

ABSTRACT
Cutaneous beta genus human papillomaviruses (β-HPVs) are suspected to promote the development of nonmelanoma skin cancer (NMSC) by destabilizing the host genome. Multiple studies have established the genome destabilizing capacities of β-HPV proteins E6 and E7 as a cofactor with UV. However, the E6 protein from β-HPV8 (HPV8 E6) induces tumors in mice without UV exposure. Here, we examined a UV-independent mechanism of HPV8 E6-induced genome destabilization. We showed that HPV8 E6 reduced the abundance of anaphase bridge resolving helicase, Bloom syndrome protein (BLM). The diminished BLM was associated with increased segregation errors and micronuclei. These HPV8 E6-induced micronuclei had disordered micronuclear envelopes but retained replication and transcription competence. HPV8 E6 decreased antiproliferative responses to micronuclei and time-lapse imaging revealed HPV8 E6 promoted cells with micronuclei to complete mitosis. Finally, whole-genome sequencing revealed that HPV8 E6 induced chromothripsis in nine chromosomes. These data provide insight into mechanisms by which HPV8 E6 induces genome instability independent of UV exposure.
IMPORTANCE Some beta genus human papillomaviruses (β-HPVs) may promote skin carcinogenesis by inducing mutations in the host genome. Supporting this, the E6 protein from β-HPV8 (8 E6) promotes skin cancer in mice with or without UV exposure. Many mechanisms by which 8 E6 increases mutations caused by UV have been elucidated, but less is known about how 8 E6 induces mutations without UV. We address that knowledge gap by showing that 8 E6 causes mutations stemming from mitotic errors. Specifically, 8 E6 reduces the abundance of BLM, a helicase that resolves and prevents anaphase bridges. This hinders anaphase bridge resolution and increases their frequency. 8 E6 makes the micronuclei that can result from anaphase bridges more common. These micronuclei often have disrupted envelopes yet retain localization of nuclear-trafficked proteins. 8 E6 promotes the growth of cells with micronuclei and causes chromothripsis, a mutagenic process where hundreds to thousands of mutations occur in a chromosome.
KEYWORDS: DNA damage, HPV, cell proliferation, genome analysis, micronuclei, mitosis
INTRODUCTION
Nonmelanoma skin cancer (NMSC) is the most prevalent type of cancer and more occurrences are noted each year (1–4). While not often deadly, the cost of treating NMSC in the United States alone is $4.8 billion annually (5). Environmental factors play an outsized role in NMSC development, with UV exposure acknowledged as the main etiological agent (6). Age and immunosuppression also contribute (7, 8). An increase in risk associated with immunosuppression indicates that an infectious agent can promote NMSC (9). Among possible infectious agents, some members of the beta genus human papillomaviruses (β-HPVs) are considered likely candidates because they promote NMSC development in people with a genetic disorder (epidermodysplasia verruciformis) or undergoing immunosuppressive therapy (10–12). However, the extent that which β-HPV infections contribute to NMSC development in the general population is unclear.
The association between β-HPV infections and NMSC is supported by in vitro systems, epidemiological studies, and animal models (13–15). These data were most compelling for a subset of β-HPVs (HPV5, HPV8, and HPV38). They also demonstrated that β-HPV infections rarely persist. However, β-HPV viral loads peak early in NMSC development before becoming dramatically reduced in NMSC cells (16). These observations caused speculation that β-HPV infections introduce tumorigenic mutations that are capable of independently driving tumorigenesis without continued viral gene expression (17, 18). In vitro and in vivo studies have shown that the E6 protein from HPV5, HPV8, and HPV38 hinder DNA repair after UV exposure, suggesting the hypothesized role of β-HPV in NMSC is feasible (19, 20). However, other studies have indicated a broader role for β-HPV proteins by showing that HPV8 E6 (8 E6) causes tumors in mice without UV exposure (21–25). This suggests that 8 E6 can introduce genome destabilization without an exogenous stimulus.
Genome integrity is also at risk during mitosis, since cellular DNA must be replicated and segregated faithfully to avoid mutations. We have shown that 8 E6 attenuates cellular responses that protect genome integrity during mitosis and increases the frequency of aneuploidy (26–28). Aneuploidy results from the unequal segregation of chromosomes suggest that 8 E6 may cause other types of genome instability caused by segregation errors. Micronuclei are one type of destabilization associated with segregation errors. These small enveloped extranuclear structures can form when a chromosome gets caught between the nuclei of forming daughter cells in a structure known as an anaphase bridge (29–31). Mutations in micronuclear DNA are common because micronuclear envelopes are unstable which limits the ability to localize cellular factors necessary for replication, transcription, nuclear protein localization, and DNA repair (32–34). It is further mutagenic for a cell to enter mitosis with unrepaired micronuclear DNA. When this occurs, the micronuclear DNA is prone to undergo chromothripsis, a phenomenon where hundreds to thousands of chromosomal rearrangements occur over a single cell cycle (35–39). If 8 E6 promotes chromothripsis, it would provide a plausible mechanism by which β-HPV infections as brief as one cell cycle could cause enough mutations to drive NMSC development. Notably, chromothripsis-like events have been identified in NMSCs, although there was no attempt to determine whether they were caused by a β-HPV infection (40, 41).
Here, we show that 8 E6 hinders chromosome segregation causing increases in anaphase bridge formation and impeding their resolution. We link this increase to a reduction of Bloom syndrome protein (BLM) and a reduced ability to localize to anaphase bridges. BLM helicase prevents anaphase bridge formation and is critical for their resolution (42, 43). Mechanistically, we provide evidence that the reduction in BLM is the result of reduced ATR-Chk1 kinase activity that stabilizes BLM by blocking cullin-3-mediated degradation (44). These data expand the significance of previous work demonstrating that 8 E6 reduces Chk1 phosphorylation (45–47). Although only a partial decrease in BLM abundance, we identified a significant impact from the BLM reduction as 8 E6 increases the frequency of segregation errors and micronuclei. We go on to characterize these micronuclei, showing that they are more likely to have disordered micronuclear envelopes and that 8 E6 makes it less likely that a cell undergoes antiproliferative responses to micronuclei (i.e., p53 accumulation, caspase activation, and senescence). 8 E6 also increases the frequency that cells with micronuclei undergo mitosis. Finally, we use whole-genome sequencing to demonstrate that 8 E6 causes chromothripsis.
RESULTS
p300-dependent BLM reduction correlates with loss of ATR and Chk1 abundance.
Because HPV8 E6 (8 E6) reduces activation of the p300-ATR-Chk1 signaling axis that facilitates BLM stabilization (45–47), we hypothesized that 8 E6 decreased BLM abundance. To evaluate this, we compared BLM abundance in vector control (iHFK LXSN) and hemagglutinin (HA)-tagged 8 E6-expressing (iHFK 8 E6) hTERT-immortalized keratinocyte cells. 8 E6 expression was confirmed by probing for the HA tag (data not shown). As expected, 8 E6 hindered the p300-ATR-Chk1 signaling axis (Fig. 1A). Consistent with our hypothesis, 8 E6 also decreased BLM abundance determined by densitometry of three independent immunoblots. To determine whether the reduced BLM required TERT immortalization, we repeated this experiment in primary foreskin keratinocyte cells (HFK LXSN and HFK 8 E6). The expression of 8 E6 in these cells was previously confirmed (27). We detected p300 by immunoblotting as further confirmation (data not shown). 8 E6 had an approximately equal ability to reduce BLM abundance in primary keratinocytes, as determined by densitometry of three independent immunoblots (Fig. 1B). These cell lines were derived from different donors, indicating that the phenotype is conserved in at least two genetic backgrounds. We took this approach of dual confirmation throughout this study to similarly probe the requirement of TERT immortalization and the conservation of phenotypes between donors.
FIG 1.

HPV8 E6 reduces BLM abundance. (A) Immunoblots of whole-cell lysates from iHFK cells expressing LXSN and 8 E6. (B) Immunoblots of whole-cell lysates HFK cells expressing LXSN and the indicated E6 genes. (C) Immunoblots of whole-cell lysates from iHFK LXSN cells treated with indicated amounts of CCS-1477 for 24 h. (D) Immunoblots of whole-cell lysates from HFK LXSN cells following 24 h of CCS-1477 treatment. (E) Representative immunoblots showing BLM abundance in iHFK cells at 0, 6, 12, 18, 24, and 30 h after 50-μg/mL cycloheximide treatment. (F) Average densitometry from three repeats of the experiment described in panel E. “ND” indicates that the half-life could not be determined by the indicated time points. BLM was quantified and normalized relative to GAPDH signal for each time point. (G) BLM mRNA expression in HFK cells measured by RT-qPCR and normalized to β-actin mRNA. Graphs depict means ± the standard errors of the mean from three independent experiments. Immunoblots are representative of at least three independent experiments. The numbers above the bands represent quantification by densitometry. Asterisks denote significant differences relative to LXSN (**, P ≤ 0.05 [Student t test]). In Δ8 E6, residues 132 to 136 were deleted from 8 E6.
8 E6 hinders the p300-ATR-Chk1 signaling axis by binding and destabilizing p300 (47), leading us to hypothesize that 8 E6 decreased BLM levels by binding p300. To test this hypothesis, we expressed a mutant 8 E6 without the p300 binding site (HFK 8 E6 Δ132-136) in primary human foreskin keratinocytes and measured BLM abundance by immunoblotting. Expression of this mutated 8 E6 in these cells was validated previously (27). BLM levels were not significantly reduced in HFK 8 E6 Δ132-136 cells compared to HFK LXSN cells. To further probe the role of p300 binding in BLM reduction, we expressed an E6 that weakly binds and does not degrade p300 (HPV38 E6) in primary foreskin keratinocytes (HFK 38 E6). The expression of 38 E6 in these cells has also been confirmed (27, 48). Consistent with the requirement for robust p300 binding, 38 E6 also did not reduce BLM abundance (Fig. 1B). Deletion of the p300 binding domain disrupts other functions of 8 E6 (23), so we verified the requirement of p300 for optimal BLM abundance by treating iHFK LXSN cells with a small molecule inhibitor of p300 (CCS-1477 [49]). CCS-1477 phosphorylation and expression of ATR and Chk1 in a dose-dependent manner (Fig. 1C) (47). CCS-1477 similarly reduced BLM abundance in these cells. Further, a similar hindrance of the p300-ATR-Chk1 signaling axis was seen when HFK cells were grown in media containing CCS-1477 (Fig. 1D). Because Chk1 increases BLM abundance by stabilizing the protein, we hypothesized that 8 E6 decreased BLM stability. To test this, we compared the impact of cycloheximide (protein synthesis inhibitor) on BLM abundance in iHFK LXSN and iHFK 8 E6 cells. This allowed us to determine that 8 E6 reduced the half-life of BLM in iHFK 8 E6 cells (Fig. 1E and F). Because p300 broadly regulates transcription, we sought to determine whether there was a p300-dependent effect on BLM transcript levels in HFK cells via RT-qPCR. 8 E6 significantly reduced BLM transcript abundance (Fig. 1G), but 8 E6 Δ132-136 did not significantly alter BLM transcript abundance.
8 E6 reduces BLM at anaphase bridges, increasing unresolved bridge frequency.
Because BLM is required for faithful chromosome segregation (42–44), we hypothesized that 8 E6 increased the frequency of segregation errors. To test this, iHFK cells were synchronized by thymidine block and released to enrich for cells in anaphase. We then used immunofluorescent microscopy to identify anaphase cells and determine whether they had segregation errors (lagging whole chromosomes, acentric chromosomes, and anaphase bridges). Both acentric chromosomes, those without centromeres, and lagging whole chromosomes both appear as nuclear material between the main groups of chromosomes being segregated into the forming daughter cells. We stained for CENP-A, a centromere marker, to distinguish between these two types of segregation errors (Fig. 2A). Anaphase bridges were identified as chromosomes connected to both main bodies of segregating DNA. 8 E6 increased the frequency of segregation errors (Fig. 2B). Most of the segregation errors caused by 8 E6 were anaphase bridges. Because BLM resolves anaphase bridges, we determined whether 8 E6 reduced BLM localization to these bridges by comparing BLM staining intensity at anaphase bridges between iHFK LXSN and iHFK 8 E6 cells (Fig. 2C). 8 E6 decreased BLM staining at chromatin bridges (Fig. 2D). To determine whether 8 E6 induces anaphase bridges via BLM reduction, we transfected iHFK cells with empty vector or BLM-GFP and compared anaphase bridge frequency approximately 72 h later. Exogenous BLM significantly decreased the frequency of anaphase bridges in 8 E6 iHFKs (Fig. 2E). We further hypothesized that 8 E6 increased the time it takes to resolve anaphase bridges. To test this, we compared anaphase bridge duration by time-lapse imaging between iHFK LXSN and iHFK 8 E6 cells expressing a mCherry-tagged H2B that allowed DNA visualization (data not shown). Although the data did not reach statistical significance (P = 0.208), anaphase bridges were more persistent in iHFK 8 E6 cells with one bridge lasting over 14 h (Fig. 2E).
FIG 2.

HPV8 E6 increases the frequency of segregation errors. (A) Images of iHFK cells stained with DAPI (white and blue) and CENP-A (green), displaying anaphases with no error (left), an anaphase bridge (left middle), acentric chromosome fragment (right middle), and a lagging whole chromosome (right), each indicated by arrows. (B) Quantification of segregation errors in iHFK cells after synchronization by thymidine block and released to enrich for mitotic cells. n = at least 220 anaphases from three experiments. (C) Representative images of BLM (red) localization to chromatin bridges stained with DAPI (white) in iHFK LXSN and 8 E6 cells. Arrows indicate the location of anaphase bridge. (D) Mean intensity of BLM staining at chromatin bridges in iHFK LXSN and 8 E6 cells after being synchronized by thymidine block and released to enrich for mitotic cells. n = at least 39 bridges from three experiments. (E) Quantification of anaphase bridges in iHFK cells after transfection with empty vector or BLM-GFP then synchronized by thymidine block and release to enrich for mitotic cells. n = at least 175 anaphases from three experiments. (F) Quantification of time-lapse anaphase bridge duration in iHFK cells. n = at least 12 bridges across five independent experiments. Each dot/square represents the duration of a separate anaphase bridge. Graphs depict means ± the standard errors of the mean from three independent experiments. Light blue asterisks denote a significant difference between iHFK LXSN and iHFK 8 E6 bridge frequency. Asterisks denote significant differences relative to LXSN unless specified by a bar (*, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001 [Student t test]).
8 E6 increases micronuclei frequency by reducing BLM availability.
Because anaphase bridges can result in the formation of micronuclei (37, 50–53), we hypothesized that 8 E6 expression increased the frequency of micronuclei. To test this, micronuclei were identified by immunofluorescence microscopy of DAPI-stained cells (Fig. 3A). Micronuclei were more common in both HFK 8 E6 and iHFK 8 E6 cells compared to their LXSN counterparts (Fig. 3B and C). We examined the p300 dependence of the increase in micronuclei by determining the micronucleus frequency in HFK 8 E6 Δ132-136 and HFK 38 E6 cells. Micronuclei occurred more frequently in both cell lines compared to HFK LXSN cells but were significantly less common than HFK 8 E6, suggesting that 8 E6 causes micronuclei through p300-independent and -dependent mechanisms.
FIG 3.

HPV8 E6 increases micronuclei frequency. (A) Representative images of DAPI (white)-stained HFK cells, one with (indicated by arrow) and one without a micronucleus. (B and C) Micronucleus frequencies in the indicated iHFK (B) and HFK (C) cells. (D) Micronucleus frequency in U2OS cells at 72 h posttransfection with BLM-GFP or pLVX-GFP. (E) Immunoblots of whole-cell lysates from HFK cells at 72 h posttransfection with control siRNA or BLM siRNA. (F) Quantification of micronucleus frequency in HFK cells at 72 h posttransfection with control siRNA or BLM siRNA (same cells as in panel E). At least 150 cells were quantified for micronucleus frequency across three independent experiments. (G) Representative images of CENP-A-positive and -negative micronuclei (indicated by arrows) stained for DAPI (white and blue) and CENP-A (green). (H and I) Percentages of micronuclei with at least one CENP-A focus iHFK (H) and HFK (I) in LXSN and β-HPV E6 cells. At least 250 micronuclei/cell line were quantified for CENP-A frequency across three independent experiments. Graphs depict means ± the standard errors of the mean (n ≥ 3). Double daggers denote significant differences relative to LXSN plus siControl (‡‡, P = P ≤ 0.01; ‡‡‡, P ≤ 0.001). Asterisks denote significant differences relative to LXSN unless specified by a bar (*, P ≤ 0.05; **, P ≤ 0.01 [Student t test]). In Δ8 E6, residues 132 to 136 were deleted from 8 E6.
To determine the extent that which reduced BLM abundance contributed to the elevated frequency of micronuclei, we exogenously expressed a green fluorescent protein (GFP)-tagged BLM or GFP alone (transfection control) in U2OS cells. We were unable to test the BLM dependence of 8 E6-induced micronucleus formation in HFK or iHFK cells since BLM confirmation via GFP expression could not be seen in 8 E6-expressing cells. We also confirmed the ability of 8 E6 to increase the prevalence of micronuclei in these cells (data not shown). Cells were fixed 72 h posttransfection and DAPI stained to detect micronuclei. This allowed the exogenous BLM to be expressed as cells pass through mitosis one to three times. We only examined cells expressing GFP to ensure that only transfected cells were considered. While 8 E6 increased micronuclei frequency in transfection control, exogenous GFP-BLM expression reduced the frequency of micronuclei in U2OS 8 E6 cells, but not in U2OS LXSN (Fig. 3D). To further validate the role of BLM in preventing micronuclei, we determined micronuclei frequency 72 h after transfecting HFK LXSN, HFK 8 E6, and HFK 8 E6 Δ132-136 cells with scramble control small interfering RNA (siRNA) or BLM siRNA. Knockdown of BLM was confirmed by immunoblotting in these cells (Fig. 3E). While transfection with control siRNA did not change the frequency of micronuclei in any cell line, BLM knockdown made micronuclei significantly more common in HFK LXSN and HFK 8 E6 Δ132-136 cells (Fig. 3F).
Micronuclei contain either whole (centric) or partial (acentric) chromosomes depending on how they form (54–57). Unrepaired DSBs lead to smaller acentric micronuclei (58), whereas chromosomal missegregation results in larger centric micronuclei (59, 60). Our group and others have shown that 8 E6 attenuates DSB repair (19). Here, we demonstrate that 8 E6 can cause anaphase bridges (Fig. 2B). Therefore, we hypothesize that 8 E6 caused both acentric and centric micronuclei. To test this, we probed for CENP-A in micronuclei by immunofluorescence microscopy (61). 8 E6 caused acentric and centric micronuclei in iHFK and HFK cells (Fig. 3E to G). Micronuclei produced by 8 E6 were on average significantly smaller than vector control (data not shown). This, along with 8 E6 micronuclei containing a DSB marker (γH2AX), is consistent with some of these micronuclei forming because of impaired DSB repair (data not shown).
8 E6 reduces lamin B1 integrity of micronuclei.
Anaphase bridges tend to produce micronuclei lacking micronuclear envelope integrity (62). We therefore hypothesized that 8 E6-induced micronuclei would more frequently have unstable micronuclear envelopes. To evaluate micronuclear envelope integrity, we used immunofluorescent microscopy to group micronuclei based on lamin B1 morphology into established categories (32). First, micronuclei were placed into two large groups based on whether lamin B1 staining displayed clear peripheral localization around the micronucleus (intact) or not (disordered). Micronuclei with intact membranes were further segregated based on whether they had a continuous (no holes; Fig. 4A, first row; see also Movie S1 in the supplemental material) or discontinuous (one or more holes; Fig. 4A, second row; see also Movie S2) lamin B1 staining. Micronuclei with disordered micronuclear membranes were also further split based on whether nuclear lamin B1 staining appeared collapsed (loss of peripheral localization and appear collapsed; Fig. 4A, third row; see also Movie S3) or was absent (“none”; Fig. 4A, fourth row; see also Movie S4). The ratio of intact to disordered micronuclear membranes in iHFK LXSN cells was similar to prior reports for other cell lines (Fig. 4B) (32, 63). In contrast, micronuclear membranes in iHFK 8 E6 cells were more often disordered, with a specific increase in micronuclei without a membrane (Fig. 4B). Disordered membranes typically prevent nuclear factors from being imported after translation in the cytoplasm (32, 51, 64). To characterize the impact of micronuclear membrane integrity on the localization of nuclear proteins, we stably transfected LXSN and 8 E6 iHFK cells with a fusion gene consisting of two red fluorescence proteins (RFPs) and a nuclear localization signal (RFP-NLS) to visualize and track micronuclear localization. We examined lamin B1 morphology and RFP staining in these cells and found RFP localized to micronuclei with intact envelopes (Fig. 4C). 8 E6 increased the proportion of RFP-positive micronuclei with discontinuous envelopes. This suggests that 8 E6 disrupts envelope integrity but retains nuclear localization for more time as the envelope begins to disappear.
FIG 4.

HPV8 E6 reduces lamin B1 integrity of micronuclei. (A) Representative images of cells with intact (continuous and discontinuous) and disordered (collapsed and none) micronuclear envelopes. DNA is stained with DAPI (blue), and nuclear membranes were detected with antibodies against lamin B1 (green). A white arrow denotes the micronucleus in the “none” images. (B) Quantification of lamin B1 micronuclear morphology in asynchronous iHFK cells. n ≥ 234 micronuclei from three independent experiments. (C) Quantification of RFP-NLS-positive micronuclei segregated by micronuclear morphology (as determined by lamin B1 staining) in iHFK cells. (D) Quantification of lamin B1 micronuclear morphology in asynchronous HFK cells. n = at least 146 micronuclei from three experiments. (E) Depicts is the results from repeating the experiment as described in for panel D in HCT 116 cells. Quantification of lamin B1 micronuclear morphology in asynchronous iHFK cells. n ≥ 200 micronuclei from three independent experiments. (F) Length of time (in minutes) that RFP-NLS-positive micronuclei in iHFK RFP-NLS cells persisted across 30 h. Red, salmon, and blue asterisks denote a significant difference from LXSN corresponding with continuous, discontinuous, and no lamin B1 morphologies, respectively. Graphs depict means ± the standard errors of the mean from three independent experiments. Asterisks denote significant differences relative to LXSN (*, P ≤ 0.05; ***, P ≤ 0.001 [Student t test]). In Δ8 E6, residues 132 to 136 were deleted from 8 E6.
To determine whether 8 E6 could reduce micronuclear membrane stability in HFK cells, we examined micronuclear membranes by immunofluorescence microscopy. While not statistically significant (P = 0.079), these data were similar to the observations in iHFK cells (Fig. 4D). To determine the extent that p300 destabilization contributed to this phenotype, we compared membrane integrity between HFK 8 E6, HFK 8 E6 Δ132-136, and HFK 38 E6 cells. This did not reach the thresholds of significance to strongly indicate that 8 E6 was decreasing membrane integrity by destabilizing p300 (Fig. 4D). We also did not find a difference in micronuclear envelope integrity when comparing HCT 116 cells with or without p300 (Fig. 4E).
8 E6 increased the frequency of RFP-NLS-positive micronuclei with discontinuous envelopes (Fig. 4C). Because discontinuous micronuclear envelopes are prone to become disordered with time (25), we hypothesized that 8 E6 increased the chance that cells lost nuclear RFP localization over time. To test this, we used time-lapse imaging to track the RFP-NLS status of micronuclei (retained or lost) for 30 h in iHFK LXSN and 8 E6 cells (see Movies S5 and S6). Instead, 8 E6 increased the retention of nuclear localization (Fig. 4F). Together, these data indicate that 8 E6 promotes nuclear localization despite also impairing micronuclear envelope stability.
8 E6 allows nuclear function in micronuclei with disrupted membranes.
Membrane integrity is critical for the proper localization of nuclear proteins that facilitate replication, transcription, and DNA damage repair. Failure to localize nuclear factors to micronuclei has been proposed to result in damage to micronuclear DNA (32, 51, 65). We used immunofluorescence microscopy to characterize these nuclear processes in the micronuclei of HFK LXSN and HFK 8 E6 cells. Histone H3 acetylated at K27 (H3K27ac) was used as a marker of transcription. Bromodeoxyuridine (BrdU) incorporation was used as a replication marker. Consistent with previous results (32), transcription and replication were reduced in micronuclei with disordered membranes in HFK LXSN cells (Fig. 5A and B). Although this interpretation of the data is most consistent with the current understanding of micronuclear biology, we cannot formally rule out the possibility that H2K27ac and BrdU staining are the result of a sequestration effect stemming from improper chromosome segregation. However, 8 E6 allowed both replication and transcription to occur significantly more frequently in micronuclei without envelopes. To evaluate the DDR in micronuclei, we exposed cells to a radiomimetic (zeocin) and assessed the formation of DSB repair foci (53bp1) 1 h afterward. Consistent with previous reports (32, 33), we found the DDR response is attenuated in micronuclei with disordered envelopes in HFK LXSN and HFK 8 E6 cells (Fig. 5C).
FIG 5.
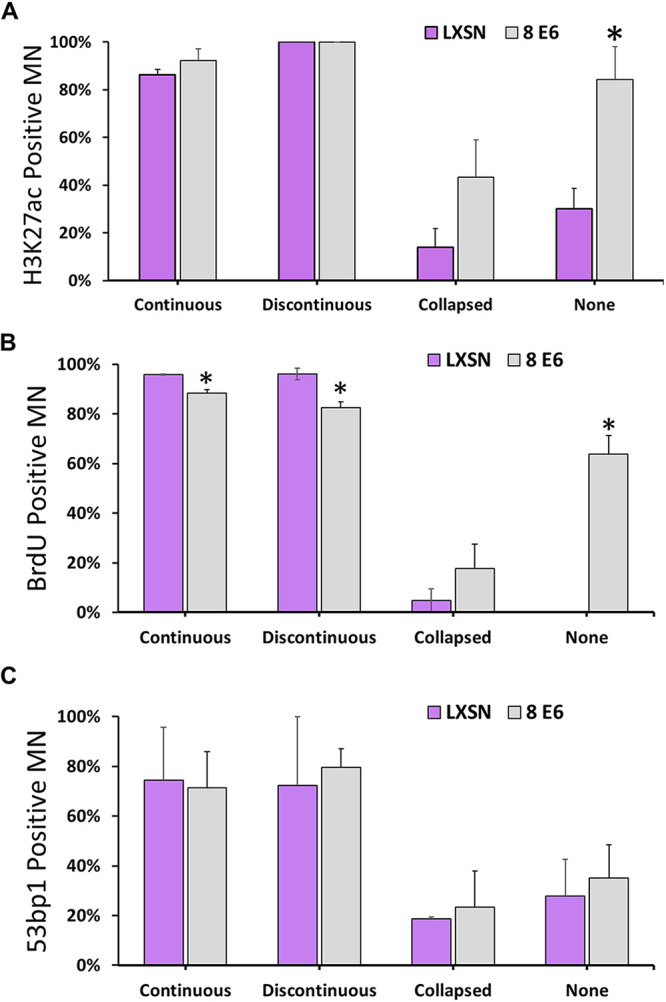
HPV8 E6 allows transcription and replication of DNA in micronuclei without membranes. (A to C) Percentages of iHFK cells with acetylated H3K27-positive (A), 53bp1-positive (B), and BrdU-positive (C) micronuclei. At least 150 micronuclei/cell line were quantified across three independent experiments. Graphs depict means ± the standard errors of the mean. Asterisks denote significant differences relative to LXSN (*, P ≤ 0.05 [Student t test]). MN, micronuclei.
8 E6 reduces apoptosis and senescence in response to micronuclei.
Micronuclei induce apoptosis and senescence (66, 67). Because HPV replication requires cellular proliferation, we hypothesized that 8 E6 and 38 E6 impeded apoptosis and senescence in response to micronuclei. Since p53 can facilitate these responses, we examined p53 staining by immunofluorescence microscopy in HFK LXSN, HFK 8 E6, HFK 8 E6 Δ132-136, and HFK 38 E6 cells that did and did not have micronuclei (Fig. 6A). The presence of a micronucleus similarly increased p53 staining intensity in HFK LXSN, HFK 8 E6, and HFK 8 E6 Δ132-136 cells (Fig. 6B). HFK 38 E6 cells had significantly increased p53 intensity compared to all other HFK cells.
FIG 6.

HPV8 E6 reduces a cell’s apoptotic and senescent response to micronuclei. (A) Representative images of HFK cells stained for p53 (red) and DNA (DAPI, white), with (indicated by white arrow) or without a micronucleus. (B) Quantification of normalized p53 intensity in HFK cells with or without micronuclei. (C) Representative images of methylated H3K9 (red)- and DAPI (white)-stained HFK cells, with (indicated by white arrow) or without a micronucleus. (D) Quantification of normalized methylated H3k9 intensity in HFK cells with or without micronuclei. At least 200 cells/cell line were quantified across three independent experiments. (E) Representative images of HFK cells stained for cleaved caspase-3 (red) and DAPI (white), with (indicated by white arrow) or without a micronucleus. (F) Quantification of normalized cleaved caspase-3 intensity in iHFK cells, with or without micronuclei. We quantified ≥550 cells/cell line across four independent experiments. (G) Quantification of cell fate imaging micronucleus-positive iHFK LaminB1-GFP H2B-RFP cells for 30 h. Light- and dark-blue asterisks denote significant differences between “live, no mitosis” and “live, mitosis” relative to LXSN, respectively. Graphs depict means ± the standard errors of the mean. Asterisks denotes significant differences relative to LXSN unless specified by a bar (*, P ≤ 0.05; **, P ≤ 0.01 [Student t test]). In Δ8 E6, residues 132 to 136 were deleted from 8 E6. MN, micronuclei.
We next examined markers of senescence (histone H3 methylated at K9 [H3K9me3]) and apoptosis (cleaved caspase-3) in these cells. H3K9me3 staining intensity was increased by micronuclei in all cell lines (Fig. 6D). However, this increase was significantly reduced in HFK 8 E6 and HFK 8 E6 Δ132-136 cells compared to HFK LXSN cells (Fig. 6C and D). Cleaved caspase-3 staining was likewise elevated in all iHFK cell lines with micronuclei (Fig. 6E and F). However, 8 E6 significantly attenuated the increase in cleaved caspase-3 staining in cells with micronuclei (Fig. 6E and F). Because 8 E6 attenuates the accumulation of apoptosis and senescent markers in cells with micronuclei, we hypothesized that 8 E6 increased the frequency that cells with micronuclei underwent mitosis. To test this, we transduced LXSN and 8 E6 iHFK cells with mCherry-tagged histone H2B to visualize DNA and used time-lapse imaging to track the fate of cells with micronuclei over 30 h. This demonstrated that 8 E6 made it significantly more likely that a cell with a micronucleus completed mitosis (Fig. 6G; see also Movies S7 and S8).
8 E6 promotes chromothripsis events.
Micronuclear DNA is at risk of chromothripsis, a highly mutagenic process where hundreds to thousands of mutations occur in a single chromosome during a single progression through the cell cycle. This can be prevented by avoiding mitosis (34, 36, 39). Because 8 E6 increases the frequency of micronuclei and promotes proliferation (Fig. 3B and 6G), we hypothesized that 8 E6 caused chromothripsis. To detect chromothripsis, we performed whole-genome sequencing on passage-matched iHFK LXSN and 8 E6 cells with sequencing depths of 53× and 75×, respectively. Raw reads were trimmed for quality and analyzed using a standard pipeline, using the vector mutations as a control (see Materials and Methods). Shatterseek software (68) was used to determine chromothripsis by analyzing copy number changes (CN) and structural variant (SV) differences between cell lines. We identified nine chromosomes with chromothripsis mutations and seven others with high numbers of structural variants (Fig. 7A and B). Significant instability was not found in eight other chromosomes. We found smaller chromosomes were less likely to show instability following mitosis but did observe a decrease in copy number changes and an increase in structural variants per nucleotide (Fig. 7C).
FIG 7.

HPV8 E6 promotes noncanonical chromothripsis. (A) Table summarizing the chromothripsis status of iHFK 8 E6 chromosomes analyzed compared to iHFK LXSN (vector-control) chromosomes. (B) Representative copy numbers and structural variations in iHFK 8 E6 cells (chromosome 2). Vertical hashed bars show deletion-like events (orange), head-to-head inversions (black), and tail-to-tail inversions (green). Copy number (CN) variations are indicated by solid black bars ranging from 1 to 14. The red arrows denote an area with Shatterseek-defined chromothripsis. (C) Total copy number variations per nucleotide (CN/N) and structural variants per nucleotide (SV/N) per chromosome in iHFK 8 E6 cells. The purple line represents the CN/N values, and the blue bars represents the SV/N values. The chromosome color corresponds to the chromothripsis status from panel A.
DISCUSSION
β-HPV research often focuses on the ability of the β-HPV proteins to facilitate the accumulation of UV-induced mutations (69, 70). However, a growing body of evidence has demonstrated that 8 E6 also hinders genome integrity independent of UV exposure (26–28). In this study, we show that 8 E6 causes anaphase bridges by reducing BLM abundance (Fig. 1 and 2). In turn, this leads to an increased frequency of micronuclei (Fig. 3). These micronuclei are less likely to have intact membranes but surprisingly retain the localization of nuclear proteins and processes (Fig. 4 and 5). It should be noted that while 8 E6 reduced micronuclear membrane integrity in both primary and TERT-immortalized HFKs, the diminished were only significant in TERT-immortalized HFKs. Thus, 8 E6 may require other mutations to cause these disruptions. 8 E6 attenuates the antiproliferative responses that typically accompany the micronuclei that it causes which allow cells to complete mitosis (Fig. 6). Our whole-genome sequencing data suggest that this culminates in significant genome destabilization via chromothripsis (Fig. 7).
The ability of HPV8 to cause chromothripsis, mitotic defects, and micronuclei are not unique among HPVs. Other members of the alpha and beta genus of HPVs can also do this (71–74). This is not a cell culture phenomenon since micronuclei are also associated with alpha HPV oncogene expression in human tissues, where they can be used to distinguish between HPV-positive and HPV-negative tumors (75, 76). HPV16 oncoprotein E7 (HPV16 E7) exacerbates HPV16 E6-induced anaphase bridges and micronuclei (73, 74, 77). HPV38 E6 and E7 together also increase the frequency of genome destabilizing anaphase bridges (71). Our data show that HPV38 E6 alone can increase the frequency of micronuclei (Fig. 3C). Further, one caveat to our data are that chromothripsis was only analyzed in one TERT-immortalized HFK cell line. This it will be important to determine the extent that immortalization is required for 8 E6-associated chromothripsis. Similarly, the extent that 8 E6 causes chromothripsis in cells derived from different donors should also be examined.
The shared ability to make mitosis more mutagenic may be linked to the requirement of proliferatively active host cells for the completion of the HPV life cycle. Albeit via differing mechanisms, HPV16, HPV38, and HPV8 all express proteins that promote cell proliferation despite inhibitory stimuli, including those associated with mitotic errors (23, 78, 79). Micronuclei can trigger these inhibitory stimuli, resulting in apoptosis and senescence (80–82). Here, we show that 8 E6 promotes growth in cells with micronuclei by reducing apoptotic and senescence signaling (Fig. 6). This is notable because it likely leads to the chromothripsis described in Fig. 7, since completing mitosis with a micronucleus puts the micronuclear DNA at risk of chromothripsis (36, 39). There is a further risk should a second mitosis occur as the micronuclear DNA often becomes mis-segregated and continue to be segregated in a micronucleus (83, 84). We also found that 8 E6 facilitated replication in micronuclei without envelopes. This, too, is expected to be deleterious for the host genome since premature chromatin condensation at DNA replication sites can result in chromosome shattering (38). We find it notable that the HPV8 genome (and the genome of other HPVs) remains relatively stable despite replicating in the genome destabilizing environment caused by HPV E6 and E7. This suggests that the virus has found mechanisms to preserve the integrity of its genome in a harsh cellular environment.
The whole-genome sequencing analysis described here followed established protocols for identifying chromothripsis with one exception. Instead of identifying mutations by comparing our sequencing to the reference human genome, we made comparisons between two passage-matched hTERT-immortalized primary cell lines. This allowed us to specifically account for host mutations in the donor cells. This modification could improve the existing knowledge of (i) HPV16-induced chromothripsis (72), (ii) mutations in cellular genes that prevent chromothripsis, or (iii) any other systems where primary cells can be used.
There are several areas of inquiry that we would like to pursue moving forward. Here, we show that reduced BLM likely leads to chromothripsis. However, BLM regulates DNA recombination, replication, and both homologous and nonhomologous double-strand break repair pathways (85). Thus, we hypothesize that the 8 E6-induced BLM reduction results in as-yet-unreported defects in DNA repair. Whether 8 E6 is actively involved in recruiting nuclear factors to micronuclei without envelopes or causes this localization indirectly is also unclear. Compared to previous chromothripsis reports, the frequency of copy number oscillations with 8 E6 expression is higher, and the changed fragment sizes are smaller (68, 86). This observation suggests that HPV8 infection may result in a recognizable pattern with distinct mutations that if identified would provide evidence that HPV8 infections are mutagenic. It is unclear whether 8 E6 will continue to promote chromothripsis and anaphase bridges when expressed in the context of the complete early region. Similarly, is p300 degradation required for 8 E6 to cause chromothripsis? Do other β-HPV E6s cause chromothripsis? Due to the cost of the analysis, we only examined chromothripsis in one 8 E6-expressing cell line. Does it occur in cells from other donors?
MATERIALS AND METHODS
Cell culture.
U2OS and HCT 116 cells were maintained in Dulbecco modified Eagle medium supplemented with 10% fetal bovine serum and penicillin-streptomycin. Primary HFK cells were derived from neonatal human foreskins. hTERT-immortalized-HFK (iHFK cells were obtained from Michael Underbrink, University of Texas Medical Branch). HFK and iHFK cells were grown in EpiLife medium supplemented with calcium chloride (60 μM), human keratinocyte growth supplement (Thermo Fisher Scientific), or keratinocyte growth medium 2 (PromoCell) supplemented with calcium chloride (60 μM), SupplementMix, and penicillin-streptomycin. HPV genes were cloned, transfected, and confirmed as previously described (27).
Plasmids and siRNAs.
2×RFP-NLS (RFP-NLS) was a gift from Emily Hatch (see reference 32) and contains mCherry-TagRFP-NLS in the Gateway vector pDONOR20. GFP-BLM was a gift from Nathan Ellis (Addgene plasmid 80070 [http://n2t.net/addgene:80070]; RRID, Addgene_80070). pLV-LaminB1-GFP/H2B-mCherry was purchased from VectorBuilder (https://en.vectorbuilder.com/vector/VB210611-1255jkx.html). (87) Control siRNA-A was purchased from Santa Cruz (sc-37007). BLM siRNA was purchased from Thermo Scientific (catalog no. 146898). We utilized 60 pmol of siRNA for knockdown experiments.
Immunoblotting.
After being washed with ice-cold phosphate-buffered saline (PBS), cells were lysed with radioimmunoprecipitation assay lysis buffer (VWR Life Science) supplemented with phosphatase inhibitor cocktail 2 (Sigma) and protease inhibitor cocktail (Bimake). A Pierce bicinchoninic acid (BCA) protein assay kit (Thermo Scientific) was used to determine protein concentration. Equal protein lysates were run on Novex 4 to 12% Tris-glycine WedgeWell mini gels (Invitrogen) and transferred to Immobilon-P membranes (Millipore). Membranes were then probed with the following primary antibodies: glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Santa Cruz Biotechnologies, catalog no. sc-47724), pATR (Thr1989; catalog no. 58014S; Cell Signaling Technology), ATR (2790S; Cell Signaling Technology), pCHK1 (Ser345; 133D3, 2348S; Cell Signaling Technology), CHK1 (2G1D5; 2360S; Cell Signaling Technology), p300 (Santa Cruz Biotechnologies, catalog no. sc-584), and BLM (2742S; Cell Signaling Technology) and, after exposure to the matching horseradish peroxidase-conjugated secondary antibody, the cells were visualized using SuperSignal West Femto maximum sensitivity substrate (Thermo Scientific).
RT-qPCR.
Cells were lysed using TRIzol (Invitrogen), and RNA was isolated using an RNeasy kit (Qiagen). Two micrograms of RNA were reverse transcribed using the iScript cDNA synthesis kit (Bio-Rad). Quantitative real-time PCR (RT-qPCR) was performed in triplicate with a TaqMan FAM-MGB gene expression assay (Applied Biosystems) on an Applied Biosystems 7500/7500 Fast real-time PCR system (Thermo Scientific). The following probes (Thermo Scientific) were used: ACTB (Hs01060665_g1) and BLM (Hs00172060_m1).
Immunofluorescence microscopy.
Cells were seeded onto either 96-well glass-bottom plates (Cellvis) or etched coverslips and grown overnight. Cells were fixed with 4% formaldehyde. Then, 0.1% Triton-X solution in PBS was used to permeabilize the cells, followed by blocking with 3% bovine serum albumin in PBS for 30 min. Cells were then incubated with the following: alpha-tubulin (Abcam, catalog no. ab18251), α-tubulin (3873S; Cell Signaling Technology), p53 (2527S; Cell Signaling Technology), BLM (a gift from Matthew Weitzman, Children’s Hospital of Philadelphia), CENP-A (3-19; GTX13939; GeneTex), lamin B1 (C-12; sc-365214; Santa Cruz Biotechnology), phospho-Histone H2AX (Ser139; 20E3; 9718S; Cell Signaling Technology), 53bp1 (4937S; Cell Signaling Technology), Histone H3 (acetyl K27, ab4729; Abcam), Tri-methyl-Histone H3 (Lys9; D4W1U; 13969T; Cell Signaling Technology), cleaved caspase-3 (Asp175; 9661S; Cell Signaling Technology). The cells were washed and stained with the following appropriate secondary antibodies: Alexa Fluor 594-goat anti-rabbit (Thermo Scientific, catalog no. A11012) and Alexa Fluor 488 goat anti-mouse (Thermo Scientific, catalog no. A11001). After washing, the cells were stained with 2 μM 4′,6-diamidino-2-phenylindole (DAPI) in PBS and visualized with a Zeiss LSM 770 or a Zeiss LSM 880 Airyscan microscope. Laser power, gain, and the pinhole remained the same between experiments to ensure accurate quantification. Background fluorescence was used as an internal control between samples. Images were analyzed using ImageJ techniques, as described previously (88).
Detection of BrdU incorporation.
Detection of BrdU was performed the same as immunofluorescent cells with the addition of a 30-min incubation with 1.5 M HCl for 30 min between Triton-X and 3% bovine serum albumin. Primary antibodies were used as indicated and stained with Alexa Fluor 594 anti-BrdU (BU1/75 [ICR1]; ab20076; Abcam). After washing, the cells were stained with 2 μM DAPI in PBS and visualized with a Zeiss LSM 770 microscope. Images were analyzed using ImageJ techniques, as described previously (88).
53bp1 detection.
Cells were seeded onto etched coverslips and grown overnight. Cells were exposed to 10 μg/mL of zeocin for 10 min and then washed. At 1 h after zeocin treatment, the cells were fixed and stained according to the immunofluorescence microscopy steps described above.
Segregation error.
Cells were arrested in G1/S phase by culture in 2 mM thymidine for 16 h, washed to release, and grown for 9 h 10 min in the absence of thymidine to enhance mitotic cells. Cells were shaken into the media, concentrated, and cytocentrifuged onto glass coverslips. Cells were fixed with 4% formaldehyde. Then, 0.1% Triton-X solution in PBS was used to permeabilize the cells, followed by blocking with 3% bovine serum albumin in PBST supplemented with 300 mM glycine for 1 h. Primary and secondary antibodies were stained for 16 h and 1 h 20 min, respectively. After washing, the cells were stained with 2 μM DAPI in PBS and visualized with a Zeiss LSM 770 microscope. Images were analyzed using ImageJ techniques, as described previously (88).
Establishment of RFP-NLS and LaminB1-GFP/H2B-RFP-expressing cells and transient expression of BLM-GFP.
RFP-NLS expressing iHFK cells were generated by transfecting with the RFP-NLS expression plasmids using Xfect transfection reagent (TaKaRa Bio, catalog no. 631317). Cells containing RFP-NLS plasmids were selected using 1 μg/mL puromycin. Drug-resistant colonies were pooled after 10 days. LaminB1-GFP/H2B-RFP-expressing iHFK cells were generated by transducing them with LaminB1-GFP/H2B-RFP lentivirus according to the manufacturer’s protocol and then selected with 1 μg/mL puromycin until mock-transfected cells ceased proliferation.
Time-lapse imaging.
Cells were plated into 6-well glass-bottom plates (Cellvis) and imaged on a BioTek LionheartFX automated microscope with a 20× air objective at 37°C and 5% CO2 for 30 h. Images were captured using Gen5 software (BioTek). Videos were cropped and adjusted for brightness and contrast using ImageJ.
Whole-genome mate-pair sequencing.
Passage-matched iHFK LXSN and 8 E6 cells were subjected to whole-genome sequencing. DNA was extracted from cells using a Qiagen high-molecular-weight DNA extraction kit, library prepped using an Illumina DNA prep library prep kit, and sequenced on an Illumina Novaseq using an SP 250PE v1.5 cartridge according to the manufacturer’s specifications. The average coverages were 53× and 75× for iHFK LXSN and 8 E6 cells, respectively. The raw reads were analyzed using a validated pipeline with some modifications (86). Most notably, mutations in 8 E6-expressing HFK cells were identified by comparison to the sequencing from vector control cells rather than the reference genome (86). Raw reads were trimmed with Trimmomatic v0.40 and aligned with BWA v0.7.17 to GenBank reference GRCh37 (RefSeq GCF_000001405.25), and copy numbers (CN) were calculated with Xcavator using default parameters (89). The alignment was subjected to structural variant (SV) calling in sVABA using a log odds ratio of 4 and quality score of >30 (90). CN and SV variants were used to determine the likelihood of chromothripsis in Shatterseek with default parameters (68). Confidence was called for each chromosome based on the number of coinciding structural and copy number variants, the location of genomic breakpoints, and the frequency of all three parameters coinciding.
Measurement of protein half-life.
iHFK LXSN and 8 E6 cells were grown in 6-well plates until reaching ~80% confluence. Cells were treated with 50 μg/mL cycloheximide for the indicated times, and whole-cell lysates for all time points were harvested together. Western blot analysis was performed as described above.
Statistical analysis.
Unless otherwise noted, statistical significance was determined by using an unpaired Student t test and was confirmed, when appropriate, by two-way analysis of variance (ANOVA) with Tukey’s correction. Only P values of <0.05 are reported as significant.
ACKNOWLEDGMENTS
We thank and acknowledge the Kansas State University College of Veterinary Medicine (KSU-CVM) Confocal Core, especially Joel Sanneman, for assisting with our immunofluorescence imaging and Michael Underbrink for providing the TERT-immortalized HFKs. We also thank Emily Hatch (Fred Hutchinson Cancer Research Center, Seattle, WA) for advice and guidance that allowed us to characterize micronuclear and micronuclear membrane biology.
We thank the Terry Johnson Basic Cancer Research Center for their support of this project. We also received support from a career development award provided by the US Department of Defense’s Congressionally Directed Medical Research Program’s Peer Reviewed Cancer Research Program (CMDRP PRCRP CA160224 [N.A.W.]) and from the National Institutes of Health (NCI R15 CA242057 01A1). Finally, the research reported here was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award P20GM130448. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Supplemental material is available online only.
Contributor Information
Nicholas A. Wallace, Email: nwallac@ksu.edu.
Lawrence Banks, International Centre for Genetic Engineering and Biotechnology.
REFERENCES
- 1.Madan V, Lear JT, Szeimies R-M. 2010. Non-melanoma skin cancer. Lancet 375:673–685. 10.1016/S0140-6736(09)61196-X. [DOI] [PubMed] [Google Scholar]
- 2.Ciążyńska M, Kamińska-Winciorek G, Lange D, Lewandowski B, Reich A, Sławińska M, Pabianek M, Szczepaniak K, Hankiewicz A, Ułańska M, Morawiec J, Błasińska-Morawiec M, Morawiec Z, Piekarski J, Nejc D, Brodowski R, Zaryczańska A, Sobjanek M, Nowicki RJ, Owczarek W, Słowińska M, Wróbel K, Bieniek A, Woźniacka A, Skibińska M, Narbutt J, Niemczyk W, Ciążyński K, Lesiak A. 2021. The incidence and clinical analysis of non-melanoma skin cancer. Sci Rep 11:4337. 10.1038/s41598-021-83502-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 71:209–249. 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 4.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. 2018. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 68:394–424. 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 5.Guy GP, Machlin SR, Ekwueme DU, Yabroff KR. 2015. Prevalence and costs of skin cancer treatment in the United States, 2002–2006 and 2007–2011. Am J Prev Med 48:183–187. 10.1016/j.amepre.2014.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fahradyan A, Howell AC, Wolfswinkel EM, Tsuha M, Sheth P, Wong AK. 2017. Updates on the management of non-melanoma skin cancer (NMSC). Healthcare 5:82. 10.3390/healthcare5040082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lubov J, Labbé M, Sioufi K, Morand GB, Hier MP, Khanna M, Sultanem K, Mlynarek AM. 2021. Prognostic factors of head and neck cutaneous squamous cell carcinoma: a systematic review. J Otolaryngol Head Neck Surg 50:54. 10.1186/s40463-021-00529-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Didona D, Paolino G, Bottoni U, Cantisani C. 2018. Nonmelanoma skin cancer pathogenesis overview. Biomedicines 6:6. 10.3390/biomedicines6010006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kiviat NB. 1999. Papillomaviruses in non-melanoma skin cancer: epidemiological aspects. 6. Semin Cancer Biol 9:397–403. 10.1006/scbi.1999.0143. [DOI] [PubMed] [Google Scholar]
- 10.Orth G. 2008. Host defenses against human papillomaviruses: lessons from epidermodysplasia verruciformis. Curr Top Microbiol Immunol 321:59–83. 10.1007/978-3-540-75203-5_3. [DOI] [PubMed] [Google Scholar]
- 11.Nunes EM, Talpe-Nunes V, Sichero L. 2018. Epidemiology and biology of cutaneous human papillomavirus. Clinics (Sao Paulo) 73:e489s. 10.6061/clinics/2018/e489s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bandolin L, Borsetto D, Fussey J, Mosto MCD, Nicolai P, Menegaldo A, Calabrese L, Tommasino M, Boscolo-Rizzo P. 2020. Beta human papillomaviruses infection and skin carcinogenesis. Rev Med Virol 30:e2104. 10.1002/rmv.2104. [DOI] [PubMed] [Google Scholar]
- 13.Tommasino M. 2017. The biology of beta human papillomaviruses. Virus Res 231:128–138. 10.1016/j.virusres.2016.11.013. [DOI] [PubMed] [Google Scholar]
- 14.Chahoud J, Semaan A, Chen Y, Cao M, Rieber AG, Rady P, Tyring SK. 2016. Association between β-genus human papillomavirus and cutaneous squamous cell carcinoma in immunocompetent individuals: a meta-analysis. JAMA Dermatol 152:1354–1364. 10.1001/jamadermatol.2015.4530. [DOI] [PubMed] [Google Scholar]
- 15.Patel AS, Karagas MR, Perry AE, Nelson HH. 2008. Exposure profiles and human papillomavirus infection in skin cancer: an analysis of 25 genus β-types in a population-based study. J Invest Dermatol 128:2888–2893. 10.1038/jid.2008.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weissenborn SJ, Nindl I, Purdie K, Harwood C, Proby C, Breuer J, Majewski S, Pfister H, Wieland U. 2005. Human papillomavirus-DNA loads in actinic keratoses exceed those in non-melanoma skin cancers. J Invest Dermatol 125:93–97. 10.1111/j.0022-202X.2005.23733.x. [DOI] [PubMed] [Google Scholar]
- 17.Hufbauer M, Akgül B. 2017. Molecular mechanisms of human papillomavirus induced skin carcinogenesis. Viruses 9:187. 10.3390/v9070187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Howley PM, Pfister HJ. 2015. Beta genus papillomaviruses and skin cancer. Virology 479–480:290–296. 10.1016/j.virol.2015.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wendel SO, Wallace NA. 2017. Loss of genome fidelity: beta HPVs and the DNA damage response. Front Microbiol 8:2250. 10.3389/fmicb.2017.02250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meyers JM, Grace M, Uberoi A, Lambert PF, Munger K. 2018. Inhibition of TGF-β and NOTCH signaling by cutaneous papillomaviruses. Front Microbiol 9:389. 10.3389/fmicb.2018.00389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Viarisio D, Müller-Decker K, Accardi R, Robitaille A, Dürst M, Beer K, Jansen L, Flechtenmacher C, Bozza M, Harbottle R, Voegele C, Ardin M, Zavadil J, Caldeira S, Gissmann L, Tommasino M. 2018. Beta HPV38 oncoproteins act with a hit-and-run mechanism in ultraviolet radiation-induced skin carcinogenesis in mice. PLoS Pathog 14:e1006783. 10.1371/journal.ppat.1006783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dong W, Kloz U, Accardi R, Caldeira S, Tong W-M, Wang Z-Q, Jansen L, Dürst M, Sylla BS, Gissmann L, Tommasino M. 2005. Skin hyperproliferation and susceptibility to chemical carcinogenesis in transgenic mice expressing E6 and E7 of human papillomavirus type 38. J Virol 79:14899–14908. 10.1128/JVI.79.23.14899-14908.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meyers JM, Uberoi A, Grace M, Lambert PF, Munger K. 2017. Cutaneous HPV8 and MmuPV1 E6 proteins target the NOTCH and TGF-β tumor suppressors to inhibit differentiation and sustain keratinocyte proliferation. PLoS Pathog 13:e1006171. 10.1371/journal.ppat.1006171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marcuzzi GP, Hufbauer M, Kasper HU, Weissenborn SJ, Smola S, Pfister H. 2009. Spontaneous tumour development in human papillomavirus type 8 E6 transgenic mice and rapid induction by UV-light exposure and wounding. J Gen Virol 90:2855–2864. 10.1099/vir.0.012872-0. [DOI] [PubMed] [Google Scholar]
- 25.Schaper ID, Marcuzzi GP, Weissenborn SJ, Kasper HU, Dries V, Smyth N, Fuchs P, Pfister H. 2005. Development of skin tumors in mice transgenic for early genes of human papillomavirus type 8. Cancer Res 65:1394–1400. 10.1158/0008-5472.CAN-04-3263. [DOI] [PubMed] [Google Scholar]
- 26.Dacus D, Cotton C, McCallister TX, Wallace NA. 2020. Beta human papillomavirus 8E6 attenuates LATS phosphorylation after failed cytokinesis. J Virol 94. 10.1128/JVI.02184-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wallace NA, Robinson K, Galloway DA. 2014. Beta human papillomavirus E6 expression inhibits stabilization of p53 and increases tolerance of genomic instability. J Virol 88:6112–6127. 10.1128/JVI.03808-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dacus D, Riforgiate E, Wallace NA. 2020. β-HPV 8E6 combined with TERT expression promotes long-term proliferation and genome instability after cytokinesis failure. Virology 549:32–38. 10.1016/j.virol.2020.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ye CJ, Sharpe Z, Alemara S, Mackenzie S, Liu G, Abdallah B, Horne S, Regan S, Heng HH. 2019. Micronuclei and genome chaos: changing the system inheritance. Genes 10:366. 10.3390/genes10050366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kisurina-Evgenieva OP, Sutiagina OI, Onishchenko GE. 2016. Biogenesis of micronuclei. Biochemistry (Mosc) 81:453–464. 10.1134/S0006297916050035. [DOI] [PubMed] [Google Scholar]
- 31.Naim V, Rosselli F. 2009. The FANC pathway and BLM collaborate during mitosis to prevent micro-nucleation and chromosome abnormalities. Nat Cell Biol 11:761–768. 10.1038/ncb1883. [DOI] [PubMed] [Google Scholar]
- 32.Hatch EM, Fischer AH, Deerinck TJ, Hetzer MW. 2013. Catastrophic nuclear envelope collapse in cancer cell micronuclei. Cell 154:47–60. 10.1016/j.cell.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Terradas M, Martín M, Hernández L, Tusell L, Genescà A. 2012. Nuclear envelope defects impede a proper response to micronuclear DNA lesions. Mutat Res 729:35–40. 10.1016/j.mrfmmm.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 34.Zhang C-Z, Spektor A, Cornils H, Francis JM, Jackson EK, Liu S, Meyerson M, Pellman D. 2015. Chromothripsis from DNA damage in micronuclei. Nature 522:179–184. 10.1038/nature14493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hatch EM, Hetzer MW. 2015. Linking micronuclei to chromosome fragmentation. Cell 161:1502–1504. 10.1016/j.cell.2015.06.005. [DOI] [PubMed] [Google Scholar]
- 36.Mammel AE, Hatch EM. 2021. Genome instability from nuclear catastrophe and DNA damage. Semin Cell Dev Biol 123:131–139. 10.1016/j.semcdb.2021.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Crasta K, Ganem NJ, Dagher R, Lantermann AB, Ivanova EV, Pan Y, Nezi L, Protopopov A, Chowdhury D, Pellman D. 2012. DNA breaks and chromosome pulverization from errors in mitosis. Nature 482:53–58. 10.1038/nature10802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Terzoudi GI, Karakosta M, Pantelias A, Hatzi VI, Karachristou I, Pantelias G. 2015. Stress induced by premature chromatin condensation triggers chromosome shattering and chromothripsis at DNA sites still replicating in micronuclei or multinucleate cells when primary nuclei enter mitosis. Mutat Res Genet Toxicol Environ Mutagen 793:185–198. 10.1016/j.mrgentox.2015.07.014. [DOI] [PubMed] [Google Scholar]
- 39.Ly P, Cleveland DW. 2017. Rebuilding chromosomes after catastrophe: emerging mechanisms of chromothripsis. Trends Cell Biol 27:917–930. 10.1016/j.tcb.2017.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Perry J, Ashford B, Thind AS, Gauthier M-E, Minaei E, Major G, Iyer NG, Gupta R, Clark J, Ranson M. 2020. Comprehensive mutational and phenotypic characterization of new metastatic cutaneous squamous cell carcinoma cell lines reveal novel drug susceptibilities. Int J Mol Sci 21:9536. 10.3390/ijms21249536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ashford BG. 2019. The mutational landscape of metastatic cutaneous squamous cell carcinoma. PhD thesis. University of Wollongong, Wollongong, Australia. [Google Scholar]
- 42.Chan K-L, North PS, Hickson ID. 2007. BLM is required for faithful chromosome segregation and its localization defines a class of ultrafine anaphase bridges. EMBO J 26:3397–3409. 10.1038/sj.emboj.7601777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ke Y, Huh J-W, Warrington R, Li B, Wu N, Leng M, Zhang J, Ball HL, Li B, Yu H. 2011. PICH and BLM limit histone association with anaphase centromeric DNA threads and promote their resolution. EMBO J 30:3309–3321. 10.1038/emboj.2011.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Petsalaki E, Dandoulaki M, Morrice N, Zachos G. 2014. Chk1 protects against chromatin bridges by constitutively phosphorylating BLM serine 502 to inhibit BLM degradation. J Cell Sci 127:3902–3908. 10.1242/jcs.155176. [DOI] [PubMed] [Google Scholar]
- 45.Snow JA, Murthy V, Dacus D, Hu C, Wallace NA. 2019. β-HPV 8E6 attenuates ATM and ATR signaling in response to UV damage. Pathogens 8:267. 10.3390/pathogens8040267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hufbauer M, Cooke J, van der Horst GTJ, Pfister H, Storey A, Akgül B. 2015. Human papillomavirus mediated inhibition of DNA damage sensing and repair drives skin carcinogenesis. Mol Cancer 14:183. 10.1186/s12943-015-0453-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wallace NA, Robinson K, Howie HL, Galloway DA. 2012. HPV 5 and 8 E6 abrogate ATR activity resulting in increased persistence of UVB induced DNA damage. PLoS Pathog 8:e1002807. 10.1371/journal.ppat.1002807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Howie HL, Koop JI, Weese J, Robinson K, Wipf G, Kim L, Galloway DA. 2011. Beta-HPV 5 and 8 E6 promote p300 degradation by blocking AKT/p300 association. PLoS Pathog 7:e1002211. 10.1371/journal.ppat.1002211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brooks N, Raja M, Young BW, Spencer GJ, Somervaille TC, Pegg NA. 2019. CCS1477: a Novel small molecule inhibitor of p300/CBP bromodomain for the treatment of acute myeloid leukaemia and multiple myeloma. Blood 134:2560–2560. 10.1182/blood-2019-124707. [DOI] [Google Scholar]
- 50.Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, Pleasance ED, Lau KW, Beare D, Stebbings LA, McLaren S, Lin M-L, McBride DJ, Varela I, Nik-Zainal S, Leroy C, Jia M, Menzies A, Butler AP, Teague JW, Quail MA, Burton J, Swerdlow H, Carter NP, Morsberger LA, Iacobuzio-Donahue C, Follows GA, Green AR, Flanagan AM, Stratton MR, Futreal PA, Campbell PJ. 2011. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 144:27–40. 10.1016/j.cell.2010.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu S, Kwon M, Mannino M, Yang N, Renda F, Khodjakov A, Pellman D. 2018. Nuclear envelope assembly defects link mitotic errors to chromothripsis. Nature 561:551–555. 10.1038/s41586-018-0534-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vietri M, Schultz SW, Bellanger A, Jones CM, Petersen LI, Raiborg C, Skarpen E, Pedurupillay CRJ, Kjos I, Kip E, Timmer R, Jain A, Collas P, Knorr RL, Grellscheid SN, Kusumaatmaja H, Brech A, Micci F, Stenmark H, Campsteijn C. 2020. Unrestrained ESCRT-III drives micronuclear catastrophe and chromosome fragmentation. Nat Cell Biol 22:856–867. 10.1038/s41556-020-0537-5. [DOI] [PubMed] [Google Scholar]
- 53.Rosin MP, German J. 1985. Evidence for chromosome instability in vivo in bloom syndrome: increased numbers of micronuclei in exfoliated cells. Hum Genet 71:187–191. 10.1007/BF00284570. [DOI] [PubMed] [Google Scholar]
- 54.Guo X, Ni J, Liang Z, Xue J, Fenech MF, Wang X. 2019. The molecular origins and pathophysiological consequences of micronuclei: new insights into an age-old problem. Mutat Res Rev Mutat Res 779:1–35. 10.1016/j.mrrev.2018.11.001. [DOI] [PubMed] [Google Scholar]
- 55.Fenech M, Knasmueller S, Bolognesi C, Holland N, Bonassi S, Kirsch-Volders M. 2020. Micronuclei as biomarkers of DNA damage, aneuploidy, inducers of chromosomal hypermutation and as sources of proinflammatory DNA in humans. Mutat Res Rev Mutat Res 786:108342. 10.1016/j.mrrev.2020.108342. [DOI] [PubMed] [Google Scholar]
- 56.Rosefort C, Fauth E, Zankl H. 2004. Micronuclei induced by aneugens and clastogens in mononucleate and binucleate cells using the cytokinesis block assay. Mutagenesis 19:277–284. 10.1093/mutage/geh028. [DOI] [PubMed] [Google Scholar]
- 57.MacDonald KM, Benguerfi S, Harding SM. 2020. Alerting the immune system to DNA damage: micronuclei as mediators. Essays Biochem 64:753–764. 10.1042/EBC20200016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Terradas M, Martín M, Genescà A. 2016. Impaired nuclear functions in micronuclei results in genome instability and chromothripsis. Arch Toxicol 90:2657–2667. 10.1007/s00204-016-1818-4. [DOI] [PubMed] [Google Scholar]
- 59.Hatch EM, Hetzer MW. 2016. Nuclear envelope rupture is induced by actin-based nucleus confinement. J Cell Biol 215:27–36. 10.1083/jcb.201603053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hashimoto K, Nakajima Y, Matsumura S, Chatani F. 2010. An in vitro micronucleus assay with size-classified micronucleus counting to discriminate aneugens from clastogens. Toxicol in Vitro 24:208–216. 10.1016/j.tiv.2009.09.006. [DOI] [PubMed] [Google Scholar]
- 61.Marsh EK, Delury CP, Davies NJ, Weston CJ, Miah MAL, Banks L, Parish JL, Higgs MR, Roberts S. 2017. Mitotic control of human papillomavirus genome-containing cells is regulated by the function of the PDZ-binding motif of the E6 oncoprotein. Oncotarget 8:19491–19506. 10.18632/oncotarget.14469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hoffelder DR, Luo L, Burke NA, Watkins SC, Gollin SM, Saunders WS. 2004. Resolution of anaphase bridges in cancer cells. Chromosoma 112:389–397. 10.1007/s00412-004-0284-6. [DOI] [PubMed] [Google Scholar]
- 63.Kneissig M, Keuper K, de Pagter MS, van Roosmalen MJ, Martin J, Otto H, Passerini V, Campos Sparr A, Renkens I, Kropveld F, Vasudevan A, Sheltzer JM, Kloosterman WP, Storchova Z. 2019. Micronuclei-based model system reveals functional consequences of chromothripsis in human cells. Elife 8:e50292. 10.7554/eLife.50292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vargas JD, Hatch EM, Anderson DJ, Hetzer MW. 2012. Transient nuclear envelope rupturing during interphase in human cancer cells. Nucleus 3:88–100. 10.4161/nucl.18954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hatch EM. 2018. Nuclear envelope rupture: little holes, big openings. Curr Opin Cell Biol 52:66–72. 10.1016/j.ceb.2018.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kwon M, Leibowitz ML, Lee J-H. 2020. Small but mighty: the causes and consequences of micronucleus rupture. Exp Mol Med 52:1777–1786. 10.1038/s12276-020-00529-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gekara NO. 2017. DNA damage-induced immune response: micronuclei provide key platform. J Cell Biol 216:2999–3001. 10.1083/jcb.201708069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cortés-Ciriano I, Lee JJ-K, Xi R, Jain D, Jung YL, Yang L, Gordenin D, Klimczak LJ, Zhang C-Z, Pellman DS, Park PJ, PCAWG Consortium . 2020. Comprehensive analysis of chromothripsis in 2,658 human cancers using whole-genome sequencing. Nat Genet 52:331–341. 10.1038/s41588-019-0576-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rollison DE, Viarisio D, Amorrortu RP, Gheit T, Tommasino M. 2019. An emerging issue in oncogenic virology: the role of beta human papillomavirus types in the development of cutaneous squamous cell carcinoma. J Virol 93:e01003-18. 10.1128/JVI.01003-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lambert PF, Münger K, Rösl F, Hasche D, Tommasino M. 2020. Beta human papillomaviruses and skin cancer. Nature 588:E20–E21. 10.1038/s41586-020-3023-0. [DOI] [PubMed] [Google Scholar]
- 71.Gabet A-S, Accardi R, Bellopede A, Popp S, Boukamp P, Sylla BS, Londoño-Vallejo JA, Tommasino M. 2008. Impairment of the telomere/telomerase system and genomic instability are associated with keratinocyte immortalization induced by the skin human papillomavirus type 38. FASEB J 22:622–632. 10.1096/fj.07-8389com. [DOI] [PubMed] [Google Scholar]
- 72.Schütze DM, Krijgsman O, Snijders PJF, Ylstra B, Weischenfeldt J, Mardin BR, Stütz AM, Korbel JO, de Winter JP, Meijer CJLM, Quint WGV, Bosch L, Wilting SM, Steenbergen RDM. 2016. Immortalization capacity of HPV types is inversely related to chromosomal instability. Oncotarget 7:37608–37621. 10.18632/oncotarget.8058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Duensing S, Münger K. 2002. The human papillomavirus type 16 E6 and E7 oncoproteins independently induce numerical and structural chromosome instability. Cancer Res 62:7075–7082. [PubMed] [Google Scholar]
- 74.Plug-DeMaggio AW, Sundsvold T, Wurscher MA, Koop JI, Klingelhutz AJ, McDougall JK. 2004. Telomere erosion and chromosomal instability in cells expressing the HPV oncogene 16E6. Oncogene 23:3561–3571. 10.1038/sj.onc.1207388. [DOI] [PubMed] [Google Scholar]
- 75.Adam ML, Pini C, Túlio S, Cantalice JCLL, Torres RA, Correia MTDS. 2015. Assessment of the association between micronuclei and the degree of uterine lesions and viral load in women with human papillomavirus. Cancer Genomics Proteomics 12:67–71. [PubMed] [Google Scholar]
- 76.Cassel APR, Barcellos RB, da Silva CMD, de Matos Almeida SE, Rossetti MLR. 2014. Association between human papillomavirus (HPV) DNA and micronuclei in normal cervical cytology. Genet Mol Biol 37:360–363. 10.1590/s1415-47572014005000010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Marullo R, Werner E, Zhang H, Chen GZ, Shin DM, Doetsch PW. 2015. HPV16 E6 and E7 proteins induce a chronic oxidative stress response via NOX2 that causes genomic instability and increased susceptibility to DNA damage in head and neck cancer cells. Carcinogenesis 36:1397–1406. 10.1093/carcin/bgv126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jackson S, Storey A. 2000. E6 proteins from diverse cutaneous HPV types inhibit apoptosis in response to UV damage. Oncogene 19:592–598. 10.1038/sj.onc.1203339. [DOI] [PubMed] [Google Scholar]
- 79.Lanfredini S, Olivero C, Borgogna C, Calati F, Powell K, Davies K-J, De Andrea M, Harries S, Tang HKC, Pfister H, Gariglio M, Patel GK. 2017. HPV8 field cancerization in a transgenic mouse model is due to Lrig1+ keratinocyte stem cell expansion. J Invest Dermatol 137:2208–2216. 10.1016/j.jid.2017.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yang H, Wang H, Ren J, Chen Q, Chen ZJ. 2017. cGAS is essential for cellular senescence. Proc Natl Acad Sci USA 114:E4612–E4620. 10.1073/pnas.1705499114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Glück S, Guey B, Gulen MF, Wolter K, Kang T-W, Schmacke NA, Bridgeman A, Rehwinkel J, Zender L, Ablasser A. 2017. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat Cell Biol 19:1061–1070. 10.1038/ncb3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Maciejowski J, Hatch EM. 2020. Nuclear membrane rupture and its consequences. Annu Rev Cell Dev Biol 36:85–114. 10.1146/annurev-cellbio-020520-120627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Soto M, García-Santisteban I, Krenning L, Medema RH, Raaijmakers JA. 2018. Chromosomes trapped in micronuclei are liable to segregation errors. J Cell Sci 131:jcs214742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.He B, Gnawali N, Hinman AW, Mattingly AJ, Osimani A, Cimini D. 2019. Chromosomes missegregated into micronuclei contribute to chromosomal instability by missegregating at the next division. Oncotarget 10:2660–2674. 10.18632/oncotarget.26853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kaur E, Agrawal R, Sengupta S. 2021. Functions of BLM helicase in cells: is it acting like a double-edged sword? Front Genet 12:634789. 10.3389/fgene.2021.634789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Voronina N, Wong JKL, Hübschmann D, Hlevnjak M, Uhrig S, Heilig CE, Horak P, Kreutzfeldt S, Mock A, Stenzinger A, Hutter B, Fröhlich M, Brors B, Jahn A, Klink B, Gieldon L, Sieverling L, Feuerbach L, Chudasama P, Beck K, Kroiss M, Heining C, Möhrmann L, Fischer A, Schröck E, Glimm H, Zapatka M, Lichter P, Fröhling S, Ernst A. 2020. The landscape of chromothripsis across adult cancer types. Nat Commun 11:2320. 10.1038/s41467-020-16134-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hu P, Beresten S, van Brabant A, Ye T-Z, Pandolfi P-P, Johnson FB, Guarente L, Ellis NA. 2001. Evidence for BLM and topoisomerase IIIα interaction in genomic stability. Hum Mol Genet 10:1287–1298. 10.1093/hmg/10.12.1287. [DOI] [PubMed] [Google Scholar]
- 88.Murthy V, Dacus D, Gamez M, Hu C, Wendel SO, Snow J, Kahn A, Walterhouse SH, Wallace NA. 2018. Characterizing DNA repair processes at transient and long-lasting double-strand DNA breaks by immunofluorescence microscopy. J Vis Exp 2018:e57653. 10.3791/57653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Magi A, Pippucci T, Sidore C. 2017. XCAVATOR: accurate detection and genotyping of copy number variants from second and third generation whole-genome sequencing experiments. BMC Genomics 18:747. 10.1186/s12864-017-4137-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wala JA, Bandopadhayay P, Greenwald NF, O’Rourke R, Sharpe T, Stewart C, Schumacher S, Li Y, Weischenfeldt J, Yao X, Nusbaum C, Campbell P, Getz G, Meyerson M, Zhang C-Z, Imielinski M, Beroukhim R. 2018. SvABA: genome-wide detection of structural variants and indels by local assembly. Genome Res 28:581–591. 10.1101/gr.221028.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1. Download jvi.01015-22-s0001.avi, AVI file, 19.4 MB (19.9MB, avi)
Movie S2. Download jvi.01015-22-s0002.avi, AVI file, 14.6 MB (14.9MB, avi)
Movie S3. Download jvi.01015-22-s0003.avi, AVI file, 17.5 MB (17.9MB, avi)
Movie S4. Download jvi.01015-22-s0004.avi, AVI file, 12.6 MB (12.9MB, avi)
Movie S5. Download jvi.01015-22-s0005.avi, AVI file, 1.2 MB (1.3MB, avi)
Movie S6. Download jvi.01015-22-s0006.avi, AVI file, 1.5 MB (1.5MB, avi)
Legends of Movies S1 to S8. Download jvi.01015-22-s0009.pdf, PDF file, 0.2 MB (170.9KB, pdf)