

Abstract
Cancer vaccines aim to direct the immune system to eradicate cancer cells. We review the essential immunologic concepts underpinning natural immunity, and highlight the multiple unique challenges faced by vaccines targeting cancer. Recent technological advances in mass spectrometry, neoantigen prediction, genetically and pharmacologically engineered mouse models, and single cell ‘omics have revealed new biology which can help to bridge this divide. We particularly focus on translationally relevant aspects, such as antigen selection and delivery and monitoring human post-vaccination responses, and encourage more aggressive exploration of novel approaches.
ETOC Blurb
For many decades, vaccines have provided effective protection against a variety of infectious diseases. Attention is now focussing on utilising the power of the immune system to target tumours through the development of cancer vaccines. This review provides a background in how the immune system responds to cancer, how these immune responses can be harnessed for cancer vaccines and highlights some of the remaining challenges.
Introduction
The goal for cancer vaccines is obvious – apply what has been done for a few infectious diseases and focus the power of human immunity on eliminating tumor cells. There are encouraging signs that this aim is achievable, but success remains elusive. Immunity has evolved over millennia to effectively respond to and prevent future pathogen attack, providing a well-tested template for vaccines for infectious diseases. However, tumors are not acute foreign events, but rather evolve from and within the host and are only apparent clinically after escape from immune pressure that controls incipient neoplasms (Dunn et al., 2002). Moreover, surviving tumor cells accumulate intrinsic defenses and co-opt other cells, including immune cells, to support escape. The resulting complex ecosystem of the immunosuppressive microenvironment impacts both innate and adaptive immune cells, can extend beyond the tumor border (van den Hout et al., 2017) and can even alter newly infiltrated cells (Li et al., 2022). Thus, therapeutic cancer vaccines, meant to treat that extant disease, face distinct challenges from prophylactic vaccines for infectious pathogens.
Undeniably a disease of mutation, tumor possess enormous molecular diversity and exquisite individuality; cancer is thus not one or a hundred, but thousands of diseases. Those mutations also bring opportunity: novel non-self-protein targets for immune recognition (‘neoantigens’). However, the vastness of that mutational landscape makes target identification a far more difficult task than for foreign infectious pathogens with relatively low genetic complexity.
Herein, we review the essential immunologic concepts underpinning productive immune responses and highlight the unique demands faced by vaccines targeting cancer. Numerous recent technological advances – from mass spectrometry and machine learning in neoantigen prediction, to genetically and pharmacologically engineered mouse models, to advances in single cell transcriptomics and genomics – have revealed new biology and point to how to bridge this divide. We particularly focus on those aspects that are translationally relevant, such as antigen selection and delivery, as well as unique challenges, like the immunosuppressive tumor immune microenvironment (TIME) and how to gather evidence for vaccine efficacy.
Natural Immunity – The starting point
Vaccines are pharmacologic manipulations of our immune system designed to achieve the physiological effect of defense and protection from a pathogen. Hence, a logical starting point is natural immunity, which has been ‘educated’ over eons by endless challenges from extant and emergent pathogens. Here, we review our current understanding of the contributing key molecular and cellular players and how these principles can be applied to cancer vaccines.
It takes a village
Perhaps the most remarkable feature of natural immunity is its ability parlay the initial events of pathogen infection into a multi-pronged, target-agnostic and target-specific attack (Figure 1). At the start, many cell types, but particularly innate immune myeloid cells, use pattern recognition receptors (PRR) to sense pathogen-derived foreign materials as well as byproducts from damaged tissue. These ‘danger signals’ (i.e. Pathogen- and Damage-Associated Molecular Patterns; PAMPs and DAMPs, respectively) trigger inflammatory signals which recruit and activate other innate immune cell types early on to broadly and non-specifically kill cells altered by infection. Importantly, PAMP/DAMP signals also trigger innate immune cells to initiate long-lasting adaptive immunity, the sine qua non for cancer vaccines.
Figure 1. Natural immunity to an intracellular pathogen.
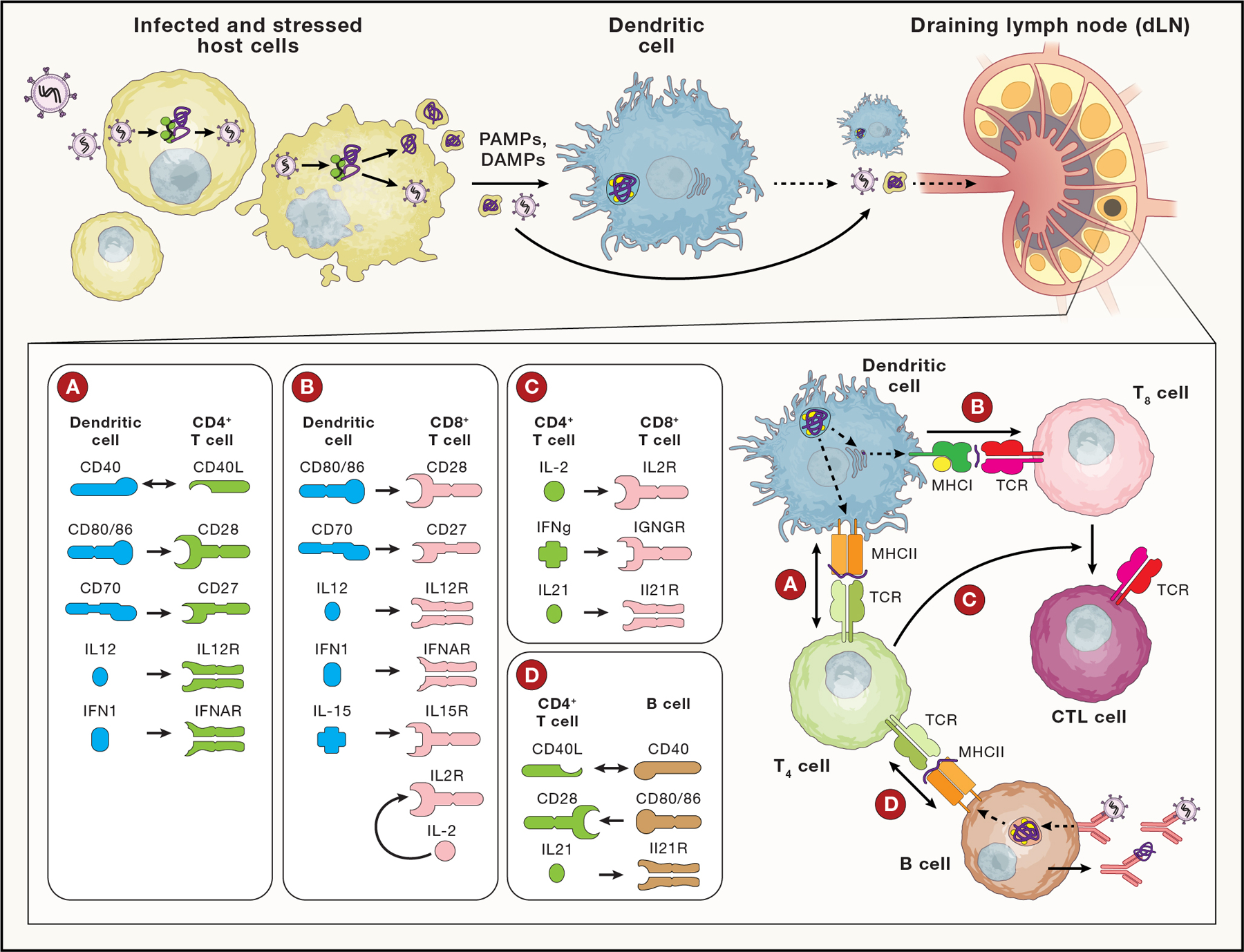
Infected and stressed host cells (top left) release PAMP/DAMPs, activating APCs, like DCs. Activated DCs mature, upregulating phagocytosis, antigen presentation and co-stimulatory molecules, and migrate to a draining lymph node (dLN); lymph drainage also transports local antigens to the dLN. The activated DCs present MHCII restricted peptides to CD4+ T cells, which can in turn license DCs (including via CD40L:CD40 signals; see Inset A) to cross-present antigen on MHCI and activate CD8+ T cells. Cytokines from DCs also shape CD4+ Helper T cell differentiation. DCs, via p:MHCI stimulation of TCR and co-stimulatory signals (Inset B) activate CD8 cells to respond, produce IL2 and IL-12 (for autocrine signaling) and differentiate into cytotoxic T lymphocytes (CTLs). CD4+ T cells potentiate CTL differentiation via IL2, maintain CTL effector function in viral infection via IL21 and induce CD8+ resident memory differentiation via IFNγ (Inset C). CD4+ T cells that recognize cognate p:MHCII on B cells, can provide co-stimulation (Inset D) to support affinity maturation, antibody class switching and plasma cell differentiation. Abbreviations: Pathogen- /Damage- associated molecular pattern (PAMP/DAMP), Dendritic cell (DC), T4 (CD4+ Helper T cell), T8 (CD8+ T cell), CTL (Cytotoxic T lymphocyte), B (B cell), p:MHC (peptide bound multi-histocompatibility complex), TCR (T Cell Receptor).
Adaptive immunity utilizes protein-protein interactions to provide its hallmark exquisite specificity in responding to epitopes from protein antigens encoded by the genome of the invading organism. PAMP/DAMP-activated myeloid lineage antigen-presenting cells (APCs, e.g. dendritic cells [DCs]) recognize and ingest dying cells and cell debris, proteolytically processing them into small peptides. A subset of those peptides will specifically bind one or more members of the large family of highly divergent major histocompatibility complexes (MHCI and MHCII; often referred to as HLAI and II, respectively, in humans) such that they are displayed on the cell surface. These MHC-epitope complexes are then recognized by the highly diverse family of T-cell receptors (TCR), each uniquely expressed on the surface of CD8+ and CD4+ T cells. Importantly, those same pathogen proteins are proteolytically processed within, and displayed on the surface of infected cells to facilitate T cell immune surveillance. Cell-cell communication, critically dependent on highly specific MHC:peptide∷TCR interactions, enables and strengthens such responses. Pathogen-specific CD4+ T cells provide critical immunologic ‘help’ to produce more effective CD8+ T cells by licensing APCs via costimulatory receptors (e.g. CD40-CD40L) and by local cytokine secretion (acting on APCs and CD8+ T cells).
Simultaneously, rare target-specific B cells scan the extracellular milieu for proteins (released from stressed/dying cells) that bind to their unique but similarly highly diverse B-cell receptor (BCR). Antigen binding stimulates B cell clonal expansion and maturation to produce one or more forms of a secreted target-specific antibody. B cells may also function as APCs, presenting processed peptides from those target (and other co-internalized) proteins on MHCII to cognate CD4+ T cells, which in turn, stimulate the presenting B cell. Although initiated in infected tissue, these cellular and molecular interactions largely occur, and are spatially and temporally enabled, within highly specialized lymph nodes (LN) (Grant et al., 2020).
Adaptive immunity thus depends on the recognition of epitope-containing protein antigens. A critical challenge is separating the wheat of desirable targets from the chaff of self. Which epitopes an individual can even ‘see’ is in part defined by the limited subset of MHC alleles encoded in their genome, amongst the hundreds that exist across the species (allele diversity across the species contributes to population-level control). Foreign- vs self-specificity arises at multiple levels beginning with central tolerance during progenitor T and B cell development, wherein TCRs and BCRs with strong reactivity to self are removed from repertoires, to peripheral tolerance, featuring target-specific Treg cells as well as varied immune dampening mechanisms that prevent response in the absence of clear danger. Additionally, immunologic history and environment (e.g. the highly diverse gut microbiome (Fluckiger et al., 2020)), and the possible reactivity of a given TCR to different MHC:peptides (Birnbaum et al., 2014) can affect the TCR profile, potentially leaving irreplaceable ‘holes’ in the repertoire.
What then is unique about cancer and cancer vaccines with respect to natural immunity? First, cancer is a disease of self, without (except for a handful of virally-induced cancers) a ‘foreign invader.’ This provides cancer major advantages: natural immunity is limited by the loss of foreign PAMPs and completely foreign antigens as surveillance tools, and by physiologic dampening mechanisms that prevent damage to self. Fortunately, cancer cell intrinsic mutations central to transformation also create neoantigen targets for the immune system, and the dysregulated growth and DNA repair processes of malignant cells lead to further ‘passenger’ mutations, unique to the cancer cell and its descendants, and provide substantially more neoantigens. Mutations in cancer thus establish an evolutionary competition with the immune system, in which the immune system sometimes wins (‘Elimination’) or generates a persistent draw (‘Equilibrium’); or the cancer eventually wins (‘Escape’) (Dunn et al., 2002). But even this Achille’s Heel can have two sides, as genetically heterogeneous tumors enhance the opportunities for immune escape by target antigen loss (Anagnostou et al., 2017). Chronic infections illuminate another important aspect of the immunity-cancer interface. Continual antigen exposure, also at play in cancer, can phenotypically shift cytolytic T cells to an ‘exhausted’ state. This shift compromises the ability of T cells to eliminate infected target cells (Philip and Schietinger, 2021), resulting in an equilibrium between viral control and tissue damage.
An immunosuppressive, tissue-specific microenvironment – The endpoint
Tumors contain not only cancerous cells but also numerous non-malignant cell types, paradoxically including, immune cells, creating a suppressive niche that protects the tumor from immune attack.
Long characterized as highly vascularized, glucose-depleted and hypoxic, the rich cellular immune features of the TIME were linked to clinical outcomes almost two decades ago and have been extensively explored since. The TIME in a primary tumor may differ from that in a metastasis, and the types of suppressive niches formed are associated with target tissue-specific-(Zagorulya et al., 2020), and tumor histology specific- patterns (Wellenstein and de Visser, 2018); this highlights both far-reaching tissue of origin consequences and the potential for diverse mechanisms in the same patient.
The TIME is composed of multiple pro-tumor cell types and even pro-tumor acellular structures (Figure 2):
Tumor-associated macrophages (TAMs): Long considered solely in tissue repair and clean-up, macrophage lineage cells are now appreciated as phenotypically heterogeneous and plastic. Among the most abundant pro-tumor cell types in many cancers (Mehta et al., 2021), TAMs are co-opted by tumors to suppress local immunity. This ‘renaissance’ lineage can promote tumor growth and metastasis via tissue remodeling by stimulating angiogenesis and producing TGFβ (to stimulate tissue growth and Treg differentiation), and via multiple secreted and cell surface factors.
Myeloid-derived suppressor cells (MDSCs): MDSCs are increasingly associated with progressing cancer. Initially defined functionally (suppressive) and morphologically (‘immature’), high dimensional technologies have now more precisely characterized them, revealing nuanced functional/molecular ‘archetypes’ driven by myeloid cell plasticity, multiple suppressive mechanisms and tissue specificity (Hegde et al., 2021).
Regulatory T cells (Tregs): Tregs have been long recognized as suppressive. Effector CD8+ to Treg cell ratio in tumors is a positive prognostic marker (Togashi et al., 2019). However, the importance of antigen-specificity - expected based on the presence of unique TCRs - awaits more definitive experiments (Moorman et al., 2021). Of note, melanoma neoantigen-specific Treg expansion has been found amongst tumor infiltrating lymphocytes (TIL) in MHCII+ melanomas (Oliveira et al., 2022), potentially representing a unique escape mechanism for MHCII-expressing tumors.
Cancer associated fibroblasts (CAFs): CAFs are the predominant non-immune cell type within tumors. Like TAMs, they are a heterogeneous and plastic population associated with both pro- or anti-tumor properties (Desbois and Wang, 2021), and even direct antigen presentation to CD4+ T cells (Huang et al., 2022). CAFs contribute to dense, T cell excluding, extracellular stroma via secreted cytokines and chemokines, and recruit suppressive cells (Tregs and MDSCs) or directly inhibit T cells.
mregDCs: While dendritic cells are typically thought to initiate immunity, this subset (discussed below) appears to have suppressive capability.
Figure 2: Immunosuppression in the Tumor Immune Microenvironment (TIME).
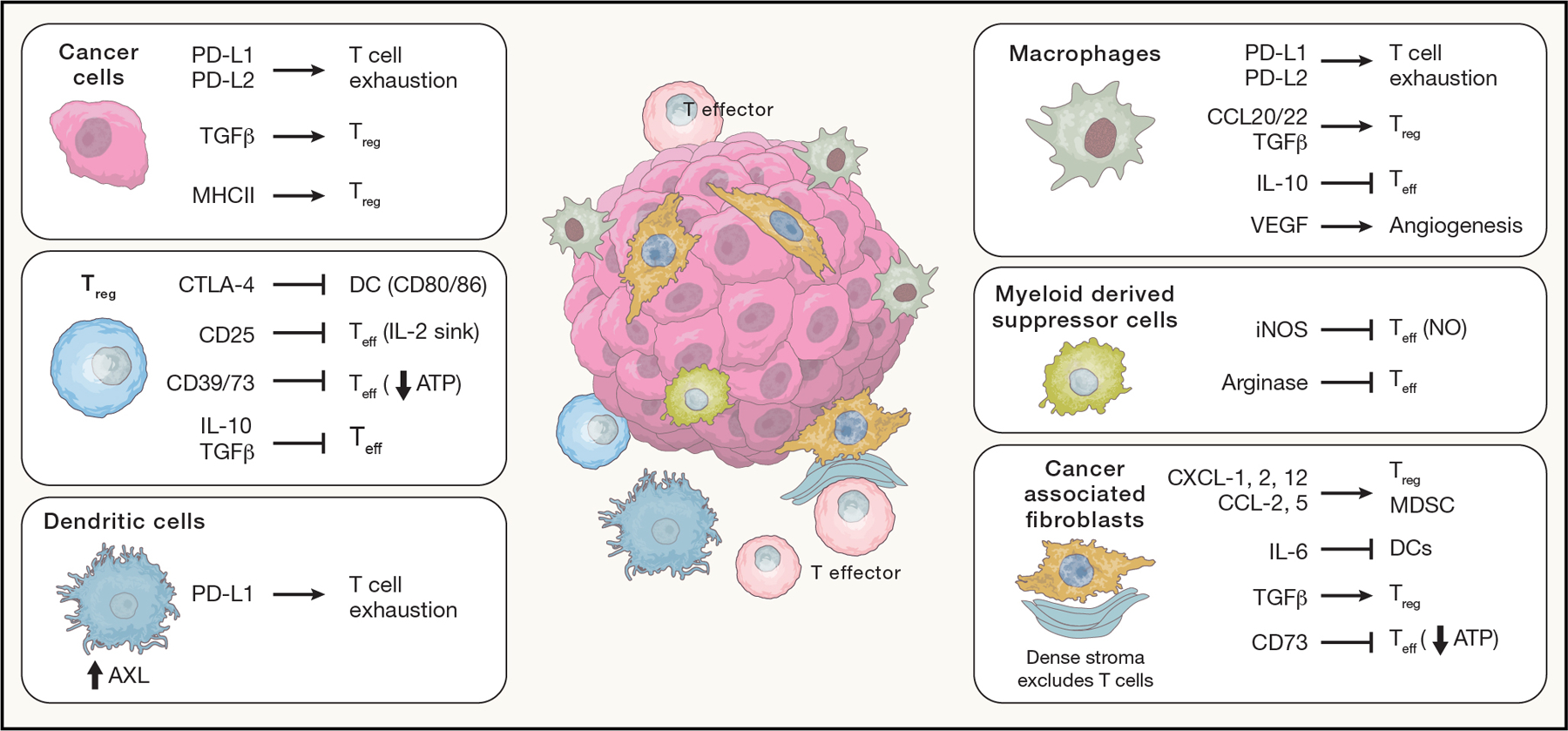
The TIME is made up of malignant cells, stomal cells and immune cells, all of which can suppress tumor directed immunity by acting directly or indirectly on effector T cells (Teff). Boxes depict select immunosuppressive mechanisms employed by these various cell types: cancer cells (Ca), regulatory T cells (Treg), dendritic cells (DC), macrophages (MΦ) and myeloid derived suppressor cells (MDSC; (Veglia et al., 2018)), and cancer associated fibroblasts (CAF).
Multiple chemokines, cytokines and other signaling molecules (e.g. VEGF) and exosomes produced by tumors are involved in the recruitment of a complex mix of cells, their conversion to a pro-tumor phenotype (Propper and Balkwill, 2022) and the structural features of the TIME. This environment creates a final challenge to immune cells, either induced physiologically or via vaccine, and must be considered in the design of any vaccine strategy.
The logistics of building the vaccine bridge
How do we train an immune system that has evolved to fight external pathogens to target a tumor that continually evolves to evade that same immune system? Using natural immunity as a blueprint, we discuss 4 inter-dependent ‘towers’ that support this bridge: cellular players, antigen, delivery to APCs, and co-therapies (Figure 3).
Figure 3: Vaccines, a bridge to cure?
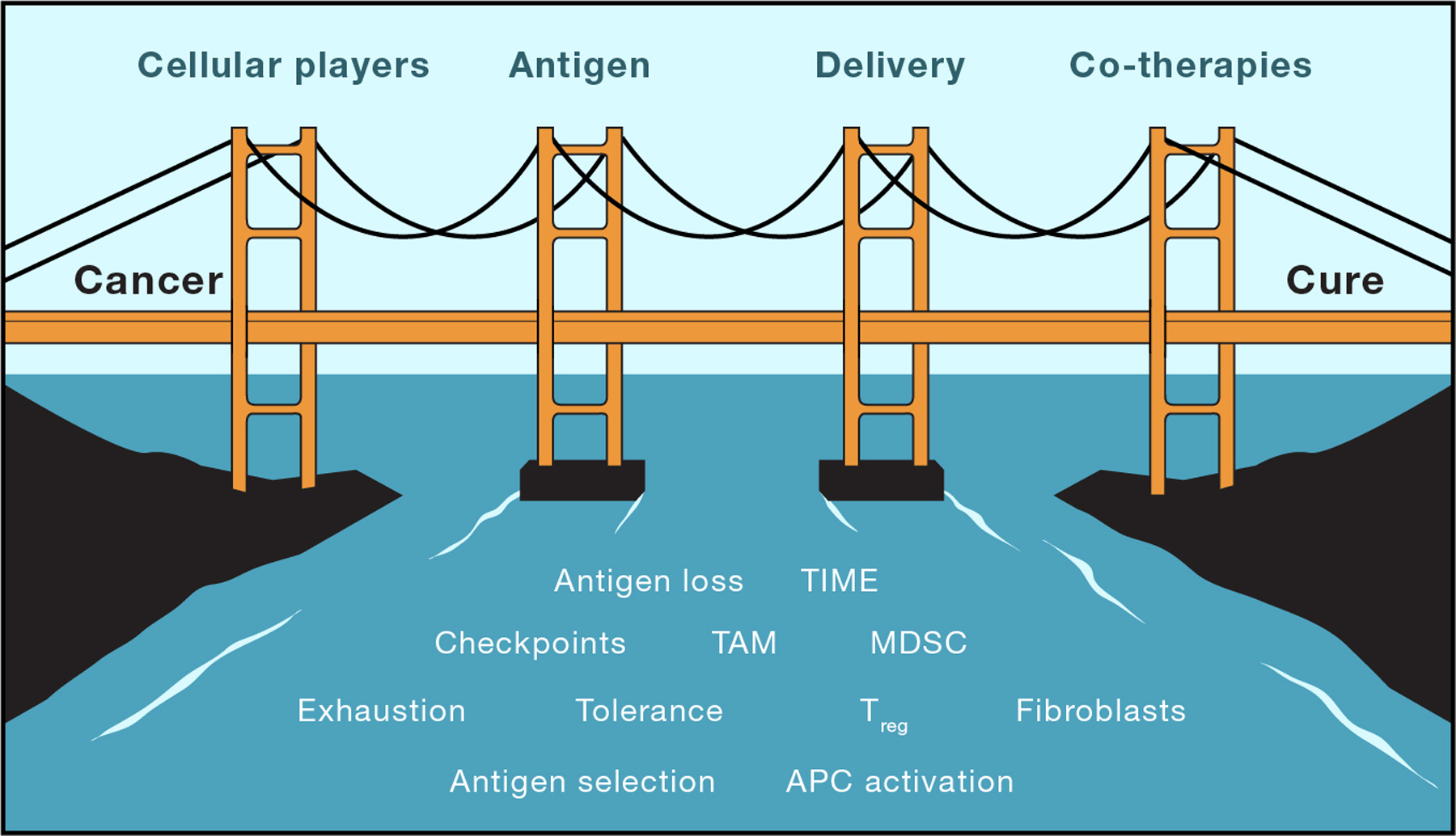
While there are many barriers to natural immune responses to cancer, we propose that focusing on four pillars (innate and adaptive immune cells, antigen targets, delivery strategies and co-therapies) will help vaccines become a bridge from cancer to immune-based cure.
LYMPHOCYTES
Cancer vaccines have generally focused on eliciting tumor-specific CD8+ cytotoxic T cells (CTLs). Indeed, CTLs are undeniably central to tumor cell killing and foundational to most immune therapies, and across many tumor types survival correlates with CD8+ TIL numbers (Reiser and Banerjee, 2016). Because of this centrality, improving the number or phenotype, and ideally both, of the CTL repertoire is a critical unmet need for the field. A key to this objective is engaging the full range of adaptive immune cells to both complement and augment CTL responses. CD4+ T cells may have cytotoxic functions (Cachot et al., 2021), and transfer of neoantigen-specific CD4+ TIL can control metastatic cholangiocarcinoma (Tran et al., 2014), either by direct cytolytic activity or licensing other immune cells. CD4+ T cells can also directly recognize MHCI presented peptides without co-receptor involvement (Oliveira et al., 2022), although how to induce this phenotype or select epitopes with this feature is currently unknown.
Perhaps more important is the role CD4+ T cells play in providing help to CD8+ CTLs. In a syngeneic tumor model, CTL-mediated tumor control and checkpoint inhibitor efficacy were dependent on CD4+ T cell recognition of an MHCII restricted neoantigen (Alspach et al., 2019). The presence of both an MHCI/CTL epitope and an MHCII/CD4+ T cell epitope in the same tumor cell (vs. in different tumor cells) was required to elicit maximal tumor-specific CTL responses, including after vaccination with irradiated tumor cells. These data support the long-standing dogma that optimal CTL stimulation by an APC requires interaction of that APC with an interferon-gamma producing CD4+ ‘helper’ Th1 T cell recognizing an MHCII restricted epitope on the same APC. This licensing by the APC requires CD40L (on the T cell)-CD40 (on the APC) interaction, and can be mimicked by an agonistic CD40 antibody (Morrison et al., 2020). Still unclear are whether epitope-intrinsic features (e.g. abundance or TCR avidity) are needed to induce effective help, or if it is determined by the local APC environment.
Simply considering helper and cytotoxic T cell responses does not reflect the tremendous progress over the last decade, driven by bulk and single cell RNA sequencing, epigenetics, and animal models, in understanding the breadth, inter-relationships and trajectories of different T cell subsets. This complex T cell biology has been reviewed in depth elsewhere (Kumar et al., 2018), but we highlight two vaccine-related features. First, T cells are phenotypically dichotomized as effector, which are short lived (weeks) and have active functionality (e.g. cytotoxicity), or memory, which persist for months to years, ready to respond to later challenge. Interconversion from memory to effector states is routine, but the reverse, less common. In cancer and chronic infection, effectors often take on an ‘exhausted’ state with limited functionality, while a subset of ‘precursor exhausted’ cells maintain the proliferative, self-renewal and differentiation potential that are critical to successful immune checkpoint blockade (ICB) therapy (Liu et al., 2021; Sade-Feldman et al., 2018). Second, memory cells have three major forms (Kok et al., 2022; Kumar et al., 2018). Effector memory (TEM) cells are found in tissues and blood and are poised to rapidly expand and gain effector functionality upon antigen encounter; Resident memory (TRM) cells are found only in tissues, and are similarly poised to rapidly expand and gain effector function. Central memory (TCM) cells are found in tissues and blood, but respond to antigen by migrating to draining LNs to proliferate extensively and provide a second round of effectors. TCM cells also can self-renew.
How vaccine approaches can proactively modulate T cell phenotypes is unclear, as are the optimal pattern and dynamics. We hypothesize that vaccine strategies that induce strong memory populations will be more efficacious for cancer than those that favor short-lived effectors. In this vein, stimulation of naïve cells to create de novo responses may bring more to the table than stimulating pre-existing, potentially exhausted effector cells.
In contrast to T cells, B cells and their interactions with T cells have been far less well-studied (Downs-Canner et al., 2022) in cancer vaccinology. B cell tumor infiltrates, especially in the context of intra- or peri-tumoral tertiary lymphoid structures, are associated with longer survival and response to ICB in patients (Schumacher and Thommen, 2022). Antibodies targeting surface receptors have been used clinically for years, and engage multiple cells types to kill cancer cells and can even induce immunogenic cell death (Pozzi et al., 2016). Further, neoantigen specific IgA can control ovarian cancer by redirecting myeloid cells to attack malignant cells (Biswas et al., 2021). B cell:T cell interactions are important in two ways. First, antibody maturation and production are enhanced in the presence of activated CD4+ T cells that recognize epitopes associated with the target protein, ensuring that antibodies are made in the context of a broader immune response (classically defined IL-4-producing Th2 T cell help). Second, B cells can support T cell function. For example, in a murine model, interactions between tumor-antigen specific B cells and tumor-antigen specific CD4+ T follicular helper cells (TFH) triggered IL-21 dependent TFH cell support of CTL differentiation and promoted ICB responsiveness (Cui et al., 2021).
For cancer vaccines to engage the full range of adaptive immune cells and maximize CTL responses, antigen structures and formats recognized and internalized by both APCs and B cells, and with the complexity to contain MHCI, MHCII and B cell epitopes, will be needed. Conversely, this argues against the reductionist approaches extensively employed in the past (i.e. inclusion of only minimal length MHCI/CD8+ epitopes or of only a few epitopes).
ANTIGENS and MHC
The complex and evolutionarily optimized cellular and molecular output of natural immunity begins with two central components, antigens and antigen-presenting cells; thus, optimizing both these parameters is warranted. Most current generations of cancer vaccines (and to reflect that current interest, our discussion here of antigens) focus on neoantigens, which are truly non-self antigens. Other antigen classes include tumor associated antigens, which are also expressed in normal tissues, and have been extensively tested with only limited success (reviewed over the years), and viral antigens, which are relevant to only a handful of virus-driven cancers; many of the principles we discuss are relevant to these antigens as well.
Neoantigen-targeting vaccines broadly require identifying translated, cancer-specific mutations and predicting those most likely to be presented in MHC molecules (still with a primary focus on MHCI, due to the well-described cytolytic role of CTLs and also out of technological necessity [see below]). The results of neoantigen vaccination have been tantalizing (Ott et al., 2017; Sahin et al., 2017), but these trials and new and more facile approaches to examining tumor and peripheral blood T cell populations for tumor-reactive cells (Danilova et al., 2018; Lam et al., 2021) show that only a small fraction of neoantigens predicted to be immunologically useful are spontaneously and/or durably immunogenic. Thus, our understanding of what constitutes an immunogenic epitope remains rudimentary. Several pressing hurdles regarding neoantigen selection remain.
First, the types and numbers of neoantigens observable in each tumor sample are currently limited by sequencing and/or bioinformatic technologies (Figure 4A). Single nucleotide variants (SNV) and small insertions/deletions (indels) in the annotated genome are now standardly detected. However, still beyond routine detection are SNVs in unannotated open reading frames (nuORFs), more complex novel ORFs (neoORFs) encoding potentially longer neoantigens from gene rearrangement/duplication transcripts, splicing variations, ‘exitrons’, and dysregulated splicing due to mutated or dysregulated splicing machinery, dysregulated translation, endogenous retroviral elements, and post-genetic changes such as A to I editing (Gupta et al., 2021; Ouspenskaia et al., 2021; Wang et al., 2021; Wu et al., 2018). Depending on the underlying defects in DNA repair and RNA splicing, conventional methods may thus miss more than half of a given tumor’s neoantigens (Wu et al., 2018). Still unanswered is if any of these classes are more effective targets than others.
Figure 4: Neoantigens, our blind spots and their presentation.
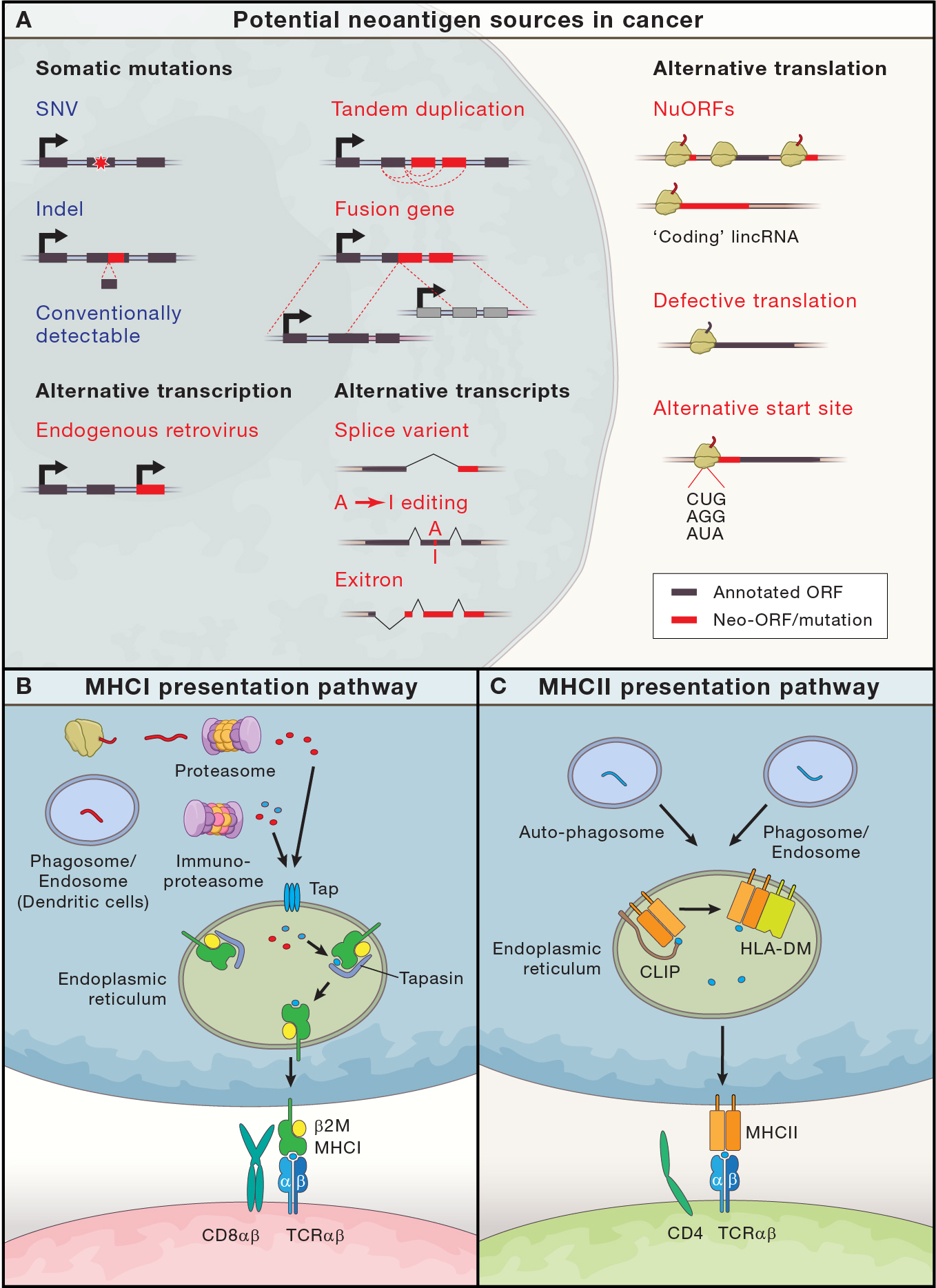
(A) Potential neoantigen sources in cancer, adapted with modifications from Gupta et. al. (Gupta et al., 2021). Conventional bioinformatic techniques efficiently detect neoantigens derived from single nucleotide variations (SNVs) and insertions/deletions (Indels) (circled in blue). Largely beyond current clinical prediction techniques (red text) are neoantigens from some somatic tandem duplication and fusion transcripts, endogenous retroviruses, transcriptional variations (alternative splicing, exitrons and A-to-I editing), and translational abnormalities (unannotated NuORFs, defective translation products and peptides from alternative start sites). (B) and (C) depict antigen presentation pathways in MHCI and MHCII respectively. (B) For MHCI, the proteasome process endogenous proteins into short peptides, which the TAP complex transports into the endoplasmic reticulum. Immune cells can diversify the peptidome by, processing proteins from phagosomes to allow exogenous peptide cross-presentation (i.e. cDC1s), and/or by immunoproteasome processing, which provides different protease specificities via replacement of three constitutive proteasome subunits with alternative subunits (Murata et al., 2018). In the ER, peptides are further processed and chaperones/editors, including Tapasin, facilitate MHCI loading with stable 8–10 aa peptides, before transport to the cell surface where T cells survey them via TCR:p:MHCI interactions that generally require CD8αβ:MHCI interactions. (C) For MHCII, proteins are processed into smaller peptides in phagosomes, auto-phagosomes (products of autophagy) and lysosomes (Roche and Furuta, 2015), which eventually fuse with the late endosome. There proteolytic cleavage of the invariant chain, CLIP (which facilitates MHCII folding during translation and resides partially in its antigen binding grove), allows loading of 13–25 amino-acid peptides on to MHCII; of note, MHCII is more promiscuous in peptide binding and peptides are considerably longer than for MHCI. p:MHCII complexes can then be edited by HLA-DM, which selects for peptides that are more stably bound to MHCII, before being transported to the cell surface for surveillance by where CD4+ T cell TCRs. In contrast to the importance of CD8 in TCR:p:MHCI interactions, TCR:p:MHCII interactions are not as dependent on CD4 recognition of MHC.
Second, determining which putative neoantigens are actually presented on MHC and if they will be immunogenic is yet unresolved. Three general approaches, ordered based on decreasing scalability, have been used to identify target neoantigens.
(A) Prediction: From in vitro binding affinities measured for thousands of short peptides across a subset of MHC class I alleles, binding prediction algorithms have been developed (Nielsen et al., 2007) and further refined by training with substantially larger mass spectrometry datasets (Reynisson et al., 2020). MHCII binding predictors have lagged due to length variability and more limited binding affinity data, but breakthroughs have come via immunopeptidomic analyses (Abelin et al., 2019). However, despite improvement, there are still limitations and unexpected findings. Across multiple trials with fundamentally different formats of neoantigen-targeting vaccines (Ott et al., 2017; Sahin et al., 2017), only ~15–30% of predicted epitopes yielded CD8+ responses, a fraction consistent with an evaluation of in vitro immunogenicity (Stronen et al., 2016), and these responses were generally weak. Moreover, systematic screening of predicted peptides has revealed limited capacity to generate spontaneous T cell responses for the majority of predicted class I and II neoantigens (Lam et al., 2021) and observations of antigens with low binding score to MHCI having efficacy in vaccine-induced tumor models highlight shortcomings in current algorithms (Ebrahimi-Nik et al., 2021). Finally, the majority of epitopes predicted for MHCI binding, when given as part of longer peptides have tended to more effectively elicit CD4+ T cell responses; this observation about the efficiency of CD4+ stimulation is still not satisfactorily explained and the functional significance remains uncertain.
(B) Observation: Identifying MHC-bound peptides on tumor cells by MS provides enormous confidence that the peptides have cleared numerous hurdles toward immunogenicity (expression, processing, transport, and MHC binding to an observable level – all of which may be modulated by the tumor to evade immune detection (Jhunjhunwala et al., 2021)). Direct immunopeptidome analysis of melanoma tissue from 5 patients identified (only) 11 bona fide MHCI neoantigens, of which 4 stimulated specific T cell responses (Bassani-Sternberg et al., 2016). Thus, this strategy is limited by low sensitivity and also by tissue availability (i.e. biopsy size), inaccessibility of MHCII presented peptides on APCs (APCs would be rare in biopsy samples) and the requirement for advanced MS/bioinformatic technology.
(C) Screening: Multiple groups have explored empiric strategies to directly test the ability of individual epitopes to stimulate patient-derived peripheral blood T cells (Danilova et al., 2018; Lam et al., 2021; NCT03633110)). For example, Lam identified responses to fewer than 10% of predicted ‘strong’ neoantigen epitopes and ~30% of positive responses came from neoantigens that would not have been selected using conventional criteria (see (A)). This particular assay also detected epitopes that inhibited baseline response (‘Inhibigens’); it remains unclear if these epitopes should be discarded. An important caveat to these strategies is that they likely only reproducibly measure pre-existing patient responses.
These models might be improved by adding other features. A better understanding of peptide processing dynamics due to (immuno)proteasome or cellular compartment in APCs (Grotzke et al., 2017) and cancer cells (Jhunjhunwala et al., 2021) may be especially important. Other avenues to consider include: peptide-MHC stability (Harndahl et al., 2011), similarity to self or to pathogens (Richman et al., 2019), and TCR∷peptide:MHC dynamics (Ebrahimi-Nik et al., 2021) (Figure 4B–C). How to weigh each feature is challenging.
Third, future iterations of vaccines will likely benefit from inclusion of epitopes to stimulate not just CD4+ and CD8+ cells, but also B cells. As previously discussed, CD4+ help is critical to efficient CTL priming, and so it follows that including both MHCI and MHCII presented peptides will improve efficacy. Likewise, tumor-specific B cells/antibodies have been shown to play an important role in suppressing cancer growth and augmenting CTL responses (Biswas et al., 2021). It remains unclear whether universal class II (e.g. PADRE, Tetanus Toxin p30) and B cell epitopes (e.g. Tetanus toxoid) (Fletcher et al., 2018; Swartz et al., 2021) are equally or less effective than similar tumor-specific epitopes and how important physical linkage is. Incorporating tumor-specific B cell epitopes, at least in peptide vaccines, may be challenging as BCRs generally survey non-linear protein epitopes (Potocnakova et al., 2016).
Fourth, in choosing antigens, we must take into account that vaccines enter the equation towards the end of a long evolutionary battle between immunity and cancer, a struggle that likely selects for clones with less spontaneously immunogenic mutations, profoundly affecting the mutational landscape (Turajlic et al., 2019). Further, the phenomenon of antigen immunodominance (Burger et al., 2021) may limit the number of neoantigens which are naturally recognized without intervention. Indeed, only a small fraction of known potential neoantigens exhibit detectable T cell responses in tumors (Kristensen et al., 2022). On the other side of this evolutionary battle, the T cell compartment no doubt bears scars as well, and tumor-immune co-existence may shape the breadth of future T cell responses. Neoantigen-specific TILs are exhausted (Caushi et al., 2021; Oliveira et al., 2021), and as has been elegantly shown in viral infections, the exhausted state leaves epigenetic scars which may be difficult or impossible to remove (Abdel-Hakeem et al., 2021; Yates et al., 2021). Similarly, ICB response in lung cancer is not associated with expansion or reinvigoration of terminally exhausted T cells, but rather with ‘clonal revival’ - the local and peripheral expansion of new and pre-existing T cell clonotypes which acquire precursor exhausted phenotype (Liu et al., 2021). Further, neoantigen-specific Tregs have been identified (Oliveira et al., 2022). These observations raise clinically relevant questions: Can vaccination reverse the predominance of exhausted cells? Are naive rather than antigen-experienced T cells a better target? Will vaccination stimulate immunosuppressive neoantigen-specific Tregs?
DELIVERY TO APCs
Just as a natural adaptive immune response grows from innate immune processing at the start of an infection, productive vaccine responses are driven by effective delivery to innate immune cells. Delivery is thus about getting the target antigens to the right innate immune cells, with the proper maturation cues to properly activate antigen-specific T cells. Two important considerations include antigen format (Box 1) and delivery. Vaccine formats have been extensively reviewed elsewhere (see (Irvine et al., 2020)), but here we highlight key cross-cutting principles: antigen presenting cell targets, adjuvants, and delivery.
Box 1: Vaccine Formats.
Protein: Initially recombinant proteins and epitope-length peptides, but now more commonly synthetic long peptides (SLPs, 15–30 amino acids). Advantages include modularity, the favoring of MHCI loading via cross-presentation by professional APCs (e.g. DCs), and the possibility of co-delivering peptide/protein domains that target DCs. Hurdles for SLPs include antigen prediction, technically difficult synthesis and solubility, the lack of intrinsic immunogenicity and difficulty uniting epitopes for T and B cells in one SLP.
mRNA: Proven during the COVID-19 pandemic with the rapid production and approval of BNT and mRNA1273 vaccines, mRNAs can encode target antigen(s) and potentially DAMPs, cytokines and co-stimulatory molecules. Certain liposome formulations of mRNAs are preferentially internalized by DCs and mRNA can intrinsically function as adjuvant – in fact, this property must be dampened for optimal efficacy. Self-replicating mRNAs offer extended antigen expression kinetics, potentially mimicking natural infection. Drawbacks include inherent instability.
DNA: The DNA platform, usually a plasmid, can encode antigen(s), DAMPs, cytokines and co-stimulatory molecules. DNA vectors tend to induce longer lasting expression than mRNA platforms, potentially augmenting responses. Hurdles include delivery, which requires electroporation or other delivery device, and inability to target to preferred cell types (professional APCs) via the typical intramuscular injection route.
Microbial Vector: Weakened or attenuated viral and bacterial vectors can be modified to express neoantigens. They are inherently immunogenic and may be engineered to encode costimulatory molecules to augment responses. Vector directed immune responses can limit effective boosting.
Cell Vectors: Most often DCs are differentiated in vitro from blood monocytes (moDCs), stimulated with synthetic PAMPs and loaded with recombinant proteins/peptides or whole tumor lysate. An advantage is that APCs can be directly loaded with antigen and readily manipulated ex vivo. The labor-intensive cell preparation, however, is limiting. Non-moDC cell types are actively being investigated as alternative cellular delivery vehicles.
In Situ: Adjuvants, oncolytic viruses, mRNAs encoding immune stimulants and activated autologous or allogeneic dendritic cells can all be injected directly into sites of tumor. This antigen agnostic strategy bypasses the need for antigen prediction and directly modifies the TIME. However, only patients with accessible disease are candidates for treatment.
Antigen-presenting cells.
Which APCs should be targeted by a vaccine? A key property of APCs for vaccine applications is antigen cross-presentation. Typically the provenance of specialized cells, such as some dendritic cells (Wculek et al., 2020), cross-presentation uniquely allows for MHCI presentation of external peptides by delivering endocytosed external antigens (e.g. from cancer cells or vaccines) to the cytoplasm for proteasomal degradation, with the peptide products transported into the endoplasmic reticulum for MHCI loading (Figure 4B). This capability complements the constitutive presentation of endogenously-produced peptides from most cells, including cancer cells, through the same cytoplasm/proteasome/MHCI pathway, enabling T cell surveillance.
Four classical DC subtypes have been defined (Box 2): conventional DC 1 (cDC1), cDC2, and plasmacytoid DCs (pDCs), all derived from common DC precursors, and monocyte-derived DCs (moDCs) (Wculek et al., 2020). Recent single cell transcriptional analyses have provided higher resolution understanding of the DC compartment (Villani et al., 2017), with functional studies revealing additional subsets (e.g. mregDCs; (Maier et al., 2020)).
Box 2. Dendritic Cells.
cDC1: cDC1s are especially effective antigen cross-presenters (Lee and Radford, 2019), express scavenging molecules like CLEC9A which facilitate dead-cell uptake and cross-priming, are central to tumor immunity in murine models (Ferris et al., 2020) and are associated via gene signatures with improved survival in multiple cancer types (Lee and Radford, 2019). Further, cDC1s are critical for priming of both CD4+ and CD8+ T cell responses in syngeneic tumor models (Ferris et al., 2020).
cDC2: cDC2s are important for priming CD4 responses (Lee and Radford, 2019). They can also efficiently prime CD4+ helper T cell responses upon Treg depletion in mouse syngeneic models, leading to tumor eradication; in keeping with this, an increased cDC2/Treg ratio in head & neck cancer is associated with increased survival following ICB therapy (Binnewies et al., 2019). Interferon gamma-stimulated cDC2 can also prime effective tumor specific CTL responses in mice by directly acquiring peptide:MHCI complexes from tumor cells – fulfilling the role of cross-presentation (Duong et al., 2021).
MoDCs: In vivo, monocyte derived DCs, driven by inflammation, may prime T cell responses after chemotherapy (Wculek et al., 2020) or viral infection. Ex vivo moDCs expansion is routinely used in cell-based vaccine delivery approaches (Nava et al., 2021), with new approaches still under investigation (Han et al., 2020). While adding complexity to vaccine production, ex vivo differentiated moDCs allow direct antigen delivery to the APC, potentially overcoming substantial hurdles compared to in vivo antigen delivery.
mregDCs: Recent studies have identified a subset of regulatory DCs which negatively regulate T cell responses to tumors via AXL-dependent PD-L1 upregulation (Maier et al., 2020); interestingly, these mregDCs can also positively regulate T cell responses upon IL4 signaling blockade. Thus, care may be needed to avoid activating the ‘wrong’ DC during vaccination and it will be important to untangle their tolerogenic vs immunogenic programs.
pDCs: Plasmacytoid DCs, although ontologically related to cDCs, are typically considered first responders which amplify type I interferon responses. However, they may present, and even cross-present, antigen, or cooperate with other cross-presenting dendritic cells to optimize responses (Fu et al., 2022).
These DC subsets differ by frequency, PRR expression, scavenging receptors, trafficking properties, and capacity for antigen (cross) presentation (Wculek et al., 2020). Human cDC1s, which are highly specialized in cross-presentation, are uniquely marked by CLEC9A and XCR1, both of which (Fossum et al., 2015; Zeng et al., 2018), in addition to DEC-205 (Bhardwaj et al., 2020), have been used as targets to efficiently deliver antigens to this rare DC subset in vivo.
Other cell types can also serve as APCs. Just as for moDCs, neutrophils (Mysore et al., 2021), T cells (Booty et al., 2022; Veatch et al., 2021), B cells (Booty et al., 2022), unfractionated human PBMCs (Booty et al., 2022), or immunoproteasome-engineered mesenchymal stem cells (Abusarah et al., 2021) can be coaxed ex vivo to effectively cross-present antigen and generate strong anti-tumor immunity as part of cell-based vaccine therapies. Further, improvements in bead-based isolation and stimulation of various DC subsets have enabled ex vivo production of blood-derived DC subsets (Hanel et al., 2021). Ex vivo cell manipulation adds substantial cost and complexity to vaccine delivery, which may be offset by the benefit of direct antigen (or antigen-encoding nucleic acid) delivery to APCs.
An open question is whether high-quality T cell responses are optimally generated through only a specific APC (e.g. cDC1s), or if combinatorial strategies to engage multiple APC subsets, potentially including B cells (Sagiv Barfi et al., 2021), would be more effective. Approaches such as the use of FLT-3 ligand to enhance natural dendritic cell numbers in the blood, and presumably therefore in key tissues, may also be required (Bhardwaj et al., 2020).
Adjuvants.
Effective antigen presentation depends upon DCs taking up antigens with activating signals to induce their maturation. For vaccination, this maturation can be driven by ‘danger signals’ (DAMPs or PAMPs; Figure 5) from adjuvants and/or co-stimulatory molecules (e.g. FLT3, CD40). PAMPs and DAMPs are recognized by similar PRRs, which have been extensively reviewed (Wicherska-Pawlowska et al., 2021). However, PAMP- and DAMP-triggered signals are not necessarily equivalent; these differences may allow unique responses to the simple presence of a pathogen vs. a pathogen that is killing host cells. For example, DCs treated with the gram-negative bacterial cell wall component lipopolysaccharide (LPS; a PAMP), or oxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphorylcholine (oxPAPC; a DAMP), both induce IL1β production via Caspase-11 mediated inflammasome activation. oxPAPC, however, protects DCs from LPS-induced pyroptosis, yielding a longer-lived “hyperactivated” DC with improved capacity to migrate to lymph nodes, induce CTL responses and control murine tumors (Zhivaki et al., 2020). Moreover, different DC subtypes may express different patterns of PRRs; for example, human cDC1s specifically express high levels of TLR3 (which recognizes the common adjuvant polyI:C). Thus, one avenue to improve cancer vaccine design will likely be based on better understanding how DAMP/PAMP combinations differentially generate unique APC phenotypes. Alternatively, DC interactions with other cells present at sites of inflammation, such as platelets, have generated effective DCs (Han et al., 2020).
Figure 5: Dendritic cells, PAMPs and DAMPs.
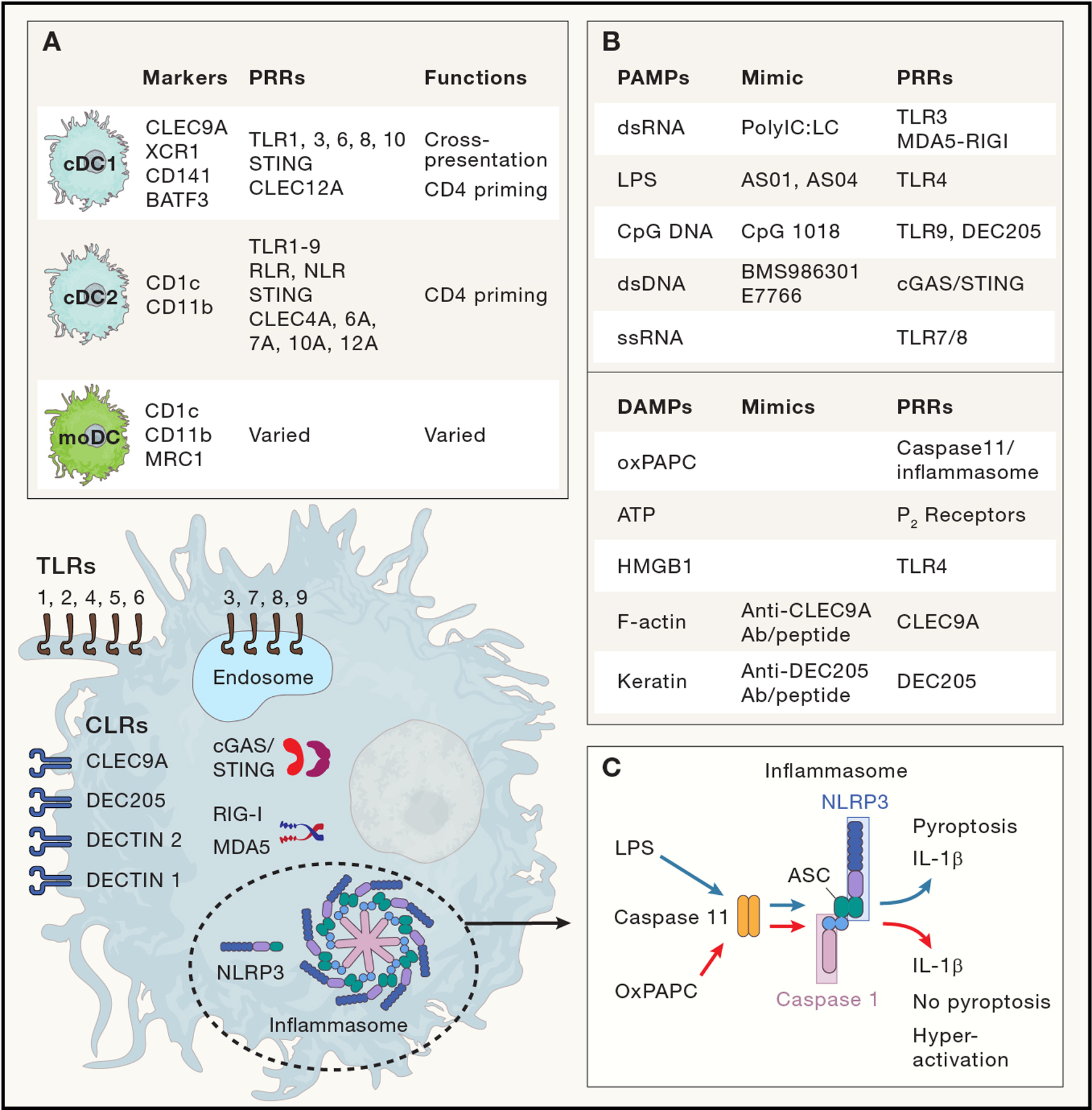
The diverse dendritic cell lineage expresses a broad array of pattern recognition receptors (PRR). PRRs include Toll like receptors (TLRs), which are present as transmembrane proteins to directly detect PAMPs externally (i.e. cell surface; TLR 1,2,4,5,6) or within endosomes (TLR 3,7,8,9; which focus on nucleic acids released from internalized pathogens) (Fitzgerald and Kagan, 2020); cell surface C-Lectin type Receptors (CLRs; (Geijtenbeek and Gringhuis, 2009)), which directly detect external PAMPs/DAMPs, and in the case of CLEC9A/DNRG1 and DEC205/CLEC13B, can facilitate phagocytosis of dead and dying cells. Intracellular PRRs, including cGAS/STING and RIG-I/MDA5, recognize intracellular nucleic acids. Nod-like receptors (NLR), including NLRP3, can participate in PAMP/DAMP sensing via interactions with caspases and the inflammasome. (Inset A) DC subsets most commonly considered in vaccine production, with lineage defining cell surface markers and transcription factors, differential PRR expression and unique functions. (Inset B) Select PAMPs/DAMPs, the PRRs that detect them, and mimics that are either commonly used as clinical vaccine adjuvants or are under pre-clinical testing. (Inset C) LPS and oxPAPC signaling via caspase-11 highlights differential processing of DAMP and PAMP signals, even via the same PRR pathway. LPS and oxPAPC both induce IL-1β production via caspase 11 and downstream NLRP3/atypical inflammasome signaling. While LPS stimulation leads to short lived, pyroptotic IL-1β-expressing DCs, OxPACP blocks pyroptosis, inducing a hyperactivated state and efficient CTL priming.
An important related consideration is consistent co-delivery of antigen and adjuvant to the same APC to overcome the potential for induction of tolerogenic responses by non-activated APCs (Toes et al., 1996). Nanoparticles containing TLR 7/8 agonists and specially modified peptide antigens elegantly solved this problem, ensuring the uptake by each APC of multiple copies of both peptide antigen and adjuvant molecules, which improved the magnitude and breadth of CD8+ responses (Lynn et al., 2020). Genetic or chemical linkage strategies have also been developed for protein antigens (Belnoue et al., 2019) and for DC-targeted RNA-based vaccines (using the intrinsic immunostimulatory properties of RNA (Kranz et al., 2016)). Solutions for in vivo delivered DNA-based vaccines are less apparent due to uncertainty and perhaps heterogeneity with which cells express the encoded antigens, although encoding of the DAMP HMBG1 has shown some promise (Fagone et al., 2011). A consistent theme is the superiority of linked antigen and adjuvant.
Schedule, boosting and route.
A relatively under-explored aspect of vaccination is how the schedule of antigen delivery impacts downstream outcomes. In natural acute infections, immunogenic antigens rise exponentially to peak levels over days to a week and are generally cleared within 2–3 weeks (Irvine et al., 2020), resulting in sterilizing immunity. These kinetics differ from chronic infection and cancer, where some level of antigen exposure continues for an extended period (months), resulting in an undesirable ‘exhausted’ T cell population which, without other intervention, is unable to mount effective responses. An exponential dosing scheme over 4–8 days, maximizes T and/or B cell responses and tumor control in murine models (Johansen et al., 2008), and has support also in non-human primates (Pauthner et al., 2017). To our knowledge, no clinical trials have yet tested this natural immunity-mimicking concept directly. However, trials using self-amplifying RNA vaccines based on alphaviruses, which replicate exponentially to increase antigen production (Melo et al., 2019) like in natural infection, are ongoing for rabies (NCT04062669), SARS-CoV-2 (NCT05132907) and cancer (as part of a heterologous prime-boost strategy (see below); NCT05141721).
Similar unresolved questions are the timing of boosting immunizations and whether a ‘heterologous prime-boost’ (different immunogenic formats for the prime and boost) provides even stronger or broader immunity. Undoubtedly, ‘homologous’ boosting is effective, at least with respect to antibody responses, as recently observed with the approved SARS-CoV-2 vaccines. For effective anti-cancer vaccines, however, where the most relevant measure may be T cell immunity, optimal timing and dosing of booster immunization (during expansion, contraction or ‘resting’ of primed cells) in humans is non-existent.
Heterologous prime-boost strategies were initially necessitated by strong anti-vector (typically a viral vector) responses on priming, which were thought to limit T cell responses following boosting. Over the years the concept has been extended beyond vector changes to include alterations which engage the immune system in different ways (adjuvants, APCs, dose kinetics, vaccine formats) and many preclinical studies (Hofer et al., 2021) have demonstrated improvements in magnitude, breadth of response and efficacy. While cancer trials are recruiting (e.g. NCT03794128), early definitive clinical outcomes are most likely to come from ongoing COVID heterologous prime-boost studies (e.g. NCT05074368, NCT04907331) although analysis of T cell responses has typically been lower priority in infectious disease studies.
Finally, the anatomical site of delivery is emerging as an important variable. In a comparison of intravenous vs subcutaneous delivery of antigen/adjuvant nanoparticles, when the doses were adjusted to yield equivalent circulating T cell numbers, tumor control was significantly better when delivered intravenously, associating with a higher proportion of TCF-1+ stem-like cells (Baharom et al., 2021). Similar differences between intravenous vs. subcutaneous delivery have been observed with some long peptides (but not all, potentially due to peptide biochemical properties) admixed with adjuvant (Sultan et al., 2019), and in a peptide-multi-adjuvanted liposome format, improved responses were shown with intraperitoneal rather than subcutaneous immunization (Korsholm et al., 2014). Finally, targeting vaccines to appropriate anatomic sites may also improve efficacy by enhancing TRM induction, as observed with mucosal targeting via albumin (Rakhra et al., 2021), and with epicutaneous administration of a modified vaccinia ankara virus vector, mimicking a ‘pox’ lesion (Pan et al., 2021).
CO-THERAPIES
Given the extensive tumor-immune evolution of ‘escaped’ tumors, we posit that vaccines will require combination with other therapies to unlock the power of the immune system. These co-therapies fall into three partially overlapping buckets.
Maximizing innate T cell priming.
Taking a cue from natural immunity, one strategy is to mimic the inter-cellular cross-talk mechanisms between the innate and adaptive immune cells that drive priming. For example, local CD40 agonist antibodies induced robust CTL responses in mice immunized with epitope-length peptides that were otherwise tolerogenic (Diehl et al., 1999), presumably by mimicking the APC licensing normally provided by CD4+ T cell help. Indeed, agonist CD40 antibodies or soluble CD40L are under renewed clinical development after initial toxicity concerns (Vonderheide, 2020). Similarly, CD27 agonist antibodies (mimicking CD70 signals from DCs) augmented peptide vaccine responses and vaccine-induced syngeneic tumor control (Riccione et al., 2018). Thus, replicating the licensing signals that helper CD4+ T cell give DCs (via CD40) or that DCs give to T cells (via CD27) can amplify vaccine responses. Another route amplifying productive APC-T cell interactions may be simply increasing APC numbers. Pretreating patients with FLT3L increased circulating DC numbers by ~30x, and augmented antibody and T cell responses (Bhardwaj et al., 2020). Collectively, these studies indicate that vaccine design can continue to mine the cytokines and co-receptors critical in natural immune responses.
Immune checkpoint blockade.
ICB targeting CTLA-4, PD-1/L1 and LAG-3 can re-invigorate (some) ‘exhausted’ T cells resulting in dramatic and durable clinical responses that have revolutionized treatment for many cancers. Although these agents may be also useful in augmenting vaccine de novo induced T cell responses, questions remain regarding when and how to combine ICB with vaccine priming. PD-1/L1 has been most extensively investigated. In mice, PD1 is required in CD8+ T cells during viral infection for effective memory T cell formation (Pauken et al., 2020). In a therapeutic murine syngeneic model, concomitant vaccination and PD-1 blockade unveiled strong T cell responses that controlled tumor growth, while PD-1 blockade beginning just 3 days prior to vaccination resulted in dysfunctional PD-1+ CD38hi T cell responses (Verma et al., 2019). Finally, treatment with anti-PD-1 prior to tumor irradiation dramatically reduced the abscopal effect in a dual flank model and shortened survival compared to treatment with anti-PD-1 after irradiation (Wei et al., 2021). These data caution against vaccination closely preceded by anti-PD-1 therapy, raising a timing conundrum, as standard-of-care for multiple diseases often begins with ≥1 years of uninterrupted ICB. Possible approaches to this timing issue are discussed below but we emphasize that to be welcomed as a routine part of combination immunotherapy, personal neoantigen vaccines need to make significant manufacturing improvements (Fritsch et al., 2020).
Short-circuiting the suppressive TIME.
Vaccine strategies almost certainly need to include mechanisms that relieve immune suppression at the tumor site, especially if treating advanced disease is envisaged since the TIME generally evolves to impede immunity. Multiple strategies to facilitate T cell function in this environment are being developed and should be considered in vaccine trials (Box 3).
Box 3: TIME management strategies.
Low dose cyclophosphamide can deplete Treg’s, a strategy exploited in DC based vaccines for ovarian cancer (Tanyi et al., 2018), among other vaccine formats. In multiple mouse models, depletion of regulatory T cells is crucial to the activity of anti-CTLA-4 therapy, although in humans this may not be the case for activity (Sharma et al., 2019), and other Treg abrogating approaches are being developed (Chen et al., 2022).
A myriad of TAM-targeted therapies are under evaluation (Allavena et al., 2021). Early (and ongoing) trials targeted the CSF-1 receptor or chemokines to block their activity and/or recruitment. More recent approaches attempt to capitalize on macrophage plasticity by re-programming them to anti-tumor phenotypes, targeting molecules like PI3K, STAT3 and IDO-1.
Specific targets for MDSC inhibition have been difficult to define, although multiple studies are examining CCR2/CXCR2 axis inhibition; this axis is a generic marker targeting inflamed tissue (Bullock and Richmond, 2021).
Targeting fibroblast activation protein (FAP), which marks immune suppressive CAFs, is most advanced clinically (Xin et al., 2021).
Cytokines or adjuvants can be targeted to tumors to modify the TIME and recruit immune cells, including IL-2 (Ren et al., 2022), STING agonist (Perera et al., 2021), CCL4 (Williford et al., 2019), mRNA encoding multiple cytokines (Hotz et al., 2021) and photodynamic therapy (Huis In ‘t Veld et al., 2021).
Outcomes
What can be expected of cancer vaccines and how should we look? We discuss four pertinent concepts for cancer vaccine trials.
Clinical Impact.
Despite an impressive increase in technical capability to monitor immune responses and more sophisticated understanding of cell phenotypes afforded by recent profiling technologies, clinical impact remains the most satisfying and trustworthy outcome. Thus, an important design variable is conducting trials in scenarios where a clinical benefit could be discernible. We posit that trials focused only on the less useful question of ‘are immune responses generated?’ should ideally be evaluated quickly (while analyzing immune correlatives thoroughly) and, if warranted, advanced to small trials that can generate a signal of clinical benefit. Simon two-phase designs can be useful to maximize the utility of recruitment.
Design and deconvolution of co-therapy trials.
In hindsight, a major flaw in the design of many past vaccine efforts was immunization alone in advanced disease with a highly evolved and suppressive TIME. Vaccines may effectively generate tumor-killing T cells (and maybe tumor-specific antibodies), but getting these warriors to the tumor, in sufficient numbers and quickly enough, and with the ability to overcome the tumor defenses is challenging as monotherapy in the setting of advanced disease. The use of co-therapies raises two important considerations: (1) how will the co-therapy affect the vaccine response – will it be complementary and synergistic or antagonistic? (2) de-convoluting effects of the combined therapy from those of the co-therapy alone. For the first question, unless the co-therapy is chosen based on a hypothesis of concomitant synergistic effects, temporally separating the vaccine and co-therapy (and recognizing that for de novo responses, priming and boosting are two critical and immunologically distinct phases) seems warranted. Since ICB is standard-of-care in many disease settings, weaving a vaccine-only phase into patient care can be challenging, arguing that less well-studied (or where immunotherapy has already failed) diseases need to be explored. Anti-PD-1 co-therapy, for example is a widely used standard-of-care, where sequencing of administration has biologic impact (discussed above). For the second question, historical controls are unreliable unless huge effect differences are observed; active control arms are preferred so that consideration of patients appropriate for the trial are made by the same investigators and contemporaneously.
Disease settings.
The other obvious approach to confronting TIME challenges is to look at early or minimal disease settings, where immunosuppression may be less well developed. One frequently pursued strategy is adjuvant therapy to prevent recurrence following surgical resection with curative intent. An important question regarding adjuvant vaccination is whether surgery sets up an immunologically compromised state and for what duration (Cheng et al., 2022); prolonged delays to adjuvant therapy are likely undesirable. Neoadjuvant vaccination potentially avoids surgery-induced immunosuppression during vaccination and provides correlative efficacy data in the form of clinical and pathological response, as observed with ICB (Menzies et al., 2021; Rozeman et al., 2021). The multiple weeks needed for personal neoantigen vaccine manufacture generally eliminates such an approach from consideration. Potentially even more desirable would be vaccinating during incipient disease, targeting, for example, ductal carcinoma in situ, pre-neoplastic colonic adenomas observed in colorectal cancer or pre-neoplastic vulvar intraepithelial neoplasia ((Finn, 2018); NCT04144023) or true prophylactic vaccination in high-risk groups (NCT04367675). In these settings, vaccine alone may be effective. Although such studies have focused to date on classic tumor-associated antigens, viral or common frameshift (Lynch Syndrome) antigens, personal neoantigens should be possible where precursor lesions portend increased risk of clonally related invasive disease even after removal (e.g. DCIS). Genes expressed in pluripotent stem cells, which overlap considerably with cancer stem cell gene programs, are another emerging source for targets (Ouyang et al., 2019).
Mouse models – the most commonly studied tumor vaccine setting – are an obvious place from which to draw inspiration. However, the complexities of a TIME established by immune pressure over years in humans vs days in mice, shaped by immune systems separated by 80 million years of evolution and by very different pathogen exposures, and typically in anatomically irrelevant sites challenge the translatability of murine models (Lal et al., 2021).
Nonetheless, such models can certainly provide a basis for hypothesis testing, especially when coupled with human correlative data, although they may only provide preclinical rationale for a subset of important questions about human biology. While the growing availability of autochthonous models may provide somewhat more translatable information, we posit that less expensive but safe approaches to more rapidly testing new concepts in humans need to be developed, fostering ‘reverse translation’ using the increasingly sophisticated suite of analytical technologies now available.
Immune correlatives.
The tools to investigate the ‘surrogate’ immunology of clinical samples have advanced dramatically over the past decade due to the generation of disruptive new technologies and advancements in existing tools. This includes prediction of peptide epitopes, epitope-specific MHC multimer availability, multiplex flow analysis (fluorophores or mass-labeled), single cell RNA sequencing, and bulk and single cell (including paired) TCR and BCR sequencing. Moreover, if tumor samples are available post-vaccination, multiplexed immunohistochemistry has advanced such that more discriminating spatial analysis can be performed (Tan et al., 2020) and molecular approaches are now being applied to FFPE sections (Dries et al., 2021). How is such correlative analysis best applied to assess vaccine activity?
Analysis of blood for circulating antigen-specific T cells, readily accomplished in various ways (tetramer staining, flow-based intracellular staining, IFNγ ELISPOT) ex vivo or after in vitro stimulation with one or many specific epitopes or total tumor lysate can reveal if responsive T cells are circulating - and ex vivo detection of CD8+ responses should be a minimum goal - but such analysis does not necessarily indicate tumor killing capability. Paradoxically, T cells may be rapidly sequestered in inflamed tissue or tumor, as response to checkpoint therapy has been associated with the disappearance of pre-existing T cell responses from the blood (Bochem et al., 2021), and hence circulating and intratumoral responses may differ. Analysis of longitudinal blood specimens can be highly informative for addressing the dynamics of immune responses.
Analysis of tumor-infiltrating T cells using single cell transcriptome and paired TCR sequencing can be even more informative, especially when T cell phenotypic properties can be linked to detailed features of antigen specificity (Caushi et al., 2021; Oliveira et al., 2021). An extensive analysis using single cell sequencing and other techniques on multiple tumor samples and bulk TCR sequencing on blood samples to follow T cell dynamics demonstrated ‘clonal revival’ (see above) of tumor-specific clones in tumor and blood in patients responding to ICB and chemotherapy (Liu et al., 2021). A demonstration that vaccine therapy increased the number, and changed the phenotype, of T cells in tumors would be highly encouraging, especially if coupled with clinical response, but surgical sample availability is often limiting. In the more typical setting of limited tumor fragments, small volume analytics like immunohistochemistry (IHC) can follow total changes in T cells and other populations. More advanced approaches are applying nucleic acid tools to interrogate cell genotype and spatial context at the single cell level in small biopsy specimens (Dries et al., 2021). Genetic analysis of tumor cells can also reveal signs of immunoediting as a marker of immune pressure (Anagnostou et al., 2017). Changes in blood or tumor TCR diversity and clonotype expansion may be tracked by bulk TCR sequencing and with more granularity by single cell paired TCR-sequencing tools, providing a snapshot of T cell dynamics if adequate samples are available. The current challenge with this approach is that confirming epitope-specificity is time/resource consuming and so is restricted in widespread use.
One approach which may optimally balance practicality with high informative content is evaluating blood samples for epitope spreading – an increase in the circulating T cells specific for tumor antigens that were not included in the vaccine (Figure 6B). Epitope spread may be the best indicator of tumor cytolysis, which in turn may provide a surrogate molecular indicator of clinical impact (Ott et al., 2020), is not technically challenging to conduct (ELISPOT, tetramer, ICS), only requires blood samples and has been detected following vaccination of patients with no clinical evidence of disease, perhaps pointing to its sensitivity (Hu et al., 2021). Finally, cell-free DNA analysis, particularly for tumor-specific mutations, is steadily emerging as a tool to quantitatively follow tumor genomes in the blood (Zou et al., 2021) and could potentially reveal target-specific immune pressure by tracking both vaccine targeted neoantigens and non-targeted mutations.
Figure 6: Measuring success.
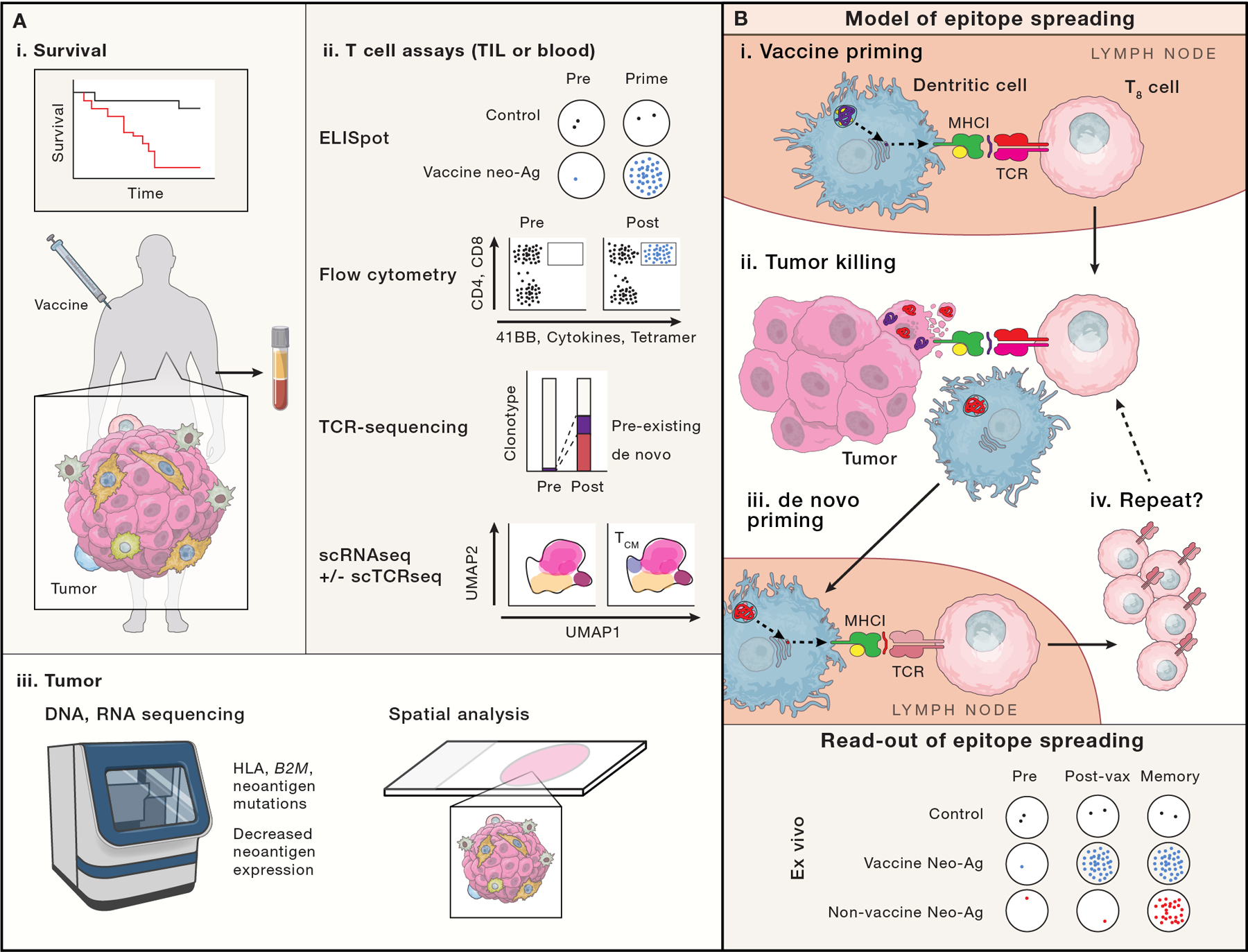
Ideally a cancer vaccine will demonstrate improved survival or progression free survival (A-i), although cancer vaccine trials are not usually powered to do so. Trials more typically focus on putative correlates of vaccine effectiveness via ex vivo analysis of T cells from the blood or tumor samples (A-ii) or analysis of tumor biopsies (A-iii). (A-ii) T cell assays include: ELISPOT, which enumerates T cell clones responsive to a defined peptide/epitope; flow cytometry based assays, which can identify antigen-specific T cells via p:MHC conjugates (e.g. tetramers), and define T cell function, activation and cytotoxic potential via cytokine, surface 41BB and CD107a exposure; bulk TCR sequencing can temporally track clonotype frequencies; single cell RNA and TCR sequencing (not broadly used in vaccine studies) may enable the linkage of clonotype changes to changes in cell state (e.g. effector, central memory, progenitor exhausted and exhausted). Linking TCR sequence to antigen specificity is not readily possible except through more extensive functional studies. (A-iii) Re-biopsy of tumor can provide evidence of vaccine effectiveness, for example, if genomic or expression analyses demonstrate loss of antigen presenting machinery or neoantigens, or decreased neoantigen expression. Newer spatial multiplexed IHC, in situ hybridization (Nanostring) and sequencing (Slide-seq), may show differences in T cell infiltration patterns (especially when paired with TCR sequencing methods). (B) Schema of epitope spreading. We propose that analysis of epitope spreading may best balance feasibility and sensitivity for vaccine efficacy. Epitope spreading is the concept that one antigen-specific immune response begets another: First, a vaccine primes a CD8+ T cell (purple T8 cell recognizing a purple neoantigen; B-i). That vaccine specific CTL migrates to the tumor (B-ii), where it recognizes and destroys a cancer cell, releasing DAMPs that stimulate a DC maturation and phagocytoses tumor cell remnants, including non-vaccine targeted neoantigens (red). The DC migrates to a draining LN (B-iii), where it presents neoantigens to T cells. A CD8+ T cell (red) with specificity for a new neoantigen is de novo primed and expanded, returning to the tumor (B-iv). B-bottom, model ELISPOT data reflecting epitope spreading is shown, with vaccine induced neoantigen responses appearing after vaccine priming, but non-vaccine neoantigen responses from epitope spreading, appearing only later.
Where to Find Help
Where can we learn lessons to accelerate the pace of tumor-immune evolution in favor of the immune system with cancer vaccines? Multiple lines of evidence support our premise of the therapeutic benefits of mimicking natural immunity.
TIL therapy – expanding T cells already present in tumors, the product of natural immunity – has provided durable benefit to late-stage patients (Kumar et al., 2021).
The oncolytic virus T-VEC, an approved therapy for accessible melanoma, mediates its effect not by direct killing, but via the immune response generated by dying, virally-infected cells – a raison d’etre of the immune system (Ferrucci et al., 2021).
Certain chemotherapies can induce immunogenic cell death (ICD), which provides sufficient DAMPs to initiate immune responses, and may be an important component of efficacy (Zitvogel et al., 2011). A host of pre-clinical and clinical studies explored parameters to optimize this interaction (Hernandez et al., 2021), culminating in the approval of anti-PD-1 in combination with first line chemotherapy for lung cancer.
The abscopal effect of radiation therapy inspired the concept of in situ vaccination, turning the tumor bed into the site of systemic immune cell priming. Directly delivering some combination of tumor cell death inducers and danger signals to the tumor, sometimes enhanced with immunotherapies, is under active preclinical and clinical investigation (Hammerich et al., 2016) and relies on generating systemic responses primed by an in situ response (Chen and Mellman, 2013).
These approaches share the theme of dying whole tumor cells (WTC) as an antigen and adjuvant source and return to one of our initial dogmas – antigen presenting cells taking up dead/dying cells to initiate natural immunity to extant antigens. Simpler than using the latest sequencing and bioinformatic tools to find the ‘best’ antigens, and less biased from incomplete knowledge, WTC vaccines rely on the ‘undefined’ antigens within a tumor. Particularly note-worthy in their preclinical and clinical resurgence are the advancement to phase II studies of a dendritic cell-AML cell fusion vaccine (Rosenblatt et al., 2016) and a promising study of a vaccine consisting of monocyte-derived dendritic cells loaded with autologous lysate from oxidized ovarian WTC (Tanyi et al., 2018). In the latter work, coupled mutation analysis enabled tracking of responses to the mutation-induced neoantigens, and demonstrated de novo induction, expansion, and TCR avidity enhancement following vaccination and a positive correlation between immunogenicity and time to progression or overall survival. These trials are encouraging, but still limited by tumor sample size and the logistics of implementation. Remarkably, despite the overwhelming abundance of self-antigens in these WTC formulations, there was no indication that auto-immunity, even as strong immunity was induced against ‘foreign’ antigen from the same immune milieu. Why is this? Central tolerance is impressively effective, but maybe less so for tissue restricted antigens (Legoux et al., 2015). Treg’s provide effective post-thymic tolerance, but depending on antigen specificity requirements, the breadth of the required Treg repertoire could be energetically unfavorable. A possible intrinsic contributor may be a limited diversity of immunogenic self-epitopes; the same evolutionary pressure that operates on tumors within a fraction of a human lifetime could have operated over eons on the ‘immunogenomic’ proteome, eliminating protein sequences that engender self-immunogenic response. Irrespective of an explanation, which certainly requires further exploration, there is little evidence that self-reactivity is an issue either as a result of natural anti-tumor immunity or with whole-cell based antigens.
Conclusions and Directions
The Golden Gate Bridge is a now timeless example of simple beauty – two towers supporting suspension cables to a roadway arching the turbulent waters of the narrow Golden Gate strait, a functional but elegant mark on an already picturesque scene. Innovative thinking and over a year of negotiating overcame an initial plan for an awkward cantilevered structure with multiple supporting trusses. By analogy, cancer vaccines, after 50 years of middling success, can stand to use more out-of-the box thinking (See Box 4 for key questions and ideas). Neoantigens have provided one such novel direction, but our ability to see through all the complexity to the other side (the right epitopes) is still clouded by gaps in knowledge, requiring still developing technologies and bioinformatic trusses (tools). An area of renewed interest is WTC vaccines, since by mimicking the stressed and dying cells that are the starting point of ‘natural immunity’, they provide access to all the antigens of the cell, at levels reflective of what is in the cell, and without any observer selection or bias, potentially providing a clearer image of a personal tumor than any technology can yet deliver.
Box 4: A box of out-of-the-box thinking.
With a long history of middling cancer vaccine success, it is time to shake things up.
Be inspired by natural immunity and look to whole tumor cell approaches to better capture the entire antigen repertoire.
Reverse the epitope information flow by using undefined whole tumor vaccines with a bigger epitope repertoire and reverse engineering to learn what makes effective epitopes.
Improve antigen target selection through better prediction of peptide processing (in APCs and cancer cells) or modelling of interactions between peptide epitopes, MHC and the TCR repertoire.
What is the best way to get antigen (and activating signals) into APCs? Consider investment in rapid ex vivo preparation of antigen presenting/delivering cells, especially allogeneic cells, to better control cell phenotype and antigen delivery.
Include mimics of innate-adaptive immune cell cross-talk in response to natural infections to maximize vaccine responses. CD40-CD40L or CD27–CD70, for example.
CTL responses remain a central challenge. Engage CD4+ T cells and B cells with antigen structures to complement and enhance CTL activity. Clarify the origin of effective helper epitopes and antigen structure. Do CD4 helper epitopes need to be tumor-specific or are universal helper epitopes sufficient?
Treg biology needs to be better understood – especially the importance of antigen specificity; Does/can vaccination inadvertently enhance/induce Tregs?
Exercise patience and humility – not to undermine vaccine potential in late-stage disease, vaccines will undeniably be more effective in early disease, which will eventually reduce the costly and difficult burden of late-stage disease. Funders, regulatory authorities and researchers need to align with this reality and opportunity.
Identify a setting - in humans - where delivery dynamics surrounding priming and boosting can be studied with several formats to determine if an optimal schedule exists.
Respect the effectiveness and uniqueness of the evolved suppressive TIME. Follow (or precede) a prime/boost vaccination cycle quickly with a ‘bucket’ trial of TIME modulators.
The importance of tumor-specific T cells to immune therapy and to durable patient survival cannot be over-emphasized. Cancer vaccines have been proven to enhance both the number of such cells and their repertoire, but there are still difficult waters to cross to arrive at the induction of truly effective T cell populations to achieve a lifeline to cure for our patients. To borrow from Joseph Strauss, the Chief Engineer for the Golden Gate Bridge, ‘When you build a [vaccine], you might build something [for the patient] for all time.’
Declaration of Interests
M.C.S. has no interests to declare. E.F.F. is an equity holder and consultant for BioNTech, an equity holder and scientific advisory board member of BioEntre, and a founder and equity holder of Dionis Therapeutics. C.J.W. is an equity holder of BioNTech. An immediate family member of C.J.W. is an advisor and equity holder for Related Sciences and receives research funding from Bristol-Myers Squibb. Patent applications have been filed that relate to the reviewed material, as follows: ‘Compositions and methods for personalized neoplasia vaccines’ (E.F.F. and C.J.W.), ‘Methods for identifying tumor specific neo-antigens’ (C.J.W.), ‘Formulations for neoplasia vaccines’ (E.F.F.), ‘Combination therapy for neoantigen vaccine’ (C.J.W. and E.F.F.), and ‘Multi-domain protein vaccine’ (E.F.F).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdel-Hakeem MS, Manne S, Beltra JC, Stelekati E, Chen Z, Nzingha K, Ali MA, Johnson JL, Giles JR, Mathew D, Greenplate AR, Vahedi G, and Wherry EJ (2021). Epigenetic scarring of exhausted T cells hinders memory differentiation upon eliminating chronic antigenic stimulation. Nat Immunol 22, 1008–1019. 10.1038/s41590-021-00975-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abelin JG, Harjanto D, Malloy M, Suri P, Colson T, Goulding SP, Creech AL, Serrano LR, Nasir G, Nasrullah Y, McGann CD, Velez D, Ting YS, Poran A, Rothenberg DA, Chhangawala S, Rubinsteyn A, Hammerbacher J, Gaynor RB, Fritsch EF, Greshock J, Oslund RC, Barthelme D, Addona TA, Arieta CM, and Rooney MS (2019). Defining HLA-II Ligand Processing and Binding Rules with Mass Spectrometry Enhances Cancer Epitope Prediction. Immunity 51, 766–779 e717. 10.1016/j.immuni.2019.08.012. [DOI] [PubMed] [Google Scholar]
- Abusarah J, Khodayarian F, El-Hachem N, Salame N, Olivier M, Balood M, Roversi K, Talbot S, Bikorimana JP, Chen J, Jolicoeur M, Trudeau LE, Kamyabiazar S, Annabi B, Robert F, Pelletier J, El-Kadiry AE, Shammaa R, and Rafei M (2021). Engineering immunoproteasome-expressing mesenchymal stromal cells: A potent cellular vaccine for lymphoma and melanoma in mice. Cell Rep Med 2, 100455. 10.1016/j.xcrm.2021.100455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allavena P, Anfray C, Ummarino A, and Andon FT (2021). Therapeutic Manipulation of Tumor-associated Macrophages: Facts and Hopes from a Clinical and Translational Perspective. Clin Cancer Res 27, 3291–3297. 10.1158/1078-0432.CCR-20-1679. [DOI] [PubMed] [Google Scholar]
- Alspach E, Lussier DM, Miceli AP, Kizhvatov I, DuPage M, Luoma AM, Meng W, Lichti CF, Esaulova E, Vomund AN, Runci D, Ward JP, Gubin MM, Medrano RFV, Arthur CD, White JM, Sheehan KCF, Chen A, Wucherpfennig KW, Jacks T, Unanue ER, Artyomov MN, and Schreiber RD (2019). MHC-II neoantigens shape tumour immunity and response to immunotherapy. Nature 574, 696–701. 10.1038/s41586-019-1671-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anagnostou V, Smith KN, Forde PM, Niknafs N, Bhattacharya R, White J, Zhang T, Adleff V, Phallen J, Wali N, Hruban C, Guthrie VB, Rodgers K, Naidoo J, Kang H, Sharfman W, Georgiades C, Verde F, Illei P, Li QK, Gabrielson E, Brock MV, Zahnow CA, Baylin SB, Scharpf RB, Brahmer JR, Karchin R, Pardoll DM, and Velculescu VE (2017). Evolution of Neoantigen Landscape during Immune Checkpoint Blockade in Non-Small Cell Lung Cancer. Cancer Discov 7, 264–276. 10.1158/2159-8290.CD-16-0828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baharom F, Ramirez-Valdez RA, Tobin KKS, Yamane H, Dutertre CA, Khalilnezhad A, Reynoso GV, Coble VL, Lynn GM, Mule MP, Martins AJ, Finnigan JP, Zhang XM, Hamerman JA, Bhardwaj N, Tsang JS, Hickman HD, Ginhoux F, Ishizuka AS, and Seder RA (2021). Intravenous nanoparticle vaccination generates stem-like TCF1(+) neoantigen-specific CD8(+) T cells. Nat Immunol 22, 41–52. 10.1038/s41590-020-00810-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassani-Sternberg M, Braunlein E, Klar R, Engleitner T, Sinitcyn P, Audehm S, Straub M, Weber J, Slotta-Huspenina J, Specht K, Martignoni ME, Werner A, Hein R, D HB, Peschel C, Rad R, Cox J, Mann M, and Krackhardt AM (2016). Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Nat Commun 7, 13404. 10.1038/ncomms13404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belnoue E, Mayol JF, Carboni S, Di Berardino Besson W, Dupuychaffray E, Nelde A, Stevanovic S, Santiago-Raber ML, Walker PR, and Derouazi M (2019). Targeting self and neoepitopes with a modular self-adjuvanting cancer vaccine. JCI Insight 5. 10.1172/jci.insight.127305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhardwaj N, Friedlander PA, Pavlick AC, Ernstoff MS, Gastman BR, Hanks BA, Curti BD, Albertini MR, Luke JJ, Blazquez AB, Balan S, Bedognetti D, Beechem JM, Crocker AS, D’Amico L, Danaher P, Davis TA, Hawthorne T, Hess BW, Keler T, Lundgren L, Morishima C, Ramchurren N, Rinchai D, Salazar AM, Salim BA, Sharon E, Vitale LA, Wang E, Warren S, Yellin MJ, Disis ML, Cheever MA, and Fling SP (2020). Flt3 ligand augments immune responses to anti-DEC-205-NY-ESO-1 vaccine through expansion of dendritic cell subsets. Nature Cancer 1, 1204–1217. 10.1038/s43018-020-00143-y. [DOI] [PubMed] [Google Scholar]
- Binnewies M, Mujal AM, Pollack JL, Combes AJ, Hardison EA, Barry KC, Tsui J, Ruhland MK, Kersten K, Abushawish MA, Spasic M, Giurintano JP, Chan V, Daud AI, Ha P, Ye CJ, Roberts EW, and Krummel MF (2019). Unleashing Type-2 Dendritic Cells to Drive Protective Antitumor CD4(+) T Cell Immunity. Cell 177, 556–571 e516. 10.1016/j.cell.2019.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birnbaum ME, Mendoza JL, Sethi DK, Dong S, Glanville J, Dobbins J, Ozkan E, Davis MM, Wucherpfennig KW, and Garcia KC (2014). Deconstructing the peptide-MHC specificity of T cell recognition. Cell 157, 1073–1087. 10.1016/j.cell.2014.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas S, Mandal G, Payne KK, Anadon CM, Gatenbee CD, Chaurio RA, Costich TL, Moran C, Harro CM, Rigolizzo KE, Mine JA, Trillo-Tinoco J, Sasamoto N, Terry KL, Marchion D, Buras A, Wenham RM, Yu X, Townsend MK, Tworoger SS, Rodriguez PC, Anderson AR, and Conejo-Garcia JR (2021). IgA transcytosis and antigen recognition govern ovarian cancer immunity. Nature 591, 464–470. 10.1038/s41586-020-03144-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochem J, Zelba H, Spreuer J, Amaral T, Gaissler A, Pop OT, Thiel K, Yurttas C, Soffel D, Forchhammer S, Sinnberg T, Niessner H, Meier F, Terheyden P, Konigsrainer A, Garbe C, Flatz L, Pawelec G, Eigentler TK, Loffler MW, Weide B, and Wistuba-Hamprecht K (2021). Early disappearance of tumor antigen-reactive T cells from peripheral blood correlates with superior clinical outcomes in melanoma under anti-PD-1 therapy. J Immunother Cancer 9. 10.1136/jitc-2021-003439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booty MG, Hlavaty KA, Stockmann A, Ozay EI, Smith C, Tian L, How E, Subramanya D, Venkitaraman A, Yee C, Pryor O, Volk K, Blagovic K, Vicente-Suarez I, Yarar D, Myint M, Merino A, Chow J, Abdeljawad T, An H, Liu S, Mao S, Heimann M, Talarico L, Jacques MK, Chong E, Pomerance L, Gonzalez JT, von Andrian UH, Jensen KF, Langer R, Knoetgen H, Trumpfheller C, Umana P, Bernstein H, Sharei A, and Loughhead SM (2022). Microfluidic Squeezing Enables MHC Class I Antigen Presentation by Diverse Immune Cells to Elicit CD8(+) T Cell Responses with Antitumor Activity. J Immunol. 10.4049/jimmunol.2100656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullock K, and Richmond A (2021). Suppressing MDSC Recruitment to the Tumor Microenvironment by Antagonizing CXCR2 to Enhance the Efficacy of Immunotherapy. Cancers (Basel) 13. 10.3390/cancers13246293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger ML, Cruz AM, Crossland GE, Gaglia G, Ritch CC, Blatt SE, Bhutkar A, Canner D, Kienka T, Tavana SZ, Barandiaran AL, Garmilla A, Schenkel JM, Hillman M, de Los Rios Kobara I, Li A, Jaeger AM, Hwang WL, Westcott PMK, Manos MP, Holovatska MM, Hodi FS, Regev A, Santagata S, and Jacks T (2021). Antigen dominance hierarchies shape TCF1(+) progenitor CD8 T cell phenotypes in tumors. Cell 184, 4996–5014 e4926. 10.1016/j.cell.2021.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cachot A, Bilous M, Liu YC, Li X, Saillard M, Cenerenti M, Rockinger GA, Wyss T, Guillaume P, Schmidt J, Genolet R, Ercolano G, Protti MP, Reith W, Ioannidou K, de Leval L, Trapani JA, Coukos G, Harari A, Speiser DE, Mathis A, Gfeller D, Altug H, Romero P, and Jandus C (2021). Tumor-specific cytolytic CD4 T cells mediate immunity against human cancer. Sci Adv 7. 10.1126/sciadv.abe3348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caushi JX, Zhang J, Ji Z, Vaghasia A, Zhang B, Hsiue EH, Mog BJ, Hou W, Justesen S, Blosser R, Tam A, Anagnostou V, Cottrell TR, Guo H, Chan HY, Singh D, Thapa S, Dykema AG, Burman P, Choudhury B, Aparicio L, Cheung LS, Lanis M, Belcaid Z, El Asmar M, Illei PB, Wang R, Meyers J, Schuebel K, Gupta A, Skaist A, Wheelan S, Naidoo J, Marrone KA, Brock M, Ha J, Bush EL, Park BJ, Bott M, Jones DR, Reuss JE, Velculescu VE, Chaft JE, Kinzler KW, Zhou S, Vogelstein B, Taube JM, Hellmann MD, Brahmer JR, Merghoub T, Forde PM, Yegnasubramanian S, Ji H, Pardoll DM, and Smith KN (2021). Transcriptional programs of neoantigen-specific TIL in anti-PD-1-treated lung cancers. Nature 596, 126–132. 10.1038/s41586-021-03752-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen BJ, Zhao JW, Zhang DH, Zheng AH, and Wu GQ (2022). Immunotherapy of Cancer by Targeting Regulatory T cells. Int Immunopharmacol 104, 108469. 10.1016/j.intimp.2021.108469. [DOI] [PubMed] [Google Scholar]
- Chen DS, and Mellman I (2013). Oncology meets immunology: the cancer-immunity cycle. Immunity 39, 1–10. 10.1016/j.immuni.2013.07.012. [DOI] [PubMed] [Google Scholar]
- Cheng X, Zhang H, Hamad A, Huang H, and Tsung A (2022). Surgery-mediated tumor-promoting effects on the immune microenvironment. Semin Cancer Biol. 10.1016/j.semcancer.2022.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui C, Wang J, Fagerberg E, Chen PM, Connolly KA, Damo M, Cheung JF, Mao T, Askari AS, Chen S, Fitzgerald B, Foster GG, Eisenbarth SC, Zhao H, Craft J, and Joshi NS (2021). Neoantigen-driven B cell and CD4 T follicular helper cell collaboration promotes anti-tumor CD8 T cell responses. Cell 184, 6101–6118 e6113. 10.1016/j.cell.2021.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danilova L, Anagnostou V, Caushi JX, Sidhom JW, Guo H, Chan HY, Suri P, Tam A, Zhang J, Asmar ME, Marrone KA, Naidoo J, Brahmer JR, Forde PM, Baras AS, Cope L, Velculescu VE, Pardoll DM, Housseau F, and Smith KN (2018). The Mutation-Associated Neoantigen Functional Expansion of Specific T Cells (MANAFEST) Assay: A Sensitive Platform for Monitoring Antitumor Immunity. Cancer Immunol Res 6, 888–899. 10.1158/2326-6066.CIR-18-0129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desbois M, and Wang Y (2021). Cancer-associated fibroblasts: Key players in shaping the tumor immune microenvironment. Immunol Rev 302, 241–258. 10.1111/imr.12982. [DOI] [PubMed] [Google Scholar]
- Diehl L, den Boer AT, Schoenberger SP, van der Voort EI, Schumacher TN, Melief CJ, Offringa R, and Toes RE (1999). CD40 activation in vivo overcomes peptide-induced peripheral cytotoxic T-lymphocyte tolerance and augments anti-tumor vaccine efficacy. Nat Med 5, 774–779. 10.1038/10495. [DOI] [PubMed] [Google Scholar]
- Downs-Canner SM, Meier J, Vincent BG, and Serody JS (2022). B Cell Function in the Tumor Microenvironment. Annu Rev Immunol. 10.1146/annurev-immunol-101220-015603. [DOI] [PubMed] [Google Scholar]
- Dries R, Chen J, Del Rossi N, Khan MM, Sistig A, and Yuan GC (2021). Advances in spatial transcriptomic data analysis. Genome Res 31, 1706–1718. 10.1101/gr.275224.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn GP, Bruce AT, Ikeda H, Old LJ, and Schreiber RD (2002). Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol 3, 991–998. 10.1038/ni1102-991. [DOI] [PubMed] [Google Scholar]
- Duong E, Fessenden TB, Lutz E, Dinter T, Yim L, Blatt S, Bhutkar A, Wittrup KD, and Spranger S (2021). Type I interferon activates MHC class I-dressed CD11b(+) conventional dendritic cells to promote protective anti-tumor CD8(+) T cell immunity. Immunity. 10.1016/j.immuni.2021.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebrahimi-Nik H, Moussa M, Englander RP, Singhaviranon S, Michaux J, Pak H, Miyadera H, Corwin WL, Keller GLJ, Hagymasi AT, Shcheglova TV, Coukos G, Baker BM, Mandoiu II, Bassani-Sternberg M, and Srivastava PK (2021). Reversion analysis reveals the in vivo immunogenicity of a poorly MHC I-binding cancer neoepitope. Nat Commun 12, 6423. 10.1038/s41467-021-26646-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagone P, Shedlock DJ, Bao H, Kawalekar OU, Yan J, Gupta D, Morrow MP, Patel A, Kobinger GP, Muthumani K, and Weiner DB (2011). Molecular adjuvant HMGB1 enhances anti-influenza immunity during DNA vaccination. Gene Ther 18, 1070–1077. 10.1038/gt.2011.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferris ST, Durai V, Wu R, Theisen DJ, Ward JP, Bern MD, Davidson J.T.t., Bagadia P, Liu T, Briseno CG, Li L, Gillanders WE, Wu GF, Yokoyama WM, Murphy TL, Schreiber RD, and Murphy KM (2020). cDC1 prime and are licensed by CD4(+) T cells to induce anti-tumour immunity. Nature 584, 624–629. 10.1038/s41586-020-2611-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrucci PF, Pala L, Conforti F, and Cocorocchio E (2021). Talimogene Laherparepvec (T-VEC): An Intralesional Cancer Immunotherapy for Advanced Melanoma. Cancers (Basel) 13. 10.3390/cancers13061383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn OJ (2018). The dawn of vaccines for cancer prevention. Nat Rev Immunol 18, 183–194. 10.1038/nri.2017.140. [DOI] [PubMed] [Google Scholar]
- Fitzgerald KA, and Kagan JC (2020). Toll-like Receptors and the Control of Immunity. Cell 180, 1044–1066. 10.1016/j.cell.2020.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher EAK, van Maren W, Cordfunke R, Dinkelaar J, Codee JDC, van der Marel G, Melief CJM, Ossendorp F, Drijfhout JW, and Mangsbo SM (2018). Formation of Immune Complexes with a Tetanus-Derived B Cell Epitope Boosts Human T Cell Responses to Covalently Linked Peptides in an Ex Vivo Blood Loop System. J Immunol 201, 87–97. 10.4049/jimmunol.1700911. [DOI] [PubMed] [Google Scholar]
- Fluckiger A, Daillere R, Sassi M, Sixt BS, Liu P, Loos F, Richard C, Rabu C, Alou MT, Goubet AG, Lemaitre F, Ferrere G, Derosa L, Duong CPM, Messaoudene M, Gagne A, Joubert P, De Sordi L, Debarbieux L, Simon S, Scarlata CM, Ayyoub M, Palermo B, Facciolo F, Boidot R, Wheeler R, Boneca IG, Sztupinszki Z, Papp K, Csabai I, Pasolli E, Segata N, Lopez-Otin C, Szallasi Z, Andre F, Iebba V, Quiniou V, Klatzmann D, Boukhalil J, Khelaifia S, Raoult D, Albiges L, Escudier B, Eggermont A, Mami-Chouaib F, Nistico P, Ghiringhelli F, Routy B, Labarriere N, Cattoir V, Kroemer G, and Zitvogel L (2020). Cross-reactivity between tumor MHC class I-restricted antigens and an enterococcal bacteriophage. Science 369, 936–942. 10.1126/science.aax0701. [DOI] [PubMed] [Google Scholar]
- Fossum E, Grodeland G, Terhorst D, Tveita AA, Vikse E, Mjaaland S, Henri S, Malissen B, and Bogen B (2015). Vaccine molecules targeting Xcr1 on cross-presenting DCs induce protective CD8+ T-cell responses against influenza virus. Eur J Immunol 45, 624–635. 10.1002/eji.201445080. [DOI] [PubMed] [Google Scholar]
- Fritsch EF, Burkhardt UE, Hacohen N, and Wu CJ (2020). Personal Neoantigen Cancer Vaccines: A Road Not Fully Paved. Cancer Immunol Res 8, 1465–1469. 10.1158/2326-6066.CIR-20-0526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu C, Zhou L, Mi QS, and Jiang A (2022). Plasmacytoid Dendritic Cells and Cancer Immunotherapy. Cells 11. 10.3390/cells11020222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geijtenbeek TB, and Gringhuis SI (2009). Signalling through C-type lectin receptors: shaping immune responses. Nat Rev Immunol 9, 465–479. 10.1038/nri2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant SM, Lou M, Yao L, Germain RN, and Radtke AJ (2020). The lymph node at a glance - how spatial organization optimizes the immune response. J Cell Sci 133. 10.1242/jcs.241828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grotzke JE, Sengupta D, Lu Q, and Cresswell P (2017). The ongoing saga of the mechanism(s) of MHC class I-restricted cross-presentation. Curr Opin Immunol 46, 89–96. 10.1016/j.coi.2017.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta RG, Li F, Roszik J, and Lizee G (2021). Exploiting Tumor Neoantigens to Target Cancer Evolution: Current Challenges and Promising Therapeutic Approaches. Cancer Discov 11, 1024–1039. 10.1158/2159-8290.CD-20-1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammerich L, Bhardwaj N, Kohrt HE, and Brody JD (2016). In situ vaccination for the treatment of cancer. Immunotherapy 8, 315–330. 10.2217/imt.15.120. [DOI] [PubMed] [Google Scholar]
- Han P, Hanlon D, Arshad N, Lee JS, Tatsuno K, Robinson E, Filler R, Sobolev O, Cote C, Rivera-Molina F, Toomre D, Fahmy T, and Edelson R (2020). Platelet P-selectin initiates cross-presentation and dendritic cell differentiation in blood monocytes. Sci Adv 6, eaaz1580. 10.1126/sciadv.aaz1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanel G, Angerer C, Petry K, Lichtenegger FS, and Subklewe M (2021). Blood DCs activated with R848 and poly(I:C) induce antigen-specific immune responses against viral and tumor-associated antigens. Cancer Immunol Immunother. 10.1007/s00262-021-03109-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harndahl M, Rasmussen M, Roder G, and Buus S (2011). Real-time, high-throughput measurements of peptide-MHC-I dissociation using a scintillation proximity assay. J Immunol Methods 374, 5–12. 10.1016/j.jim.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde S, Leader AM, and Merad M (2021). MDSC: Markers, development, states, and unaddressed complexity. Immunity 54, 875–884. 10.1016/j.immuni.2021.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez AP, Juanes-Velasco P, Landeira-Vinuela A, Bareke H, Montalvillo E, Gongora R, and Fuentes M (2021). Restoring the Immunity in the Tumor Microenvironment: Insights into Immunogenic Cell Death in Onco-Therapies. Cancers (Basel) 13. 10.3390/cancers13112821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofer T, Rossi M, Carboni S, Di Berardino Besson W, von Laer D, Wollmann G, Derouazi M, and Santiago-Raber ML (2021). Heterologous Prime-Boost Vaccination with a Peptide-Based Vaccine and Viral Vector Reshapes Dendritic Cell, CD4+ and CD8+ T Cell Phenotypes to Improve the Antitumor Therapeutic Effect. Cancers (Basel) 13. 10.3390/cancers13236107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotz C, Wagenaar TR, Gieseke F, Bangari DS, Callahan M, Cao H, Diekmann J, Diken M, Grunwitz C, Hebert A, Hsu K, Bernardo M, Kariko K, Kreiter S, Kuhn AN, Levit M, Malkova N, Masciari S, Pollard J, Qu H, Ryan S, Selmi A, Schlereth J, Singh K, Sun F, Tillmann B, Tolstykh T, Weber W, Wicke L, Witzel S, Yu Q, Zhang YA, Zheng G, Lager J, Nabel GJ, Sahin U, and Wiederschain D (2021). Local delivery of mRNA-encoded cytokines promotes antitumor immunity and tumor eradication across multiple preclinical tumor models. Sci Transl Med 13, eabc7804. 10.1126/scitranslmed.abc7804. [DOI] [PubMed] [Google Scholar]
- Hu Z, Leet DE, Allesoe RL, Oliveira G, Li S, Luoma AM, Liu J, Forman J, Huang T, Iorgulescu JB, Holden R, Sarkizova S, Gohil SH, Redd RA, Sun J, Elagina L, Giobbie-Hurder A, Zhang W, Peter L, Ciantra Z, Rodig S, Olive O, Shetty K, Pyrdol J, Uduman M, Lee PC, Bachireddy P, Buchbinder EI, Yoon CH, Neuberg D, Pentelute BL, Hacohen N, Livak KJ, Shukla SA, Olsen LR, Barouch DH, Wucherpfennig KW, Fritsch EF, Keskin DB, Wu CJ, and Ott PA (2021). Personal neoantigen vaccines induce persistent memory T cell responses and epitope spreading in patients with melanoma. Nat Med 27, 515–525. 10.1038/s41591-020-01206-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Wang Z, Zhang Y, Pradhan RN, Ganguly D, Chandra R, Murimwa G, Wright S, Gu X, Maddipati R, Muller S, Turley SJ, and Brekken RA (2022). Mesothelial cell-derived antigen-presenting cancer-associated fibroblasts induce expansion of regulatory T cells in pancreatic cancer. Cancer Cell. 10.1016/j.ccell.2022.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huis In ‘t Veld RV, Da Silva CG, Jager MJ, Cruz LJ, and Ossendorp F (2021). Combining Photodynamic Therapy with Immunostimulatory Nanoparticles Elicits Effective Anti-Tumor Immune Responses in Preclinical Murine Models. Pharmaceutics 13. 10.3390/pharmaceutics13091470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irvine DJ, Aung A, and Silva M (2020). Controlling timing and location in vaccines. Adv Drug Deliv Rev 158, 91–115. 10.1016/j.addr.2020.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhunjhunwala S, Hammer C, and Delamarre L (2021). Antigen presentation in cancer: insights into tumour immunogenicity and immune evasion. Nat Rev Cancer 21, 298–312. 10.1038/s41568-021-00339-z. [DOI] [PubMed] [Google Scholar]
- Johansen P, Storni T, Rettig L, Qiu Z, Der-Sarkissian A, Smith KA, Manolova V, Lang KS, Senti G, Mullhaupt B, Gerlach T, Speck RF, Bot A, and Kundig TM (2008). Antigen kinetics determines immune reactivity. Proc Natl Acad Sci U S A 105, 5189–5194. 10.1073/pnas.0706296105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kok L, Masopust D, and Schumacher TN (2022). The precursors of CD8(+) tissue resident memory T cells: from lymphoid organs to infected tissues. Nat Rev Immunol 22, 283–293. 10.1038/s41577-021-00590-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korsholm KS, Hansen J, Karlsen K, Filskov J, Mikkelsen M, Lindenstrom T, Schmidt ST, Andersen P, and Christensen D (2014). Induction of CD8+ T-cell responses against subunit antigens by the novel cationic liposomal CAF09 adjuvant. Vaccine 32, 3927–3935. 10.1016/j.vaccine.2014.05.050. [DOI] [PubMed] [Google Scholar]
- Kranz LM, Diken M, Haas H, Kreiter S, Loquai C, Reuter KC, Meng M, Fritz D, Vascotto F, Hefesha H, Grunwitz C, Vormehr M, Husemann Y, Selmi A, Kuhn AN, Buck J, Derhovanessian E, Rae R, Attig S, Diekmann J, Jabulowsky RA, Heesch S, Hassel J, Langguth P, Grabbe S, Huber C, Tureci O, and Sahin U (2016). Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 534, 396–401. 10.1038/nature18300. [DOI] [PubMed] [Google Scholar]
- Kristensen NP, Heeke C, Tvingsholm SA, Borch A, Draghi A, Crowther MD, Carri I, Munk KK, Holm JS, Bjerregaard AM, Bentzen AK, Marquard AM, Szallasi Z, McGranahan N, Andersen R, Nielsen M, Jonsson GB, Donia M, Svane IM, and Hadrup SR (2022). Neoantigen-reactive CD8+ T cells affect clinical outcome of adoptive cell therapy with tumor-infiltrating lymphocytes in melanoma. J Clin Invest 132. 10.1172/JCI150535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Watkins R, and Vilgelm AE (2021). Cell Therapy With TILs: Training and Taming T Cells to Fight Cancer. Front Immunol 12, 690499. 10.3389/fimmu.2021.690499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar BV, Connors TJ, and Farber DL (2018). Human T Cell Development, Localization, and Function throughout Life. Immunity 48, 202–213. 10.1016/j.immuni.2018.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lal JC, Townsend MG, Mehta AK, Oliwa M, Miller E, Sotayo A, Cheney E, Mittendorf EA, Letai A, and Guerriero JL (2021). Comparing syngeneic and autochthonous models of breast cancer to identify tumor immune components that correlate with response to immunotherapy in breast cancer. Breast Cancer Res 23, 83. 10.1186/s13058-021-01448-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam H, McNeil LK, Starobinets H, DeVault VL, Cohen RB, Twardowski P, Johnson ML, Gillison ML, Stein MN, Vaishampayan UN, DeCillis AP, Foti JJ, Vemulapalli V, Tjon E, Ferber K, DeOliveira DB, Broom W, Agnihotri P, Jaffee EM, Wong KK, Drake CG, Carroll PM, Davis TA, and Flechtner JB (2021). An Empirical Antigen Selection Method Identifies Neoantigens That Either Elicit Broad Antitumor T-cell Responses or Drive Tumor Growth. Cancer Discov 11, 696–713. 10.1158/2159-8290.CD-20-0377. [DOI] [PubMed] [Google Scholar]
- Lee YS, and Radford KJ (2019). The role of dendritic cells in cancer. Int Rev Cell Mol Biol 348, 123–178. 10.1016/bs.ircmb.2019.07.006. [DOI] [PubMed] [Google Scholar]
- Legoux FP, Lim JB, Cauley AW, Dikiy S, Ertelt J, Mariani TJ, Sparwasser T, Way SS, and Moon JJ (2015). CD4+ T Cell Tolerance to Tissue-Restricted Self Antigens Is Mediated by Antigen-Specific Regulatory T Cells Rather Than Deletion. Immunity 43, 896–908. 10.1016/j.immuni.2015.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Tuong ZK, Dean I, Willis C, Gaspal F, Fiancette R, Idris S, Kennedy B, Ferdinand JR, Penalver A, Cabantous M, Murtuza Baker S, Fry JW, Carlesso G, Hammond SA, Dovedi SJ, Hepworth MR, Clatworthy MR, and Withers DR (2022). In vivo labeling reveals continuous trafficking of TCF-1+ T cells between tumor and lymphoid tissue. J Exp Med 219. 10.1084/jem.20210749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Hu X, Feng K, Gao R, Xue Z, Zhang S, Zhang Y, Corse E, Hu Y, Han W, and Zhang Z (2021). Temporal single-cell tracing reveals clonal revival and expansion of precursor exhausted T cells during anti-PD-1 therapy in lung cancer. Nature Cancer 3, 108–121. 10.1038/s43018-021-00292-8. [DOI] [PubMed] [Google Scholar]
- Lynn GM, Sedlik C, Baharom F, Zhu Y, Ramirez-Valdez RA, Coble VL, Tobin K, Nichols SR, Itzkowitz Y, Zaidi N, Gammon JM, Blobel NJ, Denizeau J, de la Rochere P, Francica BJ, Decker B, Maciejewski M, Cheung J, Yamane H, Smelkinson MG, Francica JR, Laga R, Bernstock JD, Seymour LW, Drake CG, Jewell CM, Lantz O, Piaggio E, Ishizuka AS, and Seder RA (2020). Peptide-TLR-7/8a conjugate vaccines chemically programmed for nanoparticle self-assembly enhance CD8 T-cell immunity to tumor antigens. Nat Biotechnol 38, 320–332. 10.1038/s41587-019-0390-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier B, Leader AM, Chen ST, Tung N, Chang C, LeBerichel J, Chudnovskiy A, Maskey S, Walker L, Finnigan JP, Kirkling ME, Reizis B, Ghosh S, D’Amore NR, Bhardwaj N, Rothlin CV, Wolf A, Flores R, Marron T, Rahman AH, Kenigsberg E, Brown BD, and Merad M (2020). A conserved dendritic-cell regulatory program limits antitumour immunity. Nature 580, 257–262. 10.1038/s41586-020-2134-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta AK, Kadel S, Townsend MG, Oliwa M, and Guerriero JL (2021). Macrophage Biology and Mechanisms of Immune Suppression in Breast Cancer. Front Immunol 12, 643771. 10.3389/fimmu.2021.643771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo M, Porter E, Zhang Y, Silva M, Li N, Dobosh B, Liguori A, Skog P, Landais E, Menis S, Sok D, Nemazee D, Schief WR, Weiss R, and Irvine DJ (2019). Immunogenicity of RNA Replicons Encoding HIV Env Immunogens Designed for Self-Assembly into Nanoparticles. Mol Ther 27, 2080–2090. 10.1016/j.ymthe.2019.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menzies AM, Amaria RN, Rozeman EA, Huang AC, Tetzlaff MT, van de Wiel BA, Lo S, Tarhini AA, Burton EM, Pennington TE, Saw RPM, Xu X, Karakousis GC, Ascierto PA, Spillane AJ, van Akkooi ACJ, Davies MA, Mitchell TC, Tawbi HA, Scolyer RA, Wargo JA, Blank CU, and Long GV (2021). Pathological response and survival with neoadjuvant therapy in melanoma: a pooled analysis from the International Neoadjuvant Melanoma Consortium (INMC). Nat Med 27, 301–309. 10.1038/s41591-020-01188-3. [DOI] [PubMed] [Google Scholar]
- Moorman CD, Sohn SJ, and Phee H (2021). Emerging Therapeutics for Immune Tolerance: Tolerogenic Vaccines, T cell Therapy, and IL-2 Therapy. Front Immunol 12, 657768. 10.3389/fimmu.2021.657768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison AH, Diamond MS, Hay CA, Byrne KT, and Vonderheide RH (2020). Sufficiency of CD40 activation and immune checkpoint blockade for T cell priming and tumor immunity. Proc Natl Acad Sci U S A 117, 8022–8031. 10.1073/pnas.1918971117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata S, Takahama Y, Kasahara M, and Tanaka K (2018). The immunoproteasome and thymoproteasome: functions, evolution and human disease. Nat Immunol 19, 923–931. 10.1038/s41590-018-0186-z. [DOI] [PubMed] [Google Scholar]
- Mysore V, Cullere X, Mears J, Rosetti F, Okubo K, Liew PX, Zhang F, Madera-Salcedo I, Rosenbauer F, Stone RM, Aster JC, von Andrian UH, Lichtman AH, Raychaudhuri S, and Mayadas TN (2021). FcgammaR engagement reprograms neutrophils into antigen cross-presenting cells that elicit acquired anti-tumor immunity. Nat Commun 12, 4791. 10.1038/s41467-021-24591-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nava S, Lisini D, Frigerio S, and Bersano A (2021). Dendritic Cells and Cancer Immunotherapy: The Adjuvant Effect. Int J Mol Sci 22. 10.3390/ijms222212339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen M, Lundegaard C, Blicher T, Lamberth K, Harndahl M, Justesen S, Roder G, Peters B, Sette A, Lund O, and Buus S (2007). NetMHCpan, a method for quantitative predictions of peptide binding to any HLA-A and -B locus protein of known sequence. PLoS One 2, e796. 10.1371/journal.pone.0000796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira G, Stromhaug K, Cieri N, Iorgulescu JB, Klaeger S, Wolff JO, Rachimi S, Chea V, Krause K, Freeman SS, Zhang W, Li S, Braun DA, Neuberg D, Carr SA, Livak KJ, Frederick DT, Fritsch EF, Wind-Rotolo M, Hacohen N, Sade-Feldman M, Yoon CH, Keskin DB, Ott PA, Rodig SJ, Boland GM, and Wu CJ (2022). Landscape of helper and regulatory antitumour CD4(+) T cells in melanoma. Nature. 10.1038/s41586-022-04682-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira G, Stromhaug K, Klaeger S, Kula T, Frederick DT, Le PM, Forman J, Huang T, Li S, Zhang W, Xu Q, Cieri N, Clauser KR, Shukla SA, Neuberg D, Justesen S, MacBeath G, Carr SA, Fritsch EF, Hacohen N, Sade-Feldman M, Livak KJ, Boland GM, Ott PA, Keskin DB, and Wu CJ (2021). Phenotype, specificity and avidity of antitumour CD8(+) T cells in melanoma. Nature 596, 119–125. 10.1038/s41586-021-03704-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, Zhang W, Luoma A, Giobbie-Hurder A, Peter L, Chen C, Olive O, Carter TA, Li S, Lieb DJ, Eisenhaure T, Gjini E, Stevens J, Lane WJ, Javeri I, Nellaiappan K, Salazar AM, Daley H, Seaman M, Buchbinder EI, Yoon CH, Harden M, Lennon N, Gabriel S, Rodig SJ, Barouch DH, Aster JC, Getz G, Wucherpfennig K, Neuberg D, Ritz J, Lander ES, Fritsch EF, Hacohen N, and Wu CJ (2017). An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 547, 217–221. 10.1038/nature22991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott PA, Hu-Lieskovan S, Chmielowski B, Govindan R, Naing A, Bhardwaj N, Margolin K, Awad MM, Hellmann MD, Lin JJ, Friedlander T, Bushway ME, Balogh KN, Sciuto TE, Kohler V, Turnbull SJ, Besada R, Curran RR, Trapp B, Scherer J, Poran A, Harjanto D, Barthelme D, Ting YS, Dong JZ, Ware Y, Huang Y, Huang Z, Wanamaker A, Cleary LD, Moles MA, Manson K, Greshock J, Khondker ZS, Fritsch E, Rooney MS, DeMario M, Gaynor RB, and Srinivasan L (2020). A Phase Ib Trial of Personalized Neoantigen Therapy Plus Anti-PD-1 in Patients with Advanced Melanoma, Non-small Cell Lung Cancer, or Bladder Cancer. Cell 183, 347–362 e324. 10.1016/j.cell.2020.08.053. [DOI] [PubMed] [Google Scholar]
- Ouspenskaia T, Law T, Clauser KR, Klaeger S, Sarkizova S, Aguet F, Li B, Christian E, Knisbacher BA, Le PM, Hartigan CR, Keshishian H, Apffel A, Oliveira G, Zhang W, Chen S, Chow YT, Ji Z, Jungreis I, Shukla SA, Justesen S, Bachireddy P, Kellis M, Getz G, Hacohen N, Keskin DB, Carr SA, Wu CJ, and Regev A (2021). Unannotated proteins expand the MHC-I-restricted immunopeptidome in cancer. Nat Biotechnol. 10.1038/s41587-021-01021-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang X, Telli ML, and Wu JC (2019). Induced Pluripotent Stem Cell-Based Cancer Vaccines. Front Immunol 10, 1510. 10.3389/fimmu.2019.01510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y, Liu L, Tian T, Zhao J, Park CO, Lofftus SY, Stingley CA, Yan Y, Mei S, Liu X, and Kupper TS (2021). Epicutaneous immunization with modified vaccinia Ankara viral vectors generates superior T cell immunity against a respiratory viral challenge. NPJ Vaccines 6, 1. 10.1038/s41541-020-00265-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauken KE, Godec J, Odorizzi PM, Brown KE, Yates KB, Ngiow SF, Burke KP, Maleri S, Grande SM, Francisco LM, Ali MA, Imam S, Freeman GJ, Haining WN, Wherry EJ, and Sharpe AH (2020). The PD-1 Pathway Regulates Development and Function of Memory CD8(+) T Cells following Respiratory Viral Infection. Cell Rep 31, 107827. 10.1016/j.celrep.2020.107827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauthner M, Havenar-Daughton C, Sok D, Nkolola JP, Bastidas R, Boopathy AV, Carnathan DG, Chandrashekar A, Cirelli KM, Cottrell CA, Eroshkin AM, Guenaga J, Kaushik K, Kulp DW, Liu J, McCoy LE, Oom AL, Ozorowski G, Post KW, Sharma SK, Steichen JM, de Taeye SW, Tokatlian T, Torrents de la Pena A, Butera ST, LaBranche CC, Montefiori DC, Silvestri G, Wilson IA, Irvine DJ, Sanders RW, Schief WR, Ward AB, Wyatt RT, Barouch DH, Crotty S, and Burton DR (2017). Elicitation of Robust Tier 2 Neutralizing Antibody Responses in Nonhuman Primates by HIV Envelope Trimer Immunization Using Optimized Approaches. Immunity 46, 1073–1088 e1076. 10.1016/j.immuni.2017.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera SA, Kopinja JE, Ma Y, Muise ES, Laskey J, Chakravarthy K, Chen Y, Cui L, Presland J, Sathe M, Javaid S, Minnihan E, Ferguson H, Piesvaux J, Pan BS, Zhao S, Sharma SK, Woo HC, Pucci V, Knemeyer I, Cemerski S, Cumming J, Trotter BW, Tse A, Khilnani A, Ranganath S, Long BJ, Bennett DJ, and Addona GH (2021). STimulator of INterferon Genes Agonism Accelerates Anti-tumor Activity in Poorly Immunogenic Tumors. Mol Cancer Ther. 10.1158/1535-7163.MCT-21-0136. [DOI] [PubMed] [Google Scholar]
- Philip M, and Schietinger A (2021). CD8(+) T cell differentiation and dysfunction in cancer. Nat Rev Immunol. 10.1038/s41577-021-00574-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potocnakova L, Bhide M, and Pulzova LB (2016). An Introduction to B-Cell Epitope Mapping and In Silico Epitope Prediction. J Immunol Res 2016, 6760830. 10.1155/2016/6760830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozzi C, Cuomo A, Spadoni I, Magni E, Silvola A, Conte A, Sigismund S, Ravenda PS, Bonaldi T, Zampino MG, Cancelliere C, Di Fiore PP, Bardelli A, Penna G, and Rescigno M (2016). The EGFR-specific antibody cetuximab combined with chemotherapy triggers immunogenic cell death. Nat Med 22, 624–631. 10.1038/nm.4078. [DOI] [PubMed] [Google Scholar]
- Propper DJ, and Balkwill FR (2022). Harnessing cytokines and chemokines for cancer therapy. Nat Rev Clin Oncol. 10.1038/s41571-021-00588-9. [DOI] [PubMed] [Google Scholar]
- Rakhra K, Abraham W, Wang C, Moynihan KD, Li N, Donahue N, Baldeon AD, and Irvine DJ (2021). Exploiting albumin as a mucosal vaccine chaperone for robust generation of lung-resident memory T cells. Sci Immunol 6. 10.1126/sciimmunol.abd8003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiser J, and Banerjee A (2016). Effector, Memory, and Dysfunctional CD8(+) T Cell Fates in the Antitumor Immune Response. J Immunol Res 2016, 8941260. 10.1155/2016/8941260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Z, Zhang A, Sun Z, Liang Y, Ye J, Qiao J, Li B, and Fu YX (2022). Selective delivery of low-affinity IL-2 to PD-1+ T cells rejuvenates antitumor immunity with reduced toxicity. J Clin Invest 132. 10.1172/JCI153604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynisson B, Alvarez B, Paul S, Peters B, and Nielsen M (2020). NetMHCpan-4.1 and NetMHCIIpan-4.0: improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res 48, W449–W454. 10.1093/nar/gkaa379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riccione KA, He LZ, Fecci PE, Norberg PK, Suryadevara CM, Swartz A, Healy P, Reap E, Keler T, Li QJ, Congdon KL, Sanchez-Perez L, and Sampson JH (2018). CD27 stimulation unveils the efficacy of linked class I/II peptide vaccines in poorly immunogenic tumors by orchestrating a coordinated CD4/CD8 T cell response. Oncoimmunology 7, e1502904. 10.1080/2162402X.2018.1502904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richman LP, Vonderheide RH, and Rech AJ (2019). Neoantigen Dissimilarity to the Self-Proteome Predicts Immunogenicity and Response to Immune Checkpoint Blockade. Cell Syst 9, 375–382 e374. 10.1016/j.cels.2019.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche PA, and Furuta K (2015). The ins and outs of MHC class II-mediated antigen processing and presentation. Nat Rev Immunol 15, 203–216. 10.1038/nri3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblatt J, Stone RM, Uhl L, Neuberg D, Joyce R, Levine JD, Arnason J, McMasters M, Luptakova K, Jain S, Zwicker JI, Hamdan A, Boussiotis V, Steensma DP, DeAngelo DJ, Galinsky I, Dutt PS, Logan E, Bryant MP, Stroopinsky D, Werner L, Palmer K, Coll M, Washington A, Cole L, Kufe D, and Avigan D (2016). Individualized vaccination of AML patients in remission is associated with induction of antileukemia immunity and prolonged remissions. Sci Transl Med 8, 368ra171. 10.1126/scitranslmed.aag1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozeman EA, Hoefsmit EP, Reijers ILM, Saw RPM, Versluis JM, Krijgsman O, Dimitriadis P, Sikorska K, van de Wiel BA, Eriksson H, Gonzalez M, Torres Acosta A, Grijpink-Ongering LG, Shannon K, Haanen J, Stretch J, Ch’ng S, Nieweg OE, Mallo HA, Adriaansz S, Kerkhoven RM, Cornelissen S, Broeks A, Klop WMC, Zuur CL, van Houdt WJ, Peeper DS, Spillane AJ, van Akkooi ACJ, Scolyer RA, Schumacher TNM, Menzies AM, Long GV, and Blank CU (2021). Survival and biomarker analyses from the OpACIN-neo and OpACIN neoadjuvant immunotherapy trials in stage III melanoma. Nat Med 27, 256–263. 10.1038/s41591-020-01211-7. [DOI] [PubMed] [Google Scholar]
- Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, Lieb DJ, Chen JH, Frederick DT, Barzily-Rokni M, Freeman SS, Reuben A, Hoover PJ, Villani AC, Ivanova E, Portell A, Lizotte PH, Aref AR, Eliane JP, Hammond MR, Vitzthum H, Blackmon SM, Li B, Gopalakrishnan V, Reddy SM, Cooper ZA, Paweletz CP, Barbie DA, Stemmer-Rachamimov A, Flaherty KT, Wargo JA, Boland GM, Sullivan RJ, Getz G, and Hacohen N (2018). Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell 175, 998–1013 e1020. 10.1016/j.cell.2018.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagiv Barfi I, Czerwinski DK, and Levy R (2021). In Situ Vaccination with IL-12Fc and TLR Agonist - a Crucial Role for B Cells in Generating Anti-Tumor T Cell Immunity. Blood 138, 3514–3514. 10.1182/blood-2021-146628. [DOI] [Google Scholar]
- Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, Lower M, Bukur V, Tadmor AD, Luxemburger U, Schrors B, Omokoko T, Vormehr M, Albrecht C, Paruzynski A, Kuhn AN, Buck J, Heesch S, Schreeb KH, Muller F, Ortseifer I, Vogler I, Godehardt E, Attig S, Rae R, Breitkreuz A, Tolliver C, Suchan M, Martic G, Hohberger A, Sorn P, Diekmann J, Ciesla J, Waksmann O, Bruck AK, Witt M, Zillgen M, Rothermel A, Kasemann B, Langer D, Bolte S, Diken M, Kreiter S, Nemecek R, Gebhardt C, Grabbe S, Holler C, Utikal J, Huber C, Loquai C, and Tureci O (2017). Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 547, 222–226. 10.1038/nature23003. [DOI] [PubMed] [Google Scholar]
- Schumacher TN, and Thommen DS (2022). Tertiary lymphoid structures in cancer. Science 375, eabf9419. 10.1126/science.abf9419. [DOI] [PubMed] [Google Scholar]
- Sharma A, Subudhi SK, Blando J, Scutti J, Vence L, Wargo J, Allison JP, Ribas A, and Sharma P (2019). Anti-CTLA-4 Immunotherapy Does Not Deplete FOXP3(+) Regulatory T Cells (Tregs) in Human Cancers. Clin Cancer Res 25, 1233–1238. 10.1158/1078-0432.CCR-18-0762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stronen E, Toebes M, Kelderman S, van Buuren MM, Yang W, van Rooij N, Donia M, Boschen ML, Lund-Johansen F, Olweus J, and Schumacher TN (2016). Targeting of cancer neoantigens with donor-derived T cell receptor repertoires. Science 352, 1337–1341. 10.1126/science.aaf2288. [DOI] [PubMed] [Google Scholar]
- Sultan H, Kumai T, Nagato T, Wu J, Salazar AM, and Celis E (2019). The route of administration dictates the immunogenicity of peptide-based cancer vaccines in mice. Cancer Immunol Immunother 68, 455–466. 10.1007/s00262-018-02294-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartz AM, Congdon KL, Nair SK, Li QJ, Herndon JE 2nd, Suryadevara CM, Riccione KA, Archer GE, Norberg PK, Sanchez-Perez LA, and Sampson JH (2021). A conjoined universal helper epitope can unveil antitumor effects of a neoantigen vaccine targeting an MHC class I-restricted neoepitope. NPJ Vaccines 6, 12. 10.1038/s41541-020-00273-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan WCC, Nerurkar SN, Cai HY, Ng HHM, Wu D, Wee YTF, Lim JCT, Yeong J, and Lim TKH (2020). Overview of multiplex immunohistochemistry/immunofluorescence techniques in the era of cancer immunotherapy. Cancer Commun (Lond) 40, 135–153. 10.1002/cac2.12023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanyi JL, Bobisse S, Ophir E, Tuyaerts S, Roberti A, Genolet R, Baumgartner P, Stevenson BJ, Iseli C, Dangaj D, Czerniecki B, Semilietof A, Racle J, Michel A, Xenarios I, Chiang C, Monos DS, Torigian DA, Nisenbaum HL, Michielin O, June CH, Levine BL, Powell DJ Jr., Gfeller D, Mick R, Dafni U, Zoete V, Harari A, Coukos G, and Kandalaft LE (2018). Personalized cancer vaccine effectively mobilizes antitumor T cell immunity in ovarian cancer. Sci Transl Med 10. 10.1126/scitranslmed.aao5931. [DOI] [PubMed] [Google Scholar]
- Toes RE, Offringa R, Blom RJ, Melief CJ, and Kast WM (1996). Peptide vaccination can lead to enhanced tumor growth through specific T-cell tolerance induction. Proc Natl Acad Sci U S A 93, 7855–7860. 10.1073/pnas.93.15.7855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Togashi Y, Shitara K, and Nishikawa H (2019). Regulatory T cells in cancer immunosuppression - implications for anticancer therapy. Nat Rev Clin Oncol 16, 356–371. 10.1038/s41571-019-0175-7. [DOI] [PubMed] [Google Scholar]
- Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, Wunderlich JR, Somerville RP, Hogan K, Hinrichs CS, Parkhurst MR, Yang JC, and Rosenberg SA (2014). Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 344, 641–645. 10.1126/science.1251102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turajlic S, Sottoriva A, Graham T, and Swanton C (2019). Resolving genetic heterogeneity in cancer. Nat Rev Genet 20, 404–416. 10.1038/s41576-019-0114-6. [DOI] [PubMed] [Google Scholar]
- van den Hout M, Koster BD, Sluijter BJR, Molenkamp BG, van de Ven R, van den Eertwegh AJM, Scheper RJ, van Leeuwen PAM, van den Tol MP, and de Gruijl TD (2017). Melanoma Sequentially Suppresses Different DC Subsets in the Sentinel Lymph Node, Affecting Disease Spread and Recurrence. Cancer Immunol Res 5, 969–977. 10.1158/2326-6066.CIR-17-0110. [DOI] [PubMed] [Google Scholar]
- Veatch JR, Singhi N, Srivastava S, Szeto JL, Jesernig B, Stull SM, Fitzgibbon M, Sarvothama M, Yechan-Gunja S, James SE, and Riddell SR (2021). A therapeutic cancer vaccine delivers antigens and adjuvants to lymphoid tissues using genetically modified T cells. J Clin Invest 131. 10.1172/JCI144195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veglia F, Perego M, and Gabrilovich D (2018). Myeloid-derived suppressor cells coming of age. Nat Immunol 19, 108–119. 10.1038/s41590-017-0022-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma V, Shrimali RK, Ahmad S, Dai W, Wang H, Lu S, Nandre R, Gaur P, Lopez J, Sade-Feldman M, Yizhak K, Bjorgaard SL, Flaherty KT, Wargo JA, Boland GM, Sullivan RJ, Getz G, Hammond SA, Tan M, Qi J, Wong P, Merghoub T, Wolchok J, Hacohen N, Janik JE, Mkrtichyan M, Gupta S, and Khleif SN (2019). PD-1 blockade in subprimed CD8 cells induces dysfunctional PD-1(+)CD38(hi) cells and anti-PD-1 resistance. Nat Immunol 20, 1231–1243. 10.1038/s41590-019-0441-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villani AC, Satija R, Reynolds G, Sarkizova S, Shekhar K, Fletcher J, Griesbeck M, Butler A, Zheng S, Lazo S, Jardine L, Dixon D, Stephenson E, Nilsson E, Grundberg I, McDonald D, Filby A, Li W, De Jager PL, Rozenblatt-Rosen O, Lane AA, Haniffa M, Regev A, and Hacohen N (2017). Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 356. 10.1126/science.aah4573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonderheide RH (2020). CD40 Agonist Antibodies in Cancer Immunotherapy. Annu Rev Med 71, 47–58. 10.1146/annurev-med-062518-045435. [DOI] [PubMed] [Google Scholar]
- Wang TY, Liu Q, Ren Y, Alam SK, Wang L, Zhu Z, Hoeppner LH, Dehm SM, Cao Q, and Yang R (2021). A pan-cancer transcriptome analysis of exitron splicing identifies novel cancer driver genes and neoepitopes. Mol Cell 81, 2246–2260 e2212. 10.1016/j.molcel.2021.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, and Sancho D (2020). Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol 20, 7–24. 10.1038/s41577-019-0210-z. [DOI] [PubMed] [Google Scholar]
- Wei J, Montalvo-Ortiz W, Yu L, Krasco A, Ebstein S, Cortez C, Lowy I, Murphy AJ, Sleeman MA, and Skokos D (2021). Sequence of alphaPD-1 relative to local tumor irradiation determines the induction of abscopal antitumor immune responses. Sci Immunol 6. 10.1126/sciimmunol.abg0117. [DOI] [PubMed] [Google Scholar]
- Wellenstein MD, and de Visser KE (2018). Cancer-Cell-Intrinsic Mechanisms Shaping the Tumor Immune Landscape. Immunity 48, 399–416. 10.1016/j.immuni.2018.03.004. [DOI] [PubMed] [Google Scholar]
- Wicherska-Pawlowska K, Wrobel T, and Rybka J (2021). Toll-Like Receptors (TLRs), NOD-Like Receptors (NLRs), and RIG-I-Like Receptors (RLRs) in Innate Immunity. TLRs, NLRs, and RLRs Ligands as Immunotherapeutic Agents for Hematopoietic Diseases. Int J Mol Sci 22. 10.3390/ijms222413397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williford JM, Ishihara J, Ishihara A, Mansurov A, Hosseinchi P, Marchell TM, Potin L, Swartz MA, and Hubbell JA (2019). Recruitment of CD103(+) dendritic cells via tumor-targeted chemokine delivery enhances efficacy of checkpoint inhibitor immunotherapy. Sci Adv 5, eaay1357. 10.1126/sciadv.aay1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YM, Cieslik M, Lonigro RJ, Vats P, Reimers MA, Cao X, Ning Y, Wang L, Kunju LP, de Sarkar N, Heath EI, Chou J, Feng FY, Nelson PS, de Bono JS, Zou W, Montgomery B, Alva A, Team PSCIPCD, Robinson DR, and Chinnaiyan AM (2018). Inactivation of CDK12 Delineates a Distinct Immunogenic Class of Advanced Prostate Cancer. Cell 173, 1770–1782 e1714. 10.1016/j.cell.2018.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin L, Gao J, Zheng Z, Chen Y, Lv S, Zhao Z, Yu C, Yang X, and Zhang R (2021). Fibroblast Activation Protein-alpha as a Target in the Bench-to-Bedside Diagnosis and Treatment of Tumors: A Narrative Review. Front Oncol 11, 648187. 10.3389/fonc.2021.648187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates KB, Tonnerre P, Martin GE, Gerdemann U, Al Abosy R, Comstock DE, Weiss SA, Wolski D, Tully DC, Chung RT, Allen TM, Kim AY, Fidler S, Fox J, Frater J, Lauer GM, Haining WN, and Sen DR (2021). Epigenetic scars of CD8(+) T cell exhaustion persist after cure of chronic infection in humans. Nat Immunol 22, 1020–1029. 10.1038/s41590-021-00979-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagorulya M, Duong E, and Spranger S (2020). Impact of anatomic site on antigen-presenting cells in cancer. J Immunother Cancer 8. 10.1136/jitc-2020-001204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng B, Middelberg AP, Gemiarto A, MacDonald K, Baxter AG, Talekar M, Moi D, Tullett KM, Caminschi I, Lahoud MH, Mazzieri R, Dolcetti R, and Thomas R (2018). Self-adjuvanting nanoemulsion targeting dendritic cell receptor Clec9A enables antigen-specific immunotherapy. J Clin Invest 128, 1971–1984. 10.1172/JCI96791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhivaki D, Borriello F, Chow OA, Doran B, Fleming I, Theisen DJ, Pallis P, Shalek AK, Sokol CL, Zanoni I, and Kagan JC (2020). Inflammasomes within Hyperactive Murine Dendritic Cells Stimulate Long-Lived T Cell-Mediated Anti-tumor Immunity. Cell Rep 33, 108381. 10.1016/j.celrep.2020.108381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zitvogel L, Kepp O, and Kroemer G (2011). Immune parameters affecting the efficacy of chemotherapeutic regimens. Nat Rev Clin Oncol 8, 151–160. 10.1038/nrclinonc.2010.223. [DOI] [PubMed] [Google Scholar]
- Zou W, Yaung SJ, Fuhlbruck F, Ballinger M, Peters E, Palma JF, Shames DS, Gandara D, Jiang Y, and Patil NS (2021). ctDNA Predicts Overall Survival in Patients With NSCLC Treated With PD-L1 Blockade or With Chemotherapy. JCO Precis Oncol 5, 827–838. 10.1200/PO.21.00057. [DOI] [PubMed] [Google Scholar]