

Abstract
Owing to recent advances in proteomics analytical methods and bioinformatics capabilities there is a growing trend toward using these capabilities for the development of drugs to treat human disease, including target and drug evaluation, understanding mechanisms of drug action, and biomarker discovery. Currently, the genetic sequences of many major organisms are available, which have helped greatly in characterizing proteomes in model animal systems and humans. Through proteomics, global profiles of different disease states can be characterized (e.g. changes in types and relative levels as well as changes in PTMs such as glycosylation or phosphorylation). Although intracellular proteomics can provide a broad overview of physiology of cells and tissues, it has been difficult to quantify the low abundance proteins which can be important for understanding the diseased states and treatment progression. For this reason, there is increasing interest in coupling comparative proteomics methods with subcellular fractionation and enrichment techniques for membranes, nucleus, phosphoproteome, glycoproteome as well as low abundance serum proteins. In this review, we will provide examples of where the utilization of different proteomics-coupled enrichment techniques has aided target and biomarker discovery, understanding the drug targeting mechanism, and mAb discovery. Taken together, these improvements will help to provide a better understanding of the pathophysiology of various diseases including cancer, autoimmunity, inflammation, cardiovascular disease, and neurological conditions, and in the design and development of better medicines for treating these afflictions.
Keywords: Biomarker discovery, Drug target discovery, Exosomes, Membrane proteomics, mAb discovery
1. Introduction
With enhancements in proteomics techniques, there has been a surge in world-wide efforts aimed at large-scale protein analysis of biological samples. A global human proteome project is now underway to facilitate understanding of the relationship between physiological changes in organisms and protein abundance, subcellular localization, and functions in different tissues and environments [1]. Indeed, a large number of biomarkers for different diseases including cancer have been discovered in the past two decades using MS-based proteomics approaches [2, 3]. Proteomics has proven to be useful in cancer research because the complexity of tumorogenesis, cancer progression, tumor relapse, and metastasis may involve complex protein networks. The recent application of proteomics in cancer-related research is evident by the steadily increasing number of publications from 2000 to 2010 followed by sustained numbers through 2013, as shown in Fig. 1 (http://www.ncbi.nlm.nih.gov/pubmed). The acceleration of the use of genomics and transcriptomics in cancer research has been due to advancements in the core technologies used to sequence and analyze nucleic acid. As seen from Fig. 1, proteomics, while lower than genomics and transcriptomics, is still far exceeding other types of omics technologies, such as metabolomics, lipidomics, and glycomics. However, the recent advancements in MS technologies will hopefully accelerate the use of all of these areas in the coming years. Among the omics research community, integration of proteomics and other omics technologies such as RNA sequencing (RNA-seq) is desirable [4]. There is an ongoing effort on the integrative analysis of data types from multiple omics sources such as The Cancer Genome Atlas Project (TCGA) that integrates endometrial carcinoma data characterized by sources such as mRNA, protein, DNA methylation, and so on [5]. With increasing usage of multiple omics techniques, there is also a need for development of user-friendly tools capable of performing analysis of data from multiple omics sources [4].
Figure 1.

Number of pubmed publications per year between 2000 and 2013 containing keywords “Omics and Cancer,” “Genomics and Transcriptomics and Cancer,” “Proteomics and Cancer,” “Metabolomics and Cancer,” “Glycomics and Cancer,” and “Lipidomics and Cancer.”
The number of publications has increased over the past 10 years in part because of the need in many areas as illustrated in Fig. 2. MS-based proteomics techniques involving different ionization methods such as EI (electron ionization), ESI, and MALDI with quantification power can now be used to interpret the mechanism of action of various drugs, elucidate novel biomarkers, identify new targets, and provide antigen-specific antibody isolation. Within proteomics research, there is an increasing focus on changes in specific groups of proteins which may be present at relatively low levels. For example, secreted proteins, also referred to as the secretome, can play important roles in cell–cell communication, growth, and, as they can reflect the various stages of pathophysiological conditions [6], represent a useful source of biomarkers and potential targets to treat disease. Various proteomics approaches have been used to discover biomarkers in blood/plasma, body fluids, and tissues [7,8], exosomes [9], and conditioned media from cultured or primary cells [10].
Figure 2.
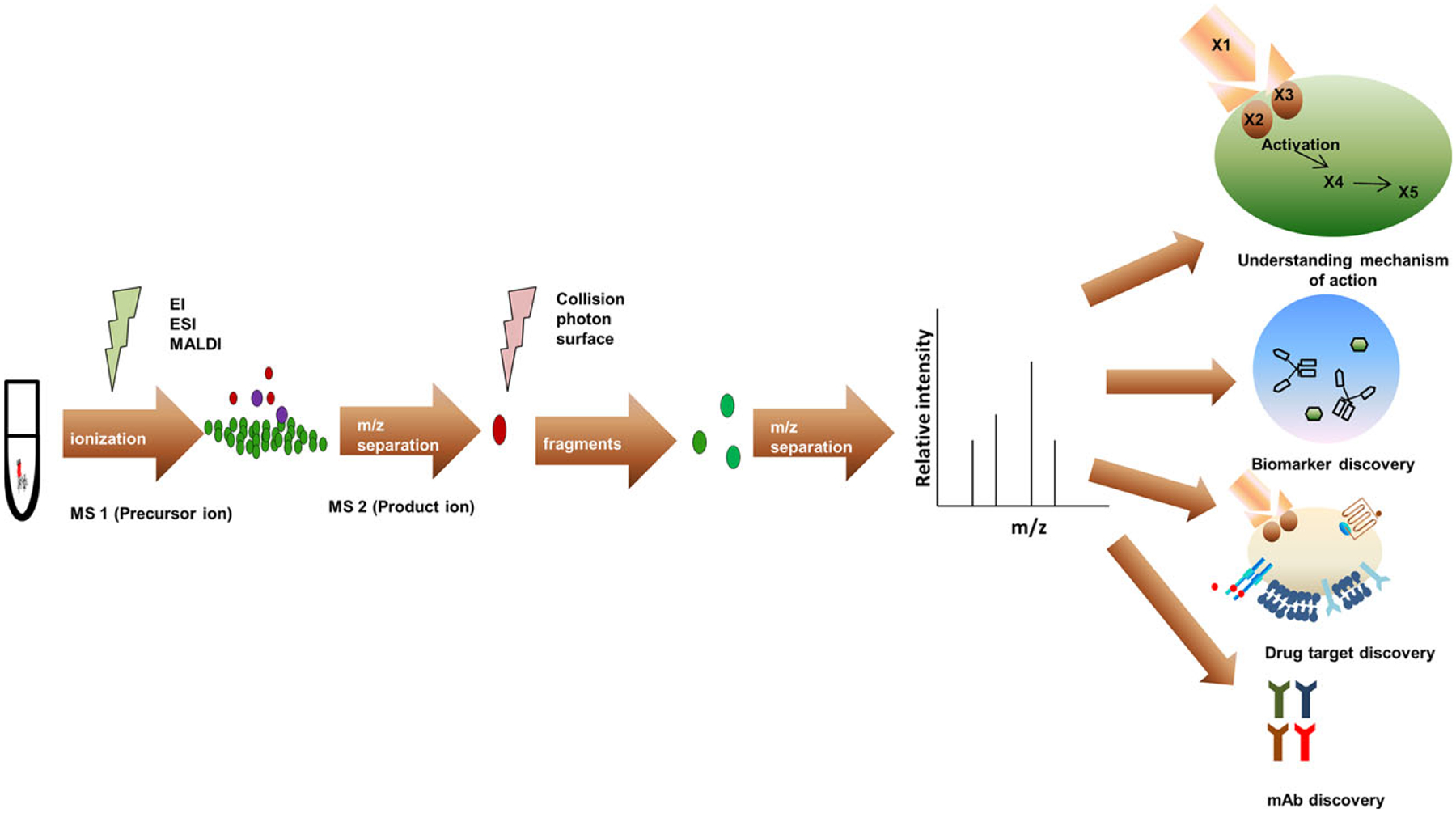
Applications of MS proteomics methodologies include understanding mechanisms of drug action, identifying novel biomarkers, elucidating drug targets, and monoclonal antibody discovery.
In addition to the secretome, post-translationally modified proteins including glycosylated [11, 12] and phosphorylated ones represent another subclass of targets since they are involved in regulating biological processes. Dysregulation of kinase signaling pathways is commonly associated with various cancers [13] and gastrointestinal stromal tumors [14]. Phosphoproteomic approaches can also assist in identifying appropriate therapeutic targets as well as elucidating drug action on pathophysiological pathways. In addition to MS [15], other high throughput proteomic technologies have been developed for studying phosphorylation including peptide arrays [16, 17], reverse-phase protein microarrays [18], and antibody arrays [19].
This review will explore the multiple identification and enrichment proteomics methods, as shown in Fig. 3, used for target discovery, biomarker discovery, and understanding the mechanism of action of drugs.
Figure 3.

Different techniques used in proteome profiling including (A) serum depletion method, (B) titanium dioxide incubation for phosphopeptides isolation, (C) glycopeptides capturing, (D) colloidal silica coupling for membrane proteins, (E) exosome isolation, and (F) secretome isolation.
2. Target discovery
Comparative quantification of low abundance and complex proteins is becoming an attractive field since it can provide novel drug targets for therapeutic intervention and to meet these developments, multiple proteomic and associated technologies are being implemented. As shown in Fig. 3, the serum depletion method reduces the complexity of a sample by removing high abundance proteins, thereby enriching the low abundance proteins. Low abundance phosphopeptides can be enriched using titanium dioxide (TiO2) incubation as a result of the high affinity of TiO2 to negatively charged phosphopeptides. Similarly, hydrazide chemistry can be used to capture glycopeptides and cationic nanoparticles such as colloidal silica can provide the high level enrichment of low abundance membrane proteins.
In particular, development of an effective strategy for membrane protein analysis is highly relevant to diseases where cell surface receptor signaling plays a vital role. For example, cell surface proteins have attracted substantial interest from cancer researchers in areas ranging from molecular diagnosis to therapy.
However, comparison of membrane proteins from normal and diseased tissues using standard extraction methods that also isolate whole cell components makes this task difficult. For this reason, recent advances in coupling proteomics methods with membrane protein enrichment protocols [20–22] are especially useful for novel target discovery.
2.1. Membrane proteomics
Membrane proteins play important roles in numerous diseases and can be potential drug targets. Various methods, as discussed below and in Table 1, have recently been developed that could potentially overcome challenges in studying membrane proteomics.
Table 1.
A summary of various methods used in membrane proteome enrichment
| Membrane enrichment method | Principle | Membrane protein percentage based on GO annotation | Transmembrane proteins percentage | Reference |
|---|---|---|---|---|
| Colloidal Silica | Electrostatic coupling between plasma membrane and cationic silica | 65% | 44% (UniProt and Human Protein Reference database) | [52] |
| Biotinylation | Affinity between biotin and avidin homologous proteins | 51% | NA | [27] |
| Ultracentrifugation | Separation based on density | 70% | NA | [24] |
| Glycoproteomics | Chemical coupling | 41% | 34% (KEGG pathways database) | [47] |
2.1.1. Ultracentrifugation techniques for membrane proteome enrichment
Membrane proteins can be enriched by the removal of cytosolic proteins under high pH conditions by using ultracentrifugation. For example, the membrane proteome of postmortem brain tissues from normal and Alzheimer’s disease cases were compared using two simultaneous ultracentrifugation procedures. The simultaneous centrifugation and treatment with high pH removed the loosely associated proteins to improve the membrane proteome quality [23].
In another application, proteome profiles of plasma membrane enriched microdomains (MD) involved in cell signaling, transport, and proliferation from human renal cell carcinoma (RCC) and adjacent normal kidney (ANK) were compared using gradient ultracentrifugation [24]. Ninety-three proteins were identified in MD isolated from RCC and 98 proteins in MD isolated from ANK. Western blot analysis indicated differentially expressed proteins such as Thy1, VDAC, and DPEP, which may represent potential biomarkers of cancer progression [24]. In another study, the apical plasma membrane proteome of a multinuclear synctiotro-phoblast was enriched using ultracentrifugation and sucrose gradient centrifugation in conjunction with 1D SDS-PAGE and ESI-MS [25]. Two hundred ninety-six nonredundant proteins were identified, of which about 60% were integral and peripheral membrane proteins [25].
2.1.2. Biotinylation coupled proteomics for cell surface proteome enrichment
Biotinylation of whole cells can be used to enrich low-abundance cell surface protein isolation by taking advantage of the high affinity of avidin, streptavidin, or neutravidin for biotin. This method works by conjugation of multiple biotin molecules to target proteins of interest to form a biotin complex. Surface biotinylation with reactive chemical derivatives of biotin followed by purification on avidin beads allows selective cell surface proteome profiling. The method often involves resuspending whole cells in a solution of a biotin derivative such as sulfo-NHS-SS-biotin. After the capture of the biotin complex on streptavidin or avidin-based supports, the biotinylated proteins can be cleaved using reducing reagents [26].Combining cell-surface protein enrichment using biotinylation and iTRAQ technology has been used to detect differentially expressed proteins in endometrial cancer and healthy cells [27]. Out of 272 overexpressed proteins, overexpression of bone marrow stromal antigen 2 (BST2) on cancer cells was investigated further. Administering a monoclonal antibody targeting BST2 (anti-BST2) resulted in growth reduction of BST2-positive endometrial cancer cells in SCID mice identifying BST2 as a possible therapeutic target [27]. In this way, biotinylation coupled with MS profiling has facilitated the identification of critical, low-abundance membrane proteins [28].
2.1.3. Glycoproteome enrichment
PTMs including glycosylation offer high diagnostic and therapeutic potential and thus glycoproteomics is another area for target identification [29–39]. However, there are many challenges to achieving efficient analysis of a complex glycoproteome containing wide concentration ranges in a complex biological environment due to the severe masking effects of highly abundant proteins and the alteration in the stoichiometric ratios of glycosylated forms of proteins. To overcome these obstacles, various enrichment strategies for glycoproteins have been developed [40]. As glycoproteins are localized on the cell surface or are secreted, N-glycoproteome enrichment methods facilitate identification and expression comparisons of membrane proteins. There are multiple glycoproteome enrichment methodologies, some of which are:
Gravity-flow columns involve use of a glycoprotein enrichment resin (such as a phenylboronic acid-based resin containing a ligand, attached to agarose beads, which binds to sugar residues of glycoproteins [41]) to capture the glycoproteins.
Cell surface capture involves treating glycoproteins of the plasma membrane by oxidizing cells with sodium-meta-periodate followed by covalent chemical labeling with biocytin hydrazide (BH), which is a biotin containing hydrazide. The BH-labeled glycopeptides can then be analyzed by LC-MS/MS for peptide identification [42].
For enrichment of N-linked glycopeptides, SPE of N-linked glycopeptides (SPEG) is highly preferred for both identification and quantification [43].The SPEG method involves proteolysis of proteins into peptides followed by coupling oxidized glycosylated peptides to hydrazide beads and removing non-glycosylated peptides. The amino-termini of glycopeptides can be labeled with succinic anhydride or other reagents followed by peptide-N-glycosidase F (PNGase F) to release N-linked glycopeptides, which can be identified and quantified by MS [44].
A highly selective glycopeptide enrichment method combining several of these glycoproteomics methods including cell surface capture and hydrazide derivatization has been applied to isolate cell surface proteins [45]. This method has several advantages over other methods as it provides complete solubilization of membrane proteins, enriches glycopeptides instead of glycoproteins that eliminates potential steric hindrance during capture, ensures robustness and tolerance to stringent washes, decreases sample loss, and offers shorter sample processing times [45]. Processing steps include cell lysis, microsomal fraction, denaturation and digestion, glycopeptide capture, LC-MS, and bioinformatics analysis. The success of this method, particularly suited to in vitro cell cultures, relies on complete digestion of the samples [45].
In another study, a modified cell surface capture strategy involved oxidizing the oligosaccharides on cell surface proteins followed by cell lysis and coupling oxidized proteins onto a hydrazide resin followed by MS analysis for quantification of glycoproteins and other cell surface proteins [46]. This strategy was tested on two biological replicates of chang liver and HepG2 human cell lines. Out of 341 identified glycoproteins, 33 exhibited significant changes in expression between the two cell lines. Western blot was used to validate the higher expression of extracellular matrix metalloproteinase inducer (EMMPRIN) and basal cell adhesion molecule in HepG2 cells, both of which are associated with the malignant potential of tumor and liver cancer [46].
Recently, a detailed large-scale LC-MS/MS data set on the membrane proteome and N-glycoproteome of the BV-2 microglia line was reported [47]. This study employed multiple strategies including crude membrane fractionation, filter-aided sample preparation (FASP)-based differential sample preparation, and N-glyco-FASP-based glycopeptide enrichment. A total of 6928 unique protein groups and 1450 unique N-glycosites and 760 unique glycoproteins were identified [47]. Among the identified glycoproteins, receptors, transporters, peptidases, and ion-binding proteins were enriched [47].
Quantitative glycoproteomics based on hydrazide chemistry was used to study cell surface and serum proteins in human serum and a prostate cancer epithelial cell line [48]. This method was found to be useful in reducing serum sample complexity with detection of 2.5 peptides per protein on average. For the prostate cancer epithelial cell line, this method could identify proteins such as HSPG2 and SSRA that were not previously identified by a 2DE gel method [48].
A targeted proteomics approach involving galactose oxidase and aniline-catalyzed oxime ligation (GAL) and periodate oxidation and aniline-catalyzed oxime ligation (PAL) was used for chemical tagging and identification of glycosylation sites on proteins of cells with an altered sialylation status [49]. Immobilized streptavidin was used to pull down the PAL and GAL labeled and biotinylated glycoproteome and the glycopeptides were released with N-glycosidase treatment to be analyzed in LC-MS/MS [49]. One hundred and eight nonredundant glycoproteins were identified by combining both methods [49].
2.1.4. Enrichment of plasma membrane proteome with cationic nanoparticles
Isolation and study of the plasma membrane proteome is especially challenging due to the low abundance and hydrophobicity of the proteins. In addition, a variety of post-translational and chemical modifications leads to a dynamic population of these proteins on the cell surface. Cationic nanoparticles synthesized from Fe3O4 (magnetite) coated with Al2O3 were used to enrich the low abundance plasma membrane proteome of human multiple myeloma RPMI 8226 cells for analysis of integral proteins and proteins from both the inner and outer surfaces [50]. Fe3O4 (magnetite) nanoparticles were synthesized and coated with Al2O3 and capture of membrane proteins was then compared to commercial aluminosilicate particles and Al2O3-coated SiO2 nanoparticles. Plasma membrane proteome enrichment provided by the Fe3O4/Al2O3 pellicles was statistically significant and all three cationic nanoparticle pellicles (Fe3O4/Al2O3, aluminosilicate, and SiO2/Al2O3) exhibited a strong enhancement of plasma membrane and transmembrane protein content relative to proteins in the whole cell lysate as classified by UniProt annotation and the web-based TMHMM tool [50]. Plasma membrane proteins represented 27.6% of the sample detected by the Fe3O4/Al2O3 pellicles method and 12% were transmembrane proteins, highest among the methods compared in this study [50].
Colloidal silica coupling relies on interactions between the negatively charged membranes to cationic colloidal particles. The method involves washing the cells with ice-cold MES-buffered saline followed by adding an ice-cold silica bead solution and incubating (for adherent cells) or gentle rocking (for suspension cells) prior to cell lysis, density gradient centrifugation, and protein purification [51].
Cationic colloidal silica particles based proteomics techniques have been utilized for the isolation of plasma membranes obtained from intact syncytiotrophoblast of human placenta [52], resulting in detection of 340 nonredundant proteins in the apical plasma membrane fraction. Proteins not previously known to be in the plasma membrane in the syncytiotrophoblast of human placenta include myosin proteins (11, 14, Ib, Id, Ie), nicalin, flotillin-1, and receptor expression enhancing protein 5 [52].
A cationic silica-polyanion (CSP) strategy was used to enrich plasma membrane from freshly isolated C57 mouse hepatocytes [53]. CSP was shown to provide a better yield compared with the biotin-avidin method. The CSP method yielded 185 nonredundant proteins while biotin-avidin isolated 49 nonredundant proteins, with 45 proteins identified by both methods [53].
A density perturbation technique modified from the silica nanoparticles subfraction strategy for in vivo studies in combination with MS was used to identify the surface proteome of liver sinusoidal endothelial cells [54]. The proteins were separated by SDS-PAGE and identified by with LC-MS/MS (GeLC-MS/MS). A total of 837 different proteins were found including 450 with membrane origin, in addition to contaminants from mitochondria, ribosomes, and the nucleus [54].
Although the progress in target discovery using proteomics has been substantial, serious challenges still remain. A large gap exists between the emerging new targets from abundant proteomics studies on various diseases and actual drug development, along with a lack of validation of specific roles played by these targets in the diseases. We believe that validation should incorporate more functional studies such as in vitro gene expression manipulation and gene knockouts along with clinical validation of identified novel targets. The recent advancements of targeted proteomics approaches such as PSAQ (protein standard for absolute quantification) and MRM techniques will hopefully accelerate both the validation and usage of proteomics targets and biomarkers in the pipeline of drug discovery, development, and clinical evaluation.
3. Biomarker discovery
Identification of both cell surface and serum markers is important for diagnostic and prognostic purposes when treating a disease. With the development of improved mass spectrometric techniques [55–60], cell surface and secreted proteins are more accessible for characterization. Analysis of cell surface protein is often made difficult by the low abundance of these proteins; however the recent application of plasma membrane enrichment [61] and glycoproteome enrichment [44,62,63] techniques have been helpful for the identification of greater numbers of potential biomarkers.
Serum biomarker discovery can be enhanced by the removal of high abundance proteins, such as albumin and immunoglobulins, so that low abundance proteins can be detected. One approach for serum depletion is to use commercially available kits such as the multiple affinity removal system (MARS—Agilent, Santa Clara, CA) spin columns or the Amersham (GE Healthcare, Piscataway, NJ) albumin and IgG removal kit [64].
Holewinski et al. tested a commercially available affinity capture reagent from Protea Biosciences and compared its efficiency and reproducibility to four other commercially available albumin depletion methods [65]. Two methods showed an albumin depletion efficiency of more than 97% for both serum and cerebrospinal fluid. Using LC-MS/MS analyses, they subsequently found 45 novel proteins in serum [65] and also showed that albumin isolation from serum can eliminate the need for fractionation to lower sample processing time [65].
Another study reported a method that coupled denaturing ultrafiltration with reverse phase fractionation and MS for characterization of low-molecular-weight proteins and peptides in serum and plasma samples [66]. The enriched peptides were analyzed by RP-LC combined with MALDI-MS/MS characterization of the analytes, resulting in identification of 250 native peptides from 50 different proteins [66].
A study focused on exploring protein biomarkers in serum from rheumatoid arthritis patients treated with infliximab, an anti-tumor necrosis factor monoclonal antibody, employed a quantitative proteomics approach using 8-plex iTRAQ labeling [67]. A combination of depletion of the most abundant serum proteins, 2D-LC fractionation, protein identification, and relative quantification with a hybrid Orbitrap mass spectrometer was used to identify 235 proteins with high confidence [67]. Fourteen proteins that were significantly abundant in nonresponder patients as compared to the responder patients were identified as potential biomarkers. Some of the proteins showing significant changes and thus representing potential biomarkers were C4B-alpha chain, complement factor H-related protein 4, mannan-binding lectin, serine protease 2, and inter alpha trypsin inhibitor heavy chain H1 and H2 [67].
A plasma-proteomics based approach employing an 8-plex iTRAQ technique was used to study the association between nutrients and proteins in the plasma of 500 subjects after immune-depletion of six high abundance proteins including albumin and transferrin using an affinity removal LC column [68]. More than 4700 nonredundant proteins were identified including more than 450 proteins identified as extracellular, secreted, membrane, or lipoprotein associated. A strong correlation between vitamin A and retinol binding protein 4 (RBP4) was observed. Overall, the method demonstrated an approach for elucidating and quantifying protein biomarkers related to the nutrition environment [68].
In another study, Miltenyi Biotech’s MACS LS column was used for proteomic analysis of human prostate cancer cell line DU145 in order to identify potential biomarkers [69]. The DU154 prostate cancer cell line was isolated into CD44+ or CD44− cells using MACS and subsequent analysis of the differential expression of proteins between CD44+ and CD44− was carried out using 2DE and LC-MS/MS. The proteins cofilin and annexin A5, associated with proliferation or metastasis in cancer, were found to be positively correlated with CD44 expression [69].
Development of glycoproteome enrichment techniques has been highly effective for biomarker discovery through a comparison of the biologically significant glycoproteins between the diseased and normal subjects. However, development of user-friendly bioinformatics tools is required in order to enhance structural and compositional analysis of glycans. For efficient screening of biomarkers, precise quantification of glycosylation site occupancy at both relative and absolute levels is required. Indeed the recent advances in high-throughput glycoproteomics should provide new leads for potential biomarker identification.
4. Understanding the mechanism of action of drugs and disease
A substantial number of biological drugs and monoclonal antibodies affect signaling and phosphorylation events where they engage cell surface receptors. For this reason, the identification and quantification of protein phosphorylation sites are important areas of study in the treatment of cancer and other diseases, such as diabetes, with biologic drugs. For example, identifying phosphotyrosine site occupancy can help in the diagnosis of these diseases [70–74] and provide a better understanding of the behavior of target cells upon treatment with drugs [75]. For this reason, a variety of phosphoproteomics techniques such as application of antiphosphotyrosine affinity chromatography, immobilized metal affinity chromatography, and TiO2 enrichment columns [72, 76] have been used to study the changes of phospho-peptide and phospho-protein states in cells.
Protein phosphorylation facilitates information transfer in cells and changes can contribute to the pathophysiology of diseases including cancer, inflammation, and metabolic disorders [13, 77–79]. One study investigated phosphorylation of serine, threonine, tyrosine, and histidine residues by protein kinases and reported a strategy using peptide arrays and motif-specific antibodies to identify and characterize substrate sequences for protein kinase A (PKA) [80]. It was found that protein kinase D and microtubule-associated protein-regulating kinase 3 can both be regulated by PKA. They also showed that the adaptor protein RIL is a PKA substrate that is phosphorylated on serine, which was predicted to regulate cell growth [80].
In order to understand the relationship between pancreatic cancer and alterations in proteomics and signaling pathways, a SILAC-based quantitative proteomics approach was employed to compare cells from three sites of metastasis (liver, lung, and peritoneum), which provided the proteome and tyrosine phosphoproteome of these tumors [81]. Approximately 42% of the proteome was found to be highly variable when compared across any two sites of metastasis in terms of receptor and signal transduction activity, suggesting that regulation of these tumors was different depending upon anatomical location [81].
Using a computational approach, in silico motif sequences were generated and compared with an experimental database—the Human Protein Reference Database (HPRD). Two hundred seven phosphotyrosine and 960 phosphoserine/threonine motifs were determined by this method [82]. The HPRD [83, 84] along with Human proteinpedia [85, 86] are centralized resources for phosphorylation site analyses and can be applied in a systems biology approach to determine the role of protein phosphorylation in protein function, cell signaling, biological processes, and their implications in the human diseases [87]. It contains experimental human phosphoproteome data from various methods including 32P-labeled ATP followed by SDS-PAGE or HPLC followed by Edman sequencing to determine the site of phosphorylation [88]; phosphoproteome enrichment and LC-MS [70, 89–94]; high throughput analysis such as SILAC combined with titanium dioxide chromatography or IMAC microspheres for phosphoproteome enrichment [95–98]; or using pTyr-100 and 4G10 phosphotyrosine antibodies to enrich the phosphotyrosine proteome [99].
More work is required for the quantification of the phosphoproteome, as currently the work is mostly focused merely on identifying the phophosites.
5. Exosomal proteome profiling
With the improvement in exosome purification strategies together with MS-based proteomics tools with higher sensitivity and mass accuracy, exosomal profiling has proven to be a useful technique in studying disease states including cancer [100–117], AIDS [118, 119], Leishmaniasis [120], age-related macular degeneration [121], and neurodegenerative diseases [122]. Interestingly, the exosomal proteome is comprised of proteins found both in membrane and cytosol as well proteins with distinct functions in different cells. Proteomics has been very useful in cataloging the exosomes (extracellular membrane vesicles) that are versatile mediators of intercellular communication, pathogenesis, and might be useful for drug and gene delivery to a variety of tissues including cerebrospinal fluid [123], hepatocytes [124], and glioblastomas [125].
Exosomes derived from several types of tumor cells such as breast adenocarcinoma [126], colorectal cancer [127, 128], mammary adenocarcinoma [129], melanoma [130,131], mesothelioma [132], and brain tumor [101, 133] have been characterized by isolation strategies including differential centrifugation, filtration, sucrose density gradient, and immunobeads, combined with MS-based proteomic strategies such as 1D and 2D LC-MS/MS and MALDI-TOF/TOF MS [134]. A comprehensive review of studies that have used proteomic methods to characterize exosomes derived from in vitro sources and biological fluids has been previously reported [135].
Various methods for exosomes’ isolation [136] include:
Ultracentrifugation: Requires centrifugation at 100 000 × g followed by resuspension in PBS and recentrifugation.
Size exclusion chromatography: Involves application of samples to a 2% agarose-based gel column and eluting isocratically with PBS.
Magnetic beads: Requires adsorption to anti-EpCAM antibodies coupled to magnetic microbeads followed by magnetic separation that results in the exosomes attached to the magnetic beads. The magnetic beads can be washed with TBS and exosomes can be extracted using a Trizol extraction protocol.
ExoQuick™ precipitation is another widely used exosomes isolation method that involves addition of ExoQuick™ precipitation solution and centrifugation. When the supernatant is aspirated, the exosomal pellet can be extracted using Trizol extraction procedures. While the mechanism of action of ExoQuick™ is not disclosed by the manufacturer, it is commonly used by some researchers [137] and can provide high yields and purity when compared to other methods [138].
In one study, ultracentrifugation and filtration techniques were used for isolating exosomes from human neuroblastoma cell lines for proteomic analysis [139]. A discrete set of molecules including tetraspanins, prominin-1 (CD133), and basigin (CD147) were found to be expressed in the exosomes that suggests the important role of exosomes in the modulation of the tumor microenvironment and indicates their potential utilization as a tumor biomarker [139].
Ultracentrifugation was used for exploring the urinary exosome in Zucker diabetic fatty rats to study diabetic nephropathy and related renal disease [140]. Two-hundred eighty-six proteins comprised mainly of membrane proteins were identified with many associated with functions such as transport, cellular communication, and cellular adhesion. A renal protein, Xaa-pro dipeptidase, which is linked with collagen breakdown was found to be upregulated and major urinary protein 1 was downregulated. These differentially regulated proteins may represent biomarkers for understanding metabolic changes during the progression of diabetes in obese diabetic mice models [140]. In another study, the proteome of exosomes shed by myeloid-derived suppressor cells (MDSC) was explored using ultracentrifugation [141]. Under an increased inflammatory response, proteins such as GTP and ATP-binding proteins (ATP-citrate synthase, ADP-ribosylation factor 1) were found to be in high abundance by MS analysis. Additionally, proinflammatory proteins S100A8 and S100A9 were found to be abundant in MDSC-derived exosomes [141].
One of the challenges in isolating low-density exosomes by ultracentrifugation is the variability between runs. Although relatively large starting volumes can be used [142], issues are still present for actual clinical samples that are low in volume. The exosomes can also be trapped on antibody-coated magnetic beads that have advantages such as immunoblotting, flow cytometry, and electron microscopy analysis of bead-exosome complexes, but this method is problematic for large sample volumes [143]. Size-exclusion chromatography, ultracentrifugation, and ExoQuick™ precipitation are not useful for preferentially isolating exosomes such as tumor-specific derived exosomes, which require immunoaffinity-based approaches [138]. In coming years, an improvement in MS-based proteomics and exosomal isolation and enrichment methods will shed light on fundamental biology questions such as the signaling pathways activated by exosomes in different diseases.
6. Secretome profiling
The secretome can be isolated from serum-starved cells or SILAC labeling can be used to quantitatively compare the secreted proteins of different cells [144]. A study on the secretome of differentiating primary adipocytes resulted in identification of 420 differentially expressed proteins including collagen triple helix repeat containing 1, cytokine receptor-like factor 1, glypican-1, hepatoma-derived growth factor, SPARC-related modular calcium-binding protein 1, SPOCK 1, and sushi repeat-containing protein [145].
In another report, SILAC-based quantitative proteomics was employed to elucidate the differences in the secretome of neoplastic and nonneoplastic gastric epithelial cells [144]. Out of 263 overexpressed proteins in cancer-driven cell lines, three novel proteins candidates—proprotein convertase subtilisin/kexin type 9 (PCSK9) with 13-fold overexpression, lectin mannose binding 2 (LMAN2) with sixfold overexpression, and PDGFA associated protein 1 (PDAP1) with 5.2-fold overexpression, represent possible biomarkers for the progression of gastric cancer [144].
7. mAb discovery
Proteomics methods are now being extensively incorporated into the mAb discovery process as a supplement to traditional hybridoma and phage display technologies. Indeed, a proteomics approach based on LC-MS/MS for identifying an antigen-specific antibody from sera and B-cell sources of immunized rabbits and mice was coupled with next-generation sequencing approaches [146]. This approach combined proteomics with enrichment of polyclonal antibodies via an affinity purification method. The enriched polyclonal antibodies were subsequently analyzed by LC-MS/MS. This integrated approach four rabbit antibodies and one mouse antibody [146]. Wine et al. also used antigen-affinity chromatography for the enrichment of antibodies followed by LC-high resolution MS/MS for discovering the antigen-specific antibody composition of a polyclonal serum response after immunization [147].
8. Bioinformatics and proteomics
With large amounts of data generated by proteomics studies, software demands and opportunities are emerging to facilitate and enhance data analysis such as: (1) compatibility with MS technology to support the data format and fragmentation patterns, (2) methods for identification of peptides and posttranslationally modified peptides, (3) application of known peptide libraries, (4) algorithms that increase speed and quality of analysis, and (5) user-friendly formats all of which will facilitate the widespread use of proteomics. Some of the most commonly used search engines for peptide and protein identification for MS-based data are Mascot [148], X!Tandem [149], SEQUEST [150], MyriMatch [http://forge.fenchurch.mc.vanderbilt.edu/scm/viewvc.php/*checkout*/trunk/doc/index.html?root=myrimatch.], and TagRecon [http://forge.fenchurch.mc.vanderbilt.edu/scm/viewvc.php/*checkout*/trunk/doc/index.html?root=tagrecon]. TagRecon allows users to identify proteins even with unexpected mutations and PTMs commonly observed in disease states [151]. After data analysis, visualization of the results is an important step toward interpretation. Most large software packages offer a variety of ways of visually displaying the data input and output, such as spectromania [152] that provides 2D, stacked chromatographic display of input spectra. It also allows a set of spectra from different packages to be merged into new and averaged spectra (with less noise). Other such visualization tools are MSight [153] and MassView [154].
A variety of bioinformatics tools are also available for biological interpretation of these high-throughput data. Tools such as KEGG (kyoto encyclopedia of genes and genomes) [155, 156], IPA (ingenuity pathway analysis—www.ingenuity.com), GO (gene ontology) [157], cytoscape [158], and DAVID (database for annotation, visualization, and integrated discovery) [159, 160] are widely used for interpretation of quantitative proteomics data. In addition to these, publically available software are present to address the localization of the proteins in a cell. For instance, TMHMM (http://www.cbs.dtu.dk/services/TMHMM-2.0/) developed by the Center for Biological Sequence Analysis in Denmark uses a hidden Markov model in order to predict transmembrane helices of the membrane proteins [161]. Phobius (http://phobius.sbc.su.se/), similar software developed at the Center for Genomics and Bioinformatics in the Karolinska Institute in Stockholm, Sweden [162], predicts signal peptides and regions of transmembrane protein sequence. In addition to Phobius, SignalP, and TargetP are two other bioinformatics tools used to predict the presence of signal peptides in a protein sequence. SignalP (http://www.cbs.dtu.dk/services/SignalP/) was developed at the Center for Biological Sequence Analysis in Denmark and is based on neural network algorithms. TargetP (http://www.cbs.dtu.dk/services/TargetP/) predicts signal peptides in protein sequences as well as their subcellular location with a success rate of 90% [163]. For identifying secreted proteins, SecretomeP 2.0 server (http://www.cbs.dtu.dk/services/SecretomeP/) uses neural networks to identify secreted proteins by nonclassical pathway i.e., the proteins not containing an N-terminal signal peptide [164]. WoLF PSORT (http://wolfpsort.org/) provides protein subcellular localization using PSORT features for prediction [165, 166]. Secreted Protein Database (SPD-http://spd.cbi.pku.edu.cn/) contains secreted proteins from human, mouse, and rat proteomes, including sequences from SwissProt, Trembl, Ensembl, and RefSeq [167].
Despite the tremendous progress in software applications, bioinformatics for MS-based proteomics still faces numerous needs such as user-friendly databases and programs, methods to reduce protein interference for quantification, tools for peptide level statistical analysis [168], and overall fully integrated software packages including capabilities provided by R (R Development Core Team 2008) [169]. With development and use of cloud-based computational resources, more proteomic data is likely to be made publically available, thereby giving open-source platforms a boost. In the future, MS instrumentation is likely to become even faster, more robust, and more accurate; equally powerful and enhanced bioinformatics approaches will be required to keep pace.
9. Conclusions
The potential of proteomics is now being realized in order to decipher the pathophysiology of various diseases and to help in the design of novel treatment strategies. Due to significant improvements in technology, powerful proteomics capabilities are emerging to provide opportunities for the discovery of new biomarkers and targets for drug discovery and development. One of the critical barriers to discovery is the challenge associated with identifying biomarkers or targets that are present in low abundance. In this review, we describe a variety of the emerging technologies that can be applied to detect and characterize lower abundance proteins that may be important in biomedicine. Different analytical techniques such as ultracentrifugation, biotinylation, colloidal silica, size exclusion chromatography, ExoQuick™ precipitation, hydrazide chemistry, and titanium dioxide incubation have been coupled with LC-MS/MS in order to achieve greater coverage of the subcellular proteome. These approaches often facilitate enrichment of the membrane proteome, glycoproteome, phosphoproteome, secretome, and exosome.
However, there is still much work needed in interpreting the enormous amounts of data generated from these improved techniques and in the development of easy to use software and robust databases. With the increasing incorporation of new high-throughput technologies, tremendous opportunities exist to expand our capabilities for discovery of new and important biomarkers and targets, all with the ultimate goal of better understanding disease and in developing better medicines to treat patients.
Abbreviations:
- CSP
cationic silica-polyanion
- FASP
filter-aided sample preparation
- GAL
galactose oxidase and aniline-catalyzed oxime ligation
- HPRD
Human Protein Reference Database
- MDSC
myeloid-derived suppressor cells
- PAL
periodate oxidation and aniline-catalyzed oxime ligation
- PKA
protein kinase A
- PDGFA
platelet-derived growth factor alpha polypeptide
- SCID
severe combined immunodeficiency
- SPARC
secreted protein, acidic, cysteine-rich
- TMHMM
transmembrane hidden Markov model
Footnotes
The authors have declared no conflict of interest.
References
- [1].Legrain P, Aebersold R, Archakov A, Bairoch A et al. , The human proteome project: current state and future direction. Mol. Cell. Proteomics 2011, 10, M111 009993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Mermelekas G, Zoidakis J, Mass spectrometry-based membrane proteomics in cancer biomarker discovery. Expert. Rev. Mol. Diagn 2014, 14, 549–563. [DOI] [PubMed] [Google Scholar]
- [3].Jimenez CR, Verheul HM, Mass spectrometry-based proteomics: from cancer biology to protein biomarkers, drug targets, and clinical applications. Am. Soc. Clin. Oncol. Educ. Book 2014, 34, e504–e510. [DOI] [PubMed] [Google Scholar]
- [4].Gomez-Cabrero D, Abugessaisa I, Maier D, Teschendorff A et al. , Data integration in the era of omics: current and future challenges. BMC Syst. Biol 2014, 8 (Suppl 2), I1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kandoth C, Schultz N, Cherniack AD, Akbani R et al. , Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Doroudgar S, Glembotski CC, The cardiokine story unfolds: ischemic stress-induced protein secretion in the heart. Trends Mol. Med 2011, 17, 207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Celis JE, Gromov P, Cabezon T, Moreira JM et al. , Proteomic characterization of the interstitial fluid perfusing the breast tumor microenvironment: a novel resource for biomarker and therapeutic target discovery. Mol. Cell Proteomics 2004, 3, 327–344. [DOI] [PubMed] [Google Scholar]
- [8].Gromov P, Gromova I, Bunkenborg J, Cabezon T et al. , Up-regulated proteins in the fluid bathing the tumour cell microenvironment as potential serological markers for early detection of cancer of the breast. Mol. Oncol 2010, 4, 65–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Raimondo F, Morosi L, Chinello C, Magni F et al. , Advances in membranous vesicle and exosome proteomics improving biological understanding and biomarker discovery. Proteomics 2011, 11, 709–720. [DOI] [PubMed] [Google Scholar]
- [10].Dowling P, Clynes M, Conditioned media from cell lines: a complementary model to clinical specimens for the discovery of disease-specific biomarkers. Proteomics 2011, 11, 794–804. [DOI] [PubMed] [Google Scholar]
- [11].Kolarich D, Lepenies B, Seeberger PH, Glycomics, glycoproteomics and the immune system. Curr. Opin. Chem. Biol 2012, 16, 214–220. [DOI] [PubMed] [Google Scholar]
- [12].Kim EH, Misek DE, Glycoproteomics-based identification of cancer biomarkers. Int. J. Proteomics 2011, 2011, 601937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hanahan D, Weinberg RA, The hallmarks of cancer. Cell 2000, 100, 57–70. [DOI] [PubMed] [Google Scholar]
- [14].Corless CL, Fletcher JA, Heinrich MC, Biology of gastrointestinal stromal tumors. J. Clin. Oncol 2004, 22, 3813–3825. [DOI] [PubMed] [Google Scholar]
- [15].Choudhary C, Olsen JV, Brandts C, Cox J et al. , Mislocalized activation of oncogenic RTKs switches downstream signaling outcomes. Mol. Cell 2009, 36, 326–339. [DOI] [PubMed] [Google Scholar]
- [16].Amanchy R, Kalume DE, Iwahori A, Zhong J et al. , Phosphoproteome analysis of HeLa cells using stable isotope labeling with amino acids in cell culture (SILAC). J. Proteome Res 2005, 4, 1661–1671. [DOI] [PubMed] [Google Scholar]
- [17].Amanchy R, Zhong J, Molina H, Chaerkady R et al. , Identification of c-Src tyrosine kinase substrates using mass spectrometry and peptide microarrays. J. Proteome Res 2008, 7, 3900–3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Gulmann C, Sheehan KM, Conroy RM, Wulfkuhle JD et al. , Quantitative cell signalling analysis reveals down-regulation of MAPK pathway activation in colorectal cancer. J. Pathol 2009, 218, 514–519. [DOI] [PubMed] [Google Scholar]
- [19].Zhong D, Liu X, Khuri FR, Sun SY et al. , LKB1 is necessary for Akt-mediated phosphorylation of proapoptotic proteins. Cancer. Res 2008, 68, 7270–7277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Schindler J, Jung S, Niedner-Schatteburg G, Friauf E et al. , Enrichment of integral membrane proteins from small amounts of brain tissue. J. Neural. Transm 2006, 113, 995–1013. [DOI] [PubMed] [Google Scholar]
- [21].Kohnke PL, Mulligan SP, Christopherson RI, Membrane proteomics for leukemia classification and drug target identification. Curr. Opin. Mol. Ther 2009, 11, 603–610. [PubMed] [Google Scholar]
- [22].Lehner I, Niehof M, Borlak J, An optimized method for the isolation and identification of membrane proteins. Electrophoresis 2003, 24, 1795–1808. [DOI] [PubMed] [Google Scholar]
- [23].Donovan LE, Higginbotham L, Dammer EB, Gearing M et al. , Analysis of a membrane-enriched proteome from postmortem human brain tissue in Alzheimer’s disease. Proteomics Clin. Appl 2012, 6, 201–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Raimondo F, Morosi L, Chinello C, Perego R et al. , Protein profiling of microdomains purified from renal cell carcinoma and normal kidney tissue samples. Mol. Biosyst 2012, 8, 1007–1016. [DOI] [PubMed] [Google Scholar]
- [25].Zhang Q, Schulenborg T, Tan T, Lang B et al. Proteome analysis of a plasma membrane-enriched fraction at the placental feto-maternal barrier. Proteomics Clin. Appl 2010, 4, 538–549. [DOI] [PubMed] [Google Scholar]
- [26].Elia G, Cell surface protein biotinylation for SDS-PAGE analysis. Methods Mol. Biol 2012, 869, 361–372. [DOI] [PubMed] [Google Scholar]
- [27].Yokoyama T, Enomoto T, Serada S, Morimoto A et al. , Plasma membrane proteomics identifies bone marrow stromal antigen 2 as a potential therapeutic target in endometrial cancer. Int. J. Cancer 2013, 132, 472–484. [DOI] [PubMed] [Google Scholar]
- [28].Qi Y, Katagiri F, Purification of low-abundance Arabidopsis plasma-membrane protein complexes and identification of candidate components. Plant J. 2009, 57, 932–944. [DOI] [PubMed] [Google Scholar]
- [29].Tian Y, Koganti T, Yao Z, Cannon P et al. , Cardiac extracellular proteome profiling and membrane topology analysis using glycoproteomics. Prot. Clin. Appl 2014,8, 595–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Liu Y, Chen J, Sethi A, Li QK et al. , Glycoproteomic analysis of prostate cancer tissues by SWATH mass spectrometry discovers N-acylethanolamine acid amidase and protein tyrosine kinase 7 as signatures for tumor aggressiveness. Mol. Cell Proteomics 2014, 13, 1753–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Baycin Hizal D, Wolozny D, Colao J, Jacobson E et al. Glycoproteomic and glycomic databases. Clin. Proteomics 2014, 11, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Yang W, Zhou JY, Chen L, Ao M et al. Glycoproteomic analysis identifies human glycoproteins secreted from HIV latently infected T cells and reveals their presence in HIV+ plasma. Clin. Proteomics 2014, 11, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Chen J, Xi J, Tian Y, Bova GS, et al. , Identification, prioritization, and evaluation of glycoproteins for aggressive prostate cancer using quantitative glycoproteomics and antibody-based assays on tissue specimens. Proteomics 2013, 13, 2268–2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Tian Y, Zhang H, Characterization of disease-associated N-linked glycoproteins. Proteomics 2013, 13, 504–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Li QK, Gabrielson E, Zhang H, Application of glycoproteomics for the discovery of biomarkers in lung cancer. Proteomics Clin. Appl 2012, 6, 244–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Almaraz RT, Tian Y, Bhattarcharya R, Tan E et al. , Metabolic flux increases glycoprotein sialylation: implications for cell adhesion and cancer metastasis. Mol. Cell Proteomics 2012, 11, M112 017558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Liu Y, Huttenhain R, Surinova S, Gillet LC et al. , Quantitative measurements of N-linked glycoproteins in human plasma by SWATH-MS. Proteomics 2013, 13, 1247–1256. [DOI] [PubMed] [Google Scholar]
- [38].Huttenhain R, Surinova S, Ossola R, Sun Z et al. , N-glycoprotein SRMAtlas: a resource of mass spectrometric assays for N-glycosites enabling consistent and multiplexed protein quantification for clinical applications. Mol. Cell Proteomics 2013, 12, 1005–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Cerciello F, Choi M, Nicastri A, Bausch-Fluck D et al. , Identification of a seven glycopeptide signature for malignant pleural mesothelioma in human serum by selected reaction monitoring. Clin. Proteomics 2013, 10, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ahn YH, Kim JY, Yoo JS Quantitative mass spectrometric analysis of glycoproteins combined with enrichment methods. Mass Spectrom. Rev 2014, doi: 10.1002/mas.21428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Xu Y, Bailey UM, Punyadeera C, Schulz BL, Identification of salivary N-glycoproteins and measurement of glycosylation site occupancy by boronate glycoprotein enrichment and liquid chromatography/electrospray ionization tandem mass spectrometry. Rapid Commun. Mass Spectrom 2014, 28, 471–482. [DOI] [PubMed] [Google Scholar]
- [42].Wollscheid B, Bausch-Fluck D, Henderson C, O’Brien R et al. , Mass-spectrometric identification and relative quantification of N-linked cell surface glycoproteins. Nat. Biotechnol 2009, 27, 378–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Baycin-Hizal D, Tian Y, Akan I, Jacobson E et al. , GlycoFly: a database of drosophila N-linked glycoproteins identified using SPEG–MS techniques. J. Proteome Res 2011, 10, 2777–2784. [DOI] [PubMed] [Google Scholar]
- [44].Tian Y, Zhou Y, Elliott S, Aebersold R et al. , Solid-phase extraction of N-linked glycopeptides. Nat. Protoc 2007, 2, 334–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Lee MC, Sun B, Glycopeptide capture for cell surface proteomics. J. Vis. Exp 2014, 87, e51349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Sun Z, Chen R, Cheng K, Liu H et al. , A new method for quantitative analysis of cell surface glycoproteome. Proteomics 2012, 12, 3328–3337. [DOI] [PubMed] [Google Scholar]
- [47].Han D, Moon S, Kim Y, Min H et al. , Characterization of the membrane proteome and N-glycoproteome in BV-2 mouse microglia by liquid chromatography-tandem mass spectrometry. BMC Genomics 2014, 15, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Zhang H, Li XJ, Martin DB, Aebersold R, Identification and quantification of N-linked glycoproteins using hydrazide chemistry, stable isotope labeling and mass spectrometry. Nat. Biotechnol 2003, 21, 660–666. [DOI] [PubMed] [Google Scholar]
- [49].Ramya TN, Weerapana E, Cravatt BF, Paulson JC Glycoproteomics enabled by tagging sialic acid- or galactose-terminated glycans. Glycobiology 2013, 23, 211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Choksawangkarn W, Kim SK, Cannon JR, Edwards NJ et al. , Enrichment of plasma membrane proteins using nanoparticle pellicles: comparison between silica and higher density nanoparticles. J. Proteome Res 2013, 12, 1134–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Kim Y, Elschenbroich S, Sharma P, Sepiashvili L et al. , Use of colloidal silica-beads for the isolation of cell-surface proteins for mass spectrometry-based proteomics. Methods Mol. Biol 2011, 748, 227–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Vandre DD, Ackerman W. E. t., Tewari A, Kniss DA et al. , A placental sub-proteome: the apical plasma membrane of the syncytiotrophoblast. Placenta 2012, 33, 207–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Li X, Jin Q, Cao J, Xie C et al. , Evaluation of two cell surface modification methods for proteomic analysis of plasma membrane from isolated mouse hepatocytes. Biochim. Biophys. Acta 2009, 1794, 32–41. [DOI] [PubMed] [Google Scholar]
- [54].Li X, Xie C, Cao J, He Q et al. , An in vivo membrane density perturbation strategy for identification of liver sinusoidal surface proteome accessible from the vasculature. J. Proteome Res 2009, 8, 123–132. [DOI] [PubMed] [Google Scholar]
- [55].Schiess R, Mueller LN, Schmidt A, Mueller M et al. , Analysis of cell surface proteome changes via label-free, quantitative mass spectrometry. Mol. Cell Proteomics 2009, 8, 624–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Zhang H, Liu AY, Loriaux P, Wollscheid B et al. , Mass spectrometric detection of tissue proteins in plasma. Mol. Cell Proteomics 2007, 6, 64–71. [DOI] [PubMed] [Google Scholar]
- [57].Gillet LC, Navarro P, Tate S, Rost H et al. , Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol. Cell Proteomics 2012, 11, O111 016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Liu Y, Huttenhain R, Collins B, Aebersold R, Mass spectrometric protein maps for biomarker discovery and clinical research. Expert. Rev. Mol. Diagn 2013, 13, 811–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Zhang H, Yi EC, Li XJ, Mallick P et al. , High throughput quantitative analysis of serum proteins using glycopeptide capture and liquid chromatography mass spectrometry. Mol. Cell Proteomics 2005, 4, 144–155. [DOI] [PubMed] [Google Scholar]
- [60].Herbrich SM, Cole RN, West KP Jr., Schulze K et al. , Statistical inference from multiple iTRAQ experiments without using common reference standards. J. Proteome Res 2013, 12, 594–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Yan M, Yang X, Wang L, Clark D et al. , Plasma membrane proteomics of tumor spheres identify CD166 as a novel marker for cancer stem-like cells in head and neck squamous cell carcinoma. Mol. Cell Proteomics 2013, 12, 3271–3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Kaji H, Saito H, Yamauchi Y, Shinkawa T et al. , Lectin affinity capture, isotope-coded tagging and mass spectrometry to identify N-linked glycoproteins. Nat. Biotechnol 2003, 21, 667–672. [DOI] [PubMed] [Google Scholar]
- [63].Zhang W, Zhou G, Zhao Y, White MA et al. , Affinity enrichment of plasma membrane for proteomics analysis. Electrophoresis 2003, 24, 2855–2863. [DOI] [PubMed] [Google Scholar]
- [64].Warder SE, Tucker LA, Strelitzer TJ, McKeegan EM et al. , Reducing agent-mediated precipitation of high-abundance plasma proteins. Anal. Biochem 2009, 387, 184–193. [DOI] [PubMed] [Google Scholar]
- [65].Holewinski RJ, Jin ZC, Powell MJ, Maust MD et al. , A fast and reproducible method for albumin isolation and depletion from serum and cerebrospinal fluid. Proteomics 2013, 13, 743–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].An Y, Goldman R, Analysis of peptides by denaturing ultrafiltration and LC-MALDI-TOF-MS. Methods Mol. Biol 2013, 1023, 13–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Ortea I, Roschitzki B, Ovalles JG, Longo JL et al. , Discovery of serum proteomic biomarkers for prediction of response to infliximab (a monoclonal anti-TNF antibody) treatment in rheumatoid arthritis: an exploratory analysis. J. Proteomics 2012, 77, 372–382. [DOI] [PubMed] [Google Scholar]
- [68].Cole RN, Ruczinski I, Schulze K, Christian P et al. , The plasma proteome identifies expected and novel proteins correlated with micronutrient status in undernourished nepalese children. J. Nutr 2013, 143, 1540–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Lee EK, Cho H, Kim CW, Proteomic analysis of cancer stem cells in human prostate cancer cells. Biochem. Biophys. Res. Commun 2011, 412, 279–285. [DOI] [PubMed] [Google Scholar]
- [70].Rush J, Moritz A, Lee KA, Guo A et al. , Immunoaffinity profiling of tyrosine phosphorylation in cancer cells. Nat. Biotechnol 2005, 23, 94–101. [DOI] [PubMed] [Google Scholar]
- [71].Luo W, Slebos RJ, Hill S, Li M et al. , Global impact of oncogenic Src on a phosphotyrosine proteome. J. Proteome Res 2008, 7, 3447–3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Iwai LK, Benoist C, Mathis D, White FM, Quantitative phosphoproteomic analysis of T cell receptor signaling in diabetes prone and resistant mice. J. Proteome Res 2010, 9, 3135–3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Watkin EE, Arbez N, Waldron-Roby E, O’Meally R et al. , Phosphorylation of mutant huntingtin at serine 116 modulates neuronal toxicity. PLoS One 2014, 9, e88284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Zhong J, Kim MS, Chaerkady R, Wu X et al. , TSLP signaling network revealed by SILAC-based phosphoproteomics. Mol. Cell Proteomics 2012, 11, M112 017764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Mitsos A, Melas IN, Siminelakis P, Chairakaki AD et al. , Identifying drug effects via pathway alterations using an integer linear programming optimization formulation on phosphoproteomic data. PLoS Comput. Biol 2009, 5, e1000591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Brill LM, Xiong W, Lee KB, Ficarro SB et al. , Phosphoproteomic analysis of human embryonic stem cells. Cell Stem Cell 2009, 5, 204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Kaminska B, MAPK signalling pathways as molecular targets for anti-inflammatory therapy—from molecular mechanisms to therapeutic benefits. Biochim. Biophys. Acta 2005, 1754, 253–262. [DOI] [PubMed] [Google Scholar]
- [78].Peifer C, Wagner G, Laufer S, New approaches to the treatment of inflammatory disorders small molecule inhibitors of p38 MAP kinase. Curr. Top Med. Chem 2006, 6, 113–149. [DOI] [PubMed] [Google Scholar]
- [79].White MF, Regulating insulin signaling and beta-cell function through IRS proteins. Can. J. Physiol. Pharmacol 2006, 84, 725–737. [DOI] [PubMed] [Google Scholar]
- [80].Smith FD, Samelson BK, Scott JD, Discovery of cellular substrates for protein kinase A using a peptide array screening protocol. Biochem. J 2011, 438, 103–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Min-Sik Kim YZ, Shinchi Yachida NV, Kumar R, Abel ML et al. , Heterogeneity of pancreatic cancer metastases in a single patient revealed by quantitative proteomics. Am. Soc. Biochem. Mol. Biol 2014, 13, 2803–2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Amanchy R, Kandasamy K, Mathivanan S, Periaswamy B et al. , Identification of novel phosphorylation motifs through an integrative computational and experimental analysis of the human phosphoproteome. J. Proteomics Bioinform 2011, 4, 22–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Peri S, Navarro JD, Kristiansen TZ, Amanchy R et al. , Human protein reference database as a discovery resource for proteomics. Nucleic Acids Res. 2004, 32, D497–D501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Peri S, Navarro JD, Amanchy R, Kristiansen TZ et al. , Development of human protein reference database as an initial platform for approaching systems biology in humans. Genome Res. 2003, 13, 2363–2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Mathivanan S, Ahmed M, Ahn NG, Alexandre H et al. , Human Proteinpedia enables sharing of human protein data. Nat. Biotechnol 2008, 26, 164–167. [DOI] [PubMed] [Google Scholar]
- [86].Kandasamy K, Keerthikumar S, Goel R, Mathivanan S et al. , Human Proteinpedia: a unified discovery resource for proteomics research. Nucleic Acids Res. 2009, 37, D773–D781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Goel R, Harsha HC, Pandey A, Prasad TS, Human Protein Reference Database and Human Proteinpedia as resources for phosphoproteome analysis. Mol. Biosyst 2012, 8, 453–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].McLachlin DT, Chait BT, Analysis of phosphorylated proteins and peptides by mass spectrometry. Curr. Opin. Chem. Biol 2001, 5, 591–602. [DOI] [PubMed] [Google Scholar]
- [89].Aebersold R, Mann M, Mass spectrometry-based proteomics. Nature 2003, 422, 198–207. [DOI] [PubMed] [Google Scholar]
- [90].Maguire PB, Wynne KJ, Harney DF, O’Donoghue NM et al. , Identification of the phosphotyrosine proteome from thrombin activated platelets. Proteomics 2002, 2, 642–648. [DOI] [PubMed] [Google Scholar]
- [91].Zheng H, Hu P, Quinn DF, Wang YK, Phosphotyrosine proteomic study of interferon alpha signaling pathway using a combination of immunoprecipitation and immobilized metal affinity chromatography. Mol. Cell Proteomics 2005, 4, 721–730. [DOI] [PubMed] [Google Scholar]
- [92].Molina H, Horn DM, Tang N, Mathivanan S et al. , Global proteomic profiling of phosphopeptides using electron transfer dissociation tandem mass spectrometry. Proc. Natl. Acad. Sci. USA 2007, 104, 2199–2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Bose R, Molina H, Patterson AS, Bitok JK et al. , Phosphoproteomic analysis of Her2/neu signaling and inhibition. Proc. Natl. Acad. Sci. USA 2006, 103, 9773–9778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Harsha HC, Molina H, Pandey A, Quantitative proteomics using stable isotope labeling with amino acids in cell culture. Nat. Protoc 2008, 3, 505–516. [DOI] [PubMed] [Google Scholar]
- [95].Olsen JV, Blagoev B, Gnad F, Macek B et al. , Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 2006, 127, 635–648. [DOI] [PubMed] [Google Scholar]
- [96].Nagaraj N, D’Souza RC, Cox J, Olsen JV et al. , Feasibility of large-scale phosphoproteomics with higher energy collisional dissociation fragmentation. J. Proteome Res 2010, 9, 6786–6794. [DOI] [PubMed] [Google Scholar]
- [97].Han G, Ye M, Liu H, Song C et al. , Phosphoproteome analysis of human liver tissue by long-gradient nanoflow LC coupled with multiple stage MS analysis. Electrophoresis 2010, 31, 1080–1089. [DOI] [PubMed] [Google Scholar]
- [98].Oyama M, Kozuka-Hata H, Tasaki S, Semba K et al. , Temporal perturbation of tyrosine phosphoproteome dynamics reveals the system-wide regulatory networks. Mol. Cell Proteomics 2009, 8, 226–231. [DOI] [PubMed] [Google Scholar]
- [99].Li H, Ren Z, Kang X, Zhang L et al. , Identification of tyrosine-phosphorylated proteins associated with metastasis and functional analysis of FER in human hepatocellular carcinoma cells. BMC Cancer 2009, 9, 366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Garnier DJN, Rak J, Extracellular vesicles as prospective carriers of oncogenic protein signatures in adult and paediatric brain tumors. Proteomics 2012, 13, 1595–1607. [DOI] [PubMed] [Google Scholar]
- [101].Kucharzewska P, Christianson HC, Welch JE, Svensson KJ et al. , Exosomes reflect the hypoxic status of glioma cells and mediate hypoxia-dependent activation of vascular cells during tumor development. Proc. Natl. Acad. Sci. USA 2013, 110, 7312–7317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Yang M, Chen J, Su F, Yu B et al. , Microvesicles secreted by macrophages shuttle invasion-potentiating microRNAs into breast cancer cells. Mol. Cancer 2011, 10, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Thomas SN, Liao Z, Clark D, Chen Y et al. , Exosomal proteome profiling: a potential multi-marker cellular phenotyping tool to characterize hypoxia-induced radiation resistance in breast cancer. Proteomes 2013, 1, 87–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Klinke DJ 2nd, Kulkarni YM, Wu Y, Byrne-Hoffman C, Inferring alterations in cell-to-cell communication in HER2+ breast cancer using secretome profiling of three cell models. Biotechnol. Bioeng 2014, 111, 1853–1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Amorim M, Fernandes G, Oliveira P, Martins-de-Souza D et al. , The overexpression of a single oncogene (ERBB2/HER2) alters the proteomic landscape of extracellular vesicles. Proteomics 2014, 14, 1472–1479. [DOI] [PubMed] [Google Scholar]
- [106].Yoon JH, Kim J, Kim KL, Kim DH et al. , Proteomic analysis of hypoxia-induced U373MG glioma secretome reveals novel hypoxia-dependent migration factors. Proteomics 2014, 14, 1494–1502. [DOI] [PubMed] [Google Scholar]
- [107].Drake RR, Kislinger T, The proteomics of prostate cancer exosomes. Expert. Rev. Proteomics 2014, 11, 167–177. [DOI] [PubMed] [Google Scholar]
- [108].Beckham CJ, Olsen J, Yin PN, Wu CH et al. , Bladder cancer exosomes contain EDIL-3/del1 and facilitate cancer progression. J. Urol 2014, 192, 583–592. [DOI] [PubMed] [Google Scholar]
- [109].Webber J, Stone TC, Katilius E, Smith BC, Gordon B et al. , Proteomics analysis of cancer exosomes using a novel modified aptamer-based array (SOMAscan) platform. Mol. Cell Proteomics 2014, 13, 1050–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Sinha A, Ignatchenko V, Ignatchenko A, Mejia-Guerrero S et al. , In-depth proteomic analyses of ovarian cancer cell line exosomes reveals differential enrichment of functional categories compared to the NCI 60 proteome. Biochem. Biophys. Res. Commun 2014, 445, 694–701. [DOI] [PubMed] [Google Scholar]
- [111].Jeppesen DK, Nawrocki A, Jensen SG, Thorsen K et al. , Quantitative proteomics of fractionated membrane and lumen exosome proteins from isogenic metastatic and nonmetastatic bladder cancer cells reveal differential expression of EMT factors. Proteomics 2014, 14, 699–712. [DOI] [PubMed] [Google Scholar]
- [112].Bijnsdorp IV, Geldof AA, Lavaei M, Piersma SR et al. , Exosomal ITGA3 interferes with non-cancerous prostate cell functions and is increased in urine exosomes of metastatic prostate cancer patients. J. Extracell Vesicles 2013, 2, doi: 10.3402/jev.v2i0.22097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Shin SJ, Smith JA, Rezniczek GA, Pan S et al. , Unexpected gain of function for the scaffolding protein plectin due to mislocalization in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 19414–19419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Camacho L, Guerrero P, Marchetti D, MicroRNA and protein profiling of brain metastasis competent cell-derived exosomes. PLoS One 2013, 8, e73790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Nyalwidhe JO, Betesh LR, Powers TW, Jones EE et al. , Increased bisecting N-acetylglucosamine and decreased branched chain glycans of N-linked glycoproteins in expressed prostatic secretions associated with prostate cancer progression. Proteomics Clin. Appl 2013, 677–689. doi: 10.1002/prca.201200134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Park JO, Choi DY, Choi DS, Kim HJ et al. , Identification and characterization of proteins isolated from microvesicles derived from human lung cancer pleural effusions. Proteomics 2013, 13, 2125–2134. [DOI] [PubMed] [Google Scholar]
- [117].Raimondo F, Morosi L, Corbetta S, Chinello C et al. , Differential protein profiling of renal cell carcinoma urinary exosomes. Mol. Biosyst 2013, 9, 1220–1233. [DOI] [PubMed] [Google Scholar]
- [118].Dominy SS, Brown JN, Ryder MI, Gritsenko M et al. , Proteomic analysis of saliva in HIV-positive heroin addicts reveals proteins correlated with cognition. PLoS One 2014, 9, e89366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Jaworski E, Saifuddin M, Sampey G, Shafagati N et al. , The use of nanotrap particles technology in capturing HIV-1 virions and viral proteins from infected cells. PLoS One 2014, 9, e96778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Hassani K, Shio MT, Martel C, Faubert D et al. , Absence of metalloprotease GP63 alters the protein content of leishmania exosomes. PLoS One 2014, 9, e95007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Kang GY, Bang JY, Choi AJ, Yoon J et al. , Exosomal proteins in the aqueous humor as novel biomarkers in patients with neovascular age-related macular degeneration. J. Proteome Res 2014, 13, 581–595. [DOI] [PubMed] [Google Scholar]
- [122].Bellingham SA, Guo BB, Coleman BM, Hill AF, Exosomes: vehicles for the transfer of toxic proteins associated with neurodegenerative diseases? Front Physiol. 2012, 3, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Chiasserini D, van Weering JR, Piersma SR, Pham TV et al. , Proteomic analysis of cerebrospinal fluid extracellular vesicles: a comprehensive dataset. J. Proteomics 2014, 106C, 191–204. [DOI] [PubMed] [Google Scholar]
- [124].Rodriguez-Suarez E, Gonzalez E, Hughes C, Conde-Vancells J et al. , Quantitative proteomic analysis of hepatocyte-secreted extracellular vesicles reveals candidate markers for liver toxicity. J. Proteomics 2014, 103, 227–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Redzic JS, Ung TH, Graner MW, Glioblastoma extracellular vesicles: reservoirs of potential biomarkers. Pharmgenomics Pers. Med 2014, 7, 65–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Koga K, Matsumoto K, Akiyoshi T, Kubo M et al. , Purification, characterization and biological significance of tumor-derived exosomes. Anticancer Res. 2005, 25, 3703–3707. [PubMed] [Google Scholar]
- [127].Choi DS, Lee JM, Park GW, Lim HW et al. , Proteomic analysis of microvesicles derived from human colorectal cancer cells. J. Proteome Res 2007, 6, 4646–4655. [DOI] [PubMed] [Google Scholar]
- [128].Ji H, Greening DW, Barnes TW, Lim JW et al. , Proteome profiling of exosomes derived from human primary and metastatic colorectal cancer cells reveal differential expression of key metastatic factors and signal transduction components. Proteomics 2013, 13, 1672–1686. [DOI] [PubMed] [Google Scholar]
- [129].Wolfers J, Lozier A, Raposo G, Regnault A et al. , Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL cross-priming. Nat. Med 2001, 7, 297–303. [DOI] [PubMed] [Google Scholar]
- [130].Craven RA, Totty N, Harnden P, Selby PJ et al. , Laser capture microdissection and two-dimensional polyacrylamide gel electrophoresis: evaluation of tissue preparation and sample limitations. Am. J. Pathol 2002, 160, 815–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Marton A, Vizler C, Kusz E, Temesfoi V et al. , Melanoma cell-derived exosomes alter macrophage and dendritic cell functions in vitro. Immunol. Lett 2012, 148, 34–38. [DOI] [PubMed] [Google Scholar]
- [132].Hegmans JP, Bard MP, Hemmes A, Luider TM et al. , Proteomic analysis of exosomes secreted by human mesothelioma cells. Am. J. Pathol 2004, 164, 1807–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Graner MW, Alzate O, Dechkovskaia AM, Keene JD et al. , Proteomic and immunologic analyses of brain tumor exosomes. FASEB J. 2009, 23, 1541–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Simpson RJ, Jensen SS, Lim JW, Proteomic profiling of exosomes: current perspectives. Proteomics 2008, 8, 4083–4099. [DOI] [PubMed] [Google Scholar]
- [135].Simpson RJ, Lim JW, Moritz RL, Mathivanan S, Exosomes: proteomic insights and diagnostic potential. Expert. Rev. Proteomics 2009, 6, 267–283. [DOI] [PubMed] [Google Scholar]
- [136].Taylor DD, Zacharias W, Gercel-Taylor C, Exosome isolation for proteomic analyses and RN A profiling. Methods Mol. Biol 2011, 728, 235–246. [DOI] [PubMed] [Google Scholar]
- [137].Van Deun J, Mestdagh P, Sormunen R, Cocquyt V et al. The impact of disparate isolation methods for extracellular vesicles on downstream RNA profiling. J. Extracell Vesicles 2014, 3, doi: 10.3402/jev.v3.24858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Taylor DD, Zacharias W, Gercel-Taylor C, Exosome isolation for proteomic analyses and RNA profiling. Methods Mol. Biol 728, 235–246. [DOI] [PubMed] [Google Scholar]
- [139].Marimpietri D, Petretto A, Raffaghello L, Pezzolo A et al. , Proteome profiling of neuroblastoma-derived exosomes reveal the expression of proteins potentially involved in tumor progression. PLoS One 2013, 8, e75054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Raimondo F, Corbetta S, Morosi L, Chinello C et al. , Urinary exosomes and diabetic nephropathy: a proteomic approach. Mol. Biosyst 2013, 9, 1139–1146. [DOI] [PubMed] [Google Scholar]
- [141].Burke M, Choksawangkarn W, Edwards N, Ostrand-Rosenberg S et al. , Exosomes from myeloid-derived suppressor cells carry biologically active proteins. J. Proteome Res 2014, 13, 836–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Street JM, Barran PE, Mackay CL, Weidt S et al. Identification and proteomic profiling of exosomes in human cerebrospinal fluid. J. Transl. Med 2012, 10, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Thery C, Amigorena S, Raposo G, Clayton A, Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. Chapter 2006, 3, Unit 3 22. [DOI] [PubMed] [Google Scholar]
- [144].Marimuthu A, Subbannayya Y, Sahasrabuddhe NA, Balakrishnan L et al. , SILAC-based quantitative proteomic analysis of gastric cancer secretome. Proteomics Clin. Appl 2013, 7, 355–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Zhong J, Krawczyk SA, Chaerkady R, Huang H et al. , Temporal profiling of the secretome during adipogenesis in humans. J. Proteome Res 2010, 9, 5228–5238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Cheung WC, Beausoleil SA, Zhang X, Sato S et al. , A proteomics approach for the identification and cloning of monoclonal antibodies from serum. Nat. Biotechnol 2012, 30, 447–452. [DOI] [PubMed] [Google Scholar]
- [147].Wine Y, Boutz DR, Lavinder JJ, Miklos AE et al. , Molecular deconvolution of the monoclonal antibodies that comprise the polyclonal serum response. Proc. Natl. Acad. Sci. USA 2013, 110, 2993–2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Perkins DN, Pappin DJ, Creasy DM, Cottrell JS, Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 1999, 20, 3551–3567. [DOI] [PubMed] [Google Scholar]
- [149].Albaum SP, Hahne H, Otto A, Haussmann U et al. A guide through the computational analysis of isotope-labeled mass spectrometry-based quantitative proteomics data: an application study. Proteome Sci 2011, 9, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Eng JK, McCormack AL, Yates JR, An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom 1994, 5, 976–989. [DOI] [PubMed] [Google Scholar]
- [151].Dasari S, Chambers MC, Slebos RJ, Zimmerman LJ et al. TagRecon: high-throughput mutation identification through sequence tagging. J. Proteome Res 2010, 9, 1716–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Zucht HD, Lamerz J, Khamenia V, Schiller C et al. , Datamining methodology for LC-MALDI-MS based peptide profiling. Comb. Chem. High Throughput Screen 2005, 8, 717–723. [DOI] [PubMed] [Google Scholar]
- [153].Palagi PM, Walther D, Quadroni M, Catherinet S et al. , MSight: an image analysis software for liquid chromatography-mass spectrometry. Proteomics 2005, 5, 2381–2384. [DOI] [PubMed] [Google Scholar]
- [154].Wang W, Zhou H, Lin H, Roy S et al. , Quantification of proteins and metabolites by mass spectrometry without isotopic labeling or spiked standards. Anal. Chem 2003, 75, 4818–4826. [DOI] [PubMed] [Google Scholar]
- [155].Kanehisa M, Goto S, Sato Y, Kawashima M et al. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 2014, 42, D199–D205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Kanehisa M, Goto S, KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Ashburner M, Ball CA, Blake JA, Botstein D et al. , Gene ontology: tool for the unification of biology. Gene Ontol. Consortium. Nat. Genet 2000, 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Cline MS, Smoot M, Cerami E, Kuchinsky A et al. , Integration of biological networks and gene expression data using Cytoscape. Nat. Protoc 2007, 2, 2366–2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Huang da W, Sherman BT, Lempicki RA, Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc 2009, 4, 44–57. [DOI] [PubMed] [Google Scholar]
- [160].Huang da W, Sherman BT, Lempicki RA, Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Krogh A, Larsson B, von Heijne G, Sonnhammer EL, Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol 2001, 305, 567–580. [DOI] [PubMed] [Google Scholar]
- [162].Kall L, Krogh A, Sonnhammer EL, A combined transmembrane topology and signal peptide prediction method. J. Mol. Biol 2004, 338, 1027–1036. [DOI] [PubMed] [Google Scholar]
- [163].Emanuelsson O, Nielsen H, Brunak S, von Heijne G, Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J. Mol. Biol 2000, 300, 1005–1016. [DOI] [PubMed] [Google Scholar]
- [164].Bendtsen JD, Jensen LJ, Blom N, Von Heijne G et al. , Feature-based prediction of non-classical and leaderless protein secretion. Protein Eng. Des. Sel 2004, 17, 349–356. [DOI] [PubMed] [Google Scholar]
- [165].Nakai K, Kanehisa M, A knowledge base for predicting protein localization sites in eukaryotic cells. Genomics 1992, 14, 897–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Horton P, Park KJ, Obayashi T, Fujita N et al. , WoLF PSORT: protein localization predictor. Nucleic Acids Res. 2007, 35, W585–W587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Chen Y, Zhang Y, Yin Y, Gao G et al. , SPD—a web-based secreted protein database. Nucleic Acids Res. 2005, 33, D169–D173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Cappadona S, Baker PR, Cutillas PR, Heck AJ et al. , Current challenges in software solutions for mass spectrometry-based quantitative proteomics. Amino Acids 2005, 43, 1087–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria. 2013. http://www.R-project.org/ [Google Scholar]