

Abstract
Growing E. coli to high densities is a common strategy for biologicals production. The process is implemented by using complex or minimal media with different feeding strategies. To understand the effect of amino acids, E. coli B and K were grown at a steady state of 0.35 h−1 in glucose minimal medium with and without amino acids, while their metabolism, protein abundance and gene expression were compared. The results showed that amino acids promoted higher acetate excretion, higher fatty acid biosynthesis (K strain), repressed glucose uptake rate, and decreased expression of proteins associated with the TCA cycle, glyoxylate shunt and amino acid biosynthesis. In presence of amino acids, E. coli K upregulated fatty acid biosynthesis and repressed more genes and proteins involved in amino acid biosynthesis than E. coli B. These findings are correlated with higher yield on glucose (Yx/s) and high specific biomass production rate (qx) in K strain in the presence of amino acids. In contrast, pre-formed precursor molecules such as amino acids did not affect fatty acid biosynthesis in E. coli B or Yx/s and qx, which were higher than those of E. coli K, suggesting that constitutive synthesis of energetically demanding precursors and higher fatty acid β-oxidation activity is key for high biomass-performer E. coli B. Both strains turned off unnecessary pathways and directed their metabolism to proteome efficient overflow metabolism likely to generate energy and provide protein to functions supporting higher growth rate.
Keywords: Proteome, Transcriptome, Overflow metabolism, Escherichia coli, Chemostat
Introduction
Growing Escherichia coli (E. coli) to high density is the preferred method for obtaining large quantities of recombinant proteins [1–3]. Since high initial glucose concentration affects bacterial growth [2,4–6], the traditional way to obtain high cell concentration is by providing glucose to the growing culture using different feeding strategies that improve glucose assimilation and reduce acetate excretion. The development of growth strategies has been accompanied by the study of central carbon metabolism, especially acetate accumulation which was found to inhibit growth and protein expression [7–11]. These studies, based on gene transcription, have shown that high glucose concentration tends to overload the central carbon metabolism and direct the accumulated acetyl-CoA towards acetate excretion [12–14]. The studies also showed that E. coli K (JM109) unlike E. coli B (BL21), when grown with high-glucose in complex medium, is less efficient in its central carbon metabolism as a result of an apparently inoperative glyoxylate shunt and slower flux through the tricarboxylic acid (TCA) cycle [14].
In addition to the glucose feeding strategy, media composition is an important consideration for growth and production. Two main types of media are routinely being used: “complex medium”, which contains yeast extract and/or tryptone, and “minimal medium”, which contains glucose, various salts and vitamins. E. coli K (MG1655) grew faster in complex medium supplemented with glucose, and repressed genes encoding for amino acid biosynthesis or ammonia assimilation [15]. When grown in minimal medium, E. coli K (MG1655) overexpressed the glyoxylate shunt-associated genes, such as aceA and aceB, compared with its expression in the complex medium [15]. E. coli B (BL21) also grew faster and excreted a higher amount of acetate in rich medium due to an unbalanced amount of central metabolic pathway enzymes [16]. Previous studies of bacterial metabolism were based on gene transcription or proteome analysis in exponential growth [15,16]; however, independent transcriptomic or proteomic analysis provides limited information for comprehensive understanding of the bacterial response to nutrients and environmental factors. Therefore, an integrated transcriptome and proteome analysis [17] and multi-omics analyses [18] of E. coli B and K were performed to obtain better insights into the bacterial metabolism. The observed differences in E. coli B can be related to enhanced replication, translation and amino acid biosynthesis, and reduced proteome degradation and flagellar motility, whereas high capabilities in E. coli K were related to cell motility, carbohydrate transport and increased tolerance to stress conditions [18].
The reported comparison of E. coli metabolism in the different nutrient environments was performed in batch cultures at non-steady state conditions. Under these conditions, E. coli grows faster on rich medium compared with minimal medium, since ribosomal translation rate and other properties of cell physiology vary with growth rate [19,20]. Thus, integrated proteome and transcriptome analysis of different E. coli strains growing at steady state should provide more accurate information. This approach is described here. E. coli K and B were grown in minimal glucose medium with and without amino acids, at a steady state and their proteomes and transcriptomes were analyzed. The results provide new perspectives and explanations for the metabolism of these two widely used E. coli strains grown to high density in a high glucose concentration, which is important for rational upstream bio-process design.
Material and methods
Bacterial strains, inoculum preparation and culture media
Escherichia coli BL21(λDE3) (F-, ompT, hsdSB (rB-, mB+), dcm, gal, (DE3), Cmr) and JM109(DE3) (endA1, recA1, gyrA96, thi, hsdR17 (rk−,mk+), relA1, supE44, Δλ−, Δ(lac-proAB), [F’, traD36, proAB, lacIqZΔM15], λDE3) were grown in continuous cultures in defined medium with the following composition: KH2PO4, 13 g/L; (NH4)2HPO4, 4 g/L; citric acid, 1.5 g/L and, where indicated, 20 g/L of casamino acids was added. The pH value was adjusted to 7.0 with 5 M NaOH prior to sterilization; after sterilization the medium was aseptically supplemented with 1 mL/L trace metal solution [21], 5 mM MgSO4, 4.5 mg/L thiamine-HCl, and 40 g/L glucose. Frozen stocks were used to inoculate overnight precultures grown at 37 °C in 100 mL of defined medium with 5 g/L of glucose.
Bacterial growth
Continuous cultures were performed at a dilution rate of 0.35 h−1 in 1.5 L laboratory fermenters (New Brunswick) at a stirrer speed of 650 rpm. Temperature was maintained at 37 °C and pH at 7 by addition of 2 M NaOH. Dissolved oxygen (dO2) concentration was measured with an oxygen electrode (Mettler Toledo, Columbus, OH) and was controlled above 20% of air saturation, as shown in Fig. 1. The working volume of the cultures was kept at 1.0 L by two peristaltic pumps, one for effluent and other for feeding. After 5 residence times, when steady state was confirmed, fermentation samples were taken for RNA isolation and determination of dry weight, metabolites and protein amounts. After withdrawal from the fermenter, samples for glucose and acetic acid assays were immediately filtered (0.2 μm pore size filter, Millipore) and kept at −20 °C until analysis. Fermentation samples for RNA analysis and protein were centrifuged at 20,800 × g for 5 min at 4 °C and the pellet was quickly frozen and stored at −80 °C. The bioreactor off gas (oxygen and carbon dioxide) composition was analyzed on-line using the BlueInOne Ferm (BlueSens, Herten, Germany) gas analyzer. The O2 transfer and CO2 production rates were calculated using an oxygen and CO2 mass balance between the inlet and outlet gas composition data, as specified in the following equations.
Fig. 1.
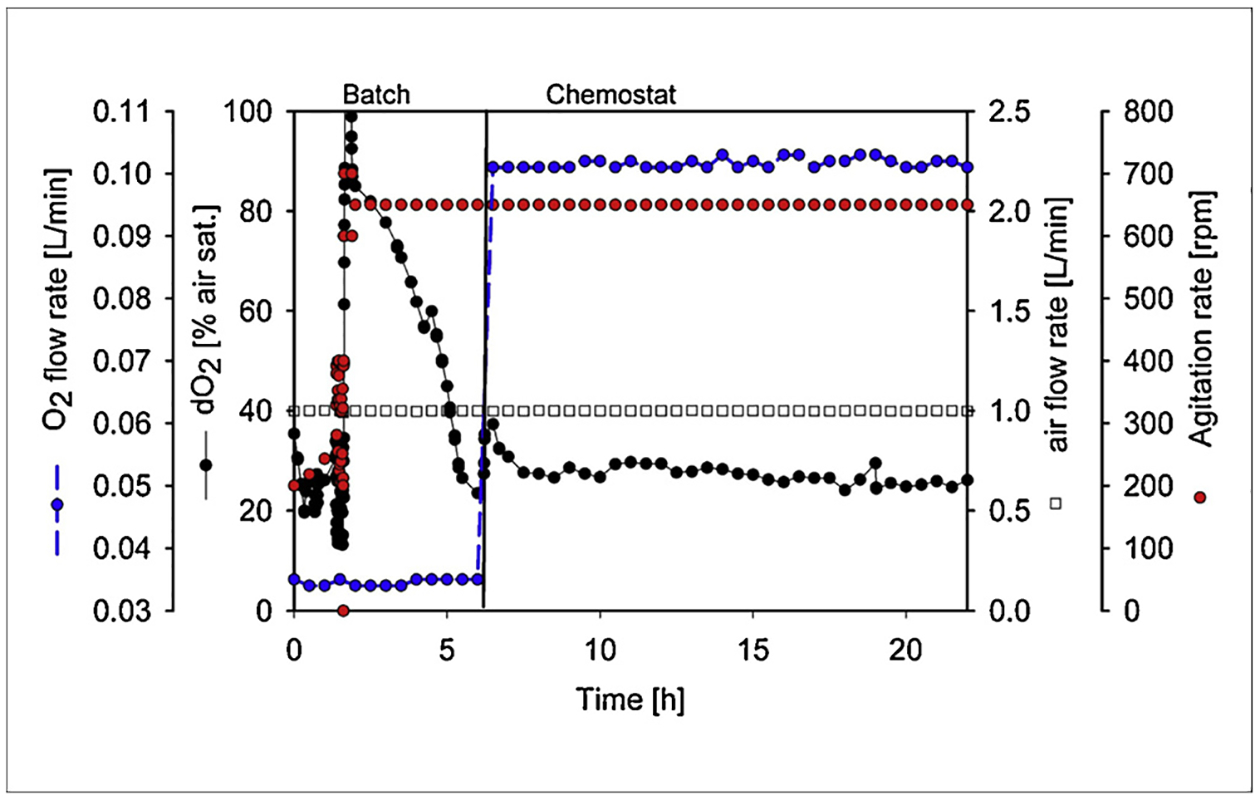
Graph showing dO2 level, oxygen flow rate, air flow rate, and agitation rate at steady-state conditions.
Analytical methods
Cell growth was followed by measuring culture Optical Density (OD) at 600 nm (Ultrospec 3000 UV/Visible spectrophotometer, Pharmacia Biotec); measurements were converted to dry cell weight per L using a calibration curve (1 optical density at 600 nm = 0.347 and 0.409 gDCW/L for BL21 with and without casamino acids; 1 OD600 nm = 0.373 and 0.403 gDCW/L for JM109 with and without casamino acids). Glucose concentration was determined by YSI 2700 Biochemistry Analyzer (YSI Instruments, Yellow Springs, OH). Acetic acid concentration was determined using the R-Biopharm AG kit (Cat. No.10 148 261 035); the determination is based on the formation of NADH measured by the increase in light absorbance at 340 nm.
Total RNA preparation and microarrays
Total RNA was isolated from the cell pellets in triplicate for each strain and each medium condition using the hot phenol method. The method requires cell pellets to be resuspended in 0.5% SDS, 20 mMNaAc and 10 mM EDTA and extracted twice with a hot acid phenol: chloroform mixture. This was followed by two extractions with phenol:chloroform isoamyl alcohol mixture. Subsequently, absolute ethanol was added to the extract, which was kept at −80 °C for 15 min. After centrifugation at 14,000 g for 15 min, the pellets were washed in 70% ethanol, resulting in purified RNA pellets. These were air dried and the RNA was resuspended in ultra-pure water (KD medical USA). The purified total RNA was then subjected to enrichment of mRNA using MICROBExpress bacterial mRNA enrichment kit (Ambion), as per manufacturer’s instructions. After this step, RNA concentration and quality were assayed using NanoDrop 2000 Spectrophotometer and Agilent 2100 Bioanalyzer as described in Kumar et al. [22]. The cDNA and GeneChip E. coli_2 array preparation and analysis were carried out as previously described [22].
Digestion, reductive methylation and anion exchange (AIX) fractionation
Three biological replicates of both the media conditions, i.e. minimal medium with and without casamino acids, were created. Serial on-column reductive methylation was adapted from Boersema et al. [23]. Pools from samples from minimal medium and from minimal medium with casamino acid were prepared by mixing equal parts of each of the samples (which had nearly the same overall cell density upon harvesting). These samples were processed by the FASP procedure [24] using the original Lys-C/Trypsin serial digestion. After harvesting peptides from the concentration device, each sample was placed on a C18 Stage tip with additional bed of POROS R2 resin calculated to have a loading capacity 5 times greater than needed for the combined protein mass of the two samples. The sample for labeling with normal methyl groups was applied and treated as described in [23], scaled down to use for serial loading onto C18 stage tips. Two serial reductive methylation treatments for each condition were used to ensure useful levels of incorporation, which was confirmed using internal controls. After both light and heavy reactions were performed, the column was washed extensively and was eluted onto SCX-Stage tip with an additional 30μL bed of Source S resin (GE). Once loaded, the resin was washed extensively and eluted first with 500 mM ammonium acetate, 20% acetonitrile in water (4 column volumes) and next with 200 mM ammonium acetate, 50% acetonitrile in water (4 column volumes). The sample was evaporated and resuspended twice in 50% acetonitrile to remove residual ammonium acetate in preparation for anion exchange (AIX) based fractionation. Three technical replicates for the dimethyl labeling were performed.
The AIX fractionation scheme described by Wisniewski et al. [25] was used except that larger bore AIX Stage tips were fashioned from the bottom simple cone portion of 1 ml sized Axygen Maxymum recovery pipette tips and only 2 Empore AIX disks were placed in the bottom of this tip and Source Q (GE) was applied to make a 50μL bed volume. Capture C18 stage tips were also additionally loaded with 20μL POROS R2 each and centrifugal forces were lowered to 1000xgwith loading at 400xg; in addition, a fourth AIX separation was performed in which each of the AIX separations was performed with 20% acetonitrile added, with pH eluted samples being nearly evaporated and resuspended in 1.6% formic acid prior to application to their respective capture tips. After capture the reversed phase stage tips were washed extensively and eluted first with 100μL of 1.6% formic acid 40% acetonitrile and then 100μL 1.6% formic acid 80% acetonitrile. The eluent was then evaporated under nitrogen and resuspended in 60μL of 1.6% formic acid.
Mass spec data collection
Samples were next submitted to the queue of an LC/MS/MS system comprising nanoscale HPLC (Waters nanoacquity in trapping configuration with 180micrometer × 20 mm Symmetry C18 trap column and 75 μm × 250 mm BEH130 C18 analytical column) interfaced to a hybrid mass spectrometer (Thermo Orbitrap Elite). Samples were applied in the trapping configuration at 10 μL/min for 10 min prior to the vent valve being closed and the analytical separation was performed at 0.15μL/min, starting at 1% acetonitrile with gradient inflections of 99% A at 5 min, 95@25, 65@160, 30@200, 40@205, 99 @210, with data collected for 240 min, with solution A being 0.1% formic acid in water and solution B being 0.1% formic acid in acetonitrile.
The mass spectrometer was configured for data dependent triggering with a parent scan with resolution setting 240 K (1e6 target, 200 ms max injection time) in the orbitrap analyzer followed by up to 20 parent ion fragmentations (2e4 target, 100 ms max injection time) in the ion trap part of the instrument. The chromatography feature was enabled with phase detection set at 30%. Charge state dependence excluded unknown and single charge states from fragmentation. The preview feature was enabled to improve sampling efficiency.
Microarray and proteomics data analysis
Microarray raw data was processed, normalized and annotated using Partek software (Partek. St. Louis, MO, USA). Principal Components Analysis was performed to determine the significant differences in the spatial distribution among the populations of different samples. The samples were divided into two groups: ‘without amino acids’ and ‘with amino acids’ for K and for B strains, resulting in four categories. The proteomics data were grouped in a similar manner. ANOVA was used to generate fold change values by comparing these two groups. This method involved estimating the geometric means of the samples in each group in order to calculate the fold change for the contrast between ‘without amino acids’ and ‘with amino acids’ of K and B strain.
A filtering criterion of > 1.5 and < −1.5-fold change was applied together with p-value < 0.05 to identify significant differentially expressed genes, both up and down-regulated. The KEGG database for the E. coli B strain (organism code ‘ebl’) and K strain (organism code ‘eco’) were integrated into the Partek Genomics Suite. Gene ontology was performed using Fisher’s exact test with the ‘Gene Set Analysis’ tool of Partek® Genomics Suite software, version 7.18 (Copyright ©; 2018 Partek Inc., St. Louis, MO, USA). Enrichment analysis was performed using Fisher’s exact test [26] with the Partek® Pathway™ tool in Partek Genomics Suite version 7.18. The pathways obtained after enrichment analysis were filtered with p-value < 0.05 and sorted based on their pathway enrichment scores. The top 20 pathways were selected from transcriptomic and proteomic enrichment, and common pathways were selected using Venn method (Supplementary File 2, S2 Table 7) in both E. coli B and K-strains. The selected pathways were analyzed with the ‘omics dashboard’ tool of EcoCyc (https://ecocyc.org/) to find the overall gene expression flow of subsystems in the enriched pathways, to predict the relative up or down-regulation of pathways [27].
Protein raw data was analyzed using MaxQuant version 1.3.0.5 [28] using a binary setting and requiring either light or heavy di-methylation of N-termini and lysine. The data was searched using protein sequences from E. coli as well as known contaminants and a list of control proteins used to monitor reaction efficiency. Default values were used except that match between runs was enabled with a 0.5 min allowable difference in elution time (which was applied after spline fitting the elution patterns).
Results
To evaluate the effect of addition of amino acids on growth characteristics, acetate production, transcription and translation in E. coli K and B, the bacteria were grown in minimal medium with and without casamino acids at steady state using chemostats. By implementing this methodology, any changes in the bacterial growth, metabolism and expression characteristics would be the result of the amino acids’ availability and not growth rate or growth phase. Bioreactor-operation parameters are seen in Fig. 1; the dissolved oxygen was kept above 20% air saturation and pH at 7.0 after 5 residence times, steady state conditions being confirming by constant biomass concentration.
Bacterial growth in defined media with and without casamino acids
Growth parameters of the E. coli B and K in chemostat culture using minimal media containing 45.3 g/L glucose with and without 20 g/L casamino acids at a dilution rate of 0.35 h−1 are shown in Table 1. Amino acids addition affected acetate yield and production rate in both strains. In E. coli K, amino acid additions increased the specific acetate production rate (qA) by 9.5-fold and glucose-based biomass yield (Yx/s) by 1.23-fold; at the same time, it decreased the specific glucose uptake rate (qs) and respiratory quotient (RQ) by 22% and 6%. A similar effect on specific acetate production rate, although somewhat lower, was observed in E. coli B, but no significant differences were observed on biomass yield, specific biomass production rate (qx) or glucose uptake rate (qs). The results confirmed that, compared with E. coli K, E. coli B is high performer; its biomass concentration and glucose-based biomass yield, with or without amino acids were higher. Both strains showed higher specific acetate formation rate (qA) and higher acetate yield per cell weight (YA/x) in medium containing amino acids.
Table 1.
Concentrations of biomass, residual glucose, and acetate in the bioreactor. Glucose uptake rate (qs), specific acetate production rate (qA), specific biomass production rate (qx), yield of glucose (Yx/s), acetate yield per gram of cells (YA/x) or per gram of glucose (YA/s) at steady-state growth of E. coli JM109 and BL21. Continuous cultures performed feeding medium containing 45.3 g/L glucose at a dilution rates of 0.35 h−1 with and without casamino acids. Only BL21 without CAA cultures were fed with 43.2 g/L of glucose.
| JM109 | BL21 | |||
|---|---|---|---|---|
| With AA | without | With AA | without | |
| Biomass conc. [g/L] | 9.26 ± 0.095 | 7.90 ± 0.148 | 16.15 ± 0.19 | 18.14 ± 0.403 |
| Glucose conc. [g/L] | 19.84 ± 0.295 | 19.23 ± 0.603 | 8.18 ± 0.27 | 0.14 ± 0.028 |
| Acetate conc. [g/L] | 7.58 ± 0.041 | 0.68 ± 0.129 | 4.47 ± 0.352 | 0.83 ± 0.134 |
| qs [g/g-h] | 0.951 ± 0.019 | 1.16 ± 0.011 | 0.806 ± 0.013 | 0.831 ± 0.018 |
| qA [g/g-h] | 0.286 ± 0.004 | 0.030 ± 0.006 | 0.097 ± 0.007 | 0.016 ± 0.003 |
| qx [g/g-h] | 0.128 ± 0.003 | 0.105 ± 0.001 | 0.152 ± 0.002 | 0.148 ± 0.003 |
| Yx/s [g/g] | 0.368 ± 0.007 | 0.30 ± 0.003 | 0.434 ± 0.007 | 0.42 ± 0.016 |
| YA/x [g/g] | 0.82 ± 0.011 | 0.086 ± 0.016 | 0.277 ± 0.021 | 0.046 ± 0.008 |
| YA/s [g/g] | 0.301 ± 0.002 | 0.026 ± 0.005 | 0.12 ± 0.009 | 0.019 ± 0.004 |
| Respiratory quotient | 0.935 ± 0.038 | 0.992 ± 0.005 | 1.012 ± 0.014 | 1.066 ± 0.021 |
| qO2 [mmol/g h] | 8.40 ± 0.23 | 11.55 ± 0.20 | 12.98 ± 0.080 | 9.92 ± 0.227 |
| qCO2 [mmol/g h] | 7.72 ± 0.21 | 11.45 ± 0.18 | 13.13 ± 0.16 | 10.57 ± 0.308 |
Effect of amino acids on proteomics and transcriptomics profile of E. coli B and K
Gene transcription and protein expression analyses of E. coli K and B grown in minimal medium without and with casamino acids at steady-state were performed by microarray and dimethyl-labeling based proteomics; the results are summarized by Venn diagram in Fig. 2. Microarray analysis in E. coli K grown without amino acids resulted in 330 upregulated genes (> 1.5-fold change) (p < 0.05) and 529 down-regulated genes (< −1.5-fold change), while in E. coli B, 1121 genes were upregulated (> 1.5-fold change) (p < 0.05) and 1373 were downregulated (< −1.5-fold change). Proteomics analysis in E. coli K grown without amino acids showed 532 high abundance proteins with 176 upregulated (> 1.5-fold change) and 356 downregulated (< −1.5-fold change). In E. coli B, the proteomics analysis identified 380 high abundance proteins with 242 upregulated (> 1.5-fold change) and 138 downregulated (< −1.5-fold change) in the culture medium without amino acids. In both strains growth without amino acids generated a higher number of downregulated genes compared with the number of upregulated genes. The list of up- and downregulated genes (filter fold change > 1.5 and < −1.5, p-value < 0.05) in K and B-strains is provided in Supplementary File 1. In E.coli K the ratio of the downregulated and upregulated genes was 1.6-fold, whereas at the protein level the ratio was more than 2-fold. In E. coli B the ratio between downregulated and upregulated genes was 1.2-fold, whereas at the protein level the ratio was 0.57-fold.
Fig. 2.
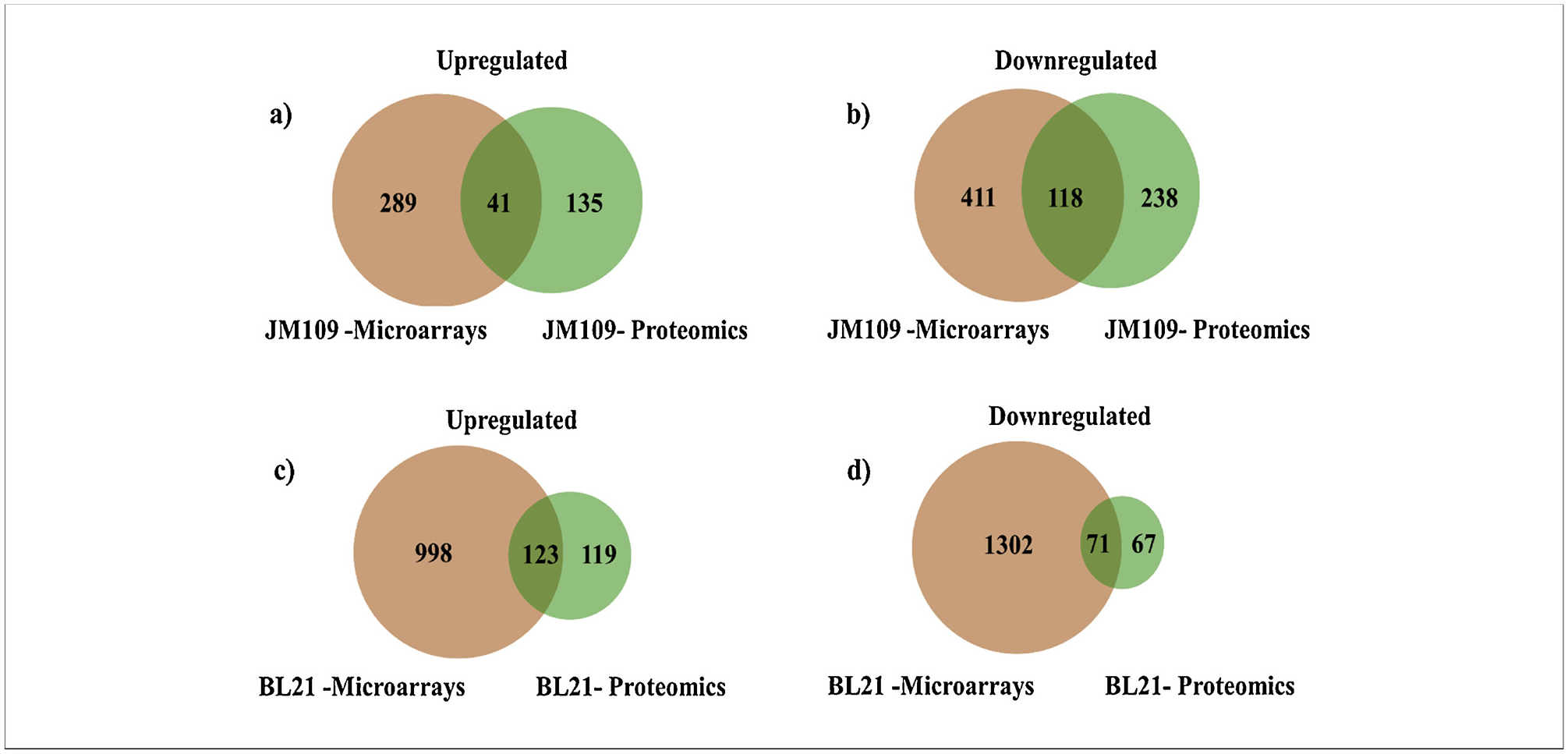
Venn diagrams showing comparison between transcriptomics and proteomics data. Analysis was performed from data obtained for bacteria grown in minimal medium without versus with casamino acids. (a) Upregulation in E. coli K; (b) downregulation in E. coli K; (c) upregulation in E. coli B; (d) downregulation in E. coli B.
Enrichment analysis of E. coli K and B at the two growth conditions using KEGG pathways
Enrichment analysis was performed to decipher the E. coli B and K omics data, in the two media compositions. The genes and proteins obtained from the ANOVA were filtered based on their fold change (> 1.5 and < −1.5) and p value 0.05. The genelist contained 859 from the K-strain and 2494 from the B-strain. The protein list contained 532 from the K-strain and 380 from the B-strain. This list containing the up and down-regulated entities was used for enrichment analysis based on Fisher’s exact test using the KEGG database in the Partek Genomics Suite. The pathways were ranked based on their enrichment scores. A p value cut-off of 0.05 identified 14 enriched pathways from B-strain (Supplementary File 2, S2 Table 1) and 13 enriched pathways from K-strain transcriptomics (Supplementary File 2, S2 Table 4). Similarly, 19 and 21 enriched pathways resulted from B and K-strain proteomics data respectively (Supplementary File 2, S2 Table 2 & 5). 7 and 8 common pathways were found among the top 20 enriched pathways of K-strain and B-strain respectively (Supplementary File 2, S2 Table 7). The relative distributions of the number of genes and proteins in the top common enriched pathways in B and K strain are shown in the Fig. 3.
Fig. 3.
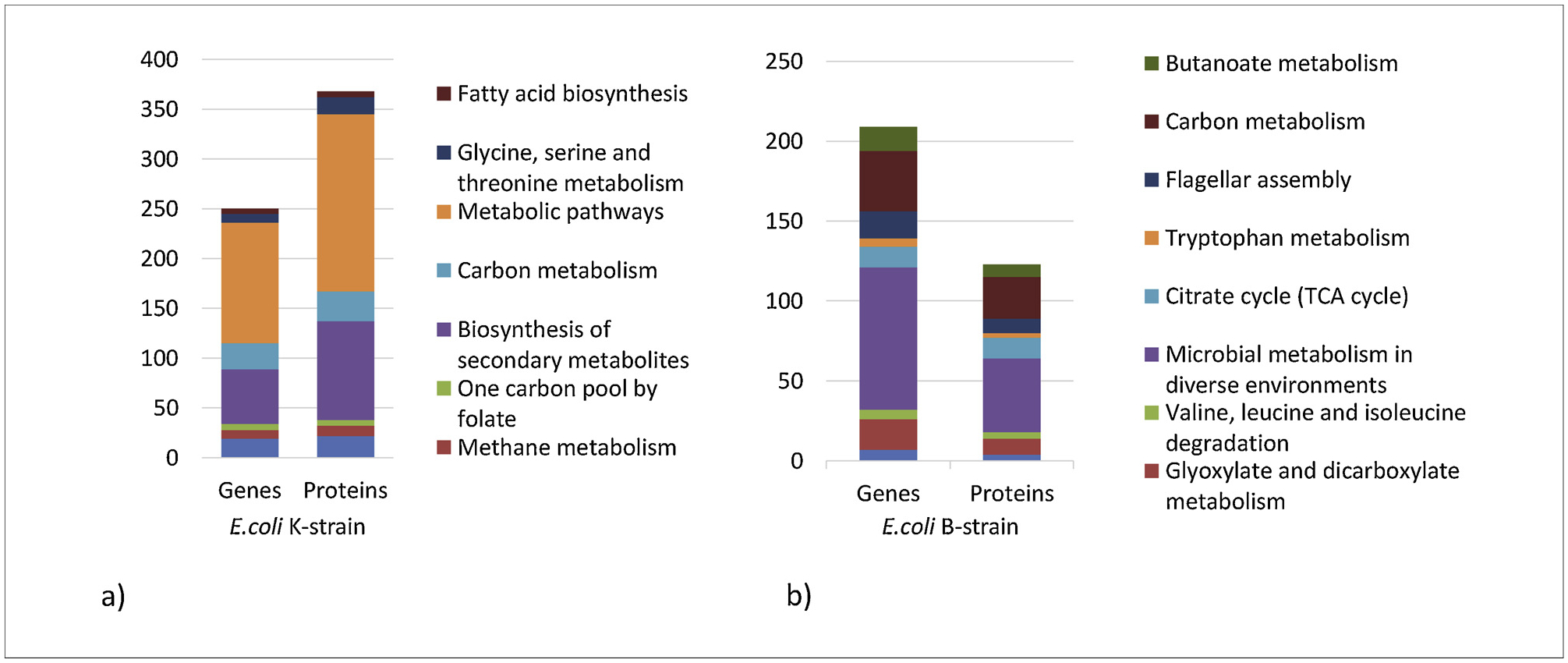
Change in the distribution of common pathways based on the number of genes and proteins involved, obtained from enrichment analysis of transcriptomics and proteomics study in (a) K-strain and (b) B-strain.
E. coli K analysis
Pathway enrichment was performed using Fisher’s exact test on the significant differentially expressed genes and proteins identified from using E. coli K grown in minimal medium without or with casamino acids. The results showed that genes and proteins associated with pyrimidine metabolism, methane metabolism, carbon metabolism, biosynthesis of secondary metabolites, glycine, serine and threonine metabolism and fatty acid biosynthesis were significantly enriched in medium without CAA (Fig. 3). From the top 20 pathways with the highest enrichment scores, 7 identified in both the transcriptomic and proteomic analysis are shown in Fig. 3(a). The up- and downregulation of major pathways were determined using overall gene expression average of subsystems in the omics dashboard, which showed that amino acids biosynthesis was up-regulated while fatty acid and lipid biosynthesis, nucleoside and nucleotide biosynthesis and secondary metabolite biosynthesis pathways were downregulated (Fig. 4). The energy generating pathways i.e. glycolysis and PPP, were down-regulated while TCA cycle was upregulated while growing without CAA.
Fig. 4.
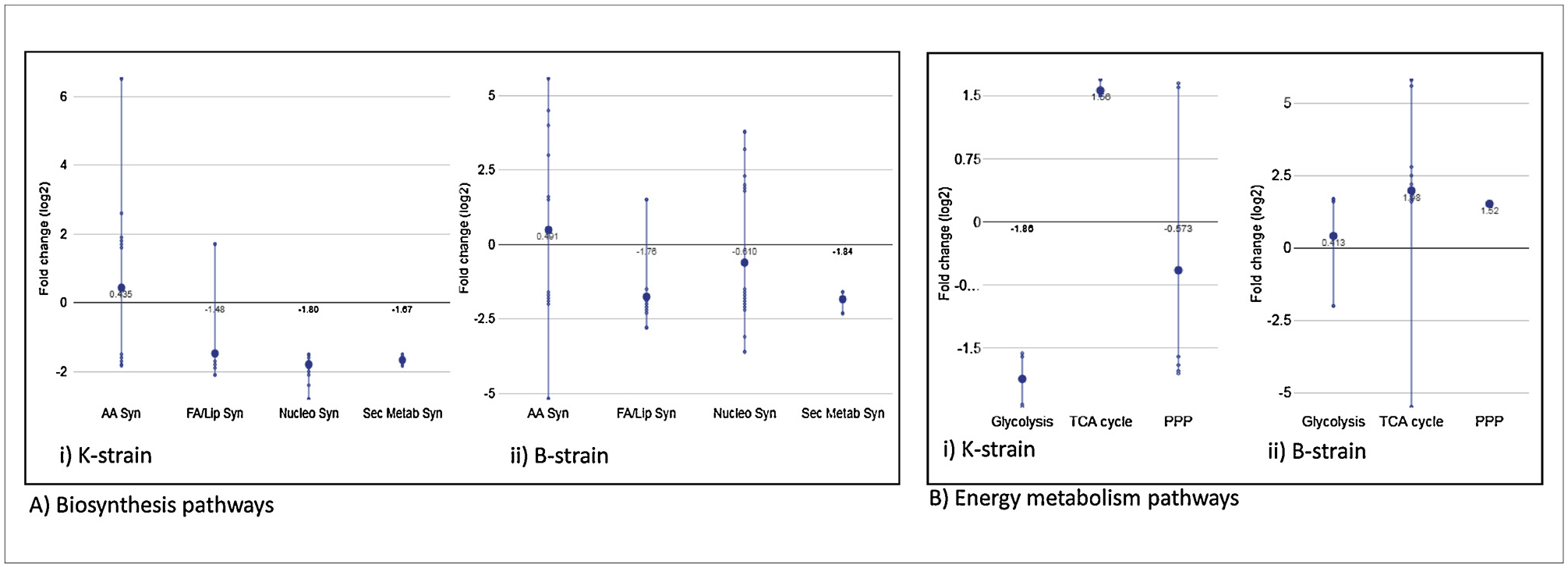
Gene expression distribution of: (A) biosynthesis pathways, i.e. amino acid biosynthesis, fatty acids and lipids biosynthesis, nucleosides and nucleotides biosynthesis and secondary metabolites biosynthesis; and (B) Energy metabolism pathways, i.e. glycolysis, TCA and PPP in (i) K-strain and (ii) B-strain. The small dots represent the fold change difference in expression between growth in medium without CAA and with CAA samples using a log2 scale. The large dot represents the average (mean) of small dots (using the EcoCyc omics dashboard).
For amino acid biosynthesis, the following pathways were enriched: a) alanine, aspartate, and glutamate; b) glycine, threonine, and aspartate; c) cysteine and methionine; d) lysine; e) arginine and proline; and f) tyrosine. P-values of the enriched pathways are shown in Supplementary File 2, S2 Tables 4 and 5. The acetate metabolism in the absence of casamino acids Pta and AckA were significantly down-regulated at both the transcriptomics and proteomics levels, Pta was downregulated 1.8-fold at the transcriptomics level and 1.9-fold at the proteomics level, and AckA was downregulated 1.7-fold at the transcriptomics level and 2.4 at the proteomics level (Supplemental File 1). The genes responsible for pyruvate conversion to acetyl-CoA, aceE and aceF, were downregulated at the protein level by 1.90 and 1.92-fold respectively, thereby decreasing the amount of acetyl-CoA available for acetate production in the medium without casamino acids supplementation. At the same time, gltA, the product of which condenses oxaloacetate and Acetyl-CoA to citrate was upregulated by 2.7-fold at the protein level, reducing further the availability of acetyl-CoA to form acetate (Fig. 5).
Fig. 5.
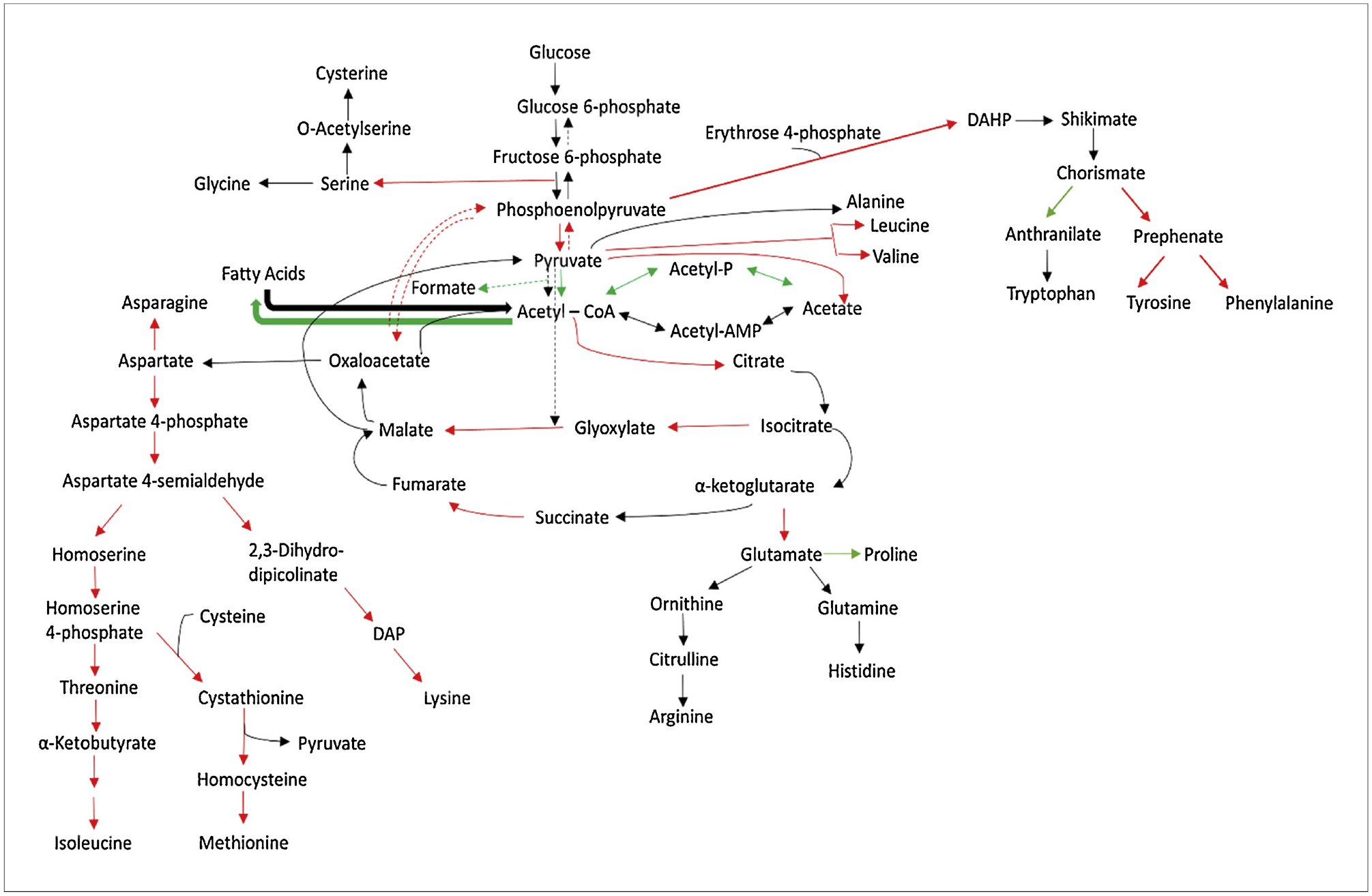
TCA cycle and associated pathways in E. coli JM109 (K strain). Red corresponds to genes with > 1.5-fold change, green corresponds to genes with < −1.5-fold change, and black corresponds to no change in gene expression. All fold changes refer to cultures without casamino acids versus those with casamino acids.
Genes related to fatty acid biosynthesis, fabA, fabB, fabF, and fabZ, were downregulated at the transcriptomics level when there were no amino acids in the medium. Similarly, purine metabolism genes xdhA and guaD were upregulated by 1.5 and 2.0-fold respectively at the transcriptomics level. Pyrimidine catabolism genes such as rutA, rutB, and rutE were upregulated by 9.2–11.9- and 15.5-fold at the transcriptomics level respectively as well as 51.5–103.7- and 126-fold at the proteomics level when medium was deprived of casamino acids. It is important to highlight that 7 of the top over-represented pathways of E. coli K obtained from transcriptomic and proteomic analysis matched, showing high correlation between protein and mRNA abundance (Supplementary File 2, S2 Table 7).
E. coli B analysis
Pathway enrichment analysis in E. coli B grown without and with amino acids showed that glycerolipid, glyoxalate and dicarboxylate metabolism, valine, leucine and isoleucine degradation, TCA cycle, tryptophan metabolism, flagellar assembly, carbon and butanoate metabolism were all enriched in both transcriptomic and proteomic analysis (Supplementary File 2, S2 Table 7). From the top 20 pathways with highest enrichment scores, 8 pathways presented in both the transcriptomic and proteomic analysis are shown in Fig. 3b. Pathway analysis in the omics dashboard showed that amino acid biosynthesis was upregulated while fatty acid, nucleoside and nucleotide biosynthesis pathways were downregulated in B-strain grown in medium without CAA (Fig. 4). The energy generating pathways of glycolysis, TCA cycle and PPP, were significantly upregulated in minimal medium without CAA.
In addition, the conversion of acetate into acetyl-CoA through ACS was significantly upregulated (32-fold and 5-fold at transcriptomic and proteomic levels) in medium without amino acids, which contributed to the lower concentration of acetate. The TCA cycle was significantly upregulated contributing further to reducing the efflux of carbon flow towards the formation of acetate.
The conversion of pyruvate to acetyl-CoA on medium lacking CAA was downregulated at the proteomic level because of a 1.5-fold lower level of AceE and 1.7-fold lower of AceF (Supplemental File 1). The availability of acetyl-CoA for conversion to acetate was further diminished by the increased expression of GltA at transcriptomic (1.7-fold) and proteomic (3.0-fold) levels, thereby increasing the TCA cycle flux and reducing acetate production flux in the medium without casamino acids. The fatty acid oxidation enzymes responsible for converting fatty acids into acetyl-CoA were upregulated when the bacterium was grown on medium lacking CAA, increasing the acetyl-CoA concentration and its utilization through the TCA cycle (Fig. 6). In the medium lacking CAA, the fadA, fadB, fadD, and fadH genes were upregulated by 7.2-, 18.8-, 6.7- and 6.9-fold respectively at the transcriptomic level and the FadA and FadB were upregulated at proteomics level by 2.9 and 4.4-fold respectively (Supplemental File 1).
Fig. 6.
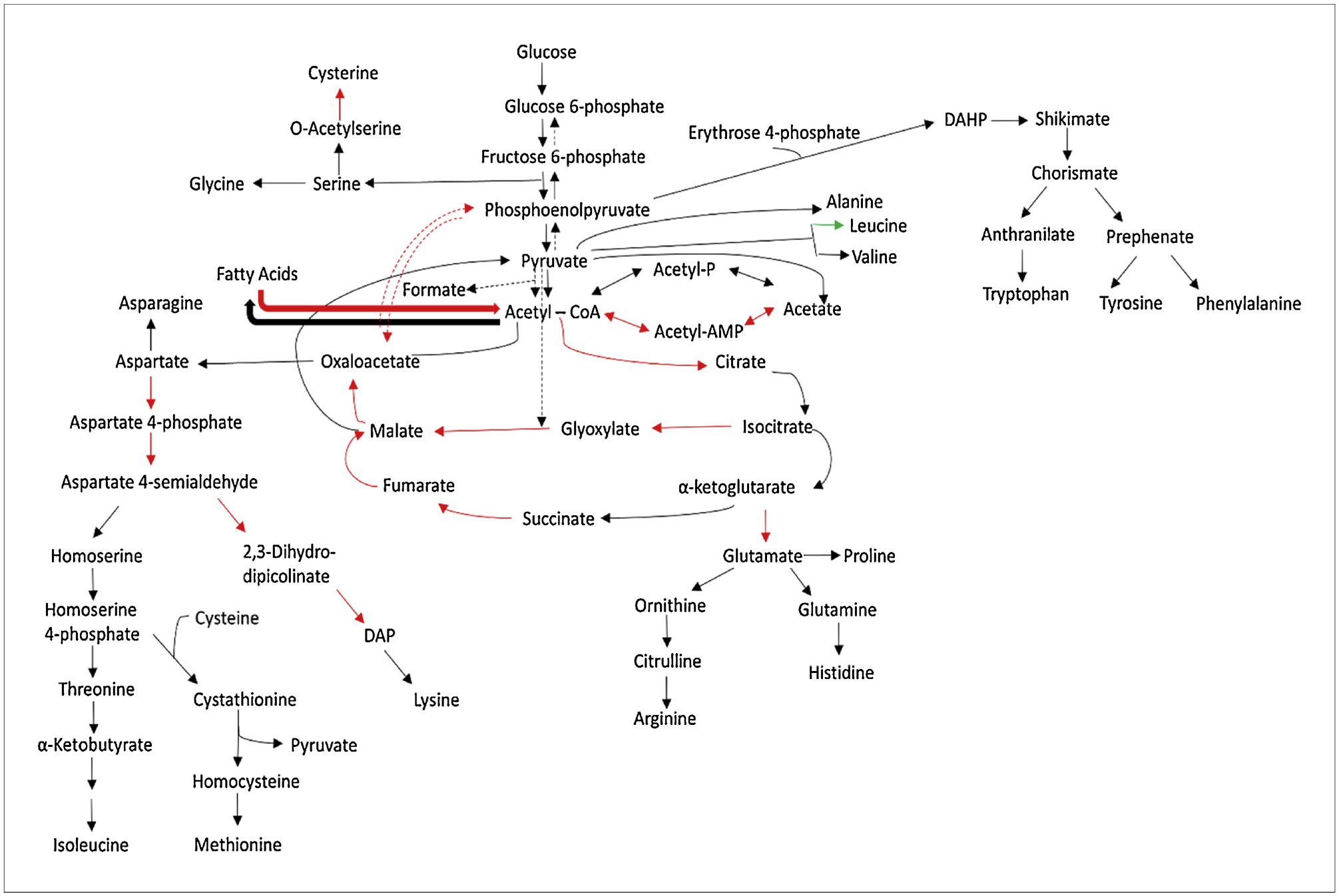
TCA cycle and associated pathways in E. coli BL21 (B strain). Red corresponds to genes with > 1.5-fold change, green corresponds to genes with < −1.5-fold change, and black corresponds to no change in gene expression. All fold changes correspond to cultures without casamino acids versus those with casamino acids.
Gene ontology (GO) analysis
To identify enriched GO terms for specific biological process a Fisher’s exact test was conducted and the results are provided in Supplementary File 3. The comparison of E. coli K growing without and with casamino acids showed differential gene expression for the GO terms for cellular amino acid biosynthetic process, amino acid transport and fatty acid biosynthetic process, whose genes were upregulated for amino acid synthesis and transport, and downregulated for fatty acid biosynthesis. The genes lysP, gadC, and cycA which are part of GO:0003333 “amino acid transmembrane transport” were upregulated by 3.6-, 3.0- and 9.4-fold respectively, at the proteomics level. This showed that not only amino acid biosynthesis is higher in E. coli K growing without amino acids, but also protein transport across the membrane. Among the other top enriched GO terms was the purine biosynthesis process, which was among the top enriched biological processes, showing correlation with KEGG analysis.
Compared with E. coli K, in E. coli B the genes associated with the enriched “TCA cycle” GO term were active and upregulated in the medium lacking casamino acids and belong to the top enriched pathways in previously discussed KEGG enrichment analysis. An active TCA cycle in E. coli B and higher conversion of acetate to acetyl-CoA leads to a low amount of residual acetate in medium without amino acids. Similar to E. coli K, amino acid transport in E. coli B was enriched. The top over-represented GO term in E. coli B grown in medium without amino acids was transmembrane transport activities carbohydrate transport, which agrees with the slight increase (3%) of specific glucose uptake rate (qs) (Table 1). Interestingly, the carbohydrate transport GO term was not enriched in E. coli K grown without amino acids, despite its higher (22%) glucose consumption rate. Both E. coli B and K showed enrichment of response to copper ion transporter in the absence of amino acids; over-representation of copper export system was expected due to its operon (cusCFBA) being induced under amino acid limitation [29].
Discussion
Addition of amino acids to E. coli K and B grown at a steady state mode in defined medium significantly affected acetate metabolism, amino acid biosynthesis, pyrimidine nucleotide degradation and fatty acid oxidation (Figs. 3–6). The presence of CAA in E. coli K cultures increased acetate excretion rate, cell yield on glucose and biomass production rate, and also decreased glucose consumption and respiration rate. For acetate excretion and glucose uptake adjustments, E. coli K modified the abundance of 859 genes as observed by microarrays, or 532 proteins observed by proteomics. In the B strain, the presence of CAA increased acetate excretion but did not modify significantly the specific biomass production rate or glucose consumption rate. Under these conditions, E. coli B modified the abundance of 2494 genes observed by microarray or 380 proteins observed by proteomics. This observation suggests that the ribosomes of E. coli B do not translate at the same efficiency as the K strain at these growth conditions.
Acetate formation rate has been shown to be directly related to E. coli growth rate [8,10]. It was also shown that E. coli BL21 grown in complex media, excreted a higher amount of acetate than in minimal media [16]. However, the cells in this study grew at different growth rates (1.6 h−1 in rich and 0.8 h−1 in defined minimal medium) and therefore it is not clear if the higher acetate production in complex medium was the result of medium composition or the higher growth rate. As demonstrated, at a constant growth rate of 0.35 h−1, the presence of amino acids promoted higher acetate excretion in both E. coli K or E. coli B strains. When grown in medium without amino acids, neither strain showed a change in the upper or lower glycolytic pathways. However, the expression of the TCA and glyoxylate cycle enzymes as well as the acetyl-CoA synthetase (ACS) were significantly higher in B, while the Pta-AckA pathway was significantly lower in K (Supplementary File 1 and Figs. 5–6), which explains the lower acetate concentration of both strains in the absence of amino acids. In addition, the increased biosynthesis of leucine, valine, arginine and glutamate in E. coli K suggests increased flux from the TCA cycle and reduced flux of metabolites leading to acetate production in medium without amino acids. Comparing with the results obtained when the growth rate was not controlled [16], it can be concluded that abundance of TCA cycle proteins in cells growing in minimal medium does not depend on the growth rate of the bacterium. The active TCA cycle together with ACS/Pta-AckA is likely to be responsible for the lower overflow in the minimal media.
The proteome allocation model described by Basan [30] proposes an elegant explanation of the overflow metabolism of E. coli grown in minimal medium with various glycolytic carbon sources, which is based on the proteome costs of energy biogenesis. Based on this concept, when the growth rate of E. coli K is higher than 0.76 h−1 and carbon uptake is high, cells maximize their growth by generating energy using the proteome-efficient fermentation pathway, and therefore most of proteome is directed towards biosynthesis required by the rapid growth. On the other hand, when the growth rate is lower than 0.76 h−1 and carbon uptake is low, the cells generate energy by the more carbon-efficient respiration pathway and the carbon influx is directed to biosynthesis and sustaining growth [30,31].
It was demonstrated that when E. coli K was grown in minimal medium, acetate excretion exhibited a threshold-linear correlation with the growth rate independent of the carbon source. In contrast with a previous report [30], we demonstrated that when amino acids are available as nitrogen source, cells excreted acetate even at a low growth rate of 0.35 h−1 (Table 1). The results presented here agree with the resource allocation theory: when grown in medium supplemented with amino acids, E. coli K show higher acetate production rate qA, and higher biomass production rate per g substrate consumed (qx) than cells grown without amino acids even at the same growth rate (Table 1). We have previously shown that amino acid supplementation increases the ability of the cells to allocate more proteins to the biosynthetic pathways as a result of higher intracellular NADPH and higher anabolic capability than when grown in minimal medium [32]. It is likely that when E. coli K cells sense nutrient-rich medium, their metabolism is directed to a proteome efficient overflow metabolism for energy generation through the less carbon efficient pathway, which was supported by the lower (0.93) respiratory quotient (Table 1). In contrast, when E. coli K cells are exposed to the nutrient-poor medium they generate energy by the more carbon-efficient respiration pathway, and more carbon flux is directed to biosynthesis, since 0.992 mol of CO2 were produced by mole of O2 consumed [30,31]. E. coli B showed higher respiratory quotients than E. coli K in the presence or absence of amino acids, demonstrating its higher biomass performance (Table 1)
Translation system
A linear relationship between translation-related proteins and specific growth rate was previously reported. E. coli cells utilize a larger fraction of their proteome to increase their translational capacity with increasing growth rate [20,31,33]. Thus, cells growing at 0.2 h−1 can devote up to 15% of proteome to their protein translational capacity and up to 25% when growing at 0.51 h−1 in minimal glucose medium [31]. In rich medium, E. coli MG1655 or BL21 grew faster and accumulated larger quantities of proteins and enzymes involved in translation, compared with cells growing in minimal glucose medium [15,16]. In contrast, the results presented here showed no changes in the amount of ribosomal proteins, translation factors or genes involved in the translation system when amino acids were supplemented at the same growth rate in minimal medium, verifying that the amount of translation-related proteins is associated with growth rate and not with the nutrients available.
Amino acid biosynthesis pathways
During growth in minimal glucose medium, cells must synthesize amino acids from glucose and ammonia, which is reflected in the expression pattern of genes and proteins associated with amino acid biosynthesis (Supplementary File 3, Fig. 4). Previous studies of E. coli BL21 or K-12 grown in minimal medium showed up regulation of 57 proteins and 67 genes involved in amino acid synthesis compared with cells grown on rich media; although these studies were not done with controlled growth rates, the outcome was consistent with the results presented here [15,16]. This was also observed in Caulobacter crescentus and Bacillus subtilis [34,35]. Since these studies were performed at growth rates of 0.46 and 1.66 h−1 in rich medium and 0.36 and 0.9 h−1 in minimal medium, and the data reported here were obtained at controlled growth rate, it can be proposed that the regulation of amino acid biosynthesis is not a function of the growth rate but of the availability of amino acids.
Transport systems
It is not surprising that, at the same growth rate, E. coli growing in minimal medium showed a higher specific glucose uptake rate (qs) than cells growing in medium supplemented with amino acids (Table 1), which agrees with the significant enrichment of the carbohydrate transport pathway in E. coli B (Enrichment score 2.21) but not in E. coli K (Enrichment score 0.015) (Supplementary File 3). It is interesting that the amino acid transport system was upregulated in minimal glucose medium although free amino acids were not available. A similar phenomenon was observed when E. coli B was grown in minimal medium [16].
Fatty acid oxidation
Fatty acids are the most energetically demanding building blocks and as a result their synthesis and degradation are tightly controlled and coordinated with growth rate and nutrient availability [36,37]. The transcription factor FadR negatively controlled the genes involved in fatty acid degradation (fadA, fadB, fadE, fadH, fadI, fadJ) through the β-oxidation pathway and positively controlled the expression of fabA, fabB, fabD, fabH and fabI, genes involved in unsaturated fatty acid biosynthesis [36,38,39].
It has been reported that lower amounts of proteins associated with fatty acid biosynthesis were detected in E. coli BL21 grown on minimal medium compared with rich medium [16,35]. The results from E. coli B described here, disagree with this information, since significant changes were observed for fatty acid biosynthesis at both the transcriptomic and the proteomic levels, which is likely to be the result of the steady growth rate. The BL21 cultures were grown at the same growth rate on medium with and without amino acids (0.35 h−1) while in the previously reported work [16], cultures on complex and defined medium were performed at different growth rates of 1.6 and 0.8 h−1, respectively. It was also reported that E. coli MG1655 (K cells) grown at non-steady-state conditions downregulated the transcript level of fatty acids biosynthesis genes, when grown on minimal medium compared with rich medium [15]. The E. coli K tested in this work (JM109) confirmed this observation, since fatty acid biosynthesis was downregulated compared with medium supplemented with amino acids (Fig. 4). These findings suggest a differential fatty acid regulation for the two popular E. coli K and B strains.
Conclusions
Addition of amino acids to minimal growth medium affects differently the transcriptome and proteome of E. coli K and B grown at steady state. Major changes were observed in pathways related to amino acid biosynthesis, acetate metabolism and fatty acid metabolism. It is not surprising that when amino acids are present, the cells turn off biosynthesis of amino acids and transport of amino acids and peptides, and direct their metabolism to proteome efficient fermentation pathways such as aerobic acetate production to generate energy, and to provide larger amount of proteins for other metabolic activities such as lipid biosynthesis, which increase the growth capabilities of the cells. A noteworthy attribute of E. coli B compared with E. coli K is its ability to synthesize fatty acids constitutively independent of amino acid availability, which explains its higher biomass production performance.
Supplementary Material
Acknowledgements
The authors would like to thank Mrs. S. Inwood for critical reading and Douglas Joubert, NIH Library Writing Center, for manuscript editing assistance.
Funding
This work was supported by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health.
Abbreviations:
- CAA
casamino acids
- CTR
carbon dioxide transfer rate (mole/L/min)
- dO2
dissolved oxygen concentration
- EDTA
ethylenediaminetetraacetic acid
- in subscript
inlet
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- OD600
optical density measured at 600 nm of wavelength
- OTR
oxygen transfer rate (mole/L/min)
- out subscript
outlet
- TCA
tricarboxylic acids
- VG
gas flow rate under standard condition of temperature and pressure (N L/min)
- VL
reactor working volume (L)
- Vm
molar volume under standard condition of temperature and pressure (N L/mole)
- yO2
oxygen molar fraction
- yCO2
carbon dioxide molar fraction
Footnotes
Appendix A. Supplementary data
Supplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.nbt.2018.10.004.
References
- [1].Chavez-Bejar MI, Balderas-Hernandez VE, Gutierrez-Alejandre A, Martinez A, Bolivar F, Gosset G. Metabolic engineering of Escherichia coli to optimize melanin synthesis from glucose. Microb Cell Fact 2013;12:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Choi JH, Keum KC, Lee SY. Production of recombinant proteins by high cell density culture of Escherichia coli. Chem Eng Sci 2006;61:876–85. [Google Scholar]
- [3].Tsuruta H, Paddon CJ, Eng D, Lenihan JR, Horning T, Anthony LC, et al. High-level production of amorpha-4,11-diene, a precursor of the antimalarial agent artemi-sinin, in Escherichia coli. PLoS One 2009;4:e4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Glazyrina J, Krause M, Junne S, Glauche F, Storm D, Neubauer P. Glucose-limited high cell density cultivations from small to pilot plant scale using an enzyme-controlled glucose delivery system. N Biotechnol 2012;29:235–42. [DOI] [PubMed] [Google Scholar]
- [5].Rinas U, Kracke-Helm H-A, Schügerl K. Glucose as a substrate in recombinant strain fermentation technology. Appl Microbiol Biotechnol 1989;31:163–7. [Google Scholar]
- [6].Tripathi N High yield production of heterologous proteins with Escherichia coli. Def Sci J 2009;59:137. [Google Scholar]
- [7].Bernal V, Castaño-Cerezo S, Cánovas M. Acetate metabolism regulation in Escherichia coli: carbon overflow, pathogenicity, and beyond. Appl Microbiol Biotechnol 2016;100:8985–9001. [DOI] [PubMed] [Google Scholar]
- [8].Eiteman MA, Altman E. Overcoming acetate in Escherichia coli recombinant protein fermentations. Trends Biotechnol 2006;24:530–6. [DOI] [PubMed] [Google Scholar]
- [9].Lara AR, Caspeta L, Gosset G, Bolívar F, Ramírez OT. Utility of an Escherichia coli strain engineered in the substrate uptake system for improved culture performance at high glucose and cell concentrations: an alternative to fed‐batch cultures. Biotechnol Bioeng 2008;99:893–901. [DOI] [PubMed] [Google Scholar]
- [10].Valgepea K, Adamberg K, Nahku R, Lahtvee P-J, Arike L, Vilu R. Systems biology approach reveals that overflow metabolism of acetate in Escherichia coli is triggered by carbon catabolite repression of acetyl-CoA synthetase. BMC Syst Biol 2010;4:166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Wolfe AJ. The acetate switch. Microbiol Mol Biol Rev 2005;69:12–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Phue JN, Noronha SB, Hattacharyya R, Wolfe AJ, Shiloach J. Glucose metabolism at high density growth of E. coli B and E. coli K: differences in metabolic pathways are responsible for efficient glucose utilization in E. coli B as determined by microarrays and Northern blot analyses. Biotechnol Bioeng 2005;90:805–20. [DOI] [PubMed] [Google Scholar]
- [13].Phue J-N, Kedem B, Jaluria P, Shiloach J. Evaluating microarrays using a semi-parametric approach: application to the central carbon metabolism of Escherichia coli BL21 and JM109. Genomics 2007;89:300–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Phue J-N, Shiloach J Transcription levels of key metabolic genes are the cause for different glucose utilization pathways in E. coli B (BL21) and E. coli K (JM109). J Biotechnol 2004;109:21–30. [DOI] [PubMed] [Google Scholar]
- [15].Tao H, Bausch C, Richmond C, Blattner FR, Conway T. Functional genomics: expression analysis of Escherichia coli growing on minimal and rich media. J Bacteriol 1999;181:6425–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Li Z, Nimtz M, Rinas U. The metabolic potential of Escherichia coli BL21 in defined and rich medium. Microb Cell Fact 2014;13:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Marisch K, Bayer K, Scharl T, Mairhofer J, Krempl PM, Hummel K, et al. A comparative analysis of industrial Escherichia coli K–12 and B strains in high-glucose batch cultivations on process-, transcriptome-and proteome level. PLoS One 2013;8:e70516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Yoon SH, Han M-J, Jeong H, Lee CH, Xia X-X, Lee D-H, et al. Comparative multi-omics systems analysis of Escherichia coli strains B and K-12. Genome Biol 2012;13:R37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].O’brien EJ, Lerman JA, Chang RL, Hyduke DR, Palsson BØ. Genome‐scale models of metabolism and gene expression extend and refine growth phenotype prediction. Mol Syst Biol 2013;9:693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Valgepea K, Adamberg K, Seiman A, Vilu R. Escherichia coli achieves faster growth by increasing catalytic and translation rates of proteins. Mol Biosyst 2013;9:2344–58. [DOI] [PubMed] [Google Scholar]
- [21].Bauer S, Shiloach J. Maximal exponential growth rate and yield of E. coli obtainable in a bench‐scale fermentor. Biotechnol Bioeng 1974;16:933–41. [DOI] [PubMed] [Google Scholar]
- [22].Kumar A, Shiloach J, Betenbaugh MJ, Gallagher EJ. The beta-3 adrenergic agonist (CL-316,243) restores the expression of down-regulated fatty acid oxidation genes in type 2 diabetic mice. Nutr Metab 2015;12:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Boersema PJ, Raijmakers R, Lemeer S, Mohammed S, Heck AJ. Multiplex peptide stable isotope dimethyl labeling for quantitative proteomics. Nat Protoc 2009;4:484. [DOI] [PubMed] [Google Scholar]
- [24].Wiśniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nat Methods 2009;6:359. [DOI] [PubMed] [Google Scholar]
- [25].Wiśniewski JR, Zougman A, Mann M. Combination of FASP and StageTip-based fractionation allows in-depth analysis of the hippocampal membrane proteome. J Proteome Res 2009;8:5674–8. [DOI] [PubMed] [Google Scholar]
- [26].Mi H, Muruganujan A, Casagrande JT, Thomas PD. Large-scale gene function analysis with the PANTHER classification system. Nat Protoc 2013;8:1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Paley S, Parker K, Spaulding A, Tomb J-F, O’Maille P, Karp PD. The Omics Dashboard for interactive exploration of gene-expression data. Nucleic Acids Res 2017;45:12113–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 2008;26:1367. [DOI] [PubMed] [Google Scholar]
- [29].Fung DKC, Lau WY, Chan WT, Yan A. Copper efflux is induced during anaerobic amino acid limitation in Escherichia coli to protect iron-sulfur cluster enzymes and its biogenesis. J Bacteriol 2013. JB. 00543–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Basan M, Hui S, Okano H, Zhang Z, Shen Y, Williamson JR, et al. Overflow metabolism in Escherichia coli results from efficient proteome allocation. Nature 2015;528:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Peebo K, Valgepea K, Maser A, Nahku R, Adamberg K, Vilu R. Proteome reallocation in Escherichia coli with increasing specific growth rate. Mol Biosyst 2015;11:1184–93. [DOI] [PubMed] [Google Scholar]
- [32].Baez A, Shiloach J. Increasing dissolved-oxygen disrupts iron homeostasis in production cultures of Escherichia coli. Antonie Van Leeuwenhoek 2017;110:115–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Nomura M Regulation of ribosome biosynthesis in Escherichia coli and Saccharomyces cerevisiae: diversity and common principles. J Bacteriol 1999;181:6857–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Hottes AK, Meewan M, Yang D, Arana N, Romero P, McAdams HH, et al. Transcriptional profiling of Caulobacter crescentus during growth on complex and minimal media. J Bacteriol 2004;186:1448–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Mäder U, Homuth G, Scharf C, Büttner K, Bode R, Hecker M. Transcriptome and proteome analysis of Bacillus subtilis gene expression modulated by amino acid availability. J Bacteriol 2002;184:4288–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Parsons JB, Rock CO. Bacterial lipids: metabolism and membrane homeostasis. Prog Lipid Res 2013;52:249–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Zhang Y-M, Rock CO. Transcriptional regulation in bacterial membrane lipid synthesis. J Lipid Res 2009;50:S115–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].My L, Achkar NG, Viala J, Bouveret E. Reassessment of the genetic regulation of fatty acid synthesis in Escherichia coli: global positive control by the dual functional regulator FadR. J Bacteriol 2015. JB. 00064–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Nunn WD, Giffin K, Clark D, Cronan J. Role for fadR in unsaturated fatty acid biosynthesis in Escherichia coli. J Bacteriol 1983;154:554–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.