

Abstract

Due to its intrinsic electrical conductivity, polyaniline (PANI) is one of the most promising conducting polymers for high-performance applications in a wide range of technological fields. However, its poor dispersibility in water and organic solvents markedly imparts its processability and electrical conductivity. Herein, we report a green and one-step approach to preparing stable colloidal dispersions of highly dispersible hybrid nanoparticles by polymerizing PANI onto chitin nanocrystals (ChNCs) as biotemplates, via initiation through the surface amino groups of ChNCs. Evidence of the grafting of PANI onto ChNCs was supported by transmission electron microscopy (TEM), as well as Raman and Fourier transform infrared (FTIR) spectroscopies. Nanocomposite films were prepared by mixing the PANI-g-ChNCs with a waterborne poly(vinyl acetate) latex dispersion followed by casting and film formation at room temperature. The mechanical properties were tested as a function of the PANI-g-ChNC content. In addition, it was shown that at a proper content of PANI in ChNCs, and over a critical loading in the PANI-g-ChNCs, a conductive film was obtained, without sacrificing the reinforcing effect of the rodlike nanofiller. As a potential application, conductive waterborne adhesives for wood were prepared and the performance of the adhesives was tested. This research provides a facile route to fabricating a new class of hybrid nanofiller from a biobased origin, stable in water and easy to mix with waterborne dispersions, combining the merits of the ChNC nanofiller with the conductivity of PANI.
1. Introduction
Electrically conducting materials that display excellent mechanical performance and a high degree of flexibility have received a great deal of scientific interest as promising and potentially sustainable materials in the field of flexible electronics and soft conductors.1 In this regard, conducting polymers, such as poly(3,4-ethylenedioxythiophene) (PEDOT),2 polypyrrole (PPY),3 polyaniline (PANI),4 and polyacetylene (PA),5 have emerged within the last few decades owing to their controllable electrical conductivity, facile processability into different forms including thin films or fibers, and low cost.5 The most frequently studied conducting polymer is PANI, given its intriguing electrical properties, high environmental stability, and, more particularly, its reversible acid/base doping/dedoping chemistry.6 Consequently, this so-called organic metal7 has extensively been exploited in multiple fields including sensing,8 metal anticorrosion,6 EMI shielding applications,9,10 biomedical fields,11 and conductive inks/paints/adhesives.12 However, despite PANI’s distinctive features, its practical applications were restricted because of its (i) highly conjugated system resulting in poor film-forming capability, (ii) limited solubility in organic solvents and infusibility due its high polarity, (iii) strong tendency to self-aggregate by hydrogen bonding, and (iv) lack of flexibility due to the rigid aromatic backbone preventing any processing of PANI in the form of thin deformable freestanding films. One alternative to taking advantage of the electrical properties of PANI while limiting its shortcomings was to embed PANI in a flexible polymer matrix with an appropriate loading to ensure the generation of a continuous conducting network through a percolating nanostructure. This particular approach has been widely adopted to produce conductive films for multiple applications including capacitors,12 supercapacitors,13,14 and biosensing.15 While this approach seemed initially promising, its processing suffered from low productivity due to the difficulty of controlling the morphology of the PANI nanostructure and the necessity to use highly polar organic solvents such as dimethyl sulfoxide (DMSO) or N-methyl-2-pyrrolidone (NMP) that are capable of dissolving PANI and the polymer matrix. These solvents are toxic and hard to completely remove from the film by evaporation. Subsequently, the design of highly conductive composites with a low amount of PANI while retaining their good processability and mechanical properties has become one of the current critical issues. Accordingly, there has been renewed interest in these composites with the emergence of multifunctional third constituents.
Prior works have reported that the addition of an organic or inorganic constituent to the polymer matrix, in combination with PANI, could provide a matrix with specific properties by lowering the percolation threshold and acting as a bio16 or hard template17 while improving the mechanical,18 thermal,19 or barrier properties of the composite. One of the most recommended strategies is the template-assisted polymerization method because of its facile, efficient, and highly controllable synthesis of conducting polymer nanostructures offering numerous advantages: (i) the nanoscale, (ii) a high electrical conductivity, (iii) a large specific surface area, and (iv) short path lengths for the transport of conducting species. Among the wide possibilities of nanoscale templates, biobased nanoparticles such as nanocelluloses, nanochitins, or starch nanoparticles have seen an increasing interest thanks to their numerous attributes including biodegradability, high stiffness, and strong reinforcing potential. For example, Wu and co-workers20 developed PANI@cellulose nanowhisker hybrid nanoparticles with a high aspect ratio and improved dispersion and then introduced them into natural rubber following a latex co-coagulation process, resulting in high-performance nanocomposites with an organized 3D hierarchical multiscale structure.
Other researchers have elaborated a series of lightweight, flexible, and conductive cellulose nanopapers through a one-step in situ polymerization of aniline on cellulose nanofibers.8 This study showed that not only these nanopapers were highly conductive (0.314 S/cm) but they were also effective green electromagnetic radiation attenuators. However, despite the tempting combination of a facile approach/biocompatibility of the nanoparticles, there are some additional parameters that must be monitored for the successful use of nanocelluloses, mainly their dispersion/aggregation into networks, particularly, in acidic conditions, due to their negative surface charge.
Chitin is the second most abundant natural polymer after cellulose, commonly found in crustacean exoskeletons, fungal cell walls, and some algae.21 It is a linear homopolymer of β-1,4-linked N-acetylglucosamine units.22,23 Aside from being nontoxic, biodegradable, and renewable, chitin is insoluble and resistant to acid, alkalis, and common organic solvents. One of its main particularities is its accessibility to produce nanoscale materials by mechanical/physical/chemical approaches. For example, rodlike chitin nanocrystals (ChNCs)24 with a high aspect ratio, low density, and high surface reactivity can be produced by an acid hydrolysis of chitin powders, generating stable colloidal suspensions.25−27 ChNCs have recently attracted a great deal of attention in the field of polymeric nanocomposite materials owing to their extremely high modulus and strength (150 GPa). Their utilization as fillers in different polymer matrices was reported to be more advantageous and efficient compared to conventional inorganic fillers.28 Despite its abundance, chitin remains among the least utilized biomass resources presumably due to its intractable structure and recalcitrance toward processing. Moreover, chitin is the only positively charged polysaccharide among all other naturally occurring biopolymers, which opens the way to wider possibilities of applications/functionalization. Regarding ChNCs, and in spite of their structural and morphological resemblance with CNCs, the hydroxyl groups in chitin are partially occupied with acetyl amine/amine, resulting in a stronger inter- and intramolecular H-bonding between the neighboring chains.29 In comparison with CNCs, ChNCs are harder to obtain by disintegration of chitin fibrils and are more chemically stable under acidic pH. In addition, thanks to the presence of the amino groups, their positively charged surface and their good stability at low pH, ChNCs should be considered as a support for the in situ polymerization of aniline. The higher chemical stability and better resistance to acidity of ChNCs than other biobased nanoparticles (NPs) would make it advantageous to explore the potential use of ChNC/PANI composites as conductive nanomaterials.
In the present work, PANI-g-ChNCs nanohybrids have been successfully prepared via in situ oxidative polymerization of the aniline monomer using chitin nanocrystals as biotemplates and ammonium persulfate as the oxidant. The polymerization was conducted in an acidic solution. The adopted approach of polymerization was easy to implement in water, straightforward, and eco-friendly. The colloidal stability and morphology of the as-prepared nanohybrids were investigated. The mechanical properties and conductivity of poly(vinyl acetate) matrices filled with pure PANI, ChNCs, and PANI-g-ChNCs were also studied. Finally, we have demonstrated that PANI-g-ChNCs could be used as electrically conductive adhesives for wood substrates, with a strength surpassing that of unfilled adhesives. To our knowledge, the hybridization grafting of ChNCs with PANI has not been reported yet.
2. Experimental Section
2.1. Materials
Aniline (≥99.0%), ammonium persulfate (APS) (≥98.0%), hydrochloric acid (HCl, 37 vol %, reagent grade), and chitin from crab shells were purchased from Sigma-Aldrich and used as received. An industrial latex based on poly(vinyl acetate) (PVAc) from Vinavil was used for the preparation of adhesive samples. Hard wood sheets (70 × 30 mm2, thickness 4 mm) were used as substrates for the lap shear test of the electrically conductive adhesive.
2.2. Preparation of Chitin Nanocrystals
Chitin nanocrystals were prepared via hydrochloric acid of crude chitin crab shells as reported previously.27 Briefly, 4 g of chitin powder was dispersed into 4 M aqueous HCl solution and remained under magnetic stirring at 95 °C for 90 min. The suspension was cooled down to 20 °C and then repeatedly washed with distilled water and subsequently centrifuged at 6000 rpm to recover ChNCs. The ChNC suspension was purified by dialysis against distilled water to remove the excess acid. A stock of ChNCs at 8 wt % solid content at pH 4–5 was obtained and used as a biotemplate for PANI-g-ChNCs. The degree of deacetylation (DD) was determined by conductometric titration according to the method described in our previous work.27
2.3. Preparation of Neat PANI and PANI-g-ChNCs
PANI and PANI-g-ChNCs were prepared following an in situ oxidative polymerization of aniline monomers in which APS was used as an oxidant and HCl as an acid dopant for PANI. Typically, aniline (0.6 g) was dissolved in 20 mL of aqueous HCl (1 M) and kept under vigorous stirring for 2 min to ensure the dissolution and protonation of the monomer. The mixture was kept in a temperature-controlled ice bath (0 °C). A solution of APS (dissolved in 1 M HCl with an APS/aniline ratio of 1.33 w/w) was gradually added to the reaction system for 30 min, and the polymerization was pursued for 3 h. For PANI-to-ChNC ratios of (6:1) and (1:1), the ChNC suspension was sonicated for 30 s in an acidic HCl solution and the corresponding amount of aniline monomer was added. The polymerization was carried out similarly as for pure PANI. For all compositions, the color of the suspensions turned dark green from the first 30 min of the reaction and the color remained green during the course of the reaction. The suspension was centrifuged at 6000 rpm and washed several times with 0.5 M HCl to remove the byproducts and remaining reagents. The pH was adjusted to 3 by adding a few drops of HCl solution to guarantee the doping of pure PANI and PANI-g-ChNC suspensions.
2.4. Fabrication of Ex Situ/In Situ PANI-g-ChNC/Latex and PANI/Latex Nanocomposites
For the ex situ method, PANI/latex and PANI-g-ChNC/latex were prepared following a two-step process. The desired amounts of PANI-g-ChNCs or PANI suspensions were sonicated for 20 s and then added to 1.5 g of latex and kept under stirring for 30 min to ensure the mixing of PANI or PANI-g-ChNCs with the latex. After that, the mixtures were cast into a Teflon mold and left to dry at room temperature for 24 h.
For the in situ procedure of the preparation of PANI/PVAc, the calculated amounts of aniline monomers (6, 13, 20, and 30%) were dispersed in 20 mL of 1 M aqueous HCl. The dispersed system was poured into a flask containing the PVAc dispersion. The reaction was kept under continuous stirring at 0 °C while adding the APS solution for 3 h. The mixture gradually turned green. Finally, the nanocomposite solution was poured into a Teflon mold and dried in an oven at 60 °C for 24 h.
2.5. Particle Morphology
Diluted ChNC and PANI-g-ChNC aqueous suspensions were sonicated for 30 s, and droplets of ca. 0.001 wt % of the well-dispersed suspensions were deposited on freshly glow-discharged carbon-coated films supported by copper TEM grids. Prior to drying, a drop of 2 wt % uranyl acetate negative stain was deposited on the specimens. After 1 min, the stain in excess was blotted with filter paper and the remaining liquid film was allowed to dry. The specimens were observed with a JEOL JEM 2100-Plus transmission electron microscope (TEM) operating at a voltage of 200 kV. The images were recorded with a Gatan Rio 16 digital camera.
2.6. Particle Size and ζ-Potential Measurements
Particle size and surface charge were measured at 25 °C by dynamic light scattering (DLS) using a Zetasizer ZS apparatus from Malvern. The concentration of pristine PANI, ChNCs, and PANI-g-ChNC suspensions was fixed at 0.2 mg/mL and pH 3–4. Each measurement was performed in triplicate, and the obtained values were averaged to obtain the mean particle size. The polydispersity index (PDI), which is dimensionless and measures the broadness of the size distribution calculated from the cumulants analysis, was also assessed.
2.7. X-ray Diffraction (XRD)
Pure PANI, ChNC, and PANI-g-ChNC powders were poured into 1 mm glass capillaries that were flame-sealed and X-rayed in a Warhus vacuum chamber using a Philips PW3830 generator operating at 30 kV and 20 mA (Ni-filtered Cu Kα radiation, λ = 0.1542 nm), during 1.5 h exposures. Two-dimensional diffraction patterns were recorded on Fujifilm imaging plates, read offline with a Fujifilm BAS 1800-II bioimaging analyzer. Diffraction profiles were calculated by rotational averaging of the 2D patterns.
2.8. Thermal Stability
The thermal stability of all samples was evaluated by thermogravimetric analysis (TGA) using a TGA 400 thermogravimetric analyzer from Perkin Elmer under an airflow in the temperature range of 50–800 °C at the heating rate of 10 °C/min.
2.9. Vibrational Spectroscopies
Fourier transform infrared (FTIR) spectroscopy was carried out on a Perkin Elmer spectrometer in the 4000–400 cm–1 range. Raman spectra were recorded on a LabRAM analytical Raman microspectrograph (Jobin-Yvon, Horiba Group, France) using a He–Ne laser source as the exciting radiation (λ = 633 nm) and an air-cooled CDD detector. The acquisition time was 100 s.
2.10. Mechanical Properties
Dynamic mechanical properties were conducted in the tension mode using a PYRIS Diamond DMA (Perkin-Elmer, Waltham, MA). The sample with dimensions of 20 mm × 5 mm × 2 mm was scanned from −20 to 100 °C at a heating rate of 2 °C/min.
2.11. Preparation of Adhesives
The adhesive for wood was prepared as follows: the appropriate amount of PANI-g-ChNC in the form of a suspension with a solid content of 2 wt % was mixed with PVAc latex, and the mixture was kept under magnetic stirring for 15 min to obtain a homogeneous viscous adhesive with a dark-green color when PANI-g-ChNC was included. A reference composition without PANI-g-ChNC was also prepared for the purpose of comparison. All adhesives tested were prepared at the same solid content (30 wt %). The formulation of the different adhesives is given in Table S1.
2.12. Fabrication and Tests of Wood Joints
Test samples for bond-strength measurement were prepared according to the NF EN 205 standard. The wood specimen with a dimension of 100 × 20 × 5 mm3 was glued on one side with the adhesives, and the two specimens were joined together, gently pressed to ensure uniform adhesive spreading, and then kept in an oven at 40 °C for 24 h, followed by compressing under a static pressure of 1 MPa at ambient temperature for 2 h. Before the bond-strength test, all specimens were conditioned at a temperature of 25 °C and relative humidity of 50% for 1 week to ensure equilibrium moisture content and reduce internal stresses in the bond joint.
2.13. Conductivity Measurements
The electric conductivities of all samples were measured via a Novocontrol Broadband dielectric spectrometer, based on an α analyzer and a Quatro temperature controller. Dielectric permittivity and conductivity data were recorded using the Win DETA impedance analysis software.
3. Results and Discussion
3.1. Morphology and Surface Charge of ChNCs and PANI-g-ChNCs
The TEM image of neat ChNCs (Figure S1) shows rodlike particles (200–800 nm in length and 20–80 nm in width) constituted by a few parallel elementary crystallites that were not separated during the preparation treatment, as was also reported for cellulose nanocrystals of various origins.30 The DLS profile showed a monomodal distribution with a hydrodynamic diameter extending from 50 to 200 nm (Figure 1A). The degree of deacetylation (DDA) of the ChNCs was 11.2% (Figure S2), meaning that about 89% of the N-acetyl groups were not hydrolyzed and 11% were in the form of amino groups. The ζ-potential of the ChNC suspension remained positive and over 25 mV at pH <6, indicating that ChNCs were positively charged within this pH domain, with enough surface charges to ensure colloidal stability by electrostatic repulsion (Figure 1B). This explains the transparency of the ChNC suspension and its stability in water for over 2 months. Within the 7–9 pH range, the ζ-potential remained lower than −7 mV, indicating early neutral particles. This evolution of the ChNC surface charge was expected due to the presence of NH2 groups on the surface of ChNCs that protonate at pH < pKNH2 – 1 (with pKNH2 ≈ 6). After in situ polymerization of aniline in the presence of ChNCs, the suspension turned to green without any sign of particle aggregation with a color intensity that increased with the aniline concentration: PANI-g-ChNCs (6:1) had a color darker than that at the ratio 1:1. Both PANI-g-ChNCs (6:1) and (1:1) suspensions remained stable at pH 3 (after sonication for 30 s) for more than 2 weeks without any sign of particle aggregation or sedimentation, which was not the case when PANI was prepared under the same conditions but without incorporation of ChNCs. The latter totally settle down after 1 day, as shown in Figure 1C,D. It is worth mentioning that the ChNC suspension remained stable and preserved nearly the same particle size distribution when left under an acidic pH of 2–3 for more than 3 h (the same as that used for the synthesis of PANI) (Figure S3). This result further demonstrates the high chemical resistance of ChNCs, which is presumably due to the stronger inter- and intramolecular H-bonding between the chitin chains, due to the presence of acetyl amine/amine groups in the chitin chains.
Figure 1.

Particle size distribution (A) and ζ-potential/mean-particle size/polydispersity index (B) of ChNC, PANI, PANI-g-ChNC (1:1), and PANI-g-ChNC (6:1) suspensions prepared by in situ polymerization. (C, D) Photos of the ChNC, PANI, PANI-g-ChNC (1:1), and PANI-g-ChNC (6:1) suspensions after a 10 s sonication (C) and a 24 h standing (D).
The particle size distribution of PANI-g-ChNCs (6:1 and 1:1) and PANI suspensions (Figure 1A) showed a monomodal distribution, peaking at 190, 280, and 1800 nm for PANI-g-ChNCs (6:1), PANI-g-ChNCs (1:1), and PANI, respectively. The PDI of the PANI-g-ChNCs remained lower than 0.25 (Figure 1B), which is indicative of a narrow distribution in the particle size. The micron size of PANI particles along with their reported density of 1.4 explains the settlement of PANI suspension over time, despite the highly positive ζ-potential (around +29 mV) (Figure 1B). For both PANI-g-ChNC suspensions (1:1 and 6:1), the ζ-potential of the particles remained over +20 mV at pH 3, which is enough to ensure colloidal stability by electrostatic repulsion. The positive ζ-potential of PANI-g-ChNCs is due to the protonation of imine groups of PANI. However, over pH 9, the deprotonation of PANI transforms the emeraldine to the pernigraniline form exempt of charges, which led to a rapid aggregation of ChNCs/PANI.
The TEM images of PANI-g-ChNCs show that the ChNCs are in contact with ellipsoidal particles presumably corresponding to polymerized PANI (Figures 2 and S2). The size of these particles ranges from 70 to 150 nm in length and from 50 to 80 nm in width for PANI-g-ChNCs (1:1) (Figures 2A and S2A,B). The long axis of the particle is often aligned with the ChNC axis. Moreover, in higher magnification images, PANI particles appear to enwrap the ChNCs (Figures 2B and S2C,D) that are not entirely covered. While a significant fraction of ChNCs is devoid of the associated PANI particle, no free PANI particles were observed, suggesting that PANI was exclusively nucleated on the surface of ChNCs. With a higher relative amount of PANI (6:1), the particles attached to the ChNCs are significantly larger, with the length ranging from 100 to 400 nm and the width from 80 to 150 nm (Figure 2C). The ChNCs are now coated with several of these PANI particles although there is still a small fraction of neat ChNCs (Figure 2D).
Figure 2.

(A–D) TEM images of negatively stained preparations of neat PANI-g-ChNCs (1:1) (A, B) and PANI-g-ChNCs (6:1) (C, D).
The first step of the mechanism involved the protonation of the amino groups of ChNCs and their oxidation by APS into the amine cation (Figure 3). Then, the polymerization was initiated by the addition of the aniline cation radical, and the growth of PANI was proceeded by chain propagation induced by the interaction of the growing oligomer radical cations with aniline radical cations until the depletion of aniline. However, the occurrence of secondary reactions including dimerization, oligomerization of aniline, or hydrolysis of imine is inevitable, which may explain why the weight ratio in PANI-g-ChNCs differs from the theoretical value based on the initial aniline/ChNC weight ratio. The specific structure of ChNCs in the presence of amino groups on the surface of the fibrils accounts for the occurrence of grafting of PANI on ChNCs ensuring an effective covalent attachment of PANI on ChNCs and colloidal stability under acidic pH. One can enquire about the preferential growth of PANI on ChNCs to the detriment of homopolymerization of PANI. One possible reason would be the higher initiation rate from ChNC-NH2+ compared to the aniline radical cation. The lower reactivity of the aniline radical cation is likely due to the delocalization of the radical across the benzene ring that stabilizes the unpaired electron, which is not the case for ChNC-NH2+, where the radical is localized on the amine function and accordingly more reactive than the conjugated radical. Moreover, the increase in size of PANI-g-ChNCs with the increment in the aniline/ChNC ratio suggests that chain propagation during the polymerization of aniline is favored compared to the initiation from the surface of ChNCs. This may explain that not all of the ChNCs were coated with PANI although the fraction of uncoated ChNCs decreased with the increase in the aniline/ChNC ratio. Work is in progress to better understand the effect of different parameters including pH, aniline/ChNC ratio, and APS concentration on the morphology of PANI-g-ChNCs. It is worth mentioning that the possible involvement of the amino groups of chitin in the grafting process of PANI was also suggested for PANI-grafted chitosan.31
Figure 3.

Mechanism of PANI synthesis and grafting onto ChNCs.
3.2. Crystallinity and Thermogravimetric Analyses
Figure 4A displays the XRD profiles of pure PANI, ChNCs, and PANI-g-ChNCs with two different weight ratios (6:1 and 1:1). The characteristic peaks at 9.5, 12.8, 19.3, 20.8, 22.3, and 26.4° correspond to the 020, 021, 110, 120, 130, and 013 reflections of α-chitin, respectively.29 Compared to α-chitin profiles found in the literature, collected from dry films and thus promoting some uniplanarity of the particles,32,33 the 013 reflection is strong, which is expected since a powder profile was recorded. Pristine PANI exhibits four broad peaks centered around 9.4, 15.1, 20.4, and 25.5°, corresponding to the (022), (020), (010), and (001) crystal planes of PANI in its emeraldine salt form.34 The peak at 25.5° is assigned to the periodic arrays perpendicular to the main chain, whereas the characteristic peaks at 9.4, 15.1, and 20.4° are attributed to the periodic channels parallel to the principal chain. The XRD profiles of both PANI-g-ChNCs were identical in terms of the peak position of PANI and chitin, but the relative peak intensities varied. This shows that PANI is still semicrystalline in the hybrid nanoparticles.
Figure 4.

XRD profiles of ChNCs, PANI, and PANI-g-ChNCs (A), TG (B), and DTG (C) curves of pure PANI, ChNCs, and PANI-g-ChNCs.
The thermogravimetric analysis (TGA) and the corresponding differential thermogravimetry (DTG) under an air atmosphere for ChNCs, PANI, and PANI-g-ChNCs are shown in Figure 4B,C. For neat ChNCs, the TGA is characterized by three stages of weight loss. The first one of about 5% is due to the evaporation of water adsorbed on ChNCs. The second one starting at 280 up to 400 °C with a weight loss around 78% is related to the degradation of the saccharide backbone of chitin, including the dehydration of saccharide rings and the depolymerization and decomposition of chitin. The third step occurring in the range of 400–600 °C with about 22% weight loss is attributed to the decomposition of the residual carbon. The TGA of PANI synthesized under similar conditions in the presence of ChNCs showed a two-step weight loss. The first one in the range of 80–400 °C is assigned to the loss of bound water and HCl dopant, while the second weight loss extending from 400 to 800 °C is due to the thermal degradation of the PANI scaffold. The TGA pattern of PANI-g-ChNCs is a combination of the TGA of ChNCs and that of PANI, with a three-stage weight loss starting with water evaporation at 100–120 °C, followed by a second loss at 200–400 °C with a lower magnitude for PANI-g-ChNCs with the highest PANI content.
Assuming that the thermal decomposition of PANI starts at 400 °C, and that 78% of ChNCs were decomposed at 400 °C, the weight fraction of appended PANI on ChNCs can be estimated. This gives about 38 and 60% for PANI-g-ChNCs (1:1) and (6:1), respectively (see calculation details in the Supporting Information), giving respective ratios of (0.15:1) and (1.5:1). This indicates that only a certain fraction of aniline was grafted on ChNCs, while the residual part remained unreactive as no free PANI was observed by TEM. More work is needed to increase the grafting efficiency without inducing the homopolymerization of aniline.
3.3. FTIR and Raman Analyses
To further understand the changes in the surface structure and the interaction between chitin and PANI, Raman spectra of PANI, ChNCs, and PANI-g-ChNCs are shown in Figure 5A. The spectrum of ChNCs is characterized by two bands at 1660 and 1621 cm–1 assigned to amide I vibrations of the amide groups within chitin. The presence of two signals in the amide I absorption band is attributed to the presence of two different types of hydrogen bonds in chitin crystals. The amide III (C–N stretching vibrations) of the chitin band is visible at 1330 cm–1. Other bands of ChNCs appear at 1100–1150 cm–1, which are assigned to the symmetric stretching vibrations of glycosidic COC groups in chitin. The intense bands at 1380 and 1115 cm–1 are relative to the bending vibration of CH2 and CO–H. The pure PANI spectrum is characterized by five main peaks at around 1590, 1470, 1330, 1260, and 1160 cm–1. The peak observed at 1590 cm–1 is related to the C=C stretching vibrations in a quinonoid ring.35 The peak with the maximum at about 1500–1470 cm–1 corresponds to the N–H deformation vibrations of the semiquinonoid structures, with the contribution of the C=N stretching vibrations in quinonoid units. The band at 1330 cm–1 is related to the C–N+• stretching vibrations of the semiquinone cation radicals in delocalized polaronic structures, which confirmed that PANI was in the form of the conducting emeraldine salt phase. The band at 1260 cm–1 is related to the benzene-ring deformation vibrations, and the band at 1160 cm–1 corresponds to the C–H bending vibrations of the benzenoid rings.36
Figure 5.

Raman spectra (A) and FTIR spectra (B) of ChNCs, PANI, and PANI-g-ChNCs.
The spectrum of PANI-g-ChNCs reveals a combination of bands of PANI and ChNCs with intensities differing according to the ratio of aniline/ChNCs used in the preparation of PANI-g-ChNCs. The most visible evolution is observed in the spectra of PANI-g-ChNCs (6:1) in the 1480–1690 cm–1 region, where the typical C=C bands of quinonoid and benzenoid in PANI are split into two bands. The band at 1470 cm–1 in PANI appears at 1490 cm–1 with another band at 1520 cm–1 in PANI-g-ChNCs (6:1), and the band at 1590 cm–1 is accompanied by another band at 1620 cm–1. The amide III band of ChNCs in PANI-g-ChNCs is also shifted by about 6 cm–1 to a lower wavenumber. The shift in the position of the amide III band presumably indicates the occurrence of interaction between the NH2/CONH groups at the surface of ChNCs and the appended PANI. Moreover, we infer the splitting of the typical bands of PANI at 1590 and 1470 cm–1 to the chemical grafting of PANI on ChNCs via the -NH2 groups of the surface of ChNCs. The better visibility of the splitting in PANI-g-ChNCs (6:1) is likely due to the increased amount of the -NH-PANI moiety as the grafting amount increased with the aniline/ChNCs ratio.
The FTIR spectra of ChNCs, PANI, and PANI-g-ChNCs are shown in Figure 5B. The main characteristic FTIR bands of ChNCs are visible in the region below 2000 cm–1, with the two absorption bands at 1657 and 1620 cm–1 assigned to amide I (C=O stretching in amide groups) and the strong band at 1556 cm–1 attributed to amide II (a combination of CNH stretching and NH bending). The bands at 1378 and 1259 cm–1 are ascribed to the C–OH and C–O stretching vibrations in the polysaccharide chain, and the strong bands at 1114, 1078, and 1026 cm–1 are typical of C–O–C glycosidic linkage. The PANI spectrum is also dominated by typical bands below 2000 cm–1. The two strong bands at 1592 and 1496 cm–1 can be assigned to the C=C stretching of the quinonoid and the benzenoid ring, respectively, while the weak peak at 1380 cm–1 is attributed to C–N stretching vibrations in the neighborhood of a quinonoid ring. The strong peak at 1306 cm–1 is ascribed to π-electron delocalization in protonated PANI.37
The evolution in the FTIR spectra of PANI-g-ChNCs was less visible than in the Raman one, with mainly a shift of about 5 cm–1 in the position of some absorption bands. The main shift was observed in amide I and amide II of ChNCs, and evolution upon grafting was also observed; the FTIR spectrum of PANI-g-ChNCs shows some shifting and lowering in band intensities, which emphasizes the electrostatic interactions between PANI and ChNCs. Small peak shifts with a remarkable decrease in intensity are detected for the amide I and II vibrations at 1652 and 1551 cm–1 for PANI-g-ChNCs due to the interaction of the two polymers. This shift suggests that interactions occur between appended PANI and ChNCs, involving the amide and NH groups.
3.4. Mechanical Properties of PANI-g-ChNCs/PVAc Nanocomposites
One of the objectives of the association of PANI with ChNCs was to use PANI-g-ChNCs as a conductive nanofiller that is easy to disperse and mix with a waterborne polymer. For this purpose, we selected a PVAc latex dispersion to prepare nanocomposite films by mixing the PANI-g-ChNCs, ChNCs, or PANI suspension with the latex, followed by the casting of the mixture until complete water evaporation and film formation. For comparison purpose, all nanocomposite films were prepared with the same nanofiller content of 13 wt %.
DMA was used to compare how the inclusion of the nanofiller affected the stiffness of the film over a wide temperature range from the glassy to the rubbery state. The trace of the storage modulus (E′) and tan δ from −20 to 120 °C is shown in Figure 6A. The neat PVAc matrix showed a glassy plateau with E′ around 1 GPa extending to about 30 °C, followed by an abrupt drop in E′ by more than 2 decades around 40 °C, followed by a rubbery plateau and a second drop in E′ by more than 2 decades around 80 °C associated with the glass-transition temperature (Tg) of poly(vinyl alcohol) (PVA), which is used as a colloidal stabilizer for the emulsion polymerization of the vinyl acetate monomer. These two transitions give rise to a maximum in the tan δ plot at 30 and 80 °C, respectively, which further confirmed the assignment of these relaxations to the glass transition.
Figure 6.

(A) Variation of storage modulus E′ and (B) tan δ with temperature for the neat PVAc matrix and nanocomposite films containing 13 wt % PANI, ChNCs, PANI-g-ChNCs (1:1) and (6:1). (C) Plot of Er vs nanofiller type at 50 and 100 °C.
In the presence of 13 wt % PANI, only a minor evolution
in both
the E′ and tan δ plots was observed,
suggesting that the inclusion of PANI at 13 wt % did not alter the
stiffness of the PVA film. In the presence of 13 wt % ChNCs, a marked
increment in the magnitude of E′ was observed,
in comparison with the matrix above the Tg of PVAc, and the drop in E′ above the Tg of PVA was modest. This effect indicated a
huge stiffening effect imparted by the inclusion of 13 wt % ChNCs
in the matrix, which is expected given the rodlike morphology of the
ChNCs and their ability to generate a percolated network at this solid
loading. In the tan δ plot, the magnitude of the Tg relaxation notably decreased in comparison
with the neat matrix, indicating a restriction in the mobility of
the macromolecular chains of PVAc and PVA in the presence of ChNCs.
In the presence of PANI-g-ChNCs, the same tendency
of E′ increases above the Tg of PVAc and PVA was observed, but with a magnitude less
pronounced than that noted for neat ChNCs. This can better be seen
in the increment in the modulus 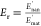 (with E′nan and E′mat the storage modulus
of the nanocomposite and unfilled matrix respectively measured at
different temperatures) vs the PANI-g-ChNC content.
As shown in Figure 6C, the highest level in Er was achieved
in the presence of ChNCs followed by PANI-g-ChNCs,
and almost no evolution in Er was observed
in the presence of PANI. This effect is the most marked at T = 50 and 100 °C, over the Tg of PVAc and PVA. The decrease in the reinforcing effect induced
by PANI-g-ChNCs in comparison with the neat ChNCs
might be due to the combination of two effects: (i) the decrease
in the magnitude of interaction through hydrogen bonding between the
PANI-g-ChNCs due to the partial coating of ChNCs
with PANI, and (ii) the partial aggregation of PANI-g-ChNCs that shifts the percolation threshold to a higher content
of the nanofiller. However, even though the presence of PANI would
reduce the possibility of interaction between the ChNCs, the inclusion
of PANI-g-ChNCs in the polymer matrix provides an
acceptable reinforcing effect above the Tg of the polymer matrix, as confirmed by the increment in Er.
(with E′nan and E′mat the storage modulus
of the nanocomposite and unfilled matrix respectively measured at
different temperatures) vs the PANI-g-ChNC content.
As shown in Figure 6C, the highest level in Er was achieved
in the presence of ChNCs followed by PANI-g-ChNCs,
and almost no evolution in Er was observed
in the presence of PANI. This effect is the most marked at T = 50 and 100 °C, over the Tg of PVAc and PVA. The decrease in the reinforcing effect induced
by PANI-g-ChNCs in comparison with the neat ChNCs
might be due to the combination of two effects: (i) the decrease
in the magnitude of interaction through hydrogen bonding between the
PANI-g-ChNCs due to the partial coating of ChNCs
with PANI, and (ii) the partial aggregation of PANI-g-ChNCs that shifts the percolation threshold to a higher content
of the nanofiller. However, even though the presence of PANI would
reduce the possibility of interaction between the ChNCs, the inclusion
of PANI-g-ChNCs in the polymer matrix provides an
acceptable reinforcing effect above the Tg of the polymer matrix, as confirmed by the increment in Er.
3.5. Electrical Conductivity of PANI-g-ChNCs/PVA Films
The electrical properties of the PVA nanocomposite containing different contents in PANI-g-ChNCs were also investigated. For the purpose of comparison, nanocomposite films prepared in the same way but using PANI synthesized following the same route as for PANI-g-ChNCs were prepared by the mixing route. The DC electrical conductivities of PVA/PANI and PVA/PANI-g-ChNC nanocomposites with various contents of PANI were recorded by a dielectric spectrometer and are summarized in Figure 7A–D.
Figure 7.

Frequency dependence of alternating current (AC) conductivity of (A) PVA/PANI blend, (B) PVA/PANI by in situ polymerization, (C) PVA/PANI-g-ChNCs (1:1), and (D) PVA/PANI-g-ChNCs (6:1).
On the one hand, the conductivity of the film prepared in the presence of PANI-g-ChNCs (1:1) remained quite low (below 10–7 S/cm) even for a high amount of nanofiller (up to 30 wt %) (Figure 7A), meaning that the film remained nonconductive even in the presence of a high loading of PANI-g-ChNCs (1:1). On the other hand, a different evolution was noted when PANI-g-ChNCs (6:1) were used (Figure 7B). The conductivity of the film increased progressively and up to 20% PANI-g-ChNCs, it reached a plateau around 0.01 S/cm (Figure 7B), which is indicative of a conductive material. The difference in the conduction property between the two grades of PANI-g-ChNCs can be seen in Figure 8A, where the conductivity was traced vs the nanofiller content. The abrupt rise in conductivity for PANI-g-ChNCs (6:1) over 10 wt % loading indicates the setup of a percolated network among PANI-g-ChNC particles with possible contacts between the grafted PANI on the ChNCs. A possible reason for the lack of conductivity in the presence of PANI-g-ChNCs (1:1) would be the lower amount of grafted PANI to create a conductive path among the grafted PANIs. In fact, with a small amount of PANI deposited on ChNCs, the PANI nanosheets on ChNCs were disconnected. As a result, the conductive pathways formed in the PVA-PVAc matrix were discontinuous. It is worth mentioning that a thin film obtained by casting a suspension of PANI-g-ChNCs (6:1) until complete evaporation of water exhibited a conductive character, with a conductivity nearly equal to that of neat PANI (Figure S4).
Figure 8.

(A) DC electrical conductivity of PANI/PVA films using simple mixing (SM) and in situ polymerization (ISP) and PVA/PANI-g-ChNC nanocomposites as a function of the PANI content. Demonstration of the conductivity through a LED lamp: neat PVA matrix (B) and PVA/PANI-g-ChNC at 20% nanofiller loading (C).
When PANI synthesized ex situ was used instead of PANI-g-ChNCs, the conductivity of the film did not exceed 10–5 S/cm, meaning that the film remained insulating over the whole composition range from 5 to 30 wt % PANI. The only possibility to achieve conduction is the synthesis of PANI in the presence of the PVA latex, followed by film formation. As shown in Figure 7C,D, a conductive film was obtained when the PANI content exceeded 20 wt %. A possible reason for the absence of conductivity when PANI was synthesized ex situ and mixed with the PVA latex is the occurrence of aggregation among the PANI particles that prevents any setup of a percolated conductive network. When PANI was polymerized in the presence of the PVA latex, the adhesion of PANI with the polymer latex might occur, improving the dispersion of PANI within the polymer latex. The strategy of the in situ synthesis of PANI in the presence of the latex was shown to be beneficial for the effective binding of PANI on the polymer latex.38
Another point worth emphasizing is the lower amount of PANI necessary to form a conductive film when PANI-g-ChNCs (6:1) were used. Indeed, if we take into account the fact that the fraction of grafted PANI in PANI-g-ChNCs (6:1) was estimated to be about 60%, then the plot of conductivity vs PANI content is shifted to lower amounts, as shown in Figure 8A. This means that conductive films were obtained in the presence of a lower amount of PANI when PANI-g-ChNCs were used, while double loading was needed when PANI prepared in situ in the presence of the latex was used. The beneficial effect for conductivity induced by the use of PANI-g-ChNCs is presumably the consequence of the reduction of the tendency of PANI to aggregate during the film formation and mixing process, allowing one to attain conduction percolation at a lower content in PANI. This effect is presumably the consequence of the rodlike morphology of ChNCs and the attachment of PANI on ChNCs, which facilitated the setup of the percolated network at a lower amount of PANI. The aptitude of rodlike nanoparticles, such as ChNCs and other biobased fibrillar nanofillers such as cellulose nanofibrils (CNFs) or nanocrystals (CNCs), to generate percolated networks over a critical content is a characteristic property of these classes of nano-objects.39 To further demonstrate the conductive character of the nanocomposite films, we performed an experiment using a LED lamp and a low-frequency generator (Figure 8B,C). Contrary to the neat matrix for which the lamp did not light up when connected to the generator, the lamp lighted up when connected with the PVA film containing 20 wt % PANI-g-ChNCs (6:1).
3.6. Application of PANI-g-ChNCs as Conductive Additives in Adhesives
One of the multiple possibilities of application of PANI-g-ChNCs is their use as reinforcing conductive nanofillers. For this purpose, we included PANI-g-ChNCs in a waterborne PVA latex and tested the composite as an adhesive for wood. The adhesive was prepared according to the recipe reported in Table S1. A reference adhesive without PANI-g-ChNCs was also prepared as a blank. Both adhesives were applied to the wood specimen, and the shear strength of the wood joints was evaluated by a tensile test. From Figure 9, it can be seen that the inclusion of PANI-g-ChNCs markedly enhanced the bond strength of the adhesive, as attested by the marked increment in the shear strength of about 150% with respect to the unfilled adhesive. Interestingly, the adhesive thin film demonstrated a conductive character as attested by the lighting of the LED lamp when connected with a low-frequency generator (Figure 9D). The enhancement in the adhesive strength is due to the reinforcing effect of PANI-g-ChNCs by increasing the stiffness of the film, as confirmed by DMA analysis. According to the latex structure, other types of conductive adhesives such as pressure-sensitive or plastic adhesives may be prepared similarly by mixing the latex with PANI-g-ChNCs and the additives if any.
Figure 9.

(A) Force vs displacement during the tensile shear test for the wood joint, and (B) shear strength at different contents of PANI-g-ChNCs (6:1) for wood joints bound with the PVA-based adhesive containing different amounts of PANI-g-ChNCs (6:1). (C) Schematic representation of the shear test specimen, and (D) conductivity demonstration of the adhesive containing 20 wt % PANI-g-ChNCs.
The use of PANI-g-ChNCs as additives to elaborate a conductive adhesive was provided as a potential application of PANI-g-ChNCs to demonstrate the merit of this hybrid nanofiller, providing a reinforcing effect along with electrical conductivity. Undoubtedly, PANI-g-ChNCs can be used in other applications such as sensors, anticorrosion materials, supercapacitors, or biomedical devices.
4. Conclusions
We reported herein a facile approach for the fabrication of highly processable stable PANI-g-ChNC suspensions containing various aniline-to-ChNC ratios (1:1 and 6:1). Evidence of the exclusive nucleation of PANI from the surface of ChNCs was provided by TEM images, where an increase in the coverage degree of ChNCs by PANI was noted at a higher aniline/ChNC ratio. Raman and FTIR analyses also confirmed the formation of PANI, presumably by chemical grafting of PANI through the amino groups on the surface of ChNCs. Thanks to the colloidal stability of PANI-g-ChNCs in water along with their nanoscale, the use of PANI-g-ChNCs as an additive in waterborne latex dispersion is easy to run, by a simple mixing route. Over a critical amount of PANI-g-ChNCs (6:1), a conductive film with a DC conductivity value of 0.01 S/cm was obtained, without loss of the reinforcing potential of the rodlike nanofiller. As a potential application of PANI-g-ChNCs, a conductive adhesive for wood was prepared by the inclusion of PANI-g-ChNCs (6:1) over 20 wt %, with a beneficial effect on the mechanical strength of the adhesive joint. Given the merits of as-prepared PANI-g-ChNCs, including the stability in an acidic solution, the colloidal stability, the conductive character, and the reinforcing effect, this new class of hybrid biobased nanofiller is promising to produce nanocomposites and for coating thin films combining the merits of the ChNC nanofiller with the attributes of PANI. It is worth pointing out that, to the best of our knowledge, this work is the first to report the hybridization of ChNCs with PANI.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.biomac.2c00635.
TEM images of ChNCs and PANI-g-ChNCs, particle size distribution of the neat ChNC suspension and after immersion at pH = 2.5 for more than 3 h, DC electrical conductivity of a PANI-g-ChNC (6:1) film, and adhesive formulation used in the present work (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Shi H. H.; Khalili N.; Morrison T.; Naguib H. E. Self-assembled nanorod structures on nanofibers for textile electrochemical capacitor electrodes with intrinsic tactile sensing capabilities. ACS Appl. Mater. Interfaces 2018, 10, 19037–19046. 10.1021/acsami.8b03779. [DOI] [PubMed] [Google Scholar]
- Groenendaal L.; Jonas F.; Freitag D.; Pielartzik H.; Reynolds J. R. Poly(3,4-ethylenedioxythiophene) and its derivatives: Past, present, and future. Adv. Mater. 2000, 12, 481–494. . [DOI] [Google Scholar]
- Machida S.; Miyata S.; Techagumpuch A. Chemical synthesis of highly electrically conductive polypyrrole. Synth. Met. 1989, 31, 311–318. 10.1016/0379-6779(89)90798-4. [DOI] [Google Scholar]
- Bhadra S.; Khastgir D.; Singha N. K.; Lee J. H. Progress in preparation, processing and applications of polyaniline. Prog. Polym. Sci. 2009, 34, 783–810. 10.1016/j.progpolymsci.2009.04.003. [DOI] [Google Scholar]
- Bekas D. G.; Hou Y.; Liu Y.; Panesar A. 3D printing to enable multifunctionality in polymer-based composites: A review. Composites, Part B 2019, 179, 107540 10.1016/j.compositesb.2019.107540. [DOI] [Google Scholar]
- Cai M.; Yan H.; Li Y.; et al. Ti3C2Tx/PANI composites with tunable conductivity towards anticorrosion application. Chem. Eng. J. 2021, 410, 128310 10.1016/j.cej.2020.128310. [DOI] [Google Scholar]
- Chu X.; Chen G.; Xiao X.; Wang Z.; Yang T.; Xu Z.; Huang H.; Wang Y.; Yan C.; Chen N.; Zhang H.; Yang W.; Chen J. Air-stable conductive polymer ink for printed wearable micro-supercapacitors. Small 2021, 17, 2100956 10.1002/smll.202100956. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Cao J.; Yang Y.; Wu X.; Zheng Z.; Zhang X.; et al. Flame-retardant, highly sensitive strain sensors enabled by renewable phytic acid-doped biotemplate synthesis and spirally structure design. Chem. Eng. J. 2019, 374, 730–737. 10.1016/j.cej.2019.05.211. [DOI] [Google Scholar]
- Gopakumar D. A.; Pai A. R.; Pottathara Y. B.; Pasquini D.; De Morais L. C.; Luke M.; Kalarikkal N.; Grohens Y.; Thomas S. Cellulose nanofiber-based polyaniline flexible papers as sustainable microwave absorbers in the X-band. ACS Appl. Mater. Interfaces 2018, 10, 20032–20043. 10.1021/acsami.8b04549. [DOI] [PubMed] [Google Scholar]
- Lakshmi K.; John H.; Mathew K. T.; Joseph R.; George K. E. Microwave absorption, reflection and EMI shielding of PU–PANI composite. Acta Mater. 2009, 57, 371–375. 10.1016/j.actamat.2008.09.018. [DOI] [Google Scholar]
- Qazi T. H.; Raia R.; Dippold D.; Roether J. E.; Schubert D. W.; Rosellini E.; Barbani N.; Boccaccini A.R.l. Development and characterization of novel electrically conductive PANI–PGS composites for cardiac tissue engineering applications. Acta Biomater. 2014, 10, 2434–2445. 10.1016/j.actbio.2014.02.023. [DOI] [PubMed] [Google Scholar]
- Xu W.; Ding Y.; Yu Y.; Jiang S.; Chen L.; Hou H. Highly foldable PANi@CNTs/PU dielectric composites toward thin-film capacitor application. Mater. Lett. 2017, 192, 25–28. 10.1016/j.matlet.2017.01.064. [DOI] [Google Scholar]
- Patil D. S.; Shaikh J. S.; Dalavi D. S.; Kalagi S. S.; Patil P. S. Chemical synthesis of highly stable PVA/PANI films for supercapacitor application. Mater. Chem. Phys. 2011, 128, 449–455. 10.1016/j.matchemphys.2011.03.029. [DOI] [Google Scholar]
- Ben J.; Song Z.; Liu X.; Lü W.; Li X. Fabrication and electrochemical performance of PVA/CNT/PANI flexible films as electrodes for supercapacitors. Nanoscale Res. Lett. 2020, 15, 151. 10.1186/s11671-020-03379-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazemi F.; Naghib S. M.; Zare Y.; Rhee K. Y. Biosensing applications of polyaniline (PANI)-based nanocomposites: A Review. Polym. Rev. 2021, 61, 553–597. 10.1080/15583724.2020.1858871. [DOI] [Google Scholar]
- Borsoi C.; Zattera A. J.; Ferreira C. A. Effect of cellulose nanowhiskers functionalization with polyaniline for epoxy coatings. Appl. Surf. Sci. 2016, 364, 124–132. 10.1016/j.apsusc.2015.12.140. [DOI] [Google Scholar]
- Qiang Z.; Liang G.; Gu A.; Yuan L. The dielectric behavior and origin of high-k composites with very low percolation threshold based on unique multi-branched polyaniline/carbon nanotube hybrids and epoxy resin. Composites, Part A 2014, 64, 1–10. 10.1016/j.compositesa.2014.04.021. [DOI] [Google Scholar]
- Tsotra P.; Friedrich K. Short carbon fiber reinforced epoxy resin/polyaniline blends: their electrical and mechanical properties. Compos. Sci. Technol. 2004, 64, 2385–2391. 10.1016/j.compscitech.2004.05.003. [DOI] [Google Scholar]
- Gu H.; Guo J.; He Q.; et al. Flame-retardant epoxy resin nanocomposites reinforced with polyaniline-stabilized silica nanoparticles. Ind. Eng. Chem. Res. 2013, 52, 7718–7728. 10.1021/ie400275n. [DOI] [Google Scholar]
- Wu X.; Lu C.; Xu H.; Zhang X.; Zhou Z. Biotemplate synthesis of polyaniline@cellulose nanowhiskers/natural rubber nanocomposites with 3D hierarchical multiscale structure and improved electrical conductivity. ACS Appl. Mater. Interfaces 2014, 6, 21078–21085. 10.1021/am505924z. [DOI] [PubMed] [Google Scholar]
- Rinaudo M. Chitin and chitosan: Properties and applications. Prog. Polym. Sci. 2006, 31, 603–632. 10.1016/j.progpolymsci.2006.06.001. [DOI] [Google Scholar]
- Perrin E.; Bizot H.; Cathala B.; Capron I. Chitin nanocrystals for pickering high internal phase emulsions. Biomacromolecules 2014, 15, 3766–3771. 10.1021/bm5010417. [DOI] [PubMed] [Google Scholar]
- Yamaguchi Y.; Nge T. T.; Takemura A.; Hori N.; Ono H. Characterization of uniaxially aligned chitin film by 2D FT-IR spectroscopy. Biomacromolecules 2005, 6, 1941–1947. 10.1021/bm0492172. [DOI] [PubMed] [Google Scholar]
- Belamie E.; Davidson P.; Giraud-Guille M. M. Structure and chirality of the nematic phase in α-chitin suspensions. J. Phys. Chem. B 2004, 108, 14991–15000. 10.1021/jp048152u. [DOI] [Google Scholar]
- Marchessault R. H.; Morehead F. F.; Walter N. M. Liquid crystal systems from fibrillar polysaccharides. Nature 1959, 184, 632–633. 10.1038/184632a0. [DOI] [Google Scholar]
- Zeng J.-B.; He Y.-S.; Li S.-L.; Wang Y.-Z. Chitin whiskers: An overview. Biomacromolecules 2012, 13, 1–11. 10.1021/bm201564a. [DOI] [PubMed] [Google Scholar]
- Ben Cheikh F.; Mabrouk A. B.; Magnin A.; Putaux J.-L.; Boufi S. Chitin nanocrystals as Pickering stabilizer for O/W emulsions: Effect of the oil chemical structure on the emulsion properties. Colloids Surf., B 2021, 200, 111604 10.1016/j.colsurfb.2021.111604. [DOI] [PubMed] [Google Scholar]
- Araki J.; Yamanaka Y.; Ohkawa K. Chitin-chitosan nanocomposite gels: reinforcement of chitosan hydrogels with rod-like chitin nanowhiskers. Polym. J. 2012, 44, 713–717. 10.1038/pj.2012.11. [DOI] [Google Scholar]
- Minke R.; Blackwell J. The structure of α-chitin. J. Mol. Biol. 1978, 120, 167–181. 10.1016/0022-2836(78)90063-3. [DOI] [PubMed] [Google Scholar]
- Elazzouzi-Hafraoui S.; Nishiyama Y.; Putaux J.-L.; Heux L.; Dubreuil F.; Rochas C. The shape and size distribution of crystalline nanoparticles prepared by acid hydrolysis of native cellulose. Biomacromolecules 2008, 9, 57–65. 10.1021/bm700769p. [DOI] [PubMed] [Google Scholar]
- Cabuk M.; Yavuz M.; Unal H. I. Electrokinetic properties of biodegradable conducting polyaniline-graft-chitosan copolymer in aqueous and non-aqueous media. Colloids Surf., A 2014, 460, 494–501. 10.1016/j.colsurfa.2014.02.053. [DOI] [Google Scholar]
- Ogawa Y.; Kimura S.; Wada M.; Kuga S. Crystal analysis and high-resolution imaging of microfibrillar α-chitin from Phaeocystis. J. Struct. Biol. 2010, 171, 111–116. 10.1016/j.jsb.2010.03.010. [DOI] [PubMed] [Google Scholar]
- Zewude D. A.; Izawa H.; Ifuku S. Optimum preparation conditions for highly individualized chitin nanofibers using ultrasonic generator. Polymers 2021, 13, 2501. 10.3390/polym13152501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H.; Chen P.; Chen W.; Liu Y. Effect of cellulose nanofibers on induced polymerization of aniline and formation of nanostructured conducting composite. Cellulose 2014, 21, 1757–1767. 10.1007/s10570-014-0189-3. [DOI] [Google Scholar]
- Pereira da Silva J. E.; de Faria D. L. A.; Córdoba de Torresi S. I.; Temperini M. L. A. Influence of thermal treatment on doped polyaniline studied by resonance Raman spectroscopy. Macromolecules 2000, 33, 3077–3083. 10.1021/ma990801q. [DOI] [Google Scholar]
- Bernard M. C.; Hugot-Le Goff A. Quantitative characterization of polyaniline films using Raman spectroscopy. Electrochim. Acta 2006, 52, 595–603. 10.1016/j.electacta.2006.05.039. [DOI] [Google Scholar]
- Ping Z. In situ FTIR–attenuated total reflection spectroscopic investigations on the base–acid transitions of polyaniline. Base–acid transition in the emeraldine form of polyaniline. J. Chem. Soc., Faraday Trans. 1996, 92, 3063–3067. 10.1039/FT9969203063. [DOI] [Google Scholar]
- Li Y.; Wang Z.; Wang Q.; Wang C.; Xue G. A Facile and Efficient Route for Coating Polyaniline onto Positively Charged Substrate. Macromolecules 2010, 43, 4468–4471. 10.1021/ma100475u. [DOI] [Google Scholar]
- Boufi S.; Kaddami H.; Dufresne A. Mechanical performance and transparency of nanocellulose reinforced polymer nanocomposites. Macromol. Mater. Eng. 2014, 299, 560–568. 10.1002/mame.201300232. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.