

Abstract
Multiple-strain (polygenomic) infections are a ubiquitous feature of Plasmodium falciparum parasite population genetics. Under simple assumptions of superinfection, polygenomic infections are hypothesized to be the result of multiple infectious bites. As a result, polygenomic infections have been used as evidence of repeat exposure and used to derive genetic metrics associated with high transmission intensity. However, not all polygenomic infections are the result of multiple infectious bites. Some result from the transmission of multiple, genetically related strains during a single infectious bite (cotransmission). Superinfection and cotransmission represent two distinct transmission processes, and distinguishing between the two could improve inferences regarding parasite transmission intensity. Here, we describe a new metric, RH, that utilizes the correlation in allelic state (heterozygosity) within polygenomic infections to estimate the likelihood that the observed complexity resulted from either superinfection or cotransmission. RH is flexible and can be applied to any type of genetic data. As a proof of concept, we used RH to quantify polygenomic relatedness and estimate cotransmission and superinfection rates from a set of 1,758 malaria infections genotyped with a 24 single nucleotide polymorphism (SNP) molecular barcode. Contrary to expectation, we found that cotransmission was responsible for a significant fraction of 43% to 53% of the polygenomic infections collected in three distinct epidemiological regions in Senegal. The prediction that polygenomic infections frequently result from cotransmission stresses the need to incorporate estimates of relatedness within polygenomic infections to ensure the accuracy of genomic epidemiology surveillance data for informing public health activities.
Keywords: genetic surveillance, malaria, cotransmission, superinfection
Significance Statement.
Accurate assessments of malaria transmission intensity are a critical component of public health surveillance and intervention campaigns. Here, we developed a metric that would determine whether multiple-strain infections resulted from multiple or single mosquito bites. This issue is relevant to public health because multiple-strain infections are frequently assumed to reflect multiple bites and high transmission rates. We show that a large fraction of multiple-strain infections in Senegal are the result of a single bite and thus not as useful for informing transmission intensity as previously believed. Future work should focus on reassessing the relationship between multiple-strain infections and transmission intensity to ensure malaria genomic surveillance produces accurate transmission assessments to public health programs.
Introduction
The past two decades have seen an increase in the collection and application of pathogen genetics in public health surveillance programs. Genomic epidemiology surveillance is now used for a wide variety of viral, bacterial, and eukaryotic pathogens (1–5). For Plasmodium falciparum, a major focus of genomic epidemiology surveillance has been to identify genetic markers associated with parasite transmission. Collectively, these surveys have revealed several genetic markers associated with parasite transmission (6–9). Chief among them is the frequency of clonal (genetically identical) parasites in the population, the frequency of multistrain (polygenomic) infections, and the number of strains per infection [commonly referred to as complexity of infection (COI)] (10–12).
Each of these metrics represents different aspects of parasite transmission. The increased frequency of clonal parasites is hypothesized to reflect decreases in transmission due to a reduction in parasite genetic diversity and a reduction in outbreeding opportunities. The frequency of polygenomic infections and their COIs are hypothesized to reflect the frequency of superinfection, or the repeated infection of individuals from multiple, infectious mosquito bites (Fig. 1A). If true, genetic metrics could augment traditional, but labor-intensive estimates of transmission, such as the entomological inoculation rate (EIR, the number of infectious mosquito bites per individual) (13, 14). To date, COI is one of the most frequently reported genetic metrics of parasite transmission (5, 12, 15–19), and a variety of statistical tools have been developed to estimate COI from various sources of genotypic data (20–22). COI is positively associated with transmission intensity and typically ranges between 1 and 5, but extreme values >10 have occasionally been reported (23).
Fig. 1.

Superinfection and cotransmission. (A) Under standard assumptions of superinfection, individuals in high-transmission settings are exposed to multiple infectious bites and have multiple opportunities for outcrossing. This results in parasite populations characterized by high proportion of polygenomic infections, high COIs, and few clones. Individuals in low-transmission settings are exposed to fewer infectious bites, resulting in fewer polygenomic infections and low COIs. Low transmission also limits outcrossing and promotes the transmission of clonal parasite strains. (B) Cotransmission weakens the relationships proposed in panel (A) because polygenomic infections can also result from a single bite. Cotransmission is initiated when a mosquito ingests multiple parasite strains from an initial polygenomic infection. The number of strains present in the initial mosquito blood feed depends on the COI and strain proportions in the initial host. These parasites undergo sexual outcrossing to produce genetically related sporozoites that are transmitted into a new host. Cotransmissions do not reflect multiple exposures and instead represent a single exposure event.
However, polygenomic infections are not always the result of superinfection. Whole genome sequencing analyses have detected genetically related parasites in both single-strain (monogenomic) and polygenomic infections (10, 23–26). These genetically related parasites are created when a mosquito vector bites a polygenomic infection and ingests multiple parasite strains that then mate and undergo sexual reproduction to complete the sexual stage of their life cycle (24, 27) (Fig. 1B). Cotransmission occurs when the mosquito vector bites a new host and transmits multiple parasite strains that are oftentimes genetically related. The genetic composition of the resulting polygenomic infection reflects the assortment and segregation of genomes during meiosis, the sampling of gametocytes by the mosquito vector, the injection of sporozoites into the human host, and the invasion of sporozoites into the liver (27). Thus, failing to accurately distinguish superinfections from cotransmissions could lead to inaccurate estimates of malaria transmission based on COI alone.
One way of distinguishing superinfection from cotransmission is to examine the relatedness of coinfecting strains within polygenomic infections. Superinfection assumes these strains are randomly drawn from the population, while cotransmission assumes they are genetically related. While several metrics have been developed to assess parasite relatedness, they have largely required whole genome sequencing and/or the establishment of genomic phase (22, 28). Here, we developed a new metric, RH, that utilizes intrahost heterozygosity to assess the relatedness of malaria infections and distinguish cotransmission from superinfection. Both superinfection and cotransmission contribute to the complexity of parasite population genetics, and distinguishing the two could lead to greater insight into how transmission and parasite genetics are linked. As a proof of concept, we used RH to quantify the genetic relatedness of parasites genotyped with a 24 single nucleotide polymorphism (SNP) barcode (29). Using RH, we show that cotransmission is prevalent in three epidemiologically distinct regions of Senegal with moderate-to-low transmission intensity.
Results
Genomic epidemiology of Kédougou, Thiès, and Richard Toll
A total of 1,758 malaria infections were collected and genotyped using a TaqMan-based molecular barcode (Fig. 2; see the “Materials and methods” section). These samples were collected from three regions of Senegal: Kédougou, Thiès, and Richard Toll. Kédougou has the highest intensity of transmission (Fig. 2B), with incidences of 300 to 500 + cases per thousand reported by the National Malaria Control Program (PNLP, Programme National de Lutte contre le Paludisme) (30–35) between 2014 and 2020. Thiès and Richard Toll have lower transmission intensity and incidences of <10 and <1 case per thousand. Based on incidence data, we expected Kédougou to have the least amount of clonal population structure, the highest proportion of polygenomic infections, and the highest COI. The allele frequencies observed for each of the assays used in the molecular barcode are presented in Fig. S1.
Fig. 2.

Epidemiology of Senegal and sampling structure. (A) Sampling of clinical isolates throughout Senegal. The white circles indicate the locations of the sampling clinics. The shaded areas denote the administrative regions that each of the clinics are in: Kédougou (red), Thiès (orange), and Richard Toll/St. Louis (blue). (B) Longitudinal incidence profiles obtained from the 2015 to 2020 annual Senegal National Malaria Control Program (PNLP, Programme National de Luttte contre le Paludisme) reports (30–35). Region-level incidences were used to represent the incidences of each clinic site. The shading represents two Poisson SDs from the mean. (C) Examples of the 24 SNP molecular barcode. Homozygous sites were denoted by A, T, C, or G. Heterozygous sites were denoted by N. Missing sites due to assay failure were denoted by X. Barcodes with two or more Ns were considered polygenomic. Longitudinal sample profiles for (D) Kédougou, (E) Thiès, and (F) Richard Toll. Gray bars indicate monogenomic samples, while colored bars indicate polygenomic samples. Black numbers and colored numbers are the monogenomic and polygenomic sample sizes for each year, respectively. Shapefiles for the map used in panel (A) were accessed from https://data.humdata.org/dataset/senegal-administrative-boundaries? under the Creative Commons Attribution for Intergovernmental Organisations (CC BY-IGO).
Polygenomic fraction and COI were elevated in Kédougou (Fig. 3 and Fig. S2A). The inverse-variance-weighted averages for polygenomic fraction (the proportion of infections polygenomic) were 0.53 (0.50, 0.57) for Kédougou, 0.27 (0.24, 0.31) for Thiès, and 0.33 (0.28, 0.38) for Richard Toll. There was no statistically significant change associated with the sampling year in Richard Toll between 2012 and 2015 (ordinary least-squares regression, slope = −0.01, P-value = 0.601 for Richard Toll). For Thiès and Kédougou, we found a small, but statistically significant increase in the polygenomic fraction from 2015 to 2020 at a rate of 0.03 (0.010, 0.04) per year (P-value = 0.01) for Thiès and 0.020 (0.001, 0.035) per year for Kédougou (P-value = 0.04).
Fig. 3.

Genetic epidemiology of Kédougou (A to C), Thiès (D to F), and Richard Toll (G to I) using the 24 SNP molecular barcode. Column 1 (A, D, andG): the proportion of monogenomic (gray) and polygenomic (colored) samples. The annotated numbers show the proportion of monogenomic samples. Column 2 (B, E, and H): The proportion of clonal haplotypes. The gray bars indicate the unique monogenomic haplotypes and the colors indicate repeated clonal haplotypes within each year, with clonal groups shaded distinctly. The annotated numbers indicate the total proportion of barcodes with at least one clone. Column 3 (C, F, and I): The abundance of clonal haplotypes in each population. Each row represents a unique clonal haplotype, and the number of samples per haplotype is indicated by the size of the circle and the number next to each circle. Connected circles indicate persistent haplotypes observed across multiple years.
As expected, we observed the least amount of clonal population structure in Kédougou (Fig. 3B and C). We detected a total of 7 clonal haplotypes in Kédougou, 23 in Richard Toll, and 38 in Thiès. We found no evidence of persistent clonal haplotypes in Kédougou (Fig. 3C). Two of the clonal haplotypes in Richard Toll and six of the clonal haplotypes in Thiès were detected in multiple years (Fig. 3F and I). Most clonal haplotypes were detected twice, but some haplotypes were detected five or more times per year in Thiès and Richard Toll.
Our point estimates for the unique monogenomic fraction were 0.95 (0.92, 0.98) for Kédougou, 0.70 (0.66, 0.75) for Thiès, and 0.86 (0.83, 0.90) for Richard Toll (Fig. 3B, E, and H and Fig. S2B). These were calculated as the inverse-variance-weighted average across all years. We found little evidence that the unique monogenomic fraction changed significantly over time in Kédougou between 2015 and 2019 or Richard Toll between 2012 and 2015 (ordinary least-squares regression, P-value >0.23). Interestingly, we detected a small increase of 0.07 (0.038, 0.102) per year (P-value = 0.004) in the unique monogenomic fraction in Thiès, but the increase was negligible when compared with the differences in the unique monogenomic fraction between populations.
THE REAL McCOIL-based estimates for polygenomic fraction (COI > 1) were 0.40 (0.44, 0.36) for Kédougou, 0.20 (0.17, 0.24) for Thiès, and 0.26 (0.22, 0.30) for Richard Toll (Fig. S2C). The average COI of polygenomic infections was 2.70 (2.53, 2.92), 2.30 (2.15, 2.45), and 2.32 (2.32, 2.48) in Kédougou, Thiès, and Richard Toll, respectively. Note that these estimates exclude monogenomic samples. However, the distribution of COI was highly skewed with most polygenomic infections: 0.60 (0.54, 0.66), 0.74 (0.66, 0.83), and 0.80 (0.75, 0.83) of the polygenomic infections in Kédougou, Thiès, and Richard Toll, respectively, estimated to have COI = 2.
Verifying the accuracy of RH for identifying cotransmission and superinfection based on samples genotyped with the 24 SNP molecular barcode
The main goal of this study was to determine if genetic metrics that measure intrahost heterozygosity and inbreeding could distinguish superinfection from cotransmission. Theoretically, an RH > 0 reflects cotransmission, an RH = 0 reflects superinfection with COI = 2, and an RH < 0 reflects superinfection with COI ≥ 3. However, it was unclear whether the 24 SNP molecular barcodes would bias RH estimates and result in errors in its interpretation. To address this, we simulated polygenomic infections genotyped with the 24 SNP molecular barcode under different hypotheses of superinfection and cotransmission ( “Materials and methods” section, Supplementary Material).
These simulations showed that an RH quantified from the 24 SNP molecular barcode could be used to distinguish superinfection from cotransmission (Fig. 4A). Superinfection with a COI = 2 resulted in a theoretical expectation (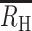 ) of zero. Superinfection with COI > 2 resulted in negative
) of zero. Superinfection with COI > 2 resulted in negative 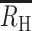 values of −0.5 for a COI = 3 infection and −1.0 for a COI = 4 infection. Cotransmission resulted in positive
values of −0.5 for a COI = 3 infection and −1.0 for a COI = 4 infection. Cotransmission resulted in positive 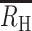 values even when the COI of the initial infection was high. For cotransmission chains originating from a COI = 2 superinfection,
values even when the COI of the initial infection was high. For cotransmission chains originating from a COI = 2 superinfection, 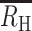 increased from 0.40 to 060 and 0.70 following the first, second, and third cotransmission event.
increased from 0.40 to 060 and 0.70 following the first, second, and third cotransmission event.
Fig. 4.

Theoretical Expectations of RH. (A) Simulated expectations of RH from 800 simulated barcodes under different cotransmission (red, “cotx”) and superinfection models (blue, “super”). The legend indicates the initial COI used. The 1x, 2x, and 3x notations indicate the expected RH values after the first, second, and third cotransmission events. The dotted red line indicates an RH of 0.3, which is the threshold used to identify cotransmission events. The doted black lines indicate an RH of 0.0, which is the expectation from a COI = 2 superinfection. Superinfection was simulated by randomly sampling monogenomic samples according to the specified COI. (B) The proportion of serial cotransmission events with COI 2 to 5 identified as cotransmission when defining cotransmission as an RH > 0.3. Shading indicates the bootstrapped 95% confidence interval. (C) The proportion of superinfection events with COI 2 to 4 identified as cotransmission when defining cotransmission as an RH > 0.3. Shading indicates the bootstrapped 95% confidence interval.
However, these simulations also showed that individual superinfection and cotransmission events could result in positive and negative RH values (Fig. 4A). To address this, we used the Bayes factor to define a classification scheme that would (1) distinguish cotransmission from superinfection (Fig. S3C), (2) identify serial cotransmission (Fig. S3D) or (3) estimate the COI of superinfections (Fig. S3E and F). Based on these results, we defined cotransmission as RH > 0.30, superinfection with COI = 2 as –0.1 < RH < 0.3, and superinfection with COI > 2 as RH < –0.1 (Table 1).
Table 1.
Classification of polygenomic infections as cotransmission or superinfection using 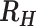 .)
.)
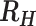
|
Classification |
|---|---|
| > 0.4 | Cotransmission |
0.3 > 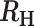 > 0.4 > 0.4 |
Likely cotransmission |
−0.1> 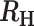 > 0.3 > 0.3 |
Superinfection, COI = 2 |
−0.1 > 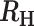 > -0.2 > -0.2 |
Superinfection, likely COI = 2 |
−0.4> 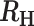 > -0.2 > -0.2 |
Superinfection, COI = 3 |
−0.6 > 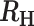 > -0.4 > -0.4 |
Superinfection, likely COI = 3 |
−0.8< 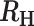 < -0.6 < -0.6 |
Superinfection, likely COI = 4 |
−0.8 < 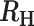
|
Superinfection, COI = 4 |
The boundaries and classification notations are based on the standards for Bayes factor classification described in ref. (36)
Of key interest was to determine whether using a threshold of RH > 0.30 accurately discriminated cotransmission from superinfection. With this threshold, the true cotransmission identification rates were 0.66 (0.63, 0.69), 0.85 (0.83, 0.87), and 0.93 (0.92, 0.95) for the first, second, and third cotransmission events following a COI = 2 superinfection (Fig. 4B). These rates decreased to 0.47 (0.44, 0.51), 0.73 (0.70, 0.76), and 0.88 (0.87, 0.91) following a COI = 5 superinfection. This threshold misidentified 0.13 (0.11, 0.15), 0.006 (0.002, 0.011), and 0.0 (0.0, 0.002) of COI = 2 to 4 superinfection events, respectively, as cotransmission (Fig. 4C).
We next evaluated the performance of this threshold using three diagnostic tests designed to evaluate how accurately a given threshold distinguishes cotransmission and serial cotransmissions from COI = 2 superinfections (Fig. S4). These tests showed that the optimum threshold differs for cotransmission and serial cotransmissions and depends on the COI of the originating polygenomic infection (Fig. S4B to D). In general, the optimal threshold is lower for cotransmission events originating from high COI infections. In the context of Senegal, where the average COI was 2, our tests show that a cotransmission threshold of RH > 0.30 is near the optimum threshold needed to accurately detect single and serial cotransmission events (Fig. S4B). Note that the true optimum is somewhat uncertain, as identifying it requires priors for the relative proportion of cotransmissions, and serial cotransmissions in the population.
R H versus FWS
We compared RH with another commonly used estimate of intrahost heterozygosity, FWS (37, 38) ( “Materials and methods” section, Supplementary Material). However, because the original definition of FWS reported in (37) required the use of reads generated from next generation sequencing data, we could only compare RH and FWS for the 2020 samples where barcode and Illumina short-read whole genome sequencing data were available for both monogenomic and polygenomic infections. Barcode-derived RH estimates were consistent with their corresponding whole genome sequencing-derived FWS estimates (R-squared = 0.66, ordinary-least squares) (Fig. S5).
To examine the theoretical differences between RH and FWS, we simulated barcode-derived RH and FWS in populations with different levels of clonal parasite sharing. For these simulations, we relied on the FWS described in (39–41) that normalizes intrahost heterozygosity with the expected, population-level heterozygosity but relaxes the requirement that heterozygosity be estimated from sequence counts. In populations with no clonal parasites, RH and FWS can both be used to identify cotransmissions and evaluate COI (Fig. S6). However, as the frequency of clones increases and the total heterozygosity of the population declines, the expected FWS values for simulated cotransmissions and superinfections shift and deviate from the expectations observed in nonclonal populations (Fig. S6). Simulated RH values were unaffected. The first cotransmission always had an expected RH of 0.5, while COI = 2 and COI = 3 superinfections always had an expected RH of 0.0 and −0.5, respectively (Fig. S6).
R H detects widespread cotransmission in Kédougou, Thiès, and Richard Toll
We next calculated RH for all the polygenomic infections collected from Kédougou, Thiès, and Richard Toll. Individual polygenomic samples had a wide range in individual RH values, with some samples with RH values close to one and some samples with negative RH values (Fig. 5A to C). Despite this variation, the average RH (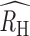 ) in each population was positive but inversely associated with regional incidence estimates. The
) in each population was positive but inversely associated with regional incidence estimates. The 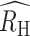 was 0.14 (0.12, 0.17) in Kédougou, 0.25 (0.23, 0.28) in Thiès, and 0.31 (0.29, 0.34) in Richard Toll. We also noted that the polygenomic infections collected from Thiès in 2018 exhibited anomalously low RH values relative to those collected between 2015 and 2017 and between 2019 and 2020. Excluding the 2018 samples resulted in an
was 0.14 (0.12, 0.17) in Kédougou, 0.25 (0.23, 0.28) in Thiès, and 0.31 (0.29, 0.34) in Richard Toll. We also noted that the polygenomic infections collected from Thiès in 2018 exhibited anomalously low RH values relative to those collected between 2015 and 2017 and between 2019 and 2020. Excluding the 2018 samples resulted in an 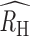 of 0.30 (0.26, 0.32) for Thiès.
of 0.30 (0.26, 0.32) for Thiès.
Fig. 5.

Polygenomic infections exhibit reduced heterozygosity. (A to C) The individual RH estimates for each of the clinical polygenomic samples collected from (A) Kédougou (red), (B) Thiès (orange), and (C) Richard Toll (blue). The open-faced square indicates the average RH of the samples in each year and the dark line is the 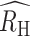 point estimate obtained for the entire region. The
point estimate obtained for the entire region. The 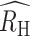 point estimate for the entire region and its associated 95% confidence interval are reported in the legend. The proportion of polygenomic infections inferred to be the result of cotransmission or superinfection with COI of two (COI = 2), three (COI = 3), or four strains (COI = 4) in Kédougou (D), Thiès (E), and Richard Toll (F), respectively . The error bars indicate two binomial SDs from the mean for each category.
point estimate for the entire region and its associated 95% confidence interval are reported in the legend. The proportion of polygenomic infections inferred to be the result of cotransmission or superinfection with COI of two (COI = 2), three (COI = 3), or four strains (COI = 4) in Kédougou (D), Thiès (E), and Richard Toll (F), respectively . The error bars indicate two binomial SDs from the mean for each category.
Based on the RH thresholds for classifying superinfection and cotransmission defined in Table 1, we found that most polygenomic infections were the result of cotransmission (Fig. 5D to F). Despite its relatively high incidence, we found that 0.43 (0.33, 0.54) of the polygenomic infections from Kédougou were the most likely result of cotransmission. This was smaller than either Thiès or Richard Toll, where the proportion was 0.53 (0.38, 0.69) and 0.52 (0.45, 0.60), respectively. These cotransmitted polygenomic infections were classified as either COI = 1 or COI = 2 by the THE REAL McCOIL (Fig. S7). Approximately half of the THE REAL McCOIL COI = 2 samples had RH values consistent with cotransmission.
These elevated RH values could also result from a technical inability to accurately detect heterozygous sites (29). From the laboratory-generated 3D7, Dd2, and TM90C6B mixtures, we knew that heterozygous site detection was reduced in polygenomic mixtures with extreme strain ratios (Supplementary Material). We adjusted the heterozygosity of each polygenomic barcode based on the strain proportions inferred from the cycle thresholds (CTs) reported by the TaqMan-based barcode assays (Supplementary Material Figs. S8 and S9). This adjustment resulted in reduced RH values but had no effect on our overall results (Fig. S10).
Discussion
To date, most genetic epidemiology analyses have ignored cotransmission and interpreted polygenomic fraction and COI under simple assumptions of superinfection. However, it is uncertain how well this assumption holds as transmission falls in moderate- or near-elimination settings where superinfection is expected to be comparatively rare. Resolving this conundrum requires collecting additional data regarding polygenomic relatedness and cotransmission rates across diverse epidemiological settings.
To enable broad-scale genetic surveillance of polygenomic relatedness and cotransmission in parasite populations, we developed a new metric, RH, that normalizes the observed polygenomic heterozygosity with the expected heterozygosity of a COI = 2 superinfection as a possible means for distinguishing superinfection from cotransmission in parasite populations. Polygenomic infections with less heterozygosity than a typical COI = 2 superinfection indicate cotransmission, while polygenomic infections with more indicate a superinfection whose COI is greater than 2.
The normalization of observed polygenomic heterozygosity to the expected heterozygosity of a COI = 2 superinfection is critical and differentiates RH from previous metrics of intrahost heterozygosity such as FWS (37–41). FWS normalizes intrahost heterozygosity to the total, population-level heterozygosity of the population 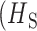 , Eq. S10). Unlike FWS, the sampling of genetic clones is excluded from RH because they are indistinguishable from COI = 1 monogenomic infections and do not directly contribute to outcrossing or cotransmission. In highly diverse populations with few genetic clones, FWS behaves similarly to RH. However, FWS estimates are sensitive to the presence of parasite clones in the population. This complicates FWS based inferences of either cotransmission or COI because these inferences would need to be recalibrated to consider the frequency of clones in each population.
, Eq. S10). Unlike FWS, the sampling of genetic clones is excluded from RH because they are indistinguishable from COI = 1 monogenomic infections and do not directly contribute to outcrossing or cotransmission. In highly diverse populations with few genetic clones, FWS behaves similarly to RH. However, FWS estimates are sensitive to the presence of parasite clones in the population. This complicates FWS based inferences of either cotransmission or COI because these inferences would need to be recalibrated to consider the frequency of clones in each population.
In contrast, RH is unaffected by the frequency of parasite clones, and its predictions remain consistent across populations with different clonalities. This property makes RH particularly relevant for examining parasite populations in historically low-transmission settings such as Southeast Asia and South America, where population fragmentation and clonal parasite populations are the norm (37, 42, 43). In fact, the frequency of parasite clones in these regions can be as extreme as the simulated populations used in Fig. S6; 60% of the P. falciparum cases collected in Quibdó, Columbia, in 2001 showed evidence of being infected by the same parasite strain (44). While the advantages of RH are clearest in low-transmission settings with highly clonal parasite populations, it can also be advantageous in moderate- and high-transmission settings to account for parasite clonality arising from falling transmission intensity or focal transmission (45).
Another advantage of RH compared to other metrics used to assess relatedness or inbreeding (22, 28, 37) is that RH does not require whole genome sequencing or that the genomes within polygenomic infections be phased. In fact, barcode-derived RH were consistent with whole genome sequence-derived estimates of Fws despite relying on a small number of SNPs present throughout the genome. This greatly expands our ability to assess superinfection and cotransmission in epidemiological settings using accessible genotyping technologies like the 24 SNP TAQman-based molecular barcodes where whole genome sequencing is unfeasible due to logistical or financial constraints.
Estimating RH from the 24 SNP molecular barcode also avoids some of the issues involving microsatellites (39). Use of an SNP barcode rather than microsatellites based on di- and tri-nucleotide repeats avoids the potential problems of replication slippage and unequal crossing over, which can alter the number of repeating units (46). SNP markers are not prone to such events, although they may change due to random mutation or gene conversion; these events are infrequent.
Using RH, we provide new evidence that cotransmission is ubiquitous among symptomatic infections reporting to clinics in three sites spread across Senegal. These results are consistent with our previous analyses of whole-genome sequences of polygenomic infections in Thiès, which also suggest that cotransmission occurs at high frequency (24). Here, we also found evidence of high rates of cotransmission in both Richard Toll and Kédougou. These results were unexpected for Kédougou, where the incidence was 300 to 500 + cases per thousand per year, as the prevailing consensus was that polygenomic infections in moderate- to high-transmission settings were the result of superinfection. However, our results are consistent with previous reports of cotransmission in polygenomic infections collected from Malawi , where transmission is high (25, 26). Altogether, our results showed that at nearly half of the sampled polygenomic infections are the result of a single infectious bite. The true cotransmission rates are likely higher, based on the theoretical misclassification rates reported in Fig. 4. Regardless, these results imply that a significant fraction of polygenomic infections result from single infectious bites and that superinfection is a rarer than expected in populations with incidences less than 500 cases per thousand per year. Whether cotransmission is equally prevalent in asymptomatic infections is a topic for future research.
Our results present a problem for genetic epidemiology analyses, as they show that a large fraction of polygenomic infections do not reflect multiple bites. Accurate EIR inference will likely require model-based approaches that incorporate all available information regarding superinfection, cotransmission, host immunity, and age (47), and any potential observation or sampling biases, such as focal transmission heterogeneity, that could affect COI estimation. An important application of RH would be to enable calibration of superinfection and cotransmission rates in future model-based inferences of EIR. Superinfection and cotransmission represent two distinct types of transmission whose impact on parasite genetics may be useful for helping genomic epidemiology models disentangle different aspects of parasite transmission. However, these models should also assess the effects of host immunity on COI as immunity could also cause intrahost heterozygosity to be depressed and be conflated with cotransmission.
Interestingly, initial genomic surveillance with the 24 SNP molecular barcode suggests that metrics of parasite clonality, COI, and polygenomic fraction did not distinguish Thiès from Richard Toll, both of which have incidences less than 10 cases per thousand per year. While this could reflect a limitation of the 24 SNP molecular, we note that whole genome sequences of polygenomic infections in Thiès also reported high cotransmission frequencies (24). Alternatively, importation from migration and migrant workers in Richard Toll could be obscuring the genetic signals associated with reduced transmission intensity (48–50). High levels of importation could explain the high unique monogenomic fraction relative in Richard Toll and the anomalously low RH values observed in Thiès during 2018. Additional analyses throughout different epidemiological clines will be valuable for determining which genetic signals are most strongly associated with transmission intensity and whether anomalies like the low RH in Thiès 2018 reflect other epidemiological factors such as migration.
To guide our understanding of epidemiology and malaria population genetics, genomic epidemiology analyses should identify and separate cotransmission from superinfection polygenomic infections. RH represents a significant advancement in malaria genomic epidemiology by explicitly measures the genetic impact of sexual reproduction and outcrossing when evaluating the relationship between malaria population genetics and transmission. Understanding the differences between these two types of polygenomic infections and their relationship with parasite transmission is relevant for future model-based estimates of parasites transmission and operational decision-making by national malaria control programs.
Materials and methods
Sample collection
Samples were collected through passive-case detection from febrile patients reporting to health posts or clinics during the malaria transmission season in Senegal (September to December), or actively detected in households in response to a case detected at the Richard Toll clinics. Patients over 6 months of age with fever within the past 24 hours of visiting the clinic with no history of antimalarial use were diagnosed with malaria using microscopy or rapid diagnostic tests (RDTs). Filter papers spotted with blood were collected from malaria-positive patients in Thiès and Kédougou. RDTs were collected from malaria-positive patients in Richard Toll as previously described (49). Ethical approval for the study was obtained (IRB Protocols: 16,330 and 17 to 1288 from Harvard T.H. Chan School of Public Health).
Molecular barcode genotyping and SNP calling
The molecular barcode consists of a series of 24 neutral SNPs spread across the malaria genome that are genotyped using a panel of TaqMan-based quantitative PCR genotyping assays. Nucleic acid material was extracted from either filter paper or RDT material and preamplified using the methods described in (49, 51). A description of the reagents and methods used for the TaqMan assays is in (29).
Criteria for calling homozygous and heterozygous sites were determined using a set of laboratory-generated mixes containing 3D7, Dd2, and TM90C6B DNA mixed with proportions ranging from 1 : 1 to 1 : 5 (Supplementary Material Table S1). These criteria were based on the normalized CTs calculated by the Applied Biosystems ViiA 7 Real-Time PCR System (v1.2). Baselines for allele 1 and allele 2 were determined by the software, and the  that determines CTs was set to 20.
that determines CTs was set to 20.
Homozygous sites were denoted by their allelic identity (A, T, C, and G) and identified as sites with (1) no heterogeneous amplification, (2) heterogeneous amplification where the difference in CT between allele 1 and allele 2 (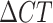 ) was greater than 8 , or (3) heterogeneous amplification, where one of the alleles had a CT > 38. For homozygous sites with heterogeneous amplification, the allelic identity was determined by the allele with the smaller CT value.
) was greater than 8 , or (3) heterogeneous amplification, where one of the alleles had a CT > 38. For homozygous sites with heterogeneous amplification, the allelic identity was determined by the allele with the smaller CT value.
Heterozygous sites were denoted as “N” and identified as sites with heterogeneous amplification and (1) a 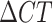 < 8 and (2) CT < 38 for both alleles. Barcodes with two or more heterozygous sites were considered polygenomic. Sites without amplification were considered missing and denoted as “X.” Barcodes with more than two missing sites were excluded from analysis. These thresholds were identified by benchmarking their accuracy in detecting heterozygous and homozygous sites using a set of lab-generated DNA mixtures generated from DNA isolated from 3D7, Dd2, and TM90C6B P. falciparum strains (Supplementary Material Figs. S11 to S13).
< 8 and (2) CT < 38 for both alleles. Barcodes with two or more heterozygous sites were considered polygenomic. Sites without amplification were considered missing and denoted as “X.” Barcodes with more than two missing sites were excluded from analysis. These thresholds were identified by benchmarking their accuracy in detecting heterozygous and homozygous sites using a set of lab-generated DNA mixtures generated from DNA isolated from 3D7, Dd2, and TM90C6B P. falciparum strains (Supplementary Material Figs. S11 to S13).
Whole genome sequencing
Monogenomic and polygenomic samples collected from 2020 were submitted for whole genome sequencing. Selective whole genome amplification was performed on extracted DNA and used to construct libraries with a NEBNext Ultra II library Kit for Illumina short-read sequencing. Variant-calling and read alignment was performed following the best practice standards set by the Pf3k consortium. Briefly, short-reads were aligned to the P. falciparum 3D7 reference genome (PlasmoDB v. 28) using BWA-mem and Picard Tools, Variants were called using HaplotypeCaller in GATK v3.5.
Analyses were carried out using a set of ∼150,000 SNPs identified from a set of 1,328 monogenomic samples obtained from the Pf3k database (52). These sites were chosen to (1) exclude the core chromosomal regions, (2) reside in nonoverlapping 2 kb windows whose average intrahost heterozygosity < 0.03, (3) be farther than seven base pairs from indels, and (4) have an average intrahost heterozygosity < 0.04 across all samples.
Inverse-variance weighting
To correct for the varying sample sizes observed in each year, we used the inverse-variance-weighted average, which is defined as
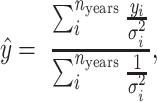 |
(1a) |
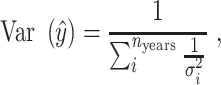 |
(1b) |
where 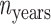 is the number of sample years,
is the number of sample years, 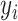 is the average of the ith year, and
is the average of the ith year, and 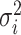 is the variance of the ith year.
is the variance of the ith year.
R H: a malaria-specific estimate of polygenomic infection heterozygosity
R H is derived from Sewall Wright's original definition for the inbreeding coefficient (53), which was defined as the correlation between alleles in uniting gametes relative to those drawn at random within a subpopulation. RH is not specific to the 24 SNP molecular barcode and can be broadly applied to SNP-based genotyping methods. RH is related to the FWS metric (37–41). Refer to the Supplementary Material for additional details regarding FWS and its differences with RH.
R H is defined as
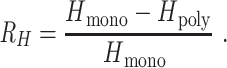 |
(2) |
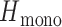 was defined by sampling monogenomic barcode pairs, summing the total number of discordant alleles between the two and dividing it by the number of comparable sites. Comparable sites were defined as those that were nonmissing and homozygous in both sampled monogenomic barcodes. As a result,
was defined by sampling monogenomic barcode pairs, summing the total number of discordant alleles between the two and dividing it by the number of comparable sites. Comparable sites were defined as those that were nonmissing and homozygous in both sampled monogenomic barcodes. As a result, 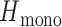 represents the expected heterozygosity of a COI = 2 superinfection and does not include the sampling of nonunique barcodes.
represents the expected heterozygosity of a COI = 2 superinfection and does not include the sampling of nonunique barcodes. 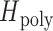 is the expected heterozygosity in polygenomic samples and defined as the number of heterozygous sites divided by the number of nonmissing sites in the polygenomic barcode.
is the expected heterozygosity in polygenomic samples and defined as the number of heterozygous sites divided by the number of nonmissing sites in the polygenomic barcode.
Evaluating RH
Individual estimates of 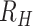 for each polygenomic sample (
for each polygenomic sample (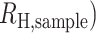 were obtained by rearranging Eq. 1 and minimizing Eq. 3 using the scipy.optimize.minimize package (v1.5.2) for Python 3 (v3.8.5) with bounds set at −1 and +1. Setting the lower bound to values below −1 did not affect our calculations.
were obtained by rearranging Eq. 1 and minimizing Eq. 3 using the scipy.optimize.minimize package (v1.5.2) for Python 3 (v3.8.5) with bounds set at −1 and +1. Setting the lower bound to values below −1 did not affect our calculations.
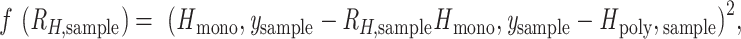 |
(3) |
where 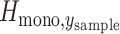 is the expected heterozygosity obtained from sampling 200 monogenomic pairs from the monogenomic barcodes present in each sample year. A sampled monogenomic pair was treated as an independent one and sampling was proportional to the frequency of each monogenomic barcode haplotype in each sample year. Monogenomic pairs consisting of identical monogenomic barcode haplotypes were excluded because these would not be recognized as polygenomic infections.
is the expected heterozygosity obtained from sampling 200 monogenomic pairs from the monogenomic barcodes present in each sample year. A sampled monogenomic pair was treated as an independent one and sampling was proportional to the frequency of each monogenomic barcode haplotype in each sample year. Monogenomic pairs consisting of identical monogenomic barcode haplotypes were excluded because these would not be recognized as polygenomic infections. 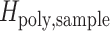 is the observed heterozygosity of the polygenomic sample. This process was repeated 200 times and the average was used as the point estimate of
is the observed heterozygosity of the polygenomic sample. This process was repeated 200 times and the average was used as the point estimate of 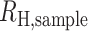 .
.
The average 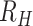 (
(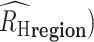 for each region was calculated through bootstrapping. Bootstrapped estimates of
for each region was calculated through bootstrapping. Bootstrapped estimates of 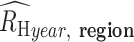 were obtained by calculating the average from 200 randomly samplied (with replacement)
were obtained by calculating the average from 200 randomly samplied (with replacement) 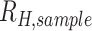 estimates observed in the region during the specified sample year. This calculation was repeated 200 times to obtain the final bootstrapped distribution of
estimates observed in the region during the specified sample year. This calculation was repeated 200 times to obtain the final bootstrapped distribution of 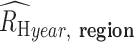 . The average and SDs of these bootstrapped distribution were then used to evaluate the inverse-variance-weighted average.
. The average and SDs of these bootstrapped distribution were then used to evaluate the inverse-variance-weighted average.
COI estimation
COI was estimated using the categorical method of THE REAL McCOIL (21) with the following parameter values: maxCOI = 25, threshold_ind = 20, threshold_site = 20, and err_method = 3. All other parameters used the default values. The median value estimated by THE REAL McCOIL was used as the point estimate of COI for each sample.
Simulating superinfection and cotransmission
Superinfection was simulated as the random sampling of unique monogenomic barcodes. Sites that were concordant across all sampled barcodes were considered homozygous. Sites that were discordant were considered heterozygous. Sites that were missing or heterozygous in the sampled monogenomic barcodes were excluded when quantifying heterozygosity and 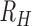 . Our superinfection simulations assume mass action and do not incorporate spatial or temporal transmission heterogeneity.
. Our superinfection simulations assume mass action and do not incorporate spatial or temporal transmission heterogeneity.
Cotransmission was simulated using a model that simulates the mating and sexual recombination of parasites as they are sampled and deposited by the mosquito vector (27). The model generates identity-by-descent maps that determine which portions of the genomes in cotransmitted strains were inherited from either of the parental strains present in the original polygenomic infection. All cotransmission chains were initiated from a superinfection with two to five randomly sampled monogenomic barcodes that were assumed to be unrelated.
The parameters used in the model were described in (27). Briefly, oocyst counts were drawn from a modified two-parameter Weibull distribution defined by
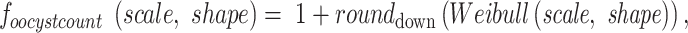 |
(4) |
where the scale = 2.5 and shape = 1. The 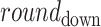 in Eq. 4 indicates that value drawn from the Weibull distribution is round down to the nearest integer. This results in a distribution whose median is 2, mean is 3, and interquartile range is between 1 and 4 . This distribution reflects the low oocyst intensities observed in naturally infected mosquitoes (54–57). Model predictions were robust to different assumptions regarding oocyst counts (Supplementary Material, Figs. S14 and S15).
in Eq. 4 indicates that value drawn from the Weibull distribution is round down to the nearest integer. This results in a distribution whose median is 2, mean is 3, and interquartile range is between 1 and 4 . This distribution reflects the low oocyst intensities observed in naturally infected mosquitoes (54–57). Model predictions were robust to different assumptions regarding oocyst counts (Supplementary Material, Figs. S14 and S15).
Infected hepatocyte counts were drawn from a lognormal distribution (mean = 1.8, SD = 0.8) whose sampled values were rounded down to the nearest integer unless the sample was less than 1 (58). In this case, the sampled value was set to 1. Uneven strain proportions were not simulated because their primary effect is on the probability of maintaining cotransmission chains (27).
Based on the identity-by-descent maps generated by the model, the barcodes of cotransmitted strains were generated by (i) determining which sites of the barcode were inherited from the first or second strain of the original infection, and (ii) copying the allelic identities of those parental barcode sites. After each cotransmission event, we sampled two unique, cotransmitted strains to generate polygenomic barcodes representing cotransmitted polygenomic infections with COI = 2.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Cory Schlesenger, Claudia R. Taccheri, and Hanamei S. Shao for their technical support in extracting DNA and molecular barcode genotyping. We thank Akanksha Khorgade for her help with the whole genome sequences. We also thank members of the Neafsey lab for their useful discussions.
Notes
Competing Interest: The authors declare no competing interest.
Contributor Information
Wesley Wong, Department of Immunology and Infectious Diseases, Harvard T.H. Chan School of Public Health, 665 Huntington Ave, Boston, MA 02115, USA.
Sarah Volkman, Department of Immunology and Infectious Diseases, Harvard T.H. Chan School of Public Health, 665 Huntington Ave, Boston, MA 02115, USA; Infectious Disease and Microbiome Program, Broad Institute, Cambridge, MA 02142, USA; College of Natural, Behavioral, and Health Sciences, Simmons University, Boston, MA 02115, USA.
Rachel Daniels, Department of Immunology and Infectious Diseases, Harvard T.H. Chan School of Public Health, 665 Huntington Ave, Boston, MA 02115, USA; Infectious Disease and Microbiome Program, Broad Institute, Cambridge, MA 02142, USA.
Stephen Schaffner, Infectious Disease and Microbiome Program, Broad Institute, Cambridge, MA 02142, USA.
Mouhamad Sy, Laboratory of Parasitology and Mycology, Aristide le Dantec Hospital, Cheikh Anta Diop University, Dakar 10200, Senegal.
Yaye Die Ndiaye, Laboratory of Parasitology and Mycology, Aristide le Dantec Hospital, Cheikh Anta Diop University, Dakar 10200, Senegal.
Aida S Badiane, Laboratory of Parasitology and Mycology, Aristide le Dantec Hospital, Cheikh Anta Diop University, Dakar 10200, Senegal.
Awa B Deme, Laboratory of Parasitology and Mycology, Aristide le Dantec Hospital, Cheikh Anta Diop University, Dakar 10200, Senegal.
Mamadou Alpha Diallo, Laboratory of Parasitology and Mycology, Aristide le Dantec Hospital, Cheikh Anta Diop University, Dakar 10200, Senegal.
Jules Gomis, Laboratory of Parasitology and Mycology, Aristide le Dantec Hospital, Cheikh Anta Diop University, Dakar 10200, Senegal.
Ngayo Sy, Laboratory of Parasitology and Mycology, Aristide le Dantec Hospital, Cheikh Anta Diop University, Dakar 10200, Senegal.
Daouda Ndiaye, Laboratory of Parasitology and Mycology, Aristide le Dantec Hospital, Cheikh Anta Diop University, Dakar 10200, Senegal.
Dyann F Wirth, Department of Immunology and Infectious Diseases, Harvard T.H. Chan School of Public Health, 665 Huntington Ave, Boston, MA 02115, USA; Infectious Disease and Microbiome Program, Broad Institute, Cambridge, MA 02142, USA.
Daniel L Hartl, Department of Organismic and Evolutionary Biology, Harvard University, Cambridge, MA 02138, USA.
Ethics Approval Statement
Informed consent was obtained from all participants.
Funding
Funding for this work was provided by the Bill & Melinda Gates Foundation (OPP1156051) to DFW, and the National Institutes of Health (5R21AI141843-02) to SKV.
Authors' Contributions
W.W., S.V., and S.S. led the analyses and computation. S.V. and R.D. were involved with sample preparation, transportation, and barcode genotyping. M.S., Y.D.N., A.S.B., A.B.D., M.A.D., J.G., and N.S., and D.N. were involved with sample collection and preparation. S.V., D.F.W., and D.L.H. were involved with project conceptualization and supervision.
Data Availability
Barcode data and analysis code are available at GitHub (https://github.com/weswong/RH_manuscript) and archived at Zenodo (doi: 10.5281/zenodo.7044606). The whole genome sequences used in this manuscript are available at the Short Read Archive (PRJNA882774).
References
- 1. Gardy JL, Loman NJ. 2017. Towards a genomics-informed, real-time, global pathogen surveillance system. Nat Rev Genet. 2017 19:1.19:9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Holmes EC, Dudas G, Rambaut A, Andersen KG. 2016. The evolution of Ebola virus: insights from the 2013–2016 epidemic. Nature. 538:193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Macklin GR, et al. 2020. Evolving epidemiology of poliovirus serotype 2 following withdrawal of the type 2 oral poliovirus vaccine. Science (1979). 368: eaba1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ribado Jv, et al. 2021. Linked surveillance and genetic data uncovers programmatically relevant geographic scale of Guinea worm transmission in Chad. PLoS Negl Trop Dis. 15:e0009609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Inzaule SC, Tessema SK, Kebede Y, Ogwell Ouma AE, Nkengasong JN. 2021. Genomic-informed pathogen surveillance in Africa: opportunities and challenges. Lancet Infect Dis. 21:e281–e289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Volkman SK, Neafsey DE, Schaffner SF, Park DJ, Wirth DF. 2012. Harnessing genomics and genome biology to understand malaria biology. Nat Rev Genet. 13:315–328. [DOI] [PubMed] [Google Scholar]
- 7. Neafsey DE, Taylor AR, MacInnis BL. 2021. Advances and opportunities in malaria population genomics. Nat Rev Genet 2021. 22:8. 22:502–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Neafsey DE, Volkman SK. 2017. Malaria genomics in the era of eradication. Cold Spring Harb Perspect Med. 7:a025544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tessema SK, et al. 2019. Applying next-generation sequencing to track falciparum malaria in sub-Saharan Africa. Malar J. 18:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Daniels RF et al. 2015. Modeling malaria genomics reveals transmission decline and rebound in Senegal. Proc Natl Acad Sci. 112:7067–7072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tusting LS, Bousema T, Smith DL, Drakeley C. 2014. Measuring changes in Plasmodium falciparum transmission: precision, accuracy, and costs of metrics. Adv Parasitol. 84:151–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fola AA et al. 2017. Higher complexity of infection and genetic diversity of Plasmodium vivax than Plasmodium falciparum across all malaria transmission zones of Papua New Guinea. Am J Trop Med Hyg. 96:630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hay SI, Rogers DJ, Toomer JF, Snow RW. 2000. AnnualPlasmodium falciparum entomological inoculation rates (EIR) across Africa: literature survey, internet access and review. Trans R Soc Trop Med Hyg. 94:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shaukat AM, Breman JG, McKenzie FE. 2010. Using the entomological inoculation rate to assess the impact of vector control on malaria parasite transmission and elimination. Malar J. 9:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Arnot D. 1998. Unstable malaria in Sudan: the influence of the dry season: Clone multiplicity of Plasmodium falciparum infections in individuals exposed to variable levels of disease transmission. Trans R Soc Trop Med Hyg. 92:580–585. [DOI] [PubMed] [Google Scholar]
- 16. Fola AA, et al. 2017. Higher complexity of infection and genetic diversity of plasmodium vivax than plasmodium falciparum across all malaria transmission zones of Papua New Guinea. Am J Trop Med Hyg. 96:630–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ndiaye T, Sy M, Gaye A, Ndiaye D. 2019. Genetic polymorphism of merozoite surface protein 1 (msp1) and 2 (msp2) genes and multiplicity of Plasmodium falciparum infection across various endemic areas in Senegal. Afr Health Sci. 19:2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Karl S, et al. 2016. Spatial effects on the multiplicity of Plasmodium falciparum infections. PLoS One. 11:e0164054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Miller RH, et al. 2017. A deep sequencing approach to estimate Plasmodium falciparum complexity of infection (COI) and explore apical membrane antigen 1 diversity. Mala J. 2017 16:1. 16:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Galinsky K, et al. 2015. COIL: a methodology for evaluating malarial complexity of infection using likelihood from single nucleotide polymorphism data. Malar J. 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chang H-H, et al. 2017. THE REAL McCOIL: a method for the concurrent estimation of the complexity of infection and SNP allele frequency for malaria parasites. PLoS Comput Biol. 13:e1005348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhu SJ, et al. 2019. The origins and relatedness structure of mixed infections vary with local prevalence of P. falciparum malaria. Elife. 8:e40845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. SC N et al. 2020. Co-transmission of related malaria parasite lineages shapes within-host parasite diversity. Cell Host Microbe. 27:93–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wong W et al. 2017. Genetic relatedness analysis reveals the cotransmission of genetically related Plasmodium falciparum parasites in Thiès, Senegal. Genome Med. 9:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nkhoma SC, Banda RL, Khoswe S, Dzoole-Mwale TJ, Ward SA. 2018. Intra-host dynamics of co-infecting parasite genotypes in asymptomatic malaria patients. Infect Genet Evol. 65:414–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nkhoma SC, et al. 2012. Close kinship within multiple-genotype malaria parasite infections. Proc R Soc London Ser B. 279:2589–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wong W, Wenger EA, Hartl DL, Wirth DF. 2018. Modeling the genetic relatedness of Plasmodium falciparum parasites following meiotic recombination and cotransmission. PLoS Comput Biol. 14:e1005923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schaffner SF, Taylor AR, Wong W, Wirth DF, Neafsey DE. 2018. hmmIBD: software to infer pairwise identity by descent between haploid genotypes. Malar J. 17:196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Daniels R, et al. 2008. A general SNP-based molecular barcode for Plasmodium falciparum identification and tracking. Malar J. 7:223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Programme National de lutte Contre le Paludisme . 2019. “Bulletin Epidemiologique Annuel 2018 Du Paludisme au Senegal.” [Google Scholar]
- 31. Programme National de lutte Contre le Paludisme . 2020. “Bulletin Epidemiologique Annuel 2019 Du Paludisme au Senegal.” [Google Scholar]
- 32. Programme National de lutte Contre le Paludisme . 2018. “Bulletin Epidemiologique Annuel 2017 Du Paludisme au Senegal.” [Google Scholar]
- 33. Programme National de lutte Contre le Paludisme . 2017. “Bulletin Epidemiologique Annuel 2016 Du Paludisme au Senegal.” [Google Scholar]
- 34. Programme National de lutte Contre le Paludisme . 2016. “Bulletin Epidemiologique Annuel 2015 Du Paludisme au Senegal.” [Google Scholar]
- 35. Programme National de Lutte Contre le Paludisme . 2015. “Bulletin Epidemiologique Annuel 2014 Du Paludisme au Senegal.” [Google Scholar]
- 36. Jeffreys H. 1961. Theory of probability. 3rd ed. New York: Oxford University Press. [Google Scholar]
- 37. Manske M, et al. 2012. Analysis of Plasmodium falciparum diversity in natural infections by deep sequencing. Nature. 487:375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Auburn S, et al. 2012. Characterization of within-host plasmodium falciparum diversity using next-generation sequence data. PLoS One. 7:e32891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Roh ME, et al. 2019. High genetic diversity of Plasmodium falciparum in the low-transmission setting of the kingdom of Eswatini. J Infect Dis. 220:1346–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Atuh NI, et al. 2021. High genetic complexity but low relatedness in Plasmodium falciparum infections from western Savannah highlands and coastal equatorial lowlands of Cameroon. Pathog Glob Health. 10.1080/20477724.2021.1953686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tessema S, et al. 2019. Using parasite genetic and human mobility data to infer local and cross-border malaria connectivity in Southern Africa. Elife. 8:e43510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Koepfli C, Mueller I. 2017. Malaria epidemiology at the clone level. Trends Parasitol. 33:974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Echeverry DF, et al. 2013. Long term persistence of clonal malaria parasite Plasmodium falciparum lineages in the Colombian Pacific region. BMC Genet. 14:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Osorio L, Todd J, Pearce R, Bradley DJ. 2007. The role of imported cases in the epidemiology of urban Plasmodium falciparum malaria in Quibdó, Colombia. Trop Med Int Health. 12:331–341. [DOI] [PubMed] [Google Scholar]
- 45. Nelson CS et al. , . 2019. High-resolution micro-epidemiology of parasite spatial and temporal dynamics in a high malaria transmission setting in Kenya. Nat Commun. 10:5615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Brandström M, Bagshaw AT, Gemmell NJ, Ellegren H. 2008. The relationship between microsatellite polymorphism and recombination hot spots in the human genome. Mol Biol Evol. 25:2579–2587. [DOI] [PubMed] [Google Scholar]
- 47. Rodriguez-Barraquer I, et al. 2018. Quantification of anti-parasite and anti-disease immunity to malaria as a function of age and exposure. Elife. 7.e35832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Littrell M, et al. 2013. Case investigation and reactive case detection for malaria elimination in northern Senegal. Malar J. 12:331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Daniels RF, et al. 2020. Genetic evidence for imported malaria and local transmission in Richard Toll, Senegal. Malar J. 19:276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Daniels R,et al. 2008. A general SNP-based molecular barcode for Plasmodium falciparum identification and tracking. Malar J 2008 7:1. 7:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mharakurwa S, et al. 2014. Pre-amplification methods for tracking low-grade Plasmodium falciparum populations during scaled-up interventions in Southern Zambia. Malar J. 13:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. MalariaGEN 2021. An open dataset of Plasmodium falciparum genome variation in 7,000 worldwide samples. Wellcome Open Res. 6:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hartl DL, Clark AG. 2007. Principles of Population Genetics. 4th Ed. New England (MA): Sinauer Associates. [Google Scholar]
- 54. Ouédraogo AL, et al. 2016. Dynamics of the human infectious reservoir for malaria determined by mosquito feeding assays and ultrasensitive malaria diagnosis in Burkina Faso. J Infect Dis. 213:90–99. [DOI] [PubMed] [Google Scholar]
- 55. Stone WJR, et al. 2013. The relevance and applicability of oocyst prevalence as a read-out for mosquito feeding assays. Sci Rep. 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gnémé A, et al. 2013. Malar J. 12:204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bompard A, et al. 2020. High Plasmodium infection intensity in naturally infected malaria vectors in Africa. Int J Parasitol. 50:985–996. [DOI] [PubMed] [Google Scholar]
- 58. Bejon P, et al. 2005. Calculation of liver-to-blood inocula, parasite growth rates, and preerythrocytic vaccine efficacy, from serial quantitative polymerase chain reaction studies of volunteers challenged with malaria sporozoites. J Infect Dis. 191:619–626. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Barcode data and analysis code are available at GitHub (https://github.com/weswong/RH_manuscript) and archived at Zenodo (doi: 10.5281/zenodo.7044606). The whole genome sequences used in this manuscript are available at the Short Read Archive (PRJNA882774).