

Abstract
Objectives: In vascular remodeling diseases, proliferation and inflammation of vascular smooth muscle cells (VSMCs) constitute the basic pathologic processes. Dehydroepiandrosterone (DHEA) exerts a protective effect on the cardiovascular system, but the molecular mechanism is unclear. Methods: The plasma DHEA was measured using enzyme-linked immunosorbent assay (ELISA) kits. The neointima hyperplasia was assessed by hematoxylin/eosin staining. MiRNA microarray analysis was used to compare the influence of Ang II and DHEA on miRNA expression profiles in VSMCs. Cell counting and MTS assay were used to evaluate the effect of Ang II, DHEA and miR-486a-3p on VSMCs proliferation. qRT-PCR was performed to detect the expression of miR-486a-3p, PCNA, IL-1β and NLRP3. Western blot analysis was performed to detect the expressions of PCNA, IL-1β and NLRP3 after miR-486a-3p was knocked down or overexpressed in VSMCs. Results: DHEA suppressed neointimal and VSMCs proliferation and inflammation. Using miRNA microarray analysis, we found that DHEA upregulated the expression of miR-486a-3p in VSMCs. Further experiments indicated that DHEA promoted miR-486a-3p expression in VSMCs and in the vascular intima. Gain- and loss-of-function experiments revealed that transfection of miR-486a-3p mimic inhibited proliferation and inflammation of VSMCs, which improved intimal hyperplasia. On the contrary, deletion of miR-486a-3p promoted VSMCs proliferation and inflammation. Furthermore, DHEA suppressed NOD-like receptor family pyrin domain containing 3 (NLRP3) expression and reduced VSMCs proliferation and inflammation. Importantly, DHEA inhibited NLRP3 expression via miR-486a-3p in VSMCs. Conclusions: DHEA inhibited VSMCs and vascular intimal proliferation and inflammation by regulating the miR-486a-3p/NLRP3 axis. Therefore, DHEA might be a candidate cardiovascular protective agent in the future.
Keywords: DHEA, miR-486a-3p, VSMCs, proliferation, inflammation
Introduction
Neointimal hyperplasia is a common pathological feature of many vascular remodeling diseases. Inflammation is accompanied by the occurrence of vascular remodeling [1]. Vascular smooth muscle cells (VSMCs) located in the middle layer of blood vessels regulate vasoconstriction and stabilize blood pressure [2]. Abnormal proliferation of VSMCs and massive extracellular matrix synthesis lead to intimal thickening and lumen narrowing, which constitute the cytopathological basis of vascular remodeling [3]. Although many studies have been performed to determine the pathophysiological mechanisms of VSMCs proliferation and inflammation, the way by which VSMCs respond to injury and coordinate these complex processes remains unknown.
The NOD-like receptor family pyrin domain containing 3 (NLRP3) inflammasome is involved in the innate immune response, mediating the activation of caspase-1 and the secretion of the pro-inflammatory cytokines IL-1β and IL-18. Abnormal activation of the NLRP3 inflammasome has been implicated in different inflammatory diseases, such as gout, diabetes, obesity, Alzheimer’s disease, and atherosclerosis [4]. Though the NLRP3 inflammasome in vascular remodeling diseases has been broadly investigated, the mechanism by which NLRP3 modulates VSMCs inflammation and proliferation has not been elucidated.
MicroRNAs (miRNAs), as negative regulators of messenger RNA (mRNA) levels, are involved in the regulation of mRNA networks during vascular remodeling. Indeed, studies have reported that some miRNAs participate in the regulation of proliferation, inflammation, and phenotypic transformation of VSMCs [5,6]. In addition, transcriptional repression, post-translational modification, and post-transcriptional regulation of microRNAs can lead to decreased NLRP3 expression and activity. A recent study has shown that the long non-coding RNA Kcnq1ot1 accelerates podocyte apoptosis by increasing NLRP3 expression through inhibiting miR-486a-3p [7]. However, whether miR-486a-3p can regulate NLRP3 expression and play a role in VSMCs proliferation and inflammation remains unclear.
Dehydroepiandrosterone (DHEA) is the most abundant steroid hormone in the human blood circulation, and it enters the blood circulation in the form of DHEA sulfate (DHEA-S) [8]. The biological effect of DHEA is not limited to its role as a precursor of steroid hormones, and it also plays unique roles in immunity, metabolism, the cardiovascular system, and lipids [9,10]. DHEA has anti-inflammatory properties. In LPS-stimulated colonic epithelial cells and macrophages, DHEA blocks p38-induced NLRP3 inflammasome activation, thereby reducing intestinal inflammation in inflammatory bowel disease (IBD) [11]. In addition, DHEA is closely related to cardiovascular disease (CVD). Observational studies have shown that plasma DHEA-S is inversely associated with CVD in men [12]. DHEA can reduce cell proliferation and promote apoptosis in VSMCs and vascular endothelial cells [13,14]. In human carotid VSMCs, DHEA reduces cell proliferation and increases apoptosis by inhibiting Akt/GSK3-β/NFAT axis [15]. In the rabbit carotid artery balloon injury model, DHEA-S significantly inhibited neointima formation after balloon injury, and its effect on VSMCs migration and proliferation was achieved by upregulating one of the cyclin dependent kinase (CDK) inhibitors p16INK4a and activating PPARα [16]. However, whether the inhibition of VSMCs inflammation and proliferation by DHEA is related to the regulation of NLRP3 and miR-486a-3p expression remains unknown.
In this study, we explored whether and how DHEA protects against inflammation and proliferation of VSMCs via modulation of the miR-486a-3p/NLRP3 axis.
Material and methods
Animal model
All animal studies were approved by the Institutional Animal Care Committee of Hebei Medical University and all efforts were made to minimize suffering. Male C57BL/6 mice were divided into three groups: unligated group, ligated + DMSO group, and ligated + DHEA group (n = 8). DHEA (3 mg/kg/d) was administered by gavage 5 days before ligation until 14 days after ligation. Animals in the ligated group received an equivalent volume of DMSO. The model of intimal hyperplasia induced by carotid ligation has been previously described [17]. After anesthesia, the mice were ligated with 6-0 suture at the left carotid artery bifurcation. In the unligated group, sutures were passed under the left carotid artery without ligation. In mimic-transfected mice, pluronic gel including oligonucleotide was applied to the outer membrane of the vessel after ligation. Then, 14 days after surgery, carotid arteries were collected for further experiments.
Serum samples
The stored plasma samples were obtained from previous human studies. The clinical study provided samples of patients without CVD (non-CVD group, n = 9) and age-matched patients with CVD (CVD group, n = 11). Demographic and baseline characteristics were shown in Table S1. Mouse plasma samples were obtained from unligated group, ligated + DMSO group, and ligated + DHEA group (n = 8).
DHEA assays
The mouse and human plasma DHEA was measured using commercially available enzyme-linked immunosorbent assay (ELISA) kits (ml024031, ml301812, Mlbio, Shanghai, China).
Morphology analysis
Carotid artery tissue was fixed in paraformaldehyde and embedded in paraffin. Sections were stained with hematoxylin and eosin. Image-Pro Plus Analyzer software was used to determine the intimal area and the intima-media (I/M) ratio. Six random fields were examined in each section.
Cell culture and treatment
Mouse aorta VSMCs (ATCC, No. Crl-2797™) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum. After 24 hours of culture in serum-free medium, various stimuli were administered. Lipo2000 was used to transfect miR-486a-3p mimic (GenePharma), miR-486a-3p inhibitor (GenePharma), miR-486a-3p control (GenePharma), si-NLRP3 (GenePharma), or si-control (GenePharma). Cells were collected after transfection for 24 h. 293A cells were cultured in DMEM supplemented with 10% fetal bovine serum. The concentrations of Ang II and DHEA that processed VSMCs were 10-7 mol/L and 10-4 mol/L.
Cell viability assay and MTS assay
VSMCs were seeded in 96-well plates for 24 h. After 24 h of culture in serum-free medium, the appropriate treatment was given. Cell activity and viability were tested according to the manufacturer’s instructions [18].
Real-time PCR
TRIzol™ (Invitrogen) was used to extract total RNA from cells or blood vessels. After the quality of RNA was determined, microRNA and mRNA were extracted according to the manufacturer’s instructions. RNU6b (U6) and GAPDH were used as internal reference genes. The PCR assay results were from 3 independent experiments. The relative quantities were calculated using the 2-ΔΔCt formula. See Table S2 for the primer sequences.
Western blot analysis
Protein was extracted from VSMCs and carotid artery tissue. The same amount of protein was separated by 10% SDS-PAGE and electrotransferred to PVDF membranes, which was then blocked at room temperature for 2 h and incubated overnight with primary antibody at 4°C. Antibodies used were as follows: anti-proliferating cell nuclear antigen (PCNA) (1:1000, ab92552, Abcam), anti-IL-1β (1:1000, ab9722, Abcam), anti-NLRP3 (1:1000, ab263899, Abcam), and anti-β-actin (1:1000, sc-47778, Santa Cruz Biotechnology). Membranes were then incubated at room temperature for 1 h with HRP-conjugated secondary antibody. Protein bands were detected by enhanced chemiluminescence (ECL) Fuazon Fx (Vilber Lourmat).
Plasmid constructs
The luciferase reporter plasmid was constructed by restriction-enzyme digestion and one-step cloning. The 3’-UTR sequence containing miR-486a-3p target sites (wild-type (wt) or mutant (mut)) in NLRP3 (Table S3) was inserted into pmir-GLO Dual-Luciferase miRNA Target Expression Vector digested by Xho1 and Sal1.
Luciferase assays
293A cells were co-transfected with miR-486a-3p mimic (or mimic control)/miR-486a-3p inhibitor (or inhibitor control) and NLRP3 reporter vector (wild-type or mutant) or empty vector for 24 h. According to the instructions, the luciferase activity was measured using a Dual-GLO Luciferase Assay System. Firefly luciferase activity was determined and normalized to Renilla luciferase activity. Six separate experiments were performed on luciferase activity.
Statistical analysis
Data are expressed as means ± standard error of the mean. The difference between two groups was analyzed by analysis of variance (ANOVA) or Student’s t-test. A valve of P < 0.05 indicated statistical significance. At least three independent experiments were conducted on all the data provided.
Results
DHEA inhibits proliferation and inflammation of the neointima and VSMCs
First of all, we detected the concentration of DHEA in the patients with or without CVD. There were no statistical differences between the two groups in age at baseline (Table S1). The plasma concentration of DHEA in the CVD group was lower than that in the non-CVD group (Figure 1A). Then we detected the plasma concentration of DHEA in mice. Consistently, after carotid artery ligation, the plasma concentration of DHEA was lower than that in the unligated group (Figure 1B). These results suggest that DHEA may be involved in the process of vascular remodeling after vascular injury. Subsequently, we examined whether DHEA has an effect on vascular proliferation and inflammation. Morphometric analysis showed that in ligated + DMSO group, the arterial wall thickness increased significantly, while the lumen area decreased by more than 90%. DHEA treatment could significantly reduce the neointimal thickness (Figure 1C). The I/M ratio and intimal area in the DHEA group were clearly smaller than those of the ligated + DMSO group (Figure 1D, 1E), indicating that DHEA could notably inhibit intimal hyperplasia. PCNA is an indicator of cell proliferation. Therefore, we measured PCNA expression in the carotid arteries. qRT-PCR and Western blotting showed that PCNA expression was remarkably higher in the ligated + DMSO group, whereas the promoting effect of carotid artery ligation was eliminated by DHEA treatment (Figure 1F, 1G). Since proliferation and inflammation are inextricably linked [19], we investigated whether the repression of neointimal hyperplasia by DHEA affects inflammation. IL-1β expression was at least 2-fold higher in the ligated vasculature. Importantly, DHEA treatment reduced the upregulation of IL-1β expression (Figure 1F, 1G). These results indicate that DHEA suppresses neointimal hyperplasia and inflammation. Elevated Ang II activates proliferation and pro-inflammatory gene expression to promote vascular remodeling. Therefore, we determined whether DHEA could inhibit Ang II-induced proliferation and inflammation of VSMCs. We treated VSMCs with 0 to 10-7 mol/L Ang II for 24 h. The presence of Ang II increased the IL-1β expression level. The maximum effect was reached at 10-7 mol/L Ang II for 24 h (Figure 1H, 1I and Figures S1 and S2). In parallel with the promotion of IL-1β expression, Ang II significantly promoted VSMCs proliferation as shown by increased PCNA expression, MTS assay and cell counting (Figure 1H, 1I and Figures S1, S2, S3, S4, S5 and S6). In other experiments, we measured the effects of DHEA on VSMCs proliferation and inflammation. Cell counting and MTS assay demonstrated that treatment with DHEA inhibited VSMCs proliferation induced by Ang II, with optimum effects at 10-4 mol/L of DHEA for 24 h (Figure 1J, 1K). In addition, DHEA could reverse the upregulation of PCNA and IL-1β expression in a concentration-dependent manner (Figure 1L and Figure S7). These results reveal that DHEA suppresses angiogenesis-related intimal hyperplasia and Ang II-induced VSMCs proliferation and inflammation.
Figure 1.
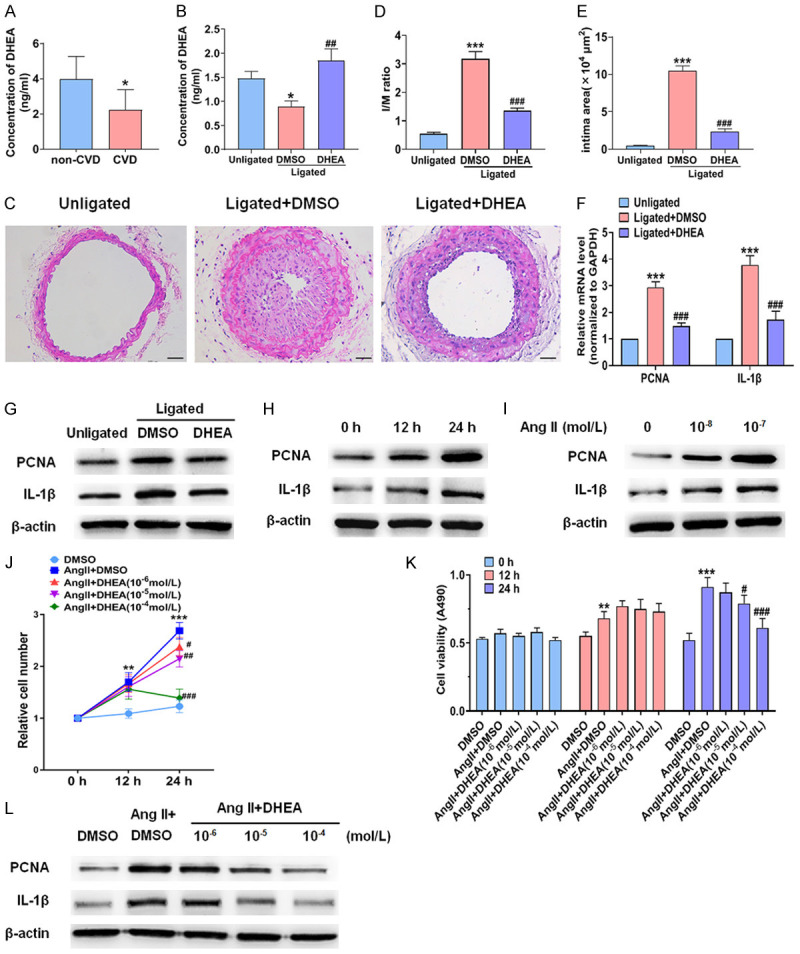
DHEA inhibits proliferation and inflammation of the neointima and VSMCs. (A) The serum DHEA concentrations in non-CVD patients and CVD patients. *P < 0.05 vs. non-CVD group. (B) DHEA concentration in serum of mice in unligated group, ligated + DMSO group, and ligated + DHEA group. *P < 0.05 vs. unligated group, ##P < 0.01 vs. ligated + DMSO group. (C) HE staining of carotid arteries was performed in unligated group, ligated + DMSO group, and ligated + DHEA group. Scale bars = 50 μm. (D, E) The I/M ratio and intimal area of carotid arteries. ***P < 0.001 vs. unligated group, ###P < 0.001 vs. Ligated + DMSO group. (F, G) qRT-PCR and Western blot analysis were performed to detect PCNA and IL-1β expression in the carotid arteries 14 days after ligation. ***P < 0.001 vs. unligated group, ###P < 0.001 vs. ligated + DMSO group. (H, I) VSMCs were incubated with Ang II (10-7 mol/L) for 0 h to 24 h (H) or various concentrations of Ang II for 24 h (I), and Western blotting was used to measure PCNA and IL-1β expression. (J, K) VSMCs were incubated with DMSO, Ang II (10-7 mol/L) plus DMSO, or Ang II (10-7 mol/L) plus different doses of DHEA for the indicated times. The Countess automated cell counter was used for cell counting and an MTS assay was performed to measure cell viability. **P < 0.01 and ***P < 0.001 vs. DMSO group, #P < 0.05, ##P < 0.01, and ###P < 0.001 vs. Ang II group. (L) VSMCs were stimulated with DMSO, Ang II (10-7 mol/L) plus DMSO, or Ang II (10-7 mol/L) plus various concentrations of DHEA for 24 h. Western blotting was used to analyze PCNA and IL-1β protein expression.
DHEA upregulates miR-486a-3p expression in VSMCs and vascular intima
Since previous studies have shown that miRNAs can regulate neointimal hyperplasia and VSMCs proliferation [3], miRNA microarray analysis was used to compare the influence of Ang II and DHEA on miRNA expression profiles in VSMCs. miRNA microarray analysis revealed many differences in miRNA expression among the three cell populations (Figure 2A). Three independent experimental samples were analyzed to determine miRNA levels. Two miRNAs were significantly upregulated in Ang II-stimulated cells but were downregulated by DHEA treatment. Moreover, five miRNAs were decreased in Ang II-treated cells and were upregulated in VSMCs stimulated with DHEA. Remarkably, miR-486a-3p expression was significantly decreased after Ang II treatment, whereas DHEA treatment increased its expression. Subsequently, we focused on whether miR-486a-3p was related to the inhibition of VSMCs proliferation and inflammation by DHEA. miR-486a-3p was decreased in Ang II-treated VSMCs. After 24 h of treatment with Ang II, miR-486a-3p expression was downregulated by approximately 50% (Figure 2B, 2C). Furthermore, we measured the effect of DHEA on miR-486a-3p expression. qRT-PCR showed that miR-486a-3p expression in cells treated with DHEA plus Ang II was notably higher than that in cells treated with Ang II alone, suggesting that DHEA abrogated the inhibitory effect of Ang II on miR-486a-3p (Figure 2D, 2E). Additionally, the expression of miR-486a-3p was lower in ligated carotid artery, whereas DHEA upregulated miR-486a-3p expression in the ligated carotid artery (Figure 2F). The above results demonstrate that DHEA promotes miR-486a-3p expression in VSMCs and neointima.
Figure 2.
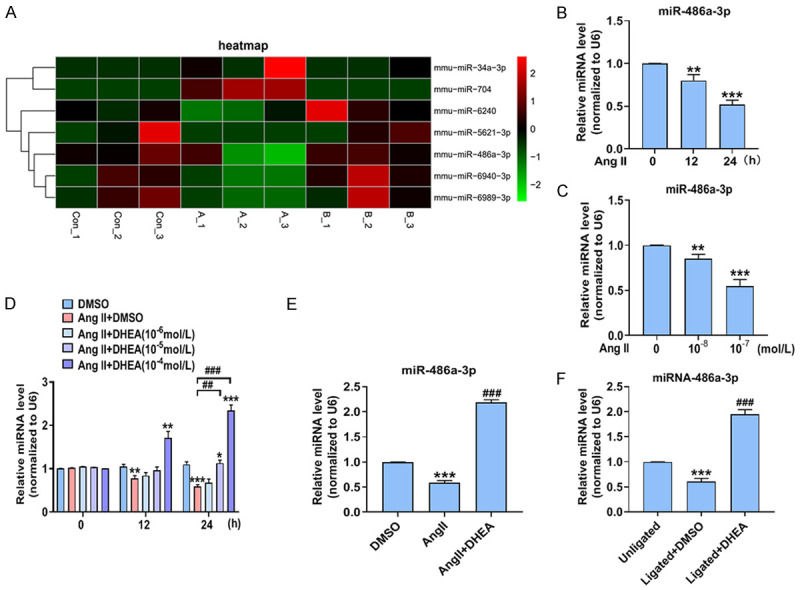
DHEA upregulates miR-486a-3p expression in VSMCs and vascular intima. (A) VSMCs were incubated with DMSO, Ang II (10-7 mol/L), or Ang II (10-7 mol/L) plus DHEA (10-4 mol/L) for 24 h. MiRNA microarray analysis was performed to compare the miRNA expression profiles. (B, C) VSMCs were treated as in Figure 1H and 1I, and qRT-PCR was performed to determine miR-486a-3p expression. **P < 0.01 and ***P < 0.001 vs. 0 h group or 0 mol/L Ang II group. (D) VSMCs were treated as in Figure 1J; qRT-PCR was used to measure miR-486a-3p expression. *P < 0.05, **P < 0.01, and ***P < 0.001 vs. DMSO group, ##P < 0.01 and ###P < 0.001 vs. Ang II group. (E) VSMCs were treated as in (A); qRT-PCR was used to determine miR-486a-3p expression. ***P < 0.001 vs. DMSO group, ###P < 0.001 vs. Ang II group. (F) qRT-PCR was performed to determine miR-486a-3p expression in the carotid arteries 14 days after ligation. ***P < 0.001 vs. unligated group, ###P < 0.001 vs. ligated + DMSO group.
DHEA decreases VSMCs proliferation and inflammation by increasing miR-486a-3p expression
To study whether miR-486a-3p is involved in VSMCs proliferation and inflammation, we performed loss- and gain-of-function experiments. First, VSMCs were transfected with miR-486a-3p control or miR-486a-3p mimic, and the results showed that the level of miR-486a-3p was increased more than 400-fold in VSMCs transfected with miR-486a-3p mimic (Figure 3A). MTS analysis and cell counting revealed that transfection of miR-486a-3p mimic significantly reduced VSMCs proliferation compared to cells transfected with miR-486a-3p control (Figure 3B, 3C). Then, we examined the influence of miR-486a-3p mimic on the expression of genes related to proliferation and inflammation. PCNA and IL-1β expression was significantly downregulated when miR-486a-3p mimic was transfected into VSMCs, whereas DHEA reversed the inhibition of PCNA and IL-1β expression (Figure 3D, 3E). The above results show that miR-486a-3p mimic markedly decreases VSMCs proliferation and inflammation and that miR-486a-3p is essential for the inhibition of VSMCs proliferation and inflammation by DHEA. To further demonstrate the effect of miR-486a-3p on VSMCs proliferation and inflammation, we treated cells with miR-486a-3p inhibitor. When transfected with miR-486a-3p inhibitor, the miR-486a-3p expression level in VSMCs was significantly reduced by at least 50% (Figure 3F). Then, MTS analysis and cell counting analysis suggested that the miR-486a-3p inhibitor promoted VSMCs proliferation (Figure 3G, 3H). In addition, we detected the influence of the miR-486a-3p inhibitor on the expression of genes related to proliferation and inflammation. When miR-486a-3p was depleted, PCNA and IL-1β expression was clearly increased, while the inhibitory effect of DHEA on these genes was significantly weakened (Figure 3I, 3J). These data indicate that DHEA inhibits VSMCs proliferation and inflammation partly through miR-486a-3p. In conclusion, miR-486a-3p mediates the inhibition of VSMCs proliferation and inflammation by DHEA.
Figure 3.
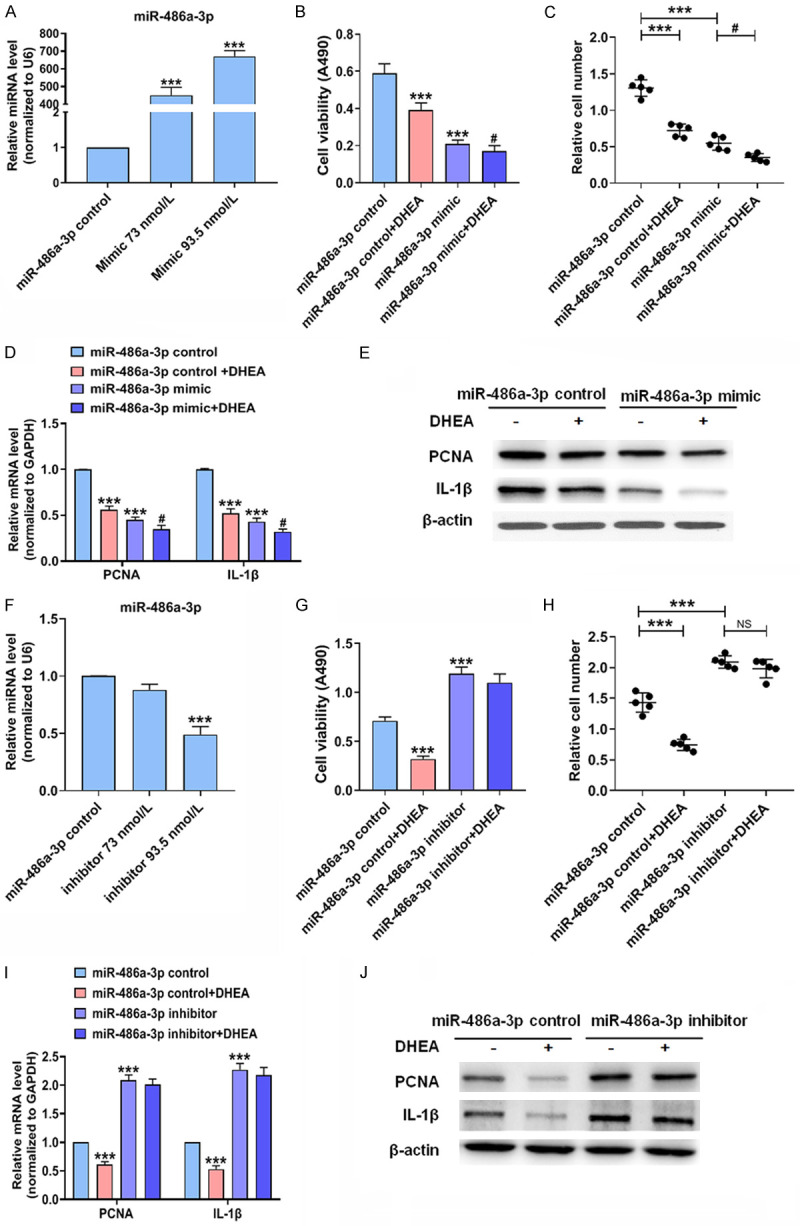
DHEA decreases VSMCs proliferation and inflammation by increasing miR-486a-3p expression. (A) VSMCs were transfected with miR-486a-3p control or mimic; qRT-PCR was used to detect miR-486a-3p. ***P < 0.001 vs. miR-486a-3p control group. (B-E) VSMCs were transfected with miR-486a-3p control or mimic for 24 h followed by stimulation with Ang II (10-7 mol/L) with or without DHEA (10-4 mol/L) for 24 h; MTS assay (B) was used to measure cell viability, and the Countess automated counter (C) was used for cell counting. The mRNA (D) and protein (E) expression of PCNA and IL-1β was measured. ***P < 0.001 vs. miR-486a-3p control group, #P < 0.05 vs. miR-486a-3p mimic group. (F) VSMCs were transfected with miR-486a-3p inhibitor or control for 24 h; qRT-PCR was performed to evaluate miR-486a-3p expression. ***P < 0.001 vs. miR-486a-3p control group. (G-J) VSMCs were transfected with miR-486a-3p inhibitor or control. After 24 h, VSMCs were incubated with Ang II (10-7 mol/L) with or without DHEA (10-4 mol/L). Cell viability assay (G), cell counting (H), qRT-PCR (I), and Western blotting (J) were performed. ***P < 0.001 vs. miR-486a-3p control group.
DHEA suppresses NLRP3 expression and decreases VSMCs proliferation and inflammation
Since miRNAs modulate gene expression at the post-transcriptional level, we combined miRNA microarray analysis, miRanda [20], and RNAhybrid [21] and found several potential target genes of miR-486a-3p associated with proliferation and inflammation. We then focused on NLRP3, which is closely related to inflammation and proliferation, and investigated whether NLRP3 is involved in the DHEA-mediated inhibition of VSMCs proliferation and inflammation. NLRP3 expression was enhanced in Ang II-treated VSMCs. The level of NLRP3 mRNA was upregulated more than 2-fold in VSMCs treated with Ang II (10-7 mol/L) for 24 h (Figure 4A-D). Subsequently, we detected NLRP3 expression in VSMCs stimulated with DHEA. DHEA significantly reduced the expression of NLRP3 (Figure 4E, 4F). In addition, NLRP3 expression was upregulated in the ligated carotid artery, whereas DHEA could abate the promoting effect of carotid artery ligation (Figure 4G, 4H). To clarify whether NLRP3 could mediate the inhibitory effect of DHEA on VSMCs proliferation and inflammation, we depleted NLRP3 and measured the expression of genes related to proliferation and inflammation. Depletion of NLRP3 clearly downregulated PCNA and IL-1β expression. More importantly, the inhibitory effect of DHEA was significantly attenuated when NLRP3 was depleted (Figure 4I, 4J). These results suggest that DHEA reduces VSMCs proliferation and inflammation through NLRP3.
Figure 4.
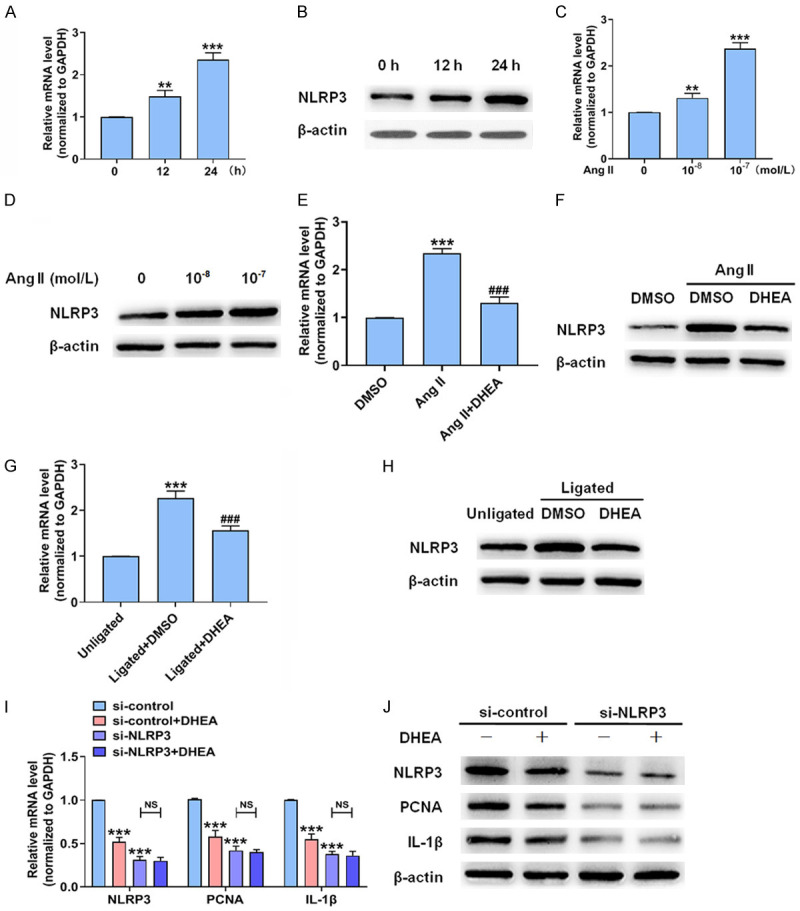
DHEA suppresses NLRP3 expression and decreases VSMCs proliferation and inflammation. (A-D) VSMCs were stimulated as in Figure 1H and 1I; qRT-PCR (A, C) and Western blotting (B, D) were performed to determine NLRP3 expression. **P < 0.01 and ***P < 0.001 vs. 0 h or 0 mol/L Ang II group. (E, F) VSMCs were treated as in Figure 2A. qRT-PCR (E) and Western blotting (F) were used to analyze NLRP3 expression. ***P < 0.001 vs. DMSO group, ###P < 0.001 vs. Ang II group. (G, H) NLRP3 expression in the carotid arteries 14 days after ligation. ***P < 0.001 vs. unligated group, ###P < 0.001 vs. ligated + DMSO group. (I, J) After transfection with si-control or si-NLRP3 for 24 h, VSMCs were treated with Ang II (10-7 mol/L) with or without DHEA (10-4 mol/L) for 24 h and then NLRP3, PCNA, and IL-1β mRNA (I) and protein (J) expression levels were detected. ***P < 0.001 vs. si-control group.
DHEA inhibits NLRP3 expression via miR-486a-3p
The above results led to a question about whether the inhibitory effect of DHEA on NLRP3 expression is mediated by miR-486a-3p, and thus, we used computer sequence analysis (miRanda and TargetScan) to search for potential matching sites of miR-486a-3p in the 3’-UTR of NLRP3. Indeed, the 3’-UTR of NLRP3 contains hypothetical binding sites for miR-486a-3p (Figure 5A). Next, we mutated the miR-486a-3p binding site in the NLRP3 3’-UTR, used miR-486a-3p mimic and inhibitor, and tested expression of the luciferase reporter gene. The results showed that in the cells with wild-type (wt)-NLRP3 3’UTR, the miR-486a-3p mimic group exhibited reduced luciferase activity by approximately 50% compared with the control group. On the contrary, in cells with a mutated NLRP3 3’UTR at the miR-486a-3p-binding site, the presence of miR-486a-3p mimic almost completely restored luciferase activity (Figure 5B). By contrast, when the miR-486a-3p inhibitor was co-transfected into the cells, luciferase activity in cells containing wt-NLRP3 3’-UTR increased significantly, while a mutation in the NLRP3 3’-UTR at the miR-486a-3p binding site counteracted this effect (Figure 5C). Furthermore, we transfected VSMCs with miR-486a-3p mimic or inhibitor and assessed NLRP3 expression. As shown in Figure 5D, when cells were transfected with miR-486a-3p mimic, NLRP3 expression was significantly downregulated, while NLRP3 expression was upregulated after transfection with miR-486a-3p inhibitor, which demonstrates that miR-486a-3p negatively modulates NLRP3 expression. To further study whether DHEA reduces NLRP3 expression via miR-486a-3p, we transfected miR-486a-3p inhibitor into VSMCs, a subset of which was stimulated with DHEA. At the protein level, the presence of miR-486a-3p inhibitor promoted NLRP3 expression, while the inhibitory effect of DHEA on NLRP3 expression was significantly weakened, suggesting that the inhibition of NLRP3 expression by DHEA was mediated by miR-486a-3p (Figure 5E). In conclusion, miR-486a-3p regulates NLRP3 expression in VSMCs by binding to the NLRP3 3’-UTR, and miR-486a-3p mediates the inhibition of NLRP3 expression by DHEA.
Figure 5.
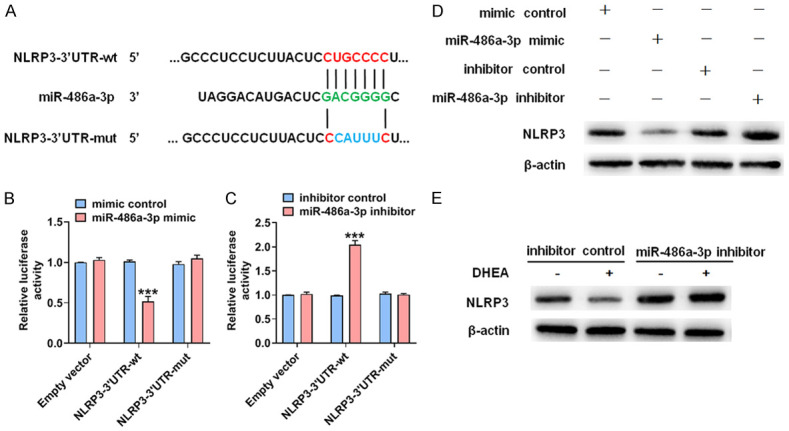
DHEA inhibits NLRP3 expression via miR-486a-3p. (A) The predicted binding site of miR-486a-3p in the 3’-UTR of NLRP3. The mutated miR-486a-3p binding site sequences were marked in blue. (B) 293A cells were co-transfected with miR-486a-3p mimic and a wt or mutant (mut) NLRP3 3’-UTR-luciferase reporter. A dual-luciferase reporter assay system was used to detect luciferase activity. ***P < 0.001 vs. mimic control group. (C) After 293A cells were transfected with miR-486a-3p inhibitor or an inhibitor control, luciferase reporter assays were performed in 293A cells transfected with the constructs containing the wt or mut NLRP3 3’-UTR. ***P < 0.001 vs. inhibitor control group. (D) VSMCs were transfected with the indicated constructs for 24 h. NLRP3 expression was detected. (E) VSMCs were treated as in Figure 3G. NLRP3 expression was measured by Western blotting.
Overexpression of miR-486a-3p reduces intimal hyperplasia and vascular inflammation
In further experiments, we evaluated the effect of miR-486a-3p mimic on the formation of vascular neointima. Fourteen days after surgery, a morphological analysis showed that the in situ transfection of endogenous miR-486a-3p could significantly reduce carotid wall thickness (Figure 6A). Compared with control mimic-transfected mice, the I/M ratio and the neointimal area were markedly decreased in the miR-486a-3p mimic-treated mice (Figure 6B, 6C). We then observed the effects of miR-486a-3p mimic on PCNA, IL-1β and NLRP3 expression in ligated carotid artery. In the control mimic-transfected mice, the expression of PCNA and IL-1β was significantly upregulated. Conversely, PCNA, IL-1β and NLRP3 expression was significantly decreased in ligated carotid artery after mice were transfected with miR-486a-3p mimic (Figure 6D, 6E). The above results demonstrate that the reduction of miR-486a-3p expression after vascular injury aggravates vascular inflammation and intimal hyperplasia and that miR-486a-3p overexpression eliminates this effect.
Figure 6.
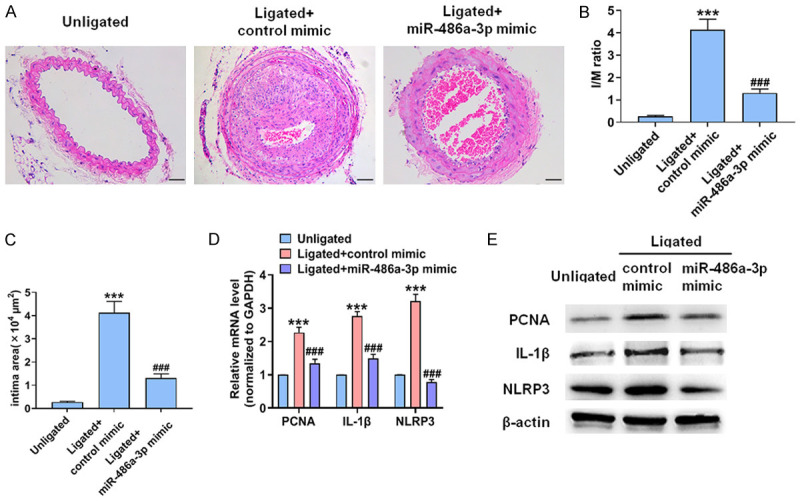
Overexpression of miR-486a-3p reduces intimal hyperplasia and vascular inflammation. (A) HE staining of carotid arteries. Scale bars = 50 μm. (B-E) I/M ratio (B), intimal area (C), the levels of PCNA, IL-1β and NLRP3 mRNA (D) and protein (E) in carotid arteries. ***P < 0.001 vs. unligated group, ###P < 0.001 vs. ligated + control mimic group.
Discussion
The biological behavior of VSMCs undergoes a series of changes including inflammation, phenotypic transformation, and proliferation after vascular injury [22]. VSMCs participate in vascular remodeling diseases by regulating proliferation, inflammation, and oxidative stress. Therefore, blocking VSMCs proliferation and inflammation is an effective way to treat vascular remodeling diseases. In this study, the major findings are: 1) DHEA inhibits proliferation and inflammation of VSMCs and neointima, 2) DHEA promotes miR-486a-3p expression in VSMCs and neointima, thus inhibiting the expression of proliferation and inflammation related genes in vitro and in vivo, 3) DHEA reduces VSMCs proliferation and inflammation by inhibiting NLRP3 expression, 4) DHEA inhibits NLRP3 expression and reduces VSMCs proliferation and inflammation via miR-486a-3p.
DHEA is the most abundant circulating hormone secreted by the adrenal cortex, and DHEA-S is converted into androgens or estrogens in peripheral tissues to play an indirect biological role. In man and woman, the concentrations of DHEA and DHEA-S gradually decrease with age [23]. In recent years, the role of DHEA in the cardiovascular system has become an attractive subject area. Multiple studies have assessed the association between DHEA/DHEA-S and CVD [24]. Epidemiological studies have demonstrated that the DHEA concentration in blood is negatively correlated with the incidence rate of atherosclerosis, fatal and nonfatal cardiovascular events, and all-cause mortality of CVD, especially in men [25]. A recent review suggested that the DHEA level in the blood circulation can predict CVD [26]. Experimental studies have revealed that DHEA inhibits VSMCs proliferation and that DHEA reverses pulmonary hypertension in part by inhibiting the Src/STAT3 axis in VSMCs [27,28]. Although the benefits of DHEA for CVD have been shown in vivo and in vitro, the specific mechanism has not been elucidated. In this study, we found that plasma DHEA concentrations in CVD patients were lower than those in non-CVD patients, and demonstrated that DHEA inhibited intimal hyperplasia and VSMCs proliferation by upregulating miR-486a-3p expression and downregulating NLRP3 expression.
A variety of danger signals can activate the NLRP3 inflammasome, including microorganisms, Ca2+ signaling, Ang II, and mitochondrial dysfunction. After NLRP3 is activated, it forms a complex with its adaptor and activates caspase-1, which processes pro-IL-1β into IL-1β, thereby triggering an inflammatory response. Multiple studies have shown that activation of the NLRP3 inflammasome accelerates vascular inflammation and promotes the progression of vascular remodeling diseases [29]. In ApoE-/- mice, silencing NLRP3 can inhibit the advancement of plaque and reduce local inflammation, thus undermining the progression of atherosclerosis [30]. In hypertensive patients, activation of the NLRP3 inflammasome accelerates VSMCs proliferation [31]. Cholesterol crystals trigger inflammation through the NLRP3 inflammasome, which induces arterial inflammation and atherosclerotic plaque instability [32]. These studies indicate that the NLRP3 inflammasome accelerates vascular remodeling diseases, which has promoted the search for agents that target this complex. A variety of drugs can inhibit the activity of inflammasomes. A recent report suggested that MCC950 inhibited the release of IL-1β from macrophages, thereby reducing atherosclerosis in ApoE mice [33]. Arglabin, an inhibitor of the NLRP3 inflammasome, suppresses inflammation and exerts anti-atherosclerotic effects in ApoE2.Ki mice fed a high-fat diet [34]. In the present study, we found that DHEA could downregulate NLRP3 expression in Ang II-treated VSMCs and in the ligated carotid artery. Furthermore, we investigated whether NLRP3 mediates the inhibitory effect of DHEA on VSMCs proliferation and inflammation. At both the mRNA and protein levels, depletion of NLRP3 markedly reduced the expression of PCNA and IL-1β. Importantly, the inhibition of VSMCs proliferation and inflammation by DHEA is significantly weakened when NLRP3 is depleted, which indicates that DHEA suppresses VSMCs proliferation and inflammation through NLRP3.
MicroRNA is a type of small, non-coding RNA that modulates gene expression through accelerating mRNA degradation or by decreasing mRNA translation at the post-transcriptional level. Studies have revealed that miRNAs participate in the proliferation and inflammation of a variety of cells. When vascular injury occurs, miR-155 promotes oxidative stress responses, proliferation, and inflammation in VSMCs [35]. According to several reports, miR-486 is involved in the growth and apoptosis of various cells. Overexpression of miR-486 facilitates the proliferation of bovine mammary epithelial cells [36]. MiR-486 can also reduce lung cancer cell proliferation and induce apoptosis [37]. However, the link between miR-486a and proliferation and inflammation in VSMCs remains unclear. Here, we performed a miRNA array and found that miR-486a-3p was obviously decreased in VSMCs treated with Ang II, while DHEA treatment increased the miR-486a-3p expression level. This finding was then verified in vivo and in vitro. In Ang II-treated VSMCs and neointima induced by carotid artery ligation, miR-486a-3p expression was clearly downregulated, whereas DHEA treatment upregulated miR-486a-3p expression. Next, we examined the relationship between miR-486a-3p and VSMCs proliferation and inflammation. Gain- and loss-of-function experiments showed that miR-486a-3p mimic suppressed VSMCs proliferation and inflammation. On the contrary, depletion of miR-486a-3p promoted proliferation and inflammation of VSMCs. In addition, miR-486a-3p mimic reduced intimal hyperplasia and vascular inflammation. These results provide sufficient evidence that miR-486a-3p inhibits the proliferation and inflammation of VSMCs.
In other experiments, we determined whether the inhibition of inflammation and proliferation of VSMCs by DHEA is related to its regulation of miR-486a-3p expression. Depletion of miR-486a-3p increased Ang II-induced VSMCs proliferation and inflammation, while DHEA treatment suppressed VSMCs proliferation and inflammation, which suggests that DHEA inhibited VSMCs proliferation and inflammation in part via miR-486a-3p. There may be other mechanisms by which DHEA inhibits VSMCs proliferation and inflammation. Importantly, we reveal that NLRP3 is the target gene of miR-486a-3p. A single miRNA can target a set of target genes. Therefore, additional research is needed to further clarify the effect of miR-486a-3p on VSMCs proliferation and inflammation.
In summary, as an NLRP3 inhibitor, DHEA inhibits VSMCs proliferation and inflammation by upregulating miR-486a-3p. Hence, DHEA may be an adjuvant in the treatment of patients with vascular remodeling diseases.
Acknowledgements
This research was supported by Hebei Provincial Government Outstanding Talent Project grant: 303-16-20-13, Hebei Province Natural Science Foundation grant: H2020206204, and Hebei Medical Science Research Project grant: 20210868.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Xu H, Du S, Fang B, Li C, Jia X, Zheng S, Wang S, Li Q, Su W, Wang N, Zheng F, Chen L, Zhang X, Gustafsson J, Guan Y. VSMC-specific EP4 deletion exacerbates angiotensin II-induced aortic dissection by increasing vascular inflammation and blood pressure. Proc Natl Acad Sci U S A. 2019;116:8457–8462. doi: 10.1073/pnas.1902119116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Frismantiene A, Philippova M, Erne P, Resink TJ. Smooth muscle cell-driven vascular diseases and molecular mechanisms of VSMC plasticity. Cell Signal. 2018;52:48–64. doi: 10.1016/j.cellsig.2018.08.019. [DOI] [PubMed] [Google Scholar]
- 3.Zhao XS, Zheng B, Wen Y, Sun Y, Wen JK, Zhang XH. Salvianolic acid B inhibits Ang II-induced VSMC proliferation in vitro and intimal hyperplasia in vivo by downregulating miR-146a expression. Phytomedicine. 2019;58:152754. doi: 10.1016/j.phymed.2018.11.014. [DOI] [PubMed] [Google Scholar]
- 4.Hoseini Z, Sepahvand F, Rashidi B, Sahebkar A, Masoudifar A, Mirzaei H. NLRP3 inflammasome: its regulation and involvement in atherosclerosis. J Cell Physiol. 2018;233:2116–2132. doi: 10.1002/jcp.25930. [DOI] [PubMed] [Google Scholar]
- 5.Guo X, Li D, Chen M, Chen L, Zhang B, Wu T, Guo R. miRNA-145 inhibits VSMC proliferation by targeting CD40. Sci Rep. 2016;6:35302. doi: 10.1038/srep35302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pan D, Liu G, Li B, Jiang J, Chen W, Li W, Zhang L, Hu Y, Xie S, Yang H. MicroRNA-1246 regulates proliferation, invasion, and differentiation in human vascular smooth muscle cells by targeting cystic fibrosis transmembrane conductance regulator (CFTR) Pflugers Arch. 2021;473:231–240. doi: 10.1007/s00424-020-02498-8. [DOI] [PubMed] [Google Scholar]
- 7.Zhang C, Gong Y, Li N, Liu X, Zhang Y, Ye F, Guo Q, Zheng J. Long noncoding RNA Kcnq1ot1 promotes sC5b-9-induced podocyte pyroptosis by inhibiting miR-486a-3p and upregulating NLRP3. Am J Physiol Cell Physiol. 2021;320:C355–C364. doi: 10.1152/ajpcell.00403.2020. [DOI] [PubMed] [Google Scholar]
- 8.Baulieu EE. Dehydroepiandrosterone (DHEA): a fountain of youth? J Clin Endocrinol Metab. 1996;81:3147–3151. doi: 10.1210/jcem.81.9.8784058. [DOI] [PubMed] [Google Scholar]
- 9.Jia X, Sun C, Tang O, Gorlov I, Nambi V, Virani SS, Villareal DT, Taffet GE, Yu B, Bressler J, Boerwinkle E, Windham BG, de Lemos JA, Matsushita K, Selvin E, Michos ED, Hoogeveen RC, Ballantyne CM. Plasma dehydroepiandrosterone sulfate and cardiovascular disease risk in older men and women. J Clin Endocrinol Metab. 2020;105:e4304–4327. doi: 10.1210/clinem/dgaa518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prall SP, Muehlenbein MP. DHEA modulates immune function: a review of evidence. Vitam Horm. 2018;108:125–144. doi: 10.1016/bs.vh.2018.01.023. [DOI] [PubMed] [Google Scholar]
- 11.Cao J, Lu M, Yan W, Li L, Ma H. Dehydroepiandrosterone alleviates intestinal inflammatory damage via GPR30-mediated Nrf2 activation and NLRP3 inflammasome inhibition in colitis mice. Free Radic Biol Med. 2021;172:386–402. doi: 10.1016/j.freeradbiomed.2021.06.025. [DOI] [PubMed] [Google Scholar]
- 12.Mitchell LE, Sprecher DL, Borecki IB, Rice T, Laskarzewski PM, Rao DC. Evidence for an association between dehydroepiandrosterone sulfate and nonfatal, premature myocardial infarction in males. Circulation. 1994;89:89–93. doi: 10.1161/01.cir.89.1.89. [DOI] [PubMed] [Google Scholar]
- 13.Williams MR, Ling S, Dawood T, Hashimura K, Dai A, Li H, Liu JP, Funder JW, Sudhir K, Komesaroff PA. Dehydroepiandrosterone inhibits human vascular smooth muscle cell proliferation independent of ARs and ERs. J Clin Endocrinol Metab. 2002;87:176–181. doi: 10.1210/jcem.87.1.8161. [DOI] [PubMed] [Google Scholar]
- 14.Altman R, Motton DD, Kota RS, Rutledge JC. Inhibition of vascular inflammation by dehydroepiandrosterone sulfate in human aortic endothelial cells: roles of PPARalpha and NF-kappaB. Vascul Pharmacol. 2008;48:76–84. doi: 10.1016/j.vph.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bonnet S, Paulin R, Sutendra G, Dromparis P, Roy M, Watson KO, Nagendran J, Haromy A, Dyck JR, Michelakis ED. Dehydroepiandrosterone reverses systemic vascular remodeling through the inhibition of the Akt/GSK3-β/NFAT axis. Circulation. 2009;120:1231–1240. doi: 10.1161/CIRCULATIONAHA.109.848911. [DOI] [PubMed] [Google Scholar]
- 16.Ii M, Hoshiga M, Negoro N, Fukui R, Nakakoji T, Kohbayashi E, Shibata N, Furutama D, Ishihara T, Hanafusa T, Losordo DW, Ohsawa N. Adrenal androgen dehydroepiandrosterone sulfate inhibits vascular remodeling following arterial injury. Atherosclerosis. 2009;206:77–85. doi: 10.1016/j.atherosclerosis.2009.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kumar A, Lindner V. Remodeling with neointima formation in the mouse carotid artery after cessation of blood flow. Arterioscler Thromb Vasc Biol. 1997;17:2238–2244. doi: 10.1161/01.atv.17.10.2238. [DOI] [PubMed] [Google Scholar]
- 18.Straszewski-Chavez SL, Visintin IP, Karassina N, Los G, Liston P, Halaban R, Fadiel A, Mor G. XAF1 mediates tumor necrosis factor-alpha-induced apoptosis and X-linked inhibitor of apoptosis cleavage by acting through the mitochondrial pathway. J Biol Chem. 2007;282:13059–13072. doi: 10.1074/jbc.M609038200. [DOI] [PubMed] [Google Scholar]
- 19.Wu W, Zhang W, Choi M, Zhao J, Gao P, Xue M, Singer HA, Jourd’heuil D, Long X. Vascular smooth muscle-MAPK14 is required for neointimal hyperplasia by suppressing VSMC differentiation and inducing proliferation and inflammation. Redox Biol. 2019;22:101137. doi: 10.1016/j.redox.2019.101137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 21.Rehmsmeier M, Steffen P, Hochsmann M, Giegerich R. Fast and effective prediction of microRNA/target duplexes. RNA. 2004;10:1507–1517. doi: 10.1261/rna.5248604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shi J, Yang Y, Cheng A, Xu G, He F. Metabolism of vascular smooth muscle cells in vascular diseases. Am J Physiol Heart Circ Physiol. 2020;319:H613–H631. doi: 10.1152/ajpheart.00220.2020. [DOI] [PubMed] [Google Scholar]
- 23.Orentreich N, Brind JL, Rizer RL, Vogelman JH. Age changes and sex differences in serum dehydroepiandrosterone sulfate concentrations throughout adulthood. J Clin Endocrinol Metab. 1984;59:551–555. doi: 10.1210/jcem-59-3-551. [DOI] [PubMed] [Google Scholar]
- 24.Teixeira CJ, Veras K, de Oliveira Carvalho CR. Dehydroepiandrosterone on metabolism and the cardiovascular system in the postmenopausal period. J Mol Med (Berl) 2020;98:39–57. doi: 10.1007/s00109-019-01842-5. [DOI] [PubMed] [Google Scholar]
- 25.Wu TT, Chen Y, Zhou Y, Adi D, Zheng YY, Liu F, Ma YT, Xie X. Prognostic value of dehydroepiandrosterone sulfate for patients with cardiovascular disease: a systematic review and meta-analysis. J Am Heart Assoc. 2017;6:e004896. doi: 10.1161/JAHA.116.004896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mannic T, Viguie J, Rossier MF. In vivo and in vitro evidences of dehydroepiandrosterone protective role on the cardiovascular system. Int J Endocrinol Metab. 2015;13:e24660. doi: 10.5812/ijem.24660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Urata Y, Goto S, Kawakatsu M, Yodoi J, Eto M, Akishita M, Kondo T. DHEA attenuates PDGF-induced phenotypic proliferation of vascular smooth muscle A7r5 cells through redox regulation. Biochem Biophys Res Commun. 2010;396:489–494. doi: 10.1016/j.bbrc.2010.04.125. [DOI] [PubMed] [Google Scholar]
- 28.Paulin R, Meloche J, Jacob MH, Bisserier M, Courboulin A, Bonnet S. Dehydroepiandrosterone inhibits the Src/STAT3 constitutive activation in pulmonary arterial hypertension. Am J Physiol Heart Circ Physiol. 2011;301:H1798–1809. doi: 10.1152/ajpheart.00654.2011. [DOI] [PubMed] [Google Scholar]
- 29.Grebe A, Hoss F, Latz E. NLRP3 inflammasome and the IL-1 pathway in atherosclerosis. Circ Res. 2018;122:1722–1740. doi: 10.1161/CIRCRESAHA.118.311362. [DOI] [PubMed] [Google Scholar]
- 30.Zheng F, Xing S, Gong Z, Mu W, Xing Q. Silence of NLRP3 suppresses atherosclerosis and stabilizes plaques in apolipoprotein E-deficient mice. Mediators Inflamm. 2014;2014:507208. doi: 10.1155/2014/507208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun HJ, Ren XS, Xiong XQ, Chen YZ, Zhao MX, Wang JJ, Zhou YB, Han Y, Chen Q, Li YH, Kang YM, Zhu GQ. NLRP3 inflammasome activation contributes to VSMC phenotypic transformation and proliferation in hypertension. Cell Death Dis. 2017;8:e3074. doi: 10.1038/cddis.2017.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Janoudi A, Shamoun FE, Kalavakunta JK, Abela GS. Cholesterol crystal induced arterial inflammation and destabilization of atherosclerotic plaque. Eur Heart J. 2016;37:1959–1967. doi: 10.1093/eurheartj/ehv653. [DOI] [PubMed] [Google Scholar]
- 33.van der Heijden T, Kritikou E, Venema W, van Duijn J, van Santbrink PJ, Slütter B, Foks AC, Bot I, Kuiper J. NLRP3 inflammasome inhibition by MCC950 reduces atherosclerotic lesion development in apolipoprotein E-deficient mice-brief report. Arterioscler Thromb Vasc Biol. 2017;37:1457–1461. doi: 10.1161/ATVBAHA.117.309575. [DOI] [PubMed] [Google Scholar]
- 34.Abderrazak A, Couchie D, Mahmood DF, Elhage R, Vindis C, Laffargue M, Matéo V, Büchele B, Ayala MR, El Gaafary M, Syrovets T, Slimane MN, Friguet B, Fulop T, Simmet T, El Hadri K, Rouis M. Anti-inflammatory and antiatherogenic effects of the NLRP3 inflammasome inhibitor arglabin in ApoE2.Ki mice fed a high-fat diet. Circulation. 2015;131:1061–1070. doi: 10.1161/CIRCULATIONAHA.114.013730. [DOI] [PubMed] [Google Scholar]
- 35.Yang Z, Zheng B, Zhang Y, He M, Zhang XH, Ma D, Zhang RN, Wu XL, Wen JK. miR-155-dependent regulation of mammalian sterile 20-like kinase 2 (MST2) coordinates inflammation, oxidative stress and proliferation in vascular smooth muscle cells. Biochim Biophys Acta. 2015;1852:1477–1489. doi: 10.1016/j.bbadis.2015.04.012. [DOI] [PubMed] [Google Scholar]
- 36.Li D, Xie X, Wang J, Bian Y, Li Q, Gao X, Wang C. MiR-486 regulates lactation and targets the PTEN gene in cow mammary glands. PLoS One. 2015;10:e0118284. doi: 10.1371/journal.pone.0118284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peng Y, Dai Y, Hitchcock C, Yang X, Kassis ES, Liu L, Luo Z, Sun HL, Cui R, Wei H, Kim T, Lee TJ, Jeon YJ, Nuovo GJ, Volinia S, He Q, Yu J, Nana-Sinkam P, Croce CM. Insulin growth factor signaling is regulated by microRNA-486, an underexpressed microRNA in lung cancer. Proc Natl Acad Sci U S A. 2013;110:15043–15048. doi: 10.1073/pnas.1307107110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.