

Abstract
Purpose
The use of ionic liquids (ILs) in drug delivery has focused attention on non-toxic IL counterions. Cationic lipids can be used to form ILs with weakly acidic drugs to enhance drug loading in lipid-based formulations (LBFs). However, cationic lipids are typically toxic. Here we explore the use of lipoaminoacids (LAAs) as cationic IL counterions that degrade or digest in vivo to non-toxic components.
Methods
LAAs were synthesised via esterification of amino acids with fatty alcohols to produce potentially digestible cationic LAAs. The LAAs were employed to form ILs with tolfenamic acid (Tol) and the Tol ILs loaded into LBF and examined in vitro and in vivo.
Results
Cationic LAAs complexed with Tol to generate lipophilic Tol ILs with high drug loading in LBFs. Assessment of the LAA under simulated digestion conditions revealed that they were susceptible to enzymatic degradation under intestinal conditions, forming biocompatible FAs and amino acids. In vitro dispersion and digestion studies of Tol ILs revealed that formulations containing digestible Tol ILs were able to maintain drug dispersion and solubilisation whilst the LAA were breaking down under digesting conditions. Finally, in vivo oral bioavailability studies demonstrated that oral delivery of a LBF containing a Tol IL comprising a digestible cationic lipid counterion was able to successfully support effective oral delivery of Tol.
Conclusions
Digestible LAA cationic lipids are potential IL counterions for weakly acidic drug molecules and digest in situ to form non-toxic breakdown products.
Supplementary Information
The online version contains supplementary material available at 10.1007/s11095-022-03305-y.
KEY WORDS: absorption, ionic liquids, lipid-based formulations, lipoaminoacids, oral bioavailability
Introduction
Ionic liquids (ILs) are typically defined as salts with melting points and glass transition temperatures below 100˚C [1–5]. The unique modular structure provided by ILs offers an interchangeable platform of cation–anion pairs [6–9] that has resulted in broad application across a range of ‘task-specific’ ionic liquids (TSILs); i.e. ILs with functionalised properties to perform specific applications [10–13]. In line with the increased general utility of ionic liquids, the use of ionic liquids to enhance drug delivery has also increased in recent years [14–21]. However, despite the wide selection of potential cation–anion pairs, the documented body of work describing IL applications remains focused on a relatively limited pool of IL counterions, especially for cations. For example, a number of studies have focussed on the potential utility of 1,3-dialkylimidazolium cations, such as 1-butyl-3-methylimidazolium (C4mim), [22–24] and 1-ethyl-3-methylimidazolium (C2mim) [25–29] and these reliably produce ILs with liquid-like properties [30, 31]. Many ILs reported in the literature are composed of organic cations comprising imidazolium or pyridinium analogues [3, 32–35]. Whilst many of these cationic counterions are effective in generating ILs, increased focus on applications in drug delivery has resulted in the need to prioritise biodegradable and non-toxic counterions. Examples of more biocompatible ILs (e.g. biodegradable surfactants and/or biocatalysts) are apparent in allied industries [36–38], but only a few studies have examined the potential for the development of ILs that are biocompatible and biodegradable in the gastrointestinal tract (GIT) and therefore safe for oral drug delivery ([39]). In contrast, many more investigations of ILs in drug delivery have focused on improvements in transdermal drug delivery where issues of acute toxicity are less severe [40–46].
In our previous studies we have focused on the use of ILs as a means to increase the lipophilicity of drug molecules and in doing so to increase drug solubility in traditional lipid based formulations (LBF) [47]. This approach requires the use of lipophilic counterions and in the case of cations paired with weakly acidic drugs, the use of cationic lipids. This however, typically raises questions around the potential toxicity of cationic lipids.
Cationic lipids are commonly utilised as transfection agents in gene delivery studies, often in combination with liposomal vehicles that can be utilised to deliver genetic payloads such as DNA, RNA and other nucleic acids into cells [48–52]. Cationic lipids possess a charged head group, generally provided by tertiary or quaternary amine groups, and a hydrophobic or lipid like tail [53]. They form complexes with negatively charged materials, assisting in eg. association and loading of (anionic) nucleic acids into delivery systems [54–57]. Cationic lipids also promote transfection due to the opposing negative surface charge of cells [58–60]. However, the ability of cationic lipids to interact strongly with most cells types has also led to toxicity concerns that ultimately restrict broader utility. For example, cells do not appear to be able to differentiate exogenous cationic lipids from other positively charged endogenous cell messengers that induce immune responses or cell death signalling pathways, such as endogenous polyamines [61–63]. Cationic lipids may also alter membrane properties and interact with negatively charged components embedded in phospholipid cell bilayers, destabilising the cell membrane [64–66].
Cationic lipids have been employed in the formation of ionic liquids for drug delivery [14, 67] although examples based on longer chain lipid-based cations, including aminoacid esters are more common in the transdermal literature than after oral administration [15, 39]. Nonetheless examples after oral administration are apparent including the use dodecylamine as a counterion for enoxaparin [68] and octadecylamine as a counterion for breviscapine [69], both employed to promote incorporation into oral lipid formulations. The latter examples, however, do not provide for biodegradation. As such, novel cationic lipids that degrade in situ to relatively benign components, and that have the potential to address the issues of toxicity surrounding ILs comprising cationic lipids, are increasingly sought.
The current studies have thus explored the potential to form cationic lipid counterions from lipoamino acids (LAA) formed via complexation of fatty alcohols with the carboxylic acid terminal of amino acids. These cationic LAAs have then been complexed with the weakly acidic poorly water-soluble drug (PWSD), tolfenamic acid (Fig. 1) to form tolfenamic acid based, active pharmaceutical ingredient (API) ionic liquids or API-ILs. In general, the cationic LAAs described herein were designed to have some similarity to endogenous lipids in order that the lipophilic ester bond in the LAA may be susceptible to digestion via lipolysis enzymes or more generic esterases in the gastrointestinal tract (GIT). This was expected to result in degradation to fatty alcohols and amino acids, which have considerable safety advantages when compared to typical cationic lipids. Amino acids are ingested through the diet, and it has been reported that aliphatic fatty alcohols (at least between 6 and 22 carbons) are generally safe for oral ingestion [70]. In addition to the API-ILs generated with a digestible LAA counterion we also generated an APL-IL with decylamine (Tol Dec) to provide a non-digestible comparator.
Fig. 1.
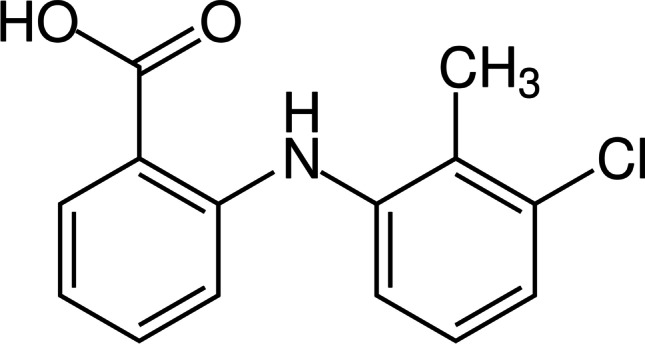
Tolfenamic acid
Materials and Methods
Alanine (Sigma, Castle Hill, NSW, Australia), phenylalanine (Fluka, Pittsburg, PA, USA), hydrogen chloride (HCl) in ether (2.0 M) (Sigma, St. Louis, Missouri, USA), ammonium dihydrogen phosphate (NH4H2PO4) was obtained from Sigma (Castle Hill, NSW, Australia). Formic acid 98 – 100% for HPLC, sodium hydroxide (NaOH) pellets, magnesium sulfate (MgSO4); solvents, diethyl ether, methanol (MeOH), chloroform (CHCl3) and acetonitrile (ACN) were obtained from Merck (Bayswater, Victoria, Australia). Capmul® MCM and Captex® 355 EP/NF were obtained from ABITEC Corporation (Janesville, Wisconsin, USA); Kolliphor EL was obtained from Sigma (Castle Hill, NSW, Australia).
Trizma® maleate (Castle Hill, NSW, Australia), calcium chloride dihydrate (CaCl2·2H2O) 99.0% (Castle Hill, NSW, Australia), 4-bromophenylboronic acid, sodium taurodeoxycholate hydrate (NaTDC) (Castle Hill, NSW, Australia) and porcine pancreatin extract (8 × USP specification activity) were purchased from Sigma (St Louis, Missouri, USA). Sodium chloride (NaCl) was purchased from Ajax FineChem (Rosedale, Auckland, New Zealand). Lipoid E PC S (lecithin for egg, ≈ 99% pure phosphatidylcholine (PC)) was obtained from Lipoid GmBH (Ludwigshafen, Germany). Tolfenamic acid (Tol) and decyl alcohol was purchased from Sigma (Castle Hill, NSW, Australia); p-toluene sulfonic acid from Fluka (Pittsburg, PA, USA) and toluene from Merck (Bayswater, Victoria, Australia). Organic solvents were used without any pre-treatment. All other chemicals and solvents were of analytical purity or high-performance liquid chromatography (HPLC).
Synthesis of cationic LAA counterions: decyl alanine ester HCl and decyl phenylalanine ester HCl
The lipoamino acid (LAA) cations (decyl alanine ester HCl and decyl phenylalanine ester HCl) were synthesized via an esterification reaction. Decyl alcohol (1.0 mmol) and the amino acid (alanine and phenylalanine respectively, 1.0 mmol) were dissolved in toluene (10 mL). p-Toluene sulfonic acid (1.3 mol eq) was added to the solution and the reaction was stirred under reflux overnight. Toluene was then removed and the crude product was dissolved in cold diethyl ether. HCl in ether solution was added (2 M, 1.0 mol eq) and the mixture cooled in an ice bath until white crystals formed. The solution was filtered to obtain the HCl salt product of the LAAs (Fig. 2).
Fig. 2.

Synthesis of Tol Dec, Tol Dec Ala and Tol Dec Phe; (i) p-TSA, toluene, reflux, 24 h then HCl, ether, 0 °C, (ii) Tolfenamic acid sodium salt, MeOH, 0 °C, 1 h
Synthesis of Tolfenamic acid ILs (Tol ILs): Tolfenamate decyl amine (Tol Dec), Tolfenamate decyl alanine ester (Tol Dec Ala) and Tolfenamate decyl phenylalanine ester (Tol Dec Phe)
Tolfenamate decyl amine (Tol Dec), Tolfenamate decyl alanine ester (Tol Dec Ala) and Tolfenamate decyl phenylalanine ester (Tol Dec Phe) were synthesized via a salt metathesis reaction. The reaction is driven by the differential solubility of the salts formed where typically the by-product, sodium chloride (NaCl) precipitates out. Equimolar amounts of the sodium salt of tolfenamic acid and the HCl salt of the cationic LAA were dissolved in methanol (MeOH) and stirred for 1 h. MeOH was then evaporated and the crude product was dissolved in chloroform (CHCl3). The organic solution was washed with water to remove the residual by-product, sodium chloride (NaCl). The solution was dried with magnesium sulfate (MgSO4) and concentrated to obtain the final product.
Formulation Preparation
Type IIIA and Type IIIB self-emulsifying drug delivery systems (SEDDS) (as defined by the Lipid Formulation Classification System [71] were freshly prepared as homogenous mixtures of lipid, surfactant and co-solvent. A Type IIIA SEDDS formulation served as a model lipid formulation and is referred to as medium chain formulation—1 (MCF-1). This was composed of 30% Captex® 355, 30% Capmul® MCM, 30% Kolliphor® EL, 10% EtOH (all % values expressed as w/w). An alternate Type IIIB medium chain lipid containing formulations with higher surfactant content was also explored and is referred to as MCF-2. This was composed of 15% Captex® 355, 15% Capmul® MCM, 60% Kolliphor® EL, 10% EtOH (all % values expressed as w/w). A Type IV LBF comprising a homogenous mixture of 50% Kolliphor® EL, 50% EtOH was also employed for the decyl phenylalanine LAA breakdown/digestion studies. This was employed to remove digestible lipids from the formulation thereby magnifying any effects of LAA digestion/breakdown. Tol ILs were dissolved in the MCFs, and concentrations of tolfenamic acid were measured by HPLC.
Solubility Studies
The solubility of tolfenamic acid and the Tol ILs was determined in the two Type III lipid formulations (described below) via a variation of the shake flask method where Tol ILs were loaded in the lipid formulations incrementally. Tol ILs were loaded into the formulations initially at 10% w/w of the mass of the LBF initially present (e.g., 100 mg of IL added to 1 g of LBF). If the Tol IL completely dissolved, an additional 10% was added to the formulation. The process was repeated until undissolved Tol IL was observed visually. Samples were incubated at 37˚C and left for a number of days until the equilibrium solubility was obtained, defined as < 5% deviation in concentration across consecutive days. Samples were taken at regular intervals and centrifuged (9800 × g, 37˚C, 10 min). The particle-free supernatant was accurately weighed (30 mg) and dissolved in acetonitrile (ACN), followed by further dilution with H2O:ACN (20:80, v/v). Samples were then analysed by HPLC. All solubility studies were performed in triplicate. To allow comparison of solubility data between Tol ILs and tolfenamic acid, results are quoted as mg/g of free acid equivalents present in solution in the vehicle.
In vitro lipolysis studies of LBF of Tol ILs
The in vitro lipid digestion testing protocol was modelled after the method published by the lipid formulation classification system (LFCS) consortium [72–74]. Digestion buffer (pH 6.5) was made up with 2 mM Trizma® maleate, 1.4 mM calcium chloride dihydrate (CaCl2·2H2O), and 150 mM sodium chloride (NaCl). To simulate the concentrations of the salts, bile, and phospholipid in the small intestine under fasted conditions a micellar solution of 3 mM sodium taurodeoxycholate (NaTDC) and 0.75 mM Lipoid E PC S was prepared in digestion buffer. Porcine pancreatin extract was prepared for digestion studies at 2400 TBU/mL (1.00 g of pancreatin mixed into 5.0 mL of cold digestion buffer). The activity of pancreatic lipase was expressed in terms of tributyrin units (TBU), where 1 TBU is the amount of enzyme that can liberate 1 mol of titratable FA from tributyrin per min. Porcine pancreatin extract with lower lipase activity was also prepared at 600 TBU/mL (0.25 g mixed into 5.0 mL of cold digestion buffer). The mixtures were centrifuged (2800 × g, 5°C, 15 min) and the supernatant collected to provide the enzyme extract.
Tolfenamic acid and Tol IL were loaded in LBFs at 90% of the equilibrium solubility obtained in the solubility studies. 1.10 g of LBF containing tolfenamic acid or Tol IL was weighed directly into a thermostatically controlled, jacketed glass reaction vessel, and 40 mL of micelle solution was added. The solution was mixed and the pH was buffered and maintained at 6.5 (pH of the small intestine) by the pH stat. Continuous mixing was maintained for 15 min to evaluate the dispersion properties of the LBF. 1 mL samples were taken at 5, 10 and 15 min. Each sample was spun down (9800 × g, 37 °C, 15 min) to separate any precipitate present and the supernatant (aqueous phase). Each phase was collected, followed by a 1 in 200 dilution with H2O:ACN, 1:1, v/v. To avoid variability in the quantity of formulation added to each digestion experiment, a control sample was taken after the formulation had been added and dispersed, but without centrifugation, to gain an accurate measure of the total (maximal) concentration of drug in the experiment. The concentration of drug in the aqueous phase digest was subsequently expressed as a % of the measured theoretical target. Digestion was initiated by the addition of 4 mL of pancreatin extract. 0.6 M NaOH solution was utilised in the pH stat to titrate liberated fatty acid and buffer the vessel solution as digestion progressed. Titrants were automatically added via the pH–stat controller and the rate of titrant addition reflected the digestibility of the LBF. 1 mL samples were taken from the dispersion/digestion media at 5, 10, 15, 30 and 60 min after initiation of digestion, and 10 µL of 4-bromophenylboronic acid was immediately added to the samples to arrest lipase activity. Digestion samples received similar treatment to samples taken prior to digestion.
In vitro studies to assess breakdown/digestibility of novel LAA counterions
The in vitro lipid digestion testing protocols detailed above were also utilised to assess the breakdown of the decyl phenylalanine ester LAA counterion. An IL formed from the phenyl LAA derivative was employed since the modified LAA possesses a UV active phenyl group and could therefore be assayed quantitatively. For these studies only, Tol Dec Phe was loaded into a Type IV LBF at 37.5% w/w of IL in the formulation i.e. 300 mg of IL in 500 mg of LBF. The required mass of Tol IL was weighed directly into clean screw-top vials, and drug-free lipid formulation was added to target mass loading. Vials were sealed, vortexed and incubated at 37°C for 24 h. The Type IV formulation was prepared as a homogenous mixture of surfactant and co-solvent (50% Kolliphor® EL, 50% EtOH) in the absence of any lipid. This was employed to remove digestible lipids from the formulation thereby magnifying any effects of LAA digestion/breakdown. In vitro digestion studies were performed as described above where samples were subsequently analysed for parent LAA (decyl phenylalanine) and the breakdown product phenylalanine.
HPLC conditions for Tol
Solubility and lipolysis samples were assayed for tolfenamic acid content via HPLC. The mobile phase comprised of 0.1% formic acid in H2O:0.1% formic acid in ACN, 20:80 (v/v). Flow rate was 1 mL/min, injection volume 50 μL, and UV detection was at 280 nm. The retention time was 2.7 min, and the concentration range of the calibration standards was 10 – 100 ug/mL. The assay was accurate and precise to within ± 10% of the theoretical concentration across 3 different concentrations.
HPLC conditions for Tol Dec Phe and Phe
Decyl phenylalanine (Dec Phe)
Lipolysis samples of Tol Dec Phe were assayed for Dec Phe content via HPLC. The mobile phase comprised of 0.1% formic acid in H2O (Mobile Phase A) and 0.1% formic acid in ACN (Mobile Phase B) and was eluted using linear gradient elution. The initial conditions were 80:20, (A/B) v/v decreasing to 40:60, v/v, between 0.00 – 1.00 min, after which the gradient remained at 40:60, v/v, between 1.00 – 3.50 min, followed by a linear gradient to 20:80, v/v, between 3.50 – 4.50 min. The mobile phase was held at 20:80, v/v, between 4.50 – 5.50 min and then immediately returned to 80:20, v/v, which then remained at that volume ratio between 5.50 – 7.50 min. The flow rate was 1 mL/min, injection volume was 50 μL, and UV absorbance was set at 214 nm. The retention time of Dec Phe was 2.5 min, and the concentration range of the calibration standards was 20 – 200 ug/mL. Standard curves were prepared by plotting peak height ratios against known concentration of standards. Calibration stock solutions of Dec Phe were prepared at a concentration of 2000 µg/mL by dissolving Dec Phe in ACN. Standard samples were prepared with H2O:ACN, 80:20, v/v. Unknown sample concentrations were calculated from the standard equation y = mx + c, as determined by linear regression of the standard curve. Assay performance was validated using standard measures of linearity, precision, and reproducibility.
Phenylalanine (Phe)
Mobile phase comprised of 0.1% formic acid in H2O (mobile phase A) and 0.1% formic acid in acetonitrile (ACN) (mobile phase B) and was eluted on a linear gradient elution. Mobile phase started at 98% v/v A between 0.0 – 4.0 min. The gradient was reduced to 20% A between 4.0 – 4.5 min, and 20% A was maintained between 4.5 – 5.0 min. The gradient was returned to 98% A between 5.0 – 6.0 min, and 98% A was maintained between 6.0 – 6.5 min. The flow rate was 1 mL/min, injection volume was 50 μL, and UV absorbance was measured at 214 nm. The retention time of Phe was 2.0 min, and the concentration range of the calibration standards was 5 – 200 µg/mL. Standard curves were prepared by plotting peak heights against known concentration of standards. Calibration stock solutions of Phe were prepared at a concentration of 2000 µg/mL by dissolving Phe in H2O. Standard samples were then prepared by dilution with H2O:ACN, 98:2 (v/v).
Unknown sample concentrations were calculated from the standard equation y = mx + c, as determined by linear regression of the standard curve. Assay performance was validated using standard measures of linearity, precision, and reproducibility.
Pharmacokinetic studies
All animal experiments were approved by the Monash Institute of Pharmaceutical Sciences Animal Ethics Committee and were conducted in accordance with the Australian and New Zealand Guidelines for the Care and Use of Animals in Research. Male Sprague Dawley rats (260–340 g) were obtained from the Monash Animal Research Platform (VIC, Australia).
One day prior to dosing, the left internal carotid artery was cannulated to facilitate blood collection as described previously [75]. Surgeries were conducted under gaseous anaesthesia using 3% isoflurane in medical carbonox (95% oxygen, 5% carbon dioxide). The cannula (a proprietary BASi Culex cannula, BASi West Lafayette, IN) was externalised by subcutaneous tunnelling to emerge at the back of the neck. Rats were placed in Raturn® metabolic cages connected to a Culex® automated blood sampler (BASi, West Lafayette, IN) to recover overnight and for the duration of the pharmacokinetic study. Animals were fasted overnight and food was returned 6 h post dose. Water was available ad libitum.
On the day of dosing animals were briefly anaesthetised with isofluorane prior to dosing via oral gavage. Animals were dosed with either i) a suspension prepared by dispersing 5.6 mg tolfenamic acid in 0.5% CMC (w/v) and 0.4% Tween 80 (w/v) in 0.5 mL of 0.9% NaCl, ii) a lipid suspension of tolfenamic acid in MCF-2 where 5.6 mg tolfenamic acid was suspended in 125 mg of MCF-2 and dispersed in 0.5 mL water immediately prior to gavage (~ 45 mg/g tolfenamic acid), or iii) 26 mg Tol Dec Ala dissolved in 125 mg of MCF-2 and dispersed in 0.5 mL water immediately prior to gavage (~ 207 mg/g IL or 110 mg/g tolfenamic acid equivalents).
Plasma samples were collected up to 15 h post-dose. At each sampling time, 220 µL of arterial blood was collected directly into heparinised borosilicate vials stored in the chilled Culex® fraction collector. Samples were centrifuged at 2000 x g for 3 min at 4°C, and 50 µL aliquots of plasma were transferred to microcentrifuge tubes and frozen until sample analysis. Tmax, Cmax and AUC(0-15 h) data were taken directly from the plasma level time curves and AUC calculated via the linear trapezoidal rule. Dose normalised AUC data were calculated using a nominal dose of 5.6 mg/rat to allow comparison of exposure at the same dose, recognising that the IL containing formulation could be administered at a higher dose due to higher solubility.
Plasma assay for tolfenamic acid
A 5 µL aliquot of internal standard (diclofenac) solution was spiked into each 50 µL plasma sample, and the samples were vortexed for 0.5 min. Acetonitrile (100 µL) was subsequently added to precipitate proteins and samples vortexed and centrifuged at 10,620 x g for 10 min at 25°C. The supernatant from each sample was collected and transferred to HPLC autosampler vials for analysis. Plasma standards were prepared by spiking aliquots (45 µL) of blank rat plasma with 5 µL of tolfenamic acid standard solution and 5 µL of internal standard solution. Plasma standards were subsequently prepared in the same manner as the plasma samples before analysis by LC–MS/MS. Mass spectrometry was performed using a Shimadzu LC–MS/MS 8050 triple quadrupole mass spectrometer (Shimadzu, Japan) coupled to an LC20AD solvent pump system and SIL 20AC HT autosampler (Shimadzu, Japan). The optimized mass spectrometry settings were: detector voltage: 1.1 kV; interface temperature: 350° C; DL temperature: 300° C; heat block: 450°C; nebulising gas flow: 1.5 L/min; drying gas flow: 6 L/min. Analytical separation was performed using a Phenomenex Kinetex 2.6 µm C8 100 Å, 50 × 2.1 mm column at a mobile phase flow rate of 0.3 mL/min. Mobile phase comprised 0.1% formic acid in both solvent A (MilliQ) and solvent B (methanol). Samples were injected onto the column and eluted using a binary gradient: 50%-97% solvent B from 0–1.65 min; 97% B from 1.65–2 min 97–50% B from 2–2.5 min and 50% B from 2.5–3 min. The retention times of tolfenamic acid and diclofenac were 2.23 and 1.99 min respectively.
Results and discussion
Solubility studies of Tol ILs in MCF
Previous studies have shown that converting poorly water soluble drugs (PWSD) into lipophilic drug-ILs can increase the drug loading capacity of lipid based formulations (LBFs) [76–78]. For PWSDs that are weak electrolytes, strong acids or bases are commonly used to provide the pairing counterions in pharmaceutical salts in order to promote complete proton transfer. As a rule of thumb complete proton transfer is expected where the ∆pKa between the acid and base counterion pairs is > 2 – 3. [79–82]. The respective pKas of Tol (pKa = 3.66) [83, 84] and the lipophilic amines employed here (pKa ~ 10) [85, 86] leads to a ∆ pKa ~ 6.4, which was expected to promote efficient proton transfer between the drug and amine. As a result, Tol ILs were expected to display more typical behaviour than for example the cinnarizine ionic liquids with fatty acid counterions we have described previously [47]. Consistent with this suggestion Tol readily formed IL with the counterions employed here and no evidence of instability or disassociation was apparent over the period of study. As synthesised the ILs produced had different physical properties. Tol Dec was isolated as a white solid, with a clearly defined melting point of 147 °C (and so sat outside the typical definition of an IL). In contrast Tol Dec Ala (and Tol Dec Phe) were isolated as yellow, viscous semi solid amorphous materials that revealed glass transition temperatures by DSC of -16 and -6 °C respectively (supplementary information).
To determine the equilibrium solubility of the Tol ILs in medium chain LBFs, Tol ILs were first loaded incrementally into MCF-1, starting at 10% mass of IL/mass of LBF and the quantity of IL added increased when complete solution was apparent. As shown in Table I, converting tolfenamic acid to Tol ILs utilising lipophilic amines generally increased drug loading capacity. Tolfenamic acid decyl amine (Tol Dec) and tolfenamic acid decyl alanine ester (Tol Dec Ala) increased the solubility of tolfenamic acid in lipid solution by 2 and 2.5 fold respectively. The greater increase in solubility provided by the Dec Ala counterion when compared to the Dec counterion may reflect the methyl side chain group on the alanine introducing steric bulk, thereby disrupting lattice formation and increasing drug loading capacity. This is consistent with the lower Tg of Tol Dec Ala when compared to the melting point of Tol Dec. Interestingly, the Tol IL with the largest side chain group, Tol Dec Phe (that was employed as a probe in the breakdown studies to allow quantification of Phe) displayed a lower solubility than the free acid, in spite of a lower Tg.
Table I.
Solubility of tolfenamic acid and Tol ILs in LBF

* M.P of Tol from [87]
The digestibility of LAA cations under simulated digestive conditions
To evaluate the digestibility of the LAA counterions, the decyl phenylalanine ester LAA was generated in order that the phenylalanine breakdown product could be readily quantified (unlike alanine that does not have a UV chromophore). To facilitate these experiments, tolfenamic acid decyl phenylalanine ester (Tol Dec Phe) was loaded into a relatively poorly digestible Type IV LBF which contains no traditional lipid to ensure that observations of the digestibility of the LAA were not complicated by parallel effects on formulation digestion. The IL loaded formulation was dispersed into simulated intestinal fluid and digestion was initiated by the addition of porcine pancreatin extract (2400 TBU/mL). Samples were routinely taken during digestion and processed to determine the concentration of Dec Phe, and the amino acid breakdown product phenylalanine (Phe).
As shown in Fig. 3. the concentrations of Dec Phe and Phe measured throughout digestion showed the expected inversely proportional relationship, i.e. breakdown of Dec Phe producing Phe. Figure 3. also reveals a very rapid rate of LAA breakdown (< 5 min) under standard simulated digestive conditions. To better illustrate the breakdown of the Dec Phe, the volume of porcine pancreatin extract added to the medium was reduced four-fold (to 600 TBU/mL, 25% w/v). As expected this reduced the rate of LAA breakdown (digestion of Dec Phe completed at 15 min). These data demonstrate the susceptibility of Dec Phe to enzymatic breakdown under digestive conditions. Since the bulky phenyl group of the Dec Phe counterion did not significantly limit enzyme approach it seems likely that Tol ILs with LAAs such as decyl alanine ester (i.e. which lack the benzyl group) as well as similar LAAs with smaller side chain groups are likely to be susceptible to enzymatic degradation. Notably the products of digestion, amino acids and fatty alcohols are classified as food additives and are expected to present no toxicity risk [88, 89].
Fig. 3.

Lipolysis of Tol Dec Phe in a Type IV formulation demonstrating the digestive breakdown of the Dec Phe counterion. The study was split into two phases, an initial dispersion phase, grey area (-15 – 0 min), and a period after initiation of digestion (0 – 60 min). Digestive breakdown of Dec Phe is illustrated by the decreasing concentration of Dec Phe upon addition of porcine pancreatin extract (> 0 min), and a proportional increase in concentrations of phenyalalnine (Phe). The dashed red line shows the theoretical maximal concentration of Dec Phe and Phe (i.e. with no digestion or complete digestion respectively)
Lipolysis studies of Tol ILs in MCF-1 (Type IIIA formulation)
Having shown that the Dec Phe LAA counterion was readily digestible, subsequent studies sought to evaluate whether LBF containing tolfenamic acid and Tol ILs based on the Dec, Dec Ala and Dec Phe LAA were able to maintain drug solubilisation during dispersion and digestion of the LBF.
On initial dispersion in the in vitro lipid digestion test, LBF containing tolfenamic acid and Tol Dec Phe resulted in good dispersion and excellent solubilisation of Tol in the aqueous phase (Fig. 4). On initiation of digestion, however, LBF containing tolfenamic acid and Tol Dec Phe generated some precipitated material, a dense oil phase (that settled above the pellet after centrifugation) and limited quantities of Tol in the aqueous phase. Interestingly, as digestion progressed the quantity of Tol in the aqueous phase increased and amounts in the dense oil phase and pellet reduced. This was particularly evident for Tol Dec Phe, where significant transfer to the aqueous phase was seen over time resulting in more than 90% of the drug being recovered in the aqueous phase after 60 min digestion. Similar patterns of distribution were seen for tolfenamic acid however, the changes were not as large.
Fig. 4.

Drug distribution of tolfenamic acid and Tol ILs in MCF during lipolysis studies (Type IIIA) (37 °C, mean ± SD, n = 3). % drug distribution of tolfenamic acid was determined via the concentration of tolfenamic acid in respective phases during dispersion (-15 – 0 min; grey section), and digestion (0 – 60 min)
In contrast, to tolfenamic acid and Tol Dec Phe, dispersion of LBF containing Tol Dec and Tol Dec Ala resulted in the immediate formation of a dense oil phase that contained most of the drug, limiting transfer to the aqueous phase. On digestion, Tol Dec loaded LBF showed behaviour very similar to that of tolfenamic acid and Tol Dec Phe, i.e. a high % of tolfenamic acid present in the dense oil phase at the onset of digestion, but subsequent transfer to the aqueous phase over time. However, unlike tolfenamic acid and Tol Dec Phe, Tol Dec resulted in increased precipitation of tolfenamic acid as digestion progressed. Similar to Tol Dec, Tol Dec Ala also formed a dense oil phase and aqueous phase during dispersion, and separated out into three phases upon digestion. Interestingly, unlike the other Tol IL, in the case of Tol Dec Ala a small quantity of oil phase remained until the end of digestion. More importantly, as the oil phase reduced, instead of drug transferring to the aqueous phase (as it did for Tol Dec and Tol Dec Phe), drug precipitated and was recovered in the pellet. The differences in phase behaviour and solubilisation over time may be related to the digestion of the counterion over time, however detailed analysis of the species present was not undertaken in these experiments.
The differing drug distribution profiles observed for the Tol ILs studied here may be due to the nature of the different Tol ILs loaded into the formulation and in particular the differences in the mass of IL loaded (due to differing solubilities). Thus, the phase behaviour of the more MCF soluble Tol ILs (such as Tol Dec Ala where > 10% w/w of mass of IL/mass of LBF was loaded), may result from the very large quantities of the (dense) IL that could be loaded into the formulation. This in turn may have driven phase separation of a drug rich oil phase during processing. Whilst a proportion of this ultimately transfers to the aqueous phase, probably due to digestion of the counterion, the large quantity of IL loaded dictates that this is incomplete. In contrast, in those ILs that are loaded at much lower levels due to lower solubility (such as Tol Dec Phe) similar processes occur, but the much lower mass of IL that must be processed dictates that transfer to the aqueous phase is more efficient. For the IL with intermediate solubility (Tol Dec) intermediate behaviour is apparent. In contrast to the IL loaded systems, tolfenamic acid behaves more typically and disperses well initially but then precipitates as digestion continues. In this case kinetic changes to the counterion due to counterion digestion do not complicate the solubilisation profile.
When the data is viewed as the concentration of tolfenamic acid in the aqueous phase (Fig. 5), rather than the proportional distribution (Fig. 4), a composite view is obtained that considers both the proportional distribution in Fig. 4 and the drug loading in Table I. These data show that the aqueous phase concentration is highest on formulation dispersion for tolfenamic acid and the Tol Dec Phe IL, in spite of the low drug loading levels. This reflects the lack of phase separation of the dense oil phase seen for Tol Dec and Tol Dec Ala. On digestion, however, the aqueous phase concentrations for the tolfenamic acid and Tol Dec Phe formulations drop rapidly such that immediately after initiation of digestion, the aqueous phase concentration of tolfenamic acid is low and similar across all formulations. As described above, over the digestion period a proportion of the dose that is initially abstracted into the dense oil phase is released to the aqueous phase. As shown in Fig. 5, the highest aqueous phase concentrations are ultimately obtained for the non-digestible Tol Dec IL in the formulation which combines reasonable solubility in the formulation with good transfer to the aqueous phase. In contrast, Tol Dec Ala which showed the highest lipid solubility, also precipitated more readily during formulation digestion and as such the aqueous phase concentrations at the end of the digestion phase were lower than formulations containing Tol Dec. For the ILs where drug loading was enhanced, and therefore where ultimate benefit was initially expected to be highest (e.g. Tol Dec Ala), aqueous phase drug concentrations were limited by the phase separation of an IL-rich oil phase. Since this occurred even during formulation dispersion (at least for the formulations where the IL were loaded at the highest levels), subsequent studies focused on examining whether the formulation characteristics could be changed to improve dispersion of the drug rich formulations.
Fig. 5.

In vitro digestion studies of Tol ILs in MCF (Type IIIA) (37°C, mean = ± SD, n = 3); tolfenamic acid (blue diamond), Tol Dec (red circle), Tol Dec Ala (green square), and Tol Dec Phe (purple square). The dashed line represents the target (100% solubilised) concentration of tolfenamic acid for each formulation. Differences in the solubility of tolfenamic acid and the different Tol ILs dictated that the drug loading of Tol ILs in MCF was different in each case and therefore the target concentrations were different in each case. Concentrations are shown during dispersion (-15 – 0 min; grey section), and digestion (0 – 60 min) of the lipolysis study
Lipolysis studies of Tol ILs in Type IIIB formulations
To improve the dispersion of LBFs containing Tol ILs at high drug loading capacity, i.e. > 10% mass of IL/mass of LBF, the composition of MCF-1 was changed. The proportion of the surfactant, Kolliphor® EL, was increased by twofold and lipid content decreased by 0.5-fold. This results in a change to a more typical Type IIIB formulation, referred to here as MCF-2 (see Table I). The equilibrium solubility of both tolfenamic acid and Tol Dec Ala was increased in MCF-2, likely as a result of the higher surfactant concentration, however the Dec Ala IL retained its solubility advantage in MCF-2, as it had in MCF-1.
The increased quantity of surfactant in MCF-2 was expected to facilitate improved dispersion, and thereby solubilisation of Tol IL. To examine this, Tol ILs were loaded into MCF-2 at molar equivalent concentrations to that employed in the lipolysis studies with MCF-1 and digested as before. The new formulation containing higher concentrations of Kolliphor® EL led to much improved solubilisation of tolfenamic acid. In contrast to the previous lipolysis studies of the Tol ILs in MCF-1, the lipolysis samples of tolfenamic acid, Tol Dec, Tol Dec Phe and Tol Dec Ala did not form a dense oil phase, presumably reflecting the ability of the higher quantities of Kolliphor® EL to better solubilise and disperse the formulation and digestion products.
As shown in Fig. 6, lipolysis samples of tolfenamic acid, Tol Dec, Tol Dec Phe and Tol Dec Ala loaded formulations revealed a translucent aqueous phase and a (limited) pellet phase on centrifugation. The highest aqueous phase concentrations were therefore obtained with Tol Dec Ala due to its higher solubility in the formulation (and therefore higher formulation loading). Comparison of the aqueous phase concentration attained (Fig. 7) relative to the data for the original formulation in Fig. 5 shows that the new MCF-2 formulations of tolfenamic acid, Tol Dec, Tol Dec Phe and Tol Dec Ala resulted in aqueous phase concentrations similar to their targeted maximal possible concentrations of Tol in the aqueous phase on dispersion. On digestion these concentrations dropped slightly, especially for Tol Dec and Tol Dec Ala, but the attained aqueous phase concentrations were significantly higher than after digestion of MCF-1. Whether this reflects concurrent digestion of the LAA counter ion remains unknown. Nonetheless, the data show that when reformulated to provide for adequate dispersion, the MCT-2 Tol Dec Ala formulation provides for the highest attainable drug concentrations after in vitro formulation digestion (Fig. 7) This reflects both higher drug loading in the formulation of Tol Dec Ala, due to higher solubility of the ionic liquid, and effective solubilisation post dispersion by virtue of using MCF-2.
Fig. 6.

Drug distribution of tolfenamic acid and Tol ILs in MCF-2 (Type IIIB) during in vitro digestion studies (37°C, mean ± SD, n = 3). % drug distribution of tolfenamic acid was determined via the concentration of tolfenamic acid in respective phases during dispersion (-15 – 0 min; grey section), and digestion (0 – 60 min)
Fig. 7.

In vitro digestion studies of Tol IL in MCF-2 (Type IIIB) formulation. Increasing the proportion of surfactant (Kolliphor® EL) in the formulation, improved formulation dispersion (-15 – 0 min; grey section), and drug solubilisation during digestion (0 – 60 min). Tol Dec and Tol Dec Ala, were loaded into the Type IIIB formulation, at mol equivalent concentrations to that employed in the lipolysis studies with the Type IIIA formulation (MCF-1). (mean ± SD; n = 3)
Oral exposure of Tolfenamic acid after administration of Tolfenamic acid and Tol Dec Ala IL in MCF-2
In light of the in vitro data indicating that the Dec Ala IL counterion was both digestible and resulted in higher drug loading capacity in LBF than Tol Dec, and that coupling this with MCF-2 (but not MCF-1) led to higher tolfenamic acid solubilisation during formulation dispersion and digestion for MCF-2 containing Tol Dec Ala compared to Tol Dec (Fig. 7), subsequent studies explored the in vivo utility of the MCF-2 – Tol Dec Ala formulation. Tolfenamic acid and Tol Dec Ala were loaded into MCF-2 at concentrations close to their maximum solubility and dosed via oral gavage to conscious rats. As a comparator, a lipid free oral suspension of tolfenamic acid was also administered at the same dose as that given in MCF-2. Plasma samples were taken over time and assayed for tolfenamic acid by HPLC–MS/MS. The plasma level time profiles are shown in Fig. 8 and the summary pharmacokinetic data summarised in Table II.
Fig. 8.

Plasma level time profiles for tolfenamic acid after oral gavage of a suspension and LBF of tolfenamic acid loaded at a dose of ~ 5.6 mg per rat in comparison to the same LBF loaded with ~ 26 mg of the more lipid soluble Tol Dec Ala IL (mean ± SE, n = 3–6). Data normalised to dose of 18.7 or 87.7 mg/kg within each dosing group to account for variation in animal weights
Table II.
Summary pharmacokinetic parameters for tolfenamic acid in plasma after oral gavage of suspension and lipid-based formulations (MCF-2) of tolfenamic acid and a lipid-based formulation (MCF-2) of Tol Dec Ala ionic liquid
| Compound | Formulation | Dose (mg/kg Tol equivalents) |
Tmax (h) | Cmax (µg/mL) | AUC (0-15 h) (µg/mL.h) | AUC (0-15 h) (µg/mL.h) normalised* |
|---|---|---|---|---|---|---|
| Tolfenamic acid | Suspension | 18.7 | 1.19 ± 0.88 | 5.3 ± 1.9 | 27.2 ± 6.85 | 27.2 ± 6.85 |
| Tolfenamic acid | LBF (MCF-2) | 18.7 | 0.33 ± 0.14 | 20.6 ± 6.0 | 30.5 ± 9.82 | 30.5 ± 9.82 |
| Tol Dec Ala IL | LBF (MCF-2) | 87.7 | 0.33 ± 0.13 | 21.7 ± 11 | 95.5 ± 46.1 | 38.8 ± 18.7 |
* Data normalised to a nominal dose of 18.7 mg/kg Tol to provide dose normalised exposure comparison.
The plasma level time data show that administration in a lipid-based formulation leads to a more rapid Tmax for formulations of Tol than administration as an oral suspension, but that no significant difference is evident in total exposure (AUC). This was expected and is consistent with previous studies employing tolfenamic acid as a model poorly water-soluble anion and likely reflects the fact that whilst the aqueous solubility of the free acid of tolfenamic acid is low, under intestinal conditions at neutral pH, where Tol is expected to be ionised, the solubility is less likely to limit absorption. Reflecting the greater lipid solubility of the Tol Dec Ala IL, LBF containing this IL could be loaded with larger quantities of drug. The larger quantities dosed resulted in a significant increase in oral exposure. In spite of the larger dose, Tol exposure after oral administration of MCF-2 containing Tol Dec Ala increased proportionally and oral bioavailability was maintained as shown by the consistency of the dose normalised AUC data across all formulations. Some evidence of double peaking was apparent after administration of the Tol Dec Ala IL. This may reflect enterohepatic recycling as this has been suggested previously [90, 91], although it was less evident in the other dosing groups. It may also reflect the administration of a higher dose, requiring longer to absorb, possibly coupled with gradual digestion of the LAA counterion.
Conclusion
Pairing tolfenamic acid, a weakly acidic drug with a cationic LAA produced a lipophilic drug-IL with improved drug loading capacity in LBF compared to the parent drug. This approach is applicable to a range of weakly acidic small molecules and is expected to improve the utility of LBF and reduce pill burden or offer reduced capsule sizes to patients. Cationic lipophilic salt counterions, however, are challenging pharmaceutically since most cationic lipids have toxicity liabilities. In the current studies LAA were therefore employed in order to introduce a digestible bond into the cationic lipid. Consistent with this hypothesis, data obtained under model intestinal conditions showed very rapid breakdown under digestion conditions to produce less toxic fatty alcohols and amino acids. Initial attempts to employ Type IIIA medium chain lipid-based formulations to formulate the LAA-based ILs were unsuccessful when loaded at the high drug levels that were possible using the ILs. However, substitution of the Type IIIA formulations with a similar Type IIIB formulation with higher surfactant levels resulted in improved performance and good solubilisation throughout the experiment. Subsequently oral bioavailability studies in rats demonstrated that the same Type III LBF containing the Tol Dec Ala IL was able to support effective oral exposure even at the higher drug loading level facilitated by IL conversion. The data suggest that cationic LAAs can serve as digestible lipid cations for the formation of highly lipid soluble ILs from weakly acidic drugs, and provides proof-of-concept for a novel series of biocompatible lipid cations suitable for in vivo use.
Supplementary Information
Below is the link to the electronic supplementary material.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions. These studies were funded by Monash University and an Australian Government Research Training Program post graduate scholarship to AL.
Declarations
Conflict of Interest statement
AL, LF, HW, HB, PS and CP are inventors on patents describing ionic liquid technologies that were developed by Monash University and assigned to Lonza or were developed by Lonza. LF, HB and HW were previous employees of Lonza.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
7/9/2022
This article was updated to fix incorrect Figure 1 in online version which was caused due to production fault.
References
- 1.Murray SM, Zimlich TK, Mirjafari A, O’Brien RA, Davis JH., Jr Thermophysical properties of imidazolium-based lipidic ionic liquids. J Chem Eng Data. 2013;58(6):1516–1522. doi: 10.1021/je301004f. [DOI] [Google Scholar]
- 2.Mallakpour S, Dinari M. Ionic liquids as green solvents: Progress and prospects. In: Mohammad A, Inamuddin D, editors. Green Solvents II. Dordrecht:Springer; 2012. pp. 1–32. 10.1007/978-94-007-2891-2_1.
- 3.Durga G, Mishra A. Ionic liquids: Industrial applications. In: Encyclopedia of Inorganic and Bioinorganic Chemistry. Wiley; 2016. pp. 1–13. 10.1002/9781119951438.eibc2434.
- 4.MacFarlane DR, Seddon KR. Ionic liquids—Progress on the fundamental issues. Aust J Chem. 2006;60(1):3–5. doi: 10.1071/CH06478. [DOI] [Google Scholar]
- 5.Ho TD, Zhang C, Hantao LW, Anderson JL. Ionic liquids in analytical chemistry: Fundamentals, advances, and perspectives. Anal Chem. 2013;86:262–285. doi: 10.1021/ac4035554. [DOI] [PubMed] [Google Scholar]
- 6.Wei Di, Ivaska A. Applications of ionic liquids in electrochemical sensors. Anal Chim Acta. 2007;607(2):126–35. doi: 10.1016/j.aca.2007.12.011. [DOI] [PubMed] [Google Scholar]
- 7.Zhang W, Mohammed A, Hines LG, Jr, Xiao D, Martinez OJ, Bartsch RA, et al. Effect of cation symmetry on the morphology and physicochemical properties of imidazolium ionic liquids. J Phys Chem B. 2011;115(20):6572–6584. doi: 10.1021/jp1115614. [DOI] [PubMed] [Google Scholar]
- 8.Shirota H, Castner EW., Jr Physical properties and intermolecular dynamics of an ionic liquid compared with its isoelectronic neutral binary solution. J Phys Chem Lett. 2005;109(42):9388–9392. doi: 10.1021/jp054664c. [DOI] [PubMed] [Google Scholar]
- 9.Dupont J. Molten salts to ionic liquids: A “nano” journey. Acc Chem Res. 2011;44(11):1223–1231. doi: 10.1021/ar2000937. [DOI] [PubMed] [Google Scholar]
- 10.Davis JH., Jr Task-specific ionic liquids. Chem Lett. 2004;33(9):1072–1077. doi: 10.1246/cl.2004.1072. [DOI] [Google Scholar]
- 11.Trivedi TJ, Rao KS, Singh T, Mandal SJ, Sutradhar N, Panda AB, et al. Task-specific, biodegradable amino acid ionic liquid surfactants. Chemsuschem. 2011;4(5):604–608. doi: 10.1002/cssc.201100065. [DOI] [PubMed] [Google Scholar]
- 12.Zhao H, Baker GA, Wagle DV, Ravula S, Zhang Q. Tuning task-specific ionic liquids for the extractive desulfurization of liquid fuel. ACS Sustainable Chemistry & Engineering. 2016;4:4771–4780. doi: 10.1021/acssuschemeng.6b00972. [DOI] [Google Scholar]
- 13.Yue C, Fang D, Liu L, Yi T-F. Synthesis and application of task-specific ionic liquids used as catalysts and/or solvents in organic unit reactions. J Mol Liq. 2011;163:99–121. doi: 10.1016/j.molliq.2011.09.001. [DOI] [Google Scholar]
- 14.Curreri AM, Mitragotri S, Tanner EEL. Recent Advances in Ionic Liquids in Biomedicines. Advanced Science. 2021;8(2004819):1–18. doi: 10.1002/advs.202004819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu X, Zhu Q, Chen Z, Wu W, Lu Y, Qi J. Ionic liquids as a useful tool for tailoring active pharmaceutical ingredients. J Control Release. 2021;338:268–283. doi: 10.1016/j.jconrel.2021.08.032. [DOI] [PubMed] [Google Scholar]
- 16.Tang J, Song H, Feng X, Yohannes A, Yao S. Ionic Liquid-Like Pharmaceutical Ingredients and Applications of Ionic Liquids in Medicinal Chemistry: Development. Status and Prospects Curr Med Chem. 2019;26(32):5947–5967. doi: 10.2174/0929867325666180605123436. [DOI] [PubMed] [Google Scholar]
- 17.Balk A, Holzgrabe U, Meinel L. “Pro et contra” ionic liquid drugs - Challenges and opportunities for pharmaceutical translation. Eur J Pharm Biopharm. 2015;94:291–304. doi: 10.1016/j.ejpb.2015.05.027. [DOI] [PubMed] [Google Scholar]
- 18.Shi Y, Zhao Z, Gao Y, Pan DC, Salinas AK, Guo J, et al. Oral delivery of sorafenib through spontaneous formation of ionic liquid nanocomplexes. J Control Release. 2020;322:602–609. doi: 10.1016/j.jconrel.2020.03.018. [DOI] [PubMed] [Google Scholar]
- 19.Zandu SK, Chopra H, Singh I. Ionic Liquids for Therapeutic and Drug Delivery Applications. Curr Drug Res Rev. 2020;12(1):26–41. doi: 10.2174/2589977511666191125103338. [DOI] [PubMed] [Google Scholar]
- 20.Griesser J, Hetényi G, Moser M, Demarne F, Jannin V. Hydrophobic ion pairing: Key to highly payloaded self-emulsifying peptide drug delivery systems. Int J Pharm. 2017;520:267–274. doi: 10.1016/j.ijpharm.2017.02.019. [DOI] [PubMed] [Google Scholar]
- 21.Balk A, Widmer T, Wiest J, Bruhn H, Rybak J-C, Matthes P, et al. Ionic liquid versus prodrug strategy to address formulation challenges. Pharm Res. 2014;32:2154–2167. doi: 10.1007/s11095-014-1607-9. [DOI] [PubMed] [Google Scholar]
- 22.Yadav JS, Reddy BVS, Reddy PN. Rhodium acetate dimer immobilized in 1-butyl-3-methylimidazolium hexfluorophosphate ionic liquid: a novel and recyclable catalytic system for the cyclopropanation of alkenes. Adv Synth Catal. 2004;346(1):53–56. doi: 10.1002/adsc.200303136. [DOI] [Google Scholar]
- 23.Kamio A, Nagaosa Y. 1-Butyl-3-methylimidazolium hexafluorophosphate ionic liquid as a new solvent for the determination of lead(II) and cadmium(II) by anodic stripping voltammetry after extraction of the iodide complexes. Analytical Sciences: The International Journal of the Japan Society for Analytical Chemistry. 2008;24(10):1363–1367. doi: 10.2116/analsci.24.1363. [DOI] [PubMed] [Google Scholar]
- 24.Carda-Broch S, Berthod A, Armstrong DW. Solvent properties of the 1-butyl-3-methylimidazolium hexafluorophosphate ionic liquid. Anal Bioanal Chem. 2003;375(2):191–199. doi: 10.1007/s00216-002-1684-1. [DOI] [PubMed] [Google Scholar]
- 25.Giammanco CH, Kramer PL, Fayer MD. Ionic liquid versus Li+ aqueous solutions: Water dynamics near bistriflimide anions. J Phys Chem B. 2016;120(37):9997–10009. doi: 10.1021/acs.jpcb.6b07145. [DOI] [PubMed] [Google Scholar]
- 26.Hosokawa T, Matsumoto K, Nohira T, Hagiwara R, Fukunaga A, Sakai S, et al. Stability of ionic liquids against sodium metal: a comparative study of 1-ethyl-3-methylimidazolium ionic liquids with bis(fluorosulfonyl)amide and bis(trifluoromethylsulfonyl)amide. J Phys Chem B. 2016;120(18):9628–9636. [Google Scholar]
- 27.Chen S, Izgorodina EI. Prediction of 1H NMR chemical shifts for clusters of imidazolium-based ionic liquids. Phys Chem Chem Phys. 2017;19(26):17411–17425. doi: 10.1039/C7CP02951A. [DOI] [PubMed] [Google Scholar]
- 28.Seo S, DeSilva MA, Brennecke JF. Physical properties and CO2 reaction pathway of 1-ethyl-3- methylimidazolium ionic liquids with aprotic heterocyclic anions. J Phys Chem B. 2014;77(8):624–626. doi: 10.1021/jp509583c. [DOI] [PubMed] [Google Scholar]
- 29.Kusuma VA, Macala MK, Baker JS, Hopkinson D. Cross-linked poly(ethylene oxide) ion gels containing functionalized imidazolium ionic liquids as carbon dioxide separation membranes. Ind Eng Chem Res. 2018;57(34):11658–11667. doi: 10.1021/acs.iecr.8b02739. [DOI] [Google Scholar]
- 30.Wilkes JS, Zaworotko MJ. Air and water stable 1-ethyl-3-methylimidazolium based ionic liquids. Chem Commun. 1992;13:965–7. 10.1039/C39920000965.
- 31.Wilkes JS. A short history of ionic liquids - from molten salts to neoteric solvents. Green Chem. 2002;4(2):73–80. doi: 10.1039/b110838g. [DOI] [Google Scholar]
- 32.Bonhote P, Dias A-P, Papageorgiou N, Kalyanasundaram K, Gratzel M. Hydrophobic, highly conductive ambient-temperature molten salts. Inorg Chem. 1996;35(5):1168–1178. doi: 10.1021/ic951325x. [DOI] [PubMed] [Google Scholar]
- 33.Fayer MD. Dynamics and structure of room temperature ionic liquids. Chem Phys Lett. 2014;616–617:259–274. doi: 10.1016/j.cplett.2014.09.062. [DOI] [Google Scholar]
- 34.Huddleston JG, Visser AE, Reichert WM, Willauer HD, Broker GA, Rogers RD. Characterisation and comparison of hydrophilic and hydrophobic room temperature ionic liquids incorporating the imidazolium cation. Green Chem. 2001;3(4):156–164. doi: 10.1039/b103275p. [DOI] [Google Scholar]
- 35.Haghbakhsh R, Raeissi S. Two simple correlations to predict viscosities of pure and aqueous solutions of ionic liquids. J Mol Liq. 2015;211:948–956. doi: 10.1016/j.molliq.2015.08.036. [DOI] [Google Scholar]
- 36.Fukaya Y, Iizuka Y, Sekikawa K, Ohno H. Bio ionic liquids: room temperature ionic liquids composed wholly of biomaterials. Green Chem. 2007;9(11):1155–1157. doi: 10.1039/b706571j. [DOI] [Google Scholar]
- 37.Rahimi M, Shafiei-Irannejad V, Safa KD, Salehi R. Multi-branched ionic liquid-chitosan as a smart and biocompatible nano-vehicle for combination chemotherapy with stealth and targeted properties. Carbohyd Polym. 2018;196:299–312. doi: 10.1016/j.carbpol.2018.05.059. [DOI] [PubMed] [Google Scholar]
- 38.Ohno H, Fukumoto K. Amino acid ionic liquids. Acc Chem Res. 2007;40(11):1122–1129. doi: 10.1021/ar700053z. [DOI] [PubMed] [Google Scholar]
- 39.Moshikur RM, Raihan CM, Wakabayashi R, Tahara Y, Moniruzzaman M, Goto M. Characterization and cytotoxicity evaluation of biocompatible amino acid esters used to convert salicylic acid into ionic liquids. Int J Pharm. 2018;546:31–38. doi: 10.1016/j.ijpharm.2018.05.021. [DOI] [PubMed] [Google Scholar]
- 40.Wang C, Zhu J, Zhang D, Yang Y, Zheng L, Qu Y, et al. Ionic liquid – microemulsions assisting in the transdermal delivery of Dencichine: Preparation, in-vitro and in-vivo evaluations, and investigation of the permeation mechanism. Int J Pharm. 2017;535(1–2):120–131. doi: 10.1016/j.ijpharm.2017.10.024. [DOI] [PubMed] [Google Scholar]
- 41.Rai R, Pandey S. Evidence of water-in-ionic liquid microemulsion formation by nonionic surfactant Brij-35. Langmuir. 2014;30:10156–10160. doi: 10.1021/la502174a. [DOI] [PubMed] [Google Scholar]
- 42.Wu X, Yu Q, Wu J, Li T, Ding N, Wu W, Lu, Y., Zhu, Q., Chen, Z., Qi, J. Ionic liquids containing ketoconazole improving topical treatment of T. Interdigitale infection by synergistic action. Int J Pharm. 2020;589:119842. 10.1016/j.ijpharm.2020.119842. [DOI] [PubMed]
- 43.Goindi S, Kaur R, Kaur R. An ionic liquid-in-water microemulsion as a potential carrier for topical delivery of poorly water soluble drug: Development, ex-vivo and in-vivo evaluation. Int J Pharm. 2015;495(2):913–923. doi: 10.1016/j.ijpharm.2015.09.066. [DOI] [PubMed] [Google Scholar]
- 44.Furukawa S-Y, Hattori G, Sakai S, Kamiya N. Highly efficient and low toxic skin penetrants composed of amino acid ionic liquids. RSC Adv. 2016;6(90):87753–87755. doi: 10.1039/C6RA16926K. [DOI] [Google Scholar]
- 45.Moniruzzaman M, Tahara Y, Tamura M, Kamiyaab N, Goto M. Ionic liquid-assisted transdermal delivery of sparingly soluble drugs. Chem Commun. 2010;46(9):1452–1454. doi: 10.1039/b907462g. [DOI] [PubMed] [Google Scholar]
- 46.Ibsen KN, Mitragotri S. Transdermal insulin delivery using choline-based ionic liquids (CAGE) J Control Release. 2018;286:137–144. doi: 10.1016/j.jconrel.2018.07.029. [DOI] [PubMed] [Google Scholar]
- 47.Lai A, Sahbaz Y, Ford L, Nguyen T-H, Haque S, Williams HD, Benameur H, Scammells PJ, Porter CJH. Stabilising disproportionation of lipophilic ionic liquid salts in lipid-based formulations. Int J Pharm. 2021;597:120292. 10.1016/j.ijpharm.2021.120292. https://www.sciencedirect.com/science/article/pii/S037851732100096X.. [DOI] [PubMed]
- 48.Hofland HEJ, Shephard L, Sullivan SM. Formation of stable cationic lipid/DNA complexes for gene transfer. PNAS. 1996;93:7305–7309. doi: 10.1073/pnas.93.14.7305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Felgner PL, Gadek TR, Holm M, Roman R, Chan HW, Wenz M, et al. Lipofection: A highly efficient, lipid-mediated DNA-transfection procedure. Proc Natl Acad Sci USA. 1987;84(21):7413–7417. doi: 10.1073/pnas.84.21.7413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fraley RT, Dellaporta SL, Papahadjopoulos D. Liposome-mediated delivery of tobacco mosaic virus RNA into tobacco protoplasts: A sensitive assay for monitoring liposome-protoplast interactions. Proc Natl Acad Sci USA. 1981;79:1859–1863. doi: 10.1073/pnas.79.6.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tranchant I, Thompson B, Nicolazzi C, Mignet N, Scherman D. Physicochemical optimisation of plasmid delivery by cationic lipids. J Gene Med. 2004;6:24–35. doi: 10.1002/jgm.509. [DOI] [PubMed] [Google Scholar]
- 52.Damen M, Groenen AJJ, van Dongen SFM, Nolte RJM, Scholte BJ, Feiters MC. Transfection by cationic gemini lipids and surfactants. Medicinal Chemistry Communications. 2018;9:1404–1425. doi: 10.1039/C8MD00249E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lonez C, Vandenbranden M, Ruysschaert J-M. Cationic liposomal lipids: From gene carriers to cell signaling. Prog Lipid Res. 2008;47(5):340–347. doi: 10.1016/j.plipres.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 54.Zelphati O, Wang Y, Kitada S, Reeds JC, Felgner PL, Corbeil J. Intracellular delivery of proteins with a new lipid-mediated delivery system. J Biol Chem. 2001;276(37):35103–35110. doi: 10.1074/jbc.M104920200. [DOI] [PubMed] [Google Scholar]
- 55.Dalkara D, Chandrashekhar C, Zuber G. Intracellular protein delivery with a dimerizable amphiphile for improved complex stability and prolonged protein release in the cytoplasm of adherent cell lines. J Control Release. 2006;116(3):353–359. doi: 10.1016/j.jconrel.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 56.Lindner LH, Brock R, Arndt-Jovin D, Eibl H. Structural variation of cationic lipids: Minimum requirement for improved oligonucleotide delivery into cells. J Control Release. 2005;110(2):444–456. doi: 10.1016/j.jconrel.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 57.Malone RW, Felgner PL, Verma IM. Cationic liposome-mediated RNA transfection. Proc Natl Acad Sci USA. 1989;86(16):6077–6081. doi: 10.1073/pnas.86.16.6077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Saxena V. Preparation and physical characterization of DNA-binding cationic liposomes. In: D’Souza GGM, editor. Liposomes methods and protocols, methods in molecular biology, vol. 1522. New York: Humana Press; 2016. p. 245–50. 10.1007/978-1-4939-6591-5_19. [DOI] [PubMed]
- 59.Eş I, Ok MT, Puentes-Martinez XE, de Toledo MAS, de Pinho Favaro MT, Cavalcanti LP, et al. Evaluation of siRNA and cationic liposomes complexes as a model for in vitro siRNA delivery to cancer cells. Colloid and Surfaces A. 2018;555:280–289. doi: 10.1016/j.colsurfa.2018.06.073. [DOI] [Google Scholar]
- 60.Liang G, Li P, Lei W. Delivery of miRNA using Fe2O3 nanoparticles capped polyethyleneimine as a nonviral carrier. Key Eng Mater. 2012;531–532:543–546. doi: 10.4028/www.scientific.net/KEM.531-532.543. [DOI] [Google Scholar]
- 61.Coburn RF. Polyamine effects on cell function: Possible central role of plasma membrane PI(4,5)P2. J Cell Physiol. 2009;221(3):544–551. doi: 10.1002/jcp.21899. [DOI] [PubMed] [Google Scholar]
- 62.Verzola D, Fama A, Villaggio B, Di Rocco M, Simonato A, D'Amato E, et al. Lysine triggers apoptosis through a NADPH oxidase-dependent mechanism in human renal tubular cells. J Inherit Metab Dis. 2012;35(6):1011–1019. doi: 10.1007/s10545-012-9468-z. [DOI] [PubMed] [Google Scholar]
- 63.Ray RM, Li C, Bhattacharya S, Naren AP, Johnson LR. Spermine, a molecular switch regulating EGFR, integrin β3, Src, and FAK scaffolding. Cell Signal. 2011;24(4):931–942. doi: 10.1016/j.cellsig.2011.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Carmona-Ribeiro AM, Ortis F, Schumacher RI, Armelin MCS. Interactions between cationic vesicles and cultured mammalian cells. Langmuir. 1996;13(8):2215–2218. doi: 10.1021/la960759h. [DOI] [Google Scholar]
- 65.Dymond MK, Attard GS. Cationic type I amphiphiles as modulators of membrane curvature elastic stress in vivo. Langmuir. 2008;24(20):11743–11751. doi: 10.1021/la8017612. [DOI] [PubMed] [Google Scholar]
- 66.Lonez C, Lensink MF, Vandenbranden M, Ruysschaert J-M. Cationic lipids activate cellular cascades Which receptors are involved? Biochimica et Biophysica Acta, General Subjects. 2009;1790(6):425–30. doi: 10.1016/j.bbagen.2009.02.015. [DOI] [PubMed] [Google Scholar]
- 67.Gamboa A, Hecht N, Soto-Bustamante E, Romero-Hasler P, Meinel L, Morales JO. Delivery of ionizable hydrophilic drugs based on pharmaceutical formulation of ion pairs and ionic liquids. Eur J Pharm Biopharm 156:203-218. 10.1016/j.ejpb.2020.09.007. [DOI] [PubMed]
- 68.Zupančič O, Grießinger JA, Rohrer J, de Sousa IP, Danninger L, Partenhauser A, et al. Development, in vitro and in vivo evaluation of a self-emulsifying drug delivery system (SEDDS) for oral enoxaparin administration. Eur J Pharm Biopharm. 2016;109:113–121. doi: 10.1016/j.ejpb.2016.09.013. [DOI] [PubMed] [Google Scholar]
- 69.Li M, Zheng Y, Shan F-Y, Zhou J, Gong T, Zhang Z-R. Development of ionic-complex-based nanostructured lipid carriers to improve the pharmacokinetic profiles of breviscapine. Acta Pharmacol Sin. 2013;34:1108–1115. doi: 10.1038/aps.2013.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Veenstra G, Webb C, Sanderson H, Belanger SE, Fisk P, Nielsen A, et al. Human health risk assessment of long chain alcohols. Ecotoxicol Environ Saf. 2009;72(4):1016–1030. doi: 10.1016/j.ecoenv.2008.07.012. [DOI] [PubMed] [Google Scholar]
- 71.Pouton CW. Formulation of poorly water-soluble drugs for oral administration: Physicochemical and physiological issues and the lipid formulation classification system. Eur J Pharm Sci. 2006;29(3–4):278–287. doi: 10.1016/j.ejps.2006.04.016. [DOI] [PubMed] [Google Scholar]
- 72.Williams HD, Sassene P, Kleberg K, Bakala-N'Goma J-C, Calderone M, Jannin V, et al. Toward the establishment of standardized in vitro tests for lipid-based formulations, part 1: Method parameterization and comparison of in vitro digestion profiles across a range of representative formulations. J Pharm Sci. 2012;101(9):3360–3380. doi: 10.1002/jps.23205. [DOI] [PubMed] [Google Scholar]
- 73.Zangenberg NH, Mullertz A, Kristensen HG, Hovgaard L. A dynamic in vitro lipolysis model I. Controlling the rate of lipolysis by continuous addition of calcium. European Journal of Pharmaceutical Sciences. 2001;14(2):115–22. doi: 10.1016/S0928-0987(01)00169-5. [DOI] [PubMed] [Google Scholar]
- 74.Sek L, Porter CJH, Kaukonen AM, Charman WN. Evaluation of the in-vitro digestion profiles of long and medium chain glycerides and the phase behaviour of their lipolytic products. J Pharm Pharmacol. 2002;54:29–41. doi: 10.1211/0022357021771896. [DOI] [PubMed] [Google Scholar]
- 75.Trevaskis NL, Hu L, Caliph SM, Han S, Porter CJH. The Mesenteric Lymph Duct Cannulated Rat Model: Application to the Assessment of Intestinal Lymphatic Drug Transport. J Vis Exp. 2015;97(52389):1–11. doi: 10.3791/52389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Williams HD, Sahbaz Y, Ford L, Nguyen T-H, Scammells PJ, Porter CJH. Ionic liquids provide unique opportunities for oral drug delivery: structure optimization and in vivo evidence of utility. Chem Commun. 2014;50(14):1688–1690. doi: 10.1039/C3CC48650H. [DOI] [PubMed] [Google Scholar]
- 77.Sahbaz Y, Nguyen T-H, Ford L, McEvoy CL, Williams HD, Scammells PJ, et al. Ionic liquid forms of weakly acidic drugs in oral lipid formulations: Preparation, characterization, in vitro digestion, and in vivo absorption studies. Mol Pharm. 2017;14(11):3669–3683. doi: 10.1021/acs.molpharmaceut.7b00442. [DOI] [PubMed] [Google Scholar]
- 78.Sahbaz Y, Williams HD, Nguyen T-H, Saunders J, Ford L, Charman' SA, et al. Transformation of poorly water-soluble drugs into lipophilic ionic liquids enhances oral drug exposure from lipid based formulations. Mol Pharm. 2015;12(6):1980–1991. doi: 10.1021/mp500790t. [DOI] [PubMed] [Google Scholar]
- 79.Haynes, W.M. (Ed.). Dissociation constants of organic acids and bases. In: CRC Handbook of Chemistry and Physics. 91th ed. Taylor and Francis, Boca Raton, pp. 42–51. 10.1201/b12286.
- 80.Childs SL, Stahly GP, Park A. The salt-co-crystal continuum: the influence of crystal structure on ionisation state. Mol Pharm. 2007;4(3):323–338. doi: 10.1021/mp0601345. [DOI] [PubMed] [Google Scholar]
- 81.Tong W-QT, Whitesell G. In situ salt screening - A useful technique for discovery support and preformulation studies. Pharmaceutical Development and Technology. 2008;3(2):215–23. doi: 10.3109/10837459809028498. [DOI] [PubMed] [Google Scholar]
- 82.Tarsa PB, Towler CS, Woollam G, Berghausen J. The influence of aqueous content in small scale salt screening - Improving hit rate for weakly basic, low solubility drugs. Eur J Pharm Sci. 2010;41(1):23–30. doi: 10.1016/j.ejps.2010.05.009. [DOI] [PubMed] [Google Scholar]
- 83.Nechipadappu SK, Trivedi DR. Structural and physicochemical characterization of pyridine derivative salts of anti-inflammatory drugs. J Mol Struct. 2017;1141:64–74. doi: 10.1016/j.molstruc.2017.03.086. [DOI] [Google Scholar]
- 84.Sun N, Avdeef A. Biorelevant pKa (37°C) predicted from the 2D structure of the molecule and its pKa at 25°C. J Pharm Biomed Anal. 2011;56(2):173–182. doi: 10.1016/j.jpba.2011.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Matulis D, Bloomfield VA. Thermodynamics of the hydrophobic effect. I. Coupling of aggregation and pKa shifts in solutions of aliphatic amines. Biophysical Chemistry. 2001;93(1):37–51. doi: 10.1016/S0301-4622(01)00207-1. [DOI] [PubMed] [Google Scholar]
- 86.Mirgorodskaya AB, Kudryavtseva LA. Aqueous solutions of geminal alkylammonium surfactants as a medium for reactions of long-chain amines. Russ J Gen Chem. 2009;79(1):42–48. doi: 10.1134/S1070363209010071. [DOI] [Google Scholar]
- 87.Wassvik CM, Johansson K, Hubatsch I. Poorly soluble marketed drugs display solvation limited solubility. J Med Chem. 2007;50(23):5858–5862. doi: 10.1021/jm0706416. [DOI] [PubMed] [Google Scholar]
- 88.CFR - Code of Federal Regulations Title 21, U.S Food & Drug Administration; 2022. https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfCFR/CFRSearch.cfm?fr=172.864. Accessed 22 May 2022.
- 89.Food Additive Status List, U.S Food & Drug Administration: 2021. https://www.fda.gov/food/food-additives-petitions/food-additive-status-list#ftnD. Accessed 22 May 2022.
- 90.Priymenko N, Ferre JP, Rascol A, Costes G, Toutain PL. Migrating motor complex of the intestine and absorption of a biliary excreted drug in the dog. J Pharmacol Exp Ther. 1993;267(3):1161–1167. [PubMed] [Google Scholar]
- 91.Pentikainen PJ, Penttila A, Neuvonen PJ, Khalifah RG, Hignite CE. Isolation, preliminary characterization and pharmacokinetics of tolfenamic acid and its metabolites. Eur J Drug Metab Pharmacokinet. 1982;7:259–267. doi: 10.1007/BF03189628. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.