

Abstract
The development of multidrug resistance in cancer chemotherapy is a major obstacle to the effective treatment of human malignant tumors. Several epidemiological studies have demonstrated that inflammation is closely related to cancer and plays a key role in the development of both solid and liquid tumors. Therefore, targeting inflammation and the molecules involved in the inflammatory process may be a good strategy for treating drug-resistant tumors. In this review, we discuss the molecular mechanisms underlying inflammation in regulating anticancer drug resistance by modulating drug action and drug-mediated cell death pathways. Inflammation alters the effectiveness of drugs through modulation of the expression of multidrug efflux transporters (e.g., ABCG2, ABCB1, and ABCC1) and drug-metabolizing enzymes (e.g., CYP1A2 and CYP3A4). In addition, inflammation can protect cancer cells from drug-mediated cell death by regulating DNA damage repair, downstream adaptive response (e.g., apoptosis, autophagy, and oncogenic bypass signaling), and tumor microenvironment. Intriguingly, manipulating inflammation may affect drug resistance through various molecular mechanisms validated by in vitro/in vivo models. In this review, we aim to summarize the underlying molecular mechanisms that inflammation participates in cancer drug resistance and discuss the potential clinical strategies targeting inflammation to overcome drug resistance.
1. Introduction
Current cancer treatments (e.g., surgery, chemotherapy, radiotherapy, targeted therapy, and immune therapy) benefit an increasing number of patients who suffer from cancer [1]. Still, the effectiveness of these strategies is limited by drug resistance, which remains a primary obstacle to the curative treatment of various cancers [2]. Resistance to anticancer drugs can be divided into two categories: intrinsic and acquired drug resistance [3, 4]. Intrinsic resistance indicates that resistance regulators are present in a large number of tumor cells prior to chemotherapy, rendering treatment ineffective. Acquired resistance may develop during treatment as a result of nongenetic changes, which enhance tumor cell survival [5, 6]. The anticancer drug resistance mechanism involves many aspects, such as deregulated drug transport, altered target proteins/receptors, and abnormal regulation of cellular metabolic pathways [7, 8]. Of note, several new cancer treatment strategies have been applied to the clinic in response to the above mechanisms.
Inflammation is an immune response to bodily injury that can fight infection and trauma by removing harmful factors and damaged tissue, thereby repairing the tissue and returning it to normal [9]. In detail, in the early or acute phase of inflammation, pathogen-associated molecular patterns (PAMPs) are recognized by tissue macrophages or mast cells, which in turn activate the secretion of inflammatory cytokines, chemokines, vasoactive amines, and other substances, thereby enhancing the immune response around the blood vessels [10, 11]. Activated inflammatory cells produce anti-inflammatory cytokines as well as pro-inflammatory cytokines [12, 13]. The former mainly includes IL-4, IL-13, IL-10, and TGF-β, and the latter mainly includes TNF-α, IL-1β, IL-2, IL-6, IL-8, IL-17, and IFN-γ. Pro-inflammatory and anti-inflammatory cytokines interact to form a complex network whose dynamic balance determines the development and outcome of inflammation [14]. Studies have shown that multiple pathways are involved in the initial regulation of the inflammatory response, such as Jak/Stat signaling [15], NF-kB signaling [16], Wnt signaling [17], and Toll-like receptor signaling [18, 19]. Many similarities have been found between inflammatory response and tumor development in regulating signaling pathways and gene expression [20, 21]. For instance, NF-κB is a transcription factor that can be activated by a variety of cytokines and plays a key role in inflammatory processes. In cancer, it is usually kept active and can regulate the cell cycle and apoptosis process by activating genes encoding related proteins, thereby inducing survival and promoting cancer progression [22].
As one of the hallmarks of cancer, multiline evidence indicates that inflammation contributes to tumorigenesis, tumor progression, metastasis, and resistance, yet its specific mechanism in most tumors remains unclear [23]. If the inflammatory response persists for too long, it may turn into chronic inflammation, which has many carcinogenic mechanisms, including inducing gene mutation, promoting angiogenesis, changing gene status, promoting cell proliferation and malignant transformation, etc [24]. It proved that inflammation plays a promoting role in the occurrence and development of tumors [25]. Nevertheless, there are few studies on the effect of inflammation on tumor drug resistance. In the current review, we suspect that inflammation plays a certain role in tumor drug resistance and focus on several aspects underlying inflammation-mediated drug resistance (Figure 1).
Figure 1.
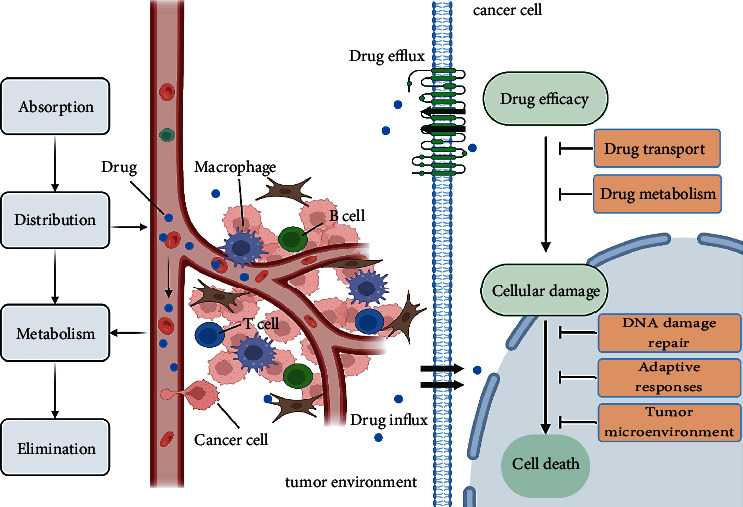
Factors affecting tumor drug resistance. Quantitative and qualitative changes in drug influx and efflux systems affect the distribution of anticancer drugs throughout the body and their accumulation in tumor cells. The drug metabolism of the body determines the efficacy of drugs. DNA damage repair, activation of pro-survival pathway or oncogenic bypass pathway, and changes in the tumor microenvironment are the main influencing factors of drug resistance.
2. Inflammation-Mediated Changes in Drug Transport and Metabolism
Abnormal activation of drug efflux is one of the important reasons for tumor chemotherapy resistance. Meanwhile, the metabolic pathways are also major routes of drug resistance due to their drug-clearing and activation functions, particularly for anti-infectives and cancer drugs [26]. Abnormal activation of tumor efflux and excessive drug metabolism will cause the drug concentration in tumor cells to be lower than the killing concentration during tumor chemotherapy, resulting in tumor cell survival [27]. Therefore, exploring the activation level of drug efflux and drug metabolism in the inflammatory state is helpful to deeply understand the molecular mechanism of drug resistance under an inflammatory state. Here we attempt to explore the effects of inflammation on drug transport and metabolism.
2.1. Inflammation and Drug Efflux Transporters
There exist 48 ATP-binding cassette (ABC) transporters that can be subdivided into seven distinct subfamilies A-G [28, 29]. These drug efflux transporters reduce the intracellular drug concentration and inhibit drug efficacy [30]. Sufficient experimental results suggest that ABC transporter family proteins are associated with drug resistance, especially multidrug resistance protein 1 (MDR1; also known as P-glycoprotein and ABCB1), MDR-associated protein 1 (MRP1; also known as ABCC1), and breast cancer resistance protein (BCRP; also known as ABCG2) [31]. Some research has found that inflammation directly or indirectly impacts drug transporters. For example, peripheral inflammatory pain changes paracellular permeability and increases the expression and activity of P-glycoprotein (P-gp) at the blood-brain barrier, leading to arduous drug uptake by the brain [32]. MDR1 expression in immune cells has been studied in recent years. The inflammatory environment can cause upregulation of MDR1 in some cells involved in innate immunity [33]. Furthermore, the expression of BCRP was significantly reduced by IL-1, IL-6, and TNF-α during acute inflammation, and IL-2 can stimulate JAK3 activation, thus phosphorylating tyrosine residues in BCRP promote their drug efflux function [34]. Endotoxin-induced systemic inflammation in rats is correlated with the apparent changes in the placental expression of several drug transporters; with the increased level of pro-inflammatory cytokines IL-6 and TNF-α, the mRNA expression of Abcc3 increased, while the expression of transporters such as Abcb1a and Abcc2 decreased [35]. Researchers found that gene polymorphisms of ABCG2 potentially affect the serum levels of pro-inflammatory markers by drug efflux in some chronic inflammatory arterial diseases, implying an interaction between inflammation and drug transporters [36]. Several endogenous compounds related to inflammation development, such as cAMP and PGs, are also substrates of MRPs, which could provide new targets for the treatment of inflammatory diseases [37]. Together, inflammatory cytokines have different effects on drug transporters. Several cytokines (e.g., IL-6, TNF-α, and IL-1) inhibit drug efflux, while IL-2 increases drug efflux. The dual roles of cytokines indicate that the effect of inflammation on tumor cell drug resistance is the result of a multifactorial interaction, which is affected by the inflammatory microenvironment, the stage of inflammation induction, and the type of inflammation. Systematic studies on tumor resistance will help continue to elucidate the molecular events of inflammation-induced drug efflux and drug metabolism.
2.2. Inflammation and Drug Metabolism
In addition to drug efflux, activation or inactivation of drugs conducted by cytochrome P450 enzymes (CYPs) have been assumed to be important molecular mechanisms underlying cancer drug resistance [38]. The effects of cancer-induced inflammation on the hepatic metabolism of anticancer drugs have been noticed in recent years. Several inflammatory states were associated with decreased expression of some hepatic CYP enzymes such as 1A, 2A, 2C, 2E, and 3A subfamilies operated by pro-inflammatory cytokines at the transcriptional level [39]. Studies have shown that IL-6-mediated pathways, especially the MAPK/ERK and PI3K/AKT signaling pathways, are critical for the downregulation of CYP enzymes during inflammation [40], whereas IL-6 inhibitor tocilizumab can upregulate the expression of CYP3A4 mRNA [41]. Furthermore, JAK inhibitor has been found to reverse the IL-6-mediated downregulated CYP1A2 and CYP3A4 mRNA levels in HepaRG and PHHS cells, suggesting a prominent role of the JAK/STAT pathway mediated by IL-6 in CYP downregulation [42]. It is noteworthy that CYP enzyme activity is differentially affected by the presence of tumor-associated inflammation. For instance, increased activity of CYP2E1 was associated with raised serum levels of IL‐6, IL‐8, and TNF‐α [43]. Beyond this exception, IL-6 predominantly reduces CYP expression and thus attenuates the biotransformation of some drugs that are metabolized through CYP enzymes. Furthermore, studies found that after the TNF-α treatment for 24 h, the induction of CYP3A4 mRNA downregulation is not the same as protein downregulation. The latter is not obvious, suggesting that post-transcriptional mechanisms are involved in the downregulation of CYP protein levels or enzyme activity by TNF-α [44, 45]. As mentioned above, the same cytokines (e.g., IL-6 and TNF-α) had opposite effects on different P450 enzymes. Since different drug-metabolizing enzymes are selective for specific drugs, the characteristics of inflammation in patients during clinical drug treatment can indicate the activation or inhibition of specific drug enzymes, thereby assessing the risk of clinical drug resistance.
3. DNA Damage Response
The DNA damage response (DDR) is a protective mechanism under physiological conditions involved in DNA repair, checkpoint activation, and transcription regulation [46, 47]. On the other hand, it can help tumor cells survive the distractions of drug treatment [48–50]. Cancer treatment usually consists of some chemotherapy drugs that rely on the induction of DNA damage. Thus, the DNA damage repair capacity of cancer cells significantly influences the efficiency of DNA-damaging medications [51]. Recent studies indicate a positive correlation between IL-17 expression and the DDR in mice after cigarette smoke exposure, and excessive damage responses, in turn, lead to increased DNA mutation rates, which contribute to the genetic instability of cancer [52]. Meanwhile, by measuring DDR markers like activation of ataxia-telangiectasia mutated kinase (ATM), researchers found that inflammation in the gastric cardia mucosa may cause accumulated DNA damage, leading to mutagenesis and chromosomal rearrangements [53]. After treating lipopolysaccharides (LPS), the increased mRNA transcription of some DNA repair enzymes like AP endonuclease and DNA glycosylase excising εA can be detected in rat intestines, which implies stimulation of the repair of oxidative DNA damage. Thus, inflammation may cause an imbalance in genome instability, more generally, to cancer development and drug resistance [54].
The DNA damage incurred during the inflammatory response triggers the activation of DNA repair pathways by stimulating a cascade of pro-inflammatory signals required for cell survival [55, 56]. Many DNA repair pathways are involved in regulating the transcription of molecules important for infection control, which implies a link between DDR and the inflammatory system [57, 58]. However, when DNA repair is not sufficient to deal with increasing DNA damage, the genes in the cells often mutate. Some cell mutations (e.g., BRAF V600E and EGFR T790M) can drive the development of tumors, causing tumor resistance. Among them, the occurrence of inflammation will accelerate this process [59]. For example, a recent study indicated the synergic effect between bacterial-driven inflammation and a mutant BRAF V600E to promote colon tumorigenesis in an enterotoxigenic Bacteroides fragilis (ETBF) murine model [60]. Similarly, the pro-inflammatory STAT3 pathway was found to be an important mechanism for EGFR T790M mutation-mediated drug resistance in NSCLC [61]. Inflammation can promote DNA repair in tumor cells, leading to tumor chemotherapy resistance. Blocking the inflammatory response is expected to act as an adjuvant therapy strategy for tumor-resistant treatment.
4. Downstream Adaptive Responses
In addition to the intake and elimination of drugs, tumor cells can also obtain drug resistance through downstream adaptive responses [62, 63]. Currently, commonly used tumor therapy drugs are chemotherapeutic drugs and targeted therapeutic agents. Chemotherapy drugs ultimately lead to tumor cell apoptosis by injuring biomolecules within cells, while targeted therapies suppress tumor progression by inhibiting key regulators in tumor survival-maintaining mechanisms [64–66]. Tumor cells can escape death through various pathways, and these mechanisms are also important reasons for tumor drug resistance, where inflammation plays an essential role (Figure 2).
Figure 2.
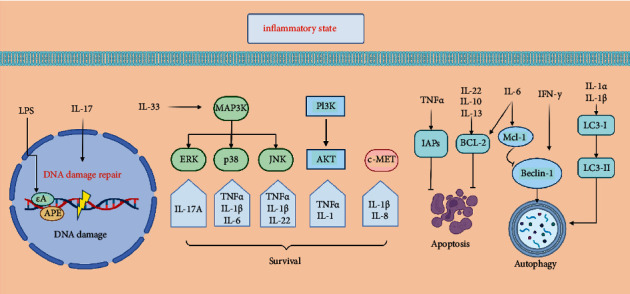
Molecular mechanisms of anticancer drug resistance. Inflammation not only aggravates DNA damage but also affects the expression of DNA repair enzymes, which leads to the instability of tumor genes and further promotes tumor drug resistance. Inflammation can regulate apoptosis and autophagy of tumor cells. For example, IL-6, 10, and 22 may inhibit the apoptosis of tumor cells by promoting the expression of BCL-2. Interferon and IL-1α/β can act on Beclin and LC3, respectively, to promote autophagy, while IL-6 can promote the binding of Mcl-1 and Beclin-1 to inhibit autophagy. Inflammatory mediators activate oncogenic bypass signaling pathways (e.g., MAPK, C-MET, or PI3K/AKT signaling pathways) leading to tumor resistance.
4.1. Evade from Apoptosis
Cancer cells can regulate the expression of some apoptosis-related proteins, such as B-cell lymphoma 2 (Bcl-2) and inhibitors of apoptosis proteins (IAPs), thus reducing their sensitivity to anticancer drugs that induce cell apoptosis and surviving the treatment [67, 68]. Bcl-2-related proteins are a large family that maintains the normal life state of cells by regulating apoptosis, which can be influenced by various cytokines associated with inflammation factors. For example, IL-13 has been found to induce Bcl-2 expression in airway epithelial cells [69, 70]. Results showed that the Bcl-2 level was increased after adding IL-6 in lymphoblast cells [71]. In addition, IL-22 activates the expression of the antiapoptotic protein Bcl-2 in renal cortex tissues [72]. Meanwhile, IL-10 has a role in protecting cells from apoptosis through upregulating Bcl-2 expression, especially in breast tumors [73]. It has worth noting that the effect of inflammation on apoptosis is not unilateral. By targeting the overexpressed Bcl-2 protein with the nanoparticle Bcl-2 inhibitor ABT-199, apoptosis of inflammatory cells and the reduction of IL-4 and IL-5 levels were observed [74]. Moreover, given that TNFα is one of the key mediators of cancer-related inflammation, at the same time, cIAP1 and cIAP2 function as key mediators of TNFα-induced the activation of NF-κB and then allow the cell to survive against external interference, which indicates the interaction between inflammation and IAPs [75, 76]. Inflammation often accompanies the whole process of tumor occurrence and development and plays a key role in the process of tumor drug resistance [10]. Targeting tumor inflammation may act as a potential strategy to promote tumor apoptosis.
4.2. Autophagy
Autophagy is an evolutionarily conserved mechanism that disposes of excessive or potentially dangerous cytosolic entities through lysosomal degradation to maintain cellular biosynthesis and viability during stress conditions [77, 78]. Therefore, this process may result in the survival of cancer cells that are undergoing anticancer drug treatment. Inflammatory cytokines, including IFN-γ, TNF-α, IL-17, and the IL-1 family, are closely related to autophagy in tumors [79]. In human melanoma cells, blocking the IL-1 pathway by IL-1α or IL-1β treatment increases autophagy-related components such as LC3-I and LC3-II, indicating an increase of autophagy [80]. The overexpression of IFN-γ upregulated Beclin-1 mRNA expression and protein levels in the stomachs of mice, indicating that IFN-γ induces autophagy in part through upregulation of Beclin-1 [81]. However, inflammatory cytokines also play a role in inhibiting autophagy. For example, IL-6 overexpression was sufficient to block autophagy by supporting Beclin-1/Mcl-1 interaction and promoting arsenic-induced cell transformation in lung carcinoma cell lines [82]. Autophagy may also modulate inflammatory cytokine release and secretion through different mechanisms such as secretion of mediators of inflammation, regulation of inflammasomes, and p62/SQSTM1 proteins [83]. Since autophagy plays a dual role in tumor survival, the regulation of autophagy by inflammation is also context dependent. Further research is critically needed to understand the crosstalk between autophagic processes and inflammation as well as the underlying molecular mechanism.
4.3. Oncogenic Bypass Pathway
Activating the oncogenic bypass signaling pathways (e.g., MAPK, c-MET, or PI3K/AKT signaling) is crucial in tumor drug resistance. Studies have found that inflammation can directly or indirectly affect signal transduction in these pathways [84, 85]. For example, the JNK and p38 MAPK signaling pathways can be activated by pro-inflammatory cytokines such as TNF-α and IL-1β, associated with anticancer drug resistance in colon and liver cancer [86]. Studies have shown that the pro-inflammatory cytokine IL-22 can downregulate Cx43 gene transcription and promote keratinocyte proliferation and migration through the JNK-dependent pathway [87]. IL-17A stimulation leads to upregulation of Plet1 expression, which contributes to tissue regeneration and colonic tumorigenesis via regulating ERK [88]. IL-33 can activate the classical MyD88/IRAK/TRAF6 module, which activates three subfamily pathways of MAPK, including ERK, p38, and JNK [89]. Interestingly, activation of the JNK pathway can promote inflammatory actions such as directly activated NF-κB by promoting IκBα degradation, which indicates a positive feedback regulation of inflammation on the JNK pathway [90]. As for cholangiocarcinoma, inflammatory mediators such as IL-6 and TNF-α activate several pathways such as JAK-STAT, p38 MAPK, and Akt, resulting in increased cell growth and survival and proliferation [91]. Moreover, the expression of three major inflammatory cytokine genes, IL-1β, IL-8, and CXCL1, was positively correlated with c-MET expression in patients with brain metastases. In addition, the MAPK pathway is the central downstream transducer between c-Met and IL-1β [92].
Furthermore, under the chronic inflammation state, multiple inflammatory factors are associated with the PI3K/Akt pathway, which plays an important role in the initiation and development of downstream inflammatory pathways. For example, studies have shown that NF-κB is triggered by PI3K/Akt by activating protein kinase C, which confirms the link between inflammation and oncogenic pathways [93]. In summary, inflammation promotes the activation of oncogenic bypass pathways in tumor cells, which may lead to targeted drug resistance. Therefore, the level of inflammation in the tumor microenvironment should be considered during clinical treatment, and combination with anti-inflammatory drugs may improve the efficacy.
5. Inflammatory Tumor Microenvironment
5.1. Oxidative Stress
Oxidative stress refers to the imbalance between the generation of free oxygen radicals and their elimination through the antioxidant defense system after being stimulated [94, 95]. As a by-product of normal metabolic processes, reactive oxygen species (ROS) are indispensable for various biological processes in normal and cancer cells [96]. Recent evidence suggests that ROS is closely related to tumor cell proliferation, heterogeneity, dormancy, and stemness, which are considered the critical requirements for tumor progression [97, 98]. However, the effects of excess ROS on tumor cells can be quite different in its anticancer properties, such as inducing apoptosis. Although most current chemotherapeutic agents increase ROS to cytotoxic levels when targeting cancer cells, exposure to ROS inevitably reduces the efficacy of chemotherapy in the long term, resulting in cancer drug resistance [99].
During inflammation, mast cells and white blood cells are recruited to the site of injury, resulting in increased release and accumulation of ROS at the site of injury due to increased oxygen uptake, leading to oxidative stress [100]. ROS promotes cancer growth and progression via different signaling pathways (PI3/Akt/mTOR, MAPK, etc.) when accumulating to a particular concentration [95]. Studies have shown that ROS-mediated drug resistance in cancer cells may be due to the activation of redox-sensitive transcription factors such as NF-κB, Nrf2, c-Jun, and HIF-1α 30471641. Nrf2, a transcription factor involved in cellular homeostasis, is responsible for targeting genes related to cellular defense and plays a crucial role in regulating REDOX homeostasis [101]. Under oxidative stress, the specific cysteine residues of KEAP1 in the KEAP1-CUL3-RBX1 complex are destroyed, thus interfering with the ubiquitination process of Nrf2, which may ultimately lead to cancer drug tolerance [102]. Inhibition of Nrf2 gene expression has been found to reverse the resistance of head and neck tumors to iron death inducers [103].
Studies have found that oxidative stress and inflammation are interdependent and interrelated processes in the inflammatory RA joint, where inflammatory cells release large amounts of ROS, leading to excessive oxidative damage. In addition, many ROS and oxidative stress products enhance the pro-inflammatory response [104].
Exposure of phagocytes to pro-inflammatory cytokines such as TNF, IFN-γ, and/or IL-1β induces the formation of the NOX2 complex, which significantly increases intracellular ROS levels [105]. What's more, as a pro-inflammatory factor, TNF mainly acts through multiple TNF receptor-mediated signaling pathways, such as NF-KB and MAPKs, to regulate inflammatory responses. It is now clear that ROS/RNS are an integral part of TNF signaling because they are closely involved in numerous feedback loops, which are part of TNFR's downstream signaling pathways. Furthermore, ROS production also creates a positive feedback loop through autocrine TNF-α-mediated inflammatory cytokine/chemokine expression, which contributes to tumor progression [106, 107]. These findings suggest that inflammation may play a role in tumor resistance by stimulating the production of ROS.
5.2. Immune Response
The immune system has a crucial role in cancer development and treatment [108, 109]. Deregulation of the immune response may cause cancer cells to evade immunogenic cancer cell death; however, the underlying molecular mechanisms involved in resistance to immunotherapy need further elucidation [109]. There is increasing evidence that immune response and systemic inflammation play an important role in tumor progression, and there is a complex interaction between them [110]. Researchers have found that tumor regulates the inflammatory environment by secreting soluble growth factors and chemokines so that inflammatory cells inhibit the anticancer T-cell response [111]. In the chronic inflammatory state, pro-inflammatory cytokines recruit immune cells such as M2, N2, and MDSCs, which are associated with suppressing immune surveillance and maintaining depleted T-cell phenotypes [112]. As a major pro-inflammatory cytokine, IL-6 plays an immunosuppressive role by inhibiting IFN-γ production and Th1 differentiation through SOCS1 induction, making CD8+T cells helpless. Studies have shown that upregulation of IL-6 activates STAT3, thereby inhibiting CD8 T-cell infiltration in non-small cell lung cancer and pancreatic cancer [113, 114]. While IL-10 has different effects on most immune cells and can inhibit the activation and effector functions of T cells, monocytes, and macrophages, it can also stimulate CD8+T cells and inhibit tumors to a certain extent [115]. The activation of MAPK signaling leads to increased VEGF and IL-8 expression, which then restrains T-cell recruitment and function [116]. Under the condition of inflammation, the metabolism of the stimulated immune cells may be disturbed, thus causing the abnormal function of the immune system [117]. Neutrophils under inflammatory conditions can affect other immune cells like T cells by producing chemokines and secreting granule contents and then promote immune paralysis of the adaptive immune system [118]. To avoid continuous inflammation, the cytokine IL-27 promotes Treg expression of T-bet and CXCR3 and triggers Tr1 cells. It has also been linked to increased expression of inhibitory receptors by T cells, antagonizing the development of Th2 and Th17 cell subsets. In addition, the observation that IL-27 induces PD-L1 and PD-1 suggests that IL-27 may be an important molecule in controlling cancer-related immune checkpoint mechanisms [119]. In summary, the relationship between tumor-associated inflammation and immune response still needs to be elucidated in many experiments. Furthermore, in-depth knowledge of the molecular mechanism of inflammation-influenced immune response could be beneficial to overcoming immunotherapy failure (Figure 3).
Figure 3.
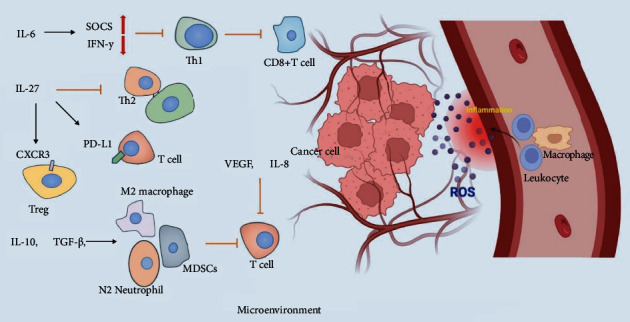
Inflammatory regulation of tumor microenvironment. In the tumor microenvironment, inflammatory factors can specifically activate or inhibit the function of immune cells, thus affecting the survivability of tumor cells. At the same time, the large amount of reactive oxygen species produced in the inflammatory site also plays a role in tumor resistance.
6. Targeting Inflammation to Overcome Drug Resistance
As mentioned in the previous discussion, inflammation can affect tumor development and drug resistance in multiple ways (Table 1). Of note, evidence increasingly suggests that drugs with anti-inflammatory properties have been shown to resist cancer. However, anti-inflammatory drugs are still in the early stages of clinical use in treating cancer due to drug risk. Here we summarize some of the current clinical advances that support the use of anti-inflammatory approaches for treating tumors (Table 2).
Table 1.
Summary of cytokine-mediated effects on drug resistance.
| Cytokines | Downstream effector | Resistance mechanism | Role | References | Potential targeted drugs |
|---|---|---|---|---|---|
| IL-6 | BCRP | Reducing drug efflux | Anti-resistant | [34] | Tocilizumab, sarilumab, clazakizumab, siltuximab [120] |
| P-gp | Reducing drug efflux | Anti-resistant | [35] | ||
| CYP1A2 and CYP3A4 | Inhibiting drug metabolism | Pro-resistant | [42] | ||
| CYP2E1 | Promoting drug metabolism | Anti-resistant | [43] | ||
| Beclin-1/Mcl-1 interaction | Inhibiting autophagy | Anti-resistant | [82] | ||
| Bcl-2 | Inhibiting apoptosis | Pro-resistant | [71] | ||
| JAK-STAT, p38 MAPK, Akt | Mediating survival signaling pathways | Pro-resistant | [91] | ||
|
| |||||
| TNF-α | BCRP | Reducing drug efflux | Anti-resistant | [34] | Adalimumab [121] |
| P-gp | Reducing drug efflux | Anti-resistant | [35] | ||
| CYP2E1 | Promoting drug metabolism | Anti-resistant | [43] | ||
| CYP3A4 | Inhibiting drug metabolism | Pro-resistant | [44] | ||
| Beclin-1, LC3 | Increasing autophagy | Pro-resistant | [79] | ||
| IAPs | Inhibiting apoptosis | Pro-resistant | [75] | ||
| JAK-STAT, p38 MAPK, Akt | Mediating survival signaling pathways | Pro-resistant | [91] | ||
|
| |||||
| IL-1 | BCRP | Inhibiting drug efflux | Anti-resistant | [34] | Anakinra, canakinumab [122] |
| LC3-I, LC3-II | Increasing autophagy | Pro-resistant | [80] | ||
| JNK, p38 MAPK, c-MET | Mediating survival signaling pathways | Pro-resistant | [86, 92] | ||
|
| |||||
| IL-22 | Bcl-2 | Inhibiting apoptosis | Pro-resistant | [72] | IL-22BP [123] |
| JNK | Mediating survival signaling pathways | Pro-resistant | [87] | ||
|
| |||||
| IL‐8 | CYP2E1 | Promoting drug metabolism | Anti-resistant | [43] | Tocilizumab [124] |
| c-MET | Mediating survival signaling pathways | Pro-resistant | [92] | ||
|
| |||||
| IL-17 | DDR | Inhibiting DNA damage | Pro-resistant | [52] | Secukinumab, ixekizumab, brodalumab [125] |
| ERK | Mediating survival signaling pathways | Pro-resistant | [88] | ||
|
| |||||
| IL-2 | BCRP | Increasing drug efflux | Pro-resistant | [34] | Ro26-4550 [126] |
| IFN-γ | Beclin-1 | Increasing autophagy | Pro-resistant | [81] | Emapalumab |
| IL-10 | Bcl-2 | Inhibiting apoptosis | Pro-resistant | [73] | AS101 [127] |
| IL-33 | JNK, p38 MAPK, ERK | Mediating survival signaling pathways | Pro-resistant | [89] | N.A. |
| IL-13 | Bcl-2 | Inhibiting apoptosis | Pro-resistant | [69] | Suplatast tosilate [128] |
Table 2.
Summary of clinical trials of anti-inflammation drugs treating drug-resistant tumors.
| Agent | Tolerated drug | Drug-resistant tumor type | Mechanism | Phase | Clinical trial number |
|---|---|---|---|---|---|
| Celecoxib | Platinum | Ovarian cancer/primary peritoneal cavity cancer | Stops the growth | II | NCT00084448 |
| Dexamethasone | Platinum | Ovarian cancer | Stops the growth | II | NCT00003449 |
| Aspirin | Osimertinib | “Non-small cell lung cancer” | Promotes cells apoptosis | I | NCT03532698 |
| Prednisone | Hormone-resistant | Prostate cancer | Stops the growth | III | NCT00110214 |
| Aspirin | EGFR-TKI | NSCLC | Promotes cells apoptosis | I | NCT03543683 |
| Dexamethasone | Most MM drugs | Multiple myeloma | Inhibits tumor metastasis | II | NCT02626481 |
| Dexamethasone | Chemotherapy | Recurrent plasma cell myeloma | Inhibits tumor metastasis | II | NCT03457142 |
Conventional anti-inflammatory drugs like NSAIDs (aspirin) have been identified as a broad-spectrum anticancer agent based on data from epidemiological studies [129–131]. Aspirin can inhibit the nuclear translocation of NF-κB, thereby inhibiting the PI3 kinase/Akt-mediated cell survival pathway and promoting cell apoptosis. In multiple myeloma cells, aspirin inhibited tumor cell proliferation and induced apoptosis by upregulating Bax and downregulating Bcl-2, changing the ratio of Bax/Bcl-2 [132]. Based on this, aspirin has been combined with other drugs in clinical trials treating drug-resistantnon-small cell cancers.
Several studies have shown that corticosteroids have significant anticancer effects both alone or in combination. As a selective COX-2 inhibitor and a nonsteroidal anti-inflammatory drug that inhibits prostaglandin production, celecoxib can induce apoptosis by activating transcriptional regulators of ER stress in hepatoma cells [133]. Dexamethasone can induce cell death in multiple myeloma mediated by miR-125b expression [134]. Other glucocorticoids, such as prednisone, its inhibition of prostate cancer growth may be due to inhibition of tumor-associated angiogenesis by reducing VEGF and IL-8 production [135].
As some infectious diseases that cause chronic inflammation have been linked to the development of cancer, anti-infective agents including antiviral, antibacterial, and antifungal drugs may play a role in cancer treatment [136–138]. Statins, which inhibit HMG-CoA reductase, also have anti-inflammatory properties and may have a beneficial effect on HCC caused by hepatitis [139].
Tumor immunotherapy is very effective in some patients, but resistance to which can also develop due to the immunosuppressive tumor microenvironment. Therefore, anti-inflammatory drugs that target immunosuppressive cells or cytokines may make tumor cells more sensitive to immunotherapy drugs and thus avoid resistance [115]. Studies found that monotherapy with COX-2 inhibitors or prostaglandin 2 (PGE2) receptor antagonists activate IFN-γ-driven transcriptional remodeling and synergy with immune checkpoint inhibitors to enhance effector T-cell accumulation in tumors [140].
As inflammatory mediators, cytokines and chemokines also mediate the tumor's response to external disturbances, which provides new targets for tumor drug therapy. For example, siltuximab and tocilizumab were used for ovarian cancer treatment as IL-6 antibodies [141]. MABp1 was used for colorectal cancer treatment as an anti-IL-1α antibody [142]. Blockade of inflammatory pathways such as TGFβ signaling by galunisertib, which inhibits the TGFβR1 kinase, has been used to treat many types of cancer [143]. Due to the inflammation network within the tumor microenvironment being complex, inhibiting one molecule can cause a cascade effect, so the safety of the targeted drugs still needs to be tested in more trials.
Finally, some natural anti-inflammatory supplements might also help control cancer and they have the advantage of fewer side effects and greater safety. Berberine, which is a plant-derived alkaloid, may have the potential to improve colorectal adenomas [144]. Likewise, vitamin C has been extensively explored for potential anticancer effects [145].
Vitamin C's anti-inflammatory and antioxidant abilities can be attributed to its ability to regulate the DNA-binding activity of NF-κB and reduce the production of inflammatory factors [146]. Dietary supplementation of vitamin C was found to result in a significant decrease in the mRNA expression of pro-inflammatory cytokines (e.g., IL-1β, IL-6, and IFN-γ) [147]. Cancer cells depend primarily on the gene expression of KRAS or BRAF to survive. Vitamin C gets inside these cancer cells and disrupts the expression of KRAS or BRAF genes. Once vitamin C has an effect, the mutation probability of cancer cells will also be greatly reduced, which reduces the resistance of targeted drugs [148].
The idea of targeting inflammation to treat drug-resistant tumors is innovative, but the molecular mechanisms behind the drugs still need extensive research and clinical trials to ensure the efficacy and safety of treatment regiments.
7. Conclusions
In this review, we summarize the recent studies on inflammation in influencing cancer drug resistance from several aspects including drug transport and metabolism, DNA damage response, downstream adaptive responses, oncogenic bypass signaling, and tumor microenvironment.
The activity of drug transporters and CYP enzymes changes in the inflammatory state [39]. Of note, different inflammatory cytokines have different effects on diverse drug transporters and CYP enzymes. Except for CYP2E1, the expression level of most liver drug enzymes is reduced in the inflammatory state, which inhibits the drug metabolism of tumors. In this regard, the mechanism of drug resistance to tumors remains to be studied. As DNA repair determines the potential of tumor cells to resist DNA-damaging drugs, the distinct roles of inflammatory cytokines in DDR emphasize the necessity to clarify the molecular mechanisms underlying the inflammation-related drug resistance. The role of multiple pro-inflammatory factors including IFN-γ, TNF-α, IL-17, and the IL-1 family in promoting drug resistance through autophagy has been confirmed by detecting the expression of autophagy-related molecules like LC3 and Beclin-1. It has also been suggested that IL-6, IL-22, and IL-10 can influence tumor drug resistance by regulating apoptosis-related proteins like BCL-2 and IAPs. Moreover, it is noteworthy that autophagy and apoptosis can also regulate inflammatory signaling pathways, thus forming a feedback regulatory pathway. We also summarized the regulatory role of some inflammatory factors in activating the oncogenic bypass signaling pathway, especially the cross-connections between NF-κB and three typical pathways including MAPK, c-MET, and PI3K/AKT signaling. In the tumor microenvironment, the production of ROS is closely related to the failure of anticancer drugs. Some targets of the oxidative stress pathway are involved in the conduction of the inflammatory pathways, and some pro-inflammatory factors such as TNFα, IFN-γ, and IL-1β can promote the production of ROS. Although some studies have found that the function of some immune cells is suppressed under chronic inflammation, the specific mechanism of inflammatory components on cancer immune escape remains to be further elucidated due to the complexity of the immune system. At last, we summarize some anti-inflammatory applications in tumor therapy and propose some hypotheses for targeting inflammation against tumor resistance. Therefore, further studies on the molecular mechanism at multiple levels behind inflammation and cancer are required to overcome cancer drug resistance.
Acknowledgments
The authors thank BioRender for helping in creating figures.
Data Availability
No data were used to support this study.
Conflicts of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as potential conflicts of interest.
Authors' Contributions
Shuaijun Lu and Yang Li contributed equally to this article.
References
- 1.Sung H., Ferlay J., Siegel R. L., et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer Journal for Clinicians . 2021;71(3):209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 2.Ward R. A., Fawell S., Floc’h N., Flemington V., McKerrecher D., Smith P. D. Challenges and opportunities in cancer drug resistance. Chem Rev . 2021;121(6):3297–3351. doi: 10.1021/acs.chemrev.0c00383. [DOI] [PubMed] [Google Scholar]
- 3.Nam A. S., Chaligne R., Landau D. A. Integrating genetic and non-genetic determinants of cancer evolution by single-cellmulti-omics. Nat Rev Genet . 2021;22(1):3–18. doi: 10.1038/s41576-020-0265-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown R., Curry E., Magnani L., Wilhelm-Benartzi C. S., Borley J. Poised epigenetic states and acquired drug resistance in cancer. Nature Reviews. Cancer . 2014;14(11):747–753. doi: 10.1038/nrc3819. [DOI] [PubMed] [Google Scholar]
- 5.Chatterjee N., Bivona T. G. Polytherapy and targeted cancer drug resistance. Trends in Cancer . 2019;5(3):170–182. doi: 10.1016/j.trecan.2019.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li B., Jiang J., Assaraf Y. G., Xiao H., Chen Z. S., Huang C. Surmounting cancer drug resistance: new insights from the perspective of N(6)-methyladenosine RNA modification. Drug Resistance Updates . 2020;53 doi: 10.1016/j.drup.2020.100720.100720 [DOI] [PubMed] [Google Scholar]
- 7.Boumahdi S., de Sauvage F. J. The great escape: tumour cell plasticity in resistance to targeted therapy. Nature Reviews Drug Discovery . 2020;19(1):39–56. doi: 10.1038/s41573-019-0044-1. [DOI] [PubMed] [Google Scholar]
- 8.Assaraf Y. G., Brozovic A., Gonçalves A. C., et al. The multi-factorial nature of clinical multidrug resistance in cancer. Drug Resistance Updates . 2019;46 doi: 10.1016/j.drup.2019.100645.100645 [DOI] [PubMed] [Google Scholar]
- 9.Rathinam V. A. K., Chan F. K. M. Inflammasome, inflammation, and tissue homeostasis. Trends in Molecular Medicine . 2018;24(3):304–318. doi: 10.1016/j.molmed.2018.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao H., Wu L., Yan G., et al. Inflammation and tumor progression: signaling pathways and targeted intervention. Signal Transduction and Targeted Therapy . 2021;6(1):p. 263. doi: 10.1038/s41392-021-00658-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zindel J., Kubes P. DAMPs, PAMPs, and LAMPs in immunity and sterile inflammation. Annual Review of Pathology: Mechanisms of Disease . 2020;15(1):493–518. doi: 10.1146/annurev-pathmechdis-012419-032847. [DOI] [PubMed] [Google Scholar]
- 12.Dong C. Cytokine regulation and function in T cells. Annu Rev Immunol . 2021;39(1):51–76. doi: 10.1146/annurev-immunol-061020-053702. [DOI] [PubMed] [Google Scholar]
- 13.Fajgenbaum D. C., June C. H., Cytokine C. H. Cytokine s. N Engl J Med . 2020;383(23):2255–2273. doi: 10.1056/NEJMra2026131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Balkwill F. Tumour necrosis factor and cancer. Nature Reviews. Cancer . 2009;9(5):361–371. doi: 10.1038/nrc2628. [DOI] [PubMed] [Google Scholar]
- 15.Villarino A. V., Kanno Y., O’Shea J. J. Mechanisms and consequences of Jak-STAT signaling in the immune system. Nat Immunol . 2017;18(4):374–384. doi: 10.1038/ni.3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Taniguchi K., Karin M. N. F.-κB. NF-κB, inflammation, immunity and cancer: coming of age. Nature Reviews. Immunology . 2018;18(5):309–324. doi: 10.1038/nri.2017.142. [DOI] [PubMed] [Google Scholar]
- 17.Li X., Xiang Y., Li F., Yin C., Li B., Ke X. WNT/β-Catenin signaling pathway regulating T cell-inflammation in the tumor microenvironment. Front Immunol . 2019;10:p. 2293. doi: 10.3389/fimmu.2019.02293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yeung Y. T., Aziz F., Guerrero-Castilla A., Arguelles S. Signaling pathways in inflammation and anti-inflammatory therapies. Current Pharmaceutical Design . 2018;24(14):1449–1484. doi: 10.2174/1381612824666180327165604. [DOI] [PubMed] [Google Scholar]
- 19.Fitzgerald K. A., Kagan J. C. Toll-like receptors and the control of immunity. Cell . 2020;180(6):1044–1066. doi: 10.1016/j.cell.2020.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Andrejeva G., Rathmell J. C. Similarities and distinctions of cancer and immune metabolism in inflammation and tumors. Cell Metabolism . 2017;26(1):49–70. doi: 10.1016/j.cmet.2017.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McFarland D. C., Jutagir D. R., Miller A. H., Breitbart W., Nelson C., Rosenfeld B. Tumor mutation burden and depression in lung cancer: association with inflammation. Journal of the National Comprehensive Cancer Network . 2020;18(4):434–442. doi: 10.6004/jnccn.2019.7374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Viatour P., Merville M. P., Bours V., Chariot A. Phosphorylation of NF-κB and IκB proteins: implications in cancer and inflammation. Trends in Biochemical Sciences . 2005;30(1):43–52. doi: 10.1016/j.tibs.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 23.Greten F. R., Grivennikov S. I. Inflammation and cancer: triggers, mechanisms, and consequences. Immunity . 2019;51(1):27–41. doi: 10.1016/j.immuni.2019.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khandia R., Munjal A. Interplay between inflammation and cancer. Advances in Protein Chemistry and Structural Biology . 2020;119:199–245. doi: 10.1016/bs.apcsb.2019.09.004. [DOI] [PubMed] [Google Scholar]
- 25.Singh N., Baby D., Rajguru J. P., Patil P. B., Thakkannavar S. S., Pujari V. B. Inflammation and cancer. Ann Afr Med . 2019;18(3):121–126. doi: 10.4103/aam.aam_56_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kirchmair J., Göller A. H., Lang D., et al. Predicting drug metabolism: experiment and/or computation? Nature Reviews. Drug Discovery . 2015;14(6):387–404. doi: 10.1038/nrd4581. [DOI] [PubMed] [Google Scholar]
- 27.McIntosh K., Balch C., Tiwari A. K. Tackling multidrug resistance mediated by efflux transporters in tumor-initiating cells. Expert Opinion on Drug Metabolism & Toxicology . 2016;12(6):633–644. doi: 10.1080/17425255.2016.1179280. [DOI] [PubMed] [Google Scholar]
- 28.Kathawala R. J., Gupta P., Ashby C. R., Jr., Chen Z. S. The modulation of ABC transporter-mediated multidrug resistance in cancer: a review of the past decade. Drug Resistance Updates . 2015;18:1–17. doi: 10.1016/j.drup.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 29.Li W., Zhang H., Assaraf Y. G., et al. Overcoming ABC transporter-mediated multidrug resistance: molecular mechanisms and novel therapeutic drug strategies. Drug Resistance Updates . 2016;27:14–29. doi: 10.1016/j.drup.2016.05.001. [DOI] [PubMed] [Google Scholar]
- 30.Wilkens S. Structure and mechanism of ABC transporters. F1000Prime Rep . 2015;7:p. 14. doi: 10.12703/p7-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Robey R. W., Pluchino K. M., Hall M. D., Fojo A. T., Bates S. E., Gottesman M. M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nature Reviews Cancer . 2018;18(7):452–464. doi: 10.1038/s41568-018-0005-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ronaldson P. T., Davis T. P. Targeting blood-brain barrier changes during inflammatory pain: an opportunity for optimizing CNS drug delivery. Therapeutic Delivery . 2011;2(8):1015–1041. doi: 10.4155/tde.11.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bossennec M., Di Roio A., Caux C., Ménétrier-Caux C. MDR1 in immunity: friend or foe? Oncoimmunology . 2018;7(12) doi: 10.1080/2162402x.2018.1499388.e1499388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mishra J., Simonsen R., Kumar N. Intestinal breast cancer resistance protein (BCRP) requires Janus kinase 3 activity for drug efflux and barrier functions in obesity. Journal of Biological Chemistry . 2019;294(48) doi: 10.1074/jbc.RA119.007758.18348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ghoneim R. H., Kojovic D., Piquette-Miller M. Impact of endotoxin on the expression of drug transporters in the placenta of HIV-1 transgenic (HIV-Tg) rats. European Journal of Pharmaceutical Sciences . 2017;102:94–102. doi: 10.1016/j.ejps.2017.03.004. [DOI] [PubMed] [Google Scholar]
- 36.Zhang D., Ding Y., Wang X., et al. Effects of ABCG2 and SLCO1B1 gene variants on inflammation markers in patients with hypercholesterolemia and diabetes mellitus treated with rosuvastatin. Eur J Clin Pharmacol . 2020;76(7):939–946. doi: 10.1007/s00228-020-02882-4. [DOI] [PubMed] [Google Scholar]
- 37.Yang A. K., Zhou Z. W., Wei M. Q., Liu J. P., Zhou S. F. Modulators of multidrug resistance proteins in the management of anticancer and antimicrobial drug resistance and the treatment of inflammatory diseases. Current Topics in Medicinal Chemistry . 2010;10(17):1732–1756. doi: 10.2174/156802610792928040. [DOI] [PubMed] [Google Scholar]
- 38.Ingelman-Sundberg M., Lauschke V. M. Can CYP inhibition overcome chemotherapy resistance? Trends in Pharmacological Sciences . 2020;41(8):503–506. doi: 10.1016/j.tips.2020.05.007. [DOI] [PubMed] [Google Scholar]
- 39.Kacevska M., Robertson G. R., Clarke S. J., Liddle C. Inflammation and CYP3A4-mediated drug metabolism in advanced cancer: impact and implications for chemotherapeutic drug dosing. Expert Opinion on Drug Metabolism & Toxicology . 2008;4(2):137–149. doi: 10.1517/17425255.4.2.137. [DOI] [PubMed] [Google Scholar]
- 40.Keller R., Klein M., Thomas M., et al. Coordinating role of RXRα in downregulating hepatic detoxification during inflammation revealed by fuzzy-logic modeling. PLoS Comput Biol . 2016;12(1) doi: 10.1371/journal.pcbi.1004431.e1004431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vet N. J., Brussee J. M., de Hoog M., et al. Inflammation and organ failure severely affect midazolam clearance in critically ill children. American Journal of Respiratory and Critical Care Medicine . 2016;194(1):58–66. doi: 10.1164/rccm.201510-2114OC. [DOI] [PubMed] [Google Scholar]
- 42.Febvre-James M., Bruyère A., Le Vée M., Fardel O. The JAK1/2 inhibitor ruxolitinib reverses interleukin-6-mediated suppression of drug-detoxifying proteins in cultured human hepatocytes. Drug Metab Dispos . 2018;46(2):131–140. doi: 10.1124/dmd.117.078048. [DOI] [PubMed] [Google Scholar]
- 43.Trousil S., Lee P., Edwards R. J., et al. Altered cytochrome 2E1 and 3A P450-dependent drug metabolism in advanced ovarian cancer correlates to tumour-associated inflammation. Br J Pharmacol . 2019;176(18):3712–3722. doi: 10.1111/bph.14776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aitken A. E., Morgan E. T. Gene-specific effects of inflammatory cytokines on cytochrome P450 2C, 2B6 and 3A4 mRNA levels in human hepatocytes. Drug Metab Dispos . 2007;35(9):1687–1693. doi: 10.1124/dmd.107.015511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dallas S., Sensenhauser C., Batheja A., et al. De-riskingbio-therapeutics for possible drug interactions using cryopreserved human hepatocytes. Current Drug Metabolism . 2012;13(7):923–929. doi: 10.2174/138920012802138589. [DOI] [PubMed] [Google Scholar]
- 46.Pilié P. G., Tang C., Mills G. B., Yap T. A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat Rev Clin Oncol . 2019;16(2):81–104. doi: 10.1038/s41571-018-0114-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Olivieri M., Cho T., Álvarez-Quilón A., et al. A genetic map of the response to DNA damage in human cells. Cell . 2020;182(2):481–496.e21. doi: 10.1016/j.cell.2020.05.040. e421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ferri A., Stagni V., Barilà D. Targeting the DNA damage response to overcome cancer drug resistance in glioblastoma. International Journal of Molecular Sciences . 2020;21(14):p. 4910. doi: 10.3390/ijms21144910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kaina B., Christmann M. DNA repair in personalized brain cancer therapy with temozolomide and nitrosoureas. DNA Repair (Amst) . 2019;78:128–141. doi: 10.1016/j.dnarep.2019.04.007. [DOI] [PubMed] [Google Scholar]
- 50.Hosoya N., Miyagawa K. Targeting DNA damage response in cancer therapy. Cancer Sci . 2014;105(4):370–388. doi: 10.1111/cas.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bouwman P., Jonkers J. The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nature Reviews. Cancer . 2012;12(9):587–598. doi: 10.1038/nrc3342. [DOI] [PubMed] [Google Scholar]
- 52.Cao C., Tian B., Geng X., et al. IL-17-Mediated inflammation promotes cigarette smoke-induced genomic instability. Cells . 2021;10(5):p. 1173. doi: 10.3390/cells10051173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lin R., Xiao D., Guo Y., et al. Chronic inflammation-related DNA damage response: a driving force of gastric cardia carcinogenesis. Oncotarget . 2015;6(5):2856–2864. doi: 10.18632/oncotarget.3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kowalczyk P., Jaworek J., Kot M., et al. Inflammation increases oxidative DNA damage repair and stimulates preneoplastic changes in colons of newborn rats. J Physiol Pharmacol . 2016;67(2):277–286. [PubMed] [Google Scholar]
- 55.Kawanishi S., Ohnishi S., Ma N., Hiraku Y., Murata M. Crosstalk between DNA damage and inflammation in the multiple steps of carcinogenesis. International Journal of Molecular Sciences . 2017;18(8):p. 1808. doi: 10.3390/ijms18081808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yousefzadeh M., Henpita C., Vyas R., Soto-Palma C., Robbins P., Niedernhofer L. DNA damage-how and why we age? Elife . 2021 doi: 10.7554/eLife.62852.e62852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nakad R., Schumacher B. DNA damage response and immune defense: links and mechanisms. Front Genet . 2016;7:p. 147. doi: 10.3389/fgene.2016.00147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fontes F. L., Pinheiro D. M. L., Oliveira A. H. S. d., Oliveira R. K. d. M., Lajus T. B. P., Agnez-Lima L. F. Role of DNA repair in host immune response and inflammation. Mutation Research/Reviews in Mutation Research . 2015;763:246–257. doi: 10.1016/j.mrrev.2014.11.004. [DOI] [PubMed] [Google Scholar]
- 59.Martínez-Jiménez F., Muiños F., Sentís I., et al. A compendium of mutational cancer driver genes. Nature Reviews. Cancer . 2020;20(10):555–572. doi: 10.1038/s41568-020-0290-x. [DOI] [PubMed] [Google Scholar]
- 60.DeStefano Shields C. E., White J. R., Chung L., et al. Bacterial-driven inflammation and mutant BRAF expression combine to promote murine colon tumorigenesis that is sensitive to immune checkpoint therapy. Cancer Discovery . 2021;11(7):1792–1807. doi: 10.1158/2159-8290.Cd-20-0770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zulkifli A. A., Tan F. H., Putoczki T. L., Stylli S. S., Luwor R. B. STAT3 signaling mediates tumour resistance to EGFR targeted therapeutics. Molecular and Cellular Endocrinology . 2017;451:15–23. doi: 10.1016/j.mce.2017.01.010. [DOI] [PubMed] [Google Scholar]
- 62.Cheng P., Levesque M. P., Dummer R., Mangana J. Targeting complex, adaptive responses in melanoma therapy. Cancer Treatment Reviews . 2020;86 doi: 10.1016/j.ctrv.2020.101997.101997 [DOI] [PubMed] [Google Scholar]
- 63.Melgar K., Walker M. M., Jones L. M., et al. Overcoming adaptive therapy resistance in AML by targeting immune response pathways. Science Translational Medicine . 2019;11(508) doi: 10.1126/scitranslmed.aaw8828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li J. Y., Chen Y. P., Li Y. Q., Liu N., Ma J. Chemotherapeutic and targeted agents can modulate the tumor microenvironment and increase the efficacy of immune checkpoint blockades. Mol Cancer . 2021;20(1):p. 27. doi: 10.1186/s12943-021-01317-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mun E. J., Babiker H. M., Weinberg U., Kirson E. D., Von Hoff D. D. Tumor-treating fields: a fourth modality in cancer treatment. Clinical Cancer Research . 2018;24(2):266–275. doi: 10.1158/1078-0432.Ccr-17-1117. [DOI] [PubMed] [Google Scholar]
- 66.Lee Y. T., Tan Y. J., Oon C. E. Molecular targeted therapy: treating cancer with specificity. European Journal of Pharmacology . 2018;834:188–196. doi: 10.1016/j.ejphar.2018.07.034. [DOI] [PubMed] [Google Scholar]
- 67.Warren C. F. A., Wong-Brown M. W., Bowden N. A. BCL-2 family isoforms in apoptosis and cancer. Cell Death & Disease . 2019;10(3):p. 177. doi: 10.1038/s41419-019-1407-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kumar S., Fairmichael C., Longley D. B., Turkington R. C. The multiple roles of the IAP super-family in cancer. Pharmacology & Therapeutics . 2020;214 doi: 10.1016/j.pharmthera.2020.107610.107610 [DOI] [PubMed] [Google Scholar]
- 69.Chand H. S., Harris J. F., Tesfaigzi Y. IL-13 in LPS-induced inflammation causes Bcl-2 expression to sustain hyperplastic mucous cells. Sci Rep . 2018;8(1):p. 436. doi: 10.1038/s41598-017-18884-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Delbridge A. R. D., Grabow S., Strasser A., Vaux D. L. Thirty years of BCL-2: translating cell death discoveries into novel cancer therapies. Nature Reviews. Cancer . 2016;16(2):99–109. doi: 10.1038/nrc.2015.17. [DOI] [PubMed] [Google Scholar]
- 71.Qin B., Zhou Z., He J., Yan C., Ding S. IL-6 inhibits starvation-induced autophagy via the STAT3/Bcl-2 signaling pathway. Sci Rep . 2015;5(1) doi: 10.1038/srep15701.15701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Xu M. J., Feng D., Wang H., Guan Y., Yan X., Gao B. IL-22 ameliorates renal ischemia-reperfusion injury by targeting proximal tubule epithelium. Journal of the American Society of Nephrology . 2014;25(5):967–977. doi: 10.1681/asn.2013060611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yang C., He L., He P., et al. Increased drug resistance in breast cancer by tumor-associated macrophages through IL-10/STAT3/bcl-2 signaling pathway. Med Oncol . 2015;32(2):p. 14. doi: 10.1007/s12032-014-0352-6. [DOI] [PubMed] [Google Scholar]
- 74.Larrayoz M., Blakemore S. J., Dobson R. C., et al. The SF3B1 inhibitor spliceostatin A (SSA) elicits apoptosis in chronic lymphocytic leukaemia cells through downregulation of Mcl-1. Leukemia . 2016;30(2):351–360. doi: 10.1038/leu.2015.286. [DOI] [PubMed] [Google Scholar]
- 75.Gyrd-Hansen M., Meier P. IAPs: from caspase inhibitors to modulators of NF-κB, inflammation and cancer. Nature Reviews. Cancer . 2010;10(8):561–574. doi: 10.1038/nrc2889. [DOI] [PubMed] [Google Scholar]
- 76.Silke J., Meier P. Inhibitor of apoptosis (IAP) proteins-modulators of cell death and inflammation. Cold Spring Harbor Perspectives in Biology . 2013;5(2) doi: 10.1101/cshperspect.a008730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chang H., Zou Z. Targeting autophagy to overcome drug resistance: further developments. J Hematol Oncol . 2020;13(1):p. 159. doi: 10.1186/s13045-020-01000-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Doherty J., Baehrecke E. H. Life, death and autophagy. Nat Cell Biol . 2018;20(10):1110–1117. doi: 10.1038/s41556-018-0201-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ge Y., Huang M., Yao Y. M. Autophagy and proinflammatory cytokines: interactions and clinical implications. Cytokine & Growth Factor Reviews . 2018;43:38–46. doi: 10.1016/j.cytogfr.2018.07.001. [DOI] [PubMed] [Google Scholar]
- 80.Qin Y., Ekmekcioglu S., Liu P., et al. Constitutive aberrant endogenous interleukin-1 facilitates inflammation and growth in human melanoma. Molecular Cancer Research . 2011;9(11):1537–1550. doi: 10.1158/1541-7786.Mcr-11-0279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tu S. P., Quante M., Bhagat G., et al. IFN-γ inhibits gastric carcinogenesis by inducing epithelial cell autophagy and T-cell apoptosis. Cancer Research . 2011;71(12):4247–4259. doi: 10.1158/0008-5472.Can-10-4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Qi Y., Zhang M., Li H., et al. Autophagy inhibition by sustained overproduction of IL6 contributes to arsenic carcinogenesis. Cancer Research . 2014;74(14):3740–3752. doi: 10.1158/0008-5472.Can-13-3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lapaquette P., Guzzo J., Bretillon L., Bringer M. A. Cellular and molecular connections between autophagy and inflammation. Mediators of Inflammation . 2015;2015:1–13. doi: 10.1155/2015/398483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gacche R. N., Assaraf Y. G. Redundant angiogenic signaling and tumor drug resistance. Drug Resistance Updates . 2018;36:47–76. doi: 10.1016/j.drup.2018.01.002. [DOI] [PubMed] [Google Scholar]
- 85.Zhao Y., Murciano-Goroff Y. R., Xue J. Y., et al. Diverse alterations associated with resistance to KRAS(G12C) inhibition. Nature . 2021;599(7886):679–683. doi: 10.1038/s41586-021-04065-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kim E. K., Choi E. J. Compromised MAPK signaling in human diseases: an update. Arch Toxicol . 2015;89(6):867–882. doi: 10.1007/s00204-015-1472-2. [DOI] [PubMed] [Google Scholar]
- 87.Hammouda M. B., Ford A. E., Liu Y., Zhang J. Y. The JNK signaling pathway in inflammatory skin disorders and cancer. Cells . 2020;9(4):p. 857. doi: 10.3390/cells9040857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zepp J. A., Zhao J., Liu C., et al. IL-17A-Induced PLET1 expression contributes to tissue repair and colon tumorigenesis. The Journal of Immunology . 2017;199(11):3849–3857. doi: 10.4049/jimmunol.1601540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pinto S. M., Subbannayya Y., Rex D. A. B., et al. A network map of IL-33 signaling pathway. J Cell Commun Signal . 2018;12(3):615–624. doi: 10.1007/s12079-018-0464-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Grynberg K., Ma F. Y., Nikolic-Paterson D. J. The JNK signaling pathway in renal fibrosis. Front Physiol . 2017;8:p. 829. doi: 10.3389/fphys.2017.00829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Labib P. L., Goodchild G., Pereira S. P. Molecular pathogenesis of cholangiocarcinoma. BMC Cancer . 2019;19(1):p. 185. doi: 10.1186/s12885-019-5391-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Xing F., Liu Y., Sharma S., et al. Activation of the c-met pathway mobilizes an inflammatory network in the brain microenvironment to promote brain metastasis of breast cancer. Cancer Research . 2016;76(17):4970–4980. doi: 10.1158/0008-5472.Can-15-3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zarneshan S. N., Fakhri S., Farzaei M. H., Khan H., Saso L. Astaxanthin targets PI3K/Akt signaling pathway toward potential therapeutic applications. Food and Chemical Toxicology . 2020;145 doi: 10.1016/j.fct.2020.111714.111714 [DOI] [PubMed] [Google Scholar]
- 94.Hayes J. D., Dinkova-Kostova A. T., Tew K. D. Oxidative stress in cancer. Cancer Cell . 2020;38(2):167–197. doi: 10.1016/j.ccell.2020.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Moloney J. N., Cotter T. G. ROS signalling in the biology of cancer. Seminars in Cell & Developmental Biology . 2018;80:50–64. doi: 10.1016/j.semcdb.2017.05.023. [DOI] [PubMed] [Google Scholar]
- 96.Yang H., Villani R. M., Wang H., et al. The role of cellular reactive oxygen species in cancer chemotherapy. J Exp Clin Cancer Res . 2018;37(1):p. 266. doi: 10.1186/s13046-018-0909-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.de Sá Junior P. L., Câmara D. A. D., Porcacchia A. S., et al. The roles of ROS in cancer heterogeneity and therapy. Longevity . 2017;2017:12. doi: 10.1155/2017/2467940.2467940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Li B., Huang Y., Ming H., Nice E. C., Xuan R., Huang C. Redox control of the dormant cancer cell life cycle. Cells . 2021;10:p. 2707. doi: 10.3390/cells10102707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cui Q., Wang J. Q., Assaraf Y. G., et al. Modulating ROS to overcome multidrug resistance in cancer. Drug Resistance Updates . 2018;41:1–25. doi: 10.1016/j.drup.2018.11.001. [DOI] [PubMed] [Google Scholar]
- 100.Reuter S., Gupta S. C., Chaturvedi M. M., Aggarwal B. B. Oxidative stress, inflammation, and cancer: how are they linked? Free Radical Biology and Medicine . 2010;49(11):1603–1616. doi: 10.1016/j.freeradbiomed.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ma Q. Role of nrf2 in oxidative stress and toxicity. Annual Review of Pharmacology and Toxicology . 2013;53(1):401–426. doi: 10.1146/annurev-pharmtox-011112-140320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wang X. J., Sun Z., Villeneuve N. F., et al. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis . 2008;29(6):1235–1243. doi: 10.1093/carcin/bgn095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Shin D., Kim E. H., Lee J., Roh J. L. Nrf2 inhibition reverses resistance to GPX4 inhibitor-induced ferroptosis in head and neck cancer. Free Radical Biology and Medicine . 2018;129:454–462. doi: 10.1016/j.freeradbiomed.2018.10.426. [DOI] [PubMed] [Google Scholar]
- 104.McGarry T., Biniecka M., Veale D. J., Fearon U. Hypoxia, oxidative stress and inflammation. Free Radical Biology and Medicine . 2018;125:15–24. doi: 10.1016/j.freeradbiomed.2018.03.042. [DOI] [PubMed] [Google Scholar]
- 105.Dupré-Crochet S., Erard M., Nüβe O. ROS production in phagocytes: why, when, and where? Journal of Leukocyte Biology . 2013;94(4):657–670. doi: 10.1189/jlb.1012544. [DOI] [PubMed] [Google Scholar]
- 106.Blaser H., Dostert C., Mak T. W., Brenner D. TNF and ROS crosstalk in inflammation. Trends in Cell Biology . 2016;26(4):249–261. doi: 10.1016/j.tcb.2015.12.002. [DOI] [PubMed] [Google Scholar]
- 107.El-Kenawi A., Ruffell B. Inflammation, ROS, and mutagenesis. Cancer Cell . 2017;32(6):727–729. doi: 10.1016/j.ccell.2017.11.015. [DOI] [PubMed] [Google Scholar]
- 108.O’Donnell J. S., Teng M. W. L., Smyth M. J. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat Rev Clin Oncol . 2019;16(3):151–167. doi: 10.1038/s41571-018-0142-8. [DOI] [PubMed] [Google Scholar]
- 109.Pérez-Ruiz E., Melero I., Kopecka J., Sarmento-Ribeiro A. B., García-Aranda M., De Las Rivas J. Cancer immunotherapy resistance based on immune checkpoints inhibitors: targets, biomarkers, and remedies. Drug Resistance Updates: Reviews and Commentaries in Antimicrobial and Anticancer Chemotherapy . 2020;53 doi: 10.1016/j.drup.2020.100718. doi. 10.1016/j.drup.2020.100718.100718 [DOI] [PubMed] [Google Scholar]
- 110.Diakos C. I., Charles K. A., McMillan D. C., Clarke S. J. Cancer-related inflammation and treatment effectiveness. The Lancet. Oncology . 2014;15(11):e493–503. doi: 10.1016/s1470-2045(14)70263-3. [DOI] [PubMed] [Google Scholar]
- 111.Elinav E., Nowarski R., Thaiss C. A., Hu B., Jin C., Flavell R. A. Inflammation-induced cancer: crosstalk between tumours, immune cells and microorganisms. Nature Reviews Cancer . 2013;13(11):759–771. doi: 10.1038/nrc3611. [DOI] [PubMed] [Google Scholar]
- 112.Sacdalan D. B., Lucero J. A. The association between inflammation and immunosuppression: implications for ICI biomarker development. OncoTargets and Therapy . 2021;14:2053–2064. doi: 10.2147/ott.S278089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Jing B., Wang T., Sun B., et al. IL6/STAT3 signaling orchestrates premetastatic niche formation and immunosuppressive traits in lung. Cancer Research . 2020;80(4):784–797. doi: 10.1158/0008-5472.Can-19-2013. [DOI] [PubMed] [Google Scholar]
- 114.Mitchem J. B., Brennan D. J., Knolhoff B. L., et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Research . 2013;73(3):1128–1141. doi: 10.1158/0008-5472.Can-12-2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Shalapour S., Karin M. Pas de Deux: control of anti-tumor immunity by cancer-associated inflammation. Immunity . 2019;51(1):15–26. doi: 10.1016/j.immuni.2019.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sharma P., Hu-Lieskovan S., Wargo J. A., Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell . 2017;168(4):707–723. doi: 10.1016/j.cell.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Gaber T., Strehl C., Buttgereit F. Metabolic regulation of inflammation. Nat Rev Rheumatol . 2017;13(5):267–279. doi: 10.1038/nrrheum.2017.37. [DOI] [PubMed] [Google Scholar]
- 118.Leliefeld P. H. C., Wessels C. M., Leenen L. P. H., Koenderman L., Pillay J. The role of neutrophils in immune dysfunction during severe inflammation. Crit Care . 2016;20(1):p. 73. doi: 10.1186/s13054-016-1250-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yoshida H., Hunter C. A. The immunobiology of interleukin-27. Annu Rev Immunol . 2015;33(1):417–443. doi: 10.1146/annurev-immunol-032414-112134. [DOI] [PubMed] [Google Scholar]
- 120.Kampan N. C., Xiang S. D., McNally O. M., Stephens A. N., Quinn M. A., Plebanski M. Immunotherapeutic interleukin-6 or interleukin-6 receptor blockade in cancer: challenges and opportunities. Current Medicinal Chemistry . 2018;25(36):4785–4806. doi: 10.2174/0929867324666170712160621. [DOI] [PubMed] [Google Scholar]
- 121.Horiuchi T., Mitoma H., Harashima S. i., Tsukamoto H., Shimoda T. Transmembrane TNF-: structure, function and interaction with anti-TNF agents. Rheumatology (Oxford) . 2010;49(7):1215–1228. doi: 10.1093/rheumatology/keq031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Malcova H., Strizova Z., Milota T., et al. IL-1 inhibitors in the treatment of monogenic periodic fever syndromes: from the past to the future perspectives. Front Immunol . 2020;11 doi: 10.3389/fimmu.2020.619257.619257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Markota A., Endres S., Kobold S. Targeting interleukin-22 for cancer therapy. Human Vaccines & Immunotherapeutics . 2018;14(8):2012–2015. doi: 10.1080/21645515.2018.1461300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Alraouji N. N., Aboussekhra A. Tocilizumab inhibits IL-8 and the proangiogenic potential of triple negative breast cancer cells. Molecular Carcinogenesis . 2021;60(1):51–59. doi: 10.1002/mc.23270. [DOI] [PubMed] [Google Scholar]
- 125.Ly K., Smith M. P., Thibodeaux Q., Reddy V., Liao W., Bhutani T. Anti IL-17 in psoriasis. Expert Review of Clinical Immunology . 2019;15(11):1185–1194. doi: 10.1080/1744666x.2020.1679625. [DOI] [PubMed] [Google Scholar]
- 126.Wilson C. G. M., Arkin M. R. Small-molecule inhibitors of IL-2/IL-2R: lessons learned and applied. Current Topics in Microbiology and Immunology . 2011;348:25–59. doi: 10.1007/82_2010_93. [DOI] [PubMed] [Google Scholar]
- 127.Kalechman Y., Gafter U., Gal R., et al. Anti-IL-10 therapeutic strategy using the immunomodulator AS101 in protecting mice from sepsis-induced death: dependence on timing of immunomodulating intervention. J Immunol . 2002;169(1):384–392. doi: 10.4049/jimmunol.169.1.384. [DOI] [PubMed] [Google Scholar]
- 128.Michitaka S., Yasuo S., Kenju H., et al. IPD-1151T (suplatast tosilate) inhibits interleukin (IL)-13 release but not IL-4 release from basophils. Japanese Journal of Pharmacology . 1999;79(4):501–504. doi: 10.1254/jjp.79.501. [DOI] [PubMed] [Google Scholar]
- 129.Algra A. M., Rothwell P. M. Effects of regular aspirin on long-term cancer incidence and metastasis: a systematic comparison of evidence from observational studies versus randomised trials. The Lancet. Oncology . 2012;13(5):518–527. doi: 10.1016/s1470-2045(12)70112-2. [DOI] [PubMed] [Google Scholar]
- 130.Rothwell P. M., Price J. F., Fowkes F. G. R., et al. Short-term effects of daily aspirin on cancer incidence, mortality, and non-vascular death: analysis of the time course of risks and benefits in 51 randomised controlled trials. The Lancet . 2012;379(9826):1602–1612. doi: 10.1016/s0140-6736(11)61720-0. [DOI] [PubMed] [Google Scholar]
- 131.Rothwell P. M., Wilson M., Price J. F., Belch J. F., Meade T. W., Mehta Z. Effect of daily aspirin on risk of cancer metastasis: a study of incident cancers during randomised controlled trials. The Lancet . 2012;379(9826):1591–1601. doi: 10.1016/s0140-6736(12)60209-8. [DOI] [PubMed] [Google Scholar]
- 132.Ma J., Cai Z., Wei H., Liu X., Zhao Q., Zhang T. The anti-tumor effect of aspirin: what we know and what we expect. Biomedicine & Pharmacotherapy . 2017;95:656–661. doi: 10.1016/j.biopha.2017.08.085. [DOI] [PubMed] [Google Scholar]
- 133.Maeng H. J., Song J. H., Kim G. T., et al. Celecoxib-mediated activation of endoplasmic reticulum stress induces de novo ceramide biosynthesis and apoptosis in hepatoma HepG2 cells. BMB Reports . 2017;50(3):144–149. doi: 10.5483/bmbrep.2017.50.3.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Murray M. Y., Rushworth S. A., Zaitseva L., Bowles K. M., Macewan D. J. Attenuation of dexamethasone-induced cell death in multiple myeloma is mediated by miR-125b expression. Cell Cycle . 2013;12(13):2144–2153. doi: 10.4161/cc.25251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Yano A., Fujii Y., Iwai A., Kageyama Y., Kihara K. Glucocorticoids suppress tumor angiogenesis and in vivo growth of prostate cancer cells. Clinical Cancer Research . 2006;12(10):3003–3009. doi: 10.1158/1078-0432.Ccr-05-2085. [DOI] [PubMed] [Google Scholar]
- 136.Okamura Y., Sugiura T., Ito T., et al. The achievement of a sustained virological response either before or after hepatectomy improves the prognosis of patients with primary hepatitis C virus-related hepatocellular carcinoma. Ann Surg Oncol . 2019;26(13):4566–4575. doi: 10.1245/s10434-019-07911-w. [DOI] [PubMed] [Google Scholar]
- 137.Zhu F., Willette-Brown J., Song N. Y., et al. Autoreactive T cells and chronic fungal infection drive esophageal carcinogenesis. Cell Host & Microbe . 2017;21(4):478–493.e7. doi: 10.1016/j.chom.2017.03.006. e477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Bullman S., Pedamallu C. S., Sicinska E., et al. Analysis of Fusobacterium persistence and antibiotic response in colorectal cancer. Science . 2017;358(6369):1443–1448. doi: 10.1126/science.aal5240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Simon T. G., Duberg A. S., Aleman S., et al. Lipophilic statins and risk for hepatocellular carcinoma and death in patients with chronic viral hepatitis: results from a nationwide Swedish population. Ann Intern Med . 2019;171(5):318–327. doi: 10.7326/m18-2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Pelly V. S., Moeini A., Roelofsen L. M., et al. Anti-inflammatory drugs remodel the tumor immune environment to enhance immune checkpoint blockade efficacy. Cancer Discovery . 2021;11(10):2602–2619. doi: 10.1158/2159-8290.Cd-20-1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.van Zaanen H. C., Lokhorst H. M., Aarden L. A., et al. Chimaeric anti-interleukin 6 monoclonal antibodies in the treatment of advanced multiple myeloma: a phase I dose-escalating study. British Journal of Haematology . 1998;102(3):783–790. doi: 10.1046/j.1365-2141.1998.00835.x. [DOI] [PubMed] [Google Scholar]
- 142.Hong D. S., Hui D., Bruera E., et al. MABp1, a first-in-class true human antibody targeting interleukin-1α in refractory cancers: an open-label, phase 1 dose-escalation and expansion study. The Lancet. Oncology . 2014;15(6):656–666. doi: 10.1016/s1470-2045(14)70155-x. [DOI] [PubMed] [Google Scholar]
- 143.Rodon J., Carducci M. A., Sepulveda-Sánchez J. M., et al. First-in-human dose study of the novel transforming growth factor-β receptor I kinase inhibitor LY2157299 monohydrate in patients with advanced cancer and glioma. Clinical Cancer Research . 2015;21(3):553–560. doi: 10.1158/1078-0432.Ccr-14-1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Chen Y. X., Gao Q. Y., Zou T. H., et al. Berberine versus placebo for the prevention of recurrence of colorectal adenoma: a multicentre, double-blinded, randomised controlled study. The Lancet Gastroenterology & Hepatology . 2020;5(3):267–275. doi: 10.1016/s2468-1253(19)30409-1. [DOI] [PubMed] [Google Scholar]
- 145.Ngo B., Van Riper J. M., Cantley L. C., Yun J. Targeting cancer vulnerabilities with high-dose vitamin C. Nature Reviews Cancer . 2019;19(5):271–282. doi: 10.1038/s41568-019-0135-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ellulu M. S., Rahmat A., Patimah I., Khaza’ai H., Abed Y. Effect of vitamin C on inflammation and metabolic markers in hypertensive and/or diabetic obese adults: a randomized controlled trial. Drug Design, Development and Therapy . 2015;9:3405–3412. doi: 10.2147/dddt.S83144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Jang I. S., Ko Y. H., Moon Y. S., Sohn S. H. Effects of vitamin C or E on the pro-inflammatory cytokines, heat shock protein 70 and antioxidant status in broiler chicks under summer conditions. Asian-Australas J Anim Sci . 2014;27(5):749–756. doi: 10.5713/ajas.2013.13852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Yun J., Mullarky E., Lu C., et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science . 2015;350(6266):1391–1396. doi: 10.1126/science.aaa5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No data were used to support this study.