] [Google Scholar]; b Sender M.; Ziegenbalg D. Light Sources for Photochemical Processes – Estimation of Technological Potentials. Chem. Ing. Tech. 2017, 89, 1159–1173. 10.1002/cite.201600191. [DOI] [Google Scholar]; c Buglioni L.; Raymenants F.; Slattery A.; Zondag S. D. A.; Noël T. Technological Innovations in Photochemistry for Organic Synthesis: Flow Chemistry, High-Throughput Experimentation, Scale-up, and Photoelectrochemistry. Chem. Rev. 2022, 122, 2752–2906. 10.1021/acs.chemrev.1c00332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For reviews, see:; a Prier C. K.; Rankic D. A.; MacMillan D. W. C. Visible light photoredox catalysis with transition metal complexes: applications in organic synthesis. Chem. Rev. 2013, 113, 5322–5363. 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Shaw M. H.; Twilton J.; MacMillan D. W. C. Photoredox catalysis in organic chemistry. J. Org. Chem. 2016, 81, 6898–6926. 10.1021/acs.joc.6b01449. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Twilton J.; Le C.; Zhang P.; Shaw M. H.; Evans R. W.; MacMillan D. W. C. The merger of transition metal and photocatalysis. Nat. Rev. Chem. 2017, 1, 0052 10.1038/s41570-017-0052. [DOI] [Google Scholar]
- For reviews, see:; a Chen J.-R.; Hu X.-Q.; Lu L.-Q.; Xiao W.-J. Visible light photoredox-controlled reactions of N-radicals and radical ions. Chem. Soc. Rev. 2016, 45, 2044–2056. 10.1039/C5CS00655D. [DOI] [PubMed] [Google Scholar]; b Kwon K.; Simons R. T.; Nandakumar M.; Roizen J. L. Strategies to Generate Nitrogen-centered Radicals That May Rely on Photoredox Catalysis: Development in Reaction Methodology and Applications in Organic Synthesis. Chem. Rev. 2022, 122, 2353–2428. 10.1021/acs.chemrev.1c00444. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Pratley C.; Fenner S.; Murphy J. A. Nitrogen-Centered Radicals in Functionalization of sp2 Systems: Generation, Reactivity, and Applications in Synthesis. Chem. Rev. 2022, 122, 8181–8260. 10.1021/acs.chemrev.1c00831. [DOI] [PubMed] [Google Scholar]; d Rivas M.; Palchykov V.; Jia X.; Gevorgyan V. Recent advances in visible light-induced C(sp3)-N bond formation. Nat. Rev. Chem. 2022, 6, 544–561. 10.1038/s41570-022-00403-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Hioe J.; Šakic D.; Vrček V.; Zipse H. The stability of nitrogen-centered radicals. Org. Biomol. Chem. 2015, 13, 157–169. 10.1039/C4OB01656D. [DOI] [PubMed] [Google Scholar]; b Davies J.; Morcillo S. P.; Douglas J. J.; Leonori D. Hydroxylamine Derivatives as Nitrogen-Radical Precursors in Visible-Light Photochemistry. Chem. – Eur. J. 2018, 24, 12154–12163. 10.1002/chem.201801655. [DOI] [PubMed] [Google Scholar]
- Crespin L. N. S.; Greb A.; Blakemore D. C.; Ley S. V. Visible-Light-Mediated Annulation of Electron-Rich Alkenes and Nitrogen-Centered Radicals from N-Sulfonylallylamines: Construction of Chloromethylated Pyrrolidine Derivatives. J. Org. Chem. 2017, 82, 13093–13108. 10.1021/acs.joc.7b02146. [DOI] [PubMed] [Google Scholar]
- Zhang P.; Le C.; MacMillan D. W. C. Silyl Radical Activation of Alkyl Halides in Metallaphotoredox Catalysis: A Unique Pathway for Cross-Electrophile Coupling. J. Am. Chem. Soc. 2016, 138, 8084–8087. 10.1021/jacs.6b04818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott K. A.; Njardarson J. T. Analysis of US FDA-Approved Drugs Containing Sulfur Atoms. Top Curr. Chem. 2018, 376, 5 10.1007/s41061-018-0184-5. [DOI] [PubMed] [Google Scholar]
- a Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 1997, 23, 3–25. 10.1016/S0169-409X(96)00423-1. [DOI] [PubMed] [Google Scholar]; b Lipinski C. A. Rule of five in 2015 and beyond: Target and ligand structural limitations, ligand chemistry structure and drug discovery project decisions. Adv. Drug Delivery Rev. 2016, 101, 34–41. 10.1016/j.addr.2016.04.029. [DOI] [PubMed] [Google Scholar]
- a Shvydkiv O.; Gallagher S.; Nolan K.; Oelgemöller M. From conventional to microphotochemistry: photodecarboxylation reactions involving phthalimides. Org. Lett. 2010, 12, 5170–5173. 10.1021/ol102184u. [DOI] [PubMed] [Google Scholar]; b Knowles J. P.; Elliott L. D.; Booker-Milburn K. I. Flow photochemistry: Old light through new windows. J. Org. Chem. 2012, 8, 2025–2052. 10.3762/bjoc.8.229. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Gilmore K.; Seeberger P. H. Continuous flow photochemistry. Chem. Rec. 2014, 14, 410–418. 10.1002/tcr.201402035. [DOI] [PubMed] [Google Scholar]; d Telmasani R.; Sun A. C.; Beeler A. B.; Stephenson C. R. J.. Flow Photochemistry in Organic Synthesis. In Flow Chemistry in Organic Synthesis, 1st ed.; Jamison T. F.; Koch G., Eds.; Thieme: Germany, 2019; pp 103–145. [Google Scholar]; e Beeler A. B.; Corning S. R. Photochemistry in Flow. Photochemistry 2015, 43, 173–190. [Google Scholar]; f Cambié D.; Bottecchia C.; Straathof N. J. W.; Hessel V.; Noël T. Applications of continuous-flow photochemistry in organic synthesis, material science, and water treatment. Chem. Rev. 2016, 116, 10276–10341. 10.1021/acs.chemrev.5b00707. [DOI] [PubMed] [Google Scholar]
- a Tucker J. W.; Zhang Y.; Jamison T. F.; Stephenson R. R. J. Visible-light photoredox catalysis in flow. Angew. Chem., Int. Ed. 2012, 51, 4144–4147. 10.1002/anie.201200961. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Nguyen J. D.; Reiß B.; Dai C.; Stephenson C. R. J. Batch to flow deoxygenation using visible light photoredox catalysis. Chem. Commun. 2013, 49, 4352–4354. 10.1039/C2CC37206A. [DOI] [PubMed] [Google Scholar]; c Elliott L. D.; Knowles J. P.; Koovits P. J.; Maskill K. G.; Ralph M. J.; Lejeune G.; Edwards L. J.; Robinson R. I.; Clemens I. R.; Cox B.; Pascoe D. D.; Koch G.; Eberle M.; Berry M. B.; Booker-Milburn K. Batch versus flow photochemistry: a revealing comparison of yield and productivity. Chem. – Eur. J. 2014, 20, 15226–15232. 10.1002/chem.201404347. [DOI] [PubMed] [Google Scholar]; d Lima F.; Kabeshov M. A.; Tran D. N.; Battilocchio C.; Sedelmeier J.; Sedelmeier G.; Schenkel B.; Ley S. V. Visible light activation of boronic esters enables efficient photoredox C(sp2)-C(sp3) cross-couplings in flow. Angew. Chem., Int. Ed. 2016, 55, 14085–14089. 10.1002/anie.201605548. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Plutschack M. B.; Pieber B.; Gilmore K.; Seeberger P. H. The hitchhiker’s guide to flow chemistry. Chem. Rev. 2017, 117, 11796–11893. 10.1021/acs.chemrev.7b00183. [DOI] [PubMed] [Google Scholar]; f Lima F.; Grunenberg L.; Rahman H. B. A.; Labes R.; Sedelmeier J.; Ley S. V. Organic photocatalysis for the radical couplings of boronic acid derivatives in batch and flow. Chem. Commun. 2018, 54, 5606–5609. 10.1039/C8CC02169D. [DOI] [PubMed] [Google Scholar]; g Griffiths O. M.; Esteves H. A.; Chen Y.; Sowa K.; May O. S.; Morse P.; Blakemore D. C.; Ley S. V. Photoredox-Catalyzed Dehydrogenative Csp3–Csp2 Cross-Coupling of Alkylarenes to Aldehydes in Flow. J. Org. Chem. 2021, 86, 13559–13571. 10.1021/acs.joc.1c01621. [DOI] [PubMed] [Google Scholar]
- a Newman S. G.; Jensen K. F. The role of flow in green chemistry and engineering. Green Chem. 2013, 15, 1456–1472. 10.1039/c3gc40374b. [DOI] [Google Scholar]; b Gutmann B.; Cantillo D.; Kappe C. O. Continuous-Flow Technology – A Tool for the Safe Manufacturing of Active Pharmaceutical Ingredients. Angew. Chem., Int. Ed. 2015, 54, 6688–6728. 10.1002/anie.201409318. [DOI] [PubMed] [Google Scholar]
- Singh R.; Lee H.-J.; Singh A. K.; Kim D.-P. Recent advances for serial processes of hazardous chemicals in fully integrated microfluidic systems. Korean J. Chem. Eng. 2016, 33, 2253–2267. 10.1007/s11814-016-0114-6. [DOI] [Google Scholar]
- Movsisyan M.; Delbeke E. I. P.; Berton J. K. E. T.; Battilocchio C.; Ley S. V.; Stevens C. V. Taming hazardous chemistry by continuous flow technology. Chem. Soc. Rev. 2016, 45, 4892–4928. 10.1039/C5CS00902B. [DOI] [PubMed] [Google Scholar]
- Grace Victoria G.; Reddy S. R. Recent advances in the synthesis of organic chloramines and their insights into health care. New. J. Chem. 2021, 45, 8386–8408. 10.1039/D1NJ01086G. [DOI] [Google Scholar]
- Cosgrove S. C.; Douglas G. E.; Raw S. A.; Marsden S. P. Continuous Flow for the Photochemical C-H Amination of Arenes. ChemPhotoChem 2018, 2, 851–854. 10.1002/cptc.201800105. [DOI] [Google Scholar]
- Pickford H. D.; Nugent J.; Owen B.; Mousseau J. J.; Smith R. C.; Anderson E. A. Twofold Radical-Based Synthesis of N,C-Difunctionalized Bicyclo[1.1.1]pentanes. J. Am. Chem. Soc. 2021, 143, 9729–9736. 10.1021/jacs.1c04180. [DOI] [PubMed] [Google Scholar]
- Gheorghe A.; Quiclet-Sire B.; Vila X.; Zard S. Z. Synthesis of substituted 3-arylpiperidines and 3-arylpyrrolidines by radical 1,4 and 1,2-aryl migrations. Tetrahedron 2007, 63, 7187–7212. 10.1016/j.tet.2007.04.091. [DOI] [Google Scholar]
- Go T.; Morimatsu A.; Wasada H.; Tanabe G.; Muraoka O.; Sawada Y.; Yoshimatsu M. Unprecedented nucleophile-promoted 1,7-S or Se shift reactions under Pummerer reaction conditions of 4-alkenyl-3-sulfinylmethylpyrroles. Beilstein J. Org. Chem. 2018, 14, 2722–2729. 10.3762/bjoc.14.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark A. J.; Collis A. E.; Fox D. J.; Halliwell L. L.; James N.; O’Reilly R. K.; Parekh H.; Ross A.; Sellars A. B.; Willcock H.; Wilson P. Atom-Transfer Cyclization with CuSO4/KBH4: A Formal “Activators Generated by Electron Transfer” Process Also Applicable to Atom-Transfer Polymerization. J. Org. Chem. 2012, 77, 6778–6788. 10.1021/jo301429a. [DOI] [PubMed] [Google Scholar]
- Trillo P.; Baeza A.; Nájera C. FeCl3.6H2O and TfOH as Catalysts for Allylic Amination Reaction: A Comparative Study. Eur. J. Org. Chem. 2012, 2012, 2929–2934. 10.1002/ejoc.201101844. [DOI] [Google Scholar]
- Liu X.; Wang Z.-S.; Zhai T.-Y.; Luo C.; Zhang Y.-P.; Chen Y.-B.; Deng C.; Liu R.-S.; Ye L.-W. Copper-Catalyzed Azide-Ynamide Cyclization to Generate α-Imino Copper Carbenes: Divergent and Enantioselective Access to Polycyclic N-Heterocycles. Angew. Chem., Int. Ed. 2020, 59, 17984–17990. 10.1002/anie.202007206. [DOI] [PubMed] [Google Scholar]
- Liu X.; Rong X.; Liu S.; Lan Y.; Liu Q. Cobalt-Catalyzed Desymmetric Isomerization of Exocyclic Olefins. J. Am. Chem. Soc. 2021, 143, 20633–20639. 10.1021/jacs.1c11343. [DOI] [PubMed] [Google Scholar]
- Soulard V.; Villa G.; Vollmar D. P.; Renaud P. Radical Deuteration with D2O: Catalysis and Mechanistic Insights. J. Am. Chem. Soc. 2018, 140, 155–158. 10.1021/jacs.7b12105. [DOI] [PubMed] [Google Scholar]
- Han X.; Nie X.; Feng Y.; Wei B.; Si C.; Lin G. Intermolecular [4+2] process of N-acyliminium ions with simple olefins for construction of functional substituted-1,3-oxazinan-2-ones. Chin. Chem. Lett. 2021, 32, 3526–3530. 10.1016/j.cclet.2021.05.003. [DOI] [Google Scholar]
- Spangler B.; Fontaine S. D.; Shi Y.; Sambucetti L.; Mattis A. N.; Hann B.; Wells J. A.; Renslo A. R. A Novel Tumor-Activated Prodrug Strategy Targeting Ferrous Iron Is Effective in Multiple Preclinical Cancer Models. J. Med. Chem. 2016, 59, 11161–11170. 10.1021/acs.jmedchem.6b01470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nóvoa L.; Trulli L.; Parra A.; Tortosa M. Stereoselective Diboration of Spirocyclobutenes: A Platform for the Synthesis of Spirocycles with Orthogonal Exit Vectors. Angew. Chem. 2021, 133, 11869–11874. 10.1002/ange.202101445. [DOI] [PubMed] [Google Scholar]
- Watson D. W.; Gill M.; Kemmitt P.; Lamont S. G.; Popescu M. V.; Simpson I. An investigation into the role of 2,6-lutidine as an additive for the RuCl3-NaIO4 mediated oxidative cleavage of olefins to ketones. Tetrahedron Lett. 2018, 59, 4479–4482. 10.1016/j.tetlet.2018.11.013. [DOI] [Google Scholar]
- Poplata S.; Bach T. Enantioselective Intermolecular [2+2] Photocycloaddition Reaction of Cyclic Enones and Its Application in a Synthesis of (−)-Grandisol. J. Am. Chem. Soc. 2018, 140, 3228–3231. 10.1021/jacs.8b01011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z.; Guan R.; Shanmugam M.; Bennett E. L.; Robertson C. M.; Brookfield A.; McInnes E. J. L.; Xiao J. Oxidative Cleavage of Alkenes by O2 with a Non-Heme Manganese catalyst. J. Am. Chem. Soc. 2021, 143, 10005–10013. 10.1021/jacs.1c05757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen M.; Bastos C. M.; Parks D.; Munoz B.. Proteasome Activity Enhancing Compounds. Int. Patent WO2020/6296A1, 2020.
- Law J. A.; Bartfield N. M.; Frederich J. H. Site-Specific Alkene Hydromethylation via Protonolysis of Titanacyclobutanes. Angew. Chem., Int. Ed. 2021, 60, 14360–14364. 10.1002/anie.202103278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fodran P.; Wallentin C.-J. Harnessing Energy-Transfer in N-Centered Radical-Mediated Synthesis of Pyrrolidines. Eur. J. Org. Chem. 2020, 2020, 3213–3218. 10.1002/ejoc.202000537. [DOI] [Google Scholar]
- Diaba F.; Puigbo G.; Bonjoch J. Synthesis of Enantiopure 1-Azaspiro[4.5]decanes by Iodoaminocyclization of Allylaminocyclohexanes. Eur. J. Org. Chem. 2007, 2007, 3038–3044. 10.1002/ejoc.200700056. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.