

Abstract

Treatment of [Ru(PPh3)(C6H4PPh2)2H][Li(THF)2] with AlMe2Cl and SnMe3Cl leads to elimination of LiCl and CH4 and formation of the heterobimetallic complexes [Ru(C6H4PPh2)2{PPh2C6H4AlMe(THF)}H] 5 and [Ru(PPh3)(C6H4PPh2)(PPh2C6H4SnMe2)] 6, respectively. The pathways to 5 and 6 have been probed by variable temperature NMR studies, together with input from DFT calculations. Complete reaction of H2 occurs with 5 at 60 °C and with 6 at room temperature to yield the spectroscopically characterized trihydride complexes [Ru(PPh2)2{PPh2C6H4AlMe}H3] 7 and [Ru(PPh2)2{PPh2C6H4SnMe2}H3] 8. In the presence of CO, 6 forms the acylated phosphine complex, [Ru(CO)2(C(O)C6H4PPh2)(PPh2C6H4SnMe2)] 9, through a series of intermediates that were identified by NMR spectroscopy in conjunction with 13CO labeling. Complex 6 undergoes addition and substitution reactions with the N-heterocyclic carbene 1,3,4,5-tetramethylimidazol-2-ylidene (IMe4) to give [Ru(IMe4)2(PPh2C6H4)(PPh2C6H4SnMe2)] 10, which converted via rare N-Me group C–H activation to [Ru(IMe4)(PPh3)(IMe4)′(PPh2C6H4SnMe2)] 11 upon heating at 60 °C and to a mixture of [Ru(IMe4)2(IMe4)′(PPh2C6H4SnMe2)] 12 and [Ru(PPh3)(PPh2C6H4)(IMe4-SnMe2)′] 13 at 120 °C.
Introduction
Heterobimetallic (HBM) complexes featuring a transition metal (TM) center in combination with a Lewis acidic s- or p-block metal (M′) continue to be the subject of considerable interest, primarily due to the ability of such species to bring about small molecule activation chemistry.1,2 The TM-Zn, -Ga and -Al complexes shown in Scheme 1 represent three recent examples in which [TM-M′] HBM complexes have been employed to bring about not only small molecule activation but also a subsequent catalytic functionalization step.
Scheme 1. Examples of Catalytic Transformations Mediated by [TM-M′] Heterobimetallic Complexes.

A commonly employed preparative route to [TM-M′] HBM complexes involves the reaction of a TM-hydride precursor with a Lewis acidic metal alkyl reagent to give a [TM-M′] product following elimination of an alkane.3 In a recent study,4 we employed such a reaction of [Ru(PPh3)3HCl] with LiMe, MgMe2, and ZnMe2 to give the bis-cyclometalated complexes5 [Ru(PPh3)(C6H4PPh2)2H][M′] (M′ = Li(THF)21, MgMe(THF)22, and ZnMe 3) shown in Scheme 2. A combination of X-ray crystallography and DFT calculations showed that the level of interaction between Ru–H and M′ increased in the order of 1 < 2 < 3 such that 1 and 2 were best considered as ruthenate anions with a group 1 or 2 countercation, whereas Zn compound 3 exhibited far more covalent character. As a result, the latter proved susceptible to reductive elimination of the hydride ligand onto one of the metalated phosphines, to yield the “dual unsaturated” isomer, [Ru(PPh3)2(C6H4PPh2)(ZnMe)] 4, which although only present in ca. 2%, allowed 3 to react with H2 at −40 °C, ca. 100 °C lower than the temperature required with either 1 and 2.
Scheme 2. Synthesis of [Ru-M′] Complexes 1 (M′ = Li(THF)2), 2 (M′ = MgMe(THF)2) and 3 (M′ = ZnMe) and Equilibrium of the Latter with 4.

Prompted by the enhanced reactivity of the [Ru–Zn] complex, we have extended our studies to [Ru-M′] complexes in which M′ = Al and Sn, on the basis that they would also exhibit strong covalent interactions with the Ru center. We now describe the synthesis and reactivity of the [Ru–Al] and [Ru–Sn] heterobimetallic complexes [Ru(C6H4PPh2)2{PPh2C6H4AlMe(THF)}H] 5 and [Ru(PPh3)(C6H4PPh2)(PPh2C6H4SnMe2)] 6.
Results and Discussion
Synthesis and Characterization of [Ru(C6H4PPh2)2{PPh2C6H4AlMe(THF)}H] and [Ru(PPh3)(C6H4PPh2)(PPh2C6H4SnMe2)]
We showed previously that the [Ru–Li] salt 1 was a convenient precursor to both 2 and 3 upon treatment with MgMeCl and ZnMeCl, respectively (Scheme 2), thanks to the relative ease of removal of the LiCl byproduct.4 Heating 1 with AlMe2Cl at 60 °C led to full conversion through to yellow [Ru(C6H4PPh2)2{PPh2C6H4AlMe(THF)}H] 5, which was isolated in 69% yield, whereas SnMe3Cl reacted with 1 at room temperature to generate deep-blue [Ru(PPh3)(C6H4PPh2)(PPh2C6H4SnMe2)] 6 in a near quantitative amount (Scheme 3). Comparison of Schemes 2 and 3 shows that while both reactions were indeed accompanied by loss of LiCl, the availability of additional M′-Me groups on moving from ZnMeCl to AlMe2Cl and SnMe3Cl allowed elimination of an extra molecule of CH4, resulting in metalation of a further phosphine ligand (vide infra).6
Scheme 3. Synthesis of [Ru(C6H4PPh2)2{PPh2C6H4AlMe(THF)}H] 5 and [Ru(PPh3)(C6H4PPh2)(PPh2C6H4SnMe2)] 6.

The X-ray crystal structure of 5 (Figure 1, Table 1) showed a Ru atom at the center of a highly distorted octahedral arrangement of ligands (e.g., P(2)–Ru(1)–C(38) = 150.40(6)°), including three metalated phosphines7 in a mer-arrangement (cf. fac-RuP3 geometry of both 1 and 2). The ruthenium and aluminum centers formed part of a 6-membered ring dimetalacycle in which the Al was attached to both a bridging hydride ligand (located and refined with an Al(1)–H(1) distance of 1.83(3) Å) and a phosphine phenyl group (Al(1)–C(2) = 1.991(3) Å).8,9 The structure of 6 (Figure 1, Table 1) contained a 5-coordinate Ru center (thus resembling 4) with a direct Ru–Sn interaction (Ru(1)–Sn(1) = 2.5686(2) Å).10 Phosphine metalation occurred onto Sn to generate a cyclostannylated phosphine ligand, which bridges across the basal and axial sites of the square pyramidal Ru complex. A related, albeit coordinatively saturated, osmium derivative, [Os(PPh3)(CO)(C6H4PPh2)(PPh2C6H4SnMe2)], has been reported by Roper to form as a minor product upon refluxing [Os(PPh3)2(CO)(SnMe3)Cl] with PPh3.11−13
Figure 1.

Molecular structures of (left) 5 and (right) 6. Ellipsoids at 30% level; all H atoms, except Ru–H–Al, omitted for clarity. In 5, the solvent and the minor disordered component have also been omitted for clarity.
Table 1. Selected Bond Lengths (Å) and Angles (deg) in 5 and 6.
| 5 | 6 | |
|---|---|---|
| Ru-PPh3 | - | 2.3504(6) |
| Ru-PPh2(C6H4Ru) | 2.3854(6), 2.3555(6) | 2.3616(7) |
| Ru-PPh2(C6H4Al) | 2.3202(6) | - |
| Ru-PPh2(C6H4Sn) | - | 2.3245(6) |
| Ru···E | (E = Al) 2.5911(7) | (E = Sn) 2.5686(2) |
| trans–P–Ru-P | 166.16(2) | 158.22(2) |
The solution NMR spectra of 5 and 6 (Figures S1–S7) were consistent with their solid-state structures.14 Thus, the 1H NMR spectrum of 5 showed a broad triplet of doublets Ru-H-Al signal at δ −6.2915 with small 2JHP splittings (12 and 6 Hz) to the three cis-phosphorus nuclei. In the 31P{1H} NMR spectrum, there were three doublets, with those at δ 70 and δ −15 assigned to the phosphines metalated onto Al and Ru respectively, based on their mutually large (trans) coupling of 266 Hz, as well as the established upfield shift associated with phosphines metalated onto a TM center and downfield shift arising from 6-membered ring phosphine chelates.16,17 The 1H NMR spectrum of 6 yielded very little in the way of diagnostic information, but the presence of high (δ 75) and low (δ −29) frequency 31P{1H} NMR signals with a large mutual 2JPP splitting of 240 Hz was consistent with the presence of cyclostannylated and cycloruthenated ligands respectively.
Pathways to Formation of 5 and 6
The very different structures of 5 and 6 led us to investigate their pathways to formation using variable temperature NMR spectroscopy. Introduction of a frozen, yellow-orange THF-d8 solution of 1 and AlMe2Cl into a precooled (193 K) NMR probe revealed the rapid formation of a 1:1 ratio of two intermediates, assigned as the structures I and II shown in Scheme 4a. Characterization of these species (Figures S8–S12), as well as the higher temperature intermediates III and IV (Scheme 4a), was based on (i) the number of 31P NMR resonances and their relative chemical shifts,16,18 (ii) the relative magnitudes of 2JPP/2JHP couplings, and (iii) 31P–1H HMQC connectivities.
Scheme 4. Proposed Structures of Intermediates in the Formation of 5 and 6 (Based on Low-Temperature NMR Studies) Are Shown in (a–c), with a Proposed Pathway from V to 6 Illustrated in (d).
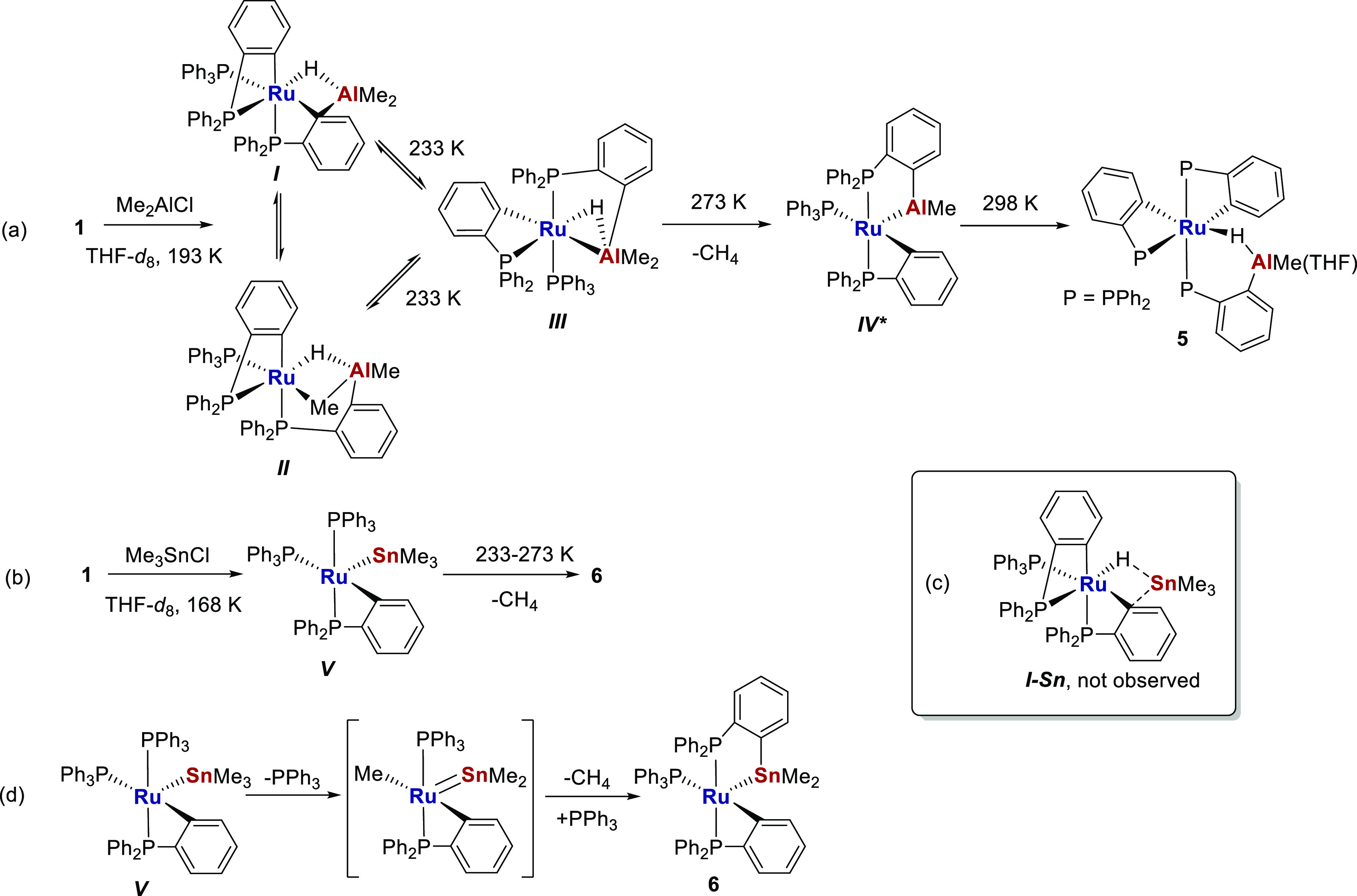
The * on IV in part (a) denotes uncertainty as to whether THF is or is not bound on Al.
Intermediate I results from substitution of the Li(THF)2 moiety in 1 by AlMe2 and was assigned based on the retention of a fac-RuP3 arrangement, comparable 31P chemical shifts to those of 1 (especially the two low frequency resonances for the metalated phosphines) and the presence of a low frequency hydride resonance (δ −10.50 cf. δ −9.62 in 1),4 attributed to the bridging Ru-H-Al interaction. Intermediate II showed a broad 1H singlet at δ −2.53, in a 3:1 ratio with doublet of doublet of doublets Ru-H-Al signal at δ −13.92, suggestive of it being an isomer with a bridging Ru-Me-Al group in place of the Ru-C6H4-Al bridge in I. The replacement of one of the low-frequency 31P signals in I by a new high frequency signal for II supported the presence of a phosphine metalated onto just Al.
Isomerization of I and II to the mer-III was seen at 233 K, while further warming (to 273 K) generated a deeper-red colored solution, consistent with formation of a coordinatively unsaturated isomer IV. This showed just a single AlMe proton resonance (cf. two resonances in III), consistent with a structure arising out of the combination of the Ru-H-Al and one of the two AlMe groups in III and subsequent reductive elimination of methane. After 1 h at 273 K, I-III had been fully consumed, and IV represented ca. 80% of the reaction mixture. A final color change from red to orange was observed at 298 K, concomitant with the formation of the final product 5 through metalation of the third phosphine ligand. After ca. 40 min at 298 K, 5 comprised ca. 65% of all species in solution.19
An analogous study of the formation of 6 (Scheme 4b; Figures S13–S16) failed to show any spectroscopic evidence for the comparable initial substitution product I–Sn (Scheme 4c). This may imply that reductive elimination of Ru–H onto RuC6H4PPh2 in such a species is very fast, supporting further the analogous behavior of [Ru–Sn] and [Ru–Zn] species. Only a single (deep-blue) intermediate was observed between 168 and 273 K, which we propose is [Ru(PPh3)2(C6H4PPh2)SnMe3] (V) based on (i) the presence of only a single SnMe resonance in the 1H NMR spectrum (cf. two signals in 6 for the diastereotopic Me groups) and (ii) the observation of a single low frequency (δ −29) 31P{1H} NMR signal for a cycloruthenated phosphine, together with two “medium” frequency signals (δ 49, 41–cf. IV) arising from two PPh3 ligands. 6 began to appear above 273 K (Figures S13–S16). Following studies by Wada20 and Roper,11b a possible pathway for the transformation of V to 6 involves Me group transfer from Sn to Ru and generation of a transient Ru stannylene intermediate, (Scheme 4d) that could generate the final cyclostannylated phosphine containing complex 6 through attack on a phosphine phenyl C–H bond, followed by elimination of methane.
Determination of the free energies of IV, V, 5, and 6 by density functional theory (DFT) calculations (BP86-D3BJ(C6H6)/BS2//BP86/BS1) were in agreement with the experimental findings. Thus, the free energy of IV was computed to be higher than that of 5 (Scheme 5; see also Supporting Information for further details). Unsurprisingly, coordination of THF stabilized both structures. 6 was calculated to be more stable than V, as well as 7.8 kcal/mol more stable than 5Sn, the Sn analogue of [Ru–Al] complex 5 (Scheme 6).
Scheme 5. Free Energies (BP86-D3BJ(C6H6)/BS2//BP86/BS1) Relative to 5 (kcal/mol).

Scheme 6. Free Energies (BP86-D3BJ(C6H6)/BS2//BP86/BS1) Relative to V (kcal/mol).

Reactivity of 5 and 6 with H2
Complex 5 showed a similar reluctance to 1 and 2 in reacting with H2 only at elevated temperature (60 °C) to yield a single product, which was characterized as the trihydride species [Ru(PPh2)2{PPh2C6H4AlMe}H3] 7 (Scheme 7) based on NMR spectroscopy (Figures S17–S22).21 We were unable to crystallize the product which decomposed in the absence of a H2 atmosphere to a mixture of species, two of which were identified as [Ru(PPh3)3(η2-H2)H2] and [Ru(PPh3)4H2].22 The fate of the aluminum was not determined.
Scheme 7. Proposed Structure of 7 from Reaction of 5 with H2 (1 atm, 60°C).

The 31P{1H} NMR spectrum of 7 exhibited three signals with JPP values indicative of a mer-RuP3 geometry. Based on the findings for 5, the high frequency (δ 74, doublet of doublets) signal was attributed to the Al-metalated phosphine, with a doublet of doublets at δ 62 and a triplet at δ 59 arising from the two PPh3 ligands. The low frequency region of the room temperature 1H NMR spectrum of 7 showed an AlMe resonance at δ −0.39, which integrated to 3 relative to three hydride signals (each of relative integral 1) at δ −8.46 (broad doublet), −8.72 (triplet of doublets) and −11.07 (doublet of triplets).23,24 The lowest frequency hydride signal was assigned to the bridging hydride Hc (Scheme 7) based on the presence of (i) a NOESY peak to the AlMe resonance and (ii) a 54 Hz 2JHP doublet splitting, indicative of a pseudo-trans PPh3 ligand. The well-resolved appearance of this signal suggests it is more closely associated with Ru than quadrupolar Al. The highest frequency hydride signal was assigned to Hb based on the presence of a NOESY peak to the AlMe signal. The signal stayed broad between 223 and 337 K (Figure S19), consistent with it being associated more with Al (i.e., Ru···H-Al).25 The magnitude of the 2JHP splittings (28 and 14 Hz) on the resonance at δ −8.72 (Ha) support it being cis to three phosphine ligands.26 No 2JHH coupling was observed on any of the hydride resonances in the 1H{31P} NMR spectrum.27
NMR spectra of the analogous reaction with D2 showed that the three hydride signals were present in the proton NMR spectrum, but all in an integral ratio of <1 relative to the AlMe resonance. This, together with broad 31P resonances, indicates that both H2/D2 addition as well as phosphine cyclometalation must be reversible, allowing H/D exchange to take place into the ortho-positions of PPh3 ligands.
In contrast to 5, [Ru–Sn] complex 6 showed behavior that aligned with [Ru–Zn] complex 3 in reacting with H2 at room temperature, to yield what we assign as the trihydride complex [Ru(PPh2)2{PPh2C6H4SnMe2}H3] 8 (Scheme 8). A gradual color change from a blue to a colorless/pale-yellow was observed when H2 was allowed to diffuse slowly through a sample of 6, although if H2 was added and the sample shaken vigorously, a colorless precipitate (which failed to redissolve in most common solvents) formed almost instantaneously. The IR spectra of the precipitate and material from solution were identical,28 implying that 8 is the product in both cases. We assume that 8 sits right on the edge of solubility and that shaking results in precipitation.
Scheme 8. Proposed Structure of 8 from the Room Temperature Reaction of 6 with H2 (1 atm).

The 1H NMR spectrum of a homogeneous solution of 8 formed upon slow diffusion of H2 exhibited a single SnMe2 resonance (cf. two different SnMe resonances for 6) of integral 6 at room temperature, along with a single, broad (fwhm = 36 Hz) hydride signal at δ −7.6 of relative integral 3 (Figures S23–S27) with a 1JHSn coupling of 184 Hz. The magnitude is suggestive of some degree of interaction between Ru-H and Sn centers,29,30 although the hydride T1 value of 390 ms (400 MHz, 298 K) would exclude any appreciable nonclassical behavior. Variable temperature NMR measurements were consistent with 8 being fluxional in solution. Thus, cooling to 223 K (THF) only broadened the hydride signal, whereas warming to 332 K resolved it into a single doublet of triplets, with JHP values of 16 and 7 Hz, indicative of the hydride ligands being cis to all three phosphorus nuclei. The fluxionality was mirrored in the 31P{1H} NMR spectrum, which comprised at low temperature of a triplet (δ 85, cyclostannylated phosphine), together with a broad singlet (δ 56, two PPh3 ligands) that resolved into a doublet upon warming to (or above) room temperature. The mutual 2JPP splitting of 98 Hz is in-between the values typically associated with trans- and cis–P–Ru−P arrangements.31
The ease with which 8 precipitated thwarted all attempts to generate single crystals suitable for X-ray crystallography, even via a solid-state transformation.32Scheme 8 shows a structure for 8 (Table S2) that is based on other group 8 metal derivatives [Ru(PR3)3(ER3′)H3] (ER3′ = SiR3′, SnR3′),29a,30,33 which all feature a common tetrahedral arrangement of Si/Sn and 3PR3 units with hydride ligands capping the Si/Sn(PR3)2 faces.
Reactivity of 6 with Lewis Bases
CO
Additional studies of small molecule reactivity focused on [Ru–Sn] precursor 6. As shown in Scheme 9, both addition and insertion of CO took place when 6 was heated under 1 atm CO at 80 °C, to ultimately form the acylated phosphine complex [Ru(CO)2(C(O)C6H4PPh2)(PPh2C6H4SnMe2)] 9, which could be isolated in 60% yield. Typically, acylated phosphine ligands are generated by oxidative addition of phosphino substituted aldehydes,34 rather than by CO insertion into a M–aryl bond,35 although the latter route does have precedence with ruthenium.35b
Scheme 9. Spectroscopically Detected Intermediates VI–VIII in the Formation of 9.

The X-ray structure (Figure 2) of 9 revealed an octahedral coordination sphere with a trans-arrangement of the P atoms of the stannylated and acylated phosphines, leaving the -SnMe2 and -C(O)(aryl) groups trans to the two carbonyl ligands. As a result of this geometry, the Ru–Sn (2.6879(2) Å) and Ru–P (2.3869(6) Å) distances of the stannylated phosphine were significantly longer than in 6. The Ru–C(O) distance (2.129(2) Å) was comparable to that in [Ru(PPh3)(CO)2(C(O)C6H4PPh2)H] (2.110(1) Å).34c In the 31P NMR spectrum, there was only a minor change in the chemical shift of the stannylated phosphine relative to 6, whereas the phosphine metalated onto Ru moved ca. 100 ppm to higher frequency as a result of CO insertion (Figures S32–S35).35b
Figure 2.
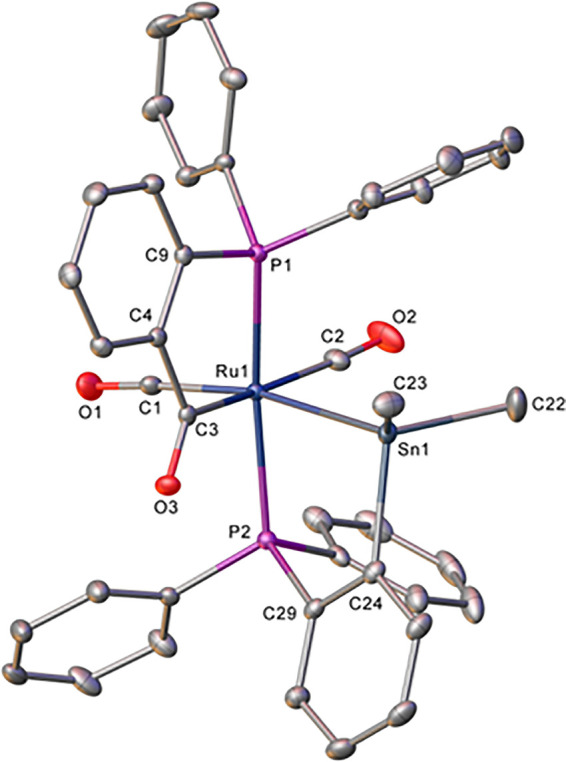
Molecular structure of 9. Ellipsoids are shown at 30% level with all H atoms omitted for clarity. Selected bond lengths (Å) and angles (deg): Ru1–P1 2.3214(6), Ru1–P2 2.3869(6), Ru1–C1 1.920(2), Ru1–C2 1.942(2), Ru1–C3 2.129(2), Ru1–Sn1 2.6879(2), P1–Ru1–P2 170.79(2).
In situ NMR measurements, in conjunction with 13CO labeling, revealed a series of intermediates on the pathway to 9 (Scheme 9; Figures S37–S42). Thus, shaking 6 with 13CO (1 atm) brought about an instantaneous change in color at room temperature from blue to yellow, concomitant with formation of the 18-electron CO addition species VI. This was identified by the appearance of three doublet of doublet of doublet 31P resonances, each with a cis-sized 2JPC coupling (8–11 Hz) to a single 13CO ligand, which resonated in the 13C{1H} NMR spectrum at δ 207 as a doublet of triplets. Isomerization of VI occurred overnight at room temperature to yield VII, which exhibited one metalated phosphorus signal with a much greater 2JPSn splitting (970 Hz vs 180 Hz), consistent with a change in orientation to trans P–Ru–SnMe2. There was also a small amount of the dicarbonyl species VIII (identified on the basis of two multiply coupled high frequency signals in the 13C{1H} NMR spectrum), which increased in intensity upon heating at 80 °C, leaving it as the main product in solution after 1 h. Further heating converted VIII to the final product 9. No signals attributable to IX (Scheme 9), a “Ru-SnPhos” analogue of [Ru(ZnPhos)(CO)3], which we have shown to be the product formed when a mixture of [Ru(PPh3)3HCl] and LiCH2TMS/ZnMe2 was heated under CO,36 were observed at any point in the overall reaction.
1,3,4,5-Tetramethylimidazol-2-ylidene (IMe4)
Treatment of 6 with ca. 3 equiv of the N-heterocyclic carbene 1,3,4,5-tetramethylimidazol-2-ylidene (IMe4) led to full consumption of the starting material over the course of ca. 1 h to form the coordinatively saturated product [Ru(IMe4)2(PPh2C6H4)(PPh2C6H4SnMe2)] 10 (Scheme 10), which was isolated as an orange microcrystalline solid in 55% yield. The 31P{1H} NMR spectrum of 10 displayed doublets at both high (δ 69) and low (δ −36) frequency, consistent with retention of both the cyclostannylated and cycloruthenated phosphines, although the magnitude of 2JPP (18 Hz) now implied they were in a cis-configuration (Figures S43–S48). Two inequivalent IMe4 ligands were evident from the appearance of four NMe and four NCMe resonances in the 1H NMR spectrum and the presence of two 13C carbenic resonances (δ 191, 2JCP = 86 and 16 Hz; δ 200, 2JCP = 8 and 2 Hz).
Scheme 10. Synthesis of bis-IMe4 Complex 10 and Formation of 11–13 upon Heating.

As shown in Figure 3 and Table 2, X-ray crystallography revealed that the Ru center in 10 was significantly distorted from regular octahedral. Accommodation of the two IMe4 ligands caused a reduction (relative to 6) in the bite angles of both the cyclostannylated (84.371(17)° to 80.092(15)°) and cycloruthenated (67.93(7)° to 66.42(6)° respectively) phosphines, as well as an acute trans–P–Ru−Sn angle (155.047(15)°). Incorporation of a (phosphine) ligand trans to tin increased the Ru–Sn distance to 2.6345(2) Å from the value of 2.5686(2) Å in 6.
Figure 3.

Molecular structures of one of the molecules in the asymmetric unit of (left) 10 and (right) 11. Ellipsoids at 30% level. All H atoms and solvent have been omitted for clarity in 10, while all hydrogens, with the exception of those attached to C1, have been omitted in 11.
Table 2. Selected Bond Lengths (Å) and Angles (deg) in the IMe4 Complexes 10–13.
| 10 | 11 | 12 | 13 | |
|---|---|---|---|---|
| Ru-CIMe4 | 2.121(2), 2.132(2) | 2.119(3) | 2.109(2), 2.087(3) | - |
| Ru-CIMe4′ | - | 2.091(3) | 2.089(3) | 2.032(3) |
| Ru-CH2 | - | 2.246(3) | 2.224(3) | - |
| Ru-PC6H4 | 2.4073(6) | - | - | 2.3445(9) |
| Ru-C6H4P | 2.132(2) | - | - | 2.084(3) |
| Ru-PC6H4Sn | 2.3451(5) | 2.3162(8) | 2.3267(6) | - |
| Ru-PPh3 | - | 2.3296(7) | - | 2.3118(8) |
| Ru-Sn | 2.6345(2) | 2.6435(3) | 2.6604(3) | 2.5223(4) |
| CIMe4-Ru-CIMe4 | 88.50(8) | - | - | - |
| CIMe4′-Ru-Sn | - | 99.05(9) | 96.38(8) | 81.25(11) |
Heating 10 at 60 °C in THF or benzene brought about metalation of one of the IMe4 ligands to give [Ru(IMe4)(PPh3)(IMe4)′(PPh2C6H4SnMe2)] (11, Scheme 10). The 31P{1H} NMR spectrum showed replacement of the low frequency signal for the cycloruthenated phosphine in 10 by a resonance at δ 54, arising from a Ru-PPh3 resulting from reductive elimination of Ru–H (resulting from IMe4 activation) onto the Ph2PC6H4Ru ligand. The 1H NMR spectrum exhibited a total of seven carbene methyl resonances, and also showed two doublets of doublets at δ 2.42 and 2.22 (each of relative integral 1) for the diastereotopic protons of the Ru-CH2 arm (Figures S49–S51).
NMR monitoring of the reaction indicated that optimum conversion of the starting material (ca. 80–85%) occurred over ca. 2 h at 60 °C to yield 11 as the main reaction product, although always alongside a number of other, smaller, unidentifiable species, which became more prominent with longer heating. While we were therefore unable to isolate 11 as an analytically pure material, a combination of multinuclear NMR spectra and a crystal structure determination (achieved by picking of a single crystal) identified 11 unequivocally.
In contrast to the well-known metalation of NHCs bearing N-aryl or bulky N-alkyl substituents,37 C–H activation of N-methylated carbenes is restricted to a very small number of examples,38 most likely because of the severe structural constraints imposed by forming a four-membered ring metalacycle. These structural impositions are apparent in the X-ray structure of 11 (Figure 3), which shows a dramatically tilted carbene ring with very different N(1)–C(2)–Ru(1) and N(2)–C(2)–Ru(1) angles (99.3(2)° and 156.1(3)° respectively, Δ = 56.8°). The C1–Ru–C2 angle subtended at Ru (63.36(12)°) is more similar to that in [Os(PiPr3)2(CO)(IMe2)′Cl] (63.01(16)/63.11(16)°) reported by Esteruelas38b than [TptBu,MeYb(IMe4)(IMe4)′] (55.4(2)°) described by Ferrence et al.,38a most likely due to the presence of both the bigger lanthanide and the very different ligand coordination spheres.
When 10 was heated to 120 °C in toluene, very different activation chemistry of the carbene took place with the IMe4 ligands from two molecules of 10 undergoing redistribution to give a mixture of the six-coordinate, tris-carbene product [Ru(IMe4)2(IMe4)′(PPh2C6H4SnMe2)] 12 and five-coordinate, monocarbene species [Ru(PPh3)(PPh2C6H4)(IMe4-SnMe2)′] 13 (Scheme 10). As for 11, we were able to manually separate yellow crystals of 12 and purple crystals of 13 to allow their structural characterization, but were unable to separate enough clean material for elemental analyses or measurement of pristine NMR spectra. Purple 13 was more obvious to identify and manually separate, resulting in NMR spectra that were typically cleaner than those of 12 (Figures S52–S59).
The structure of 12, which is shown in Figure 4 (metrics in Table 2), displayed a cis-arrangement of two intact IMe4 ligands, one of which was trans to the carbenic carbon of the third, metalated IMe4. The difference in the two Ru–C–N angles (Δ = 57.2°) showed that this was even more distorted in terms of coordination than that in 11, although the bite angle did not change (C(1)–Ru(2)–C(7) = 63.47(11)°). A cyclostannylated phosphine occupied the last two coordination sites of the highly distorted (e.g., C(7)–Ru(2)–Sn(1) = 159.35(8)°) octahedral Ru coordination sphere. In accord with the structure, the 1H and 13C NMR spectra showed separate resonances for each of the 11 inequivalent NMe and NCMe groups.
Figure 4.

Molecular structures of one of the molecules in the asymmetric unit in (left) 12 and (right) 13. Ellipsoids at 30% level. In 12, the minor disordered component and hydrogens, with the exception of those attached to C7, have been omitted for clarity. In 13, the minor disordered component and hydrogens, with the exception of those attached to C6, have been omitted for clarity.
The most striking feature of 13 (Figure 4, Table 2) was the formation of a novel chelating stannylcarbene ligand, attached to Ru at the apical and equatorial positions of a distorted square pyramid through very short Ru–C and Ru–Sn bond lengths (Ru(2)–C(1) = 2.032(3) Å, Ru(2)–Sn(1) = 2.5223(4) Å). While metal-bound NHCs with p-block functionalized N-substituents are quite common, they are typically preformed prior to either addition onto or substitution at a metal center,39 as opposed to through a bond activation reaction as seen here.40 We are unaware of any examples of bidentate NHC-Sn ligands prepared by any route,41 although Tilley has recently described a bidentate P–Sn ligand arising from C–H activation of an Fe-PMeiPr2 ligand onto Sn.42
Summary and Conclusions
The synthesis and reactivity of the heterobimetallic PPh3-derived [Ru–Al] and [Ru–Sn] complexes [Ru(C6H4PPh2)2{PPh2C6H4AlMe(THF)}H] 5 and [Ru(PPh3)(C6H4PPh2)(PPh2C6H4SnMe2)] 6 has been described in a study that represents a continuation of our ongoing research line, in which we attempt to describe and rationalize the effects of M′ on heterobimetallic Ru-main group metal M′ complexes. In conjunction with previous studies on [Ru–Li], [Ru–Mg] and [Ru–Zn] systems 1–3 (Scheme 1), we can conclude that
-
1.
The nature of M′ strongly affects both the structure and reactivity of such heterobimetallic complexes, with ate-type chemistry predominant in the case of more “ionic” M′ metals such as Li, Mg and Al, whereas more “covalent” behavior is observed for M′ = Sn and Zn, with direct Ru–Sn (and Ru–Zn) bonds prevalent (Scheme 11).
-
2.
Reactivity toward H2 provides a means to discriminate ate-[Ru-M′] from bonded [Ru-M′] type complexes; the former react sluggishly, while the latter react instantaneously, as a result of the presence of a coordinatively unsaturated Ru center.
-
3.
Combining (reversibly) cyclometalated, and substitutionally labile, Ru-PPh3 ligands and an increasing number of M′-Me groups across M′ = Li, Mg, Zn, Al, and Sn provides a valuable route to heterobimetallic [Ru-M′] containing new ligand frameworks (e.g., chelating stannylcarbene ligand in 13) with potential for further interesting small molecule reactivity.
Scheme 11. Representation of the Two Extremes of [Ru-M′] Complexes Arsing in Our Work.

Experimental Section
General Comments
All manipulations were carried out at room temperature under argon using standard Schlenk, high vacuum, and glovebox techniques using dry and degassed solvents. C6D6, C6D5CD3, and THF-d8 were vacuum transferred from potassium. NMR spectra were recorded at 298 K (unless otherwise stated) on Bruker Avance 400 and 500 MHz NMR spectrometers and referenced as follows: C6D6 (1H, δ 7.16; 13C, δ 128.0), C6D5CD3 (1H, δ 2.09), THF-d8 (1H, δ 3.58; 13C, δ 25.3). 31P{1H} spectra were referenced externally to 85% H3PO4 and 119Sn to SnMe4. IR spectra were recorded on a Nicolet Nexus spectrometer and UV–vis spectra on a PerkinElmer Lambda 35 spectrometer. Elemental analyses were performed by Elemental Microanalysis Ltd., Okehampton, Devon, U.K. [Ru(PPh3)3HCl]·toluene,43 [Ru(PPh3)(C6H4PPh2)2H][Li(THF)2] (1)4 and IMe4,44 were prepared according to literature methods. Prior to use, [Ru(PPh3)3HCl]·toluene was dried under high vacuum and ground to a fine powder affording a material with ca. 1 molecule of toluene per Ru (based on 1H NMR analysis). IMe4 was purified by sublimation. LiCH2TMS was used as a colorless solid obtained upon cooling a commercial 1.0 M solution in pentane at −32 °C, separating the resulting colorless crystals by decantation in a glovebox and drying under vacuum. AlMe2Cl (1.0 M solution in hexane, Merck) and SnMe3Cl (Merck) were used as received.
[Ru(C6H4PPh2)2{PPh2C6H4AlMe(THF)}H] 5
AlMe2Cl (135 μL of a 1.0 M solution in hexane, 0.135 mmol) was added to an agitated suspension of [Ru(PPh3)(C6H4PPh2)2H][Li(THF)2] (1, 94 mg, 0.09 mmol) and PPh3 (47 mg, 0.18 mmol)45 in benzene (1.5 mL), and the reaction mixture was heated at 60 °C for 1 h. After the mixture was cooled to room temperature, the precipitate of LiCl was separated by cannula filtration and the filtrate reduced to dryness. The residue was dissolved in THF (2 mL), layered with hexane (2 mL), and left to crystallize at −20 °C (3 days). The yellow crystalline product was separated by decantation, washed with hexane (2 × 1 mL), and dried under vacuum. Yield: 71 mg (69%; contains ca. 3 molecules of THF per Ru based on 1H NMR analysis; Figure S1). 1H NMR (500 MHz, C6D6): δ 8.32 (t, J = 9.0 Hz, 2H, Ar), 7.90 (d, J = 6.9 Hz, 1H, Ar), 7.84–7.74 (m, 4H, Ar), 7.50 (t, J = 6.9 Hz, 1H, Ar), 7.40 (br s, 1H, Ar), 7.30–7.14 (m, 7H, Ar; partially overlapped with residual C6D5H), 7.05−6.99 (m, 4H, Ar), 6.93 (br m, 2H, Ar), 6.88–6.78 (m, 5H, Ar), 6.75–6.61 (m, 10H, Ar), 6.45 (t, J = 7.9 Hz, J = 2.3 Hz, 2H, Ar), 6.06 (t, J = 9.0 Hz, 2H, Ar), 5.83 (br m, 1H, Ar), 3.56 (m, 11H, THF), 1.40 (m, 11H, THF), −1.00 (s, 3H, AlMe), −6.30 (m, 1H, RuH). 31P{1H} NMR (202 MHz, C6D6): δ 70.0 (dd, 2JPP = 266 Hz, 2JPP = 23 Hz), −15.4 (dd, 2JPP = 266 Hz, 2JPP = 30 Hz), −25.9 (dd, 2JPP = 30 Hz, 2JPP = 23 Hz). Anal. Calcd. for C55H46AlP3Ru·2.75THF (1126.1): C 70.38, H 6.09. Found: C 70.49, H 6.28.
[Ru(PPh3)(C6H4PPh2)(PPh2C6H4SnMe2)] 6
A THF suspension (10 mL) of [Ru(PPh3)3HCl]·toluene (509 mg, 0.50 mmol) was treated with LiCH2TMS (97 mg, 1.03 mmol) and stirred for 30 min a J. Young resealable ampule to afford an orange solution. A solution of SnMe3Cl (100 mg in 3 mL C6H6, 0.50 mmol) was added dropwise over ca. 3 min with stirring (the vial containing the SnMe3Cl solution was washed with C6H6 (2 × 1 mL), and the washings added to the reaction). The resulting dark blue solution was stirred (2 h), and the volatiles were then removed under vacuum. The residual blue oil was treated with 20 mL hexane and 10 mL of benzene to precipitate LiCl. The suspension was cannula filtered, the residue washed with hexane (5 mL), and the combined filtrate and washings were concentrated under vacuum to yield a blue oil. Recrystallization from benzene/hexane (1:2 ratio) at room temperature for 24 h, and then at −20 °C for 72 h, afforded 6 as dark blue crystals, which were separated, washed with hexane (2 × 1 mL), and dried under vacuum to give 520 mg of product (94% yield). 6 is present in solution together with ca. 5% of a minor isomer, which we propose to have the structure 6′ shown in Figure S4. Data for 6: 1H NMR (500 MHz, THF-d8): δ 7.74 (t, J = 6.5 Hz, 1H, Ar), 7.66 (t, J = 8.4 Hz, 2H, Ar), 7.58 (d, J = 6.9 Hz (JHSn = 25.0 Hz), 1H, Ar), 7.47 (t, J = 8.8 Hz, 2H, Ar), 7.34–7.16 (m, 9H (partially overlaps with C6H6), Ar), 7.14–6.99 (m, 5H, Ar), 6.95–6.84 (m, 14H, Ar), 6.77–6.66 (m, 2H, Ar), 6.64 (t, J = 7.3 Hz, 1H, Ar), 6.55 (t, J = 7.5 Hz, 2H, Ar), 6.40–6.28 (m, 4H, Ar), 0.40 (s (2JHSn = 41 Hz), 3H, SnMe), −1.10 (s (2JHSn = 46 Hz), 3H, SnMe). 31P{1H} NMR (202 MHz, THF-d8): δ 74.8 (dd, 2JPP = 241 Hz, 2JPP = 16 Hz (2JPSn = 150 Hz)), 39.9 (dd, 2JPP = 25 Hz, 2JPP = 16 Hz (2JPSn = 76 Hz)), −28.5 (dd, 2JPP = 241 Hz, 2JPP = 25 Hz (2JPSn = 148 Hz)). 119Sn{1H} NMR (187 MHz, THF-d8): δ 21.4 (td, 2JSnP = 150 Hz, 2JSnP = 78 Hz). UV/vis (toluene, nm): λmax = 600 (ε = 2080 dm3 mol–1 cm–1), 486 (ε = 1890 dm3 mol–1 cm–1). Anal. Calcd. for C56H49P3RuSn·C6H6 (1112.8): C 66.92, H 4.98. Found: C 68.36, H 5.17. Repeated attempts at analysis gave consistently a high%C value, which might be attributable to an incorrect formulation for 6′. Selected NMR data for 6′. 1H NMR (500 MHz, THF-d8): δ 0.11 (s (2JHSn = 47 Hz), 3H, SnMe), −0.20 (s (2JHSn = 44 Hz), 3H, SnMe). 31P{1H} NMR (202 MHz, THF-d8): δ 50.1 (dd, 2JPP = 243 Hz, 2JPP = 17 Hz), 37.4 (dd, 2JPP = 27 Hz, 2JPP = 17 Hz), 3.5 (dd, 2JPP = 243 Hz, 2JPP = 27 Hz).
Variable Temperature NMR Study of the Formation of 5
AlMe2Cl (52 μL of 1.0 M hexane solution, 0.05 mmol) was vacuum transferred into a J. Young resealable NMR tube containing a THF-d8 (0.5 mL) solution of 1 (11 mg, 0.01 mmol). The yellow-orange solution was maintained at 193 K prior to insertion into a precooled (193 K) NMR spectrometer. 1H, 31P{1H}, 1H{31P}, and 1H–31P HMQC NMR spectra acquired over the temperature range of 193–298 K (Figures S8–S12) showed the formation of intermediates I–IV. Selected 1H NMR data for I. 1H NMR (400 MHz, THF-d8, 193 K): δ −10.49 (ddd, 2JHP = 45 Hz, 20 Hz, 5 Hz, 1H, RuH).4631P{1H} NMR (162 MHz, THF-d8, 193 K): δ 46.6 (br), −27.3 (br), −28.5 (t, 2JPP = 21 Hz). 1H NMR (400 MHz, THF-d8, 233 K): δ −10.18 (ddd, 2JHP = 49.7, 2JHP = 21.8 Hz, 2JHP = 8.0 Hz, 1H, RuH). 31P{1H} NMR (162 MHz, THF-d8, 233 K): δ 51.1 (t, 2JPP = 21 Hz), −22.6 (t, 2JPP = 18 Hz), −30.9 (t, 2JPP = 21 Hz). Selected 1H NMR data for II. 1H NMR (400 MHz, THF-d8, 193 K): δ −0.58 (br t, 3JHP = 7 Hz, 3H, RuMe), −13.92 (apparent dd, 2JHP = 47 Hz, 2JHP = 19 Hz, 1H, RuH).5331P{1H} NMR (162 MHz, THF-d8, 193 K): δ 51.1 (br), 50.3 (br), −33.2 (t, 2JPP = 19 Hz). 1H NMR (400 MHz, THF-d8, 233 K): δ −0.80 (RuMe, overlapped with AlMe signals, based on 31P HMQC), −13.73 (ddd, 2JHP = 49.7 Hz, 2JHP = 19.4 Hz, 2JHP = 6.9 Hz, 1H, RuH). 31P{1H} NMR (162 MHz, THF-d8, 233 K): δ 56.5 (t, 2JPP = 21 Hz), 48.6 (t, 2JPP = 17 Hz), −35.5 (dd, 2JPP = 23 Hz, 2JPP = 17 Hz). Selected 1H NMR data for III. 1H NMR (400 MHz, THF-d8, 233 K): δ 5.51 (t, J = 7.8 Hz, 2H, Ar), −1.00 (s, 3H, AlMe), −2.02 (s, 3H, AlMe), −12.20 (dt, 2JHP = 56.9 Hz, 2JHP = 18.4 Hz, 1H, RuH). 31P{1H} NMR (162 MHz, THF-d8, 233 K): δ 56.7 (dd, 2JPP = 252 Hz, 2JPP = 25 Hz), 48.7 (dd, 2JPP = 252 Hz, 2JPP = 22 Hz), −36.1 (t, 2JPP = 24 Hz). Selected 1H NMR data for IV. 1H NMR (400 MHz, THF-d8, 273 K): δ −0.45 (s, 3H, AlMe). 31P{1H} NMR (162 MHz, THF-d8, 273 K): δ 67.4 (dd, 2JPP = 228 Hz, 2JPP = 17 Hz), 46.9 (dd, 2JPP = 26 Hz, 2JPP = 17 Hz), −31.0 (dd, 2JPP = 228 Hz, 2JPP = 26 Hz).
Variable Temperature NMR Study of the Formation of 6
SnMe3Cl (2.5 mg, 0.012 mmol, in 0.1 mL THF-d8) was injected into a J. Young resealable NMR tube containing a frozen THF-d8 (0.4 mL) solution of 1 (11.5 mg, 0.011 mmol). The reaction mixture was warmed until the THF melted (165 K), at which point the color changed from yellow-orange to dark blue. The blue solution was inserted into a precooled (193 K) NMR spectrometer and 1H, 31P{1H} and 1H{31P} NMR spectra were acquired over the range 193–233 K. A single species assigned as V (Scheme 4) was observed up to 273 K, at which point, the final product 6 was also observed (Figures S13–S16). Selected NMR data for V. 1H NMR (400 MHz, THF-d8, 193 K): δ −0.21 (s (2JHSn = 38 Hz), 9H, SnMe3). 31P{1H} NMR (162 MHz, THF-d8, 193 K): δ 48.6 (dd, 2JPP = 241 Hz, 2JPP = 14 Hz (2JPSn = 177 Hz)), 40.6 (dd, 2JPP = 24 Hz, 2JPP = 14 Hz (2JPSn = 155 Hz)), −36.1 (dd, 2JPP = 241 Hz, 2JPP = 24 Hz (2JPSn = 198 Hz)).
[Ru(PPh3)2(PPh2C6H4AlMe)H3] 7
A C6D6 (0.5 mL) solution of 5 (10 mg, 0.009 mmol) in a J. Young resealable NMR tube was freeze–pump–thaw degassed (×3) and placed under 1 atm H2. Heating at 60 °C for 2 h resulted in complete conversion (based on 31P{1H} NMR spectroscopy) to [Ru(PPh3)2(PPh2C6H4AlMe)H3] 7, which was characterized by 1H and 31P{1H} NMR spectroscopy. Selected NMR data for 7: 1H NMR (500 MHz, C6D6): δ −0.39 (s, 3H, AlMe), −8.46 (br d, 1H, Ru···H-Al), −8.72 (td, 2JHP = 28.2 Hz, 2JHP = 13.9 Hz, 1H, Ru-H), −11.07 (dt, 2JHP = 53.5 Hz, 2JHP = 23.3 Hz, 1H, Ru-H···Al). 31P{1H} NMR (202 MHz, C6D6): δ 73.5 (dd, 2JPP = 240 Hz, 2JPP = 28 Hz), 62.1 (dd, 2JPP = 240 Hz, 2JPP = 23 Hz), 58.8 (br t, 2JPP = 25 Hz). Exposure of the sample to vacuum for 2 h followed by redissolution of the residue in C6D6 revealed complete degradation of 7 and appearance of a number of resonances in both the 1H and 31P NMR spectra (Figure S22), including signals for both [Ru(PPh3)3(η2-H2)H2] (1H: δ −7.08 (s); 31P{1H}: δ 57.7 (s)) and [Ru(PPh3)4H2] (1H: δ −10.13 (m); 31P{1H}: δ 49.3 (t), 41.1 (t)).25 The fate of the Al metal was not established.
[Ru(PPh3)2(PPh2C6H4SnMe2)H3] 8
A C6D6 (0.5 mL) or THF-d8 (0.5 mL) solution of 6 (12 mg, 0.011 mmol) in a J. Young resealable NMR tube was placed under 1 atm of H2, which was then allowed to slowly diffuse through the sample to yield a pale yellow-colorless homogeneous solution of [Ru(PPh3)2(PPh2C6H4SnMe2)H3] 8. This was characterized by 1H and 31P{1H} variable temperature NMR spectroscopy. 1H NMR (500 MHz, C6D6): δ 8.0 (d, JHH = 7.0 Hz (JHSn = 32 Hz), 1H, Ar), 7.51 (dd, JHH = 7.8, JHH = 4.5 Hz, 1H, Ar), 7.39 (t, JHH = 8.9 Hz, 4H, Ar), 7.28 (t, JHH = 8.8 Hz, 12H, Ar), 7.02 (t, J = 7.3 Hz, 1H, Ar), 6.94 (t, JHH = 7.6 Hz, 6H, Ar), 6.88–6.79 (m, 18H, Ar), −0.50 (s (2JHSn = 47 Hz), 6H, SnMe), −7.51 (br m (2JHSn = 183 Hz), 3H, RuH). 31P{1H} NMR (162 MHz, C6D6): δ 84.7 (t, 2JPP = 99 Hz (2JPSn = 124 Hz)), 56.6 (d, 2JPP = 99 Hz (2JPSn = 95 Hz)). 1H NMR (500 MHz, THF-d8): δ 7.89 (d, JHH = 7.3 Hz (JHSn = 33 Hz), 1H, Ar), 7.39 (m, 1H, Ar), 7.13 (t, JHH = 7.2 Hz, 7H, Ar), 7.08 (t, JHH = 9.0 Hz, 3H, Ar), 7.04–6.89 (m, 23H, Ar), 0.05 (s (2JHSn = 48 Hz), 6H, SnMe), −7.91 (br m (2JHSn = 179 Hz), 3H, RuH). 31P{1H} NMR (162 MHz, THF-d8): δ 85.1 (t, 2JPP = 97 Hz (2JPSn = 127 Hz)), 56.2 (d, 2JPP = 97 Hz (JPSn = 89 Hz)). Selected 1H NMR (500 MHz, THF-d8, 332 K): δ 0.04 (s (2JHSn = 48 Hz), 6H, SnMe), −7.95 (dt, 2JHP = 16.4 Hz, 2JHP = 7.4 Hz (2JHSn = 180 Hz), 3H, RuH). 31P{1H} NMR (162 MHz, THF-d8, 332 K): δ 84.0 (t, 2JPP = 96 Hz (JPSn = not determined)), 54.6 (d, 2JPP = 96 Hz (JPSn ∼ 96 Hz)). IR (KBr, cm–1): 1967 (νRuHSn), 1746 (νRuHSn).
The formation of 8 could also be performed in the solid-state. Stirring a microcrystalline sample of 6 (15 mg, 0.013 mmol) under 1 atm of H2 in a J. Young resealable ampule for 24 h brought about a color change from purple to off-white. Conversion to 8 was proven by IR spectroscopy (Figure S30). In an attempt to prepare an isolable derivative of 8, an excess of pyridine (50 μL, 0.49 mmol) was added to a C6D6 (0.5 mL) solution of 6 (10 mg, 0.022 mmol) to give [Ru(NC5H5)(C6H4PPh2)(PPh2C6H4SnMe2)] (Figure S31), assigned from the appearance of two doublets (δ 80.1 (d, 2JPP = 285 Hz), −26.1 (d, 2JPP = 285 Hz)) in the 31P{1H} NMR spectrum. Addition of 1 atm H2 to the crude sample rapidly yielded 31P{1H} NMR signals of 8 at ca. δ 84 and 55.
[Ru(CO)2(C(O)C6H4PPh2)(PPh2C6H4SnMe2)] 9
A benzene (2 mL) solution of 6 (111 mg, 0.10 mmol) was placed under CO (1 atm), and the solution was stirred at 80 °C for 4 h. The resulting yellow solution was filtered through a pad of Celite. The pad was washed with 1 mL C6H6 and the combined filtrate and washings layered with hexane (6 mL). An initial batch of yellow crystals of 9 were formed. Treatment with additional hexane (6 mL) and cooling to −32 °C for 24 h afforded yellow crystalline needles. The yellow solids were combined, washed with hexane (2 × 1 mL) and dried under vacuum. Yield: 54 mg (63%). 1H NMR (400 MHz, C6D6): δ 8.30–8.23 (m, 2H, Ar), 7.89–7.79 (m, 3H, Ar), 7.74 (d, J = 7.3 Hz (JHSn = 26 Hz), 1H, Ar), 7.58–7.46 (m, 3H, Ar), 7.42 (t, J = 7.2 Hz, 1H, Ar), 7.27–6.87 (m, 18H, Ar), 0.24 (s (2JHSn = 41 Hz), 3H, SnMe), −0.24 (s (2JHSn = 43 Hz), 3H, SnMe). 31P{1H} NMR (162 MHz, C6D6): δ 66.9 (d, 2JPP = 210 Hz (2JP-117Sn = 140 Hz, 2JP-119Sn = 146 Hz)), 64.2 (d, 2JPP = 210 Hz (2JP-117Sn = 134, 2JP-119Sn = 140 Hz)). 13C{1H} NMR (126 MHz, C6D6): δ 202.9 (t, 2JCP = 8 Hz, Ru-CO), 200.0 (t, 2JCP = 10 Hz, Ru-CO), 158.1 (d, JCP = 40 Hz, Ar), 155.5 (dd, JCP = 61 Hz, JCP = 4 Hz, Ar), 140.7 (d, JCP = 42 Hz, Ar), 140.1 (d, JCP = 4 Hz, Ar), 139.6 (br m, Ar), 139.3 (br m, Ar), 138.0 (dd, JCP = 42 Hz, JCP = 4 Hz, Ar), 136.2 (d, JCP = 24 Hz, Ar), 135.5 (d, JCP = 9 Hz, Ar), 134.8 (dd, JCP = 39 Hz, JCP = 3 Hz, Ar), 132.9 (d, JCP = 13 Hz, Ar), 132.6 (d, JCP = 9 Hz, Ar), 132.5 (d, JCP = 9 Hz, Ar), 131.7 (d, JCP = 13 Hz, Ar), 131.6 (d, JCP = 11 Hz, Ar), 130.6 (d, JCP = 9 Hz, Ar), 130.4 (d, JCP = 5 Hz, Ar), 130.1 (s, Ar), 129.9 (s, Ar), 129.4 (s, Ar), 128.7 (d, JCP = 11 Hz, Ar), 122.0 (d, JCP = 17 Hz, Ar), −5.8 (s, SnMe), −8.2 (s, SnMe). 119Sn{1H} NMR (187 MHz, C6D6): δ 115.3 (t, 2JSnP = 143 Hz). IR (KBr, cm–1): 2010 (νCO), 1966 (νCO), 1963 (νCO), 1954 (νCO), 1596 (νC(O)C6H4), 1568 (νC(O)C6H4). IR (C6D6, cm–1): 2008 (νCO), 1961 (νCO). Anal. Calcd. for C41H34O3P2RuSn (856.4): C 57.50, H 4.00. Found: C 57.88, H 4.09.
Variable Temperature/13CO NMR Study of the Formation of 9
A C6D6 (0.5 mL) solution of 6 (11 mg, 0.010 mmol) was placed under 1 atm 13CO. Upon shaking, an instantaneous color change from dark blue to yellow took place. 1H, 31P{1H} and 13C{1H} NMR spectroscopy was used to follow the progress of the reaction and allow characterization of intermediates VI–13CO, VII–13CO, and VIII–13CO, initially over 20 h at room temperature, and then at 80 °C. The reaction was repeated using 12CO (1 atm) with 10 mg 6 in 0.5 mL C6D5CD3 to afford spectra of non-13CO labeled VI, VII and VIII. VI–13CO. Selected 1H NMR (400 MHz, C6D6): δ 0.67 (s (2JHSn = 29 Hz), 3H, SnMe), −0.25 (s (2JHSn = 35 Hz), 3H, SnMe). 31P{1H} NMR (162 MHz, C6D6): δ 76.1 (ddd, 2JPP = 241 Hz, 2JPP = 15 Hz, 2JPC = 11 Hz), 40.2 (ddd, 2JPP = 24 Hz, 2JPP = 15 Hz, 2JPC = 8 Hz), −35.2 (ddd, 2JPP = 241 Hz, 2JPP = 24 Hz, 2JPC = 10 Hz). Selected 13C{1H} NMR (101 MHz, C6D6): δ 206.5 (td, 2JCP = 11 Hz, 2JCP = 8 Hz, Ru-CO). VI: 31P{1H} NMR (202 MHz, C6D5CD3): δ 76.1 (dd, 2JPP = 241 Hz, 2JPP = 15 Hz (2JPSn = 174 Hz)), 40.2 (dd, 2JPP = 23 Hz, 2JPP = 15 Hz (2JPSn = 163 Hz)), −35.2 (dd, 2JPP = 241 Hz, 2JPP = 24 Hz (2JPSn = 182 Hz)). VII–13CO. Selected 1H NMR (400 MHz, C6D6): δ 0.62 (s (2JHSn = 39 Hz), 3H, SnMe), 0.18 (s (2JHSn = 39 Hz), 3H, SnMe). 31P{1H} NMR (162 MHz, C6D6): δ 58.9 (ddd, 2JPP = 254 Hz, 2JPP = 18 Hz, 2JPC = 9 Hz), 43.4 (ddd, 2JPP = 254 Hz, 2JPP = 28 Hz, 2JPC = 15 Hz), −33.7 (ddd, 2JPP = 28 Hz, 2JPP = 18 Hz, 2JPC = 4 Hz). Selected 13C{1H} NMR (101 MHz, C6D6): δ 207.3 (ddd, 2JCP = 15 Hz, 2JCP = 9 Hz, 2JCP = 4 Hz, Ru-CO). VII. 31P{1H} NMR (202 MHz, C6D5CD3): δ 58.9 (dd, 2JPP = 254 Hz, 2JPP = 18 Hz (2JPSn = 173 Hz)), 43.4 (dd, 2JPP = 254 Hz, 2JPP = 28 Hz (2JPSn = 196 Hz)), −33.7 (dd, 2JPP = 28 Hz, 2JPP = 18 Hz (2JPSn = 970 Hz)). VIII–13CO. Selected 1H NMR (400 MHz, C6D6): δ 0.57 (s (2JHSn obscured by overlap with other signals), 3H, SnMe), −0.64 (s (2JHSn = 44 Hz), 3H, SnMe). 31P{1H} NMR (162 MHz, C6D6): δ 69.5 (ddd, 2JPP = 227 Hz, 2JPC = 9 Hz, 2JPC = 7 Hz), −29.2 (dt, 2JPP = 227 Hz, 2JCP = 9 Hz). Selected 13C{1H} NMR (101 MHz, C6D6): δ 202.8 (m, Ru-CO), 200.4 (td, 2JCP = 9 Hz, 2JCC = 3 Hz, Ru-CO). VIII. 31P{1H} NMR (202 MHz, C6D5CD3): δ 69.5 (d, 2JPP = 228 Hz (2JPSn = 143 Hz)), −29.2 (d, 2JPP = 228 Hz (2JPSn = 141 Hz)).
[Ru(IMe4)2(C6H4PPh2)(PPh2C6H4SnMe2)] 10
IMe4 (17 mg, 0.13 mmol) was added to an agitated blue solution of 6 (54 mg, 0.048 mmol) in benzene (3 mL). The resulting yellow-orange solution was stirred for 1 h and then treated with hexane (3 mL) and left to crystallize for 24 h. The yellow-orange crystals of product were separated and dried under vacuum. Yield: 29 mg (55%). 1H NMR (500 MHz, THF-d8): δ 7.65–7.58 (m, 1H, Ar), 7.37–7.58 (m, 3H, Ar, overlapped with C6H6), 7.19–7.13 (m, 1H, Ar), 7.06–6.95 (m, 5H, Ar), 6.93–6.40 (m, 17H, Ar), 6.36 (ddd, J = 9.1 Hz, J = 7.1 Hz, J = 1.5 Hz, 1H, Ar), 3.83 (s, 3H, NMe), 3.73 (s, 3H, NMe), 3.16 (s, 3H, NMe), 2.43 (s, 3H, NMe), 2.11 (s, 3H, NCMe), 2.00 (s, 3H, NCMe), 1.91 (s, 3H, NCMe), 1.39 (s, 3H, NCMe), 0.54 (s (2JHSn = 24 Hz), 3H, SnMe), −0.23 (s (2JHSn = 23 Hz), 3H, SnMe). 31P{1H} NMR (162 MHz, THF-d8): δ 69.1 (d, 2JPP = 18 Hz (2JP-119Sn = 257 Hz, 2JP-117Sn = 220 Hz)), −36.4 (d, 2JPP = 18 Hz (2JP-119Sn = 1311 Hz, 2JP-117Sn = 1254 Hz)). Selected 13C{1H} NMR (101 MHz, THF-d8): δ 199.9 (dd, 2JCP = 8 Hz, 2JCP = 2 Hz, RuCNHC), 191.0 (dd, 2JCP = 86 Hz, 2JCP = 16 Hz, RuCNHC), 178.5 (d, 2JCP = 17 Hz, RuCAr), 41.2 (dd, 4JCP = 9 Hz, 4JCP = 6 Hz, NMe), 38.7 (s, NMe), 35.9 (d, 4JCP = 2 Hz, NMe), 35.7 (d, 4JCP = 8 Hz, NMe), 10.0 (s, NCMe), 9.9 (s, NCMe), 9.7 (s, NCMe), −0.8 (d, 3JCP = 9 Hz, SnMe), −3.5 (dd, 3JCP = 11 Hz, 3JCP = 3 Hz, SnMe). 119Sn{1H} NMR (187 MHz, THF-d8): δ 51.0 (dd, 2JSnP = 1315 Hz, 2JSnP = 243 Hz). Anal. Calcd. for C52H58N4P2RuSn·2C6H6 (1177.0): C 65.31, H 5.99, N, 4.76. Found: C 65.22, H 6.12, N, 4.91.
[Ru(IMe4)(PPh3)(IMe4′)(PPh2C6H4SnMe2)] 11
A THF-d8 solution of 10 (37 mg, 0.034 mmol) in a J. Young resealable NMR tube was heated at 60 °C. Monitoring by 1H and 31P NMR spectroscopy showed ca. 85% conversion through to 11 after 135 min. The reaction was pumped to dryness and the residue dissolved in a minimum amount of benzene and layered with hexane to give 21 mg of orange/yellow product comprised ca. 90% 11, which was spectroscopically characterized. Selected 1H NMR (500 MHz, THF-d8): δ 3.22 (s, 3H, NMe), 2.98 (s, 3H, NMe), 2.77 (s, 3H, NMe), 2.42 (dd, JHH = 3.3 Hz (second J coupling obscured by overlap of signal with N-Me of side product), 1H, NCHH), 2.22 (dd, J = 7.4 Hz, J = 3.3 Hz, 1H, NCHH), 1.75 (s, 3H, NCMe), 1.73 (s, 3H, NCMe), 1.47 (s, 3H, NCMe), 0.23 (s (2JHSn = 18 Hz), 3H, SnMe), −0.02 (s (2JHSn = 23 Hz), 3H, SnMe). 31P{1H} NMR (202 MHz, THF-d8): δ 81.4 (d, 2JPP = 294 Hz (2JPSn = 204 Hz)), 54.9 (d, 2JPP = 294 Hz (2JPSn = 218 Hz)). Selected 13C{1H} NMR (101 MHz, THF-d8): δ 192.4 (t, 2JCP = 13 Hz, RuCNHC), 169.3 (dd, 1JCP = 64 Hz, 3JCP = 3 Hz, RuPPh2C), 163.1 (t, 2JCP = 14 Hz, RuCNHC), 38.1 (s, NMe), 36.8 (s, NMe), 32.8 (s, NMe), 21.9 (t, 2JCP = 9 Hz, RuCH2), 10.1 (s, NCMe), 9.8 (s, NCMe), 8.8 (s, NCMe), 6.1 (s, NCMe), 2.6 (s (1JCSn = 46 Hz), SnMe), 1.0 (s, SnMe).
[Ru(IMe4)2(IMe4′)(PPh2C6H4SnMe2)] 12 and [Ru(PPh3)(IMe4′-SnMe2)(C6H4PPh2)] 13
A J. Young resealable NMR tube containing a C6D5CD3 (0.5 mL) solution of 9 (40 mg, 0.034 mmol) was heated at 120 °C and conversion to 12 and 13 monitored by 1H and 31P{1H} NMR spectroscopy. The reaction was stopped after 1 h, concentrated and layered with hexane to afford 15 mg of a mixture of yellow (12) and purple (13) crystals. These were separated manually to allow NMR characterization and to isolate single crystals suitable for X-ray crystallography. Selected 1H NMR data for 12. (500 MHz, THF-d8): δ 3.89 (s, 3H, NMe), 3.37 (s, 3H, NMe), 3.36 (s, 3H, NMe), 2.84 (s, 3H, NMe), 2.73 (s, 3H, NMe), 2.09 (s, 3H, NCMe), 2.05 (s, 3H, NCMe), 1.97 (s, 3H, NCMe), 1.84 (s, 3H, NCMe), 1.80 (s, 3H, NCMe), 1.16 (s, 3H, NCMe), 0.42 (s (2JHSn = 12 Hz), 3H, SnMe), 0.33 (s (2JHSn = 14 Hz), 3H, SnMe). 31P{1H} NMR (162 MHz, THF-d8): δ 76.5 (s). Selected 13C{1H} NMR (101 MHz, THF-d8): δ 202.8 (d, 2JCP = 11 Hz, RuCNHC), 198.6 (d, 2JCP = 116 Hz, RuCNHC), 170. 0 (d, 2JCP = 18 Hz, RuCNHC), 168.9 (d, 1JCP = 78 Hz, PC6H4Sn), 39.3 (s, NMe), 37.3 (d, 4JCP = 4 Hz, NMe), 36.1 (s, NMe), 34.2 (s, NMe), 32.4 (s, NMe), 23.2 (d, 2JCP = 6 Hz, RuCH2), 10.4 (s, NCMe), 10.1 (s, NCMe), 9.9 (s, NCMe), 9.8 (s, NCMe), 6.6 (s, NCMe), −1.5 (d, 3JCP = 5 Hz, SnMe), −1.7 (s, SnMe). 119Sn{1H} NMR (187 MHz, THF-d8): δ 63.6 (d, 2JSnP = 195 Hz). Selected NMR data for 13. 1H NMR (400 MHz, THF-d8): δ 7.61–7.53 (m, 2H, Ar), 7.34–7.24 (m, 5H, Ar), 7.22–7.10 (m, 11H, Ar), 7.08–7.00 (m, 6H, Ar), 6.97–6.86 (m, 2H, Ar), 6.84–6.74 (m 2H, Ar), 6.51 (m, 1H, Ar), 2.70 (d, 2JHH = 11.2 Hz (2JHSn = 32 Hz), 1H, NCHH), 2.55 (s, 3H, NMe), 2.18 (d, 2JHH = 11.2 Hz (2JHSn = 14 Hz), 1H, NCHH), 2.12 (s, 3H, NMe), 2.01 (s, 3H, NMe), −0.20 (s (2JHSn = 41 Hz), 3H, SnMe), −0.59 (s (2JHSn = 44 Hz), 3H, SnMe). 31P{1H} NMR (162 MHz, THF-d8): δ 48.5 (d, 2JPP = 21 Hz (2JPSn = 145 Hz)), −22.6 (d, 2JPP = 21 Hz (2JPSn = 124 Hz)). 13C{1H} NMR (101 MHz, THF-d8): δ 196.3 (dd, 2JCP = 79 Hz, 2JCP = 7 Hz, RuCNHC), 183.5 (dd, 2JCP = 66 Hz, 2JCP = 5 Hz, RuCAr), 158.3 (dd, 1JCP = 41 Hz, 3JCP = 3 Hz, PAr), 142.5 (d, 1JCP = 25 Hz, P-Cipso), 140.9 (d, 1JCP = 25 Hz, P-Cipso), 139.5 (d, JCP = 14 Hz, PAr), 137.5 (dd, JCP = 14 Hz, JCP = 3 Hz, PAr), 134.5 (d, JCP = 13 Hz, PAr), 134.2 (d, JCP = 12 Hz, PAr), 133.5 (d, JCP = 11 Hz, PAr), 129.4 (s, PAr), 129.0 (s, PAr), 128.9 (d, JCP = 14 Hz, PAr), 128.6 (s, PAr), 128.5 (d, JCP = 8 Hz, PAr), 127.1 (s, NCMe), 125.7 (d, JCP = 8 Hz, PAr), 124.0 (s, NCMe), 123.5 (d, JCP = 8 Hz, PAr) 34.6 (d, 3JCP = 6 Hz, NCH2), 38.4 (s, NMe), 34.5 (s, NMe), 10.7 (s, NCMe), 9.0 (s, NCMe), −5.4 (d, 3JCP = 4 Hz, SnMe), −6.4 (d, 3JCP = 3 Hz, SnMe). 119Sn{1H} NMR (187 MHz, THF-d8): δ 41.5 (dd, 2JSnP = 145 Hz, 2JSnP = 128 Hz).
X-ray Crystallography
Data for 5, 9, 10, and 13 were collected on an Agilent Xcalibur diffractometer (using a Mo Kα radiation) while the 6, 11, and 12 data sets were obtained using an Agilent SuperNova instrument and a Cu Kα source (Table S1). All experiments were conducted at 150 K, solved by employing either the solution program native to Olex247 or SHELXT.48 Refinements were conducted using SHELXL49 via the Olex2 interface. Convergence of the models was largely unremarkable and only exceptional points of note will be outlined herein. In particular, the asymmetric unit in 5 was seen to contain one molecule of the organometallic complex and two molecules of THF. The hydride ligand in the main feature was located and refined without restraints, while C58 from the ligated THF was modeled to take account of 55:45 disorder. One of the guest THF molecules also resolved satisfactorily into two disordered components (60:40 ratio) with the inclusion of some distance and ADP restraints in the final least-squares. The second solvent moiety was readily identifiable as a THF, but disorder was messy, and it prevailed beyond two fractions. As such, this was ultimately treated using the solvent mask algorithm in Olex2, and an allowance for the same was made in the formula as presented.
In 10, the asymmetric unit was noted to comprise one molecule of the Sn–Ru complex, one full-occupancy molecule of benzene and another benzene moiety which was modeled to take account of 72:28 disorder. Each component of the latter was treated as a rigid hexagon in the refinement. The highest residual electron density peaks in the difference Fourier map are at chemically insignificant distances from atoms in the main feature. Indeed, they may point toward some very minor disorder, at a level which negates modeling.
The hydrogen atoms attached to C1 in 11 were located and refined subject to being a common distance from the parent atom. The highest residual electron density peak is located at a chemically insignificant distance from Sn1. One molecule of the organometallic complex and a region of solvent correspond to the asymmetric unit in the structure of 12. The hydrogen atoms attached to C7 were located and refined freely. Analysis of the electron density indicated 5% disorder of the tin center (at location Sn1a) and this was accounted for in the model. However, no effort was made to model the necessary 5% disorder of the phosphine ligand attached the main group metal, as it would be imprudent to invest in location of 5% disorder for first row elements with the expectation of credibility. The solvent moiety was very disordered and was ultimately treated using the solvent mask algorithm available in Olex2, with an allowance made for the presence of one molecule of toluene, per asymmetric unit, in the formula as presented.
In 13, the hydrogen atoms attached to C6 were located and refined freely. The electron density indicated 9% disorder of the tin center (at location Sn1a) and this was accounted for in the model. However, (using similar rationale to that employed for 6) no effort was made to model the necessary 9% disorder of the phosphine ligand attached the minor tin component. Distance and ADP restraints were included for the minor tin component.
Computational Methodology
DFT calculations were run with Gaussian 16 (C.01).50 The Al, P, Ru and Sn centers were described with the Stuttgart RECPs and associated basis sets,51 and the 6-31G** basis set was used for all other atoms (BS1).52 A polarization function was also added to Al (ζd = 0.190), P (ζd = 0.387) and Sn (ζd = 0.180).53 Initial BP86 optimizations54 were performed using the ‘grid = ultrafine’ option, with all stationary points being fully characterized via analytical frequency calculations as minima. All energies were recomputed with a larger basis set (BS2) featuring 6-311++G** basis sets on all atoms, with the exception of Ru and Sn which employed the basis set aug-cc-pVTZ-PP. Corrections for the effect of solvent (benzene: ε = 2.2706; THF: ε = 7.4257) solvent were employed using the polarizable continuum model and BS1.55 Single-point dispersion corrections to the BP86 results employed Grimme’s D3 parameter set with Becke-Johnson damping as implemented in Gaussian.56 Natural Bonding Orbital (NBO 3.1)57 analyses were performed on the BP86/BS1 optimized geometries at the BP86/BS2 level.
Acknowledgments
This project has received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No. 792674 (to F.M.M.) and EPSRC (grant EP/T019743/1 to A.F.P., Doctoral Training Award to C.J.I.). This research made use of the Balena and Anatra High Performance Computing (H.P.C.) Services at the University of Bath. We thank Dr Nasir Rajabi for preliminary studies. The authors gratefully acknowledge the University of Bath’s Research Computing Group (doi.org/10.15125/b6cd-s854) for their support in this work. We thank Dr. Mary Mahon for X-ray assistance and Dr. Dan Pantos for access to his UV–vis spectrometer.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.organomet.2c00344.
Author Present Address
† F.M.M.: Laboratory of Organic Chemistry, Wageningen University, Stippeneng 4, 6708 WE Wageningen, The Netherlands
The authors declare no competing financial interest.
Supplementary Material
References
- a For reviews, see:Bouhadir G.; Bourissou D. Complexes of ambiphilic ligands: Reactivity and catalytic applications. Chem. Soc. Rev. 2016, 45, 1065–1079. 10.1039/C5CS00697J. [DOI] [PubMed] [Google Scholar]; b Cammarota R. C.; Clouston L. J.; Lu C. C. Leveraging molecular metal-support interaction for H2 and N2 activation. Coord. Chem. Rev. 2017, 334, 100–111. 10.1016/j.ccr.2016.06.014. [DOI] [Google Scholar]; c Butler M. J.; Crimmin M. R. Magnesium, zinc, aluminium and gallium hydride complexes of the transition metals. Chem. Commun. 2017, 53, 1348–1365. 10.1039/C6CC05702K. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Campos J. Bimetallic cooperation across the Periodic Table. Nat. Rev. Chem. 2020, 4, 696–702. 10.1038/s41570-020-00226-5. [DOI] [PubMed] [Google Scholar]; e Takaya J. Catalysis using transition metal complexes featuring main group metal and metalloid compounds as supporting ligands. Chem. Sci. 2021, 12, 1964–1981. 10.1039/D0SC04238B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a For recent examples, see:Steinhoff P.; Paul M.; Schroers J. P.; Tauchert M. E. Highly efficient palladium-catalysed carbon dioxide hydrosilylation employing PMP ligands. Dalton Trans. 2019, 48, 1017–1022. 10.1039/C8DT03777A. [DOI] [PubMed] [Google Scholar]; b Takaya J.; Ogawa K.; Nakaya R.; Iwasawa N. Rhodium-catalyzed chemoselective hydrosilylation of nitriles to an imine oxidation level enabled by a pincer-type group 13 metallylene ligand. ACS Catal. 2020, 10, 12223–12228. 10.1021/acscatal.0c02779. [DOI] [Google Scholar]; c Seki R.; Hara N.; Saito T.; Nakao Y. Selective C-O bond reduction and borylation of aryl ethers catalyzed by a rhodium-aluminum heterobimetallic complex. J. Am. Chem. Soc. 2021, 143, 6388–6394. 10.1021/jacs.1c03038. [DOI] [PubMed] [Google Scholar]
- a Fryzuk M. D.; McConville D. H.; Rettig S. J. Reactions of the electron-rich binuclear hydride complexes [{Pri2P(CH2)xPPri2}Rh]2(μ-H)2 (x = 2 or 3) with ZnR2 and MgR’2. Organometallics 1993, 12, 2152–2161. 10.1021/om00030a027. [DOI] [Google Scholar]; b Molon M.; Gemel C.; Fischer R. A. From AlCp*- and GaCp*-ligated ruthenium hydrides to zinc-rich heterometallic complexes. Eur. J. Inorg. Chem. 2013, 2013, 3616–3622. 10.1002/ejic.201300317. [DOI] [Google Scholar]; c Riddlestone I. M.; Rajabi N. A.; Lowe J. P.; Mahon M. F.; Macgregor S. A.; Whittlesey M. K. Activation of H2 over the Ru-Zn bond in the transition metal-Lewis acid heterobimetallic species [Ru(IPr)2(CO)ZnEt]+. J. Am. Chem. Soc. 2016, 138, 11081–11084. 10.1021/jacs.6b05243. [DOI] [PubMed] [Google Scholar]
- Miloserdov F. M.; Rajabi N. A.; Lowe J. P.; Mahon M. F.; Macgregor S. A.; Whittlesey M. K. Zn-Promoted C-H reductive elimination and H2 activation via a dual unsaturated Ru-Zn intermediate. J. Am. Chem. Soc. 2020, 142, 6340–6349. 10.1021/jacs.0c01062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole-Hamilton D. J.; Wilkinson G. Ortho-metallated triphenylphosphine(2-diphenylphosphinophenyl) and related complexes of ruthenium(II): Interaction of chlorohydridotris(triphenylphosphine)ruthenium(II) with methyl and trimethylsilylmethyl alkylating agents (Li, Mg, Zn); hydrido methyl and trimethylsilylmethyl complexes. J. Chem. Soc., Dalton Trans. 1977, 797–804. 10.1039/dt9770000797. [DOI] [Google Scholar]
- It is worth noting that the reaction of [Ru(PPh3)3Cl2] with AlMe3, in which alkane elimination is not possible, proceeds in arene solvents to generate [(η6-arene)Ru(Me)(PPh3)2][AlCl2Me2].Fang X. G.; Watkin J. G.; Scott B. L.; John K. D.; Kubas G. J. One-pot synthesis of (η6-arene)bis(triphenylphosphine)(methyl)ruthenium(II) cations. X-ray structures of [(η6-C6H6)Ru(Me)(PPh3)2][AlCl2Me2] and the η5-thiophene analogue. Organometallics 2002, 21, 2336–2339. 10.1021/om0200860. [DOI] [Google Scholar]
- Bennett M. A.; Bhargava S. K.; Ke M.; Willis A. C. Complexes of platinium(II), platinium(IV), rhodium(III) and iridium(III) containing orthometallated triphenylphosphine. J. Chem. Soc., Dalton Trans. 2000, 3537–3545. 10.1039/b004908p. [DOI] [Google Scholar]
- Examples of metalation onto Al are rare.; a Buchin B.; Gemel C.; Kempter A.; Cadenbach T.; Fischer R. A. Reaction of iron and ruthenium halogenide complexes with GaCp* and AlCp*: Insertion, Cp* transfer reactions and orthometallation. Inorg. Chim. Acta 2006, 359, 4833–4839. 10.1016/j.ica.2006.07.021. [DOI] [Google Scholar]; b Sircoglou M.; Bouhadir G.; Saffon N.; Miqueu K.; Bourissou D. A zwitterionic gold(I) complex from an ambiphilic diphosphino-alane ligand. Organometallics 2008, 27, 1675–1678. 10.1021/om800101s. [DOI] [Google Scholar]; c Courtemanche M.-A.; Larouche J.; Legare M.-A.; Bi W.; Maron L.; Fontaine F.-G. A tris(triphenylphosphine)aluminum ambiphilic precatalyst for the reduction of carbon dioxide with catecholborane. Organometallics 2013, 32, 6804–6811. 10.1021/om400645s. [DOI] [Google Scholar]; d Cowie B. E.; Tsao F. A.; Emslie D. J. H. Synthesis and platinium complexes of an alane-appended 1,1′-bis(phosphino)ferrocene ligand. Angew. Chem., Int. Ed. 2015, 54, 2165–3169. 10.1002/anie.201410828. [DOI] [PubMed] [Google Scholar]
- The Ru···Al distance is 2.5911(7) Å, which compares to values for the sum of the covalent radii of 2.51 and 2.67 Å by Pyykkö and Alvarez respectively.; a Pyykkö P.; Atsumi M. Molecular single-bond covalent radii for elements 1–118. Chem.—Eur. J. 2009, 15, 186–197. 10.1002/chem.200800987. [DOI] [PubMed] [Google Scholar]; b Cordero B.; Gómez V.; Platero-Prats A. E.; Revés M.; Echeverría J.; Cremades E.; Barragán F.; Alvarez S. Covalent radii revisited. Dalton Trans. 2008, 2832–2838. 10.1039/b801115j. [DOI] [PubMed] [Google Scholar]
- This is comparable to the distance found in other Ru-stannyl species.; a Clark G. R.; Flower K. R.; Roper W. R.; Wright L. J. Coordinatively unsaturated stannyl complexes of ruthenium(II) via a new synthetic route involving vinylstannanes: Molecular-structure of Ru(SnMe3)Cl(CO)(PPh3)2. Organometallics 1993, 12, 259–260. 10.1021/om00026a006. [DOI] [Google Scholar]; b Aarnts M. P.; Stufkens D. J.; Oskam A.; Fraanje J.; Goubitz K. Syntheses, structures and spectroscopic properties of novel inorganometallic complexes Ru(E)(E’)(CO)2(iPr-DAB) (E = Cl, E’ = SnPh3, PbPh3; E = Me, E’ = SnPh3, PbPh3; E = SnPh3, E’ = SnPh3, SnMe3, GePh3; E = PbPh3, E’ = PbPh3, PbMe3, GePh3; iPr-DAB = N,N’-diisopropyl-1,4-diaza-1,3-butadiene). Inorg. Chim. Acta 1997, 256, 93–105. 10.1016/S0020-1693(96)05423-0. [DOI] [Google Scholar]
- a Lu G. L.; Mohlen M. M.; Rickard C. E. F.; Roper W. R.; Wright L. J. A cyclic osmastannyl complex, Os(κ2(Sn,P)-SnMe2C6H4PPh2)(κ2-S2CNMe2(CO)(PPh3) derived from the osmastannol complex, Os(SnMe2OH)(κ2-S2CNMe2(CO)(PPh3)2. Inorg. Chim. Acta 2005, 358, 4145–4155. 10.1016/j.ica.2004.12.059. [DOI] [Google Scholar]; b Lu G. L.; Rickard C. E. F.; Roper W. R.; Wright L. J. Metallacyclic complexes with ortho-stannylated triphenylphosphine ligands, LnOs(κ2(Sn,P)-SnMe2C6H4PPh2), derived from thermal reactions of the five-coordinate complex, Os(SnMe3)Cl(CO)(PPh3)2. J. Organomet. Chem. 2005, 690, 4114–4123. 10.1016/j.jorganchem.2005.05.048. [DOI] [Google Scholar]
- Roper W. R.; Wright L. J. Similarities and contrasts between silyl and stannyl derivatives of ruthenium and osmium. Organometallics 2006, 25, 4704–4718. 10.1021/om060526d. [DOI] [Google Scholar]
- a For other examples of stannylated phosphines, see:Wächtler E.; Wahlicht S.; Privér S. H.; Bennett M. A.; Gerke B.; Pöttgen R.; Brendler E.; Gericke R.; Wagler J.; Bhargava S. K. Tin(IV) compounds with 2-C6F4PPh2 substituents and their reactivity toward palladium(0): Formation of tin palladium complexes via oxidative addition. Inorg. Chem. 2017, 56, 5316–5327. 10.1021/acs.inorgchem.7b00410. [DOI] [PubMed] [Google Scholar]; b Krebs K. M.; Freitag S.; Maudrich J. J.; Schubert H.; Sirsch P.; Wesemann L. Coordination chemistry of stannylene-based Lewis pairs - insertion into M-Cl and M-C bonds. From base stabilized stannylenes to bidentate ligands. Dalton Trans. 2018, 47, 83–95. 10.1039/C7DT04044J. [DOI] [PubMed] [Google Scholar]; c Salazar-Díaz J. J.; Muñoz-Hernández M. A.; Rufino-Felipe E.; Flores-Alamo M.; Ramirez-Solís A.; Montiel-Palma V. Bi- and tridentate stannylphosphines and their coordination to low-valent platinum. Dalton Trans. 2019, 48, 15896–15905. 10.1039/C9DT03317C. [DOI] [PubMed] [Google Scholar]
- The low frequency region of the 1H NMR spectrum, together with 31P{1H} NMR data, revealed that 6 was present alongside ca. 5% of a minor species, which we assign as 6’ with the proposed structure shown in Figure S4 on the basis of the 31P chemical shifts and coupling constants.
- T1 = 340 ms in C6D6 (400 MHz, 298 K).
- Garrou P. E. ΔR Ring contributions to 31P NMR parameters of transition-metal-phosphorus chelate complexes. Chem. Rev. 1981, 81, 229–266. 10.1021/cr00043a002. [DOI] [Google Scholar]
- a For downfield shifts in P,Si and P,Sn derived phosphines, see:MacInnis M. C.; MacLean D. F.; Lundgren R. J.; McDonald R.; Turculet L. Synthesis and reactivity of platinum group metal complexes featuring the new pincer-like bis(phosphino)silyl ligand [κ3-(2-Ph2PC6H4)2SiMe]− ([PSiP]): Application in the ruthenium-mediated transfer hydrogenation of ketones. Organometallics 2007, 26, 6522–6525. 10.1021/om7009528. [DOI] [Google Scholar]; b Kameo H.; Ishii S.; Nakazawa H. Facile synthesis of rhodium and iridium complexes bearing a [PEP]-type ligand (E = Ge or Sn) via E-C bond cleavage. Dalton Trans. 2012, 41, 11386–11392. 10.1039/c2dt30996c. [DOI] [PubMed] [Google Scholar]
- a Garrou P. E. Ring contributions to phosphorus-31 chemical shifts of transition metal-phosphorus chelate complexes. Inorg. Chem. 1975, 14, 1435–1439. 10.1021/ic50148a055. [DOI] [Google Scholar]; b Mohr F.; Privér S. H.; Bhargava S. K.; Bennett M. A. Ortho-metallated transition metal complexes derived from tertiary phosphine and arsine ligands. Coord. Chem. Rev. 2006, 250, 1851–1888. 10.1016/j.ccr.2005.10.003. [DOI] [Google Scholar]
- As detailed in the Experimental Section, full conversion to 5 was achieved by heating 1 in the presence of excess AlMe2Cl, which was used to ensure there was no interference from the released LiCl.
- Wada H.; Tobita H.; Ogino H. Intramolecular aromatic C-H bond activation by a silylene ligand in a methoxy-bridged bis(silylene)-ruthenium complex. Organometallics 1997, 16, 3870–3872. 10.1021/om970408d. [DOI] [Google Scholar]
- DFT calculations (BP86-D3BJ(C6H6)/BS2//BP86/BS1) showed 7 to be exergonic by 20.0 kcal/mol with respect to 5.
- Samouei H.; Miloserdov F. M.; Escudero-Adán E. C.; Grushin V. V. Solid-state structure and solution reactivity of [(Ph3P)4Ru(H)2] and related Ru(II) complexes used in catalysis: A reinvestigation. Organometallics 2014, 33, 7279–7283. 10.1021/om5010572. [DOI] [Google Scholar]
- T1 values of 243–299 ms were measured at room temperature (500 MHz, 298 K) consistent with the presence of terminal hydrides.
- Other [TM(H)xAl] systems have been reported which instead show only a single hydride resonance due to high fluxionality.; a Lau S.; White A. J. P.; Casely I. J.; Crimmin M. R. Tunable binding of dinitrogen to a series of heterobimetallic hydride complexes. Organometallics 2018, 37, 4521–4526. 10.1021/acs.organomet.8b00340. [DOI] [Google Scholar]; b Escomel L.; Del Rosal I.; Maron L.; Jeanneau E.; Veyre L.; Thieuleux C.; Camp C. Strongly polarized irdiumδ−-aluminiumδ+ pairs: Unconventional reativity patterns including CO2 cooperative reductive cleavage. J. Am. Chem. Soc. 2021, 143, 4844–4856. 10.1021/jacs.1c01725. [DOI] [PubMed] [Google Scholar]; c Escomel L.; Soulé N.; Robin E.; Del Rosal I.; Maron L.; Jeanneau E.; Thieuleux C.; Camp C. Rational preparation of well-defined multinuclear iridium-aluminium polyhydride clusters and comparative reactivity. Inorg. Chem. 2022, 61, 5715–5730. 10.1021/acs.inorgchem.1c03120. [DOI] [PubMed] [Google Scholar]
- Lee D. H.; Suzuki H.; Moro-oka Y. Preparation of novel aluminohydride complexes of ruthenium(II). J. Organomet. Chem. 1987, 330, C20–C22. 10.1016/S0022-328X(00)99060-0. [DOI] [Google Scholar]
- It is worth comparing these data to those of [Ru(PCy3)2(H)2(Mes-BDIAlH2)] (Mes-BDIAlH2 = {((2,4,6-Me3C6H2)NCMe)2CH}AlH2) which shows four different hydride resonances at low temperature, but an absence of coupling from any of these to phosphorus.Hooper T. N.; Lau S.; Chen W. Y.; Brown R. K.; Garçon M.; Luong K.; Barrow N. S.; Tatton A. S.; Sackman G. A.; Richardson C.; White A. J. P.; Cooper R. I.; Edwards A. J.; Casely I. J.; Crimmin M. R. The partial dehydrogenation of aluminium dihydrides. Chem. Sci. 2019, 10, 8083–8093. 10.1039/C9SC02750E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An further, interesting comparison to 7 is provided by [Ru(P2P3tBu)(AlH4)H] (P2P3tBu = P(CH2CH2PtBu2)3) which exhibits sharp, JHP/JHH coupled resonances for the terminal Ru-H and one of the bridging Ru-H-Al signals, but a broad, unresolved signal for the second Ru-H-Al.Gilbert-Wilson R.; Field L. D.; Bhadbhade M. Ruthenium hydrides containing the superhindered polydentate polyphosphine ligand P(CH2CH2PtBu2)3. Inorg. Chem. 2014, 53, 12469–12479. 10.1021/ic501895s. [DOI] [PubMed] [Google Scholar]
- The IR spectra (Figures S28−S30) showed two absorption bands at 1967 and 1750 cm–1, both of which were assigned to νRu-H on the grounds that they disappeared when H2 was replaced by D2.
- a Schubert U.; Gilbert S.; Mock S. Transition-metal silyl complexes. 43. Preparation and fluxionality of the complexes FeH3(PPh2R’)3ER3 (E = Si, Sn; R’ = Et, nBu). Chem. Ber. 1992, 125, 835–837. 10.1002/cber.19921250413. [DOI] [Google Scholar]; b Khaleel A.; Klabunde K. J.; Johnson A. Photochemical synthesis of new (η6-arene)Cr-hydrido stannyl and (η6-arene)Cr-bis-stannyl complexes. Ligand effects on the Sn-H interaction in the hydrido stannyl compounds. J. Organomet. Chem. 1999, 572, 11–20. 10.1016/S0022-328X(98)00917-6. [DOI] [Google Scholar]; c Rummelt S. M.; Radkowski K.; Roşca D.-A.; Fürstner A. Interligand interactions dictate the regioselectivity of trans-hydrometalations and related reactions catalyzed by Cp*RuCl. Hydrogen bonding to a chloride ligand as a steering principle in catalysis. J. Am. Chem. Soc. 2015, 137, 5506–5519. 10.1021/jacs.5b01475. [DOI] [PubMed] [Google Scholar]
- A much smaller splitting is observed upon oxidative addition of a Sn-H bond. See, for example:Buil M. L.; Espinet P.; Esteruelas M. A.; Lahoz F. J.; Lledós A.; Martínez-Ilarduya J. M.; Maseras F.; Modrego J.; Oñate E.; Oro L. A.; Sola E.; Valero C. Oxidative addition of group 14 element hydrido compounds to OsH2(η2-CH2=CHEt)(CO)(PiPr3)2: Synthesis and characterization of the first trihydrido-silyl, trihydrido-germyl, and trihydrido-stannyl derivatives of osmium(IV). Inorg. Chem. 1996, 35, 1250–1256. 10.1021/ic9509591. [DOI] [PubMed] [Google Scholar]
- Pregosin P. S.NMR in Organometallic Chemistry; Wiley-VCH: Weinheim, 2012. [Google Scholar]
- We found that 8 could be formed upon stirring a microcrystalline sample of 5 under H2 (ESI) in a solid-state reaction, although efforts to generate X-ray quality crystals of the former through a single crystal to single crystal transformation of 5 failed as a result of loss of crystallinity from the sample over the course of the reaction. This is most likely due to the significant changes that take place in the coordination geometry around the Ru center. In an alternative approach, we set out to prepare a more soluble derivative of 8. However, although substitution of pyridine into 6 gave [Ru(NC5H5)(C6H4PPh2)(PPh2C6H4SnMe2)], treatment of a crude sample of this material (i.e., one still retaining the eliminated PPh3) with H2 resulted in recoordination of PPh3 and formation of 8, which then precipitated from solution.
- a Gilbert S.; Knorr M.; Mock S.; Schubert U. Transition-metal silyl complexes. 50. Synthesis, structure and reactivity of trihydrosilyl and trihydrostannyl complexes L3FeH3(ER3) (E = Si, Sn). J. Organomet. Chem. 1994, 480, 241–254. 10.1016/0022-328X(94)87125-6. [DOI] [Google Scholar]; b Hübler K.; Hübler U.; Roper W. R.; Schwerdtfeger P.; Wright L. J. The nature of the metal-silicon bond in M(SiR3)H3(PPh3)3 (M = Ru, Os) and the crystal structure of Os{Si(N-pyrrolyl)3}H3(PPh3)3. Chem.—Eur. J. 1997, 3, 1608–1616. 10.1002/chem.19970031010. [DOI] [Google Scholar]; c Dioumaev V. K.; Procopio L. J.; Carroll P. J.; Berry D. H. Synthesis and reactivity of silyl ruthenium complexes: The importance of trans effects in C-H activation, Si-C bond formation, and dehydrogenative coupling of silanes. J. Am. Chem. Soc. 2003, 125, 8043–8058. 10.1021/ja035131p. [DOI] [PubMed] [Google Scholar]
- a Klein H. F.; Lemke U.; Lemke M.; Brand A. 2-diphenylphosphinobenzaldehyde as chelating ligand in trimethylphosphine complexes of cobalt and nickel. Organometallics 1998, 17, 4196–4201. 10.1021/om980462+. [DOI] [Google Scholar]; b Ciganda R.; Garralda M. A.; Ibarlucea L.; Mendicute C.; Pinilla E.; Torres M. R. Dehydrogenation of hydridoirida-β-diketones in methanol: The selective formation of mono- and dinuclear acyl complexes. Eur. J. Inorg. Chem. 2010, 2010, 3167–3173. 10.1002/ejic.201000250. [DOI] [Google Scholar]; c Benhamou L.; César V.; Lugan N.; Lavigne G. Snapshot of a chelation-assisted C-H/alkyne coupling: A ruthenium complex caught in the act of C-C bond formation. Organometallics 2007, 26, 4673–4676. 10.1021/om7005003. [DOI] [Google Scholar]
- a McKinney R. J.; Hoxmeier R.; Kaesz H. D. Intramolecular metalation with methylmanganese and methylrhenium carbonyl complexes. IV. Primary metalation products derived from the thermolysis of tetracarbonylmethyltriphenylphosphinemanganese and related derivatives. J. Am. Chem. Soc. 1975, 97, 3059–3064. 10.1021/ja00844a023. [DOI] [Google Scholar]; b Bennett M. A.; Clark A. M.; Contel M.; Rickard C. E. F.; Roper W. R.; Wright L. J. Cyclometallated complexes of ruthenium and osmium containing the o-C6H4PPh2 ligand. J. Organomet. Chem. 2000, 601, 299–304. 10.1016/S0022-328X(00)00086-3. [DOI] [Google Scholar]
- Miloserdov F. M.; Isaac C. J.; Beck M. L.; Burnage A. L.; Farmer J. C. B.; Macgregor S. A.; Mahon M. F.; Whittlesey M. K. Impact of the novel Z-acceptor ligand bis{(ortho-diphenylphosphino)phenyl}zinc (ZnPhos) on the formation and reactivity of low-coordinate Ru(0) centers. Inorg. Chem. 2020, 59, 15606–15619. 10.1021/acs.inorgchem.0c01683. [DOI] [PubMed] [Google Scholar]; See also:; Fukuda K.; Harada T.; Iwasawa N.; Takaya J. Facile synthesis and utilization of bis(o-phosphinophenyl)zinc as isolable PZnP-pincer ligands enabled by boron-zinc double transmetallation. Dalton Trans. 2022, 51, 7035–7039. 10.1039/D2DT01222G. [DOI] [PubMed] [Google Scholar]
- For a recent review of C–H activation reactions (as well as other bond cleavage processes) in NHC ligands, see:Nicholls T. P.; Williams J. R.; Willans C. E. Reactivities of N-heterocyclic carbenes at metal centers. Adv. Organomet. Chem. 2021, 75, 245–329. 10.1016/bs.adomc.2021.01.006. [DOI] [Google Scholar]
- a Ferrence G. M.; Arduengo A. J. III; Jockisch A.; Kim H.-J.; McDonald R.; Takats J. Reaction of tetramethylimidazol-2-ylidene with (TptBu,Me)YbE(thf) (E = I, CH2SiMe3): Simple adduct and a hydrocarbyl tethered carbene ligand. J. Alloys Compd. 2006, 418, 184–188. 10.1016/j.jallcom.2005.09.091. [DOI] [Google Scholar]; b Esteruelas M. A.; Gay M. P.; Oñate E. Conceptual extension of the degradation-transformation of N-heterocyclic carbenes: Unusual rearrangements on osmium. Organometallics 2018, 37, 3412–3424. 10.1021/acs.organomet.8b00110. [DOI] [Google Scholar]; c During the preparation of this manuscript, the Braunschweig group reported C–H activation of the N–Me group in IMe4 in a di-aluminium system.Dhara D.; Fantuzzi F.; Härterich M.; Dewhurst R. D.; Krummenacher I.; Arrowsmith M.; Pranckevicius C.; Braunschweig H. Stepwise reduction of a base-stabilised ferrocenyl aluminium(III) dihalide for the synthesis of a structurally-diverse dialane species. Chem. Sci. 2022, 13, 9693–9700. 10.1039/D2SC02783F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Salem H.; Schmitt M.; Herrlich U.; Kühnel E.; Brill M.; Nägele P.; Bogado A. L.; Rominger F.; Hofmann P. Bulky N-phosphinomethyl-functionalized N-heterocyclic carbene chelate ligands: Synthesis, molecular geometry, electronic structure, and their ruthenium alkylidene complexes. Organometallics 2013, 32, 29–46. 10.1021/om300487r. [DOI] [Google Scholar]; b Tan G. W.; Enthaler S.; Inoue S.; Blom B.; Driess M. Synthesis of mixed silylene-carbene chelate ligands from N-heterocyclic silylcarbenes mediated by nickel. Angew. Chem., Int. Ed. 2015, 54, 2214–2218. 10.1002/anie.201409739. [DOI] [PubMed] [Google Scholar]; c Rull S. G.; Rama R. J.; Álvarez E.; Fructos M. R.; Belderrain T. R.; Nicasio M. C. Phosphine-functionalized NHC Ni(II) and Ni(0) complexes: Synthesis, characterization and catalytic properties. Dalton Trans. 2017, 46, 7603–7611. 10.1039/C7DT01805C. [DOI] [PubMed] [Google Scholar]
- For a recent example of the latter, see:Cybulski M. K.; Beattie N. A.; Macgregor S. A.; Mahon M. F.; Whittlesey M. K. Unexpected vulnerability of DPEphos to C-O activation in the presence of nucleophilic metal hydrides. Chem.—Eur. J. 2020, 26, 11141–11145. 10.1002/chem.202001685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An inverse process involving the formal addition of a Sn-NHC bond at ML2 centres (M = Ni, Pd, Pt; L = PR3, COD) has been reported.Sindlinger C. P.; Weiß S.; Schubert H.; Wesemann L. Nickel-triad complexes of a side-on coordinating distannene. Angew. Chem., Int. Ed. 2015, 54, 4087–4091. 10.1002/anie.201411025. [DOI] [PubMed] [Google Scholar]
- Handford R. C.; Nesbit M. A.; Smith P. W.; Britt R. D.; Tilley T. D. Versatile Fe-Sn bonding interactions in a metallostannylene system: Multiple bonding and C-H bond activation. J. Am. Chem. Soc. 2022, 144, 358–367. 10.1021/jacs.1c10144. [DOI] [PubMed] [Google Scholar]
- Schunn R. A.; Wonchoba E. R.; Wilkinson G. Chlorohydridotris(triphenylphosphine)ruthenium(II). Inorg. Synth. 1972, 13, 131–134. 10.1039/J19680003143. [DOI] [Google Scholar]
- Ansell M. B.; Roberts D. E.; Cloke F. G. N.; Navarro O.; Spencer J. Synthesis of an (NHC)2Pd(SiMe3)2 complex and catalytic cis-bis(silyl)ations of alkynes with unactivated disilanes. Angew. Chem., Int. Ed. 2015, 54, 5578–5582. 10.1002/anie.201501764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- During our initial studies to prepare 5, 31P NMR spectroscopy showed the formation of signals consistent with unwanted bis-phosphine products, together with a resonance for free PPh3. Thus, additional, free phosphine was added to to a solution of 1 prior to treatment with AlMe2Cl to enhance the synthesis of 5.
- The broadness of the peaks, together with the low intensity, prevented more accurate values of J from being determined.
- Dolomanov O. V.; Bourhis L. J.; Gildea R. J.; Howard J. A. K.; Puschmann H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. 10.1107/S0021889808042726. [DOI] [Google Scholar]
- Sheldrick G. M. SHELXT - Integrated space-group and crystal structure determination. Acta Cryst., Sect. A: Found. Adv. 2015, A71, 3–8. 10.1107/S2053273314026370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldrick G. M. Crystal structure refinement with SHELXL. Acta Cryst., Sect. C: Struct. Chem. 2015, C71, 3–8. 10.1107/S2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Petersson G. A.; Nakatsuji H.; Li X.; Caricato M.; Marenich A. V.; Bloino J.; Janesko B. G.; Gomperts R.; Mennucci B.; Hratchian H. P.; Ortiz J. V.; Izmaylov A. F.; Sonnenberg J. L.; Williams-Young D.; Ding F.; Lipparini F.; Egidi F.; Goings J.; Peng B.; Petrone A.; Henderson T.; Ranasinghe D.; Zakrzewski V. G.; Gao J.; Rega N.; Zheng G.; Liang W.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Throssell K.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M. J.; Heyd J. J.; Brothers E. N.; Kudin K. N.; Staroverov V. N.; Keith T. A.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A. P.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Millam J. M.; Klene M.; Adamo C.; Cammi R.; Ochterski J. W.; Martin R. L.; Morokuma K.; Farkas O.; Foresman J. B.; Fox D. J.. Gaussian 16, Rev. C.01; Gaussian, Inc.; Wallingford, CT, 2016.
- Andrae D.; Häußermann U.; Dolg M.; Stoll H.; Preuß H. Energy-adjusted ab initio pseudopotentials for the second and third row transition elements. Theor. Chim. Acta 1990, 77, 123–141. 10.1007/BF01114537. [DOI] [Google Scholar]
- a Hehre W. J.; Ditchfield R.; Pople J. A. Self-consistent molecular orbital methods. XII. Further extensions of Gaussian-type basis sets for use in molecular orbital studies of organic molecules. J. Chem. Phys. 1972, 56, 2257–2261. 10.1063/1.1677527. [DOI] [Google Scholar]; b Hariharan P. C.; Pople J. A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 1973, 28, 213–222. 10.1007/BF00533485. [DOI] [Google Scholar]
- Höllwarth A.; Böhme M.; Dapprich S.; Ehlers A. W.; Gobbi A.; Jonas V.; Köhler K. F.; Stegmann R.; Veldkamp A.; Frenking G. A set of d-polarization functions for pseudo-potential basis sets of the main group elements Al-Bi and f-type polarization functions for Zn, Cd, Hg. Chem. Phys. Lett. 1993, 208, 237–240. 10.1016/0009-2614(93)89068-S. [DOI] [Google Scholar]
- a Becke A. D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. 10.1103/PhysRevA.38.3098. [DOI] [PubMed] [Google Scholar]; b Perdew J. P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822–8824. 10.1103/PhysRevB.33.8822. [DOI] [PubMed] [Google Scholar]
- Tomasi J.; Mennucci B.; Cammi R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3094. 10.1021/cr9904009. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Ehrlich S.; Goerigk L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. 10.1002/jcc.21759. [DOI] [PubMed] [Google Scholar]
- Glendening E. D.; Reed A. E.; Carpenter J. E.; Weinhold F.. NBO 3.1; Theoretical Chemistry Institute, University of Wisconsin: Madison, 1990.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.