

Abstract
Pancreatic ductal adenocarcinoma (PDA) is an aggressive metastatic cancer with a very low survival rate. This tumor is hypovascularized and characterized by severe hypoxic regions, yet these regions are not impeded by the oxidative stress in their microenvironment. PDA’s high resilience raises the need to find new effective therapeutic targets. This study investigated the suitability of methionine aminopeptidase 2 (MetAp2), a metallopeptidase known to play an important role in tumor progression, as a new target for treating PDA. In our examination of patient-derived PDA tissues, we found that MetAp2 is highly expressed in metastatic regions compared with primary sites. At the cellular level, we found that the basal expression levels of MetAp2 in pancreatic cancer cells were higher than its levels in endothelial cells. Pancreatic cancer cells showed a significant suppression of proliferation in a dose-dependent manner upon exposure to TNP-470, a selective MetAp2 inhibitor. In addition, a significant reduction in glutathione (GSH) levels - known for its importance in alleviating oxidative stress - was detected in all treated cells, suggesting a possible anti-cancer activity mechanism that would be feasible for treating highly hypoxic PDA tumors. Furthermore, in an orthotopic pancreatic cancer murine model, systemic oral treatment with a MetAp2 inhibitor significantly reduced tumors’ growth. Taken together, our findings indicate that MetAp2 enhances tumor sensitivity to hypoxia and may provide an effective target for treating hypoxic tumors with high expression levels of MetAp2.
Keywords: PDA, hypoxia, metastasis, MetAp2, GSH
Introduction
Pancreatic ductal adenocarcinoma (PDA) is an aggressive malignant cancer with an overall median survival rate of less than one year after diagnosis [1]. Most patients are in an advanced and metastatic stage by the time of diagnosis, mainly due to the late initiation of clinical symptoms. Among diagnosed patients, only 10-15% are eligible for surgical resection. Still, most patients relapse in spite of adjuvant systemic therapies that, up to date, mainly target primary sites [2]. Despite the massive amount of research, there has been very modest progress in the therapeutic options available for metastatic disease [3].
In addition to the inherent resistance of pancreatic cancer cells, the specific tumor microenvironment (TME) in PDA has a central role in limiting the effect of treatments in pancreatic cancer and in promotes tumor progression to become metastatic [4]. PDA is characterized by an abundant extracellular matrix (ECM) and desmoplastic fibrotic stroma. The ECM’s component accumulation - specifically collagen, hyaluronic acid, etc. [5] - and the rapid proliferation of cancer cells results in a deformed pancreas architecture, giving rise to abnormal blood and lymphatic vessel structures [6,7]. Due to this, PDA is hypovascularized and characterized by severe hypoxic regions [8] which are closely correlated with tumor aggressiveness, progression and poor prognosis [9]. The hypoxic microenvironment and the high adjustability of the tumor cells to these harsh conditions raise the need to identify effective therapeutic targets for such conditions.
One might be methionine aminopeptidases (MetAp). These are metallopeptidases that selectively catalyze the removal of the N-terminal methionine from newly synthesized proteins, constituting an important step in protein maturation and proper function [10]. There are two main MetAp isoforms in eukaryotes, MetAp1 and MetAp2 [11], that are essential for cellular growth and viability [12]. The exact differential physiological roles are still not completely understood [13]. However, the significant growth inhibition observed in cells sensitive to MetAp2 inhibition and the identification of MetAp2 as a specific target of antiangiogenic drugs, such as fumagillin and its analogues, has attracted more attention to MetAp2 than to MetAp1 [14,15]. MetAp2 is overexpressed in many forms of cancer. Several reports indicate that MetAp2 plays an important role in different tumors’ growth. Fumagillin and its derivatives inhibit the proliferation, not only of endothelial cell lines, but also of a subset of cancer cell lines [16]. The higher concentration of this enzyme in tumor cells suggests they have a considerable dependence on it for their functioning and proliferation. Accordingly, targeting this enzyme to inhibit tumor cell growth might be an effective approach to treat cancer [17].
It is well known that the high metabolic demands of tumors are due to their rapid growth. Another possible therapeutic target might be glutathione (GSH), also known as L-γ-glutamyl-L-cysteinylglycine, a common tripeptide that functions as an important intracellular radical scavenger. It protects cells against reactive oxygen species (ROS), as well as against many toxins and drugs [18,19]. GSH is one of the major regulators of cancer progression and its response to therapy [20]. Shifts in GSH metabolism often accompany tumor development, allowing it to alleviate the increased oxidative stress [21]. Since it was found that glutathione redox homeostasis is altered by MetAp2 inhibition [22] together with the findings of enhanced GSH levels in pancreatic carcinoma and its essential role in cell proliferation and resistance [23], we aimed in this work to study the effect of MetAp2 inhibition in PDA, a highly hypoxic tumor. In light of MetAp2’s important role in cancer progression, we hypothesize that MetAp2 is highly expressed in metastatic tumors relative to primary regions.
Collectively, our data suggest that MetAp2 enhances tumor sensitivity to hypoxia and therefore may be a valid therapeutic target for metastatic tumors, thereby fulfilling the unmet clinical need of PDA treatment at late stages of the disease, when it is commonly diagnosed.
Materials and methods
Compliance with ethical standards
The study complies with the Declaration of Helsinki. The study protocol was approved by the Institutional Ethics Committee (0346-12, 04-02-2013). All experiments described in this paper were performed according to the guidelines and regulations of the approved IRB protocols. Human patient-derived pancreatic cancer cells were obtained from pancreatic adenocarcinoma tumor biopsies approved by the Institutional Review Board (IRB)/Ethics (Helsinki) committee of the Hadassah Medical Center (#920051034, and 0628-14-HMO). All subjects gave their informed consent.
Six-week-old Foxn1 nu male mice were purchased from Jackson Laboratory (Bar Harbor, ME, USA). Animal studies were reviewed and approved by the Animal Care and Use Committee of Beth Israel Deaconess Medical Center, Boston, MA (protocol number 070-2016). Mice were housed at a maximum of 5 mice per cage in a pathogen-free facility with unlimited access to sterile water and chow. Daily welfare evaluations and animal sacrifices were carried out according to the Committee guidelines.
Human pancreatic adenocarcinoma immunofluorescence staining
Paraffinized patient-derived tissue sections containing microarrays were purchased from BioConsult LTD. First, the microarrays were deparaffinized by a multi-step method. The tissue-containing slides were baked in a dry 60°C oven for 30 min. A series of 3 xylene incubations (5 min each), followed by two 100% ethanol (10 min), one 96% ethanol (10 min), and 70% ethanol (5 min) incubations were performed. Slides were washed while shaking for 10 min with DDW and for another 10 min with 0.1% tween 20 (diluted in PBS). Heat-induced epitope retrieval (HIER) was performed by heating 20 min in a TRIS/EDTA buffer solution (1.21 g Tris base, 0.37 g EDTA, 0.5 ml tween 20 in 100 ml DDW, diluted 1:10 with DDW before use). Tissue section blocking was performed by incubation for 30 minutes in 3% normal goat serum (VE-S-1000, Vector Labs) and 3% normal rabbit serum (VE-S-5000, Vector Labs). Blocking solution was removed and first Antibody diluted in 3% normal goat or rabbit serum was placed on the slides for overnight incubation in 4°C. The Antibodies were diluted as follows: anti-fibroblasts (BCL271, Merck) -1:25, anti-CD31 (ab28364, Abcam) -1:50, anti-collagen-IV (ab6586, Abcam) -1:500, anti-hyaluronic acid (ab53842, Abcam) -1:250 and anti-MetAp2 (Ab134124, Abcam, Cambridge, UK) -1:50. After incubation, the slides were washed three times with tween diluted in PBS and incubated for 2 h with a secondary Antibodies diluted in 3% goat serum or in rabbit serum. All secondary Antibodies were diluted in a ratio of 1:200. The slides were washed three times with tween diluted in PBS and then incubated with DAPI (1:1,000) for nuclei staining. After three washes with PBS, the mounting media was applied on the slides and samples were visualized using a fluorescent microscope (Olympus IX-73). Tissue staining analysis was performed using ImageJ analysis software. The fluorescently dyed tissue intensities were evaluated for the different markers by measuring the pixels stained per area, followed by normalizing the number of cells represented by blue DAPI nuclei staining. The markers included: fibroblasts, CD31, collagen IV and hyaluronic acid. The data was divided into 4 main groups: normal tissue, normal tissue adjacent to the tumor (NAT), primary tissue (stages I-IV) and metastatic tissues. n=7-50.
Cell culture
Human pancreatic cancer cell lines BxPC-3, PANC-1 and AsPC-1 (ATCC, Manassas, Virginia, USA), HUVECs (Lonza, Walkersville, MD, USA) and PancOH7 (obtained from Dr. Dipak Panigrahy’s lab [24]) were characterized and mycoplasma free (EZ-PCR mycoplasma test kit [Biological Industries, Beit HaEmek, Israel]) before use. All cells were kept in a humidified incubator at 37°C with 5% CO2. PANC-1 and PancOH7 were maintained in DMEM (Life Technologies, Carlsbad, California, USA) and supplemented with 10% FCS and penicillin/streptomycin (P/S). BxPC-3 and AsPC-1 cells were maintained in RPMI-1640 (Life Technologies) and supplemented with 10% FCS and P/S. HUVECs were cultured in a medium supplemented with the PeproGrow-MacroV kit (ENDO-BM & GS-MacroV, PeproTech) and P/S. PC cells (patient-derived pancreatic cancer cells) were isolated from pancreatic cancer tissue by digesting the tissue for 30 min at 37°C with 5% CO2 in DMEM/F12 medium (Life Technologies) and supplemented with P/S and 0.14 Wunsch units/mL of LiberaseTM Research Grade (Roche Diagnostics, Basel, Switzerland). A stop reaction medium (DMEM/F12 supplemented with 15% FCS and P/S) was used to stop the digestion, after which the digested tissue was filtered through a cell strainer. Epithelial cells were isolated using the sedimentation technique (as previously described [25]) and maintained in DMEM/F12 medium supplemented with 10% FCS, 1% Glutamine, EGF, Insulin, HEPES and P/S.
Western blot
A RIPA buffer in a protease inhibitor cocktail (Sigma Aldrich, St. Louis City, Missouri, USA, S8820) was used for 30 min on ice in order to cause cell lysis. Cell lysates were then centrifuged, and the supernatant was collected. BCA Protein Assay kit (PierceTM, Thermo Fisher Scientific, Cambridge, Massachusetts, USA) was used to determine the protein content. A 12.5% Tris-glycine SDS-PAGE was used to separate the proteins (15 μg protein) that were next transferred onto a polyvinylidene difluoride membrane (Millipore Corporation, Billerica, MA, USA). Membranes were blocked for 2 h and then incubated with anti-MetAp2 antibodies or anti-MetAp1 antibodies, Ab134124 or Ab185540 (Abcam, Cambridge, UK), respectively, overnight at 4°C in TBST containing 5% BSA. Next, the membranes were washed three times in TBST and incubated with a 1:5,000 dilution of goat anti-rabbit secondary antibody conjugated to horseradish peroxidase for 1 h (Ab97080, Abcam). β-actin or cofilin, Ab49900 or Ab124979 (Abcam), respectively, were used as the loading control. Original Western Blot images are shown in Figure S3.
Activity assay
BxPC-3, PANC-1, AsPC-1, PancOH7 and PC cells were counted and then centrifuged and resuspended in an appropriate volume of cold RIPA buffer containing a protease inhibitor cocktail to obtain an equivalent cell number in the cell homogenates. The Eppendorf containing the cells was placed in ice and a probe sonicator (Sonic Ruptor 400, OMNI International) was used to disrupt them. Centrifugation at 15,000 RPM for 10 min at 4°C was used to remove insoluble cellular components. Next, the Bradford protein assay was conducted (using BSA as the standard) [26] in order to determine the protein content of the supernatant. 5 μg of protein and the substrate L-Met-AMC (Sancta Cruz Biotechnology) in an assay buffer (containing 50 mM HEPES, 0.1 mM CoCl2, 100 mM NaCl and 1 mg/mL PEG 6,000, in a final volume of 100 μL) were used to test the enzymatic activity of MetAp. Fluorescence was measured every minute and 30 sec or every minute and 45 sec for MetAp1 or MetAp2, respectively, for ~40 min at 25°C, using a plate reader (Synergy HT Multi-Mode Microplate Reader, BioTek). n=3.
Activity assay following MetAp2 inhibition
BxPC-3, PancOH7, AsPC-1 and PANC-1 cells were cultured in 10×10 cm petri dishes, left to grow to 80-90% confluency, washed with cold PBS and scraped on ice using a protease inhibitor-free RIPA buffer. A total of three dishes were scraped from each cell line using the same buffer, which was transferred from one plate to the other. Centrifugation at 15,000 RPM for 10 min at 4°C was done to remove insoluble cellular components. The protein content of the supernatant was determined as previously mentioned. The enzymatic assay was performed using 5 µg of protein per sample, as described previously. The samples were incubated with TNP-470 (O-Chloroacetylcarbamoy fumagillol [MedChem Partners, Lexington, MA, USA]) for 15 min at RT before adding the substrate. An increase of fluorescence (due to substrate degradation during the enzymatic assay) was measured every 20 sec for 1 h at 25°C using a plate reader, as previously mentioned. n=3.
Viability assay
BxPC-3, PancOH7, AsPC-1 and PANC-1 cells were seeded in 96-well plates (2,000 cells/well). After 24 h, the cells were exposed to a range of TNP-470 concentrations (0-5 μM) for either 24 (BxPC-3 and PancOH7 cells) or 48 h (AsPC-1 and PANC-1 cells) to detect viability. MTT (Sigma Aldrich, St. Louis City, Missouri, USA) was added (0.5 mg/mL) into each well and incubated at 37°C and 5% CO2 for 3 h. Their absorbance was measured at 570 nm, using a plate reader (Wallac 1420 VICTOR plate reader, Perkin-Elmer Life Sciences, USA). n=5.
Quantification of oxidized and reduced glutathione
The Quantification kit for oxidized and reduced glutathione was purchased from Sigma-Aldrich (item no. 38185, Milan, Italy). Quantification of glutathione was determined in AsPC-1, BxPC-3, PancOH7 and PC cells, with and without treatment with TNP-470 10 μM, according to Akerboom and Sies’s assay [27], following the manufacturer’s guidelines. Cells were harvested by centrifugation. The different forms of glutathione were extracted in a buffer containing sulfosalicylic acid. Glutathione reductase’s enzymatic reaction was used for quantification. Oxidized glutathione was specifically quantified by treating the cell lysate with 2-vinylpyridine. Fluorescence was measured every 2 min and 40 sec for ~35 min at 25°C using a plate reader (Synergy HT Multi-Mode Microplate Reader, BioTek). n=3.
Reactive oxygen species detection assay
The reactive oxygen species (ROS) detection cell-based assay kit was purchased from Cayman Chemical (item no. 601520, Michigan, USA). 2,7-Dichlorofluoroscin Diacetate (DCFDA) fluorescent probe was used for the detection of ROS generation in PancOH7 and AsPC-1 cells according to the manufacturer’s instructions. ROS generation was determined in cells with and without TNP-470 1 μM treatment and with and without additional rescue treatment with N-acetyl-L-cysteine 5 mM (NAC) purchased from Sigma-Aldrich. The pancreatic cancer cells were seeded 5,000 cells/well in 96-well plates incubated at 37°C and 5% CO2 at either 10% O2 (normoxia) or 1% O2 (hypoxia chamber) for 72 h post addition of treatments. After 72 h, DCFDA was added (10 μM final concentration) into each well and incubated at 37°C and 5% CO2 for 1 h. Cell fluorescence intensity was measured using a 485 nm excitation and a 535 nm emission filter using a plate reader (Spark 10M multimode microplate reader, Tecan, Switzerland). n=4.
Proliferation assay
PancOH7 and AsPC-1 cells were seeded in 96-well plates (5,000 cells/well). After 24 h, the cells were exposed to the same conditions as mentioned previously in the ROS detection assay. WST1 (Cayman Chemical) was added into each well and incubated at 37°C and 5% CO2 for 3 h. Their absorbance was measured at 450 nm using Wallac 1420 VICTOR plate reader. n=4.
Murine orthotopic tumors
Pancreatic orthotopic tumors were developed in Foxn1 nude mice by injecting 1×105 PancOH7 cells directly into their pancreases with a 30-G needle. Treatment was initiated on the day of tumor cell injection. Oral TNP-470 (mPEG-PLA-TNP-470) was prepared as previously mentioned [28] and was given 30 mg/kg equivalent q.o.d. Control mice received empty mPEG-PLA carriers. Mice were weighed and observed daily throughout the experiment. On day 28, the mice were sacrificed, and the pancreatic tumors, ascites, livers and spleens were surgically removed and weighed. n=5-7 mice/group.
Data analysis and statistics
Studies carried out on two groups were analyzed using the unpaired two-tailed Student’s t-test. Studies containing more than three groups were analyzed using a one-way analysis of variance (ANOVA), and significant differences were determined using Tuckey’s multiple comparison post-test. Differences were considered statistically significant for P<0.05. Results are presented as the mean ± SEM. Statistical data was analyzed on GraphPad Prism 8 (www.graphpad.com, San Diego, California, USA) and all experiments had at least 2-3 independent replicates unless otherwise specified.
Results
Expression of Metap2 and CD31 in human pancreatic cancer patient samples
Human pancreatic tumor tissues were obtained from patients in different stages of the disease for immunohistology examination and expression of MetAp2 as well as detection of endothelium (indicated by CD31 expression). Immunofluorescent staining of the tissue sections of normal tissue, normal adjacent to the tumor (NAT), stage I-IV tissues (referred to as “primary tissue”) and metastatic tissue showed both MetAp2 and CD31 expression (Figure 1A).
Figure 1.

Tissue samples obtained from human pancreatic-cancer patients expressed high levels of MetAp2 and CD31. A. Immunofluorescent staining of MetAp2 (green), CD31 (red) and DAPI (Blue) revealed. B, C. Higher expression of MetAp2 and CD31 in metastatic cancer organs versus the primary sites. Samples were imaged using a fluorescent microscope (Olympus IX-73). Analysis of tissue staining was performed by using the ImageJ analysis program. The fluorescently dyed tissue staining intensities were assessed for the different markers by an evaluation of pixels stained per area, followed by normalizing the number of cells represented by blue DAPI nuclei staining. The data was divided into 4 main groups: normal tissue, normal tissue adjacent to the tumor (NAT), primary tissue (stages I-IV) and metastatic tissue. n=7-50. *P<0.05 and ***P<0.001, compared with normal tissues. Results are presented as mean ± SEM. Scale bar =100 μm.
Tissue staining analysis was performed using ImageJ analysis software. The fluorescently dyed tissue intensities were evaluated for the different markers by measuring the pixels stained per area, followed by normalization for the number of cells represented by blue DAPI nuclei staining (explained in more detail in the Materials and Methods section). MetAp2 expression levels were ~25% higher in pancreatic cancer tissues obtained from patients with stage I and II of the disease compared with normal tissues and NAT (Figure S2). Importantly, MetAp2 levels were significantly higher in tissues resected from patients with stage III and IV of the disease, with over 50% more expression of MetAp2 compared with normal tissues. Furthermore, metastatic tissue sites expressed significantly higher levels of MetAp2 compared with primary sites in patients with cancer stages I or II and III or IV, displaying >70% and >40% higher levels, respectively. Collectively, metastatic tissues expressed >60% higher levels of MetAp2 compared with primary tissue sites (Figure 1B). In correlation with many studies [29-31], a notable enhancement in CD31 expression (indicating higher angiogenesis) was observed in metastatic tissues and late stage tumors compared with normal tissues (Figure 1C).
MetAp1 and MetAp2 expression and activity in HUVECs, pancreatic cancer cell lines and patient-derived pancreatic cancer cells
Immunoblotting was used to measure MetAp1 and MetAp2 protein expression at the cell-level of endothelial cells, pancreatic cancer cell lines and patient-derived pancreatic cancer cells (PC cells). Since the exact physiological roles of MetAp1 and MetAp2 are not completely understood and they can compensate for one another [13], the basal cellular protein levels (baseline expression in the activated state of cells grown in optimal growth conditions) of MetAp1 and MetAp2 were measured (Figure 2).
Figure 2.
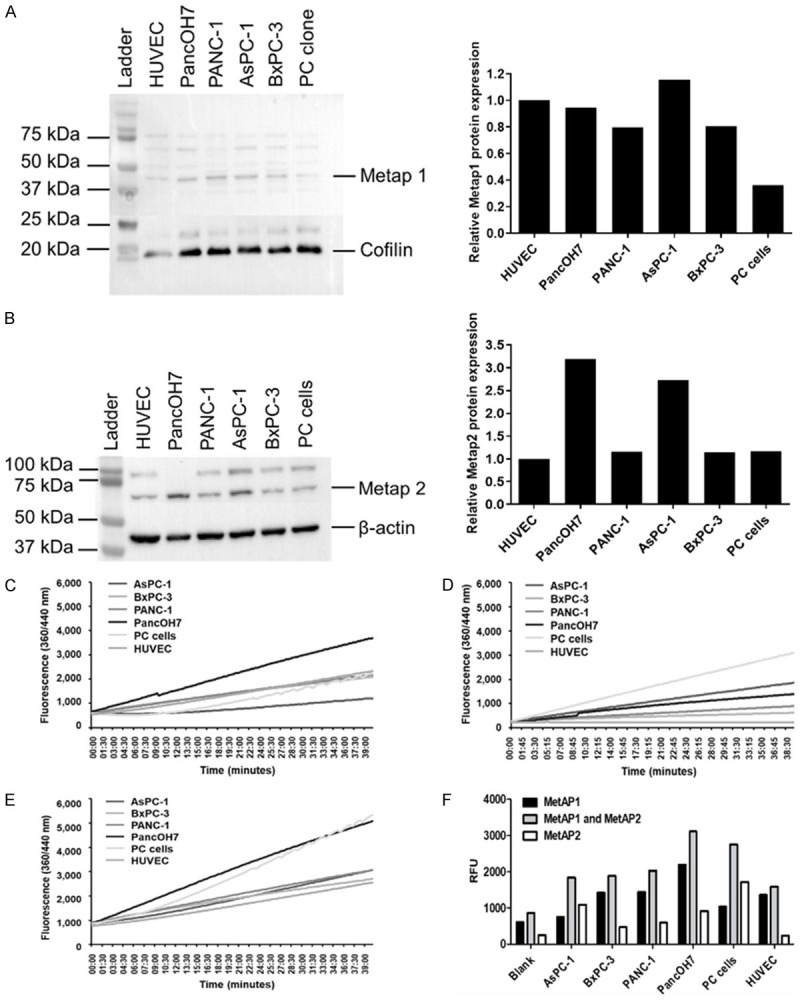
Basal MetAp1 and MetAp2 activity and expression in pancreatic-cancer cell lines, patient-derived pancreatic cancer cells and endothelial cells. (A, B) Western blot analyses for determining the expression of (A) MetAp1 and (B) MetAp2 in all cell lines. MetAp2 enzyme levels in pancreatic-cancer cell lines and patient-derived pancreatic cancer cells are higher than the levels in HUVECs. n=3. (C) Measurement of the basal activity of MetAp1, (D) MetAp2 and (E). MetAp1 and MetAp2 in HUVECs, PancOH7, PANC-1, AsPC-1, BxPC-3 and PC cells. The reaction was monitored for ~40 min by fluorescent quantification of the cleaved substrate. (F) Measurement of MetAp1’s and MetAp2’s activity after 20 min. PancOH7 cells showed the highest activity of MetAp1 and MetAp2 compared with the other cells. Results are representative and were normalized to the total cell number. n=2.
Considering that MetAp1 and MetAp2 are ubiquitously expressed in endothelial cells [32], all results were compared to human umbilical vein endothelial cells’ (HUVECs) expression and activity levels of MetAp1 and MetAp1. All cells showed comparable levels of MetAp1 and MetAp2, while AsPC-1 displayed higher levels as opposed to PC cells, which showed lower MetAp1 levels. In addition, PancOH7 and AsPC-1 cells exhibited higher levels of MetAp2 compared with the other cells (Figure 2A, 2B). Since enzyme activity does not always correspond to the actual protein levels, the basal activity of MetAp1 and MetAp2 was measured separately and jointly (Figure 2C-E). In Figure 2F, it can be seen that MetAp1 and MetAp2 activity at 20 min is highest in PancOH7 and PC cells, while the other cells display lower and comparable enzyme activity.
Effect of MetAp2’s inhibition on its activity and proliferation of pancreatic cancer cell lines and in vivo effects of MetAp2’s inhibition
In order to inhibit MetAp2’s basal activity, we used a specific fumagillin analogue, TNP-470, that functions as a specific MetAp2 antagonist [33]. MetAp2’s basal activity in PancOH7 cells was measured for 1 h after treatment with 0-5 μM TNP-470 (Figure 3A). Five μM of TNP-470 reduced MetAp2 activity in PancOh7 cells by >35% over the course of 1 h.
Figure 3.
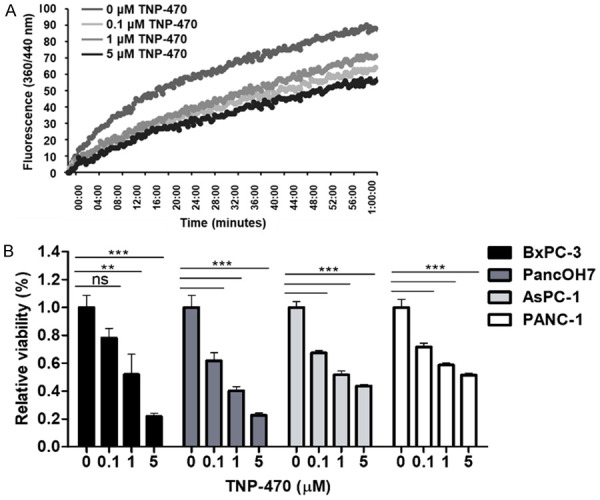
Biochemical inhibition of MetAp2 activity and cell viability using TNP-470. A. MetAp2 enzymatic activity in PancOH7 cells after TNP-470 0-5 µM treatment. The enzymatic activity was tested with 5 μg of protein, using L-Met-AMC as a substrate. The addition of 5 μM TNP-470 treatment inhibits MetAp2’s enzymatic activity by >35% over the course of 1 h. n=2. B. TNP-470 was added to pancreatic-cancer cell lines and MTT was performed after 24 h of treatment for the BxPC-3 and PancOH7 cells, and after 48 h of treatment for AsPC-1 and PANC-1 cells. A dose-dependent effect was observed, where higher concentrations of the MetAp2 inhibitor induced lower viability. The absorbance was measured at 570 nm using a plate reader (Wallac 1420 VICTOR plate-reader, Perkin-Elmer Life Sciences, USA). n=5. ns, not significant, **p<0.01, ***p<0.001 compared with non-treated control cells. Results are presented as mean ± SEM.
Since tumor progression involves several cellular activities, such as cell proliferation, we conducted a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay to assess the effect of MetAp2 inhibition on pancreatic cancer cell proliferation (Figure 3B). BxPC-3, PancOH7, AsPC-1 and PANC-1 cells were treated with 0, 0.1, 1 and 5 μM of TNP-470 and showed a significant dose-dependent reduction in proliferation following a 24-48 h incubation period. A reduction of ~40%, 65% and 88% in PancOH7 cell proliferation was observed with 0.1, 1 and 5 μM TNP-470 treatment, respectively.
We next studied the inhibition effects of MetAp2 on tumor growth in vivo. We induced orthotopic pancreatic cancer in Foxn1 nude mice by injecting PancOH7 cells directly into their pancreases. Oral TNP-470 (mPEG-PLA-TNP-470) was prepared as previously mentioned [28] and was administered 30 mg/kg equivalent q.o.d. Pancreatic tumors treated with oral TNP-470 showed a significant reduction in their weight of ~37% compared with those of the non-treated mice that received empty mPEG-PLA carriers (Figure S4A, S4B). In addition, treated mice showed lower ascites volume and spleen weight compared with control mice, while both groups presented similar liver weights, indicating the nontoxic effect (Figure S4C).
Effect of MetAp2’s inhibited activity on glutathione and ROS levels
Since PDA is hypovascularized and characterized by severe hypoxic regions [8], combined with the findings of elevated levels of GSH in pancreatic carcinoma and its important role in cell proliferation [23], we next studied MetAp2’s inhibition effects on GSH levels in AsPC-1, BxPC-3, PancOH7 and PC cells. Interestingly, GSH levels were significantly reduced while GSSG levels were markedly elevated in all the pancreatic cancer cells tested (Figure 4). The glutathione ratio (GSH/GSSG) in cells treated with 10 μM TNP-470 was 57%, 63%, 84% and 97% lower than control non-treated PancOH7, PC, BxPC-3 and AsPC-1 cells, respectively (Figure 4B). In order to further evaluate the effect of MetAp2 inhibition on the redox state of cells, ROS levels in PancOH7 and AsPC-1 cells were determined. The addition of 1 μM TNP-470 induced a 10% increase in ROS in PancOH7 cells, while in AsPC-1 cells the increase in ROS was significantly higher (over 40%) compared with non-treated control cells (Figure S5A). This effect correlated with the reduction of >30% in cell proliferation (Figure S5B). However, ROS levels did not increase significantly compared to control non-treated cells when the cells were grown in hypoxic conditions, though the addition of NAC treatment (serving as an antioxidant) reduced the oxidative stress in AsPC-1 and PancOH7 cells by ~30% and ~50%, respectively. Interestingly, the addition of NAC to AsPC-1 cells grown in hypoxic conditions significantly increased their proliferation by >55% and >45%, with and without TNP-470 treatment, respectively, compared to control non-treated cells, while the proliferation of PancOH7 cells was not significantly affected (Figure S5).
Figure 4.
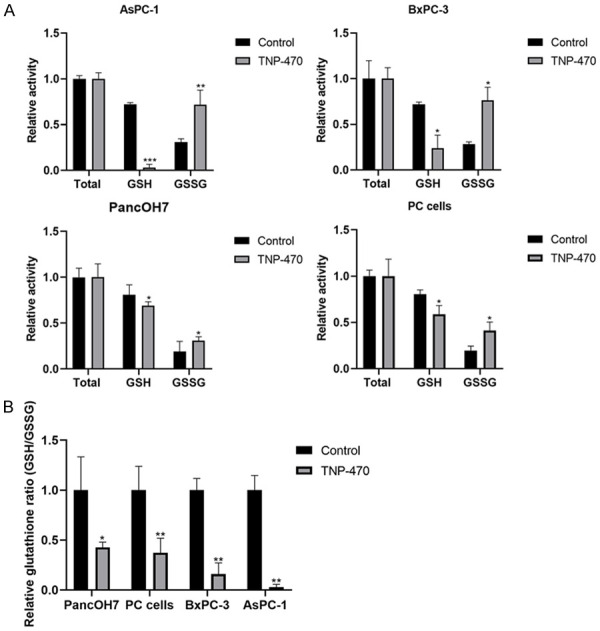
Inhibition of MetAp2 activity influences glutathione levels. Quantification of glutathione was determined in AsPC-1, BxPC-3, PancOH7 and PC cells, with and without TNP-470 10 μM treatment. A. Results of total glutathione, GSH and GSSG activity are displayed after 17 min. B. GSH/GSSG ratio in AsPC-1, BxPC-3, PancOH7 and PC cells, with and without TNP-470 10 μM treatment. Inhibition of MetAp2 activity significantly reduces GSH levels and enhances GSSG levels. n=3. *P<0.1, **P<0.01, ***P<0.001 compared with non-treated control cells. Results are presented as mean ± SEM.
Discussion
PDA is the fourth leading cause of cancer-related deaths worldwide [34]. In the US, the number of PDA cases is expected to more than double in the near future [35]. The aggressive nature of this cancer is due to its pronounced ability to progress and metastasize [36]. Approximately 80% of all diagnosed patients are already present with locally advanced or metastatic disease [37] and 75% of all patients die of metastatic disease within 5 years of intended curative surgery, showing no evidence of metastasis upon resection [38]. To date, chemotherapy regimens are based on 5-fluorouracil or gemcitabine and they only prolong survival time by months; that is to say, the increase in the overall survival rate remains poor [39].
This emphasizes the critical unmet clinical need to develop novel approaches that address not just the primary PDA tumor, but that mainly target the metastatic PDA cells, possibly leading to higher survival rates. Most research has focused on MetAp2’s role in angiogenesis, and in our recently published paper, we found its involvement in lymphangiogenesis [40]. However, its role in metastatic cell biology has not been sufficiently explored.
In this study, we demonstrate the high expression levels of MetAp2 in PDA, thus providing a possible target for the metastatic disease. Histological samples from human pancreatic cancer patients showed a significantly higher expression of MetAp2 in metastatic samples (>60%) compared with primary tissue sites (Figures 1B and S2), supporting our hypothesis that it is a valid target in metastasis. It is well known that MetAp2 plays an important role in the proliferation of endothelial cells [41] - as can be indicated by CD31’s high expression levels and co-localization with MetAp2 in primary and metastatic PDA in comparison to normal tissues (Figure 1) - however, its levels in pancreatic cancer cells are yet to be determined. We found that MetAp2 was highly expressed in patient-derived cancer cells and pancreatic cancer cell lines compared with HUVECs (Figure 2). Interestingly, patient-derived cancer cells displayed the highest MetAp2 activity. This may be explained by the increase in ratio of oxidized to reduced MetAp2 in stressed tumor cells due to the enhanced production of ROS, causing a shift in the substrate specificity of MetAp2. Another work showed that MetAp2 activity is controlled by the redox state of the allosteric disulfide bond [42]. Moreover, MetAp2 activity was found to be susceptible to biochemical inhibition by TNP-470 in a dose dependent manner in PancOh7 cells (Figure 3A).
Next, we aimed to investigate MetAp2’s effect on pancreatic cancer cell functionality. We found that the pancreatic cancer cells’ proliferation was significantly impaired in a dose-dependent manner when MetAp2 was inhibited. Even with the lowest concentration used, 0.1 μM, a significant reduction in proliferation was observed in all cell lines (22-40%). These findings indicate the potential of MetAp2 as a target in impeding PDA growth and progression.
A hallmark of PDA is its desmoplastic nature [6], characterized in patient-derived tissue specimens by high fibrotic tissue surrounded by an altered ECM (Figure S1), leading, when combined with rapid abnormal vessel formation [7], to limited oxygen availability and severe hypoxic regions [8]. Elevated GSH levels were found in various human cancer tissues, such as breast, colon and lungs [43-45]. A different work found that pancreatic cancer cells displayed high levels of GSH and it plays an important role in cell proliferation [23]. Interestingly, we found that MetAp2 inhibition led to a significant change in glutathione ratio (GSH/GSSG) levels in patient-derived and pancreatic cancer cell lines, displaying a significant reduction in GSH levels and increased GSSG levels compared with non-treated cells. MetAp2 in endothelial cells is known to regulate their proliferation through cell cycle arrest in the late G1 phase [46]. However, MetAp2 is also known to be involved in the stabilization of eIF-2α in its phosphorylated state, thus having an important impact on protein synthesis and cell growth [47]. A different work revealed the essential role of eIF2α signaling in promoting the survival of resistant hypoxic cells by inducing GSH synthesis and protection against ROS produced in hypoxic environments [48]. It should be noted that AsPC-1 and BxPC-3 cells showed a higher change in their glutathione ratio compared with PacnOH7 and PC cells, which may be explained by the existence of alternative oxygen stress mediators in the latter cells. Interestingly, AsPC-1 cells showed a higher significant increase in their ROS levels than the rise observed in PancOH7 cells when treated with the MetAp2 inhibitor, and accordingly the addition of NAC as an antioxidant rescue treatment significantly increased the proliferation of AsPC-1 cells cultured in hypoxic conditions in which ROS levels are elevated [49]. This, combined with in-depth in vivo chemical analysis would be interesting avenues to explore in the future. Our results, together with other work - demonstrates that glutathione homeostasis is altered by MetAp2 inhibition - [22] suggest MetAp2 may be an effective target for treating the severely hypoxic PDA.
To determine whether our in vitro findings were also expressed in vivo, we utilized an orthotopic cancer animal model to investigate the effect of MetAp2 inhibition on pancreatic tumor growth. To develop local tumors, PancOH7 cells were injected directly into the pancreases of Foxn1 nude mice. Treatment with an oral MetAp2 inhibitor led to a significant reduction in the pancreatic tumors’ weights (37%), compared with non-treated mice (Figure S4). In addition, treated mice displayed lower ascites volume compared with non-treated mice, in keeping with our previous findings of reduced ascites volume in mice treated with oral TNP-470 [28]. This decrease in volume may be a consequence of reduced edema as a result of the anti-vascular leakage activity of the MetAp2 inhibitor. This can also explain the reduction in spleen weight in the treated group compared with the control group, implying a decrease in inflammation [50]. Since the liver is a major detoxification and metabolic organ, changes in its weight may be indicative of a toxic effect. Therefore, the previous results, combined with the comparable liver weights in both mice groups, suggest the nontoxic and effective treatment of MetAp2 inhibition. It would be interesting to further test the exact in vivo mechanism of MetAp2 inhibition in metastatic tumor models to validate these results.
In our previous study, we showed the preventative effect of an orally available form of TNP-470 in melanoma liver metastases, mainly attributed to its anti-angiogenic activity [28]. In our recently published work, we suggested the involvement of MetAp2 in lymphangiogenesis and the potential of preventing a lymphatic pro-metastatic process upon inhibition of MetAp2 [40]. Our collective work, combined with our recent findings of elevated expression of MetAp2 in PDA metastatic sites and similar findings in human colorectal carcinoma tissues, with a specific increase of MetAp2 in the invasive component that suggests its association with metastatic tumor progression [51], together advocate MetAp2 as a potentially effective target for treating not just PDA, but also other metastatically progressed solid tumors.
This study is the first to show the high expression levels of MetAp2 in PDA metastatic regions, possibly providing a new target for treating this highly devastating disease. Inhibition of MetAp2 significantly reduced proliferation at the cell level and also in vivo, in an orthotopic pancreatic tumor model. Additionally, GSH levels were markedly reduced in pancreatic cancer cells treated with a MetAp2 inhibitor, suggesting a possible mechanism of anti-cancer activity and its suitability for treating highly hypoxic PDA tumors. These results offer a new treatment target for the deadly PDA disease and possibly other tumors with high expression levels of MetAp2.
Acknowledgements
This study was supported by the Israel Cancer Association (ICA) (No. 0394691), the Israel Foundation of Science (ISF) (No. 0394883) and the Israel Ministry of Science and Technology (MOST) (grant agreement #0394906). The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Hidalgo M. Pancreatic cancer. N Engl J Med. 2010;362:1605–1617. doi: 10.1056/NEJMra0901557. [DOI] [PubMed] [Google Scholar]
- 2.Klaiber U, Hackert T, Neoptolemos JP. Adjuvant treatment for pancreatic cancer. Transl Gastroenterol Hepatol. 2019;4:27. doi: 10.21037/tgh.2019.04.04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Spadi R, Brusa F, Ponzetti A, Chiappino I, Birocco N, Ciuffreda L, Satolli MA. Current therapeutic strategies for advanced pancreatic cancer: a review for clinicians. World J Clin Oncol. 2016;7:27–43. doi: 10.5306/wjco.v7.i1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA. The pancreas cancer microenvironment. Clin Cancer Res. 2012;18:4266–4276. doi: 10.1158/1078-0432.CCR-11-3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hosein AN, Brekken RA, Maitra A. Pancreatic cancer stroma: an update on therapeutic targeting strategies. Nat Rev Gastroenterol Hepatol. 2020;17:487–505. doi: 10.1038/s41575-020-0300-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mahadevan D, Von Hoff DD. Tumor-stroma interactions in pancreatic ductal adenocarcinoma. Mol Cancer Ther. 2007;6:1186–1197. doi: 10.1158/1535-7163.MCT-06-0686. [DOI] [PubMed] [Google Scholar]
- 7.Neesse A, Michl P, Frese KK, Feig C, Cook N, Jacobetz MA, Lolkema MP, Buchholz M, Olive KP, Gress TM, Tuveson DA. Stromal biology and therapy in pancreatic cancer. Gut. 2011;60:861–868. doi: 10.1136/gut.2010.226092. [DOI] [PubMed] [Google Scholar]
- 8.Koong AC, Mehta VK, Le QT, Fisher GA, Terris DJ, Brown JM, Bastidas AJ, Vierra M. Pancreatic tumors show high levels of hypoxia. Int J Radiat Oncol Biol Phys. 2000;48:919–922. doi: 10.1016/s0360-3016(00)00803-8. [DOI] [PubMed] [Google Scholar]
- 9.Gupta N, Park JE, Tse W, Low JK, Kon OL, McCarthy N, Sze SK. ERO1a promotes hypoxic tumor progression and is associated with poor prognosis in pancreatic cancer. Oncotarget. 2019;10:5970–5982. doi: 10.18632/oncotarget.27235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jackson R, Hunter T. Role of methionine in the initiation of haemoglobin synthesis. Nature. 1970;227:672–676. doi: 10.1038/227672a0. [DOI] [PubMed] [Google Scholar]
- 11.Arfin SM, Kendall RL, Hall L, Weaver LH, Stewart AE, Matthews BW, Bradshaw RA. Eukaryotic methionyl aminopeptidases: two classes of cobalt-dependent enzymes. Proc Natl Acad Sci U S A. 1995;92:7714–7718. doi: 10.1073/pnas.92.17.7714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sawanyawisuth K, Wongkham C, Pairojkul C, Saeseow OT, Riggins GJ, Araki N, Wongkham S. Methionine aminopeptidase 2 over-expressed in cholangiocarcinoma: potential for drug target. Acta Oncol. 2007;46:378–85. doi: 10.1080/02841860600871061. [DOI] [PubMed] [Google Scholar]
- 13.Li X, Chang YH. Amino-terminal protein processing in Saccharomyces cerevisiae is an essential function that requires two distinct methionine aminopeptidases. Proc Natl Acad Sci U S A. 1995;92:12357–12361. doi: 10.1073/pnas.92.26.12357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shusterman S, Maris JM. Prospects for therapeutic inhibition of neuroblastoma angiogenesis. Cancer Lett. 2005;228:171–179. doi: 10.1016/j.canlet.2005.01.049. [DOI] [PubMed] [Google Scholar]
- 15.Sin N, Meng L, Wang MQW, Wen JJ, Bornmann WG, Crews CM. The anti-angiogenic agent fumagillin covalently binds and inhibits the methionine aminopeptidase, MetAP-2. Proc Natl Acad Sci U S A. 1997;94:6099–6103. doi: 10.1073/pnas.94.12.6099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Selvakumar P, Lakshmikuttyamma A, Dimmock JR, Sharma RK. Methionine aminopeptidase 2 and cancer. Biochim Biophys Acta - Rev Cancer. 2006;1765:148–154. doi: 10.1016/j.bbcan.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 17.Wang J, Sheppard GS, Lou P, Kawai M, BaMaung N, Erickson SA, Tucker-Garcia L, Park C, Bouska J, Wang YC, Frost D, Tapang P, Albert DH, Morgan SJ, Morowitz M, Shusterman S, Maris JM, Lesniewski R, Henkin J. Tumor suppression by a rationally designed reversible inhibitor of methionine aminopeptidase-2. Cancer Res. 2003;63:7861–9. [PubMed] [Google Scholar]
- 18.Meister A. Glutathione deficiency produced by inhibition of its synthesis, and its reversal; applications in research and therapy. Pharmacol Ther. 1991;51:155–194. doi: 10.1016/0163-7258(91)90076-x. [DOI] [PubMed] [Google Scholar]
- 19.Ketterer B. Protective role of glutathione and glutathione transferases in mutagenesis and carcinogenesis. Mutat Res. 1988;202:343–361. doi: 10.1016/0027-5107(88)90197-2. [DOI] [PubMed] [Google Scholar]
- 20.Kennedy L, Sandhu JK, Harper ME, Cuperlovic-Culf M. Role of glutathione in cancer: from mechanisms to therapies. Biomolecules. 2020;10:1–27. doi: 10.3390/biom10101429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Traverso N, Ricciarelli R, Nitti M, Marengo B, Furfaro AL, Pronzato MA, Marinari UM, Domenicotti C. Role of glutathione in cancer progression and chemoresistance. Oxid Med Cell Longev. 2013;2013:972913. doi: 10.1155/2013/972913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Frottin F, Bienvenut WV, Bignon J, Jacquet E, Vaca Jacome AS, Van Dorsselaer A, Cianferani S, Carapito C, Meinnel T, Giglione C. MetAP1 and MetAP2 drive cell selectivity for a potent anti-cancer agent in synergy, by controlling glutathione redox state. Oncotarget. 2016;7:63306–63323. doi: 10.18632/oncotarget.11216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schnelldorfer T, Gansauge S, Gansauge F, Schlosser S, Beger HG, Nussler AK. Glutathione depletion causes cell growth inhibition and enhanced apoptosis in pancreatic cancer cells. Cancer. 2000;89:1440–1447. [PubMed] [Google Scholar]
- 24.Sulciner ML, Serhan CN, Gilligan MM, Mudge DK, Chang J, Gartung A, Lehner KA, Bielenberg DR, Schmidt B, Dalli J, Greene ER, Gus-Brautbar Y, Piwowarski J, Mammoto T, Zurakowski D, Perretti M, Sukhatme VP, Kaipainen A, Kieran MW, Huang S, Panigrahy D. Resolvins suppress tumor growth and enhance cancer therapy. J Exp Med. 2018;215:115–140. doi: 10.1084/jem.20170681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lanari C, Lüthy I, Lamb CA, Fabris V, Pagano E, Helguero LA, Sanjuan N, Merani S, Molinolo AA. Five novel hormone-responsive cell lines derived from murine mammary ductal carcinomas: in vivo and in vitro effects of estrogens and progestins. Cancer Res. 2001;61:293–302. [PubMed] [Google Scholar]
- 26.Walker JM, Hammond JBW, Kruger NJ. New Protein Techniques. Humana Press; 2003. The Bradford Method for Protein Quantitation; pp. 25–32. [Google Scholar]
- 27.Akerboom TP, Sies H. Assay of glutathione, glutathione disulfide, and glutathione mixed disulfides in biological samples. Methods Enzymol. 1981;77:373–382. doi: 10.1016/s0076-6879(81)77050-2. [DOI] [PubMed] [Google Scholar]
- 28.Benny O, Fainaru O, Adini A, Cassiola F, Bazinet L, Adini I, Pravda E, Nahmias Y, Koirala S, Corfas G, D’Amato RJ, Folkman J. An orally delivered small-molecule formulation with antiangiogenic and anticancer activity. Nat Biotechnol. 2008;26:799–807. doi: 10.1038/nbt1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pang RW, Poon RT. Clinical implications of angiogenesis in cancers. Vasc Health Risk Manag. 2006;2:97–108. doi: 10.2147/vhrm.2006.2.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Folkman J. The role of angiogenesis in tumor growth. Semin Cancer Biol. 1992;3:65–71. [PubMed] [Google Scholar]
- 31.Nishida N, Yano H, Nishida T, Kamura T, Kojiro M. Angiogenesis in cancer. Vasc Health Risk Manag. 2006;2:213–219. doi: 10.2147/vhrm.2006.2.3.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang J, Lou P, Henkin J. Selective inhibition of endothelial cell proliferation by fumagillin is not due to differential expression of methionine aminopeptidases. J Cell Biochem. 2000;77:465–73. [PubMed] [Google Scholar]
- 33.Kruger EA, Figg WD. TNP-470: an angiogenesis inhibitor in clinical development for cancer. Expert Opin Investig Drugs. 2000;9:1383–96. doi: 10.1517/13543784.9.6.1383. [DOI] [PubMed] [Google Scholar]
- 34.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7–34. doi: 10.3322/caac.21551. [DOI] [PubMed] [Google Scholar]
- 35.Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the united states. Cancer Res. 2014;74:2913–2921. doi: 10.1158/0008-5472.CAN-14-0155. [DOI] [PubMed] [Google Scholar]
- 36.Orth M, Metzger P, Gerum S, Mayerle J, Schneider G, Belka C, Schnurr M, Lauber K. Pancreatic ductal adenocarcinoma: biological hallmarks, current status, and future perspectives of combined modality treatment approaches. Radiat Oncol. 2019;14:1–20. doi: 10.1186/s13014-019-1345-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li D, Xie K, Wolff R, Abbruzzese JL. Pancreatic cancer. Lancet. 2004;363:1049–1057. doi: 10.1016/S0140-6736(04)15841-8. [DOI] [PubMed] [Google Scholar]
- 38.Gillen S, Schuster T, Meyer Zum Büschenfelde C, Friess H, Kleeff J. Preoperative/neoadjuvant therapy in pancreatic cancer: a systematic review and meta-analysis of response and resection percentages. PLoS Med. 2010;7:e1000267. doi: 10.1371/journal.pmed.1000267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Conroy T, Desseigne F, Ychou M, Bouché O, Guimbaud R, Bécouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, de la Fouchardière C, Bennouna J, Bachet JB, Khemissa-Akouz F, Péré-Vergé D, Delbaldo C, Assenat E, Chauffert B, Michel P, Montoto-Grillot C, Ducreux M. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364:1817–1825. doi: 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- 40.Esa R, Steinberg E, Dror D, Schwob O, Khajavi M, Maoz M, Kinarty Y, Inbal A, Zick A, Benny O. The role of methionine aminopeptidase 2 in lymphangiogenesis. Int J Mol Sci. 2020;21:5148. doi: 10.3390/ijms21145148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yeh JJ, Ju R, Brdlik CM, Zhang W, Zhang Y, Matyskiela ME, Shotwell JD, Crews CM. Targeted gene disruption of methionine aminopeptidase 2 results in an embryonic gastrulation defect and endothelial cell growth arrest. Proc Natl Acad Sci U S A. 2006;103:10379–10384. doi: 10.1073/pnas.0511313103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chiu J, Wong JW, Hogg PJ. Redox regulation of methionine aminopeptidase 2 activity. J Biol Chem. 2014;289:15035–43. doi: 10.1074/jbc.M114.554253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Perry RR, Mazetta JA, Levin M, Barranco SC. Glutathione levels and variability in breast tumors and normal tissue. Cancer. 1993;72:783–787. doi: 10.1002/1097-0142(19930801)72:3<783::aid-cncr2820720325>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 44.Berger SJ, Gosky D, Zborowska E, Willson JK, Berger NA. Sensitive enzymatic cycling assay for glutathione: measurements of glutathione content and its modulation by buthionine sulfoximine in vivo and in vitro in human colon cancer. Cancer Res. 1994;54:4077–83. [PubMed] [Google Scholar]
- 45.Cook JA, Pass HI, Iype SN, Friedman N, DeGraff W, Russo A, Mitchell JB. Cellular glutathione and thiol measurements from surgically resected human lung tumor and normal lung tissue. Cancer Res. 1991;51:4287–94. [PubMed] [Google Scholar]
- 46.Wang J, Tucker LA, Stavropoulos J, Zhang Q, Wang YC, Bukofzer G, Niquette A, Meulbroek JA, Barnes DM, Shen J, Bouska J, Donawho C, Sheppard GS, Bell RL. Correlation of tumor growth suppression and methionine aminopetidase-2 activity blockade using an orally active inhibitor. Proc Natl Acad Sci U S A. 2008;105:1838–1843. doi: 10.1073/pnas.0708766105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Datta B. MAPs and POEP of the roads from prokaryotic to eukaryotic kingdoms. Biochimie. 2000;82:95–107. doi: 10.1016/s0300-9084(00)00383-7. [DOI] [PubMed] [Google Scholar]
- 48.Rouschop KM, Dubois LJ, Keulers TG, Van Den Beucken T, Lambin P, Bussink J, Van Der Kogel AJ, Koritzinsky M, Wouters BG. PERK/eIF2α signaling protects therapy resistant hypoxic cells through induction of glutathione synthesis and protection against ROS. Proc Natl Acad Sci U S A. 2013;110:4622–4627. doi: 10.1073/pnas.1210633110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tafani M, Sansone L, Limana F, Arcangeli T, De Santis E, Polese M, Fini M, Russo MA. The interplay of reactive oxygen species, hypoxia, inflammation, and sirtuins in cancer initiation and progression. Oxid Med Cell Longev. 2016;2016:3907147. doi: 10.1155/2016/3907147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Satchi-Fainaro R, Mamluk R, Wang L, Short SM, Nagy JA, Feng D, Dvorak AM, Dvorak HF, Puder M, Mukhopadhyay D, Folkman J. Inhibition of vessel permeability by TNP-470 and its polymer conjugate, caplostatin. Cancer Cell. 2005;7:251–261. doi: 10.1016/j.ccr.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 51.Selvakumar P, Lakshmikuttyamma A, Kanthan R, Kanthan SC, Dimmock JR, Sharma RK. High expression of methionine aminopeptidase 2 in human colorectal adenocarcinomas. Clin Cancer Res. 2004;10:2771–2775. doi: 10.1158/1078-0432.ccr-03-0218. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.