

Abstract
Aims:
Vascular endothelial growth factor (VEGF) and pathologic angiogenesis have been demonstrated to play a pathogenic role in the development and progression of inflammatory bowel disease. Thus, we hypothesized that the potent anti-angiogenic factor endostatin might play a beneficial role in experimental ulcerative colitis (UC).
Main methods:
We used three animal models of UC: (1) induced by 6% iodoacetamide (IA) in rats, or (2) by 3% dextran sulfate sodium (DSS) in matrix metalloproteinase-9 (MMP-9) knockout (KO) and wild-type mice, and (3) interleukin-10 (IL-10) KO mice. Groups of MMP-9 KO mice with DSS-induced UC were treated with endostatin or water for 5 days.
Key findings:
We found concomitant upregulation of VEGF, PDGF, MMP-9 and endostatin in both rat and mouse models of UC. A positive correlation between the levels of endostatin or VEGF and the sizes of colonic lesions was seen in IA-induced UC. The levels and activities of MMP-9 were also significantly increased during UC induced by IA and IL-10 KO. Deletion of MMP-9 decreased the levels of endostatin in both water- and DSS-treated MMP-9 KO mice. Treatment with endostatin significantly improved DSS-induced UC in MMP-9 KO mice.
Significance:
1) Concomitantly increased endostatin is a defensive response to the increased VEGF in UC, 2) MMP-9 is a key enzyme to generate endostatin which may modulate the balance between VEGF and endostatin during experimental UC, and 3) endostatin treatment plays a beneficial role in UC. Thus, antiangiogenesis seems to be a new therapeutic option for UC.
Keywords: Endostatin, Matrix metalloproteinase-9, VEGF, Ulcerative colitis
Introduction
Endostatin is a 20 kDa fragment cleaved from collagen XVIII by enzymes such as matrix metalloproteinases (MMP), particularly MMP-9 (Ferreras et al., 2000; Nilsson and Dabrosin, 2006). Endostatin is a leading member of endogenous inhibitors of angiogenesis with potent anti-tumor activity (O’Reilly et al., 1997) that controls genes that regulate angiogenesis. An analysis of 90% of the human genome revealed that human endostatin downregulates signaling pathways in the human microvascular endothelium that modulate angiogenic factors, including vascular endothelial growth factor (VEGF), basic fibroblast growth factor (bFGF), hepatocyte growth factor (HGF), hypoxia-induced factor-1α (HIF-1α), and tumor necrotic factor-α (TNF-α) receptor, and upregulates many anti-angiogenic genes, including those encoding thrombospondin-1, HIF-1α-inhibitor, maspin and others (Abdollahi et al., 2004). Endostatin also blocks motility (Neskey et al., 2008) and induces apoptosis of endothelial cells (Dhanabal et al., 1999).
Folkman (2006) suggested that endostatin suppresses mainly pathologic (abnormal) angiogenesis and has little or no activity against normal wound healing or reproduction. Pathologic or abnormal angiogenesis is a major factor in the pathogenesis of many chronic and inflammatory diseases (e.g., cancer, diabetic retinopathy, atherosclerosis, and rheumatoid arthritis) (Carmeliet, 2003; Folkman, 1995; Yin et al., 2002). Administration of endostatin protein inhibited many tumors in animal models and patients, often with a “U-shape” dose–response curve (Folkman, 2006). Treatment with endostatin reduced blood vessel density within the synovial tissues and improved signs of TNF-induced inflammatory arthritis in mice (Yin et al., 2002). Therapy with endostatin inhibited inflammatory-associated angiogenesis in acute cyclophosphamide-induced cystitis (Beecken et al., 2004).
Recent clinical and experimental findings indicate that inflammatory bowel disease (IBD) pathogenesis is associated with excessive and uncontrolled angiogenesis (Chidlow et al., 2006; Danese et al., 2006; Scaldaferri et al., 2009; Spalinger et al., 2000), upregulation and imbalance of angiogenic growth factors (e.g., VEGF, bFGF, and PDGF) (Sandor et al., 2006; Griga et al., 1998; Kanazawa et al., 2001; Tolstanova et al., 2009; Wiercinska-Drapalo et al., 2007). Blockade of angiogenesis by treatment with neutralizing anti-VEGF antibody, soluble VEGF receptor-1 or a potent inhibitor of angiogenesis thalidomide was beneficial in IBD pathogenesis (Scaldaferri et al., 2009; Tolstanova et al., 2009; Vasiliauskas et al., 1999).
Despite numerous clinical and experimental data on the expression and the role of angiogenic factors, surprisingly little is known about the role of anti-angiogenic factors in IBD pathogenesis. In our previous study we showed concomitant upregulation of VEGF and anti-angiogenic factors endostatin and angiostatin in the early stages of trinitrobenzene sulfonic acid- and iodoacetamide-induced colitis (Sandor et al., 2006). Nevertheless, data are missing on the expression of anti-angiogenic factors in advanced stages of IBD, when chronic inflammation and growth of new blood vessels are extensive.
In the present study, using models of chemically induced colitis and spontaneously developing colitis in IL-10 knockout (KO) mice, we tested the hypothesis that endostatin might play an important role in IBD pathogenesis and its levels may be associated with the stages of IBD, in connection with the rate-limiting enzyme MMP-9 in endostatin generation.
Materials and methods
Animals
Female Sprague–Dawley rats (170–200 g) were purchased from Harlan Sprague–Dawley (San Diego, CA). Male IL-10 KO mice on a C57BL/6J background and wild-type (WT) C57BL/6J were purchased from Jackson Laboratory (Bar Harbor, ME). These animals were housed in the animal research facility at the VA Medical Center in Long Beach, CA under standard environmental conditions. Part of the studies was performed using MMP-9 KO mice and WT FVB background littermates which were bred and housed in an animal research facility of Emory University, Atlanta, Georgia. WT and MMP-9 KO littermates used in the study were between 6 and 8 weeks old at the beginning of the experimental protocol. All animals had an unlimited access to Purina chow and tap water. These studies were approved by the Subcommittee for Animal Studies of the R&D Committee of the VA Medical Center in Long Beach, CA and Emory University, Atlanta, Georgia.
Iodoacetamide-induced UC
This model is characterized by sequential changes in the colon, e.g., depletion of antioxidant glutathione, increased alkylation of protein sulfhydryls (SH) leading to cell death and mucosal necrosis followed by the development of erosions, ulcers and infiltration of neutrophils as early as 6–12 h (Sandor et al., 1994; Satoh et al., 1997a). Thus, this experimental UC was induced in rats by the SH alkylator iodoacetamide. 0.1 ml of 6% iodoacetamide (Sigma; St. Louis, MO) dissolved in 1% methylcellulose (Sigma; St. Louis, MO) or the vehicle 1% methylcellulose was given to rats once by enema (7 cm from anus) via rubber catheter Nelaton S-8 (Rüsch, Germany). Rats were euthanized 0.5, 1, 2, 6, and 24 h, and 3 and 7 days after intracolonic administration of iodoacetamide by carbon dioxide inhalation. During autopsy 7 cm of the distal colon was removed for morphologic examination. At autopsy, the area of colonic lesions (e.g., extent of tissue damage) and loss of rugae (as an indicator of recently healed, regenerated mucosa) were measured in mm2 in the two largest diameters and subsequently quantified by computerized planimetry coupled with stereomicroscopy. Colon wet weight (mg/100 g body weight) and colon thickness (scale: 0 = none, 1 = mild, 2 = moderate, 3 = severe) were also measured. Parameters such as loss of rugae, colon wet weight and colon thickness reflected the severity of UC.
Dextran sodium sulfate (DSS)-induced UC
UC was induced in age- and sex-matched male and female WT and MMP-9 KO littermates following the continuous administration of 3% DSS (MW 50000; ICN Biochemicals, Aurora, OH) in drinking water for 6 days. Age-matched male and female WT and MMP-9 KO littermates receiving tap water served as controls. Mice were observed daily and evaluated for changes in body weight and development of clinical symptoms. On the 7th day mice were killed by carbon dioxide inhalation. The distal colon (2 cm) was removed and processed for histologic examination and protein isolation.
Treatment with endostatin
Recombinant mouse endostatin was obtained from the Biotechnology Unit, NIH. MMP-9 KO mice with DSS-induced colitis were treated with either water or recombinant mouse endostatin (2 mg/100 g, s.c.) twice a day for 5 days starting from the 2nd day of DSS ingestion. Body weights, stool consistency, and the presence of occult/gross blood by a guaiac test (Hemoccult Sensa; Beckman Coulter, Fullerton, CA) were monitored daily in each mouse. Colitis was quantified with a clinical score by using the parameters of weight loss, stool consistency, and fecal blood as described previously (Castaneda et al., 2005). Mice were euthanized by carbon dioxide/hypothermia on the 7th day when colonic samples were processed for biochemical and histologic evaluation.
Light microscopy
Sections of the colon were fixed in 10% buffered formalin, embedded in paraffin, cut at 5 μm, stained with hematoxylin and eosin and coded for blind light microscopic evaluation of colonic lesions. Histologic slides were examined by two experienced pathologists who were unaware of the treatment.
Western blotting
Total proteins (100 μg) which were extracted from colonic mucosa in a lysis buffer containing protease inhibitors (Sigma; St. Louis, MO) were processed routinely for Western blotting as described previously (Tolstanova et al., 2009). The primary antibodies were used against VEGF, PDGF (1:500; Santa Cruz Biotech. Biothech.; Santa Cruz, CA), endostatin (1:250; LabVision, Fremont, CA), angiostatin (1:500; Abcam, Cambridge, MA), and MMP-9 (1–500; Thermo Fisher Scientific, Fremont, CA). The loading controls were performed by using a mouse monoclonal antibody to GAPDH (1:2000; EnCor Biotech.; Alachua, FL). Every Western blot was repeated at least two times using protein from three animals/group. The density of Western blot bands was measured by the Eagle Eye II (Stratagene, Austin, TX) and expressed in arbitrary units.
MMP gelatin zymography
Enzymatic activities of MMP-9 in the colon were detected by electrophoretic zymography under non-reducing condition. Briefly, 100 μg of total proteins extracted from each tissue sample was electrophoresed at room temperature in 8% SDS-PAGE gels containing gelatin (1 mg/ml) for 4 h with 125 V. MMP activity in the polyacrylamide gels was restored by removal of SDS by gentle shaking at room temperature in Triton X-100 (2.5%) for 30 min followed by incubation at 37 °C overnight in Zymogram Developing Buffer (50 mM Tris base, 50 mM Tris acid, 0.2 mM NaCl, 5 mM CaCl2 and 0.02 mM Brij). Substrate gels were stained in Coomassie Brilliant Blue R-250 in methanol–acetic acid–water (50:10:40), and destained in the same solution without dye. Proteolytic MMP activity was visualized as clear bands of digested regions. The molecular weight of the enzyme was determined by comparison with gelatinase zymography standard for human MMP-9 (CHEMICON International, Inc.; Billerica, MA) on the same gel.
Enzyme-linked immunosorbent assay (ELISA)
Rat VEGF ELISA kit for cell and tissue lysate (Raybiotech, Inc.; Norcross, GA) and human endostatin DuoSet ELISA Development kit (R&D Systems; Minneapolis, MN) were used according to the manufacturer’s instruction for the measurement of VEGF and endostatin in colonic mucosa. We calculated the concentration as a ratio of endogenous VEGF and endostatin vs. total proteins (pg/mg).
Statistical analysis
Quantitative results were expressed as mean±SD. The statistical significance was determined by the non-parametric Mann–Whitney U-test or Student’s t-test where appropriate. Correlations were performed using Spearman’s rank correlation. P values of <0.05 were considered statistically significant.
Results
Levels of endostatin, VEGF and PDGF in rat colonic tissue during iodoacetamide-induced UC
Western blots showed concomitantly increased levels of endostatin, VEGF and PDGF at different time points of iodoacetamide-induced UC (Fig. 1A). During the subacute and chronic stages of UC, levels of VEGF, PDGF and endostatin started to decrease, but still were higher than in control rats. Our quantitative sandwich enzyme immunoassay for endostatin and VEGF confirmed our Western blotting data. Moreover, we found a positive correlation (r=0.45, p<0.05) between concentrations of endostatin and VEGF at different time points in iodoacetamide-induced UC (Fig. 1B).
Fig. 1.
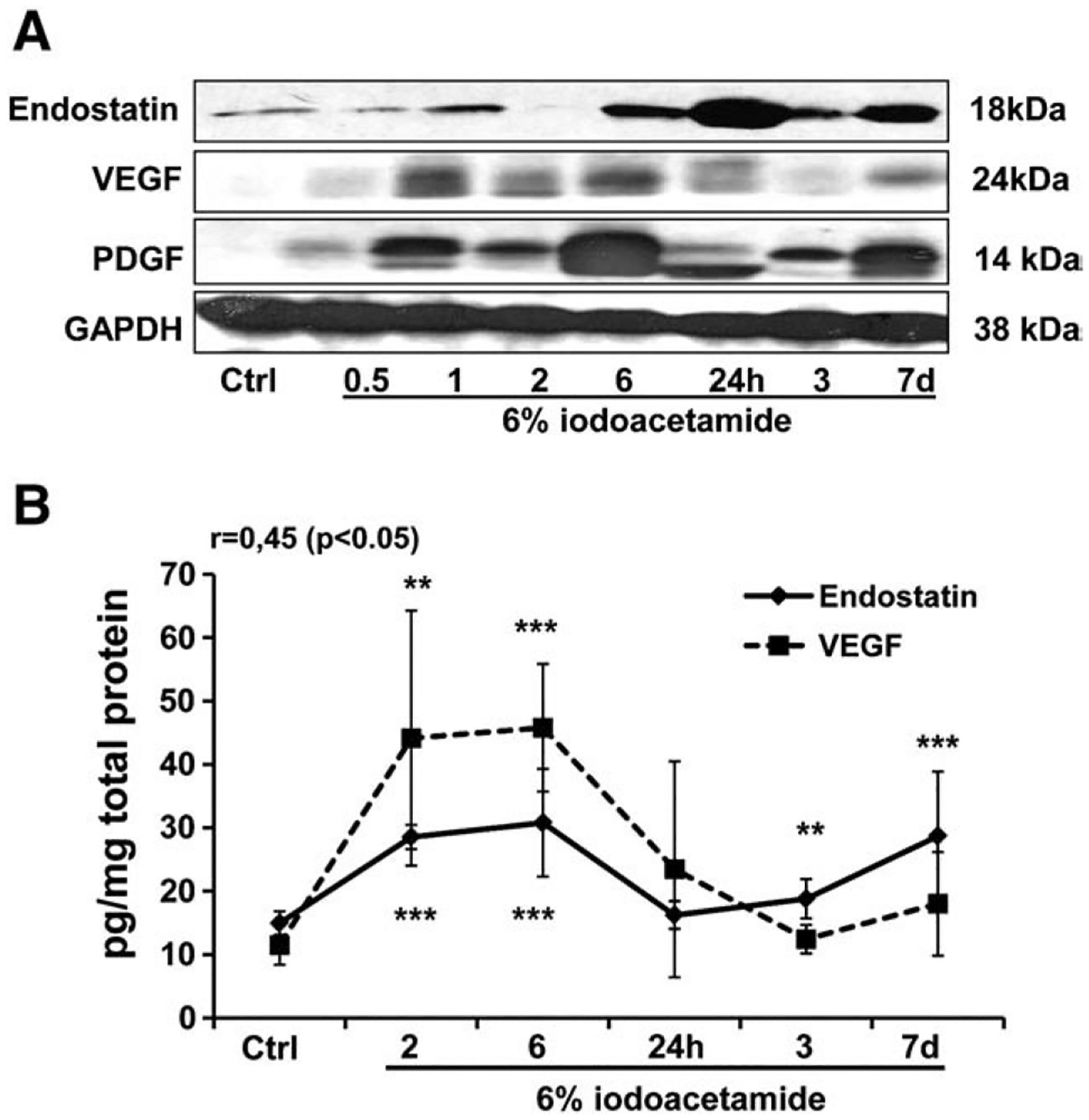
Time-dependent changes in endostatin, VEGF and PDGF protein levels in rat colonic tissue during iodoacetamide-induced UC. Protein levels in colonic tissue were determined by (A) Western blot and (B) ELISA. GAPDH levels were used as loading controls. Assays were repeated 3 times with highly reproducible results using protein from 3 rats/group. **p<0.01; ***p<0.001 vs. control (Ctrl).
Correlation between colonic levels of endostatin or VEGF and size of colonic lesions during iodoacetamide-induced UC
Iodoacetamide-induced UC is characterized by macroscopically well-demarcated lesions in distal colon in 5–7 days. We took this advantage and measured levels of endostatin and VEGF in the group of rats killed on the 7th day after iodoacetamide enema with range of colonic lesion sizes from 53 mm2 to 736 mm2 (average 289.6± 212.2 mm2, n=14). Levels of endostatin and VEGF were elevated and positively correlated with size of colonic lesions (Fig. 2A). Spearman’s (r) correlation coefficients were 0.77 (p<0.001) and 0.55 (p<0.038) for endostatin and VEGF, respectively (Fig. 2B). We didn’t find a positive correlation between levels of another anti-angiogenic factor angiostatin and size of colonic lesions (data not shown).
Fig. 2.
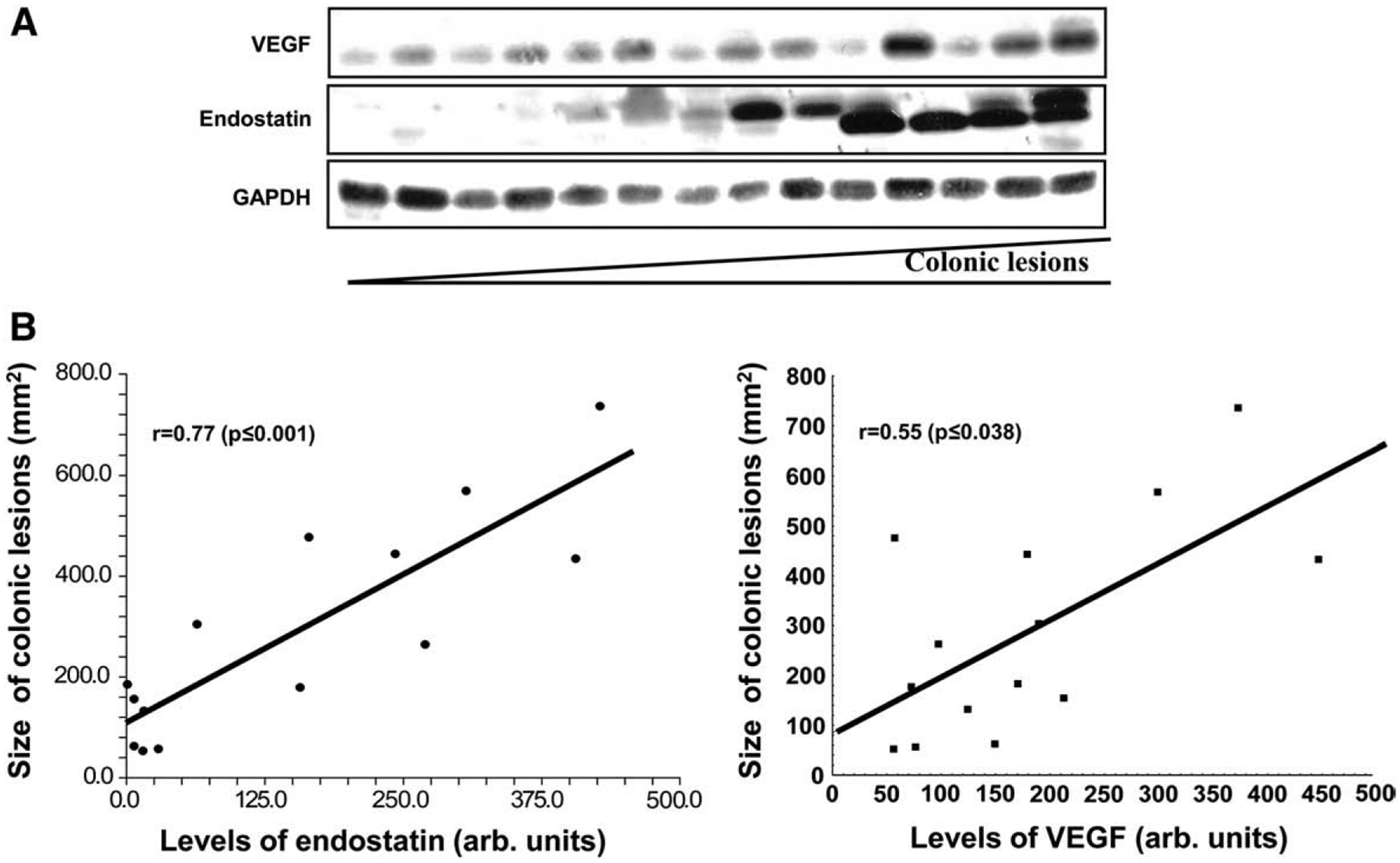
The correlation between colonic levels of endostatin, VEGF and size of colonic lesions during iodoacetamide-induced UC. (A) Protein levels in colonic tissue were determined by Western blot. Samples distributed from left to right according to size of colonic lesions from the smallest to greatest. GAPDH levels were used as loading controls. (B) Linear regression report and Spearman’s rank correlation coefficient. The density of Western blot bands was measured by the Eagle Eye II (Stratagene, Austin, TX) and presented in arbitrary units. Assays were repeated 3 times with highly reproducible results using protein from 3 rats/group.
Levels of endostatin and VEGF, PDGF in colonic tissue of IL-10 KO mice
Unlike chemically induced models of UC, IL-10 KO mice spontaneously develop chronic UC with age-dependent progression. As Fig. 3A demonstrates, colonic levels of endostatin were increased in IL-10 KO mice vs. WT, e.g., endostatin levels in 18 week old IL-10 KO mice were higher in comparison to 12 week old IL-10 KO mice. We observed a similar pattern with VEGF and PDGF levels (Fig. 3B).
Fig. 3.
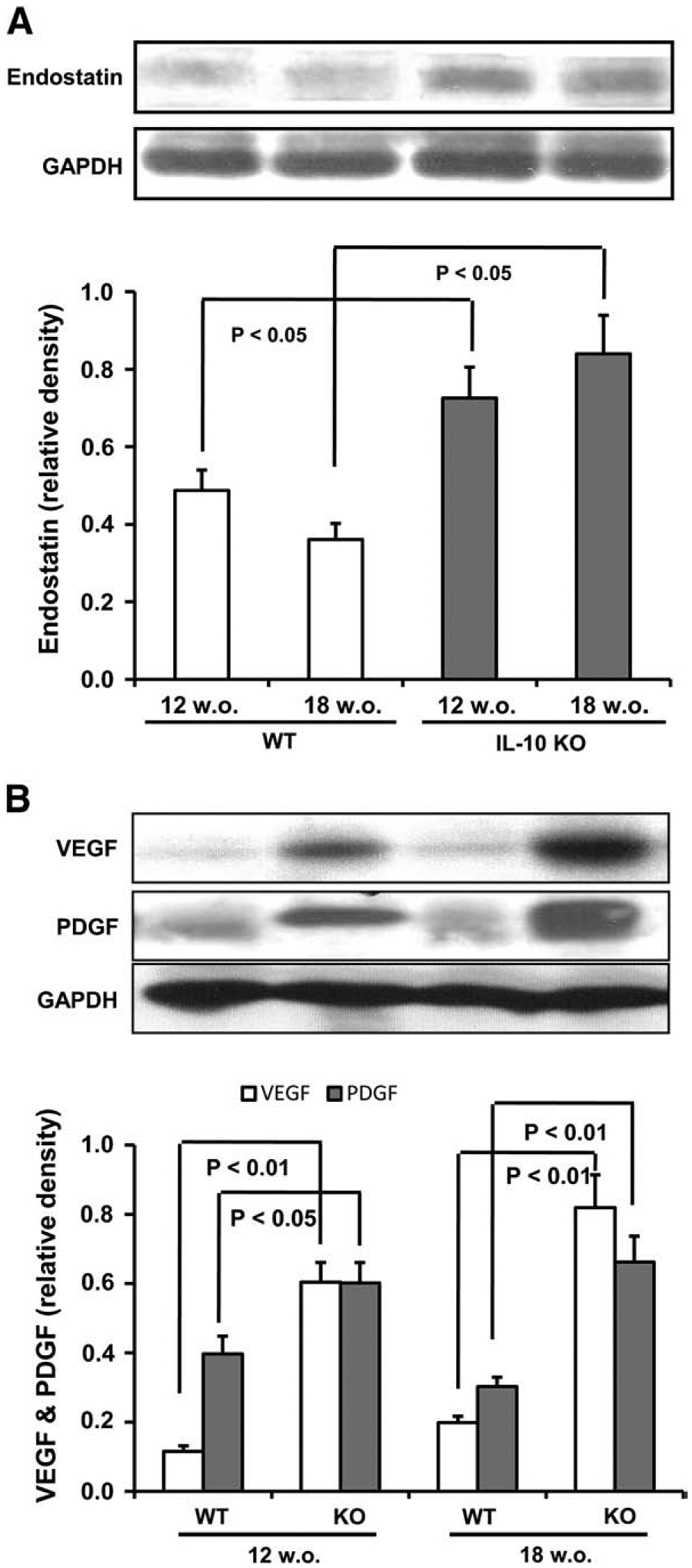
Changes in endostatin (A), VEGF and PDGF (B) protein levels in colonic tissue from IL-10 KO mice spontaneously developing chronic UC and WT mice on a C57BL/6J background. Protein levels in colonic tissue were determined by Western blot. GAPDH levels were used as loading controls. Assays were repeated 3 times with highly reproducible results using protein from 3 mice/group.
Activity of MMP-9 in colonic tissue during iodoacetamide-induced UC in rats and IL-10 KO mice
Endostatin is generated by enzymatic cleavage of NC1 domain of type XVIII collagen. Since MMP-9 belongs to gelatinase, we performed zymography to detect the gelatinolytic activity of MMP-9 in the colon of rats with iodoacetamide-induced UC and in IL-10 KO mice. Two forms of 92 kDa (monomer) and 220 kDa (dimer) MMP-9 were detected. Interestingly, in control rats and mice a slight activity of both forms was present. Iodoacetamide enema triggered time-dependent elevation of MMP-9 activity in the rat colon, associated with disease progression (Fig. 4A). IL-10 KO mice of 12 and 18 weeks had an increased activity of monomeric and dimeric MMP-9 forms vs. WT control. In addition, this enhanced activity was more profound in 18 vs. 12 week old IL-10 KO mice (Fig. 4B). Western blotting also showed an upregulation of MMP-9 protein expression in colonic tissue during iodoacetamide-induced UC in rats (Fig. 4A) and IL-10 KO mice (Fig. 4B).
Fig. 4.
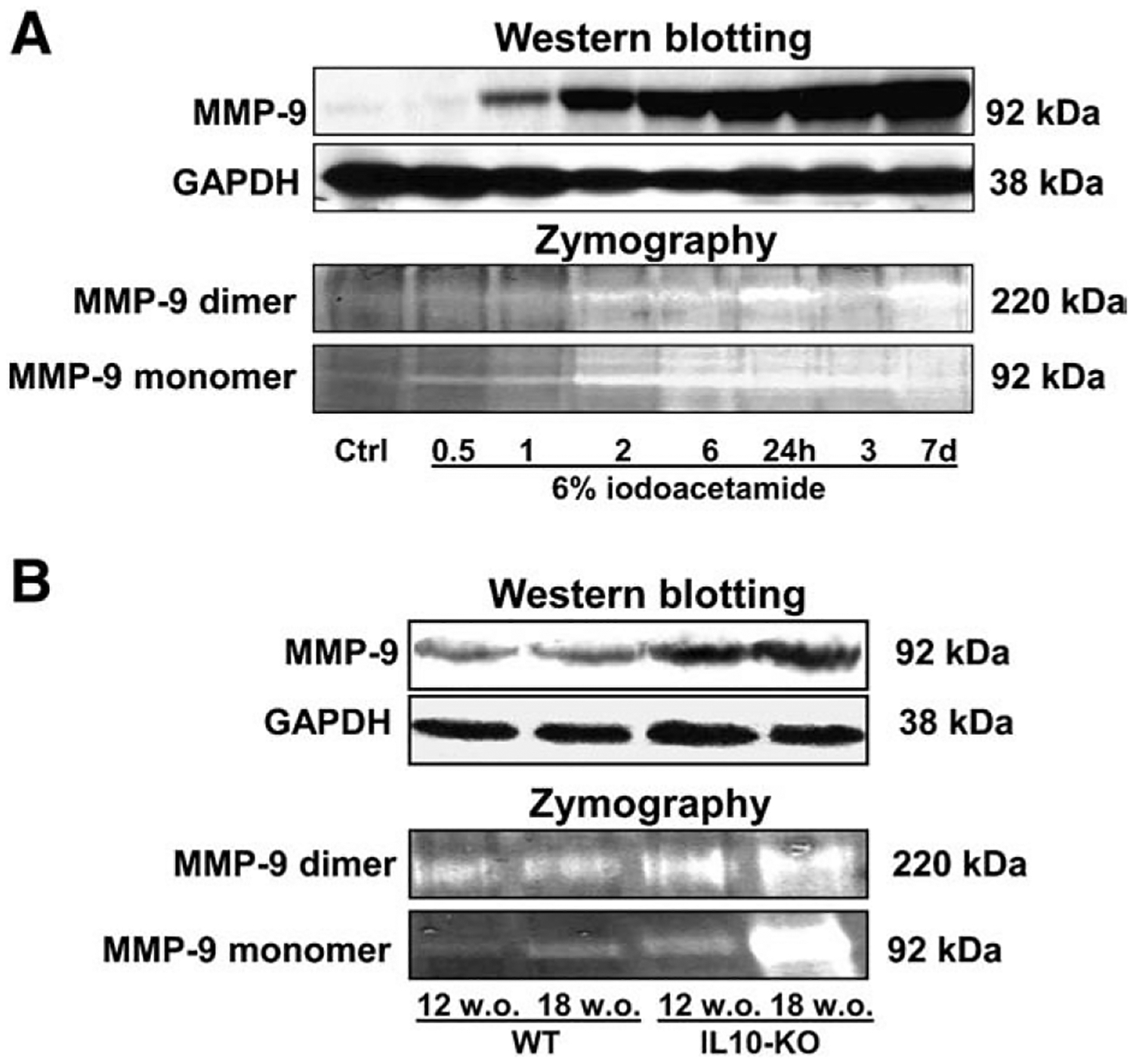
Proteolytic activity of MMP-9 by gelatin zymography and MMP-9 protein expression in colonic tissue during iodoacetamide-induced UC in rats (A) and IL-10 KO mice (B). Gelatinolytic bands of MMP-9 are represented as white bands. Protein levels in colonic tissue were determined by Western blot. GAPDH levels were used as loading controls. Assays were repeated three times with highly reproducible results using protein from 3 animals/group.
Levels of endostatin, VEGF and PDGF in colonic tissue of MMP-9 KO mice
To get further confirmation on the role of MMP-9 in endostatin generation in pathogenesis of experimental UC, we measured the levels of endostatin, VEGF and PDGF in MMP-9 KO mice vs. WT controls. Representative Western blotting bands (Fig. 5A) and their densitometric analysis (Fig. 5B, C, and D) are shown. Water-treated WT mice had significantly higher levels of endostatin vs. water-treated MMP-9 KO mice (p=0.002). Protein extracts from DSS-treated WT mice showed an abundant elevation of endostatin levels (p=0.024 vs. water-treated WT). The levels of endostatin in DSS-treated MMP-9 KO mice were lower than in DSS-treated WT mice (p=0.006) and were not different from water-treated MMP-9 KO mice (Fig. 5B).
Fig. 5.
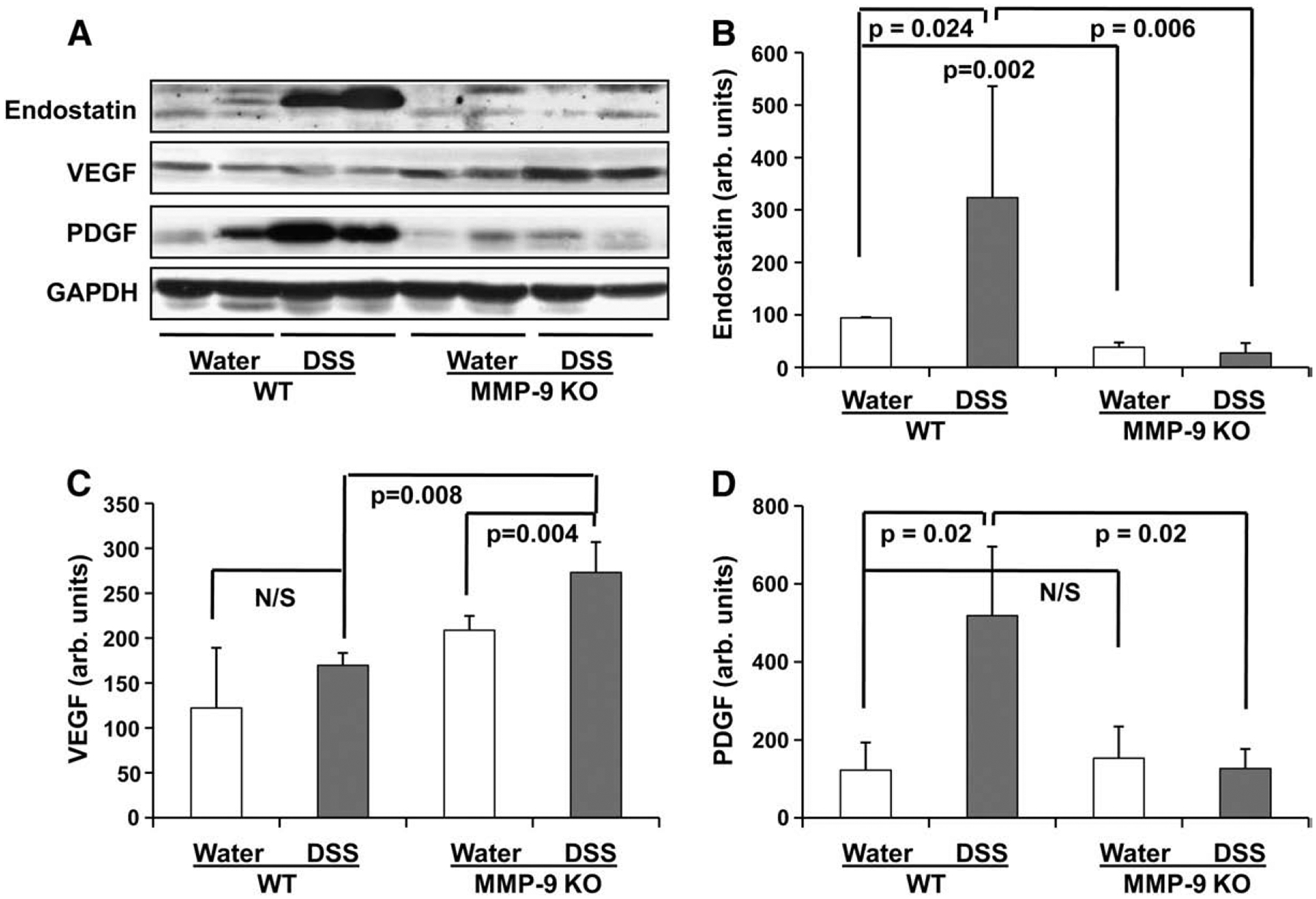
Levels of endostatin, angiostatin, VEGF and PDGF in colonic tissue of wild-type (WT) and MMP-9 KO mice with DSS-induced UC and water-ingested control. Protein levels in colonic tissue were determined by Western blot. A: Representative Western blot bands of endostatin, VEGF, and PDGF; B: Western blot density of endostatin; C: Western blot density of VEGF; D: Western blot density of PDGF. GAPDH levels were used as loading controls. Assays were repeated three times with highly reproducible results.
Since, we found a concomitant upregulation of endostatin, VEGF and PDGF during iodoacetamide-induced UC in rats (Fig. 1A) and IL-10 KO mice (Fig. 3) it was interesting to check the levels of these factors in MMP-9 KO mice with DSS-induced UC. Levels of VEGF were even higher in DSS-treated MMP-9 KO mice than in DSS-treated WT mice (p=0.008). Moreover, we didn’t find significant changes of VEGF levels in WT mice after DSS ingestion, but it was significantly increased in DSS- vs. water-treated MMP-9 KO mice (p=0.004) (Fig. 5C). We found that levels of PDGF were significantly decreased in MMP-9 KO mice vs. WT mice during DSS-induced UC (p=0.02) and similar to the pattern of endostatin levels. PDGF in water-treated WT and MMP-9 KO mice were unchanged (Fig. 5D).
Effect of endostatin on clinical and morphologic features of experimental UC
Dose–response studies showed that endostatin anti-angiogenic and anti-tumor efficacy are biphasic and operate over a U-shaped curve, i.e., levels of endostatin that are too high or too low are inactive (Folkman, 2006). Because of decreased endogenous endostatin levels in MMP-9 KO mice, we used this model to investigate the effect of endostatin administration on UC healing. Daily treatment with endostatin significantly increased body weight (p<0.01) and clinical score (p<0.001) of MMP-9 KO mice with DSS-induced UC vs. group of MMP-9 KO mice treated with saline (Fig. 6A and B). Moreover, at the end of experiments all mice from the saline-treated group had very loose bloody stool while mice from endostatin-treated group had only a tinge of blood and no diarrhea. Histologic evaluation of colons revealed extensive erosions and ulcers with abundant inflammatory cell infiltration in MMP-9 KO mice ingesting DSS and treated with saline while mice treated with endostatin have only submucosal edema and small superficial erosions (Fig. 6C).
Fig. 6.
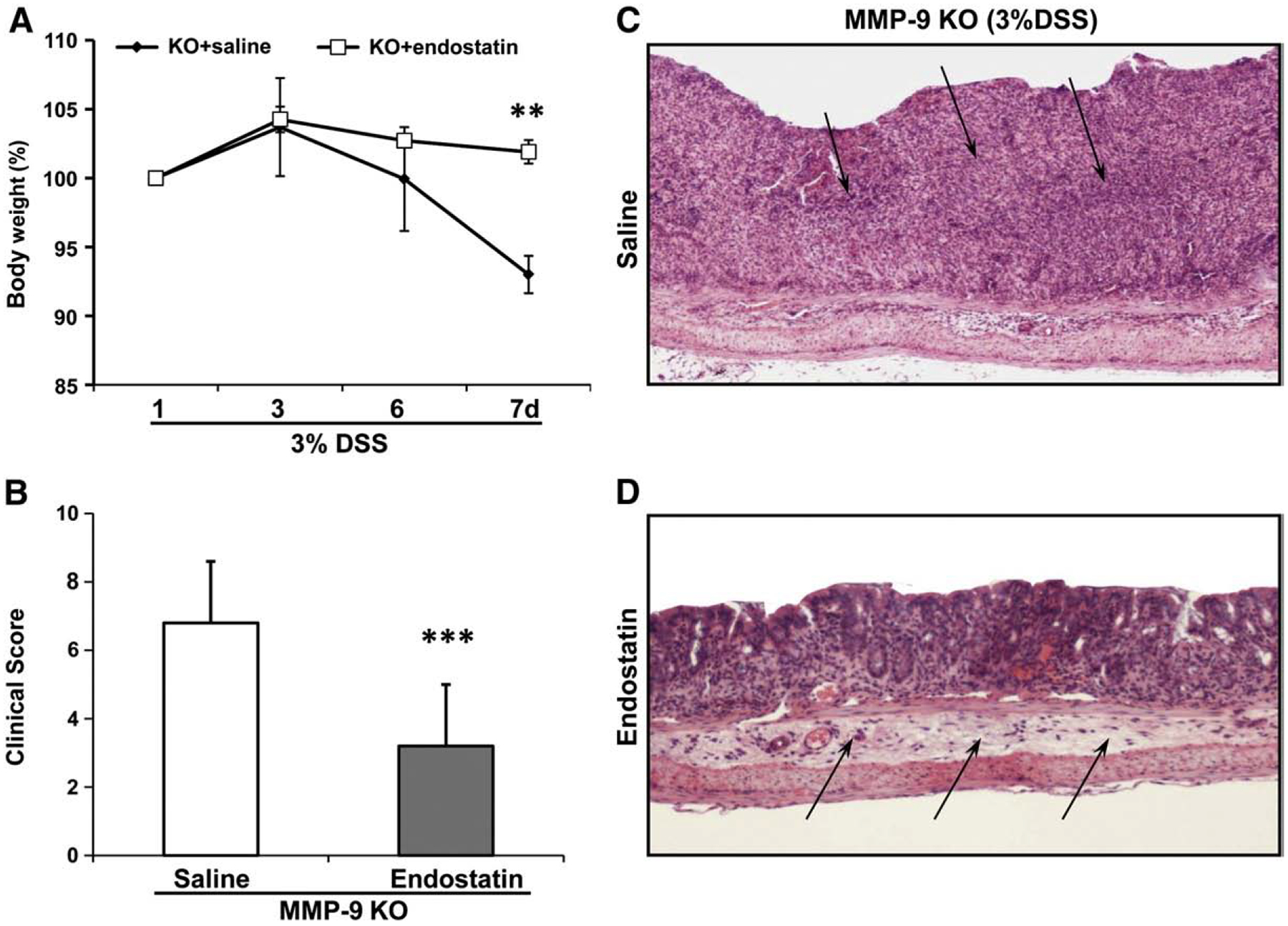
Effect of daily treatment with endostatin (2 mg/100 g, s.c.) on DSS-induced UC in MMP-9 KO mice. (A) Changes in body weight. Mean body weight on the 1st day represents 100%. (B) Clinical score. Results are expressed as mean±SD; n = 5 mice/group. ** p < 0.001; *** p < 0.0001 vs. control (saline). (C, D) Histologic changes in the colon during DSS-induced UC in MMP-9 KO mice treated with saline (×40) or endostatin (×100) (H&E staining). (C) Mice after DSS ingestion show that the normal colonic mucosa is almost completely replaced by massive inflammatory cell infiltration and early granulation tissue (arrows). (D) Endostatin-treated mice with DSS revealed intact normal mucosa and moderate submucosal edema (arrows).
Discussion
In the present study, we demonstrated for the first time the concomitantly increased levels of potent anti-angiogenic factor endostatin and angiogenic factors VEGF and PDGF not only in the acute stages but also in the chronic stages of experimental UC. Moreover, the levels of both factors were elevated and positively correlated with the severity of colonic lesions. Colonic activity of gelatinase MMP-9, which controls endostatin generation from collagen XVIII, was significantly increased with disease progression in chemically induced UC and during spontaneously developing UC in IL-10 KO mice. We demonstrated a key role of MMP-9 in endostatin generation during experimental UC: the depletion of MMP-9 in MMP-9 KO mice significantly decreased levels of endostatin vs. WT littermates in control groups as well as during DSS-induced UC. Treatment with endostatin significantly improved signs of experimental UC in MMP-9 KO mice.
IBD is a chronic disease which is characterized by sustained inflammation and tissue remodeling. Excessive/abnormal angiogenesis with increased microvascular density, distorted vasculature, increased permeability and thrombogenic potential plays a role in IBD pathogenesis (Chidlow et al., 2006; Danese et al., 2006; Scaldaferri et al., 2009; Spalinger et al., 2000). Moreover, abnormal angiogenesis facilitates the migration of inflammatory cells to the site of mucosal injury leading to the perpetuation of chronic inflammation (Chidlow et al., 2006), and anti-inflammatory therapy with anti-TNF-α antibody decreased angiogenesis (Di Sabatino et al., 2004).
Endostatin is a potent inhibitor of pathologic, inflammatory-associated angiogenesis. Treatment with endostatin-expressing lentiviral vector reduced blood vessel density within the synovial tissues and inflammation during TNF-induced inflammatory arthritis in mice (Yin et al., 2002). Endostatin treatment also suppressed airway hyperresponsiveness during asthma in the murine model which was associated with downregulation of monocyte chemoattractant protein 1, macrophage inflammatory protein 1a, interferon-inducible protein 10 and E-selectin (Suzaki et al., 2005). Furthermore, endostatin promoted integrity of the retinal endothelial barrier in vitro and in vivo by preventing VEGF-mediated alteration of tight junction (Brankin et al., 2005; Campbell et al., 2006).
In the present study, we showed a significant upregulation of endostatin in rats with chemically induced UC at the early and advanced stages of disease as well as in IL-10 KO mice with spontaneously developing chronic colitis. Endostatin is generated by proteinases, particularly MMP-9 from the non-collagenous domain of collagen XVIII. Elevated activity of MMP-9 was found in patients with IBD (Baugh et al., 1999). Clinical data and experimental studies have shown that MMP are the predominant proteinases in the gut mucosa during IBD (von Lampe et al., 2000; Salmela et al., 2002). In the present study, we detected increased activity and protein expression of MMP-9 during different stages of chemically induced UC and in IL-10 KO mice with spontaneously developing UC. We found that MMP-9 plays an important role in endostatin generation during UC. Depletion of MMP-9 in MMP-9 KO mice resulted in significantly decreased levels of endostatin in the control group and during DSS-induced UC. Levels of another anti-angiogenic factor angiostatin, which can also be enzymatically generated from plasminogen by MMP-9 (Patterson and Sang, 1997) were not changed in MMP-9 KO mice vs. WT (data not shown). Moreover, treatment with adenovirus vector carrying the human genes for MMP-9 significantly decreased tumor growth rate and microvessel area which was associated with significantly increased levels of endostatin in vivo, whereas VEGF levels were unaffected (Bendrik et al., 2008).
Feldman et al. (2000, 2001) showed that plasma endostatin levels were elevated and correlated with VEGF levels in colorectal cancer patients with liver metastases and clear cell renal carcinoma patients. Furthermore, elevated endostatin levels were associated with disease progression and bad prognosis (Feldman et al., 2000, 2001; Guan et al., 2003). We also showed that elevation of endostatin levels in rats with chemically induced UC and IL-10 KO mice was accompanied by upregulation of potent angiogenic growth factors VEGF and PDGF. Furthermore, we found a positive correlation between size of colonic lesions and levels of endostatin and VEGF during experimental UC. Recent study on IBD patients showed elevated serum levels of pro-angiogenic factors angiogenin, and angiopoietin-2. Moreover, higher endostatin levels were recorded in UC patients with extensive colitis (Oikonomou et al., in press).
Concomitant upregulation of VEGF, PDGF and endostatin allowed us to speculate on their co-regulation. Comparison studies of MMP-9 KO (decreased endostatin levels) vs. WT (increased endostatin levels) DSS-treated mice revealed significantly downregulated levels of PDGF in MMP-9 KO mice similar to the pattern of endostatin levels. To our knowledge this is the first report on the possible role of endostatin in PDGF expression. Further investigations are needed to explain the molecular mechanisms. Since PDGF is an important factor for newly formed vessels stabilization, maturation by pericytes coverage (Lindahl et al., 1997) and facilitation of physiologic angiogenesis (McCarty et al., 2007), concomitant upregulation of PDGF and endostatin might play a positive role in IBD pathogenesis. An opposite situation was observed for VEGF. Levels of VEGF were even higher in MMP-9 KO than in WT mice with DSS-induced UC. It is unlikely that MMP-9 inhibits VEGF levels, since treatment with MMP-9 didn’t affect VEGF levels (Bendrik et al., 2008). Upregulation of VEGF in MMP-9 KO mice might be the result of decreased endostatin levels. Abdollahi et al. (2005) showed that microvascular endothelial cells exposed to endostatin had decreased levels of VEGF expression by 67%. Furthermore, in our previous study we found that treatment with neutralizing anti-VEGF antibody decreased endostatin and VEGF levels in colonic tissue of rats with iodoacetamide-induced UC (Tolstanova et al., 2007). Thus, VEGF might affect endostatin levels as well, for instance via protease activation, e.g., MMP-9 (Belotti et al., 2008).
We and others showed surprising effects of different angiogenic factors on IBD healing. Diminished VEGF activity by neutralizing anti-VEGF antibody or treatment with soluble VEGFR-1 ameliorated experimental UC (Tolstanova et al., 2009; Scaldaferri et al., 2009) vs. the accelerated healing of experimental duodenal ulcers by VEGF and delayed ulcer healing by neutralizing anti-VEGF antibody (Szabo et al., 1998a). Depletion of angiopoietin-2 attenuated leukocyte infiltration, inflammation as well as blood and lymphatic vessel density during experimental UC (Ganta et al., 2010). At the same time administration of bFGF, PDGF or HGF accelerated the healing of both experimental UC (Kojima et al., 2007; Szabo and Sandor, 1996; Szabo et al., 1999; Thatch et al., 2009) and chronic duodenal ulcers (Szabo et al., 1996, 1998b; Satoh et al., 1997b). Thus, we postulate that VEGF stimulates both normal angiogenesis (e.g., in duodenal ulceration) and excessive/pathologic angiogenesis in IBD. Because of the potent anti-angiogenic effect of endostatin we assumed that endostatin might be beneficial for IBD. In present study, treatment with endostatin in a dose which had anti-angiogenic and anti-tumor effects in vivo (Folkman, 2006) significantly improved clinical and histologic signs of DSS-induced UC in MMP-9 KO mice. These results are supported by other studies showing that the potent inhibitor of angiogenesis thalidomide attenuates IBD in patients (Plamondon et al., 2007; Vasiliauskas et al., 1999) and in animal models (Prakash et al., 2008). Furthermore, deletion of thrombospondin-1 in mice, which is upregulated by endostatin (Abdollahi et al., 2004), evoked more severe experimental UC vs. WT mice (Punekar et al., 2008). So we assumed that the elevation of endostatin levels during IBD might be a defensive response to upregulated VEGF and excessive/pathologic angiogenesis. However, because of the well known “U-shape” effects of endostatin in tumor regulation, detailed dose–response experiments in other UC models are planned for our future studies.
Conclusions
The elevated levels of anti-angiogenic factor endostatin may be beneficial for accelerating the healing of experimental UC. MMP-9 plays a key role to generate endostatin and may modulate a balance between VEGF and endostatin during experimental UC. Anti-angiogenesis seems to be a new therapeutic option for UC.
Acknowledgements
We thank Loc Trinh of NIH Biotechnology Unit for producing and purifying recombinant mouse endostatin. This work was supported by the Department of Veterans Affairs, Veterans Health Administration Merit Review [Grant VAMR0710-580].
Footnotes
Conflict of interest statement
None.
References
- Abdollahi A, Hahnfeldt P, Maercker C, Grone HJ, Debus J, Ansorge W, et al. Endostatin’s antiangiogenic signaling network. Mol Cell 2004;13(5):649–63. [DOI] [PubMed] [Google Scholar]
- Abdollahi A, Hlatky L, Huber PE. Endostatin: the logic of antiangiogenic therapy. Drug Resist Updat 2005;8(1–2):59–74. [DOI] [PubMed] [Google Scholar]
- Baugh MD, Perry M, Hollander A, Davies DR, Cross SS, Lobo AJ, et al. Matrix metalloproteinases levels are elevated in inflammatory bowel disease. Gastroenterology 1999;117(4):814–22. [DOI] [PubMed] [Google Scholar]
- Beecken WD, Engl T, Blaheta R, Bentas W, Achilles EG, Jonas D, et al. Angiogenesis inhibition by angiostatin, endostatin and TNP-470 prevents cyclophosphamide induced cystitis. Angiogenesis 2004;7(1):69–73. [DOI] [PubMed] [Google Scholar]
- Belotti D, Calcagno C, Garofalo A, Caronia D, Riccardi E, Giavazzi R, et al. Vascular endothelial growth factor stimulates organ-specific host matrix metalloproteinase-9 expression and ovarian cancer invasion. Mol Cancer Res 2008;6(4):525–34. [DOI] [PubMed] [Google Scholar]
- Bendrik C, Robertson J, Gauldie J, Dabrosin C. Gene transfer of matrix metalloproteinase-9 induces tumor regression of breast cancer in vivo. Cancer Res 2008;68(9): 3405–12. [DOI] [PubMed] [Google Scholar]
- Brankin B, Campbell M, Canning P, Gardiner TA, Stitt AW. Endostatin modulates VEGF-mediated barrier dysfunction in the retinal microvascular endothelium. Exp Eye Res 2005;81(1):22–31. [DOI] [PubMed] [Google Scholar]
- Campbell M, Collery R, McEvoy A, Gardiner TA, Stitt AW, Brankin B. Involvement of MAPKs in endostatin-mediated regulation of blood-retinal barrier function. Curr Eye Res 2006;31(12):1033–45. [DOI] [PubMed] [Google Scholar]
- Carmeliet P Angiogenesis in health and disease. Nat Med 2003;9(6):653–60. [DOI] [PubMed] [Google Scholar]
- Castaneda FE, Walia B, Vijay-Kumar M, Patel NR, Roser S, Kolachala VL, et al. Targeted deletion of metalloproteinase 9 attenuates experimental colitis in mice: central role of epithelial-derived MMP. Gastroenterology 2005;129(6):1991–2008. [DOI] [PubMed] [Google Scholar]
- Chidlow JH Jr, Langston W, Greer JJ, Ostanin D, Abdelbaqi M, Houghton J, et al. Differential angiogenic regulation of experimental colitis. Am J Pathol 2006;169(6): 2014–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danese S, Sans M, Motte CDL, Graziani C, West G, Phillips MH, et al. Angiogenesis as a novel component of inflammatory bowel disease pathogenesis. Gastroenterology 2006;130(7):2060–73. [DOI] [PubMed] [Google Scholar]
- Dhanabal M, Volk R, Ramchandran R, Simons M, Sukhatme VP. Cloning, expression, and in vitro activity of human endostatin. Biochem Biophys Res Commun 1999;258(2): 345–52. [DOI] [PubMed] [Google Scholar]
- Di Sabatino A, Ciccocioppo R, Benazzato L, Sturniolo GC, Corazza GR. Infliximab downregulates basic fibroblast growth factor and vascular endothelial growth factor in Crohn’s disease patients. Aliment Pharmacol Ther 2004;19(9):1019–24. [DOI] [PubMed] [Google Scholar]
- Feldman AL, Alexander HR Jr, Bartlett DL, Kranda KC, Miller MS, Costouros NG, et al. A prospective analysis of plasma endostatin levels in colorectal cancer patients with liver metastases. Ann Surg Oncol 2001;8(9):741–5. [DOI] [PubMed] [Google Scholar]
- Feldman AL, Tamarkin L, Paciotti GF, Simpson BW, Linehan WM, Yang JC, et al. Serum endostatin levels are elevated and correlate with serum vascular endothelial growth factor levels in patients with stage IV clear cell renal cancer. Clin Cancer Res 2000;6(12):4628–34. [PubMed] [Google Scholar]
- Ferreras M, Felbor U, Lenhard T, Olsen BR, Delaissé J. Generation and degradation of human endostatin proteins by various proteinases. FEBS Lett 2000;486(3):247–51. [DOI] [PubMed] [Google Scholar]
- Folkman J Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med 1995;1(1):27–31. [DOI] [PubMed] [Google Scholar]
- Folkman J Antiangiogenesis in cancer therapy—endostatin and its mechanisms of action. Exp Cell Res 2006;312(5):594–607. [DOI] [PubMed] [Google Scholar]
- Ganta VC, Cromer W, Mills GL, Traylor J, Jennings M, Daley S, et al. Angiopoietin-2 in experimental colitis. Inflamm Bowel Dis 2010;16(6):1029–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griga T, Tromm A, Spanger J, May B. Increased serum level of vascular endothelial growth factor in patients with inflammatory bowel disease. Scand J Gastroenterol 1998;33(5):504–8. [DOI] [PubMed] [Google Scholar]
- Guan KP, Ye HY, Yan Z, Wang Y, Hou SK. Serum levels of endostatin and matrix metalloproteinase-9 associated with high stage and grade primary transitional cell carcinoma of the bladder. Urology 2003;61(4):719–23. [DOI] [PubMed] [Google Scholar]
- Kanazawa S, Tsunoda T, Onuma E, Majima T, Kagiyama M, Kikuchi K. VEGF, basic-FGF, and TGF-β in Crohn’s disease and ulcerative colitis: a novel mechanism of chronic intestinal inflammation. Am J Gastroenterol 2001;96(3):822–8. [DOI] [PubMed] [Google Scholar]
- Kojima T, Watanabe T, Hata K, Nagawa H. Basic fibroblast growth factor enema improves experimental colitis in rats. Hepatogastroenterology 2007;54(77): 1373–7. [PubMed] [Google Scholar]
- Lindahl P, Johansson BR, Levéen P, Betsholtz C. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science 1997;277(5323):242–5. [DOI] [PubMed] [Google Scholar]
- McCarty MF, Somcio RJ, Stoeltzing O, Wey J, Fan F, Liu W, et al. Overexpression of PDGF-BB decreases colorectal and pancreatic cancer growth by increasing tumor pericyte content. J Clin Investig 2007;117(8):2114–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neskey DM, Ambesi A, Pumiglia KM, McKeown-Longo PJ. Endostatin and anastellin inhibit distinct aspects of the angiogenic process. J Exp Clin Cancer Res 2008;27:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson UW, Dabrosin C. Estradiol and tamoxifen regulate endostatin generation via matrix metalloproteinase activity in breast cancer in vivo. Cancer Res 2006;66(9): 4789–94. [DOI] [PubMed] [Google Scholar]
- Oikonomou KA, Kapsoritakis AN, Kapsoritaki AI, Manolakis AC, Tiaka EK, Tsiopoulos FD, et al. Angiogenin, angiopoietin-1, angiopoietin-2, and endostatin serum levels in inflammatory bowel disease. Inflamm Bowel Dis in press. [DOI] [PubMed] [Google Scholar]
- O’Reilly MS, Boehm T, Shing Y, Fukai N, Vasios G, Lane WS, et al. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell 1997;88(2):277–85. [DOI] [PubMed] [Google Scholar]
- Patterson BC, Sang QA. Angiostatin-converting enzyme activities of human matrilysin (MMP-7) and gelatinase B/type IV collagenase (MMP-9). J Biol Chem 1997;272 (46):28823–5. [DOI] [PubMed] [Google Scholar]
- Plamondon S, Ng SC, Kamm MA. Thalidomide in luminal and fistulizing Crohn’s disease resistant to standard therapies. Aliment Pharmacol Ther 2007;25(5):557–67. [DOI] [PubMed] [Google Scholar]
- Prakash O, Medhi B, Saikia UN, Pandhi P. Effect of different doses of thalidomide in experimentally induced inflammatory bowel disease in rats. Basic Clin Pharmacol Toxicol 2008;103(1):9–16. [DOI] [PubMed] [Google Scholar]
- Punekar S, Zak S, Kalter VG, Dobransky L, Punekar I, Lawler JW, et al. Thrombospondin 1 and its mimetic peptide ABT-510 decrease angiogenesis and inflammation in a murine model of inflammatory bowel disease. Pathobiology 2008;75(1):9–21. [DOI] [PubMed] [Google Scholar]
- Salmela MT, MacDonald TT, Black D, Irvine B, Zhuma T, Saarialho-Kere U, Pender SL. Upregulation of matrix metalloproteinases in a model of T cell mediated tissue injury in the gut: analysis by gene array and in situ hybridisation. Gut 2002;51(4): 540–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandor ZS, Nagata M, Kusstatscher S, Szabo S. New animal model of ulcerative colitis associated with depletion of glutathione and protein SH alkylation. Gastroenterology 1994;106(5):A766. [Google Scholar]
- Sandor ZS, Deng XM, Khomenko T, Tarnawski AS, Szabo S. Altered angiogenic balance in ulcerative colitis: a key to impaired healing? Biochem Biophys Res Commun 2006;350(1):147–50. [DOI] [PubMed] [Google Scholar]
- Satoh H, Sato F, Takami K, Szabo S. New ulcerative colitis model induced by SH blockers in rats and the effects of antiinflammatory drugs on the colitis. Jpn J Pharmacol 1997a;73(4):299–309. [DOI] [PubMed] [Google Scholar]
- Satoh H, Shino A, Sato F, Asano S, Murakami I, Inatomi N, et al. Role of endogenous basic fibroblast growth factor in the healing of gastric ulcers in rats. Jpn J Pharmacol 1997b;73(4):59–71. [DOI] [PubMed] [Google Scholar]
- Scaldaferri F, Vetrano S, Sans M, Arena V, Straface G, Stigliano E, et al. VEGF-A links angiogenesis and inflammation in inflammatory bowel disease pathogenesis. Gastroenterology 2009;136(2):585–95. [DOI] [PubMed] [Google Scholar]
- Spalinger J, Patriquin H, Miron MC, Marx G, Herzog D, Dubois J, et al. Doppler US in patients with Crohn disease: vessel density in the diseased bowel reflects disease activity. Radiology 2000;217(3):787–91. [DOI] [PubMed] [Google Scholar]
- Szabo S, Sandor Z, Vincze A, Kusstatscher S, Sakoulas G. Role of basic fibroblast growth factor (bFGF) and platelet-derived growth factor (PDGF) in ulcer healing. Dig Dis Sci 1996;41:438. [Google Scholar]
- Szabo S, Sandor Z. Basic fibroblast growth factor and PDGF in GI diseases. Baillières Clin Gastroenterol 1996;10(1):97–112. [DOI] [PubMed] [Google Scholar]
- Szabo S, Vincze A, Sandor Z, Jadus M, Gombo Z, Pedram A, et al. Vascular approach to gastroduodenal ulceration: new studies with endothelins and VEGF. Dig Dis Sci 1998a;43:40S–5S. [PubMed] [Google Scholar]
- Szabo S, Sandor ZS, Al-Bassam J, Singh G, Vincze A. Despite different etiologies, virtually identical changes in local bFGF, PDGF and VEGF levels in duodenal ulceration and ulcerative colitis indicate similarities in healing. Gastroenterology 1998b;114 (suppl 5):G1228. [Google Scholar]
- Szabo S, Gombos Z, Sandor Z. Growth factors in gastrointestinal diseases. BioDrugs 1999;12(1):27–41. [DOI] [PubMed] [Google Scholar]
- Suzaki Y, Hamada K, Sho M, Ito T, Miyamoto K, Akashi S, et al. A potent antiangiogenic factor, endostatin prevents the development of asthma in a murine model. J Allergy Clin Immunol 2005;116(6):1220–7. [DOI] [PubMed] [Google Scholar]
- Thatch KA, Mendelson KG, Haber MM, Schwartz MZ. Growth factor manipulation of intestinal angiogenesis: a possible new paradigm in the management of inflammatory bowel disease. J Surg Res 2009;156(2):245–9. [DOI] [PubMed] [Google Scholar]
- Tolstanova G, Deng X, Khomenko T, Chen L, Sandor Z, Fox J, et al. Relationship between angiogenic and anti-angiogenic factors in the pathogenesis of inflammatory bowel disease. Gut 2007;56:A50. [Google Scholar]
- Tolstanova G, Khomenko T, Deng X, Chen L, Tarnawski A, Ahluwalia A, et al. Neutralizing anti-vascular endothelial growth factor (VEGF) antibody reduces severity of experimental ulcerative colitis in rats: direct evidence for the pathogenic role of VEGF. J Pharmacol Exp Ther 2009;328(3):749–57. [DOI] [PubMed] [Google Scholar]
- Vasiliauskas EA, Kam LY, Abreu-Martin MT, Hassard PV, Papadakis KA, Yang H, et al. An open-label pilot study of low-dose thalidomide in chronically active, steroid-dependent Crohn’s disease. Gastroenterology 1999;117(6):1278–87. [DOI] [PubMed] [Google Scholar]
- von Lampe B, Barthel B, Coupland SE, Riecken EO, Rosewicz S. Differential expression of matrix metalloproteinases and their tissue inhibitors in colon mucosa of patients with inflammatory bowel disease. Gut 2000;47(1):63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiercinska-Drapalo A, Jaroszewicz J, Parfieniuk A, Lapinski TW, Rogalska M, Prokopowicz D. Pigment epithelium-derived factor in ulcerative colitis: possible relationship with disease activity. Regul Pept 2007;140(1–2):1–4. [DOI] [PubMed] [Google Scholar]
- Yin G, Liu W, An P, Li P, Ding I, Planelles V, et al. Endostatin gene transfer inhibits joint angiogenesis and pannus formation in inflammatory arthritis. Mol Ther 2002;5(5 Pt 1):547–54. [DOI] [PubMed] [Google Scholar]