

Summary:
Microorganisms are detected in multiple cancer types, including in putatively sterile organs, but the contexts in which they influence oncogenesis or anti-tumor responses in humans remain unclear. We recently developed Single-cell Analysis of Host-Microbiome Interactions (SAHMI), a computational pipeline to recover and denoise microbial signals from single-cell sequencing of host tissues. Here, we use SAHMI to interrogate tumor-microbiome interactions in two human pancreatic cancer cohorts. We identify somatic-cell associated bacteria in a subset of tumors and their near absence in nonmalignant tissues. These bacteria predominantly pair with tumor cells, and their presence associates with cell-type specific gene expression and pathway activities, including cell motility and immune signaling. Modeling results indicate that tumor-infiltrating lymphocytes closely resemble T-cells from infected tissues. Finally, using multiple independent datasets, a signature of cell-associated bacteria predicts clinical prognosis. Collectively, tumor-microbiome crosstalk may modulate tumorigenesis in pancreatic cancer with implications for clinical management.
eTOC Blurb
Ghaddar et al. use the computational pipeline SAHMI (Single-cell Analysis of Host-Microbiome Interactions) to probe the microbiome in pancreatic cancer. They identify a subset of tumors with bacteria that associate with key cancer hallmarks, immune activity, and prognosis.
Graphical Abstract
Introduction
The microbiome contributes to both human health and disease, including oncogenesis. While it is uncertain whether the healthy pancreas harbors its own microbiome, emerging evidence indicates that bacteria and fungi can translocate to the pancreas and induce local and systemic changes that promote the development of pancreatic ductal adenocarcinoma (PDA) (Vitiello et al., 2019; Wei et al., 2019). Microbiota products alter gene regulation (Yoshimoto et al., 2013) and lead to DNA damage (Öğrendik, 2017), stimulate pattern recognition receptors that potentiate mutant KRAS signaling (Ochi et al., 2012; Zambirinis et al., 2013), and can induce both inflammation and immunosuppression (Aykut et al., 2019; Pushalkar et al., 2018; Seifert et al., 2016; Zambirinis et al., 2015). Microbiota within PDA also may confer resistance to therapies, including deactivating gemcitabine via microbial cytidine deaminase (Geller et al., 2017), while antibiotic-induced reduction of the gut microbiome may increase sensitivity to immune checkpoint inhibitors (Pushalkar et al., 2018; Sethi et al., 2018; Thomas et al., 2018). Moreover, tumors from long-term PDA survivors harbor increasingly diverse microbiota (Riquelme et al., 2019) as well as tumor neoantigens with homology to microbial peptides (Balachandran et al., 2017).
Despite being increasingly implicated in pancreatic cancer, several barriers have limited the systematic investigation of the tumor-associated microbiome in PDA patients (Sethi et al., 2019). First, many intestinal microbes are difficult to culture (Suau et al., 1999). Second, microbiome composition can differ vastly between patients (De Filippo et al., 2010; Nguyen et al., 2015), and there are few model systems that can sufficiently recapitulate tumor-microbiome interactions in humans (Mallapaty, 2017; Saluja and Dudeja, 2013). Third, the possibility of sample contamination post-surgery complicates data interpretation (de Goffau et al., 2018; Zinter et al., 2019). Recent studies have discovered cancer-type specific microbial signatures in The Cancer Genome Atlas (TCGA) (Poore et al., 2020) and tumor-specific intracellular bacteria through 16S bacterial ribosomal DNA sequencing (16S-rDNA-seq) profiling of hundreds of primary human tumors (Nejman et al., 2020). However, these studies analyzed genomic data from bulk tissue samples, which do not capture microbial-somatic cell enrichments, associations with cell-type specific activities, or microbial contributions to inter-cellular communication networks. In particular, PDA is characterized by a fibrotic stroma comprising the majority of tumor volume, which makes disentangling cellular relationships difficult by bulk profiling (Moffitt et al., 2015). Here, we use SAHMI (Single-cell Analysis of Host-Microbiome Interactions) to examine tumor-microbiome relationships in pancreatic cancer at single cell resolution using genomic approaches.
Results
Detection and validation of metagenomic reads in scRNA-seq data
We recently developed SAHMI, a pipeline to systematically recover and denoise microbial signal in human clinical tissues and to assess host-microbiome interactions at single-cell resolution (Fig. 1A, STAR Methods). Briefly, SAHMI first concurrently maps single-cell RNA sequencing (scRNA-seq) reads to the host (e.g. human) and to reference microbial genomes using Kraken2Uniq, a k-mer based taxonomic profiler (Breitwieser et al., 2018; Wood et al., 2019). A series of filters next remove low quality reads and assignments. As previously demonstrated on known infection samples (Ghaddar et al., 2022), SAHMI identifies true microbial reads by analyzing the relationship between the number of total and unique assigned k-mers per taxon across scRNA-seq barcodes in an individual sample and across samples in a study, which we name as the k-mer Correlation Test (kCT). SAHMI identifies false positive taxonomic assignments and contaminants by comparing individual taxa counts to their distributions in negative control samples using a quantile test, for which SAHMI provides an extensive sterile cell line microbiome reference dataset that can be used in the absence of matched controls (Cell Line Quantile Test, CLQT). Denoised microbial profiles can then be jointly analyzed with host cellular transcriptomes, and microbes can be paired with individual somatic cells that share the same scRNA-seq cell barcode. SAHMI thus enables the systematic analysis of host-microbiome ecosystems at single-cell resolution.
Figure 1. Detection and validation of the PDA microbiome.
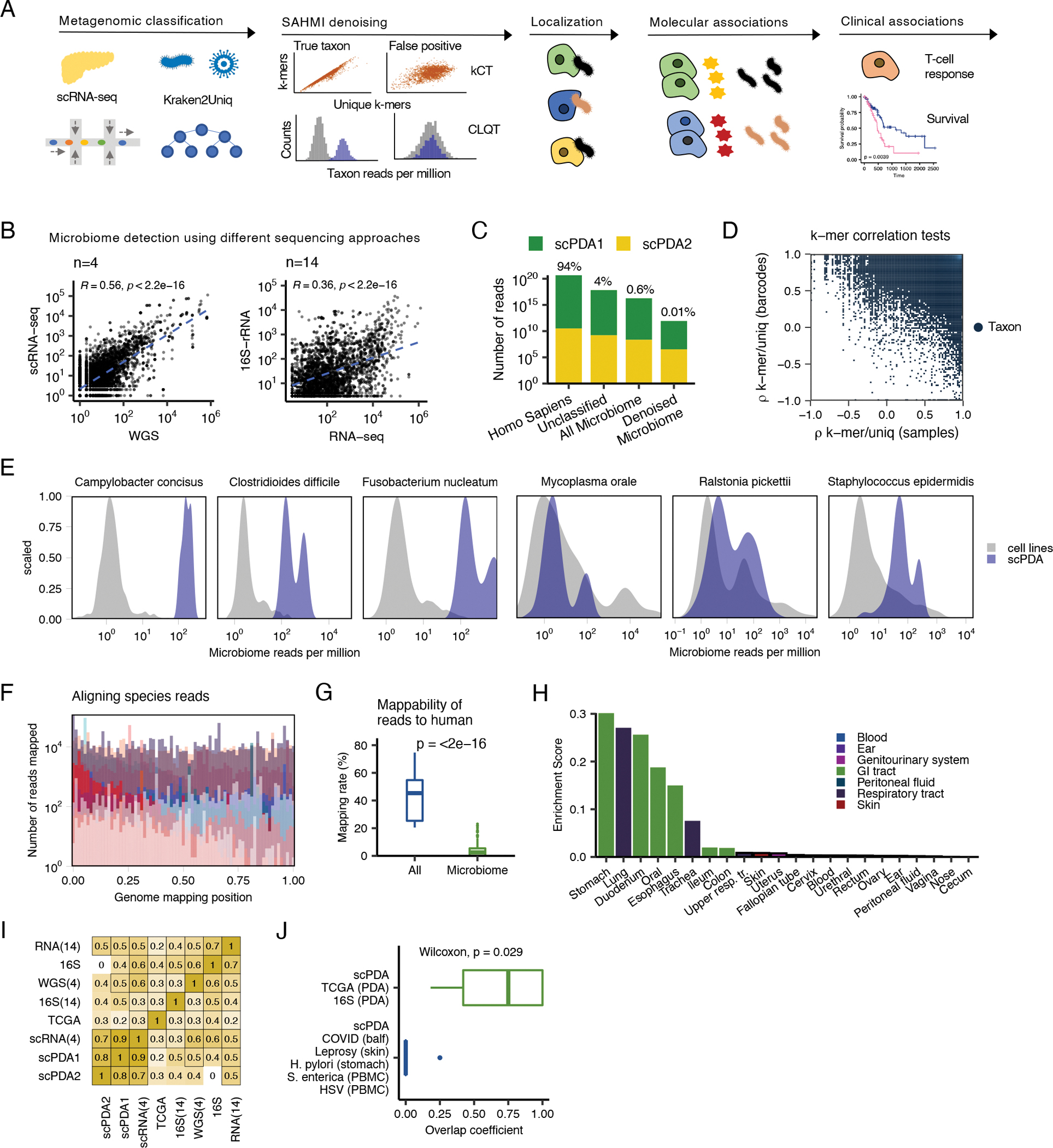
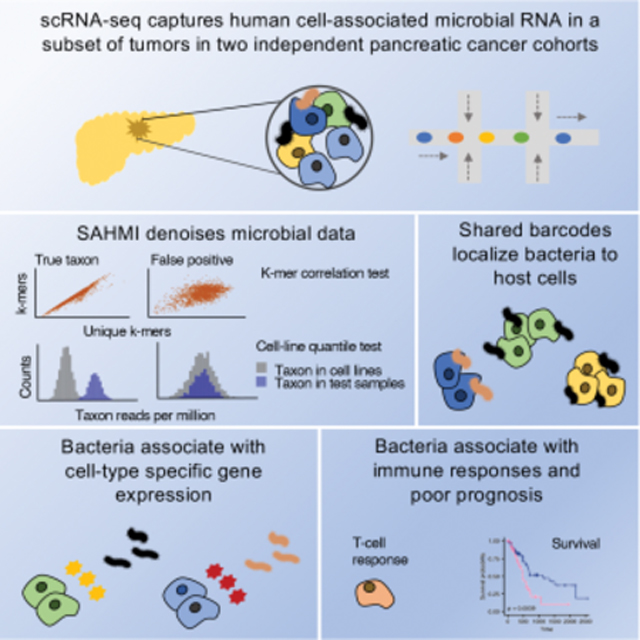
(A) Study design. See also Table S1. scRNA-seq, single-cell RNA sequencing; kCT, k-mer correlation tests; CLQT, cell line quantile test (B) Scatter plots showing microbial genus and species counts correlations from pancreatic tumor sequenced with different technologies. Blue dashed line, line of best fit; Spearman correlation test; WGS, whole genome sequencing. (C) Stacked bar plot showing the classification of reads from scPDA1-2 by SAHMI. (D). Scatter plot of the k-mer correlation tests for all detected species in scPDA1-2. Each point represents a species. x-axis, species Spearman correlation value between the number of total vs. unique k-mers assigned to a given species across samples; y-axis, Spearman correlation value between the number of total vs. unique k-mers assigned to a given species across barcodes. True species have significant correlations in both measures. (E) Example normalized counts density plots comparing reads per million for select species in detected in scPDA1-2 to the same species detected in thousands of cell-line experiments that serve as a negative control. The left three plots are species detected above the contamination and noise threshold. The right three plots are contaminants. (F) Overlaid histograms of genome mapping positions of reads resolved to the species level in scPDA1-2 showing that reads map to locations throughout their respective genome. Each color represents an individual species. Mapping positions are scaled per species. See also Fig. S1B. (G) Boxplots comparing the percent of reads mappable to the human genome for all reads vs. microbiome reads. Boxplots show median (line), 25th and 75th percentiles (box) and 1.5xIQR (whiskers). Points represent outliers; Wilcoxon testing. (H) Bar plot indicating the body location enrichment score for the genera identified in scPDA1-2. (I) Heatmap of Spearman correlations of bacterial genus counts from pancreatic tumor from multiple studies and sequencing technologies. RNA-seq(14) and 16S-rDNA-seq(14), 14 in-house samples sequenced with total RNA-seq and 16S-rDNA-seq; WGS(4) and scRNA-seq(4), 4 in-house samples profiled with single-cell RNA and whole genome sequencing. 16S, pancreas tumors from (Nejman et al., 2020).; TCGA, pancreas tumor from (Poore et al., 2020). (J) Boxplot showing overlap coefficients comparing genera from scPDA1-2 to genera from other PDA studies (green) or from other tissue types and diseases (blue). Boxplots are as in (G); Wilcoxon testing. See also Figure S1 and Table S1.
Here, we showcase the utility of SAHMI for studying host-microbiome interactions at single-cell resolution in PDA. First, we asked whether microbial sequences could be detected in human PDA tumors using scRNA-seq and how their profiles compared to those from different sequencing approaches. We analyzed 4 tumors by scRNA-seq and whole genome sequencing (WGS) and 14 tumor and normal adjacent tissues by RNA-seq and 16S-rDNA-seq. Bacteria were detected in all samples, and their profiles measured using the two sequencing technologies were significantly correlated (Spearman correlations, p<2.2e-16, Fig. 1B).
We next applied SAHMI to two large independent scRNA-seq PDA cohorts, hereon referred to as scPDA1 (Peng et al., 2019) and scPDA2 (Steele et al., 2020). Combined, these included data for 41 PDA tumors samples and 14 normal pancreatic tissues (Table S1). The normal samples were from patients who underwent pancreas surgery but did not have malignant pancreatic tumors. Within each cohort, all samples were processed similarly, and when clustering data using uniform manifold approximation and projection (UMAP), we observed no batch effects amongst samples (Fig. S1A), mitigating concerns of differential contamination within a study. These pancreatic tissues had 100–1500 million total sequencing reads per sample; on average, Kraken2Uniq classified 94% of reads as human (standard deviation=4%), 4% as unclassified (SD=3%), and it resolved 0.6% to the microbial genus level (SD=0.2%) across a total of 1,962 genera and 7,236 species (Fig. 1C).
We used multiple benchmarking and validation steps to denoise the data and ensure that our final observations were not due to artifacts or contamination. First, we applied the SAHMI k-mer Correlation Test (kCT), which identifies true taxa as having significant correlations between the number of reads and the numbers of total and unique k-mers across barcodes and samples. We observed a wide range of correlation values (Fig. 1D), with only a minority of taxa [2565 (28%) of 9198] meeting significance criteria in all correlations.
Second, given the absence of physical negative controls, we applied the SAHMI Cell-Line Test (CLQT) to identify likely false positives taxonomic assignments and contaminant species. This test compares taxon frequencies to their distribution found across thousands of sterile RNA sequencing runs from around the world involving >1000 human cell lines from normal and diseased tissues. The CLQT clearly distinguished 38 taxa in scPDA1 and 2 above the noise threshold and identified contaminants in the data (Fig. S1B). The significant taxa, such as the gastrointestinal tract species Campylobacter concisus, Clostridioides difficile, and Fusobacterium nucleatum had microbial frequencies greater than the 95th percentile in the reference dataset, whereas most taxa, including common contaminants such as Mycoplasma orale, Ralstonia picketti, and Staphylococcus epidermidis, were detected at levels consistent with the cell line data (Fig. 1E). As such, only a minority of reads (mean 0.01%) and taxa (mean 2%) per sample passed all SAHMI denoising criteria and were retained for further analysis (see STAR Methods for details; Fig. 1C). As an additional measure, we examined our data for common contaminants recently identified (Poore et al., 2020); 229 of the 419 reported contaminant genera were initially detected, but none passed our filtering measures. These denoising steps thus identified taxa in scPDA1 and 2 at higher frequencies than expected by chance and for which diverse RNA was present.
Third, we validated reads for taxa resolved to the species level by mapping them using a well-validated RNA-seq aligner (Dobin et al., 2013). For each species, we extracted its reads and aligned them to its reference genome. Across all samples and species, >90% of reads mapped (see STAR Methods) to locations throughout the genome. Mapped reads did not exclusively bias towards specific regions, indicating that they were not artifacts (Fig. 1F, Fig. S1C). While some mapping positions were over-represented, the mapping patterns were consistent with those previously observed for verified pathogens in clinical tissues analyzed by scRNA-seq (Ghaddar et al., 2022).
Fourth, although we excluded human reads during the initial taxonomic classification steps, we attempted to map the denoised microbial reads to the human transcriptome using a third mapping algorithm (Patro et al., 2017). Only a median of 2.5% of SAHMI microbiome reads per species were mappable to the human transcriptome; this was significantly less than the mapping rates for whole samples when all reads were included (Wilcoxon p<2e-16, Fig. 1G), indicating that SAHMI enriched for nonhuman reads.
Fifth, to assess whether the likely ecological sources of PDA microbes were expected, we calculated body site enrichment scores of the scPDA genera using mBodyMap (see STAR Methods), a curated human microbiome database containing more than 60,000 sequencing runs across 22 body sites (Jin et al., 2022). The ecological sources of scPDA taxa were predominantly from organs throughout the gastrointestinal (GI) tract, in addition to microbes found in the respiratory tract or blood (Wilcoxon p<2e-16, Fig. 1H). These results are expected given that the pancreas has direct contact with the GI tract via the pancreatic duct (and respiratory organisms are ingested as well), and it is consistent with literature linking GI bacteria to pancreatic disease (Adolph et al., 2019; Thomas and Jobin, 2020).
Sixth, we compared microbial profiles from scPDA1 and 2 to PDA microbiomes profiled using other technologies. These included (1) n=4 tumors we profiled with dual scRNA-seq and WGS, (2) n=7 tumors and n=7 adjacent normal pancreas we profiled with dual 16S-rDNA-seq and RNA-seq, (3) RNA-seq of PDA from TCGA (Poore et al., 2020), and (4) 16S-rDNA-seq of PDA from a recent large-scale study (Nejman et al., 2020) – for a total of 327 pancreatic samples sequenced with four different technologies. We correlated the number of reads (or normalized counts) assigned to each genus across all studies and found significant agreement between scRNA-seq and other technologies (Fig. 1I). In particular, the correlations between the scRNA-seq data were highly significant (mean Spearman ρ=0.78, mean p=1e-5), indicating that scRNA-seq profiles are reproducible, and correlations between scRNA-seq and 16S-rDNA data were comparable to those between RNA-seq and 16S-rDNA, suggesting that scRNA-seq does not introduce significant bias in read capture compared to bulk RNA-seq. These comparisons to positive controls indicate that SAHMI quantitatively identified relevant taxa in scPDA.
Seventh, we used SAHMI to analyze different scRNA-seq samples not expected to have predominant lower GI tract microbiomes that could serve as negative controls, and we compared the overlap in their detected microbiome to that from scPDA1 and 2. We analyzed data from bronchoalveolar lavage fluid from patients with coronavirus-19, skin samples from patients with leprosy, gastric epithelium from patients with Helicobacter pylori infection, and data from peripheral blood mononuclear cells infected with herpes simplex virus or Salmonella enterica (Ghaddar et al., 2022). In each study, SAHMI identified the known pathogen in the infected samples and its absence in controls. Next, we computed the overlap in microbial taxa between scPDA and the other PDA microbiome studies (Nejman et al., 2020; Poore et al., 2020) and compared it to the overlap in taxa between scPDA and these negative control samples. As expected, the overlap in taxa was significantly greater (Wilcoxon p=0.029) between the scPDA and PDA data, despite being profiled with different technologies (Fig. 1J). The mean overlap coefficient with PDA data was 0.67. In contrast, the only taxa that overlapped with scPDA from the infection studies were Clostridium and Helicobacter pylori, both of which were from the gastric samples. These results validate the ability of SAHMI to identify the appropriate tissue-present taxa.
Taken together, these benchmark and validation analyses demonstrate that the microbiome we identified in PDA through scRNA-seq is consistent with recent literature. Specific microbial reads map throughout their respective genomes and are present at frequencies greater than expected for contamination. SAHMI identified ecologically relevant taxa in PDA that had negligible overlap with taxa from other tissues and disease states.
Cell-associated bacteria are present in a subset of pancreatic tumors
After all data processing steps (see STAR Methods for details), we detected microbial reads in 48 (87%) of 55 pancreatic samples tested and identified 19 bacterial genera that were present in both scPDA1 and 2. These genera had a mean of 29,498 unique k-mer sequences (range: 1010–210,105) (Fig. 2A). Sequencing depth is a common confounder in metagenomics; however, after data denoising, there was no correlation between the number of somatic and microbial gene counts (Spearman, scPDA1: p=0.11; scPDA2: p=0.82, Fig. S2A). Microbial reads were detected on 96,135 (scPDA1) and 479,834 (scPDA2) molecular barcodes, with tumors generally having increased total microbial counts compared to normal samples (Wilcoxon, scPDA1: p=0.015; scPDA2: p=0.089, Fig. 2B). A subset of barcodes also tagged somatic cellular RNA (scPDA1: 8.7%; scPDA2: 8.9%), providing evidence that those bacteria were co-localized with a host cell. When we counted cell-associated bacterial reads across samples, we observed that cell-associated bacteria were found in appreciable amounts in only a subset of tumors and in nearly none of the nonmalignant samples (18/41 tumors, 1/14 nonmalignant tissues; Fisher test combined p-value: p=0.009; Fig. 2C). Barcode pairing of host cells and bacteria suggests that these microbes are associated with somatic cells but does not indicate whether they are intra- or extra-cellular; however, imaging data indicate that most detected bacteria in pancreatic tumors are intracellular (Nejman et al., 2020). The significantly increased presence of cell-associated bacteria in a subset of scPDA1 and 2 tumors thus suggests a distinct (bacterial cell-associated) tumor state. This finding is consistent with a previous report detecting intracellular bacteria in a subset (68%) of PDA tumors tested under stringent experimental conditions to control for contamination (Nejman et al., 2020).
Figure 2. A subset of tumors has cell-associated bacteria.
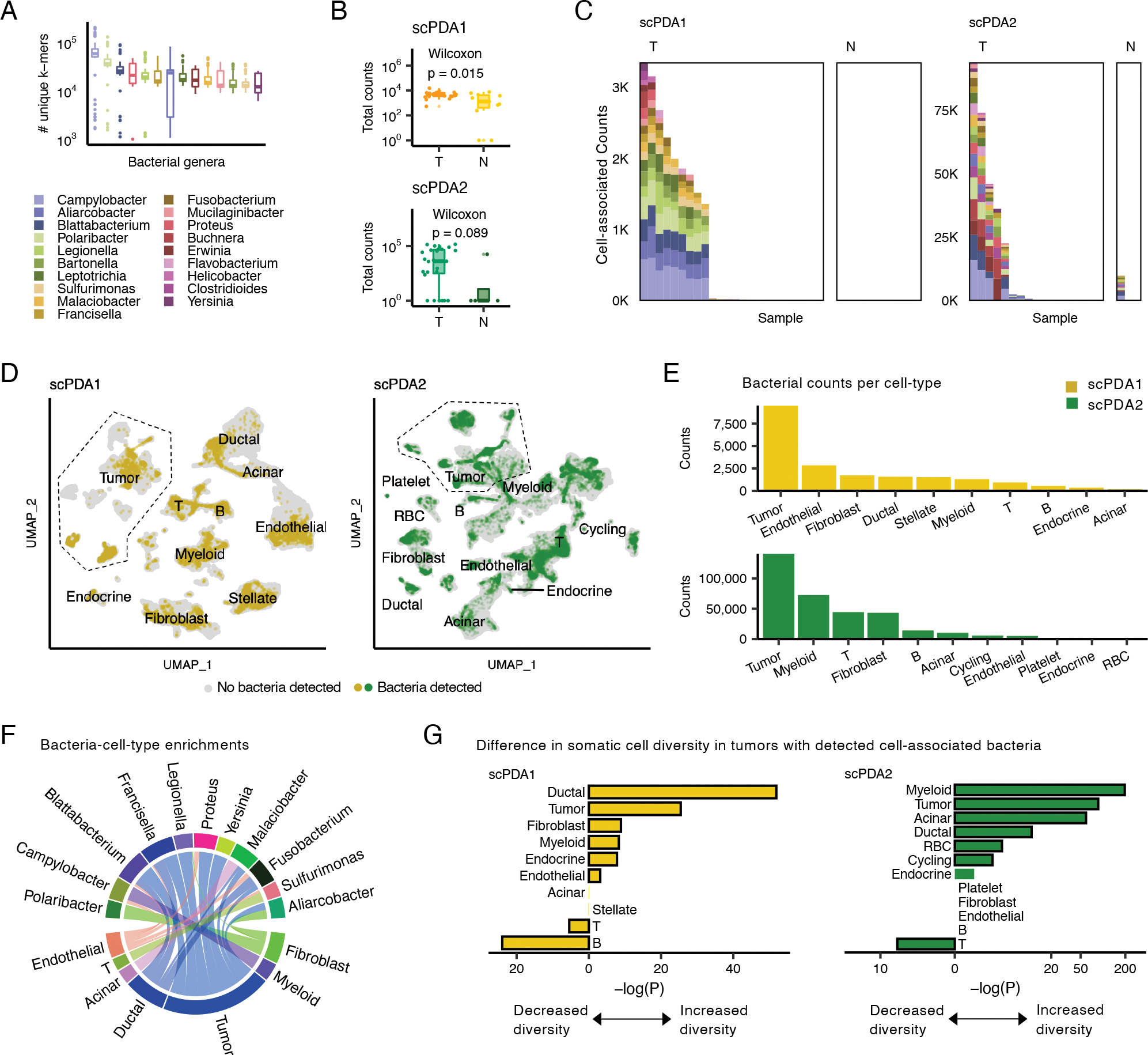
(A) Boxplots showing the number of unique k-mers assigned to each genus detected in scPDA1-2. Boxplots show median (line), 25th and 75th percentiles (box) and 1.5xIQR (whiskers); points represent outliers. (B) Boxplots (top: scPDA1, bottom: scPDA2) comparing the total number of bacterial counts per sample in T (tumor) vs. N (normal) samples. Boxplots as in (A). (C) Profiles of cell-associated bacterial counts in scPDA1 and 2. Stacked bar plots showing the number of counts and genus composition for each sample. K, thousand; T, tumor samples; N, normal samples. (D) Uniform manifold approximation and project (UMAP) plots of host somatic cells for scPDA1 (n=57,530 cells) and scPDA2 (n=59,473 cells). Clusters are labeled by cell type. Cells are colored yellow (scPDA1) or green (scPDA2) if they share a barcode with bacteria. (E) Bar plots of the number of bacterial counts associated with each cell type in scPDA1-2. (F) Circos-plot of significant (p<5e-3) bacteria-cell-type enrichments identified at the single-barcode level by Wilcoxon testing and shared in scPDA1-2. Ribbon width correlates with enrichment strength. (G) Bar plots showing the adjusted Wilcoxon p-value comparing the transcriptional diversity of bacteria-associated cells to unassociated cells for each cell type. See also Figure S2.
We did not detect any cell-associated bacteria in 13 of 14 non-malignant samples. We also analyzed a third scRNA-seq study of 13 healthy human pancreas samples and found no microbes in those tissues (Baron et al., 2016); thus, in total, only one of 27 pancreatic samples without malignancy was positive for cell-associated bacteria, compared to 44% of the PDAs. However, scRNA-seq data of paired tumor and normal adjacent tissue will be required to further address the specificity of the cell-associated bacteria in the tumors compared to normal tissues.
The scPDA1 and 2 tumors with cell-associated infections harbored 8–19 bacterial genera, many of which are associated with oral or gastrointestinal pathology. The most common bacteria in both cohorts were Campylobacter spp, which is known to cause gastrointestinal and systemic inflammation (Janssen et al., 2008). Other bacteria included Fusobacterium nucleatum, which is strongly associated with tumorigenesis in colorectal cancer (Sethi et al., 2019), Leptotrichia spp., an oral microbe previously associated with pancreatic cancer (Torres et al., 2015), and Clostridioides difficile, a gut pathogen associated with pancreas pathology (Adejumo et al., 2019). Although we included fungal and viral genomes during mapping, none passed our denoising criteria.
When we visualized somatic cell data with UMAP, we observed that the distribution of bacteria was not uniform across cell-types (Fisher test, p=4e-5) and that bacteria-associated cells generally clustered together within their cell type, indicating shared, broad gene expression changes compared to unassociated cells (Fig. 2D). Bacteria were found with all cell types, consistent with imaging of tumors showing intracellular bacteria in both malignant and immune cells (Geller et al., 2017; Nejman et al., 2020). Tumor cells had the greatest total number of associated bacterial counts (Fig. 2E). Using Wilcoxon testing, we identified 27 bacteria-cell-type specific enrichments that were shared across both scPDA1 and 2 (Wilcoxon, all p<5e-3; Fig. 2F). Tumor cells again had both the greatest number and strongest bacterial enrichments. Although immune cells co-localized with bacteria (Fig. 2E), we found only 3 bacteria-immune cell enrichments, suggesting that immune cells had non-specific interactions with intra-tumoral bacteria. These data indicate that a subset of pancreas tumors have an altered microbiome with possible bacterial tropism towards malignant tumor cells.
Bacteria associate with cell-type specific diversity and activities
Co-clustering of bacteria-associated host cells (Fig. 2D) raises the possibility of transcriptional changes in host cells in response to the presence of bacteria. We first asked whether the presence of cell-associated bacteria associated with the diversity of somatic cell states in the tumor. We calculated the Shannon diversity of each cell’s transcriptional profile, and then for each cell-type, we compared diversity values of cells from tumors with or without detected cell-associated bacteria. In both scPDA1 and 2, samples with cell-associated bacteria had significantly increased diversity in their tumor and myeloid cells and decreased diversity in their T cell populations (Fig. 2G).
We next investigated whether the presence of bacteria were themselves associated with host cell-type specific gene expression. Using Wilcoxon testing, bacterial and host cell barcode pairing allowed us to identify cell-type specific genes that were differentially expressed in cells associated with specific bacteria. There was no significant difference in expression for the vast majority of genes tested (Fig. 3A); however, a subset of genes was significantly altered in the same direction in both scPDA1 and 2. Of cells localized with bacteria, tumor cells had the greatest number of differentially expressed genes (Fig. 3B). Despite bacteria co-localizing with many immune cells (Fig. 2E), we did not identify any common transcriptional changes in lymphoid cells co-localized with bacteria and only 15 differentially expressed genes (DEG) in myeloid cells. In total, 571 unique DEGs were identified (Table S2), with the most commonly affected genes across all cell types and bacteria including keratin, mucin, and trefoil genes (Fig. 3C).
Figure 3. Bacteria associate with cell-type specific activities.
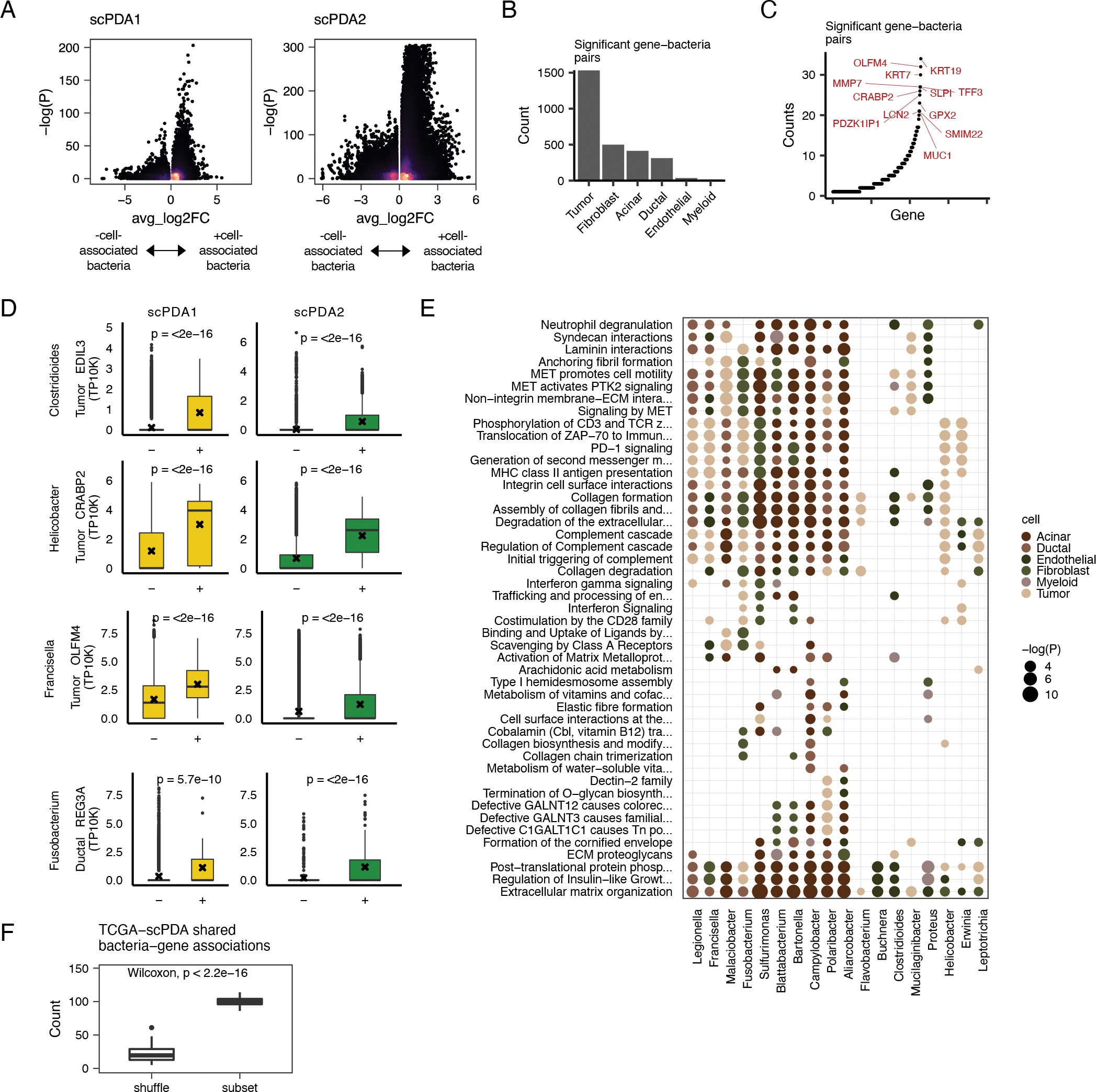
(A) Volcano plots for bacteria-cell-type specific gene associations. Wilcoxon tests are run comparing gene expression values between somatic cells with or without co-localized bacteria. X-axis, average log2 fold change in normalized expression in cells with vs. without co-localized bacteria. Y-axis, Wilcoxon test FDR-corrected p-value. Color is scaled to point density, black=low, yellow=high. (B) Bar plot showing the number of significant gene-bacteria pairs per cell type for significant pairs shared by scPDA1-2. Significant genes are were identified as having an FDR-corrected p-value<0.05 in the analysis from Fig. 3A and the a fold change in the same direction in scPDA1 and 2. (C) Ranked scatter plot showing the number of times each significant gene was differentially expressed, either with different bacteria or in different cell types. Significant genes were identified as in the same manner as in Fig. 3A–B. (D) Boxplots comparing gene expression values in cells with (+) or without (−) a specific co-localized bacterial genus. Boxplots show median (line), 25th and 75th percentiles (box) and 1.5xIQR (whiskers); points represent outliers; ‘x’ denotes the mean value. (E) Dot plot showing the Reactome pathways enriched by the differentially expressed genes shared by scPDA1-2. Dots are colored by cell-type and size-scaled by p-value. Data is shown for pathways with FDR-corrected p<0.05 (hypergeometric test). (F) Boxplot comparing the number of shared bacteria-gene associations between scPDA1-2 and TCGA data. Associations are calculated from subsampled and sample-label shuffled data. Wilcoxon testing. See also Table S2 and Table S3.
Many of the strongest bacteria-associated DEGs in scPDA1 and 2 have been previously associated with PDA or microbiome-related inflammation (Fig. 3D). For example, tumor cells co-localized with Clostridioides difficile in scPDA1 and 2 had significantly increased expression of EDIL3 (Wilcoxon, p<2e-16), an integrin ligand involved in angiogenesis that is implicated in both sterile and microbial-induced inflammation in multiple tissues (Hajishengallis and Chavakis, 2019) as well as tumor growth and poor prognosis in PDA (Jiang et al., 2016). Tumor cells in scPDA1 and 2 co-localized with Helicobacter pylori had significantly increased expression of CRABP2 (Wilcoxon, p<2e-16), an oncogene upregulated in an in vivo model of oral candidiasis and cancer (Máté et al., 2022). CRABP2 also enhances pancreatic cancer cell motility (Yu et al., 2016). Francisella spp. associated with increased OLFM4 expression in tumor cells in scPDA1 and 2 (Wilcoxon, p<2e-16). OLFM4 is a stem cell marker associated with microbiota-induced accelerated epithelial regeneration (Abo et al., 2020; Lee et al., 2018) as well as poor prognosis in pancreatic cancer (Ohkuma et al., 2020). Normal ductal epithelial cells in scPDA1 and 2 co-localized with Fusobacterium nucleatum had increased expression of REG3A (Wilcoxon, p<2e-16), a gene strongly involved in regulation of host-microbiota interplay (Zhang et al., 2019a) and which also promotes acinar to ductal metaplasia, a common precursor to pancreatic cancer (Zhang et al., 2021). These findings and others detailed in Table S2 indicate that microbiota identified by SAHMI may be associated with critical growth and inflammatory processes in PDA.
To more broadly examine the biological processes associated with cell-associated bacteria, we performed Reactome pathway gene set enrichment analysis using the cell-type and bacteria-specific DEGs (Fig. 3E). In general, the presence of cell-associated bacteria was associated with increased activity of multiple pathways related to cell motility, extracellular matrix interaction, and immune signaling in tumor cells and in normal epithelium and stroma. Such pathways included MET-PTK2 signaling, consistent with the finding that oral pathogens promote cancer aggressivity via integrin/FAK signaling (Kamarajan et al., 2020), and integrin and non-integrin membrane-extracellular matrix interactions, consistent with growing evidence of microbial disruption of tissue integrity and cancer risk (Alfano et al., 2016). Upregulated immune pathways included those related to the complement cascade and PD-1 signaling, consistent with the observation that pathogenic fungi promote PDA via lectin-induced activation of the complement cascade (Aykut et al., 2019) and with evidence that the gut microbiome modulates response to anti-PD1 therapy (Gopalakrishnan et al., 2018). These and other pathway data detailed in Table S3 thus further associate microbiota in PDA with cancer hallmark activities.
To validate our observations, we compared the bacteria-gene associations in scPDA1 and 2 to those found in the TCGA pancreatic cancer cohort (Poore et al., 2020). We identified the 12/19 genera that were in common between scPDA1 and 2 and TCGA and correlated their normalized counts in TCGA with corresponding gene expression values from the same samples. We identified 100 significant bacteria-gene correlations that overlapped with scPDA1 and 2 bacteria-cell-type-specific DEGs (9.5% of all possible comparisons). This overlap was significantly greater than expected by chance when we repeated the procedure using subsampled vs. sample label shuffled data (Wilcoxon, p<2e-16; Fig. 3F), and it is noteworthy given between-patient differences in microbial compositions and limited genomic coverage of the TCGA data. Collectively, these observations learned at single cell resolution consistently associate microbiota with key cancer-related cellular processes in individual cell-types in the tumor-microenvironment.
Tumors with cell-associated bacteria have activated T-cells
Although we did not identify many shared differentially expressed genes in individual T-cells paired with bacteria, we asked whether there was a difference in overall T-cell subtypes found in tumors with or without cell-associated bacteria. We integrated T-cell data from scPDA1 and 2 and identified regulatory, memory, effector, and natural killer T-cells based on canonical subtype markers (Fig. 4A–B, Fig. S2B). T-cells from tumors with cell-associated bacteria were more likely to have an activated phenotype (i.e. natural killer T, effector T) and were less likely to have a regulatory phenotype (Fisher test, p=1e-4, Fig. 4A). We found that, as a population, T-cells from tumors with cell-associated bacteria compared to those without had multiple differentially expressed genes that were shared across both scPDA cohorts (Fig. 4C) and that were enriched in several relevant pathways (Fig. 4D). Pathways upregulated in T-cells from tumors with cell-associated bacteria included PD-1 signaling, consistent with our finding that individual tumors cells paired with bacteria also had increased PD-1 signaling, and response to intracellular infection (both hypergeometric test, adjusted p=0.04). Downregulated pathways included FOXO-mediated transcription and interferon gamma signaling, consistent with these tumors having relatively fewer regulatory T-cells (Fig. 4A, 4D). These results are consistent with recent reports that intra-tumoral bacteria induce immune infiltration and antitumor responses (Pushalkar et al., 2018; Riquelme et al., 2019).
Figure 4. T-cell subtypes in tumors with cell-associated bacteria.
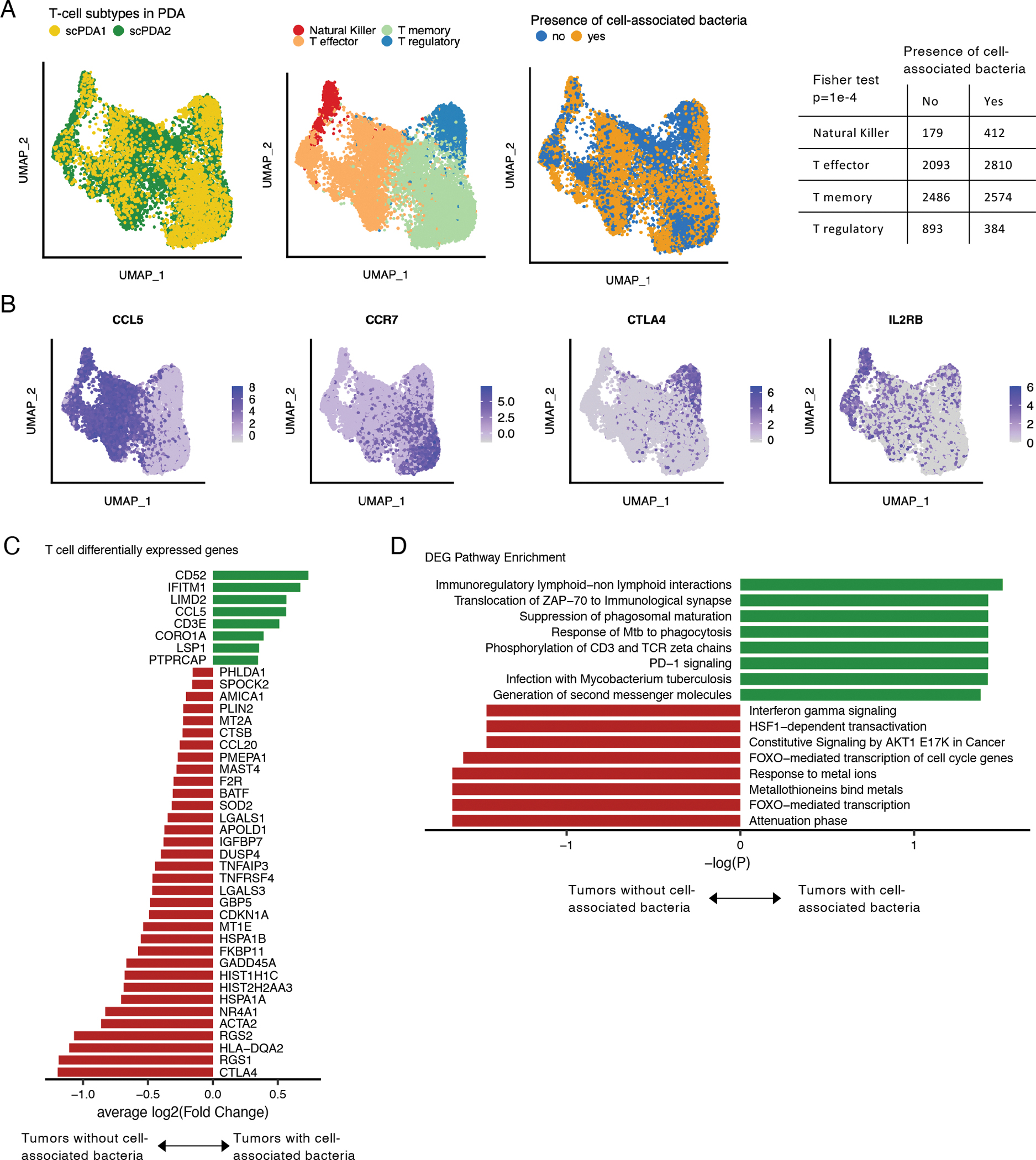
(A) Uniform manifold and projection (UMAP) plots of T-cells from scPDA1 and 2 after batch correction. Left: colored by study; middle: colored by major T-cell subtype; right: colored by the presence of absence of cell-associated bacteria in the same tumor sample. Table: counts of T-cell subtype in tumors with or without cell-associated bacteria. (B) Uniform manifold approximation and project (UMAP) plots of batch-corrected T-cell data from scPDA1 and 2 colored by normalized expression of selected T-cell subtype markers. (C) Significantly differentially expressed genes shared in scPDA1-2 (Wilcoxon tests) in T-cells from tumors with or without cell-associated bacteria. Bar length represents the mean log2(Fold change). (D) Reactome pathway enrichment for the differentially expressed genes from (C). See also Figure S2.
The majority of PDA T-cells are predicted to have infection-related transcriptional profiles
The extensive bacteria-immune co-localization and association with immune-related signaling in both scPDA cohorts suggests that the microbiome influences the PDA immune response. We asked if we could determine the target of individual T-cells in the tumor microenvironment. To do this, we constructed a random forest model to distinguish between T-cells responding to known infections from those responding to other microbiome poor tumors. First, we trained a model to classify T-cells as having either an infection microenvironment reaction (IMER) or tumor microenvironment reaction (TMER) using T-cells sampled from patients with sepsis (Reyes et al., 2020) or from tumors known to have low microbiome burden based on profiling from TCGA (Poore et al., 2020) and 16S-rDNA-seq (Nejman et al., 2020). We then tested the model on >100,000 cells taken from each of five cancer types with similarly known low microbiome burden and from three datasets representing either bacterial or fungal infection or stimulation (Fig. 5A). The model performed exceptionally well in classifying T-cell microenvironment reaction, i.e. it accurately distinguished between T-cells with microbial vs. tumor stimulation (AUC=0.98, Fig. 5B). Next, we used this model to identify IMER vs. TMER T-cells in scPDA1 and 2. The vast majority of T-cells in both PDA cohorts were classified as having an IMER pattern (scPDA1: 90% IMER; scPDA2: 92% IMER; Fig. 5C). This result indicates that T cells in PDA are transcriptionally more similar to T-cells from microenvironments with infection rather than the other tumors used in model testing (Fig. 5A). This finding suggests an explanation as to why pancreatic tumors have high levels of inflammation and respond poorly to immune therapy (Feng et al., 2017).
Figure 5. T-cell microenvironment reaction and survival model.
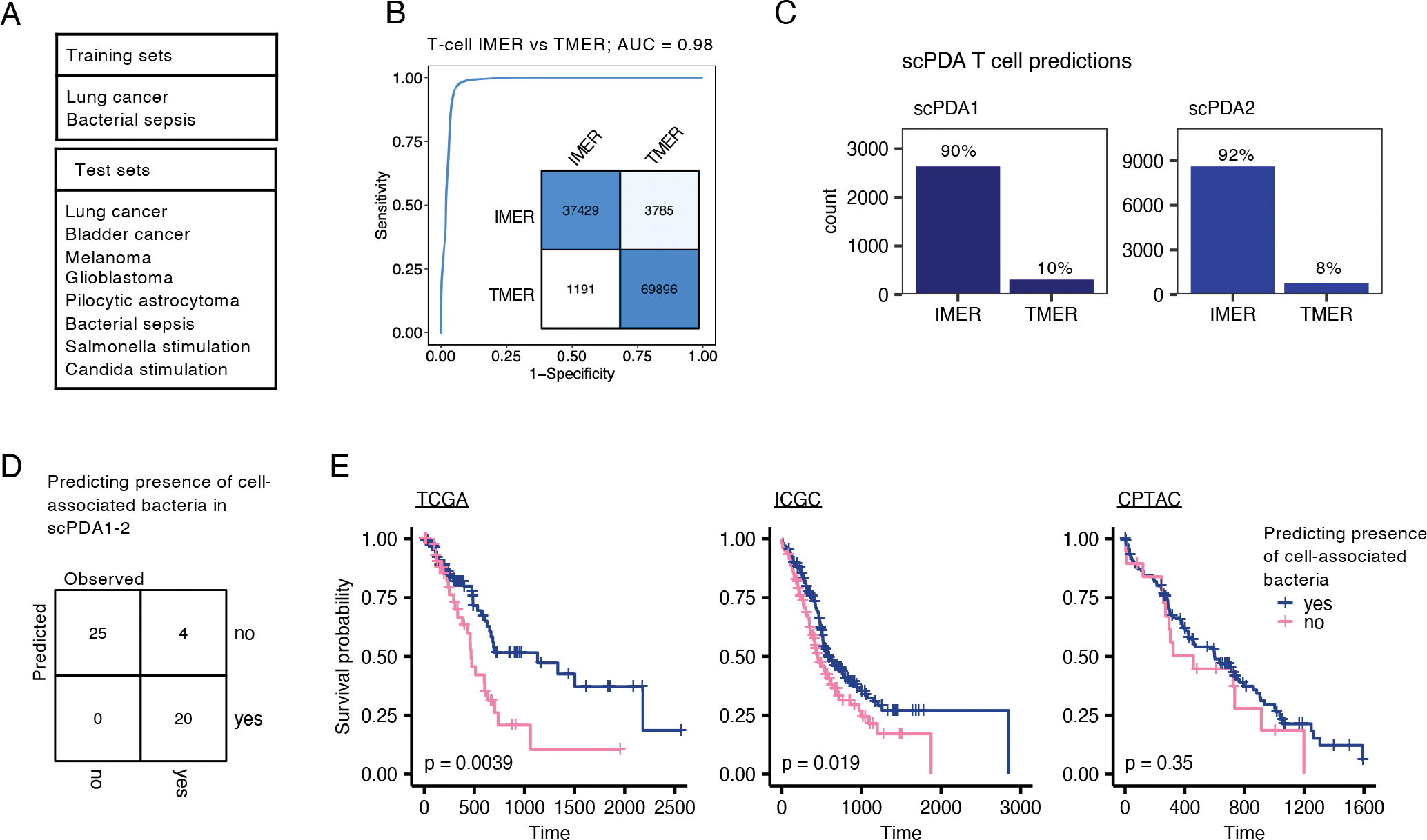
(A) Training and test datasets used to create a random forest model to distinguish between T-cells infection vs. tumor microenvironment reaction based on their gene expression profiles. (B) ROC curve indicating exceptional model performance on test datasets; AUC, area under the curve, IMER, infection-microenvironment reaction, TMER, tumor-microenvironment reaction. Inset: Confusion matrix of model assignments; rows, predicted, columns, true values. (C) Bar-plot of predicted T-cell microenvironment reaction in scPDA1 and 2. (D) Development of a classification model to predict the presence of cell-associated bacteria in a tumor using 7 bulk gene expression values. Confusion matrix showing classification accuracy of the model on scPDA1 and 2. (E) Kaplan-Meier plots of TCGA, ICGC, and CPTAC PDA cohorts stratified by predicted presence of cell-associated bacteria. P-values are determined by Cox proportional hazards models.
Microbiome characteristics stratify patient survival
Finally, we investigated whether intra-tumoral microbial signatures correlated with overall survival. Since a large single-cell PDA cohort with survival data currently does not exist, we developed a model that classified tumors with and without cell-associated bacteria based on their bulk mRNA expression and then used this model to predict infection status in pancreatic tumors from TCGA (Raphael et al., 2017), International Cancer Genomics Consortium (ICGC) (Hudson (Chairperson) et al., 2010), and CPTAC (Cao et al., 2021). Briefly, we first created pseudo-bulk gene expression profiles from the scPDA cohorts by summating gene counts across all cells in a sample. We identified the most differentially expressed genes (TLL1, CHRDL1, PSCA, DKK1, KLRC2, MEOX1, ADAMTS5) between tumors with and without cell-associated bacteria and used them to develop a gradient-boosted tree model to classify bacterial status. The model accurately distinguished between tumors with or without cell-associated bacteria in both scPDA1 and 2 (Fig. 5D). We then used the model to predict the infection status in TCGA, ICGC, and CPTAC PDA tumors and tested the relationship with patient survival using univariate Cox proportional hazards models. In TCGA and ICGC, presence of predicted cell-associated bacteria was associated with significantly decreased overall survival (TCGA: Hazard Ratio [HR] = 2.1, 95% Confidence Interval [CI]: 1.4–3.6, p = 0.006; ICGC: HR = 1.5, 95% CI: 1.1–2.0, p = 0.019; Fig. 5E). The same trend was observed in the CPTAC dataset, although with a smaller effect size and sample size (HR = 1.3, 95% CI: 0.8–2.3, p = 0.35). These results collectively suggest that the tumor microbiome may have clinical relevance in a subset of pancreatic cancer patients.
Discussion
In this work, we used SAHMI to analyze tumor-microbiome interactions at single-cell resolution in two independent pancreatic cancer studies. Our findings are validated by consistent observations in both of datasets as well as corroboration with other PDA cohorts sequenced using four differing technology platforms. We demonstrated the systematic detection of relevant gastrointestinal tract microbes in these tissues and the exclusion of contaminants and false positives. Further, we identified host cell-associated bacteria in a subset of tumors and their near absence in normal pancreas tissues. These cell-associated bacteria paired with most cell types identified in the single-cell analyses but were dominantly localized to tumor cells. Their presence associated with cell-type specific cancer-related processes, including cell motility, extracellular matrix interaction, complement cascade, and PD-1 signaling. Our T-cell microenvironment reaction model predicted that the majority of PDA T-cells have infection-related transcriptional responses. Finally, we developed a model to predict a tumor’s infection status; model predictions indicate that patients with tumors with cell-associated bacteria have significantly decreased survival. In total, our results provide evidence that intra-tumoral bacteria either reflect or influence the trajectory of tumor growth; either possibility has clinical utility.
SAHMI detects microbial nucleic acids captured in single-cell experiments, but further work is necessary to ascertain their cellular localization. A subset of microbes might be ‘free’ in the tissue microenvironment, intracellular, cell-surface-associated, or less likely, introduced during sample preparation. We observed overall decreased microbial load and nearly no cell-associated microbes in normal pancreatic tissues; this could possibly reflect the high nuclease activity in acinar cells, the predominant cell-type in healthy pancreatic tissue. That microorganisms may be intracellular seems surprising, but it is consistent with electron-microcopy findings indicating the presence of primarily intracellular bacteria in tumors (Nejman et al., 2020). Although we do not have imaging data from the two scPDA studies analyzed in our manuscript, we find similar extent and types of interactions in both studies. Furthermore, we found that bacteria-cell interactions occur only in a subset of tumors, consistent with a recent report (Nejman et al., 2020), and are largely absent in normal pancreas tissue. These observations provide evidence that bacteria-cell interactions occur in vivo. However, future studies using matched tumor and normal tissues will be necessary to further address the specificity of cell-associated bacteria to tumors tissues compared to the healthy pancreas.
It may seem surprising that sufficient microbial content is captured during scRNA-seq despite the resistance of microbial cell walls to the detergents used to lyse eukaryotic cells and despite the lack of known microbial polyadenylation. However, when we previously benchmarked SAHMI on multiple scRNA-seq datasets of diverse tissue types and known infections, we found that SAHMI successfully identified the known pathogen, and that pathogen reads were significantly increased in infected samples in proportion to pathogen load (Ghaddar et al., 2022). Several possibilities, as identified in the literature, may explain the quantitative capture of microbial reads by scRNA-seq: (1) RNA capture could occur by priming on internal adenine-rich sites (Hrdlickova et al., 2017), (2) prokaryotic transcripts may have more polyadenylation than previously believed (Hajnsdorf and Kaberdin, 2018; Maes et al., 2017), (3) microbes may alter their envelopes or become cell wall-deficient, akin to L-form switching (Chikada et al., 2021; Mickiewicz et al., 2019; Nejman et al., 2020), and (4) non-polyadenylated mammalian sequences such as those from noncoding genes or regions far from the transcript 3’ end are routinely captured in scRNA-seq (Wang et al., 2021). Additionally, the scientific and clinical utility of microbial analyses using poly-A selected human RNA sequencing has been shown (Poore et al., 2020; Westermann and Vogel, 2021). Understanding the origin and mechanism of capture of microbial sequences in single-cell workflows is an important direction for future work. Nonetheless, given our multiple validation analyses and findings identified only by using scRNA-seq, we believe that SAHMI represents a substantial advance in our ability to study host-microbiome interactions.
Although we refrain from inferring causality from correlation, our observations generate testable hypotheses regarding tumor-microbiome hologenomic evolution in which crosstalk amongst microbes and tumor, immune, and stromal cells can potentially modulate tumorigenesis and anti-tumor responses. Tumors of long-term survivors of pancreatic cancer produce neoantigens that share homology with microbial peptides (Balachandran et al., 2017). Unlike in immunotherapy-responsive cancer-types, our data provide evidence that a majority of infiltrating lymphocytes in PDA have transcriptional profiles resembling T-cells in infection-associated microenvironments; this finding could help explain the lack of efficacy of immune checkpoint inhibitors in PDA (Feng et al., 2017). If PDA-infiltrating T-cells mostly display IMER, then tumor neoantigens with homology to microbial peptides may increase susceptibility to anti-tumor immune responses. However, microbiota in tumors, or tumors expressing microbial antigens, may also contribute to the characteristic immunosuppression in PDA by attracting regulatory T-cells and then polarizing macrophages toward immunosuppressive phenotypes (Pushalkar et al., 2018; Vitiello et al., 2019). The relationship between tumor neoantigens with microbial mimicry and anti-tumor responses may reflect an equilibrium balancing between immune recognition and neoantigen expression dynamics. Overall, our observations regarding T-cell global transcriptomic reactions have important implications for immunotherapy; differential therapeutic targeting of IMER or TMER T-cells could have clinical utility. Mechanistic studies will be needed to confirm or refute these possibilities.
Finally, the signature of cell-associated bacteria derived using SAHMI could predict patients at risk of poor survival, consistent with the observations that antibiotic reductions of bacterial numbers enhances gemcitabine efficacy in humans (Imai et al., 2019) and improves PDA responses to checkpoint inhibitors (CPI) in mice (Pushalkar et al., 2018). Although broad-spectrum antibiotic treatment has been associated with reduced response to CPI in humans (Lurienne et al., 2020; Pinato et al., 2019a; Thompson et al., 2017), CPI response can be influenced by microbiome composition (Pinato et al., 2019b) and microbial effects can be modulated by microbial transfer (Gopalakrishnan et al., 2018; Routy et al., 2018; Sivan et al., 2015). While our model predicts worse outcomes for infected tumors, others have reported increased intra-tumoral bacterial diversity in long-term survivors of pancreatic cancer (Riquelme et al., 2019). This dichotomy may reflect differences in technological platforms (bulk mRNA/single-cell mRNA/16S rDNA) and sample processing (fresh/frozen/formalin fixed paraffin embedded), or that only a subset of microbes promotes tumor growth. As such, higher overall diversity may suppress the effects of the more pathogenic subset and confer a survival advantage. Further studies will be necessary to resolve this point.
Our observations at single cell resolution confirm many known tumor-microbiome associations identified using bulk genomic data, model systems, or targeted experiments (Aykut et al., 2019; Nejman et al., 2020; Poore et al., 2020; Pushalkar et al., 2018; Sethi et al., 2019; Vitiello et al., 2019). Nonetheless, our study has important limitations. First, although we took several in silico measures to minimize contamination and low-quality data, these cannot replace gold-standard microbiology practices, including sterile processing, sterile-certified reagents, negative blanks of reagents, and multiple-sample pooling as ‘positive’ controls (Eisenhofer et al., 2019; Poore et al., 2020). While contaminants should not drive paired comparisons from identically processed samples, they may limit interpretation of smaller studies or inter-study comparisons. Second, our conservative data processing filters select for microbial taxa that are broadly present, but they may eliminate individual-specific, region-specific, low-abundance, or difficult-to-detect taxa. Third, nonspecific RNA capture may affect the rate of detection of specific taxa and impact overall microbial profiles. Fourth, we cannot determine whether detected microbial nucleic acids come from live, lysed, intra-, or extracellular microorganisms.
As with prior studies (Nejman et al., 2020; Poore et al., 2020) our data also do not establish direct causal relationships between microorganisms and the tumor microenvironment, i.e. whether microbes are oncogenic or pro-inflammatory, or whether they represent infections of established tumors. We cannot assess the role of environmental exposure or prior medical intervention (e.g. preoperative biliary stenting that maintains communication between the small intestine and pancreas, or antibiotic use) in selecting the observed microbial profiles in human samples. Additional studies will be necessary to directly test causation. Regardless, SAHMI creates opportunities to examine patterns of human-microbiome interactions from single-cell sequencing data without the need for additional experimental modifications, generating testable hypotheses about host-microbiome relationships at multiple levels. This framework is not tumor-specific and can be applied to study a variety of tissues and disease states, as well as other infectious agents such as viruses, fungi, or helminths.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further data and code requests should be directed to and will be fulfilled by the Lead Contact, Subhajyoti De (subhajyoti.de@rutgers.edu)
Materials availability
This study did not generate new unique reagents
Data and code availability
Single-cell RNA-seq, whole genome sequencing, and total RNA-seq data are deposited to dbGaP: phs003035.v1. Processed single-barcode microbiome data can be accessed via https://github.com/sjdlabgroup/SAHMI. Publicly available datasets are available via their respective accession codes (scPDA1: GSA: CRA001160, scPDA2: dbGaP: phs002071). Accession numbers are listed in the Key Resource Table.
Benchmarking of SAHMI is fully described in (Ghaddar et al., 2022). SAHMI source code, examples, and user-guides are available via https://github.com/sjdlabgroup/SAHMI under release SAHMI v1.0 and DOI: 10.5281/zenodo.7017103. DOIs are also listed in the Key Resource Table.
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-CA19-9 clone CA19.9/1390R | Novus | Cat#NBP2-54585APC |
| Biological samples | ||
| Fresh human tumor samples | This paper | See deposited data section |
| Chemicals, peptides, and recombinant proteins | ||
| Collagenase | Gibco | Cat#17100-017 |
| DNaseI | Sigma | Cat#DN25 |
| Hyalaronidase | Sigma | Cat#H3506 |
| ACK lysing buffer | Gibco | Cat#A10492-01 |
| Critical commercial assays | ||
| Qiagen blood and cell culture DNA mini kit | Qiagen | 13323 |
| QIAamp Fast DNA Tissue Kit | Qiagen | 51404 |
| RNeasy Mini Kit | Qiagen | 74104 |
| High Sensitivity dsDNA kit | Invitrogen | Q32851 |
| RNA High Sensitivity kit | Invitrogen | Q32852 |
| Deposited data | ||
| Genomic data | This paper | dbGaP: phs003035.v1 |
| Processed data | This paper | https://github.com/sjdlabgroup/SAHMI |
| scPDA1 scRNA-seq counts data | Peng et al., 2019 | GSA: CRA001160 |
| scPDA2 scRNA-seq counts data | Steele et al., 2020 | dbGaP: phs002071 |
| TCGA microbiome genus level summarized read counts | Poore et al., 2020 | ftp://ftp.microbio.me/pub/cancer_microbiome_analysis/ |
| 16S rDNA-seq of pancreatic tumors | Nejman et al., 2020 | DOI: 10.1126/science.aay9189 |
| TCGA PAAD normalized bulk RNA-seq and clinical data | Raphael et al., 2017 | https://tcga-data.nci.nih.gov/docs/publications/ |
| ICGC pancreatic cancer normalized bulk RNA-seq and clinical data | Hudson et al., 2010 | https://dcc.icgc.org/repositories |
| CPTAC3 pancreatic cancer normalized bulk RNA-seq and clinical data | Cao et al., 2021 | dbGaP: phs001287.v1.p1 |
| scRNA-seq of normal pancreas tissue | Baron et al., 2016 | GEO: GSE84133 |
| Software and algorithm | ||
| SAHMI (v1.0) | This paper, and Ghaddar et al. 2022 | https://github.com/sjdlabgroup/SAHMI DOI: 10.5281/zenodo.7017103 |
| R (v3.6.1) | R CRAN | https://www.r-project.org/ |
| Kraken2Uniq (v2) | Wood et al., 2019 | https://github.com/DerrickWood/kraken2 |
| STAR (v2.7.3) | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| Salmon (v1.9.0) | Patro et al., 2017 | https://github.com/COMBINE-lab/salmon |
| Seurat | Stuart et al., 2019 | https://cran.r-project.org/web/packages/Seurat/index.html |
| Tidyverse (v1.3.2) | R CRAN | https://www.tidyverse.org/ |
| clusterProfiler | Yu et al., 2016 | https://guangchuangyu.github.io/software/clusterProfiler/ |
| ReactomePA | Yu et al., 2016 | https://yulab-smu.top/biomedical-knowledge-mining-book/reactomepa.html |
| Survminer | R CRAN | https://github.com/kassambara/survminer |
| RTCGA | Bioconductor | https://www.bioconductor.org/packages/release/bioc/html/RTCGA.html |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Deidentified human tumor and non-malignant tissue samples subjected to genomic profiling were collected with written informed consent and ethics approval by the Rutgers Cancer Institute of New Jersey Institutional Review Board under protocol no. Pro2019002924 (PI: De). Patient metadata for these samples are available in Table S1.
METHOD DETAILS
SAHMI pipeline for detection and denoising of microbial sequences from scRNA-seq data
SAHMI (Single-cell Analysis of Host-Microbiome Interactions) is a statistical pipeline to identify true microbes and denoise contaminants and false positives from scRNA-seq of mammalian host tissues. We previously reported the full details and benchmarking of SAHMI (Ghaddar et al., 2022). This manuscript details the first application of SAHMI to study the tumor microbiome. We summarize key analytical components here.
Metagenomic classification of scRNA-seq data.
SAHMI is relies on the output of existing taxonomic classifiers. While it is designed to build on the output of Kraken2Uniq, the pipeline is applicable for any k-mer based classifier. It is essential that at this step that host reads are excluded or accounted for either prior to or during metagenomic classification (Ghaddar et al., 2022).
k-mer correlation test selects for true microbes.
SAHMI selects for true taxa by analyzing four correlations, as these are all shown to be strongly positive for true species. These are the correlations between (1) total number of minimizers (maximally informative k-mer groupings utilized by Kraken) vs. the number of unique minimizers across samples in a study, (2) the total number of minimizers vs. total number of reads across samples in a study, (3) the total number of reads vs. the number of unique minimizers across samples in a study, and (4) the total number of k-mers vs. the number of unique k-mers across barcodes in an individual sample. While minimizers are computationally more efficient than k-mers, in the absence of minimizer data the same metrics can be computed for k-mers.
Cell-line quantile test identifies contaminants and false positives.
the cell-line test is based on the pattern that contaminants appear in higher frequencies in negative control samples. In the absence of matched controls, cell line microbiome data can serve as a substitute, as any taxa detected in these are contaminants or false positives. SAHMI includes a microbiome reference from publicly available RNA-seq data for >2500 samples involving >1500 healthy and diseased human cell lines from >400 data providers from around the world. For each taxon in a test sample, SAHMI compares the fraction of microbiome reads assigned to the taxon [i.e. taxon counts/sum(all bacterial, fungal, viral counts), in reads per million] to the microbiome fraction assigned to the taxon in all cell line experiments. SAHMI tests whether the taxon microbial fraction in the test sample is greater than the 95th percentile (by default) of the taxon’s microbiome fraction distribution in cell line data using a one-sample quantile test.
Quantitation of microbes and joint analysis with somatic data.
Given a list of taxa to keep, SAHMI filters, extracts, and demultiplexes the relevant reads (Ghaddar et al., 2022). Parent classifications of each cell barcode’s microbiome is performed. A taxa by barcode matrix can be constructed at the desired resolution, and sparse barcodes can optionally be filtered. The scRNA-seq cellular barcodes can be used to pair microbes to somatic cells. Downstream analyses can occur at the sample or barcode level.
Cohort selection and metagenomic classification
This study primarily analyzed data from two scRNA-seq datasets: scPDA1 (Peng et al., 2019) and scPDA2 (Steele et al., 2020). scPDA1 contained data for 24 human pancreatic ductal adenocarcinomas (PDA) and 11 control pancreas tissues. scPDA2 contained data for 17 PDA tumor samples and 3 normal pancreas tissues. In both studies, normal tissues came from patients with non-pancreatic tumors or nonmalignant pancreatic lesions. All analyses from this study were replicated in these two independent cohorts. All tumor and non-malignant pancreas data from both studies were included. There was no randomization or blinding or blinding during data analysis and sample size estimation or power computations were not applicable. All tissues were obtained during pancreatectomy or pancreatoduodenectomy (Table S1). The samples were checked for batch effects at the levels of sample and somatic cell type clusters. The cohorts had 100–1000 million reads per sample, of which a substantial proportion did not map to the human genome, and these reads were used for metagenomic analyses. We also obtained data on microbial genera classified from bulk-RNA sequencing of pancreatic adenocarcinoma (PAAD) from TCGA (Poore et al., 2020) (selecting counts and normalized expression values of TCGA genera passing all decontamination steps), and genera classified from 16S-rDNA sequencing of pancreatic cancer in a recent large-scale study (Nejman et al., 2020) (normalized expression of genera passing all filters except the multi-study filter). Metagenomic classification was done using Kraken2Uniq (Breitwieser et al., 2018; Wood et al., 2019) with the RefSeq bacterial, fungal, and viral databases.
In-house patient samples, 16S-rDNA-seq, and experimental details
Biopsy samples were obtained for patients undergoing surgery for pancreatic ductal adenocarcinoma at Rutgers Robert Wood Johnson University Hospital and the University of Rochester Medical Center under an IRB approved protocol. Tumor tissue was minced and subsequently dissociated by enzymatic digestion (200 units/ml Collagenase type I, 60 units/ml hyaluronidase, and 100 μg/ml DNAse I, dissolved in DMEM media without serum) for 30 min at 37°C. Single cell suspensions were made by passing the digested tissue through a 100 um filter. Recovered cells were centrifuged at 500×g for 5 minutes and washed with Phosphate-buffered saline (PBS) containing 0.5% bovine serum albumin (BSA). Washed cells were pelleted. To eliminate red blood cells (RBC), cell pellets were resuspended in ACK lysing buffer (Thermofisher/Gibco) for 5 minutes at room temperature, and then pelleted. Cells were again washed in PBS/0.5% BSA, pelleted, and finally resuspended in PBS/0.5% BSA. For FACS isolation of CA19+ cells, cells were treated with a rabbit monoclonal antibody against CA19 (Novus NBP2-54585), which was conjugated to APC. The antibody treatment was for 30 minutes at room temperature, and then cells were pelleted by microcentrifugation at 500×g for 5 minutes. After washing in PBS/0.5%BSA and repelleting, the cells were resuspended in PBS/0.5%BSA. DAPI was added to indicate viability. For the Rutgers University sample, FACS was performed at the Rutgers Cancer Institute of New Jersey using a BD Biosciences Influx High Speed Cell Sorter, and scRNAseq was performed by Genewiz (Piscataway, NJ). For the Rochester sample, FACS was performed at the University of Rochester Medical Center using a BD FACSAria II, and scRNAseq was performed by the Genomics Research Center at the University of Rochester Medical Center.
Tumor DNA was extracted from tissue sample using Qiagen Genomic-tips and Qiagen blood and cell culture DNA mini kit. Briefly, tissue sample was minced and homogenized mechanically using Qiagen TissueRuptor II in lysis buffer, supplemented with Qiagen Proteinase K and incubated overnight at 50 °C. Thereafter, genomic DNA was purified following QIAGEN Genomic-tips procedure according to the manufacturer’s instructions. DNA yield was determined fluorometrically using the High Sensitivity dsDNA kit (Invitrogen) on Qubit 3.0 Fluorometer and DNA purity was estimated spectrophotometrically by measuring absorbance on QuickDrop (Molecular Devices).Sequencing libraries were generated by Novogene with library size of 350 bp. Whole-genome sequences of samples were generated onNovaseq PE150 platform by Novogene using paired-end sequencing strategy (2×150 bp) at a depth of 90 G raw data per sample. Microbiome calling from whole genome sequencing data was done using Kraken2 with the RefSeq bacterial, fungal, and viral databases.
For 16S Amplicon Metagenomics sequencing, DNA was extracted from tissue samples using QIAamp Fast DNA Tissue Kit (Qiagen) and for mRNA sequencing, RNA was extracted from tissue samples using RNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions. DNA and RNA yield was determined fluorometrically using the High Sensitivity dsDNA kit (Invitrogen) and RNA High Sensitivity kit (Invitrogen) respectively on Qubit 3.0 Fluorometer and purity was estimated spectrophotometrically by measuring absorbance on QuickDrop (Molecular Devices). For Amplicon Sequencing PCR amplification on target regions (amplicons), sequencing libraries preparation and sequencing was performed by Novogene. Novaseq 6000 PE250 platform was used to sequence the library with 0.05 M raw reads per sample. Library preparation and sequencing for mRNA sequencing was performed by Novogene using NovaSeq PE150 platform with 6 G raw data per sample.
Metagenomic denoising and benchmarking analyses
We validated the scPDA1-2 microbiome by multiple methods. First, we implemented the k-mer correlation tests and kept genera and species that had positive correlation values in all metrics with p<0.001 in both scPDA1 and 2. We also filtered for taxa with >100 reads per million Kraken-classified microbiome reads, >100 total reads, >1000 unique minimizers, >100 cell-associated counts. Second, we used the cell-line test to identify contaminants and false positives and kept only genera and species present at a level >95th percentile of what is found in the cell line negative control reference. Third, for reads resolved to the species level, we used STAR to align reads to their respective genome. STAR (Dobin et al., 2013) was used with the following parameters: alignIntronMax=1, outFilterScoreMinOverLread=0.1, outFilterMatchNminOverLread=0.1. Biomartr was used to retrieve reference genomes (Drost and Paszkowski, 2017). The fraction of reads mapped reported includes uniquely mapped and multi-mapped reads. Fourth, for denoised microbiome reads, we utilized a third RNS-seq mapper, Salmon (Patro et al., 2017), to map tumor and microbiome reads to the human genome. Salmon was run with the default parameters and minScoreFraction=0.8. Fifth, we utilized the MBodyMap (Jin et al., 2022) to compute body-site enrichment scores for the scPDA microbiome. We defined a body-site enrichment score for each body site (ESbody-site) as:
To calculate enrichment score probability values, we randomly shuffled the body-site label 100 times and recalculated the enrichment scores. P-values comparing the true vs. shuffled enrichment scores were calculated using one-sample Wilcoxon test. Sixth, we compared total genus counts in scPDA1-2 to genus counts from other PDA studies sequenced with different technologies. These included genera classified from bulk-RNA sequencing of the TCGA pancreatic cancer (TCGA-PAAD)(Poore et al., 2020), from 16S-rDNA sequencing of pancreatic cancer (Nejman et al., 2020), and our in-house 16S and total RNA-seq samples. Spearman correlations were run comparing total genus counts across all studies for pairwise complete genera. Seventh, we compared the overlap in detected genera between scPDA and other tissue not expected to have a similar microbiome. We analyzed scRNA-seq data from patient samples with the following clinically verified infections: Mycobacterium leprae (skin)(Ma et al., 2021), Helicobacter pylori (stomach)(Zhang et al., 2019b), and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2, bronchoalveolar lavage fluid)(Liao et al., 2020), in addition to data from scRNA-seq experiments with the following pathogens introduced: Candida albicans (human PBMCs)(Muñoz et al., 2019), Salmonella enterica (human PBMCs)(Saliba et al., 2016), and human alpha herpesvirus 1 (human PBMCs)(Wyler et al., 2019). The overlap coefficient of any genera between two sets was calculated as overlap (X, Y) = intersect(X, Y)/min(|X|, |Y|).
scRNA-seq data processing
All scRNA-seq data processing was done using Seurat (Stuart et al., 2019) with default parameters. In brief, data were TP10K normalized per cell and 2600 highly variable genes common to scPDA1 and 2 were identified using the FindVariableGenes function. Principle component analysis was performed separately using this data and uniform manifold approximation and projection plots were created using the first 50 principle components and default parameters. Cell-type annotations for scPDA1 were used from the original study. For scPDA2, Louvain clustering of cells with a resolution parameter of 0.5 was done using the top 50 principle components. Differentially expressed genes per cluster were identified via Wilcoxon testing as implemented in the FindAllMarkers function from Seurat. Cell-types were subsequently annotated by examining differentially expressed genes in each cluster. For the merged T-cell analysis, batch correction was done using the Seurat “Integration” method with default parameters, i.e. by identifying common variable genes across both datasets, identifying integration ‘anchors’, and running the IntegrateData function.
Association between microbes and cells, cellular processes, and diversity
Associations between microbes and cells was done using the FindAllMarkers function from Seurat testing for the relationship between normalized bacterial counts and cell-type pairing. All differential gene expression was done using the Seurat FindAllMarkers function with default parameters. Reactome pathway analysis was done using ReactomePA (Yu and He, 2016) with default parameters. Transcriptome diversity was computed for each cell using its cell-type’s top 500 most variable genes (FindVariableFeatures function). Diversity values were computed using the vegan package (https://github.com/vegandevs/vegan) and were compared across cell-types using Wilcoxon tests. Validation of bacteria-gene associations was done using the TCGA pancreatic cancer cohort (Poore et al., 2020). The bacterial genera and genes that had significant associations in both scPDA1-2 were subsetted from the TCGA RNA and microbiome datasets. Spearman correlations between these gene and genera in TCGA were computed and the number of significant correlations that were consistent with the associations found in scPDA1-2 was recorded. This was repeated 100 times for subsampled data vs. sample-label shuffled data, and the distributions of the numbers of scPDA-TCGA shared associations were compared using Wilcoxon tests.
T-cell microenvironment reaction analysis
A random forest model was trained and validated to classify IMER vs TMER T-cells based on their gene expression profiles. The model was trained using single-cell RNA sequencing data of T-cells isolated from peripheral blood mononuclear cells from patients with bacterial sepsis (https://singlecell.broadinstitute.org/single_cell; SCP548) or from primary lung adenocarcinomas (E-MTAB-6149), which were previously shown to have low microbiome burden(Nejman et al., 2020; Poore et al., 2020). Processed gene expression data were analyzed using Seurat(Stuart et al., 2019); cells were clustered based on transcriptomic profiles, and T-cells were identified using known markers(Nirmal et al., 2018). The FindAllMarkers function from Seurat was used to identify ~500 genes differentially expressed in T-cells from lung cancer and sepsis patients. We subsampled 1000 T-cells from each study and used the rank order of the ~500 differentially expressed genes to train a random forest model to classify tumor-reactive or microbe-reactive T-cells. We then validated the model using the remaining T-cells from the lung cancer and sepsis studies, as well as 6 other datasets with either known microbial stimulation or cancer with low-microbiome burden: bladder cancer (GSE149652), melanoma (GSE120575), glioblastoma (GSE131928), pilocytic astrocytoma (SCP271), Salmonella stimulation (GSM3855868), and Candida stimulation (https://eqtlgen.org/candida.html). Given the model’s exceptional accuracy in classifying over 100,000 T-cells from new datasets, we used it to predict T-cell microenvironment reaction from the two scPDA cohorts.
Survival modeling
Samples were divided into two groups based on presence (100–10,000 counts) or absence (<100 counts) of cell-associated bacteria. Next, for both scPDA cohorts, pseudo-bulk expression profiles for each sample was created by summing the read counts for each gene across all cells, and a Wilcoxon test was used to identify differentially expressed genes (p<0.0001) between samples with or without cell-associated bacteria. The resulting sample by gene data was used to train a gradient boosted tree classifier (Chen and Guestrin, 2016) to predict presence or absence of cell-associated bacteria using the following parameters: eval_metric = ‘auc’, num_class = 2, colsample_bynode=0.5, colsample_bytree=0.5. This model was then used to predict microbiome diversity in the TCGA, ICGC, and CPTAC3 pancreatic cancer cohorts based on their gene ranks. We stratified patients by their predicted cell-associated and used the survminer package (https://github.com/kassambara/survminer/) to test the relationship with survival and to plot Kaplan-Meier curves.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analyses were performed using R version 3.6.1 (https://www.r-project.org/). All p-values were false-discovery rate (fdr)- corrected for multiple hypothesis using the p.adjust function with method= “fdr”, unless otherwise stated. The ggpubr package (https://github.com/kassambara/ggpubr) was used to compare group means with nonparametric tests and to perform multiple hypothesis correction for statistics that are noted in figures. P-values reported as <2.2×10−16 result from reaching the calculation limit for native R statistical test functions and indicate values below this number, not a range of values. Data processing relied heavily on the Tidyverse v1.3.2 R packages (https://www.tidyverse.org/).
Supplementary Material
Table S1. Clinical characteristics of scPDA samples, Related to Figure 1.
Table S2. Bacteria-cell-gene associations, Related to Figure 3.
Table S3. Bacteria-cell-pathway associations, Related to Figure 3.
Highlights.
SAHMI enables systematic microbial detection from single-cell sequencing
A subset of tumors and no normal tissues have somatic-cell associated bacteria
Bacteria associate with cancer hallmarks and immune activity
Presence of cell-associated bacteria predicts worse prognosis
Acknowledgments
B.G. acknowledges National Center for Advancing Translational Sciences, Rutgers Clinical and Translational Science Award [TL1TR003019]. A.B. acknowledges New Jersey Commission for Cancer Research Fellowship [COCR23PDFOO]. S.D. acknowledges funding support from the National Institute of Health [R01GM129066, R21CA248122, and 5P30CA072720]. M.J.B. acknowledges funding support from National Institute of Health [U01 AI22285]. The authors acknowledge the Office of Advanced Research Computing (OARC) at Rutgers, The State University of New Jersey for providing access to the Amarel cluster URL: https://it.rutgers.edu/oarc. The funders had no role in study design, interpretation of results, and publication.
Footnotes
Declaration of Interests
MJB declares that he serves on the Scientific Advisory Board of Micronoma, Inc. BG and SD have jointly filed PCT patent applications PCT/US2022/025829 and PCT/US2022/025832.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abo H, Chassaing B, Harusato A, Quiros M, Brazil JC, Ngo VL, Viennois E, Merlin D, Gewirtz AT, Nusrat A, et al. (2020). Erythroid differentiation regulator-1 induced by microbiota in early life drives intestinal stem cell proliferation and regeneration. Nat. Commun. 11, 513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adejumo AC, Adejumo KL, and Pani LN (2019). Risk and Outcomes of Clostridium difficile Infection With Chronic Pancreatitis. Pancreas 48. [DOI] [PubMed] [Google Scholar]
- Adolph TE, Mayr L, Grabherr F, Schwärzler J, and Tilg H (2019). Pancreas-Microbiota Cross Talk in Health and Disease. Annu. Rev. Nutr. 39, 249–266. [DOI] [PubMed] [Google Scholar]
- Alfano M, Canducci F, Nebuloni M, Clementi M, Montorsi F, and Salonia A (2016). The interplay of extracellular matrix and microbiome in urothelial bladder cancer. Nat. Rev. Urol. 13, 77–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aykut B, Pushalkar S, Chen R, Li Q, Abengozar R, Kim JI, Shadaloey SA, Wu D, Preiss P, Verma N, et al. (2019). The fungal mycobiome promotes pancreatic oncogenesis via activation of MBL. Nature 574, 264–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balachandran VP, Luksza M, Zhao JN, Makarov V, Moral JA, Remark R, Herbst B, Askan G, Bhanot U, Senbabaoglu Y, et al. (2017). Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 551, S12–S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron M, Veres A, Wolock SL, Faust AL, Gaujoux R, Vetere A, Ryu JH, Wagner BK, Shen-Orr SS, Klein AM, et al. (2016). A Single-Cell Transcriptomic Map of the Human and Mouse Pancreas Reveals Inter- and Intra-cell Population Structure. Cell Syst. 3, 346–360.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitwieser FP, Baker DN, and Salzberg SL (2018). KrakenUniq: confident and fast metagenomics classification using unique k-mer counts. Genome Biol. 19, 198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L, Huang C, Cui Zhou D, Hu Y, Lih TM, Savage SR, Krug K, Clark DJ, Schnaubelt M, Chen L, et al. (2021). Proteogenomic characterization of pancreatic ductal adenocarcinoma. Cell 184, 5031–5052.e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, and Guestrin C (2016). XGBoost: A Scalable Tree Boosting System. ArXiv. [Google Scholar]
- Chikada T, Kanai T, Hayashi M, Kasai T, Oshima T, and Shiomi D (2021). Direct Observation of Conversion From Walled Cells to Wall-Deficient L-Form and Vice Versa in Escherichia coli Indicates the Essentiality of the Outer Membrane for Proliferation of L-Form Cells. Front. Microbiol. 12, 537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drost H-G, and Paszkowski J (2017). Biomartr: genomic data retrieval with R. Bioinformatics 33, 1216–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenhofer R, Minich JJ, Marotz C, Cooper A, Knight R, and Weyrich LS (2019). Contamination in Low Microbial Biomass Microbiome Studies: Issues and Recommendations. Trends Microbiol. 27, 105–117. [DOI] [PubMed] [Google Scholar]
- Feng M, Xiong G, Cao Z, Yang G, Zheng S, Song X, You L, Zheng L, Zhang T, and Zhao Y (2017). PD-1/PD-L1 and immunotherapy for pancreatic cancer. Cancer Lett. 407, 57–65. [DOI] [PubMed] [Google Scholar]
- De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S, Collini S, Pieraccini G, and Lionetti P (2010). Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. 107, 14691 LP – 14696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geller LT, Barzily-rokni M, Danino T, Jonas OH, Shental N, Nejman D, Gavert N, Zwang Y, Cooper ZA, Shee K, et al. (2017). Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science (80-. ). 1160, 1156–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaddar B, Blaser MJ, and De S (2022). Denoising sparse microbial signals from single-cell sequencing of mammalian host tissues. BioRxiv. doi: 10.1101/2022.06.29.498176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Goffau MC, Lager S, Salter SJ, Wagner J, Kronbichler A, Charnock-Jones DS, Peacock SJ, Smith GCS, and Parkhill J (2018). Recognizing the reagent microbiome. Nat. Microbiol. 3, 851–853. [DOI] [PubMed] [Google Scholar]
- Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, Prieto PA, Vicente D, Hoffman K, Wei SC, et al. (2018). Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science (80-. ). 359, 97–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajishengallis G, and Chavakis T (2019). DEL-1-Regulated Immune Plasticity and Inflammatory Disorders. Trends Mol. Med. 25, 444–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajnsdorf E, and Kaberdin VR (2018). RNA polyadenylation and its consequences in prokaryotes. Philos. Trans. R. Soc. B Biol. Sci. 373, 20180166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrdlickova R, Toloue M, and Tian B (2017). RNA-Seq methods for transcriptome analysis. Wiley Interdiscip. Rev. RNA 8, 10.1002/wrna.1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson (Chairperson) TJ, Anderson W, Aretz A, Barker AD, Bell C, Bernabé RR, Bhan MK, Calvo F, Eerola I, Gerhard DS, et al. (2010). International network of cancer genome projects. Nature 464, 993–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai H, Saijo K, Komine K, Otsuki Y, Ohuchi K, Sato Y, Okita A, Takahashi M, Takahashi S, Shirota H, et al. (2019). Antibiotic therapy augments the efficacy of gemcitabine-containing regimens for advanced cancer: a retrospective study. Cancer Manag. Res. 11, 7953–7965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen R, Krogfelt KA, Cawthraw SA, van Pelt W, Wagenaar JA, and Owen RJ (2008). Host-pathogen interactions in Campylobacter infections: the host perspective. Clin. Microbiol. Rev. 21, 505–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang S-H, Wang Y, Yang J-Y, Li J, Feng M-X, Wang Y-H, Yang X-M, He P, Tian G-A, Zhang X-X, et al. (2016). Overexpressed EDIL3 predicts poor prognosis and promotes anchorage-independent tumor growth in human pancreatic cancer. Oncotarget 7, 4226–4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin H, Hu G, Sun C, Duan Y, Zhang Z, Liu Z, Zhao X-M, and Chen W-H (2022). mBodyMap: a curated database for microbes across human body and their associations with health and diseases. Nucleic Acids Res. 50, D808–D816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamarajan P, Ateia I, Shin JM, Fenno JC, Le C, Zhan L, Chang A, Darveau R, and Kapila YL (2020). Periodontal pathogens promote cancer aggressivity via TLR/MyD88 triggered activation of Integrin/FAK signaling that is therapeutically reversible by a probiotic bacteriocin. PLOS Pathog. 16, e1008881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y-S, Kim T-Y, Kim Y, Lee S-H, Kim S, Kang SW, Yang J-Y, Baek I-J, Sung YH, Park Y-Y, et al. (2018). Microbiota-Derived Lactate Accelerates Intestinal Stem-Cell-Mediated Epithelial Development. Cell Host Microbe 24, 833–846.e6. [DOI] [PubMed] [Google Scholar]
- Liao M, Liu Y, Yuan J, Wen Y, Xu G, Zhao J, Cheng L, Li J, Wang X, Wang F, et al. (2020). Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 26, 842–844. [DOI] [PubMed] [Google Scholar]
- Lurienne L, Cervesi J, Duhalde L, de Gunzburg J, Andremont A, Zalcman G, Buffet R, and Bandinelli P-A (2020). NSCLC Immunotherapy Efficacy and Antibiotic Use: A Systematic Review and Meta-Analysis. J. Thorac. Oncol. 15, 1147–1159. [DOI] [PubMed] [Google Scholar]
- Ma F, Hughes TK, Teles RMB, Andrade PR, de Andrade Silva BJ, Plazyo O, Tsoi LC, Do T, Wadsworth MH, Oulee A, et al. (2021). The cellular architecture of the antimicrobial response network in human leprosy granulomas. Nat. Immunol. 22, 839–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maes A, Gracia C, Innocenti N, Zhang K, Aurell E, and Hajnsdorf E (2017). Landscape of RNA polyadenylation in E. coli. Nucleic Acids Res. 45, 2746–2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallapaty S (2017). Gnotobiotics: getting a grip on the microbiome boom. Lab Anim. (NY). 46, 373–377. [DOI] [PubMed] [Google Scholar]
- Máté V, Jemima H, Nóra I, Róbert A, Dávid R, Éva V, Balázs S, Márton H, Renáta T, Attila S, et al. (2022). Candida albicans Enhances the Progression of Oral Squamous Cell Carcinoma In Vitro and In Vivo. MBio 13, e03144–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mickiewicz KM, Kawai Y, Drage L, Gomes MC, Davison F, Pickard R, Hall J, Mostowy S, Aldridge PD, and Errington J (2019). Possible role of L-form switching in recurrent urinary tract infection. Nat. Commun. 10, 4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffitt RA, Marayati R, Flate EL, Volmar KE, Loeza SGH, Hoadley KA, Rashid NU, Williams LA, Eaton SC, Chung AH, et al. (2015). Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 47, 1168–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz JF, Delorey T, Ford CB, Li BY, Thompson DA, Rao RP, and Cuomo CA (2019). Coordinated host-pathogen transcriptional dynamics revealed using sorted subpopulations and single macrophages infected with Candida albicans. Nat. Commun. 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nejman D, Livyatan I, Fuks G, Gavert N, Zwang Y, Leore T, Rotter-maskowitz A, Weiser R, Mallel G, Gigi E, et al. (2020). The human tumor microbiome is composed of tumor type-specific intra-cellular bacteria. Science (80-. ). 980, 973–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TLA, Vieira-Silva S, Liston A, and Raes J (2015). How informative is the mouse for human gut microbiota research? Dis. Model. &Amp; Mech. 8, 1 LP – 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nirmal AJ, Regan T, Shih BB, Hume DA, Sims AH, and Freeman TC (2018). Immune Cell Gene Signatures for Profiling the Microenvironment of Solid Tumors. Cancer Immunol. Res. 6, 1388 LP – 1400. [DOI] [PubMed] [Google Scholar]
- Ochi A, Nguyen AH, Bedrosian AS, Mushlin HM, Zarbakhsh S, Barilla R, Zambirinis CP, Fallon NC, Rehman A, Pylayeva-Gupta Y, et al. (2012). MyD88 inhibition amplifies dendritic cell capacity to promote pancreatic carcinogenesis via Th2 cells. J. Exp. Med. 209, 1671–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Öğrendik M (2017). Periodontal Pathogens in the Etiology of Pancreatic Cancer. Gastrointest. Tumors 3, 125–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkuma R, Yada E, Ishikawa S, Komura D, Ishizaki H, Tamada K, Kubota Y, Hamada K, Ishida H, Hirasawa Y, et al. (2020). High expression of olfactomedin-4 is correlated with chemoresistance and poor prognosis in pancreatic cancer. PLoS One 15, e0226707–e0226707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patro R, Duggal G, Love MI, Irizarry RA, and Kingsford C (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J, Sun BF, Chen CY, Zhou JY, Chen YS, Chen H, Liu L, Huang D, Jiang J, Cui GS, et al. (2019). Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Nat. Cell Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinato DJ, Howlett S, Ottaviani D, Urus H, Patel A, Mineo T, Brock C, Power D, Hatcher O, Falconer A, et al. (2019a). Association of Prior Antibiotic Treatment With Survival and Response to Immune Checkpoint Inhibitor Therapy in Patients With Cancer. JAMA Oncol. 5, 1774–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinato DJ, Gramenitskaya D, Altmann DM, Boyton RJ, Mullish BH, Marchesi JR, and Bower M (2019b). Antibiotic therapy and outcome from immune-checkpoint inhibitors. J. Immunother. Cancer 7, 287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poore GD, Kopylova E, Zhu Q, Carpenter C, Fraraccio S, Wandro S, Kosciolek T, Janssen S, Metcalf J, Song SJ, et al. (2020). Microbiome analyses of blood and tissues suggest cancer diagnostic approach. Nature 579, 567–574. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Pushalkar S, Hundeyin M, Daley D, Zambirinis CP, Kurz E, Mishra A, Mohan N, Aykut B, Usyk M, Torres LE, et al. (2018). The pancreatic cancer microbiome promotes oncogenesis by induction of innate and adaptive immune suppression. Cancer Discov. 8, 403–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raphael BJ, Hruban RH, Aguirre AJ, Moffitt RA, Yeh JJ, Stewart C, Robertson AG, Cherniack AD, Gupta M, Getz G, et al. (2017). Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 32, 185–203.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes M, Filbin MR, Bhattacharyya RP, Billman K, Eisenhaure T, Hung DT, Levy BD, Baron RM, Blainey PC, Goldberg MB, et al. (2020). An immune-cell signature of bacterial sepsis. Nat. Med. 26, 333–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riquelme E, Zhang Y, Zhang L, Montiel M, Zoltan M, Dong W, Quesada P, Sahin I, Chandra V, San Lucas A, et al. (2019). Tumor Microbiome Diversity and Composition Influence Pancreatic Cancer Outcomes. Cell 178, 795–806.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillère R, Fluckiger A, Messaoudene M, Rauber C, Roberti MP, et al. (2018). Gut microbiome influences efficacy of PD-1–based immunotherapy against epithelial tumors. Science (80-. ). 359, 91 LP – 97. [DOI] [PubMed] [Google Scholar]
- Saliba AE, Li L, Westermann AJ, Appenzeller S, Stapels DAC, Schulte LN, Helaine S, and Vogel J (2016). Single-cell RNA-seq ties macrophage polarization to growth rate of intracellular Salmonella. Nat. Microbiol. 2, 1–8. [DOI] [PubMed] [Google Scholar]
- Saluja AK, and Dudeja V (2013). Relevance of Animal Models of Pancreatic Cancer and Pancreatitis to Human Disease. Gastroenterology 144, 1194–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert L, Werba G, Tiwari S, Giao Ly NN, Alothman S, Alqunaibit D, Avanzi A, Barilla R, Daley D, Greco SH, et al. (2016). The necrosome promotes pancreatic oncogenesis via CXCL1 and Mincle-induced immune suppression. Nature 532, 245–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethi V, Kurtom S, Tarique M, Lavania S, Malchiodi Z, Hellmund L, Zhang L, Sharma U, Giri B, Garg B, et al. (2018). Gut Microbiota Promotes Tumor Growth in Mice by Modulating Immune Response. Gastroenterology 155, 33–37.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethi V, Vitiello GA, Saxena D, Miller G, and Dudeja V (2019). The Role of the Microbiome in Immunologic Development and its Implication For Pancreatic Cancer Immunotherapy. Gastroenterology 156, 2097–2115.e2. [DOI] [PubMed] [Google Scholar]
- Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, Benyamin FW, Lei YM, Jabri B, Alegre M-L, et al. (2015). Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 350, 1084–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele NG, Carpenter ES, Kemp SB, Sirihorachai VR, The S, Delrosario L, Lazarus J, Amir E. ad D., Gunchick V, Espinoza C, et al. (2020). Multimodal mapping of the tumor and peripheral blood immune landscape in human pancreatic cancer. Nat. Cancer 1, 1097–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Stoeckius M, Smibert P, Satija R, Stuart T, Butler A, Hoffman P, Hafemeister C, et al. (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suau A, Bonnet R, Sutren M, Godon J-J, Gibson GR, Collins MD, and Doré J (1999). Direct Analysis of Genes Encoding 16S rRNA from Complex Communities Reveals Many Novel Molecular Species within the Human Gut. Appl. Environ. Microbiol. 65, 4799 LP – 4807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas RM, and Jobin C (2020). Microbiota in pancreatic health and disease: the next frontier in microbiome research. Nat. Rev. Gastroenterol. Hepatol. 17, 53–64. [DOI] [PubMed] [Google Scholar]
- Thomas RM, Gharaibeh RZ, Gauthier J, Beveridge M, Pope JL, Guijarro MV, Yu Q, He Z, Ohland C, Newsome R, et al. (2018). Intestinal microbiota enhances pancreatic carcinogenesis in preclinical models. Carcinogenesis 39, 1068–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J, Szabo A, Arce-Lara C, and Menon S (2017). Antibiotic Use Is Associated with Inferior Survival for Lung Cancer Patients Receiving PD-1 Inhibitors. J. Thorac. Oncol. 12, S1998. [Google Scholar]
- Torres PJ, Fletcher EM, Gibbons SM, Bouvet M, Doran KS, and Kelley ST (2015). Characterization of the salivary microbiome in patients with pancreatic cancer. PeerJ 3, e1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitiello GA, Cohen DJ, and Miller G (2019). Harnessing the Microbiome for Pancreatic Cancer Immunotherapy. Trends in Cancer 5, 670–676. [DOI] [PubMed] [Google Scholar]
- Wang X, He Y, Zhang Q, Ren X, and Zhang Z (2021). Direct Comparative Analyses of 10X Genomics Chromium and Smart-seq2. Genomics, Proteomics, and Bioinformatics. 19, 253–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei MY, Shi S, Liang C, Meng QC, Hua J, Zhang YY, Liu J, Zhang B, Xu J, and Yu XJ (2019). The microbiota and microbiome in pancreatic cancer: More influential than expected. Mol. Cancer 18, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermann AJ, and Vogel J (2021). Cross-species RNA-seq for deciphering host–microbe interactions. Nat. Rev. Genet. 22, 361–378. [DOI] [PubMed] [Google Scholar]
- Wood DE, Lu J, and Langmead B (2019). Improved metagenomic analysis with Kraken 2. Genome Biol. 20, 257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyler E, Franke V, Menegatti J, Kocks C, Boltengagen A, Praktiknjo S, Walch-Rückheim B, Bosse J, Rajewsky N, Grässer F, et al. (2019). Single-cell RNA-sequencing of herpes simplex virus 1-infected cells connects NRF2 activation to an antiviral program. Nat. Commun. 10, 4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, Iwakura Y, Oshima K, Morita H, Hattori M, et al. (2013). Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 499, 97–101. [DOI] [PubMed] [Google Scholar]
- Yu G, and He Q-Y (2016). ReactomePA: an R/Bioconductor package for reactome pathway analysis and visualization. Mol. BioSyst. 12, 477–479. [DOI] [PubMed] [Google Scholar]
- Yu S, Parameswaran N, Li M, Wang Y, Jackson MW, Liu H, Xin W, and Zhou L (2016). CRABP-II enhances pancreatic cancer cell migration and invasion by stabilizing interleukin 8 expression. Oncotarget 8, 52432–52444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambirinis CP, Ochi A, Barilla R, Greco S, Deutsch M, and Miller G (2013). Induction of TRIF- or MYD88-dependent pathways perturbs cell cycle regulation in pancreatic cancer. Cell Cycle 12, 1153–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambirinis CP, Levie E, Nguy S, Avanzi A, Barilla R, Xu Y, Seifert L, Daley D, Greco SH, Deutsch M, et al. (2015). TLR9 ligation in pancreatic stellate cells promotes tumorigenesis. J. Exp. Med. 212, 2077–2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Corredor ALG, Messina-Pacheco J, Li Q, Zogopoulos G, Kaddour N, Wang Y, Shi B, Gregorieff A, Liu J, et al. (2021). REG3A/REG3B promotes acinar to ductal metaplasia through binding to EXTL3 and activating the RAS-RAF-MEK-ERK signaling pathway. Commun. Biol. 4, 688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M-Y, Wang J, and Guo J (2019a). Role of Regenerating Islet-Derived Protein 3A in Gastrointestinal Cancer. Front. Oncol. 9, 1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Yang M, Zhang Y, Xiao S, Lai X, Tan A, Du S, and Li S (2019b). Dissecting the Single-Cell Transcriptome Network Underlying Gastric Premalignant Lesions and Early Gastric Cancer. Cell Rep. 27, 1934–1947.e5. [DOI] [PubMed] [Google Scholar]
- Zinter MS, Mayday MY, Ryckman KK, Jelliffe-Pawlowski LL, and Derisi JL (2019). Towards precision quantification of contamination in metagenomic sequencing experiments. Microbiome 7, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Clinical characteristics of scPDA samples, Related to Figure 1.
Table S2. Bacteria-cell-gene associations, Related to Figure 3.
Table S3. Bacteria-cell-pathway associations, Related to Figure 3.
Data Availability Statement
Single-cell RNA-seq, whole genome sequencing, and total RNA-seq data are deposited to dbGaP: phs003035.v1. Processed single-barcode microbiome data can be accessed via https://github.com/sjdlabgroup/SAHMI. Publicly available datasets are available via their respective accession codes (scPDA1: GSA: CRA001160, scPDA2: dbGaP: phs002071). Accession numbers are listed in the Key Resource Table.
Benchmarking of SAHMI is fully described in (Ghaddar et al., 2022). SAHMI source code, examples, and user-guides are available via https://github.com/sjdlabgroup/SAHMI under release SAHMI v1.0 and DOI: 10.5281/zenodo.7017103. DOIs are also listed in the Key Resource Table.
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-CA19-9 clone CA19.9/1390R | Novus | Cat#NBP2-54585APC |
| Biological samples | ||
| Fresh human tumor samples | This paper | See deposited data section |
| Chemicals, peptides, and recombinant proteins | ||
| Collagenase | Gibco | Cat#17100-017 |
| DNaseI | Sigma | Cat#DN25 |
| Hyalaronidase | Sigma | Cat#H3506 |
| ACK lysing buffer | Gibco | Cat#A10492-01 |
| Critical commercial assays | ||
| Qiagen blood and cell culture DNA mini kit | Qiagen | 13323 |
| QIAamp Fast DNA Tissue Kit | Qiagen | 51404 |
| RNeasy Mini Kit | Qiagen | 74104 |
| High Sensitivity dsDNA kit | Invitrogen | Q32851 |
| RNA High Sensitivity kit | Invitrogen | Q32852 |
| Deposited data | ||
| Genomic data | This paper | dbGaP: phs003035.v1 |
| Processed data | This paper | https://github.com/sjdlabgroup/SAHMI |
| scPDA1 scRNA-seq counts data | Peng et al., 2019 | GSA: CRA001160 |
| scPDA2 scRNA-seq counts data | Steele et al., 2020 | dbGaP: phs002071 |
| TCGA microbiome genus level summarized read counts | Poore et al., 2020 | ftp://ftp.microbio.me/pub/cancer_microbiome_analysis/ |
| 16S rDNA-seq of pancreatic tumors | Nejman et al., 2020 | DOI: 10.1126/science.aay9189 |
| TCGA PAAD normalized bulk RNA-seq and clinical data | Raphael et al., 2017 | https://tcga-data.nci.nih.gov/docs/publications/ |
| ICGC pancreatic cancer normalized bulk RNA-seq and clinical data | Hudson et al., 2010 | https://dcc.icgc.org/repositories |
| CPTAC3 pancreatic cancer normalized bulk RNA-seq and clinical data | Cao et al., 2021 | dbGaP: phs001287.v1.p1 |
| scRNA-seq of normal pancreas tissue | Baron et al., 2016 | GEO: GSE84133 |
| Software and algorithm | ||
| SAHMI (v1.0) | This paper, and Ghaddar et al. 2022 | https://github.com/sjdlabgroup/SAHMI DOI: 10.5281/zenodo.7017103 |
| R (v3.6.1) | R CRAN | https://www.r-project.org/ |
| Kraken2Uniq (v2) | Wood et al., 2019 | https://github.com/DerrickWood/kraken2 |
| STAR (v2.7.3) | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| Salmon (v1.9.0) | Patro et al., 2017 | https://github.com/COMBINE-lab/salmon |
| Seurat | Stuart et al., 2019 | https://cran.r-project.org/web/packages/Seurat/index.html |
| Tidyverse (v1.3.2) | R CRAN | https://www.tidyverse.org/ |
| clusterProfiler | Yu et al., 2016 | https://guangchuangyu.github.io/software/clusterProfiler/ |
| ReactomePA | Yu et al., 2016 | https://yulab-smu.top/biomedical-knowledge-mining-book/reactomepa.html |
| Survminer | R CRAN | https://github.com/kassambara/survminer |
| RTCGA | Bioconductor | https://www.bioconductor.org/packages/release/bioc/html/RTCGA.html |