

SUMMARY
The intestinal mucosa exists in a state of “physiologic hypoxia,” where oxygen tensions are markedly lower than those in other tissues. Intestinal epithelial cells (IECs) have evolved to maintain homeostasis in this austere environment through oxygen-sensitive transcription factors, including hypoxia-inducible factors (HIFs). Using an unbiased chromatin immunoprecipitation (ChIP) screen for HIF-1 targets, we identify autophagy as a major pathway induced by hypoxia in IECs. One important function of autophagy is to defend against intracellular pathogens, termed “xenophagy.” Analysis reveals that HIF is a central regulator of autophagy and that in vitro infection of IECs with Salmonella Typhimurium results in induction of HIF transcriptional activity that tracks with the clearance of intracellular Salmonella. Work in vivo demonstrates that IEC-specific deletion of HIF compromises xenophagy and exacerbates bacterial dissemination. These results reveal that the interaction between hypoxia, HIF, and xenophagy is an essential innate immune component for the control of intracellular pathogens.
Graphical abstract
In brief
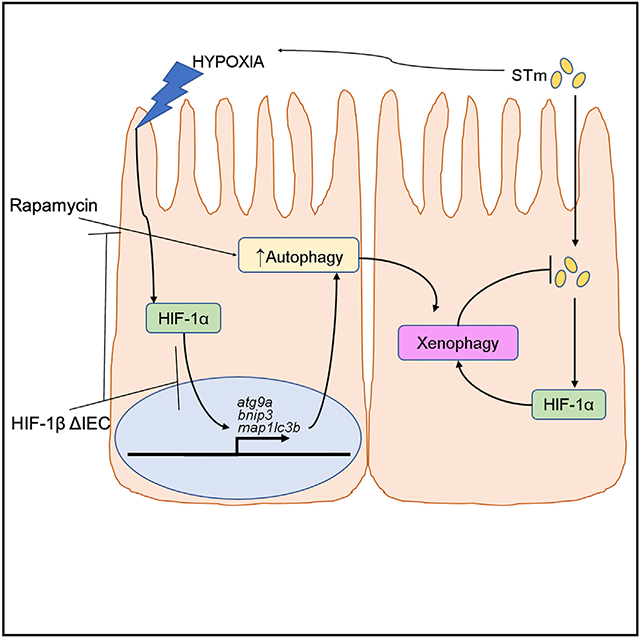
Dowdell et al. show that hypoxia, through stabilization of HIF-1α, activates autophagy in intestinal epithelial cells (IECs). Further, the model invasive bacterium Salmonella Typhimurium stabilizes HIF in IECs to trigger anti-bacterial autophagy (xenophagy). This mechanism demonstrates an essential mucosal innate immune response for control of invasive pathogens.
INTRODUCTION
Inflammatory bowel disease is a family of disorders characterized by chronic, relapsing inflammation of the gastrointestinal tract (Jairath and Feagan, 2020). The prevalence of inflammatory bowel disease (IBD) varies worldwide and corresponds roughly with the degree of industrialization; as a result, developed countries show the highest rates of IBD—greater than 1 in 200 in the USA and Europe—but with accelerating prevalence rates in the developing world due to burgeoning industrialization (Molodecky et al., 2012). IBD is currently subdivided into two major classes, Crohn’s disease (CD) and ulcerative colitis (UC), and although each demonstrates differing presentations and susceptibility factors, all forms of IBD are lifelong conditions with no cure that imparts a reduced quality of life (Cleynen et al., 2016). Although environmental factors (Ananthakrishnan et al., 2018) and genetics (Cleynen et al., 2016) have been implicated, the driving forces for IBD remain unclear, and the precise molecular mechanisms of IBD pathophysiology have yet to be elucidated.
One of the first genetic polymorphisms identified to confer susceptibility to IBD was in the gene nod2/card15, which encodes a cytoplasmic sensor to muramyl dipeptide (MDP; a peptidoglycan component) (Hugot et al., 2001; Ogura et al., 2001). These polymorphisms are most commonly found in the MDP-sensing domain of NOD2, resulting in a nonfunctional protein unable to detect intracellular bacteria and a corresponding aberrant inflammatory response (Girardin et al., 2003; Gutierrez et al., 2002; Yamamoto and Ma, 2009). Notably, NOD2 is expressed in distinct intestinal epithelial lineages, including Paneth cells—a cell type important for host-microbe interactions in the intestine (Ogura et al., 2003). Consequently, lack of functional NOD2 results in elevated levels of commensal bacteria and a compromised ability to kill intestinal pathogens such as Salmonella Typhimurium (STm) and Listera monocytogenes (Petnicki-Ocwieja et al., 2009). The mechanism of increased IBD susceptibility imparted by nod2 mutations was later found to be dependent on (macro)autophagy, a highly conserved eukaryotic system in which cytoplasmic constituents are targeted to and degraded in the lysosome (Yang and Klionsky, 2010). Autophagy acts on numerous intracellular substrates, such as misfolded proteins and organelles, but can also target intracellular pathogens in a process termed “xenophagy” (Dikic and Elazar, 2018). Defects in NOD2 impair the ability for autophagy proteins, notably ATG16L1, to localize to the plasma membrane at the site of bacterial entry, preventing an adequate xenophagic response and permitting intracellular bacterial survival (Travassos et al., 2010). ATG16L1 is an essential autophagy protein that, like NOD2, has been found to be mutated at a higher frequency in patients with IBD patients (Hampe et al., 2007; Mizushima et al., 2003; Rioux et al., 2007). The lack of functional ATG16L1, in turn, compromises autophagy and the xenophagic defense against intracellular bacteria(Homer et al., 2010; Kuballa et al., 2008). The integration of NOD2 and ATG16L1 as an intracellular xenophagic pathway was further confirmed by the discovery that the protein IRGM, itself also an IBD risk factor, orchestrates the interaction between NOD2 and ATG16L1 to promote anti-bacterial xenophagy (Brest et al., 2011; Chauhan et al., 2015; Parkes et al., 2007). Although it is currently understood that xenophagy plays an essential role in intestinal homeostasis and IBD pathogenesis, the regulation of the autophagic factors necessary for efficient bacterial clearance is not as clearly defined and the mechanisms of xenophagy regulation in vivo are unclear.
The intestinal tract is noteworthy in that it exists in a state of “physiologic hypoxia”—that is, it maintains homeostasis at oxygen tensions far below that of ambient air (Taylor and Colgan, 2017). Adaptation to this austere environment is mediated through the transcription factor family hypoxia-inducible factor (HIF), which is composed of three oxygen-labile subunits (HIF-1α, −2α, and −3α) and an oxygen-insensitive nuclear binding partner (HIF-1β/ARNT) (Colgan et al., 2020). These factors, members of the Per-ARNT-Sim family of basic helix-loop-helix transcription factors, modulate gene expression of a large number of direct targets including those responsible for a metabolic shift toward glycolysis and proteins involved in maintenance of epithelial integrity and regulation of innate immune function (Colgan et al., 2016; Dengler et al., 2014; Glover et al., 2016). Recent work has also shed light on the importance of HIF signaling in the regulation of autophagy. HIF-1α has been shown to regulate hypoxia-induced autophagy in mouse embryonic fibroblasts through BNIP3 and BNIP3L/NIX; likewise, the essential autophagy protein ATG9A, necessary for intestinal barrier/tight junction biogenesis, was found to regulated by HIF/hypoxia in intestinal epithelial cells (IECs) (Bellot et al., 2009; Dowdell et al., 2020; Saitoh et al., 2009).
Given the physiologic state of hypoxia in the intestine and considering the demonstrated regulation of autophagy by HIF/hypoxia, it follows that HIF-driven autophagy/xenophagy may regulate host-microbe interactions at the intestinal mucosa. In this study, we demonstrate an essential role for HIF in the regulation of autophagy in the intestinal epithelium. Further, we show that HIF/hypoxia regulates anti-bacterial autophagy (xenophagy) in IECs both in vitro and in vivo and, conversely, that intestinal bacteria regulate HIF and HIF target gene expression in the intestinal epithelium. Finally, we show that HIF is protective during infection of IECs with the model invasive pathogen STm using both in vitro and in vivo models. The results presented herein shed light on the role of HIF in regulating host-microbe interactions at the intestinal mucosa and provide insight into the xenophagic mechanisms dysregulated in IBD, informing the development of the next generation of IBD therapeutics.
RESULTS
HIF drives autophagy gene expression in IECs
Previous work suggests that HIF drives autophagy in a variety of cell types, including epithelial cell lineages (Bellot et al., 2009; Dowdell et al., 2020). Using our previously reported chromatin immunoprecipitation (ChIP)-chip screen (Glover et al., 2013) in which Caco-2 IECs were subjected to hypoxia, immunoprecipitated DNA was obtained using either anti-HIF-1α or anti-HIF-2α antibodies following specific fixation protocols. ChIP-enriched DNA was then hybridized to a custom microarray comprising a genome-wide set of predicted transcription start site flanking sequences at 50 bp resolution. From this dataset, we asked whether autophagy targets would be enriched in HIF-precipitated samples. Principal-component analysis (PCA) demonstrated that autophagy genes grouped well together with known HIF target genes, including pdk1 and hmox1 (Figure 1A). Further, we observed that autophagy and HIF-regulated genes clustered together separately from housekeeping genes such as tuba1 and rps27l, suggesting a distinct HIF-directed autophagic transcriptional program induced by hypoxia. Autophagy and HIF target genes showed distinct enrichment in ChIP-chip analysis as visualized by heatmap analysis (Figure 1B), whereas no such enrichment was observed for a variety of housekeeping genes. Notably, several autophagy genes (e.g., atg9a, atg5) demonstrated increased enrichment in HIF-1α-precipitated samples versus HIF-2α counterparts (Figure 1B), suggesting a distinct transcriptional response guided by a specific HIF isoform. We then sought to validate our ChIP-chip analysis through qPCR analysis of hypoxia-treated Caco-2 cells. Analysis of this cell line indicated up-regulation of a variety of targets genes implicated in both autophagy and xenophagy, including irgm, map1lc3a, and bnip3 (Figure 1C), in agreement with our ChIP-chip observations. Finally, we sought to confirm our observations using nontransformed cells, as previous observations have indicated that differences in autophagy can exist between normal and cancer IECs (Groulx et al., 2012). Validation via hypoxia treatment of murine colonoids from wild-type C57BL/6J mice resulted in the significant induction of a subset cohort of autophagy genes compared with normoxia-treated controls (Figure 1D), indicating that our initial observations were not due to a cancer cell phenotype and suggesting that expression of autophagy genes are regulated by hypoxia and, more specifically, by HIF.
Figure 1. Autophagy genes are HIF targets.

(A) Principal-component analysis (PCA) of Caco-2 IECs analyzed by ChIP-chip demonstrates association of autophagy genes with hypoxia genes, in contrast with housekeeping genes.
(B) Heatmap of genes plotted in (A) shows conserved patterns between autophagy and hypoxia targets. Sample names at bottom of columns represent biological replicates.
(C) Validation of ChIP-chip results by qPCR in Caco-2 IECs demonstrates induction of autophagy genes under hypoxic conditions.
(D) Analysis of gene expression in primary murine colon-derived organoids by qPCR shows significant increase in autophagy gene expression by hypoxia. *p < 0.05, **p < 0.01 by t test. ChIP-chip results reflect three biological replicates per sample group. qPCR results reflect at least three biological replicates.
Autophagic flux is enhanced by hypoxia in IECs
Given the finding that autophagy genes appear to be HIF targets in IECs, we next sought to examine whether autophagic flux itself is regulated by hypoxia in IECs. Autophagic flux describes “the entire process of autophagy over a period of time” and more accurately reflects the biological process of autophagy in living systems than autophagic gene expression alone (Klionsky et al., 2021). To do so, we first exposed Caco-2 IECs plated on glass coverslips to hypoxia (1% O2, 24 h), then fixed and stained them against the autophagosome-associated proteins LC3 and p62/SQSTM1. Observation of stained cells by indirect immunofluorescence (IF) indicated an increased degree of LC3 and p62 puncta formation and co-localization in hypoxia-treated cells versus normoxia-treated controls (Figure 2A), a finding suggestive of increased autophagic flux. We then assessed LC3 and p62 abundance in IECs by western blot (WB) with and without hypoxia treatment for varying lengths of time. We observed a time-dependent increase in the band intensity of LC3-I (upper band) to LC3-II (lower band) conversion in hypoxia-treated cells, as well as a corresponding decrease in p62 band intensity, compared with normoxia-treated controls (Figure 2B). These results indicate that molecular markers of autophagic flux, namely LC3 lipidation and p62 degradation (Jiang and Mizushima, 2015), are both enhanced by hypoxia and suggest that autophagic flux is enhanced by hypoxia. We investigated this further by treating tandem-fluorescent LC3 (tf-LC3)-expressing Caco-2 cells with hypoxia and imaging by IF. As previously reported, tf-LC3 is an mRFP-GFP-LC3 fusion protein that demonstrates both green and red fluorescence in the cytoplasm but, due to acidic quenching of GFP, demonstrates only red fluorescence after autophagosome-lysosome fusion (Kimura et al., 2007). In addition, tf-LC3 shows increased puncta formation during heightened autophagic flux (similar to wild-type [WT] LC3), resulting in an increase in yellow puncta (autophagosomes) and red puncta (autolysosomes) during certain stresses, such as starvation (Kuma et al., 2017). To this end, we observed an increase in both yellow and red puncta in hypoxia-treated, tf-LC3-expressing Caco-2 cells versus normoxic controls (Figure 2C), indicating an active increase in autophagic flux in these cells during hypoxia treatment. Finally, treatment of murine C57BL/6J colonoids with hypoxia resulted in stabilization of HIF-1α and induction of BNIP3 (Figure 2D), a protein previously shown to be important for hypoxia-related autophagy (Bellot et al., 2009), further demonstrating that hypoxia induces autophagy through HIF in IECs and that this induction is not limited to cancer cell lines.
Figure 2. Hypoxia induces autophagy in IECs.

(A) Incubation of Caco-2 IECs in either normoxia or hypoxia (1%O2, 24 h), followed by immunofluorescence staining, reveals increased LC3 and SQSTM1/p62 puncta in hypoxia-treated cells.
(B) Measurement of LC3 and p62 by western blot in Caco-2 IECs during time course incubation at normoxia or hypoxia.
(C) Caco-2 IECs transfected with tf-LC3 followed by incubation at normoxia or hypoxia. The increase in red puncta (bottom panels) demonstrates increased autophagic flux in hypoxia-treated cells.
(D) Caco-2 IECs incubated at hypoxia (Hx) show increased expression of HIF-1α and BNIP3 compared with normoxic (Nx) controls. Results are representative of at least two separate experiments. Microscopy images at 1,000×; scale bar: 10 μm.
Infection of IECs by STm stabilizes HIF and activates HIF signaling
Previous findings indicate that bacteria can regulate HIF signaling both in vitro and in vivo in a variety of cell types (Hartmann et al., 2008; Peyssonnaux et al., 2005). As HIF plays an indispensable role in autophagy, and given that regulation of autophagy appears to be governed in an HIF-1α-dependent fashion, we sought to examine the role of bacterial regulation of HIF-1α-driven autophagy in vitro. We first examined this by infecting Caco-2 cells with STm, a model invasive bacterium used extensively in the study of xenophagy (Birmingham et al., 2006; Mimouna et al., 2014; Tattoli et al., 2012), in a dose-response manner by treating at varying multiplicities of infection (MOIs) and in a time course fashion at a set MOI. 300 μM CoCl2 was used as a positive control for HIF stabilization, as previously described (Epstein et al., 2001). We observed that HIF-1α was stabilized by infection with STm in a time- and dose-dependent manner (Figure 3A), indicating bacterial regulation of HIF as reported previously and confirming that STm stabilizes HIF in our model system. We further examined expression of HIF target genes by qPCR to assess whether stabilized HIF-1α was transcriptionally active. We found that expression of HIF target genes involved in autophagy was induced by STm (Figure 3B), suggesting regulation of the HIF transcriptional repertoire through bacterial infection.
Figure 3. Infection of IECs by Salmonella Typhimurium stabilizes HIF and induces HIF signaling.

(A) Caco-2 cells were infected with Salmonella Typhimurium (STm) at a set MOI for the given length of time or for a constant time with variable MOIs, then whole-cell lysates were prepared and analyzed for HIF by western blot. Uninfected cells and CoCl2-treated cells were used as negative and positive controls for HIF stabilization, respectively.
(B) Expression of HIF-regulated autophagy genes in HeLa cells infected with STm were measured by qPCR. *p < 0.05, **p < 0.01 by t test. Western blot results are representative of at least two separate experiments. qPCR results reflect at least three biological replicates per group.
HIF stabilization by STm through oxygen consumption
We next sought to examine the mechanism by which STm stabilizes HIF in IECs in vitro. Previous reports have documented HIF stabilization in a variety of cell lines by pathogenic bacteria, though the mechanism(s) of this stabilization are unclear (Cane et al., 2010; Hartmann et al., 2008; Legendre et al., 2011; Mimouna et al., 2014; Peyssonnaux et al., 2005, 2008; Sharma et al., 2011; Werth et al., 2010). While one study reported the secretion of siderophores as one possible mechanism for bacterial-mediated HIF stabilization (Hartmann et al., 2008), we suspected alternative mechanisms and asked whether oxygen consumption was important for HIF stabilization due to STm. Using an oxygen-sensing microplate system previously utilized by our group to measure in vitro oxygen tensions and consumption (Campbell et al., 2014; Kelly et al., 2015), we found that treatment of IECs with STm resulted in a dramatic decrease in oxygen levels to well below the threshold usually considered to stabilize and activate HIF after an initial O2 equilibration/stabilization period (Figure 4A) (Koh and Powis, 2012). In contrast, uninfected IECs stabilized at a nonhypoxic oxygen tension. Crucially, hypoxia was not observed in control wells containing buffer alone or buffer + STm. To corroborate these results, HeLa cells were transiently transfected with a plasmid containing luciferase driven by 5× serial hypoxia-response elements (HREs) derived from vegf (HRE-Luc); this construct has been shown to demonstrate luciferase induction during HIF stabilization (Walton et al., 2018). HeLa cells were utilized, as they are an excellent model cell line for Salmonella infection, including in studies of autophagy, and are highly transferable (Steele-Mortimer, 2008; Tattoli et al., 2012). Infection of HeLa cells containing HRE-Luc with STm resulted in a significant increase in luciferase activity versus uninfected cells (Figure 4B), indicating that STm drives activity from HREs during infection of epithelial cells through stabilization of HIF.
Figure 4. STm induces cellular Hx in vitro.

(A) Caco-2 IECs, STm, or a combination of both were incubated at 37°C on Transwell inserts in an Oxodish oxygen measurement system. HBSS+ was used as the assay buffer and as the negative control.
(B) HeLa cells transfected with a plasmid containing luciferase under Hx-response element (HRE) control were infected with STm, then luciferase activity was measured. Both Oxodish and luciferase experiments utilized at least six biological replicates per group.
HIF and hypoxia are protective during in vitro infection of IECs with STm
Given that STm stabilizes HIF and drives expression of HIF-regulated genes, including autophagy genes, and as autophagy plays an essential role in the defense against intracellular pathogens (xenophagy) (Huang and Brumell, 2014), we asked whether HIF-regulated autophagy was protective during infection of IECs with STm. To investigate this, we generated stable HIF knockdown (KD) IEC lines through delivery of short hairpin RNA (shRNA) via lentiviral transduction. These lines were validated by incubating with or without the HIF stabilizing agent CoCl2 and assessing HIF isoforms through WB; results indicate successful depletion of the targeted HIF isoforms in the various cell lines relative to cells expressing nontargeting shRNA (Figure 5A). Next, we infected these cell lines with STm and measured bacterial replication with or without hypoxia treatment, a metric that has been previously used to show increased susceptibility to Salmonella in autophagy-deficient cells (Birmingham et al., 2006). Bacterial replication was quantified by infecting IECs with STm, then treating them with the membrane-impermeable antibiotic gentamicin to restrict extracellular growth of bacteria. This results in only intracellular bacteria remaining viable, with differences in bacterial replication reflected in changes in viable colony-forming units (CFUs) following collection of cell lysates and plating of serial dilutions (Steele-Mortimer, 2008). Quantification of intracellular CFUs demonstrated that hypoxia resulted in a decrease in intracellular growth of STm in control cells, a phenotype that was abolished in HIF KD cells and suggestive of HIF-driven xenophagy (Figure 5B). No differences in cell survival were observed between experimental groups, and, as each group was infected at an equal MOI, these results indicate that the observed differences in intracellular CFUs were likely due to variations in xenophagy. Interestingly, HIF-1α KD cells demonstrated significantly elevated levels of bacterial replication compared with control IECs; however, all HIF KD cell lines showed no benefit from hypoxia incubation, demonstrating that the protective effects of hypoxia with regards to restricting intracellular growth of STm were mediated by HIF. We then confirmed these observations by infecting WT IECs with STm in the presence of compounds previously shown to enhance (rapamycin [RAP]; through inhibition of mTOR) or inhibit (3-methyladenine) autophagy. As expected, an increase in autophagic flux via treatment with RAP was protective by reducing the intracellular burden of STm, which was phenocopied by treatment with hypoxia (Figure 5C). Further, inhibition of autophagy by 3-methyladenine was detrimental and enhanced intracellular STm levels. These results recapitulate previous findings in the nonintestinal HeLa cell line HeLa and in peritoneal macrophages, validating our observations that autophagy positively regulates STm infection in vitro (Owen et al., 2014; Tattoli et al., 2012). Taken together, these results indicate that xenophagy is a critical mechanism by which intracellular STm replication is controlled in IECs and that hypoxia (through HIF) is protective through regulating anti-STm xenophagy.
Figure 5. HIF/Hx promotes clearance of intracellular STm by xenophagy.

(A) HIF isoforms were knocked down in IECs using lentiviral-delivered shRNA. CoCl2 treatment was used to stabilize HIF for analysis of knockdown efficiency.
(B) Knockdown of HIF isoforms impairs Hx-mediated reduction of intracellular STm. *p < 0.05 by t test.
(C) Intracellular burden of STm in IECs is reduced by either incubation at Hx or treatment with the autophagy activator RAP. Conversely, inhibition of autophagy with 3-methyladenine (3-MA) increases intracellular proliferation of STm. MOI = 10. Results are representative of at least two separate experiments; in the case of STm infection, at least three biological replicates were used per experimental group.
Regulation of autophagy by HIF and activation of HIF by STm in vivo
Next, we sought to address whether our observations in vitro held true in animal models. To do so, we first utilized mice expressing a luciferase construct fused to the oxygen-dependent degradation domain (ODD) from HIF-1α (ODD-Luc). These mice show luciferase activity only under conditions that permit HIF stabilization; as such, the ODD-Luc fusion protein acts as a faithful reporter for hypoxia/HIF stabilization and has been used in vivo by our group for this purpose (Campbell et al., 2014). Following infection of ODD-Luc mice with STm, we observed enhanced luciferase activity in the terminal ileum, cecum, and proximal colon compared with uninfected ODD-Luc controls, indicating increased HIF stabilization (Figure 6A). These results recapitulate our in vitro results demonstrating induction of HIF signaling by STm infection (Figures 3B and 4B). Next, we asked what role HIF signaling plays in the xenophagic defense against STm in vivo. To investigate this, we first generated mice selectively lacking Hif-1β in the intestinal epithelium (Hif-1β ΔIEC) by crossing mice with a floxed Hif-1β allele to mice expressing Cre recombinase under the Villin promoter, which shows high specificity for expression in the intestinal epithelium (Maunoury et al., 1992). Hif-1β ΔIEC mice were used as opposed to Hif-1α ΔIEC mice due to the potential redundancy and/or compensation by Hif-2α, as has been noted before (Koh and Powis, 2012). The strategy behind the generation, and the subsequent genotype validation, of Hif-1β ΔIEC mice has been described elsewhere by our group, and, notably, these mice show no defects in gross intestinal development (Glover et al., 2013). Examination of autophagy in epithelial-enriched intestinal scrapings of Hif-1β ΔIEC mice reveals a striking defect in autophagic flux, evidenced by the diminished conversion of LC3-I to LC3-II and increased levels of p62, compared with littermate controls expressing Hif-1β (i.e., Cre− littermates) (Figure 6B). These findings indicate an in vivo dependence of autophagy on HIF signaling and indicate that its absence impairs normal autophagic flux. Next, we sought to examine expression of representative target genes in the colonic epithelium of Hif-1β-expressing and Hif-1β ΔIEC mice. As expected, Hif-1β ΔIEC mice show significantly diminished colonic Hif-1β expression in the colonic epithelium relative to littermate controls, as well as diminished expression of the prototypic HIF targets Pgk1 and Ckb (Figure 6C) (Glover et al., 2013). However, Hif-1β ΔIEC mice also demonstrated significantly lower expression of Atg9 in the colonic epithelium; as Atg9 is an essential autophagy gene (Saitoh et al., 2009), this indicates that normal levels of autophagy are compromised at the transcriptional level by the loss of Hif-1β and that HIF signaling is an inherently important component for autophagy in the intestinal epithelium. Given these observations, we then asked whether HIF-regulated autophagy would be protective in vivo during STm infection (i.e., through xenophagy) as it was observed to be in vitro. Hif-1β ΔIEC mice and littermate controls were infected with STm through established protocols (Bellot et al., 2009; Conway et al., 2013), after which mice were euthanized and samples collected. Although no difference was observed between HIF-1β ΔIEC mice and controls with regards to extracellular colonization by STm (i.e., fecal samples) (Figure 6D), HIF-1β ΔIEC mice demonstrated a significantly higher dissemination of STm to extraintestinal sites, including liver and spleen, as well as a reduced colon length—a metric that is directly correlated with the severity of intestinal inflammation (Figure 6E) (Garcia-Hernandez et al., 2021). These results demonstrate that HIF regulates autophagy in the normal intestinal epithelium in vivo, similar to what is observed in vitro, and that HIF-driven autophagy is protective during STm infection by limiting extraintestinal spread of pathogens and limiting inflammation.
Figure 6. HIF signaling is essential for in vivo control of STm by xenophagy.

(A) Intragastric infection of ODD-Luc mice with STm increases HIF stabilization (luciferase activity) in the cecum. Quantification of cecal radiance shown in plot below images (n = 4 per group, p < 0.01). Scale bar: 1 cm.
(B) Hif1bΔIEC mice demonstrate defects in autophagy in the intestinal epithelium by LC3 and p62 by western blot analysis of intestinal scrapings.
(C) Hif1bΔIEC mice show reduced intestinal levels of the HIF targets Pgk1 and Ckb, as well as the HIF-driven autophagy protein Atg9, by qPCR analysis. *p < 0.05 by t test.
(D) Deletion of intestinal epithelial HIF-1β (Hif1bΔIEC) in vivo does not change levels of colonization following infection with STm in the ileum, cecum, or colon. Results are normalized to wild-type (+/+) controls. N = 5 mice per group.
(E) Deletion of intestinal HIF-1β (Hif1bΔIEC) in vivo increases dissemination of STm to liver and spleen following intragastric infection and provokes increased intestinal inflammation as measured by shortened colon length. Results are normalized to wild-type (+/+) controls. N = 5 mice per group, where *p < 0.025 by t test.
HIF-xenophagy axis regulates susceptibility to Salmonella infection in vivo
Finally, we asked whether pharmacologic modulation of the HIF-xenophagy axis in HIF-1β ΔIEC mice could rescue the phenotypes of defective xenophagy and susceptibility to STm infection (Figure 6). To do so, we treated HIF-1β ΔIEC mice and littermate controls either with the mTOR inhibitor (autophagy activator) RAP or vehicle daily beginning 3 days prior to infection using a formulation and dosage scheme previously demonstrated to achieve robust induction of autophagy (Johnson et al., 2013). We chose to use RAP in order to clarify the underlying mechanism by which HIF-dependent autophagy regulates host-microbial interactions. Preliminary experiments in WT C57BL/6J mice indicated robust activation of autophagy in colonic IECs using this treatment regimen (Figure S1). With this approach, we then infected mice with STm as done before and assessed the extent of disease severity. RAP treatment did not influence colonization of mice (Figure S2); however, we did observe significantly diminished extracellular dissemination of STm to the liver in Hif-1β+/+ mice treated with RAP compared with vehicle-treated controls (Figure 7A), suggesting that activation of autophagy via RAP was protective through limiting systemic spread of STm. Further, we saw no such protection in RAP-treated HIF-1β ΔIEC mice, suggesting that the observed protection by RAP in Hif-1β+/+ mice was due to activation of autophagy specifically in the intestinal epithelium. In agreement with these data, we observed significantly lower levels of pro-inflammatory tissue cytokines in RAP-treated Hif-1β+/+ mice (Figure 7B), indicating attenuated intestinal inflammation during RAP administration. In contrast, HIF-1β ΔIEC mice showed elevated levels of tissue cytokines versus vehicle-treated controls, demonstrating a more severe state of intestinal inflammation in the HIF-1β ΔIEC+ RAP cohort. Finally, blinded histopathological analysis (Figure 7C) of cecal tissue from RAP-treated Hif-1β+/+ mice showed a decrease in the “epithelial integrity” subscore relative to vehicle-treated controls, indicative of a more intact epithelium with less severe erosion and ulceration (Figure 7D, top) (Barthel et al., 2003). By contrast, RAP-treated HIF-1β ΔIEC mice showed a significantly higher epithelial integrity histopathology score versus vehicle controls, which is indicative of more severe epithelial deterioration (Figure 7D, bottom). These data demonstrate that the protective effect of RAP was lost in the absence of epithelial Hif-1β, resulting in more severe damage to the intestinal epithelium during infection. Taken together, these results show that HIF is protective during STm infection through regulating autophagy/xenophagy in IECs and that the protective pharmacological activation of autophagy is lost in the absence of epithelial HIF signaling.
Figure 7. Treatment of mice with autophagy agonist RAP ameliorates STm-induced inflammation in a HIF-1β-dependent manner.

(A) Measurement of liver STm CFUs in mice treated either with DMSO or RAP. Data presented as CFUs per organ, normalized to DMSO-treated controls.
(B) Expression of pro-inflammatory cytokines in terminal ilea of mice infected with STm and treated with either DMSO or RAP. Data represented as fold expression of RAP-treated mice versus DMSO-treated controls.
(C) Histopathology of Salmonella-infected, RAP-treated Hif1b+/+ or Hif1bΔIEC mice was evaluated by H&E staining and microscopy. Data presented as total final score for indicated parameters.
(D) Representative histopathologic images of cecal tissue from Hif1bfl/fl (top) or Hif1bΔIEC (bottom) mice administered RAP in combination with STm. Note loss of epithelial integrity in Hif1bΔIEC. For all experiments, n = 9–10 mice per experimental group.
All statistics calculated by one-way ANOVA (A) or unpaired t test (B and C). *p < 0.05, **p < 0.01, ***p < 0.005 in all panels. Microscopy images at 400×; scale bar: 50 μm.
DISCUSSION
Previous studies have highlighted the varied mechanisms by which HIF influences intestinal homeostasis; these include regulation of creatine energetics, expression of pro-barrier genes/factors, and regulating the cellular response to “beneficial” microbiota-derived metabolites (e.g., SFCAs) (Glover et al., 2013; Karhausen et al., 2004; Kelly et al., 2015). Similarly, in vivo activation of HIF signaling has been demonstrated to be protective in murine models of colitis (Cummins et al., 2008; Robinson et al., 2008), and as such, pharmacological activation of HIF is a promising strategy for the development of IBD therapeutics (Manresa and Taylor, 2017). However, the mechanism by which HIF signaling promotes intestinal homeostasis and resilience are not well understood; indeed, HIF has been shown to directly regulate dozens of target genes (Dengler et al., 2014) and possibly hundreds more indirectly, resulting in many possible complementary pathways through which HIF is protective during intestinal inflammation. The studies presented here provide insight into an underexplored area of HIF-assisted innate immunity that adds to the arsenal of the mucosa in defense of potentially invasive microbes.
One biochemical pathway shown to be associated with HIF is (macro)autophagy, a highly conserved eukaryotic pathway in which cytoplasmic organelles, proteins, etc., are enveloped in characteristic double-membraned autophagosomes, then targeted to the lysosome for degradation (Mehrpour et al., 2010). Autophagy occurs in every cell in the body, and, accordingly, defects in autophagy underlie the molecular pathology of a diverse collection of diseases such as neurodegenerative disorders (e.g., Alzheimer’s disease) (Wong and Cuervo, 2010) and cardiomyopathies (e.g., Danon disease) (Gottlieb and Mentzer, 2013) and the development and treatment of various cancers (Mathew et al., 2007). IBD is no different, and it is theorized that defects in anti-bacterial autophagy, or xenophagy, underlie the pathogenesis of IBD based on previously characterized IBD susceptibility genes (Lassen and Xavier, 2017). Although anecdotal evidence exists for the use of autophagy activating compounds in the treatment of IBD (Dumortier et al., 2008; Massey et al., 2008), no FDA-approved therapy currently exists that leverages autophagy to treat IBD.
The present study presents evidence that autophagy is regulated by HIF in the intestinal epithelium and that this regulation, through activation of xenophagy, is protective both in vitro and in vivo against the prototypic invasive pathogen STm. Further, we present evidence that exogenous stimulation of autophagy via pharmacological intervention is beneficial during STm infection through stimulating xenophagy in an HIF-dependent manner. These findings, collectively considered, demonstrate an underappreciated role for HIF signaling in regulating xenophagy in the intestinal epithelium. They are in accord with previous studies that show an essential role for autophagy in the defense against invasive pathogens and the maintenance of commensal microorganisms (Birmingham et al., 2006; Conway et al., 2013; Petnicki-Ocwieja et al., 2009). Further, these results expand upon the already well-established role of HIF in maintaining intestinal epithelial homeostasis, indicating a unique function for HIF in vivo through the regulation of protective xenophagy (Glover et al., 2013; Karhausen et al., 2004). Finally, results presented herein suggest that modulation of the HIF-autophagy axis may be beneficial in the context of intestinal inflammation through regulating host-microbe interactions at the intestinal epithelium. Interestingly, we observed that the beneficial effect of RAP was lost in Hif-1β ΔIEC mice, suggesting a bi-directional mode of communication between the HIF and autophagy pathways. Although our data do support a conclusion that HIF drives autophagy in the intestinal epithelium, one possibility is that the loss of baseline autophagy resulting from HIF-1β knockout (Figures 6B and 6C) causes the intestine to become less sensitive to autophagy induction mediated by RAP. This would suggest that a dependency exists between autophagy and HIF signaling for regulation of host-microbe interactions in the gut.
Although the work presented here demonstrates a distinct role for the regulation of xenophagy by HIF, observations regarding the HIF-xenophagy axis have been described before in more limited contexts. One study previously suggested a role for HIF-1α in the xenophagic response to adherent-invasive Escherichia coli (AIEC) in IECs (Mimouna et al., 2014). However, that mechanism was found to be dependent on the specific AIEC receptor CEACAM6 (Barnich et al., 2007), and, as such, it is unclear how broadly applicable these findings are to intestinal host-microbe interactions. In particular, STm appears to recognize a diverse array of cell-surface receptors including GP2, MUC1, and CD209 (Hase et al., 2009; Li et al., 2019; Ye et al., 2019). This suggests that the observations made in this study are more broadly applicable toward a range of enteric microorganisms, and current studies in our group are focused on defining the role of HIF-driven autophagy/xenophagy in response to a wider variety of commensal, opportunistic (pathobionts), and pathogenic microbes. Additionally, a recent study found that HIF-1α appears not to contribute toward the host defense against STm in vivo (Robrahn et al., 2022). Although the authors of this study did observe STm-induced HIF-1α stabilization in the intestinal epithelium, epithelial-specific loss of HIF-1α did not appear to worsen outcomes during STm typhlitis/colitis. One explanation for the discrepancy between this study and the results published herein is the potential that HIF-2α can compensate for the loss of HIF-1α in the setting of HIF-driven xenophagy. HIF-2α has been previously shown to be induced by the loss of HIF-1α in a compensatory manner (Carroll and Ashcroft, 2006), which could obscure any phenotype resulting from the loss of HIF-1α alone. It is also notable that, in addition to the xenophagic response described here, HIF-1-mediated signaling influences other epithelial antimicrobial defenses, including the transcriptional regulation of defensins (Kelly et al., 2013). Our studies done in vivo rely on the loss of HIF-1β, abolishing overall HIF signaling and allowing for interrogation of the role of HIF in general to intestinal epithelial xenophagy.
Although intact HIF-driven xenophagy is essential for the control of invasive bacteria, STm, as well as several other intracellular bacteria, have evolved strategies in which they can evade host xenophagic responses to establish productive infections (Huang and Brumell, 2014). In STm, these strategies include reactivation of mTOR following infection by targeting LKB1, SIRT1, and AMPK to the lysosome for degradation, suppressing autophagy, and promoting intracellular bacterial survival (Ganesan et al., 2017; Tattoli et al., 2012). In addition, Shigella flexneri evades xenophagy through the secretion of IcsB, a virulence factor that masks the bacterial autophagy target IcsA/VirG on the bacterium’s surface and thereby prevents recognition by host ATG proteins (Ogawa et al., 2005). Expression of ActA by Listeria monocytogenes similarly “hides” the bacteria from host xenophagy by recruiting host cytoskeletal proteins and camouflaging itself as an organelle (Yoshikawa et al., 2009). Further, Mycobacterium tuberculosis can suppress the induction of autophagy altogether in macrophages through secretion of the factor Eis, which induces interleukin-10 (IL-10) expression and activates PI3K/Akt/mTOR signaling (Duan et al., 2016). These findings underscore the observation that pathogenic bacteria and their host cells exist in a sort of molecular “tug of war”—mechanisms that the eukaryotic host use to control infection are simultaneously antagonized by their targeted microorganisms.
Our results suggest that pharmacological intervention targeting the HIF-autophagy axis is a viable strategy for attenuation of intestinal inflammation, particularly that stemming from an infectious source. This observation has particular importance in the current “post-antibiotics” era, in which antimicrobial resistance (AMR) is a major health crisis and is responsible for an estimated 4.95 million deaths in 2019 alone (Antimicrobial Resistance, 2022). Further, one review of medical data from 2012 to 2017 found that >20% of USA hospitalizations were due to multi-drug-resistant (MDR) bacteria, representing over 41.6 million hospitalizations in that 6 year span and highlighting the severe burden that MDR microbes place on the USA healthcare system (Jernigan et al., 2020). The crisis of AMR is complicated by a dearth of therapeutics currently in development as well as wide-spread improper use of existing antimicrobials, which further contributes toward the prevalence of AMR (Ardal et al., 2020; Kardas et al., 2005). By mobilizing the epithelium’s innate defenses against invasive pathogens, activation of the HIF-autophagy axis would promote clearance of pathogens without encouraging consequent AMR. Such an approach has already been demonstrated to be beneficial in vitro against STm (Tattoli et al., 2012) and M. tuberculosis (Gutierrez et al., 2004), and our findings suggest that such an approach is applicable toward in vivo systems. In addition, in vivo stabilization of HIF has been demonstrated to be protective during chemical models of colitis (Cummins et al., 2008; Robinson et al., 2008), and, as these are highly dependent on the presence and constitution of the gut microbiota (Gkouskou et al., 2014), it is possible that one mode by which HIF stabilizer confers protection is by activating HIF-regulated autophagy/xenophagy to maintain epithelial homeostasis. We are currently exploring the role for pharmacological agents in regulating the HIF-autophagy axis as a means of regulating host-microbial interactions, including those involving MDR bacteria and other clinically relevant microbes.
Taken together, we have found that HIF controls autophagy in the intestinal epithelium and that this interaction promotes xenophagy in a protective fashion during infection by invasive bacteria. These findings reveal a distinctive homeostatic innate immune mechanism for HIF in the intestine. Likewise, this work lays the ground-work for future studies seeking to modulate host-microbial interactions through clinical intervention via the HIF-autophagy axis.
Limitations of the study
Although our group has strived to be comprehensive and discerning with regards to experimental design and interpretation, we do acknowledge some limitations of the current study. Specifically, our group utilizes STm as a model intracellular organism, and it is unclear as to the broad applicability of these findings toward other enteric microbes. The universality of HIF activation by prokaryotes is an area under active investigation by our group. Further, we acknowledge that our mouse model is deficient in both Hif-1α and Hif-2α signaling through deletion of Hif-1β. Although current evidence suggests that HIF-1α is the primary factor through which HIF-driven xenophagy is activated, we cannot fully exclude a role for HIF-2α in the bacterial activation of autophagy/xenophagy. Further studies will seek to elucidate the specific roles of HIF isoforms in governing the HIF-regulated autophagic response to intestinal bacteria. Lastly, the murine model of Salmonella used relies on ablation of the intestinal microbiota through streptomycin pretreatment to diminish colonization resistance. The role of the commensal microbiota in regulating HIF-driven autophagy, therefore, could not be interrogated, and its role in regulating intestinal autophagy is an area of active investigation by our group.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Sean Colgan (sean.colgan@cuanschutz.edu).
Materials availability
This study did not generate unique reagents.
Data and code availability
Section 1: Data
This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table.
Section 2: Code
This paper does not report original code.
Section 3:
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-ACTB | Abcam | Cat#ab8227 |
| anti-LC3B | Sigma-Aldrich | Cat#L7543 |
| anti-p62/SQSTM1 | Abcam | Cat#ab56416 |
| anti-HIF1A | BD | Cat#610959 |
| anti-HIF1B/ARNT | BD | Cat#611079 |
| anti-HIF2A | Novus Biologicals | Cat#NB100-122 |
| anti-BNIP3 | Abcam | Cat#ab109362 |
| anti-TUBB | Abcam | Cat#ab6046 |
| anti-Mouse IgG, HRP-conjugated | MP Biomedical | Cat#0855550 |
| anti-Rabbit IgG, HRP-conjugated | MP Biomedical | Cat#0855676 |
| anti-p62 | MBL | Cat#M162 |
| anti-LC3 | Novus Biological | Cat#NB100-2220 |
| anti-LC3 | Cell Signaling Technology | Cat#2775S |
| Bacterial and virus strains | ||
| Salmonella enterica serovar Typhimurium strain SL1344 | Lab stock | DSM 24522 |
| Chemicals, peptides, and recombinant proteins | ||
| Rapamycin | Thermo-Fisher | Cat#J62473 |
| Streptomycin sulfate | Sigma-Aldrich | Cat#S6501 |
| Phoshate buffered saline (PBS) | Thermo-Fisher | Cat#10010023 |
| LB-Miller broth | BD | Cat#244610 |
| Agar | BD | Cat#214010 |
| Glycerol | Sigma-Aldrich | Cat#G5516-500ML |
| IMDM | Corning | Cat#10-016-CV |
| GlutaMAX | Thermo-Fisher | Cat#35050061 |
| Penicillin/streptomycin | Thermo-Fisher | Cat#15140122 |
| Bovine calf serum | Cytiva | Cat#SH30072.03 |
| TRIzol reagent | Thermo-Fisher | Cat#15596026 |
| iScript Supermix reagent | Bio-Rad | Cat#1708841 |
| 2× Power SYBR Green | Thermo-Fisher | Cat#4367659 |
| SDS-PAGE sample buffer | Bio-Rad | Cat#1610747 |
| HALT Protease Inhibitor | Thermo-Fisher | Cat#78438 |
| Clarity Max ECL reagent | Bio-Rad | Cat#1705062 |
| ProLong Gold Anti-Fade reagent | Thermo-Fisher | Cat#P36930 |
| Critical commercial assays | ||
| Oxodish oxygen sensor plates | Presens | Cat#OD24 |
| EZ-10 DNAaway RNA Miniprep Kit | Bio Basic | Cat#BS88136 |
| Dual Luciferase Assay kit | Promega | Cat#E1960 |
| FuGENE HD | Promega | Cat#E2311 |
| Deposited data | ||
| HIF-1α and HIF-2α ChIP-chip profiling in intestinal epithelial cells | NCBI GEO | Accession#GSE43108 |
| Experimental models: Cell lines | ||
| HeLa | Lab stock | RRID:CVCL_0030 |
| C2BBe1 | Lab stock | RRID:CVCL_1096 |
| Colonoids | Healthy C57BL/6 mice | Generated in-house according to established protocols |
| Experimental models: Organisms/strains | ||
| Hif1b-flox/villin-cre | Lab colony | Previously generated (PMID# 24248342) |
| C57BL/6J wild-type | Lab colony | RRID:IMSR_JAX:000664 |
| ODD-luciferase | Lab colony | Previously described (PMID# 24412613) |
| Oligonucleotides | ||
| Primers | This publication | See Table S1 for sequences |
| anti-HIF1A shRNA | Functional Genomics Facility (Aurora, CO) | Accession#TRCN0000003811 |
| anti-HIF1B shRNA | Functional Genomics Facility (Aurora, CO) | Accession#TRCN0000003819 |
| anti-HIF2A shRNA | Functional Genomics Facility (Aurora, CO) | Accession#TRCN0000003806 |
| Recombinant DNA | ||
| pGL4.22-VEGF-HRE::dLUC | Chi Van Dang, via Addgene | RRID:Addgene_128096 |
| tf-LC3 | Tamotsu Yoshimori, via Addgene | RRID:Addgene_21074 |
| Software and algorithms | ||
| LinRegPCR | Untergasser et al. (PMID# 34433408) | n/a |
| ClustVis | Metsalu and Vilo (PMID# 25969447) | n/a |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mammalian and bacterial cell culture
C2BBe1 is a colorectal adenocarcinoma cell line described previously by our group (Alexeev et al., 2021; Dowdell et al., 2020; Glover et al., 2013). HeLa is a cervical adenocarcinoma cell line with high transfection efficiency. Caco-2 and HeLa cells were maintained in IMDM supplemented with 1× GlutaMAX, 1× Penicillin/Streptomycin, and 10% heat-inactivated bovine calf serum. Murine colonic organoids (“colonoids”) were prepared from healthy, wild-type C57BL/6J mice and maintained using standard protocols (Miyoshi and Stappenbeck, 2013). When necessary, antibiotics were omitted from cell culture media to prevent killing during bacterial treatments. Mammalian cell lines were maintained at 37°C, 5% CO2 in humidified incubators decontaminated regularly. Hypoxia incubations were performed in a humidified controlled atmosphere incubation chamber (Coy Laboratory Products, Inc.) fed with a feed gas of 94% N2, 5% CO2, and 1% O2 and kept at 37°C. All cell lines used were tested for mycoplasma at least once a month using established protocols (Young et al., 2010). Stable knockdown of HIF isoforms were performed via lentiviral transduction according to previously established protocols (Cartwright et al., 2020; Wang et al., 2020) using the TRCN accession numbers given in the Key Resources Table. Plasmids containing shRNA for lentiviral packaging were purchased from the Functional Genomics Core Facility (CU Anschutz, Aurora, CO).
Salmonella enterica subsp. enterica serovar Typhimurium strain SL1344 (STm) was maintained as glycerol stocks (15% (v/v) glycerol) at −80°C and struck weekly onto fresh plates for preparation of liquid cultures. STm was regularly propagated in LB-Miller broth or on LB-Miller agar, each containing 100 μg/mL streptomycin. Subcultures of STm were prepared by inoculating 20 mL of LB-Miller broth (without antibiotics) in a 250 mL baffled shaker flask with 600 μL of an overnight (16–18 h at 37°C, 250 RPM) culture and incubating for approximately 3.5 h at 37°C, 250 RPM. 1 mL of sub-culture was then pelleted at 8,000 × g for 2 min, at which point 900 μL was removed and replaced with 900 μL of sterile PBS, pH 7.4. The bacterial pellet was then gently resuspended and used immediately.
Mouse lines and Salmonella infections
All animal work was performed according to protocols reviewed and approved by the University of Colorado Institutional Animal Care and Use Committee (IACUC). All mice used were between 8 – 12 weeks old and consisted of a mix of male and female littermates. C57BL/6J “wild-type” mice and mice with IEC-specific deletion of Hif1b have been described previously (Glover et al., 2013). Specifically, mice containing a floxed-Hif1b allele were crossed with mice expressing Villin-driven Cre recombinase, resulting in selective loss of Hif1b expression in the intestinal epithelium (Glover et al., 2013). Villin-cre mice used were of the B6.Tg (Vil1-cre)997Gum/J background from Jackson Laboratories (Strain# 004586) with specific transgene expression in the gastrointestinal tract (Madison et al., 2002). Experiments using the streptomycin pretreatment model of STm in vivo infection were performed as described elsewhere (Conway et al., 2013). Briefly, mice were gavaged with 20 mg/mouse streptomycin sulfate in order to abolish colonization resistance, then 24 h later infected orally with STm SL1344. Mice were euthanized 48 h post-infection for collection of tissues. Where indicated, mice were treated daily by intraperitoneal injection with either 8 mg/kg RAP or vehicle control prepared with an equivalent volume of DMSO (Johnson et al., 2013). Histopathological scoring was performed by a trained pathologist in a blinded fashion using established metrics for scoring Salmonella-induced typhlitis/colitis (Barthel et al., 2003).
ODD-luciferase (“ODD-Luc”) mice express luciferase fused C-terminally to the oxygen-dependent degradation domain (ODD) from HIF-1α, resulting in degradation of luciferase under normoxia and, conversely, stabilization under hypoxic conditions. Use of ODD-Luc in intestinal inflammation assays, including in situ imaging of tissue luciferase activity, has been previously described by our group (Campbell et al., 2014).
METHOD DETAILS
Chromatin immunoprecipitation-microarray (ChIP-chip) and analysis
ChIP-chip data used in this paper has been reported elsewhere and is accessible in the Gene Expression Omnibus (GSE43108). Principal component analysis (PCA) of ChIP-chip data sets was performed using the freely available webtool ClustVis (https://biit.cs.ut.ee/clustvis/) (Metsalu and Vilo, 2015).
RNA isolation, cDNA synthesis, and quantitative PCR (qPCR)
Total RNA was prepared from in vitro cell culture samples by lysis and purification using TRIzol reagent. Total RNA was prepared from murine intestinal tissue using an EZ-10 DNAaway RNA Miniprep Kit. Both approaches were performed according to the manufacturers’ instructions, with the resulting RNA quantified using a NanoDrop One instrument (Thermo Scientific). Complementary DNA (cDNA) was synthesized from purified total RNA using iScript Supermix reagent. qPCR was performed using 2× Power SYBR Green master mix on either an Applied Biosystems 7300 (96-well) or 7900HT (384-well) instrument. Primers used for qPCR are given in Table S1. Analysis of qPCR data was performed using LinRegPCR (Untergasser et al., 2021).
Immunoblotting
Samples were first collected by lysing in either RIPA buffer (50 mM Tris-Cl, pH 8.0; 150 mM sodium chloride; 1% (v/v) Triton X-100; 0.5% (w/v) sodium deoxycholate; 0.1% (w/v) sodium dodecyl sulfate (SDS)) or 1× SDS-PAGE sample buffer. In either case, HALT protease inhibitor cocktail and EDTA were included at 1× and 0.5 mM final concentrations, respectively (Thermo# 78438). In vivo samples were collected as done before by scraping intestinal tissue with a clean scalpel and transferring the liberated epithelium to a fresh microcentrifuge tube (Kao et al., 2017; Zheng et al., 2017). Samples were sonicated as needed to reduce viscosity. Samples were analyzed by SDS-PAGE using standard techniques (Brunelle and Green, 2014) and transferred to PVDF for blotting. Blots were blocked in 5% (w/v) nonfat dry milk and probed with antibodies listed in the Key Resources Table. Blots were developed using Clarity Max ECL reagent and imaged using a Bio-Rad ChemiDoc MP instrument.
In vitro infection of IECs
For the observation of HIF stabilization and induction of HIF target genes, epithelial cells were infected by direct addition of bacterial subcultures at a given multiplicity of infection (MOI) based on previously described protocols (Hartmann et al., 2008). For the enumeration of intracellular colony forming units (CFUs) as a metric of intracellular replication, cells were infected briefly with STm then treated with the membrane-impermeable antibiotic gentamicin to kill extracellular bacteria (while intracellular bacteria remain protected) as described elsewhere (Wrande et al., 2016).
Oxodish measurement of in vitro oxygen concentrations
Use of Oxodish sensor plates for real-time in vitro oxygen consumption has been previously described (Kelly et al., 2015). Briefly, Caco-2 cells were grown to confluence on Transwell semi-permeable inserts (0.4μm, Corning# 3470). Cells were then washed and placed into Hanks’ Balanced Salt Solution containing Ca2+/Mg2+ and supplemented with 10mM HEPES (HBSS+) (Cartwright et al., 2021). Cells were then incubated on an Oxodish sensor plate with or without apical STm, and the percent O2 of the surrounding buffer was measured over time. Note that due to the high altitude (>5000 ft.) of the laboratory where experiments were performed, fully oxygenated air is less than the ~21% observed at sea level. Control wells included HBSS+ only, as well as wells containing HBSS+ + bacteria without IECs.
In vitro measurement of HIF-driven luciferase
HeLa cells were transiently transfected with pGL4.22-VEGF-HRE:dLUC (Walton et al., 2018) using FuGENE HD transfection reagent according to the manufacturer’s instructions. 24 h after transfection, cells were infected with STm for six hours then lysed using a Dual Luciferase Assay Kit and analyzed on 96-well plates in triplicate using a Promega GloMax 96-well luminometer.
Immunofluorescence
Caco-2 cells were plated onto sterile glass coverslips in 24-well plates and treated with/without hypoxia. In some cases, Caco-2 cells expressed tf-LC3 (Kimura et al., 2007). Cells were fixed using 4% (w/v) paraformaldehyde, then permeabilized using either 0.1% saponin or 0.1% Triton X-100 and stained with primary and secondary antibodies described in Key Reagents Table. Cells were then protected using ProLong Anti-Fade reagent and imaged using a Zeiss Axio Imager A1 microscope. Visible puncta were manually quantified in a blinded fashion and, afterwards, experimental groups were compared.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical tests were performed using either Microsoft Excel or GraphPad Prism. Details on statistical analyses, including tests performed, can be found in the figure legends.
Supplementary Material
Highlights.
Autophagy genes are HIF targets in intestinal epithelial cells
S. Typhimurium induces HIF stabilization and transcriptional activity
HIF signaling is protective against Salmonella both in vitro and in vivo
Xenophagy response to Salmonella infection is dependent on IEC HIF activity
ACKNOWLEDGMENTS
This work was supported by NIH (grants DK050189, DK103712, DK104713, DK095491, and AI007405), the Crohn’s and Colitis Foundation, and by the Veterans Administration (awards BX002182 and BX005710).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111409.
REFERENCES
- Alexeev EE, Dowdell AS, Henen MA, Lanis JM, Lee JS, Cartwright IM, Schaefer REM, Ornelas A, Onyiah JC, Vögeli B, and Colgan SP (2021). Microbial-derived indoles inhibit neutrophil myeloperoxidase to diminish bystander tissue damage. FASEB J. 35, e21552. 10.1096/fj.202100027R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ananthakrishnan AN, Bernstein CN, Iliopoulos D, Macpherson A, Neurath MF, Ali RAR, Vavricka SR, and Fiocchi C (2018). Environmental triggers in IBD: a review of progress and evidence. Nat. Rev. Gastroenterol. Hepatol 15, 39–49. 10.1038/nrgastro.2017.136. [DOI] [PubMed] [Google Scholar]
- Antimicrobial Resistance Collaborators; Ikuta KS, Sharara F, Swetschinski L, Robles Aguilar G, Gray A, Han C, Bisignano C, Rao P, Wool E, et al. (2022). Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. Lancet 399, 629–655. 10.1016/S0140-6736(21)02724-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardal C, Balasegaram M, Laxminarayan R, McAdams D, Outterson K, Rex JH, and Sumpradit N (2020). Antibiotic development - economic, regulatory and societal challenges. Nat. Rev. Microbiol 18, 267–274. 10.1038/s41579-019-0293-3. [DOI] [PubMed] [Google Scholar]
- Barnich N, Carvalho FA, Glasser AL, Darcha C, Jantscheff P, Allez M, Peeters H, Bommelaer G, Desreumaux P, Colombel JF, and Darfeuille-Michaud A (2007). CEACAM6 acts as a receptor for adherent-invasive E. coli, supporting ileal mucosa colonization in Crohn disease. J. Clin. Invest 117, 1566–1574. 10.1172/JCI30504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barthel M, Hapfelmeier S, Quintanilla-Martínez L, Kremer M, Rohde M, Hogardt M, Pfeffer K, Rüssmann H, and Hardt WD (2003). Pretreatment of mice with streptomycin provides a Salmonella enterica serovar Typhimurium colitis model that allows analysis of both pathogen and host. Infect. Immun 71, 2839–2858. 10.1128/IAI.71.5.2839-2858.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouysségur J, and Mazure NM (2009). Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell Biol 29, 2570–2581. 10.1128/MCB.00166-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birmingham CL, Smith AC, Bakowski MA, Yoshimori T, and Brumell JH (2006). Autophagy controls Salmonella infection in response to damage to the Salmonella-containing vacuole. J. Biol. Chem 281, 11374–11383. 10.1074/jbc.M509157200. [DOI] [PubMed] [Google Scholar]
- Brest P, Lapaquette P, Souidi M, Lebrigand K, Cesaro A, Vouret-Craviari V, Mari B, Barbry P, Mosnier JF, Hébuterne X, et al. (2011). A synonymous variant in IRGM alters a binding site for miR-196 and causes deregulation of IRGM-dependent xenophagy in Crohn’s disease. Nat. Genet 43, 242–245. 10.1038/ng.762. [DOI] [PubMed] [Google Scholar]
- Brunelle JL, and Green R (2014). One-dimensional SDS-polyacrylamide gel electrophoresis (1D SDS-PAGE). Methods Enzymol. 541, 151–159. 10.1016/B978-0-12-420119-4.00012-4. [DOI] [PubMed] [Google Scholar]
- Campbell EL, Bruyninckx WJ, Kelly CJ, Glover LE, McNamee EN, Bowers BE, Bayless AJ, Scully M, Saeedi BJ, Golden-Mason L, et al. (2014). Transmigrating neutrophils shape the mucosal microenvironment through localized oxygen depletion to influence resolution of inflammation. Immunity 40, 66–77. 10.1016/j.immuni.2013.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cane G, Ginouvès A, Marchetti S, Buscà R, Pouysségur J, Berra E, Hofman P, and Vouret-Craviari V (2010). HIF-1alpha mediates the induction of IL-8 and VEGF expression on infection with Afa/Dr diffusely adhering E. coli and promotes EMT-like behaviour. Cell Microbiol. 12, 640–653. 10.1111/j.1462-5822.2009.01422.x. [DOI] [PubMed] [Google Scholar]
- Carroll VA, and Ashcroft M (2006). Role of hypoxia-inducible factor (HIF)-1alpha versus HIF-2alpha in the regulation of HIF target genes in response to hypoxia, insulin-like growth factor-I, or loss of von Hippel-Lindau function: implications for targeting the HIF pathway. Cancer Res. 66, 6264–6270. 10.1158/0008-5472.CAN-05-2519. [DOI] [PubMed] [Google Scholar]
- Cartwright IM, Curtis VF, Lanis JM, Alexeev EE, Welch N, Goldberg MS, Schaefer REM, Gao RY, Chun C, Fennimore B, et al. (2020). Adaptation to inflammatory acidity through neutrophil-derived adenosine regulation of SLC26A3. Mucosal Immunol. 13, 230–244. 10.1038/s41385-019-0237-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartwright IM, Dowdell AS, Lanis JM, Brink KR, Mu A, Kostelecky RE, Schaefer REM, Welch N, Onyiah JC, Hall CHT, et al. (2021). Mucosal acidosis elicits a unique molecular signature in epithelia and intestinal tissue mediated by GPR31-induced CREB phosphorylation. Proc. Natl. Acad. Sci. USA 118, e2023871118. 10.1073/pnas.2023871118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan S, Mandell MA, and Deretic V (2015). IRGM governs the core autophagy machinery to conduct antimicrobial defense. Mol. Cell 58, 507–521. 10.1016/j.molcel.2015.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleynen I, Boucher G, Jostins L, Schumm LP, Zeissig S, Ahmad T, Andersen V, Andrews JM, Annese V, Brand S, et al. (2016). Inherited determinants of Crohn’s disease and ulcerative colitis phenotypes: a genetic association study. Lancet 387, 156–167. 10.1016/S0140-6736(15)00465-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colgan SP, Campbell EL, and Kominsky DJ (2016). Hypoxia and mucosal inflammation. Annu. Rev. Pathol 11, 77–100. 10.1146/annurev-pathol-012615-044231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colgan SP, Furuta GT, and Taylor CT (2020). Hypoxia and innate immunity: keeping up with the HIFsters. Annu. Rev. Immunol 38, 341–363. 10.1146/annurev-immunol-100819-121537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway KL, Kuballa P, Song JH, Patel KK, Castoreno AB, Yilmaz OH, Jijon HB, Zhang M, Aldrich LN, Villablanca EJ, et al. (2013). Atg16l1 is required for autophagy in intestinal epithelial cells and protection of mice from Salmonella infection. Gastroenterology 145, 1347–1357. 10.1053/j.gastro.2013.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins EP, Seeballuck F, Keely SJ, Mangan NE, Callanan JJ, Fallon PG, and Taylor CT (2008). The hydroxylase inhibitor dimethyloxalylglycine is protective in a murine model of colitis. Gastroenterology 134, 156–165. 10.1053/j.gastro.2007.10.012. [DOI] [PubMed] [Google Scholar]
- Dengler VL, Galbraith M, and Espinosa JM (2014). Transcriptional regulation by hypoxia inducible factors. Crit. Rev. Biochem. Mol. Biol 49, 1–15. 10.3109/10409238.2013.838205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikic I, and Elazar Z (2018). Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol 19, 349–364. 10.1038/s41580-018-0003-4. [DOI] [PubMed] [Google Scholar]
- Dowdell AS, Cartwright IM, Goldberg MS, Kostelecky R, Ross T, Welch N, Glover LE, and Colgan SP (2020). The HIF target ATG9A is essential for epithelial barrier function and tight junction biogenesis. Mol. Biol. Cell 31, 2249–2258. 10.1091/mbc.E20-05-0291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan L, Yi M, Chen J, Li S, and Chen W (2016). Mycobacterium tuberculosis EIS gene inhibits macrophage autophagy through up-regulation of IL-10 by increasing the acetylation of histone H3. Biochem. Biophys. Res. Commun 473, 1229–1234. 10.1016/j.bbrc.2016.04.045. [DOI] [PubMed] [Google Scholar]
- Dumortier J, Lapalus MG, Guillaud O, Poncet G, Gagnieu MC, Partensky C, and Scoazec JY (2008). Everolimus for refractory Crohn’s disease: a case report. Inflamm. Bowel Dis 14, 874–877. 10.1002/ibd.20395. [DOI] [PubMed] [Google Scholar]
- Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O’Rourke J, Mole DR, Mukherji M, Metzen E, Wilson MI, Dhanda A, et al. (2001). C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 107, 43–54. 10.1016/s0092-8674(01)00507-4. [DOI] [PubMed] [Google Scholar]
- Ganesan R, Hos NJ, Gutierrez S, Fischer J, Stepek JM, Daglidu E, Krönke M, and Robinson N (2017). Salmonella Typhimurium disrupts Sirt1/AMPK checkpoint control of mTOR to impair autophagy. PLoS Pathog. 13, e1006227. 10.1371/journal.ppat.1006227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Hernandez V, Neumann PA, Koch S, Lyons R, Nusrat A, and Parkos CA (2021). Systematic scoring analysis for intestinal inflammation in a murine dextran sodium sulfate-induced colitis model. J. Vis. Exp 10.3791/62135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, Thomas G, Philpott DJ, and Sansonetti PJ (2003). Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J. Biol. Chem 278, 8869–8872. 10.1074/jbc.C200651200. [DOI] [PubMed] [Google Scholar]
- Gkouskou KK, Deligianni C, Tsatsanis C, and Eliopoulos AG (2014). The gut microbiota in mouse models of inflammatory bowel disease. Front. Cell. Infect. Microbiol 4, 28. 10.3389/fcimb.2014.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover LE, Bowers BE, Saeedi B, Ehrentraut SF, Campbell EL, Bayless AJ, Dobrinskikh E, Kendrick AA, Kelly CJ, Burgess A, et al. (2013). Control of creatine metabolism by HIF is an endogenous mechanism of barrier regulation in colitis. Proc. Natl. Acad. Sci. USA 110, 19820–19825. 10.1073/pnas.1302840110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover LE, Lee JS, and Colgan SP (2016). Oxygen metabolism and barrier regulation in the intestinal mucosa. J. Clin. Invest 126, 3680–3688. 10.1172/JCI84429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb RA, and Mentzer RM Jr. (2013). Autophagy: an affair of the heart. Heart Fail. Rev 18, 575–584. 10.1007/s10741-012-9367-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groulx JF, Khalfaoui T, Benoit YD, Bernatchez G, Carrier JC, Basora N, and Beaulieu JF (2012). Autophagy is active in normal colon mucosa. Autophagy 8, 893–902. 10.4161/auto.19738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, and Deretic V (2004). Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 119, 753–766. 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- Gutierrez O, Pipaon C, Inohara N, Fontalba A, Ogura Y, Prosper F, Nunez G, and Fernandez-Luna JL (2002). Induction of Nod2 in myelomonocytic and intestinal epithelial cells via nuclear factor-kappa B activation. J. Biol. Chem 277, 41701–41705. 10.1074/jbc.M206473200. [DOI] [PubMed] [Google Scholar]
- Hampe J, Franke A, Rosenstiel P, Till A, Teuber M, Huse K, Albrecht M, Mayr G, De La Vega FM, Briggs J, et al. (2007). A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat. Genet 89, 207–211. 10.1038/ng1954. [DOI] [PubMed] [Google Scholar]
- Hartmann H, Eltzschig HK, Wurz H, Hantke K, Rakin A, Yazdi AS, Matteoli G, Bohn E, Autenrieth IB, Karhausen J, et al. (2008). Hypoxia-independent activation of HIF-1 by enterobacteriaceae and their siderophores. Gastroenterology 134, 756–767. 10.1053/j.gastro.2007.12.008. [DOI] [PubMed] [Google Scholar]
- Hase K, Kawano K, Nochi T, Pontes GS, Fukuda S, Ebisawa M, Kadokura K, Tobe T, Fujimura Y, Kawano S, et al. (2009). Uptake through glycoprotein 2 of FimH(+) bacteria by M cells initiates mucosal immune response. Nature 462, 226–230. 10.1038/nature08529. [DOI] [PubMed] [Google Scholar]
- Homer CR, Richmond AL, Rebert NA, Achkar JP, and McDonald C (2010). ATG16L1 and NOD2 interact in an autophagy-dependent antibacterial pathway implicated in Crohn’s disease pathogenesis. Gastroenterology 139, 1630–1632. 10.1053/j.gastro.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, and Brumell JH (2014). Bacteria-autophagy interplay: a battle for survival. Nat. Rev. Microbiol 12, 101–114. 10.1038/nrmi-cro3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugot JP, Chamaillard M, Zouali H, Lesage S, Cézard JP, Belaiche J, Almer S, Tysk C, O’Morain CA, Gassull M, et al. (2001). Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 411, 599–603. 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- Jairath V, and Feagan BG (2020). Global burden of inflammatory bowel disease. Lancet. Gastroenterol. Hepatol 5, 2–3. 10.1016/S2468-1253(19)30358-9. [DOI] [PubMed] [Google Scholar]
- Jernigan JA, Hatfield KM, Wolford H, Nelson RE, Olubajo B, Reddy SC, McCarthy N, Paul P, McDonald LC, Kallen A, et al. (2020). Multi-drug-resistant bacterial infections in U.S. Hospitalized patients, 2012-2017. N. Engl. J. Med 382, 1309–1319. 10.1056/NEJMoa1914433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang P, and Mizushima N (2015). LC3- and p62-based biochemical methods for the analysis of autophagy progression in mammalian cells. Methods 75, 13–18. 10.1016/j.ymeth.2014.11.021. [DOI] [PubMed] [Google Scholar]
- Johnson SC, Yanos ME, Kayser EB, Quintana A, Sangesland M, Castanza A, Uhde L, Hui J, Wall VZ, Gagnidze A, et al. (2013). mTOR inhibition alleviates mitochondrial disease in a mouse model of Leigh syndrome. Science 342, 1524–1528. 10.1126/science.1244360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao DJ, Saeedi BJ, Kitzenberg D, Burney KM, Dobrinskikh E, Battista KD, Vázquez-Torres A, Colgan SP, and Kominsky DJ (2017). Intestinal epithelial ecto-5’-nucleotidase (CD73) regulates intestinal colonization and infection by nontyphoidal Salmonella. Infect. Immun 85, e01022–16. 10.1128/IAI.01022-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kardas P, Devine S, Golembesky A, and Roberts C (2005). A systematic review and meta-analysis of misuse of antibiotic therapies in the community. Int. J. Antimicrob. Agents 26, 106–113. 10.1016/j.ijantimicag.2005.04.017. [DOI] [PubMed] [Google Scholar]
- Karhausen J, Furuta GT, Tomaszewski JE, Johnson RS, Colgan SP, and Haase VH (2004). Epithelial hypoxia-inducible factor-1 is protective in murine experimental colitis. J. Clin. Invest 114, 1098–1106. 10.1172/JCI21086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly CJ, Zheng L, Campbell EL, Saeedi B, Scholz CC, Bayless AJ, Wilson KE, Glover LE, Kominsky DJ, Magnuson A, et al. (2015). Cross-talk between microbiota-derived short-chain fatty acids and intestinal epithelial HIF augments tissue barrier function. Cell Host Microbe 17, 662–671. 10.1016/j.chom.2015.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura S, Noda T, and Yoshimori T (2007). Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 3, 452–460. 10.4161/auto.4451. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Abdel-Aziz AK, Abdelfatah S, Abdellatif M, Abdoli A, Abel S, Abeliovich H, Abildgaard MH, Abudu YP, Acevedo-Arozena A, et al. (2021). Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 17, 1–382. 10.1080/15548627.2020.1797280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh MY, and Powis G (2012). Passing the baton: the HIF switch. Trends Biochem. Sci 37, 364–372. 10.1016/j.tibs.2012.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuballa P, Huett A, Rioux JD, Daly MJ, and Xavier RJ (2008). Impaired autophagy of an intracellular pathogen induced by a Crohn’s disease associated ATG16L1 variant. PLoS One 3, e3391. 10.1371/journal.pone.0003391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuma A, Komatsu M, and Mizushima N (2017). Autophagy-monitoring and autophagy-deficient mice. Autophagy 13, 1619–1628. 10.1080/15548627.2017.1343770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassen KG, and Xavier RJ (2017). Genetic control of autophagy underlies pathogenesis of inflammatory bowel disease. Mucosal Immunol. 10, 589–597. 10.1038/mi.2017.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legendre C, Mooij MJ, Adams C, and O’Gara F (2011). Impaired expression of hypoxia-inducible factor-1alpha in cystic fibrosis airway epithelial cells - a role for HIF-1 in the pathophysiology of CF? J. Cyst. Fibros 10, 286–290. 10.1016/j.jcf.2011.02.005. [DOI] [PubMed] [Google Scholar]
- Li X, Bleumink-Pluym NMC, Luijkx YMCA, Wubbolts RW, van Putten JPM, and Strijbis K (2019). MUC1 is a receptor for the Salmonella SiiE adhesin that enables apical invasion into enterocytes. PLoS Pathog. 15, e1007566. 10.1371/journal.ppat.1007566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madison BB, Dunbar L, Qiao XT, Braunstein K, Braunstein E, and Gumucio DL (2002). Cis elements of the villin gene control expression in restricted domains of the vertical (crypt) and horizontal (duodenum, cecum) axes of the intestine. J. Biol. Chem 277, 33275–33283. 10.1074/jbc.M204935200. [DOI] [PubMed] [Google Scholar]
- Manresa MC, and Taylor CT (2017). Hypoxia inducible factor (HIF) hydroxylases as regulators of intestinal epithelial barrier function. Cell. Mol. Gastroenterol. Hepatol 3, 303–315. 10.1016/j.jcmgh.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massey DCO, Bredin F, and Parkes M (2008). Use of sirolimus (rapamycin) to treat refractory Crohn’s disease. Gut 57, 1294–1296. 10.1136/gut.2008.157297. [DOI] [PubMed] [Google Scholar]
- Mathew R, Karantza-Wadsworth V, and White E (2007). Role of autophagy in cancer. Nat. Rev. Cancer 7, 961–967. 10.1038/nrc2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maunoury R, Robine S, Pringault E, Léonard N, Gaillard JA, and Louvard D (1992). Developmental regulation of villin gene expression in the epithelial cell lineages of mouse digestive and urogenital tracts. Development 115, 717–728. 10.1242/dev.115.3.717. [DOI] [PubMed] [Google Scholar]
- Mehrpour M, Esclatine A, Beau I, and Codogno P (2010). Overview of macroautophagy regulation in mammalian cells. Cell Res. 20, 748–762. 10.1038/cr.2010.82. [DOI] [PubMed] [Google Scholar]
- Metsalu T, and Vilo J (2015). ClustVis: a web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 43, W566–W570. 10.1093/nar/gkv468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimouna S, Bazin M, Mograbi B, Darfeuille-Michaud A, Brest P, Hofman P, and Vouret-Craviari V (2014). HIF1A regulates xenophagic degradation of adherent and invasive Escherichia coli (AIEC). Autophagy 10, 2333–2345. 10.4161/15548627.2014.984275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi H, and Stappenbeck TS (2013). In vitro expansion and genetic modification of gastrointestinal stem cells in spheroid culture. Nat. Protoc 8, 2471–2482. 10.1038/nprot.2013.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Kuma A, Kobayashi Y, Yamamoto A, Matsubae M, Takao T, Natsume T, Ohsumi Y, and Yoshimori T (2003). Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J. Cell Sci 116, 1679–1688. 10.1242/jcs.00381. [DOI] [PubMed] [Google Scholar]
- Molodecky NA, Soon IS, Rabi DM, Ghali WA, Ferris M, Chernoff G, Benchimol EI, Panaccione R, Ghosh S, Barkema HW, and Kaplan GG (2012). Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology 142, 46–54.e42, quiz e30. 10.1053/j.gastro.2011.10.001. [DOI] [PubMed] [Google Scholar]
- Kelly CJ, Glover LE, Campbell EL, Kominsky DJ, Ehrentraut SF, Bowers BE, Bayless AJ, Saeedi BJ, and Colgan SP (2013). Fundamental role for HIF-1α in constitutive expression of human β defensin-1. Mucosal Immunol. 6, 1110–1118. 10.1038/mi.2013.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa M, Yoshimori T, Suzuki T, Sagara H, Mizushima N, and Sasakawa C (2005). Escape of intracellular Shigella from autophagy. Science 307, 727–731. 10.1126/science.1106036. [DOI] [PubMed] [Google Scholar]
- Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, Britton H, Moran T, Karaliuskas R, Duerr RH, et al. (2001). A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature 411, 603–606. 10.1038/35079114. [DOI] [PubMed] [Google Scholar]
- Ogura Y, Lala S, Xin W, Smith E, Dowds TA, Chen FF, Zimmermann E,Tretiakova M, Cho JH, Hart J, et al. (2003). Expression of NOD2 in Paneth cells: a possible link to Crohn’s ileitis. Gut 52, 1591–1597. 10.1136/gut.52.11.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen KA, Meyer CB, Bouton AH, and Casanova JE (2014). Activation of focal adhesion kinase by Salmonella suppresses autophagy via an Akt/mTOR signaling pathway and promotes bacterial survival in macrophages. PLoS Pathog. 10, e1004159. 10.1371/journal.ppat.1004159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkes M, Barrett JC, Prescott NJ, Tremelling M, Anderson CA, Fisher SA, Roberts RG, Nimmo ER, Cummings FR, Soars D, et al. (2007). Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn’s disease susceptibility. Nat. Genet 30, 830–832. 10.1038/ng2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petnicki-Ocwieja T, Hrncir T, Liu YJ, Biswas A, Hudcovic T, Tlaskalova-Hogenova H, and Kobayashi KS (2009). Nod2 is required for the regulation of commensal microbiota in the intestine. Proc. Natl. Acad. Sci. USA 106, 15813–15818. 10.1073/pnas.0907722106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyssonnaux C, Boutin AT, Zinkernagel AS, Datta V, Nizet V, and Johnson RS (2008). Critical role of HIF-1alpha in keratinocyte defense against bacterial infection. J. Invest. Dermatol 128, 1964–1968. 10.1038/jid.2008.27. [DOI] [PubMed] [Google Scholar]
- Peyssonnaux C, Datta V, Cramer T, Doedens A, Theodorakis EA, Gallo RL, Hurtado-Ziola N, Nizet V, and Johnson RS (2005). HIF-1alpha expression regulates the bactericidal capacity of phagocytes. J. Clin. Invest 115, 1806–1815. 10.1172/JCI23865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A, Green T, Kuballa P, Barmada MM, Datta LW, et al. (2007). Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat. Genet 30, 596–604. 10.1038/ng2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson A, Keely S, Karhausen J, Gerich ME, Furuta GT, and Colgan SP (2008). Mucosal protection by hypoxia-inducible factor prolyl hydroxylase inhibition. Gastroenterology 134, 145–155. 10.1053/j.gastro.2007.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robrahn L, Dupont A, Jumpertz S, Zhang K, Holland CH, Guillaume J, Rappold S, Roth J, Cerovic V, Saez-Rodriguez J, et al. (2022). Stabilization but No functional influence of HIF-1alpha expression in the intestinal epithelium during Salmonella Typhimurium infection. Infect. Immun 90, e0022221. 10.1128/iai.00222-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh T, Fujita N, Hayashi T, Takahara K, Satoh T, Lee H, Matsunaga K, Kageyama S, Omori H, Noda T, et al. (2009). Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc. Natl. Acad. Sci. USA 106, 20842–20846. 10.1073/pnas.0911267106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma M, Machuy N, Böhme L, Karunakaran K, Mäurer AP, Meyer TF, and Rudel T (2011). HIF-1alpha is involved in mediating apoptosis resistance to Chlamydia trachomatis-infected cells. Cell Microbiol. 13, 1573–1585. 10.1111/j.1462-5822.2011.01642.x. [DOI] [PubMed] [Google Scholar]
- Steele-Mortimer O (2008). Infection of epithelial cells with Salmonella enterica. Methods Mol. Biol 431, 201–211. 10.1007/978-1-60327-032-8_16. [DOI] [PubMed] [Google Scholar]
- Tattoli I, Sorbara MT, Vuckovic D, Ling A, Soares F, Carneiro LAM, Yang C, Emili A, Philpott DJ, and Girardin SE (2012). Amino acid starvation induced by invasive bacterial pathogens triggers an innate host defense program. Cell Host Microbe 11, 563–575. 10.1016/j.chom.2012.04.012. [DOI] [PubMed] [Google Scholar]
- Taylor CT, and Colgan SP (2017). Regulation of immunity and inflammation by hypoxia in immunological niches. Nat. Rev. Immunol 17, 774–785. 10.1038/nri.2017.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travassos LH, Carneiro LAM, Ramjeet M, Hussey S, Kim YG, Magalhães JG, Yuan L, Soares F, Chea E, Le Bourhis L, et al. (2010). Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat. Immunol 11, 55–62. 10.1038/ni.1823. [DOI] [PubMed] [Google Scholar]
- Untergasser A, Ruijter JM, Benes V, and van den Hoff MJB (2021). Web-based LinRegPCR: application for the visualization and analysis of (RT)-qPCR amplification and melting data. BMC Bioinf. 22, 398. 10.1186/s12859-021-04306-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton ZE, Patel CH, Brooks RC, Yu Y, Ibrahim-Hashim A, Riddle M, Porcu A, Jiang T, Ecker BL, Tameire F, et al. (2018). Acid suspends the circadian clock in hypoxia through inhibition of mTOR. Cell 174, 72–87.e32. 10.1016/j.cell.2018.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang RX, Lee JS, Campbell EL, and Colgan SP (2020). Microbiota-derived butyrate dynamically regulates intestinal homeostasis through regulation of actin-associated protein synaptopodin. Proc. Natl. Acad. Sci. USA 117, 11648–11657. 10.1073/pnas.1917597117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werth N, Beerlage C, Rosenberger C, Yazdi AS, Edelmann M, Amr A, Bernhardt W, von Eiff C, Becker K, Schäfer A, et al. (2010). Activation of hypoxia inducible factor 1 is a general phenomenon in infections with human pathogens. PLoS One 5, e11576. 10.1371/journal.pone.0011576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong E, and Cuervo AM (2010). Autophagy gone awry in neurodegenerative diseases. Nat. Neurosci 13, 805–811. 10.1038/nn.2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrande M, Andrews-Polymenis H, Twedt DJ, Steele-Mortimer O, Porwollik S, McClelland M, and Knodler LA (2016). Genetic determinants of Salmonella enterica serovar Typhimurium proliferation in the cytosol of epithelial cells. Infect. Immun 84, 3517–3526. 10.1128/IAI.00734-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto S, and Ma X (2009). Role of Nod2 in the development of Crohn’s disease. Microbes Infect. 11, 912–918. 10.1016/j.micinf.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, and Klionsky DJ (2010). Eaten alive: a history of macroautophagy. Nat. Cell Biol 12, 814–822. 10.1038/ncb0910-814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye C, Li Q, Li X, Park CG, He Y, Zhang Y, Wu B, Xue Y, Yang K, Lv Y, et al. (2019). Salmonella enterica serovar Typhimurium interacts with CD209 receptors to promote host dissemination and infection. Infect. Immun 87, e00100–19. 10.1128/IAI.00100-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa Y, Ogawa M, Hain T, Yoshida M, Fukumatsu M, Kim M, Mimuro H, Nakagawa I, Yanagawa T, Ishii T, et al. (2009). Listeria monocytogenes ActA-mediated escape from autophagic recognition. Nat. Cell Biol 11, 1233–1240. 10.1038/ncb1967. [DOI] [PubMed] [Google Scholar]
- Young L, Sung J, Stacey G, and Masters JR (2010). Detection of Mycoplasma in cell cultures. Nat. Protoc 5, 929–934. 10.1038/nprot.2010.43. [DOI] [PubMed] [Google Scholar]
- Zheng L, Kelly CJ, Battista KD, Schaefer R, Lanis JM, Alexeev EE, Wang RX, Onyiah JC, Kominsky DJ, and Colgan SP (2017). Microbial-derived butyrate promotes epithelial barrier function through IL-10 receptor-dependent repression of claudin-2. J. Immunol 199, 2976–2984. 10.4049/jimmunol.1700105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Section 1: Data
This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table.
Section 2: Code
This paper does not report original code.
Section 3:
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-ACTB | Abcam | Cat#ab8227 |
| anti-LC3B | Sigma-Aldrich | Cat#L7543 |
| anti-p62/SQSTM1 | Abcam | Cat#ab56416 |
| anti-HIF1A | BD | Cat#610959 |
| anti-HIF1B/ARNT | BD | Cat#611079 |
| anti-HIF2A | Novus Biologicals | Cat#NB100-122 |
| anti-BNIP3 | Abcam | Cat#ab109362 |
| anti-TUBB | Abcam | Cat#ab6046 |
| anti-Mouse IgG, HRP-conjugated | MP Biomedical | Cat#0855550 |
| anti-Rabbit IgG, HRP-conjugated | MP Biomedical | Cat#0855676 |
| anti-p62 | MBL | Cat#M162 |
| anti-LC3 | Novus Biological | Cat#NB100-2220 |
| anti-LC3 | Cell Signaling Technology | Cat#2775S |
| Bacterial and virus strains | ||
| Salmonella enterica serovar Typhimurium strain SL1344 | Lab stock | DSM 24522 |
| Chemicals, peptides, and recombinant proteins | ||
| Rapamycin | Thermo-Fisher | Cat#J62473 |
| Streptomycin sulfate | Sigma-Aldrich | Cat#S6501 |
| Phoshate buffered saline (PBS) | Thermo-Fisher | Cat#10010023 |
| LB-Miller broth | BD | Cat#244610 |
| Agar | BD | Cat#214010 |
| Glycerol | Sigma-Aldrich | Cat#G5516-500ML |
| IMDM | Corning | Cat#10-016-CV |
| GlutaMAX | Thermo-Fisher | Cat#35050061 |
| Penicillin/streptomycin | Thermo-Fisher | Cat#15140122 |
| Bovine calf serum | Cytiva | Cat#SH30072.03 |
| TRIzol reagent | Thermo-Fisher | Cat#15596026 |
| iScript Supermix reagent | Bio-Rad | Cat#1708841 |
| 2× Power SYBR Green | Thermo-Fisher | Cat#4367659 |
| SDS-PAGE sample buffer | Bio-Rad | Cat#1610747 |
| HALT Protease Inhibitor | Thermo-Fisher | Cat#78438 |
| Clarity Max ECL reagent | Bio-Rad | Cat#1705062 |
| ProLong Gold Anti-Fade reagent | Thermo-Fisher | Cat#P36930 |
| Critical commercial assays | ||
| Oxodish oxygen sensor plates | Presens | Cat#OD24 |
| EZ-10 DNAaway RNA Miniprep Kit | Bio Basic | Cat#BS88136 |
| Dual Luciferase Assay kit | Promega | Cat#E1960 |
| FuGENE HD | Promega | Cat#E2311 |
| Deposited data | ||
| HIF-1α and HIF-2α ChIP-chip profiling in intestinal epithelial cells | NCBI GEO | Accession#GSE43108 |
| Experimental models: Cell lines | ||
| HeLa | Lab stock | RRID:CVCL_0030 |
| C2BBe1 | Lab stock | RRID:CVCL_1096 |
| Colonoids | Healthy C57BL/6 mice | Generated in-house according to established protocols |
| Experimental models: Organisms/strains | ||
| Hif1b-flox/villin-cre | Lab colony | Previously generated (PMID# 24248342) |
| C57BL/6J wild-type | Lab colony | RRID:IMSR_JAX:000664 |
| ODD-luciferase | Lab colony | Previously described (PMID# 24412613) |
| Oligonucleotides | ||
| Primers | This publication | See Table S1 for sequences |
| anti-HIF1A shRNA | Functional Genomics Facility (Aurora, CO) | Accession#TRCN0000003811 |
| anti-HIF1B shRNA | Functional Genomics Facility (Aurora, CO) | Accession#TRCN0000003819 |
| anti-HIF2A shRNA | Functional Genomics Facility (Aurora, CO) | Accession#TRCN0000003806 |
| Recombinant DNA | ||
| pGL4.22-VEGF-HRE::dLUC | Chi Van Dang, via Addgene | RRID:Addgene_128096 |
| tf-LC3 | Tamotsu Yoshimori, via Addgene | RRID:Addgene_21074 |
| Software and algorithms | ||
| LinRegPCR | Untergasser et al. (PMID# 34433408) | n/a |
| ClustVis | Metsalu and Vilo (PMID# 25969447) | n/a |