

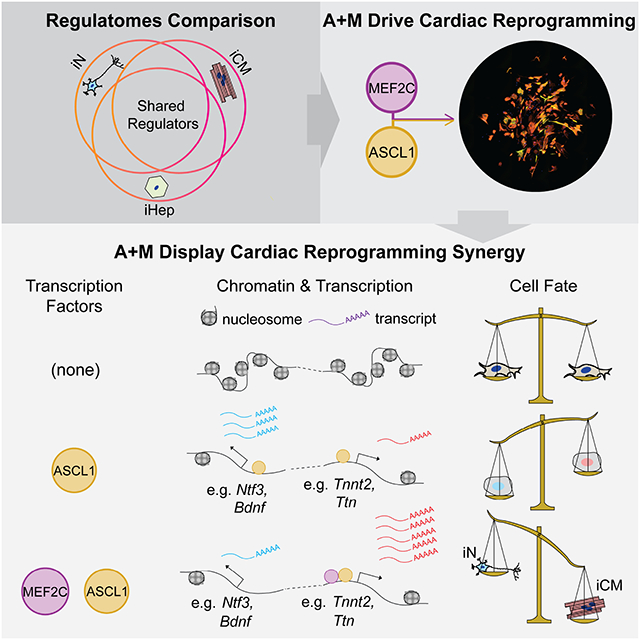
Summary:
Direct reprogramming has revolutionized the fields of stem cell biology and regenerative medicine. However, the common mechanisms governing how reprogramming cells undergo transcriptome and epigenome remodeling (i.e., regulatome remodeling) have not been investigated. Here, by characterizing early changes in the regulatome of three different types of direct reprogramming, we identify lineage-specific features as well as common regulatory transcription factors. Of particular interest, we discover that the neuronal factor Ascl1 possesses cross-lineage potential; together with Mef2c, it drives efficient cardiac reprogramming towards a mature iCM phenotype. Through ChIP-seq and RNA-seq, we find that MEF2C drives the shift in ASCL1 binding away from neuronal genes towards cardiac genes, guiding their co-operative epigenetic and transcription activities. Together, these findings demonstrate the existence of common regulators of different direct reprogramming and argue against the premise that TFs possess only lineage-specific capabilities for altering cell fate – the basic premise used to develop direct reprogramming approaches.
Keywords: Direct reprogramming, epigenetics, regulatome, Ascl1, Mef2c, cell fate conversion, neuronal reprogramming, cardiac reprogramming
Graphical Abstract
eTOC
By characterizing the epigenome of direct reprogramming processes, Qian and colleagues discovered common features of direct reprogramming. Particularly, Ascl1, the neuron reprogramming factor, activates a unique set of cardiac genes. Together with Ascl1, Mef2c efficiently induces iCM through driving Ascl1 binding away from neuronal loci to further activate cardiac program.
Introduction
Direct reprogramming is the process of converting one somatic cell type to another without transitioning through an intermediate pluripotent state (Wang et al., 2021a). Reprogramming is typically achieved by exogenous expression of one or more transcription factors (TFs) that, developmentally, are necessary for the acquisition of terminal cell fates (Ieda et al., 2010; Sekiya and Suzuki, 2011; Vierbuchen et al., 2010). Unlike development – in which master regulatory TFs drive differentiation of stem and progenitor cells – somatic cells (usually fibroblasts) are the starting cell type in direct reprogramming. Somatic cells constitute an atypical environment; their epigenetic state and endogenous molecular milieu are not specifically primed for differentiation nor trans-differentiation, suggesting that direct reprogramming has its own molecular mechanisms, distinct from development or other biological contexts.
Recent studies into the early stages of direct reprogramming have led to important insights regarding cell plasticity, fate determination, and epigenetics (Horisawa et al., 2020; Liu et al., 2017; Stone et al., 2019; Treutlein et al., 2016; Vierbuchen et al., 2017; Wapinski et al., 2013; Zhou et al., 2019). Shortly after induction, reprogramming causes broad, lineage-directed changes in chromatin and gene expression. As reprogramming progresses, the cells navigate fate “decisions” whereby they either continue towards acquiring the target cell fate or divert towards a different one (Treutlein et al., 2016; Zhou et al., 2019). Finally, when reprogramming is successful, it suppresses fibroblast identity regardless of the target cell fate. Initiating and driving these changes are reprogramming TFs; they have pioneer capabilities to open closed chromatin, lineage specificity to activate genes relevant to the target cell fate, and suppressive functions to repress non-target cell fates and silence fibroblast-specific genes (Liu et al., 2016; Zaret and Carroll, 2011). These common features of direct reprogramming suggest the existence of shared, uncharacterized mechanisms of transcriptome and epigenome remodeling that hold true across various types of direct reprogramming.
Here, we characterize and compare the re-patterning of the transcriptome as well as the enhancer and super-enhancer landscapes – i.e., the regulatome – in the early stages of direct reprogramming of induced neurons (iN), induced hepatocytes (iHep), and induced cardiomyocytes (iCM), representing derivatives of the three germ layers. We find that lineage-specific enhancer acquisition and gene expression requires not only the lineage-specific reprogramming factors but also the activities of AP-1 and TEAD factors in concert with reprogramming TFs. Based on our discovery of a common cardiac program in iN and iCM reprogramming and the enrichment of Ascl1’s motif in activated iCM enhancers, we demonstrate the cross-lineage potential of Ascl1, a neuron-specific transcription factor, as an iCM reprogramming factor. Interestingly, Ascl1 acts more potently as a pioneer factor for iCM reprogramming and can replace the canonical iCM factors Gata4 and Tbx5, forming a highly efficient two-factor combination with Mef2c. Mechanistically, we find that cardiac reprogramming with Ascl1 and Mef2c traverses different lineage branch points to repress neuronal and fibroblast identity. Furthermore, when co-expressed, these two factors shift their genomic targets away from a pro-neural program towards a pro-cardiac program.
Results
Characterization of global regulatome dynamics during iN, iHep, and iCM reprogramming
To explore both common and distinct regulatory mechanisms underlying iN, iHep, and iCM reprogramming, we utilized established reprogramming cocktails to generate the corresponding target cells (Fig. 1A–C) and performed both mRNA-seq and H3K27Ac ChIP-seq three days after initiating direct reprogramming. The genes upregulated in each type of reprogramming were associated with the molecular and functional features of the target cell types, indicating successful cell fate conversion (Supplemental Fig. 1A). Although most upregulated genes from one type of reprogramming did not show significantly increased expression in the other two types of reprogramming, subsets of activated genes exhibited elevated expression in multiple types of reprogramming (Fig. 1D–F). Most interestingly, the genes upregulated in both iN and iCM reprogramming are predominately related to muscle development, whereas genes activated in both iHep and iN cells are involved in immune response, and genes upregulated in both iCM and iHep are related to peptidase activity, kidney morphogenesis, cholesterol storage, and thermogenesis. These data suggest that cell fate changes induced by reprogramming are partly ambiguous; this may be caused by promiscuous binding of TFs to genes which they do not normally bind during development (Soufi et al., 2015; Treutlein et al., 2016).
Figure 1. Characterization of regulatome dynamics during iN, iHep and iCM reprogramming.
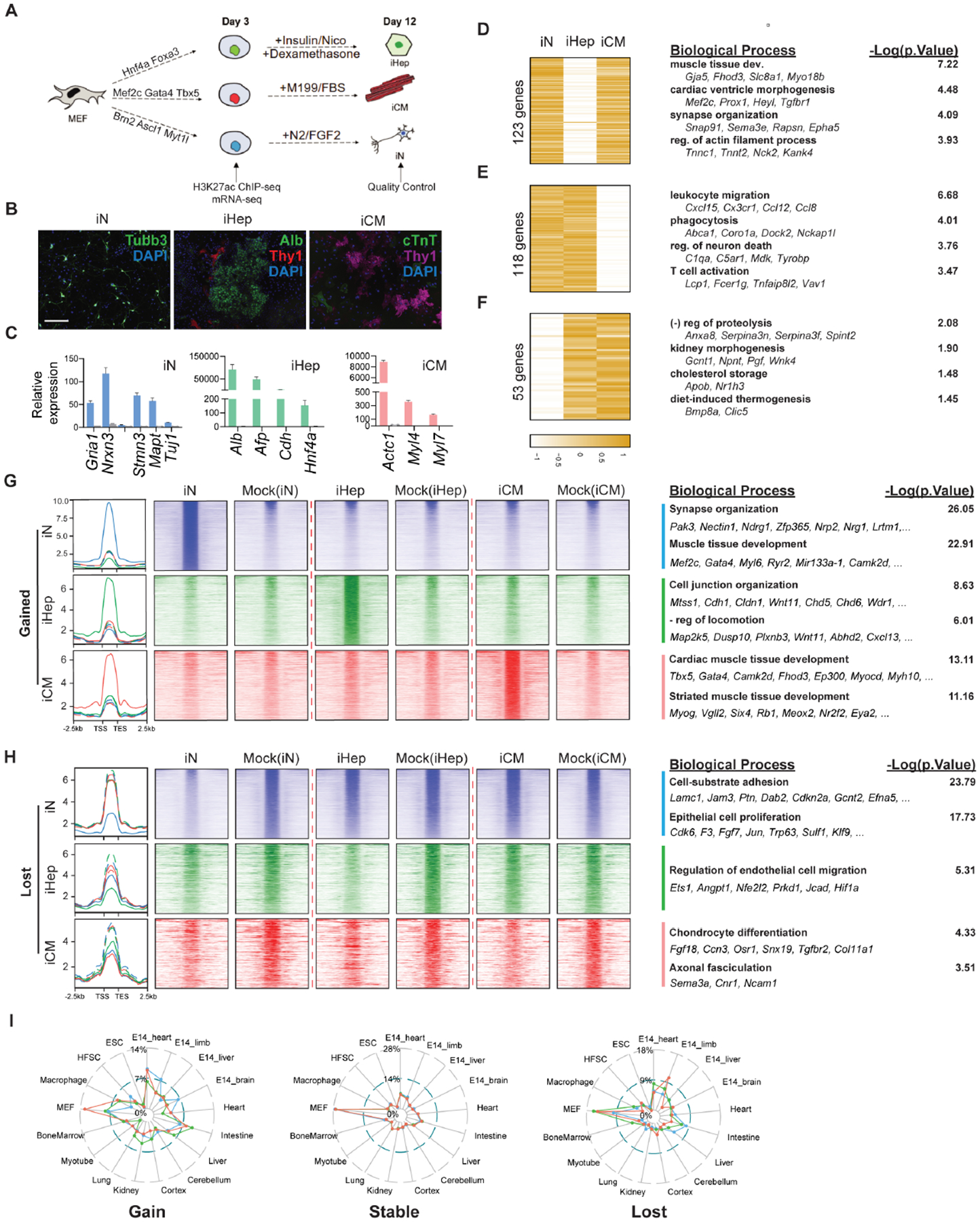
A. Schematic of the experiment design.
B. Immunofluorescence images showing successful reprogramming of iN, iHep, and iCM by their expression of lineage-specific markers. Scale bar: 275 μm
C. Lineage-specific gene expression of iN (blue), iHep (green), and iCM(red) at Day 12 post-infection. Error bars represent standard deviation. n=3.
D-F. Shared upregulated genes between iN (D), iHep (E) or iN (F) and iCM reprogramming. Gene ontology is shown to the right. Heatmap units are log2(fold change in gene expression) of reprogramming vs control. Color scale is row Z-score.
G-H. H3K27Ac signal of enhancers gained (G) and lost (H) during iN (blue), iHep (green), and iCM (red) reprogramming. The signal of mock infection control was on the right of the corresponding reprogramming. Select significant biological processes are shown together with representative genes to the right.
I. The percent overlap between the super enhancers gained, maintained (i.e., stable), or lost during reprogramming versus super enhancers of different cell types/tissues in the dbSUPER database. Color key: blue = iN; green = iHep; red = iCM.
See also Figure S1.
With this TF promiscuity hypothesis in mind, we characterized the reconfiguration of active enhancers and promoters during iN, iHep, and iCM reprogramming using our H3K27Ac ChIP-seq data. Principal component analysis, sample distance computation, and a higher number of differentially enriched H3K27Ac peaks in iN reprogramming all suggest that iN undergoes more dramatic enhancer remodeling than iHep or iCM (Supplemental Fig. 1B–D). We then sought to identify shared epigenetic changes by comparing the enhancers with significantly increased or decreased H3K27Ac across all three types of reprogramming (Fig. 1 G, H). Gene ontology (GO) analysis revealed that the biological processes associated with gained enhancers were mostly related to the molecular and functional characteristics of the target cell types, suggesting that these enhancers were largely cell-type specific – with one exception: in addition to enhancers involved in regulation of neuronal processes, we observed an enrichment of muscle-related enhancers in iN reprogramming – consistent with our transcriptome analysis and a previous report (Fig. 1D).(Treutlein et al., 2016) Analysis of lost enhancers across the three types of reprogramming suggests that each process deactivated fibroblast enhancers to varying degrees, with iN reprogramming exhibiting the broadest loss of H3K27Ac signal (Fig. 1H). These results suggest that remodeling of the enhancer landscape in iN is broad and partially directed towards muscle fate.
Following our analysis of gene expression and enhancer dynamics, we asked if changes to the super enhancers (SEs) in iN, iHep and iCM showed similar trends (Supplemental Fig. 1G). SEs are large clusters of enhancers in close genomic proximity with unusually high levels of transcription factor binding (Whyte et al., 2013). Moreover, SEs tend to be cell-type specific (Hnisz et al., 2013). As expected, most of the SE changes were found to be unique to each cell type (Supplemental Fig. 1H). The SEs gained during iCM reprogramming overlapped primarily with embryonic heart and mouse embryonic fibroblast (MEF) SEs (Fig. 1I). The SEs gained in iHep overlapped SEs found in adult liver and intestine, in line with the previously reported hepatic and intestinal potential of iHeps.(Morris et al., 2014) Similarly as the gained enhancers, in iN, the gained SEs overlapped primarily with SEs in embryonic muscle tissues (heart and limb), and secondarily with embryonic brain. This result suggests that iHep and iCM are gaining SEs related to their germ layer (intestine/liver and heart, respectively), while iN are not only gaining neuron-related SEs but also muscle-related SEs.
Integrated analysis reveals non-cocktail TFs that regulate diverse types of direct reprogramming
Following our enhancer and SE analyses, we next analyzed TF motif dynamics within H3K27Ac-marked cis-regulatory elements (CREs, i.e., enhancers and promoters) gained during reprogramming. The binding motifs for most of the reprogramming factors were significantly enriched in the gained CREs, suggesting reprogramming factors play a crucial role in reconfiguring the regulatome (Supplemental Fig. 2A). The motifs for Gata4 and Tbx5, however, were not significantly enriched in the gained CREs of iCM. In addition to enrichment of the binding motifs for reprogramming TFs, there was significant overrepresentation of the binding motifs for non-cocktail factors, such as TEAD and bZIP family TFs, in the gained CREs for all three types of direct reprogramming (Supplemental Fig. 2B). We reasoned that non-cocktail TFs critical for reprogramming should co-occupy gained CREs with reprogramming TFs; thus, we compared the co-occurrence of the binding motifs of reprogramming TFs and non-cocktail TFs in CREs gained during direct reprogramming to the motif co-occurrences present at genomic background levels (Supplemental Fig. 2C). For reprogramming TFs specifically, the binding motifs for Ascl1, Hnf4a, Foxa3, and Mef2c co-occurred significantly with the motifs of a variety of non-cocktail TFs (Fig. 2A). Among these non-cocktail TFs, motifs for TFs in the bZIP and TEAD families co-occurred with the motifs of reprogramming TFs in all three types of direct reprogramming (Fig. 2A).
Figure 2. Integrated analysis reveals non-cocktail TFs that regulate diverse types of direct reprogramming.
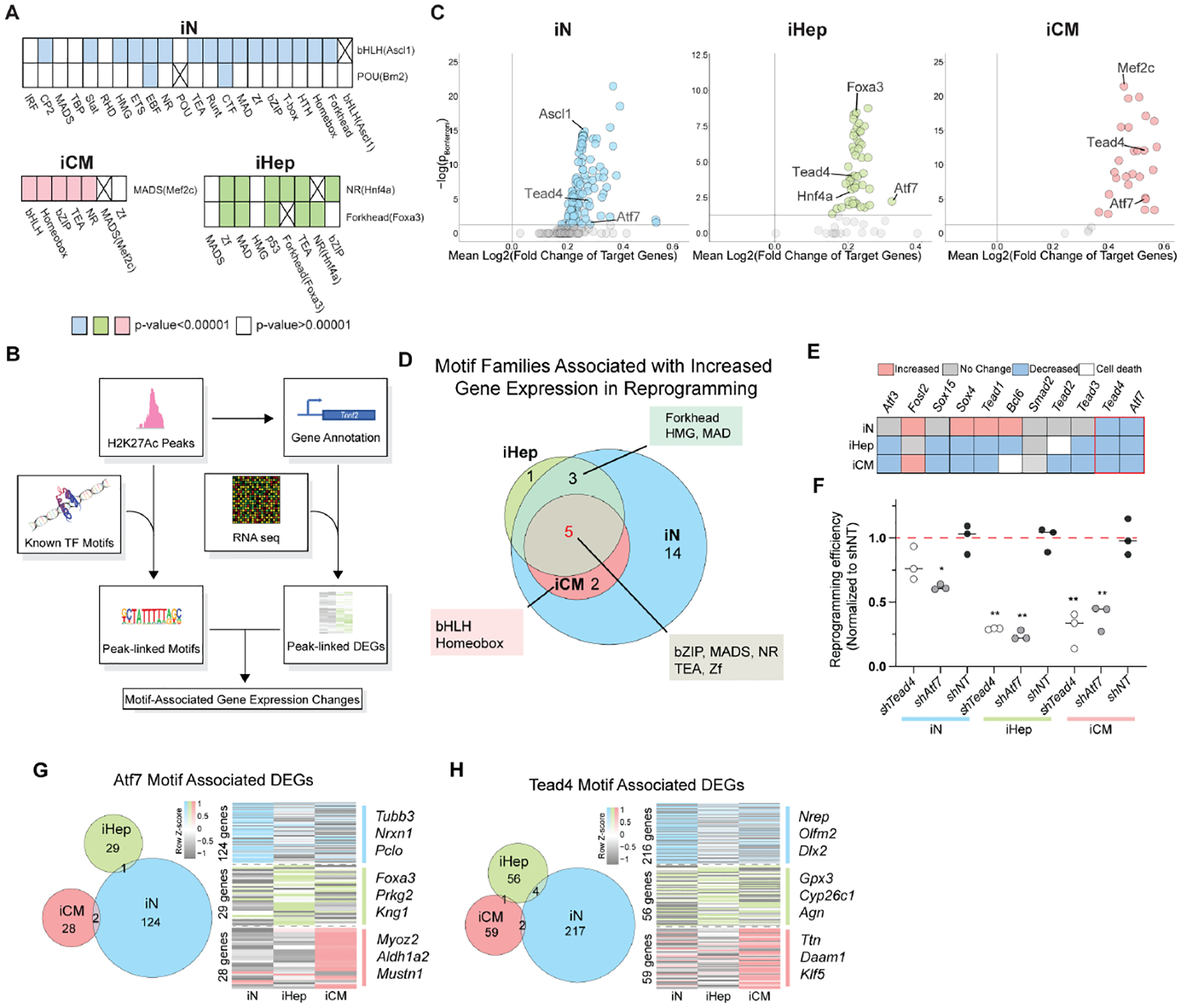
A. The significant co-occurrence of reprogramming factors’ motifs with non-cocktail motifs. An “X” marks boxes of identical motifs.
B. Diagram illustrating analysis steps to integrate H3K27Ac ChIP-seq and mRNA-seq data.
C. Scatter plot of enriched motifs in iN, iHep, and iCM and their associated gene expression changes. Gray dots fall below the corrected p value cutoff of 0.05.
D. Venn diagram showing the overlap of motif families associated with increased gene expression in all three types of reprogramming.
E. RNAi screen to determine the effect of knocking down (KD) non-cocktail TF family members on reprogramming efficiency: increased (red), decreased (blue), no change (grey), or cell death (white).
F. The reprogramming efficiencies when Tead4 (white) or Atf7 (grey) is knocked down during iN, iHep, and iCM conversion. n=3. Error-bars represent the standard deviation. The student’s t-test was used as the statistical test. * p< 0.05. ** p<0.01.
G-H. (Left) The overlap of the differentially expressed genes (DEGs) associated with an Atf7 (G) or Tead4 (H) motif in a gained H2K27Ac peak between all three types of reprogramming. (Right) DEGs associated with an Atf7 (G) or Tead4 (H) motif.
See also Figure S2.
Next, to ascertain the functional importance of both reprogramming and non-cocktail TFs in direct reprogramming, we associated enriched TF motifs with changes in expression of nearby genes by integrating our H3K27Ac ChIP-seq and RNA-seq datasets (Fig. 2B). Through this analysis, we found that – except for Brn2 – each reprogramming factor’s motif that was present in gained CREs (i.e., Ascl1, Mef2c, Hnf4α, Foxa3) had a significant association with increased gene expression, further supporting the principal that reprogramming factors are the primary drivers of early epigenetic and transcriptional reprogramming (Fig. 2C). Additionally, across the three types of reprogramming, we found the binding motifs for TEAD and bZIP factors were associated with increased gene expression (Fig. 2D). Forkhead and HMG binding motifs were associated with increased gene expression in iN and iHep, and bHLH and Homeobox binding motifs were associated with increased gene expression in both iCM and iN. To test whether these non-cocktail TFs are functionally important for iN, iHep, and/or iCM reprogramming, we performed a loss-of-function screen using RNAi to knock down these TFs individually in each type of reprogramming (Fig. 2E; Supplemental Fig. 2E). Knockdown of either Atf7 (bZIP family) or Tead4 (TEAD family) resulted in a significant decrease in the efficiency of all three types of direct reprogramming (Fig. 2F). To determine if Atf7 or Tead4 are potentially regulating a common set of genes among iN, iHep, and iCM, we compared the list of genes with Atf7 or Tead4 motifs in the gained CREs of each type of reprogramming. Surprisingly, there was little to no overlap among these genes (Fig. 2G, H). Instead, activation of genes specific to the lineage of each type of reprogramming were associated with the presence of Atf7 and Tead4 motifs, suggesting that these non-cocktail TFs regulate cell-type specific gene expression in a reprogramming factor-dependent manner.
Discovery of the cross-lineage potential of Ascl1
Intriguingly, after reconstructing motifs de novo from the gained enhancers in each type of reprogramming, we found significant enrichment of a motif closely resembling that of Ascl1 in iCM enhancers (Fig. 3A; Supplemental Fig. 2D). Additionally, gained iN enhancers with an Ascl1 motif were associated with muscle-related genes (Fig. 3B) whose expression levels were increased during iN reprogramming (Fig. 3C). Considering these data, we reasoned that Ascl1 may have cardiogenic potential in the context of iCM reprogramming. When Ascl1 is overexpressed alone in fibroblasts (Wapinski et al., 2013), ASCL1 binds to genes involved in both muscle and neuronal development (Fig. 3D), binding that coincides with increased expression of cardiac genes (Fig. 3E, F). To further evaluate the cardiogenic potential of Ascl1, we analyzed previously published RNA-seq datasets (Hashimoto et al., 2019) and compared the transcriptome of fibroblasts overexpressing Ascl1, GMT (abbreviation for Gata4/Mef2c/Tbx5), or GMT plus Hand2 (GHMT), using mouse neonatal cardiomyocytes as a reference (Fig. 3G). Ascl1 activates a cardiac gene set similar to that activated by GMT or GHMT – genes involved in cardiomyocyte development and contraction (Fig. 3G). Conversely, another set of cardiac genes related to ion transportation and metabolism is not activated by Ascl1 alone, suggesting other cardiogenic factors need to be co-expressed with Ascl1 for cardiac reprogramming.
Figure 3. Discovery of the cross-lineage potential of Ascl1.
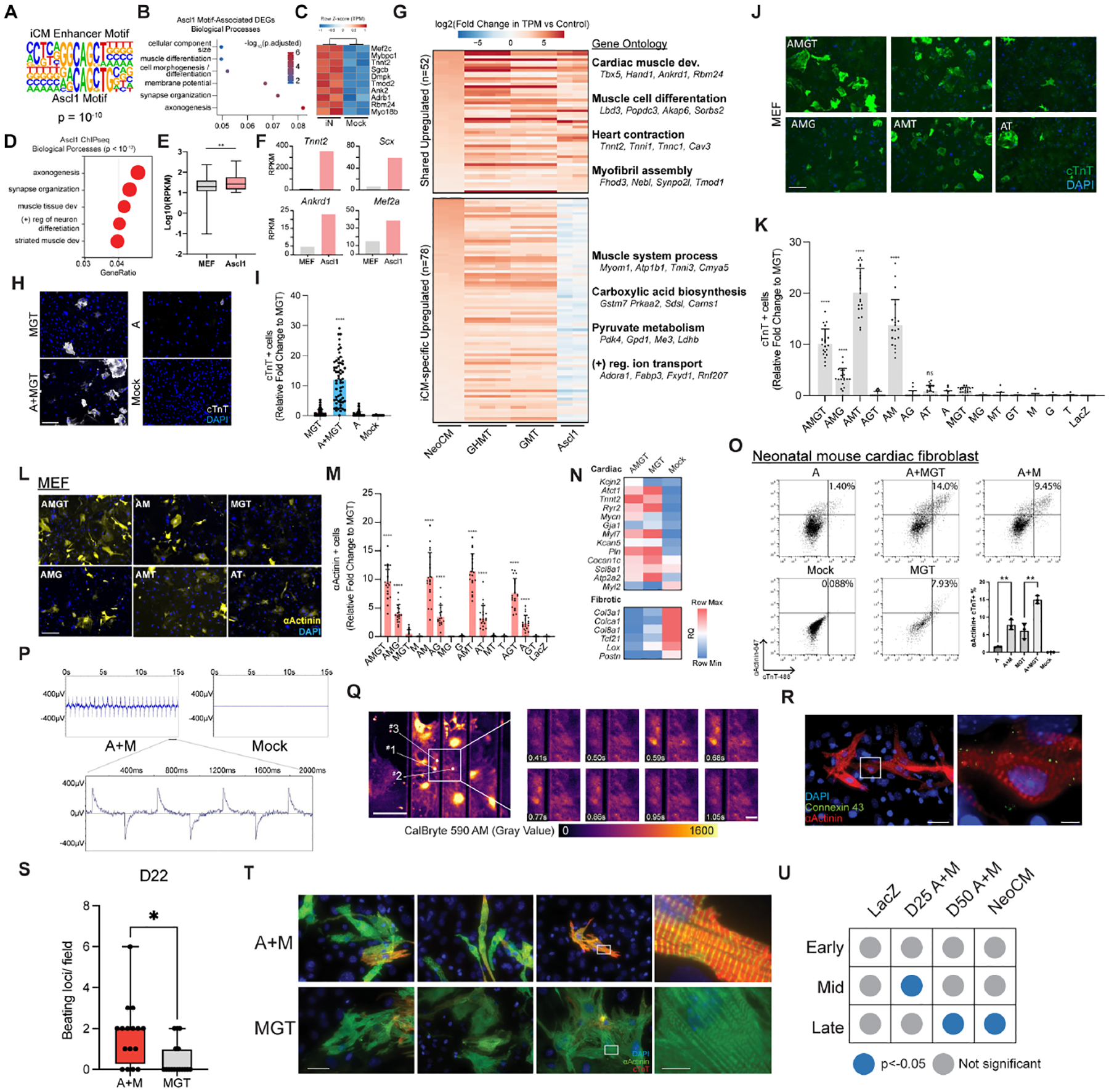
A. The alignment of motif constructed de novo from H3K27Ac-marked enhancers gained during iCM reprogramming with Ascl1’s motif.
B. Enriched biological processes among DEGs in iN reprogramming associated with an Ascl1 motif within a gained H3K27Ac peak.
C. Gene expression of muscle-related genes associated with an Ascl1 motif in iN reprogramming
D. Enriched biological processes among Ascl1’s target genes when overexpressed in MEF.
E. The expression of Ascl1’s target genes without and with Ascl1 overexpression in MEF.
F. Expression of representative genes from GO term “muscle tissue development” in MEF with and without Ascl1 overexpression. Y-axis represents the RPKM (per Million mapped reads) based on RNA-seq of Ascl1-infected fibroblast.
G. The expression of shared upregulated cardiomyocyte (CM) genes and iCM-specific upregulated CM genes (i.e. genes upregulated in GHMT, GMT, but not in fibroblasts overexpressing Ascl1) from a meta-analysis of published RNA-seq libraries. Enriched biological processes and their representative genes are shown at the right of the heatmap.
H. cTnT+ cells on Day 12 post infection of neonatal mouse cardiac fibroblasts with MGT ± Ascl1. cTnT staining is shown in white. DNA is shown in blue. Scale bar: 275 μm.
I. The automated quantification of (H). 40 images were analyzed for each treatment. Error bars represent standard deviation. The student’s t-test was performed between A+MGT and MGT. ****, p < 0.0001.
J-M. cTnT+ (J-K) or αActinin+ (L-M) cells on Day 12 post infection of MEF with all combinations of Ascl1, Mef2c, Gata4, and Tbx5. Green: cTnT; yellow: αActinin; blue:nuclei. Scale bar: 275 μm. (K) shows automated quantification of (J). (M) shows the automated quantification of (L). 20 images were analyzed for each treatment. Error bars represent standard deviation. The student’s t-test was performed between each TF combination and MGT. ****, p<0.0001; ns, p > 0.05.
N. Relative expression of cardiac and fibroblast genes by qRT-PCR on Day 12 post infection of MEF infected with MGT ± Ascl1.
O. The percentage of cTnT+/αActinin+ cells on Day 12 iCMs reprogrammed with different combinations of MGT, Ascl1, and Mef2c. n=3. (Bottom right) Quantification of flow cytometry results. Error bars represent standard deviation. The student’s t-test was performed to compare samples indicated by brackets. **, p< 0.01.
P. Field potential recordings by microelectrode array from iCMs reprogrammed with Ascl1 and Mef2c (A+M). Mock infection with lacZ was used as a negative control. (Bottom) A zoomed-in view of the field potential oscillations generated by A+M sample.
Q. Fluorescence images of CalBryte 590 signal from iCMs reprogrammed with A+M. Scale bar: 300 μm. (Right) Zoomed-in frames showing the flux of intracellular calcium. Scale bar: 100 μm.
R. The presence of gap junction protein Connexin 43 on Day 22 A+M reprogrammed iCMs. Red: αActinin; green: Connexin 43; blue: nuclei. Scale bar: 70 μm. (Right) Zoomed-in view. Scale bar: 10 μm.
S. Beating loci per field of A+M or MGT reprogrammed Day 22 iCMs. n=4. 4 fields were analyzed per sample. The student’s t-test was performed between A+M and MGT. *, p < 0.05.
T. The sarcomere structure of Day 12 A+M or MGT iCMs. Green: αActinin; red: cTnT; blue: nuclei. Scale bar: 70 μm. (Right) Zoomed-in view. Scale bar: 10 μm.
U. Enrichment of early, mid, and late embryonic heart development genes at different stages of A+M reprogramming. Features showing significant enrichment (p<0.05) were shown in blue dot. Published NeoCM RNA-seq was included as a reference control.
See also Figure S3.
The unexpected cardiogenic potential of Ascl1 prompted us to test whether Ascl1 can be used as a cardiac reprogramming factor, either to augment MGT or as a part of a new cocktail. Strikingly, the addition of Ascl1 to MGT resulted in over a ten-fold increase in the percentage of cTnT+ cells (Fig. 3H, 3I; Supplemental Fig. 3A). Next, we sought to reduce the number of TFs by systematically testing every combination of Ascl1 (A), Mef2c (M), Gata4 (G), and Tbx5 (T). We found that the combinations of A+M+G+T, A+M+G, A+M+T, and A+M induced higher percentages of cTnT+ and αActinin+ cells than M+G+T (Fig. 3J–M; Supplemental Fig. 3B, C). This suggests that Mef2c is necessary for the increase in reprogramming efficiency caused by Ascl1. Furthermore, the number of cardiac genes being activated was increased when Ascl1 was added to any combination of the other factors (Fig. 3N; Supplemental Fig. 3D). Altogether, these data suggest that A+M is an effective minimal cardiac reprogramming cocktail.
Next, we comprehensively characterized A+M reprogramming with an array of molecular and functional assays. A+M induced a substantial number of αActinin+/cTnT+ iCMs compared to Ascl1 alone and to MGT (Fig. 3O; Supplemental Fig. 3E). After culturing iCM reprogrammed from mouse cardiac fibroblasts with A+M for 28 days, we observed spontaneously contracting cells (Supplemental Movie 1), field potential oscillations (Fig. 3P), intracellular calcium oscillations (Supplemental Movie 2 and 3; Fig. 3Q; Supplemental Fig. 3F–H), and membrane localization of gap junction protein (Fig, 3R). After extended culture of both A+M and MGT reprogrammed cells, A+M induced more beating loci per field compared to MGT (Fig. 3S). These A+M induced iCMs showed organized sarcomere structure with longer sarcomere length and larger cell length/width ratio compared to those generated by MGT (Fig. 3T; Supplemental Fig. 3I, J). To further investigate the maturation state of A+M reprogrammed iCM, we performed RNA-seq experiment and determined the enrichment of genes related to the early, mid, and late stage of embryonic heart development(Uosaki et al., 2015) in A+M reprogramming cells. Day 50 A+M induced iCMs, similar to the endogenous neonatal cardiomyocytes, showed a significant enrichment of late-embryonic-heart genes (Fig. 3U). We also tested A+M reprogramming in H9-derived human fibroblasts. Adding ASCL1 to MGT plus miR-133 resulted in a significantly higher proportion of cells expressing both αActinin and cTnT (Supplemental Fig. 3K–N). The A+M minimum cocktail plus miR-133 reprogrammed human fibroblasts into iCM with high efficiency, activating cardiac genes and suppressing fibroblasts genes (Supplemental Fig. 3O). The human iCM reprogrammed with A+M plus miR-133 also adopted a rod-like shape with a banded sarcomere structure (Supplemental Fig. 3P). To determine whether both factors contribute to cardiac reprogramming efficiency, we tested the effect of independently varying the exogenous level of ASCL1 and MEF2C expression (Supplemental Fig. 3Q–S). Increasing the level of expression of either factor led to a corresponding increase in the percentage of reprogrammed cells. Then we further tested whether varying the ratio of ASCL1 and MEF2C influences the reprogramming outcomes, such as the relative proportions of iCMs versus iNs, Although Tuj1+ cells appeared at a low percentage, with higher ASCL1 to MEF2C ratio, we saw a decrease of αActinin + iCM to Tuj1+ iN ratio (Supplemental Fig. 3T). Finally, we also tested whether Ascl1 is required for the initial stage of reprogramming or the entire process to generate iCMs using an inducible TetON Ascl1 vector. We observed the highest reprogramming efficiency was achieved by continuous doxycycline exposure and thus continuous Ascl1 expression, suggesting Ascl1 is necessary for the entire process (Supplemental Fig. 3U). Taken together, these results demonstrate that Ascl1 and Mef2c are sufficient to induce cardiac reprogramming without Gata4 or Tbx5. The continuous expression and a fine balance of Ascl1 and Mef2c dosage are critical for a successful iCM conversion.
Successful A+M reprogramming terminates in a more mature iCM phenotype
To characterize the transdifferentiation induced by A+M and how it compares to that induced by MGT, the most widely used iCM cocktail, we performed single-cell multi-omics at Day 3 of reprogramming to simultaneously profile gene expression and chromatin accessibility (Supplemental Fig. 4A–D). After bioinformatically removing non-relevant cell types (e.g. epicardial cells, monocytes), we annotated each cluster as fibroblast (Fib), induced fibroblast (iFib), or induced cardiomyocyte (iCM) (Fig. 4A, C) (Liu et al., 2017; Zhou et al., 2019). Notably, in A+M reprogramming, we identified an additional type of iCM state, iCM_4, that has stronger activation of cardiac sarcomere (Myl2, Myl4, Ttn, Tnnt2, Myh6, Myh7, Actc1), ion-channel (Ryr2, Camk2d), and transcription factor genes (Nkx2–5) (Fig. 4B). The other iCM clusters either do not express these genes or express them at a lower level. Notably, this cell state has no analogous cell state in MGT reprogramming (Fig. 4D). Furthermore, iCM_4 has the highest expression of Ascl1, suggesting that with Mef2c, it induces more complete reprogramming at this early time point than MGT (Fig. 4B).
Figure 4. Successful A+M reprogramming terminates in a more mature cardiomyocyte-like fate.
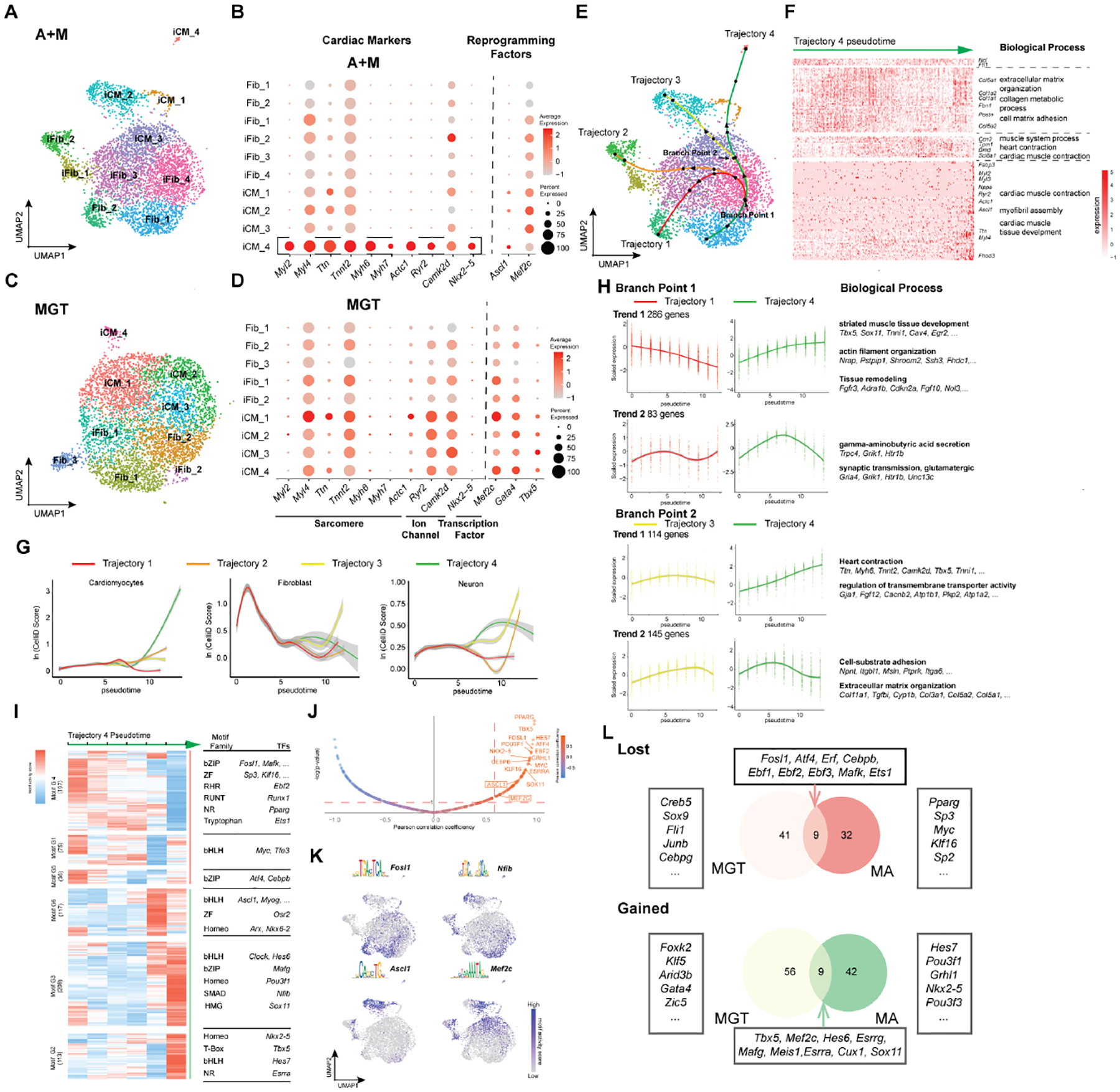
A. UMAP visualization of A+M single-cell multi-omics library at Day 3 of reprogramming.
B. Dot plot showing expression of key cardiac genes and reprogramming factors for each cell cluster in A+M reprogramming.
C. UMAP visualization of MGT single-cell multi-omics library at Day 3 of reprogramming.
D. Dot plot showing expression of key cardiac genes and reprogramming factors for each cell cluster in MGT reprogramming.
E. Trajectory analysis of A+M reprogramming based on pseudotime. Each trajectory was separated into 5 pseudotime intervals (marked by black dots). Two branch points were identified along the successful reprogramming trajectory (Trajectory 4), marked by the arrows.
F. Relative expression of differentially expressed genes (DEGs) along trajectory 4. Significantly enriched biological processes along with representative genes are right to the heatmap.
G. CelliD score for Cardiomyocytes (left), Fibroblast (middle), and Neuron (right) along each pseudotime trajectory. Gray areas indicate confidence intervals.
H. Different trends of gene expression at each branch point along trajectory 4 pseudotime. Average gene expression over pseudotime along each reprogramming trajectory is plotted as a smoothed solid line. Biological processes enriched in each gene cluster are shown on the right along with representative genes.
I. Motif enrichment scores for different transcription factors (from JASPAR) along trajectory 4. The trajectory was divided into six pseudotime bins. Motifs were clustered as motifs lost (red line) and gained (green line) during A+M reprogramming. In each motif group, enriched motif families and their representative TFs are shown to the right of the heatmap.
J. Pearson correlation between motif enrichment score and expression of its corresponding TF along trajectory 4 pseudotime. Y-axis is the log transformation of the motif enrichment p-value calculated using chromVar. Only motifs with a Pearson correlation coefficient larger than 0.6 and -log(p-value) larger than 1 (indicate with the red dashed lines) were considered active motifs.
K. Motif activity score of representative motifs from clustering in (I) superimposed onto UMAP visualization of single-cell clustering.
L. The overlap of gained or lost motifs between A+M and MGT reprogramming, comparing trajectory 4 of A+M reprogramming to trajectory 1 of MGT reprogramming.
See also Figure S4.
To further understand the reprogramming progression and the cell fate decisions involved, we reconstructed the pseudo-temporal cell state trajectories within the single-cell libraries using Slingshot (Fig. 4E) (Street et al., 2018). A+M reprogramming possesses four possible trajectories (Fig. 4E) while, consistent with our previous findings (Zhou et al., 2019), MGT reprogramming possesses only two (Supplemental Fig. 4F). Next, we used CelliD to quantify the cells’ cardiac-, neuron-, and fibroblast-like molecular features along the pseudotime trajectories of A+M reprogramming (Fig. 4G) (Cortal et al., 2021). Trajectory 1 is characterized by a transient loss of fibroblast identity early in pseudotime, followed by a return to the fibroblast state at the end of the trajectory. Trajectory 2 features progression to a neuron-like state. Trajectory 3 exhibits continuous activation of neuron genes and maintains a high-level expression of fibroblast genes. Finally, trajectory 4 was reconstructed from cells that gradually lose fibroblast identity, transiently activate neuronal features, and eventually acquire cardiac features (Fig. 4G). The ultimate loss of neuron identity on trajectory 4 is consistent with the loss of neuron genes expression in iCM_4 (Supplementary Fig. 4E). Seeing that it terminated in activation of cardiomyocyte features, we further characterized trajectory 4 by identifying genes which are differentially expressed along the trajectory and performing unsupervised hierarchical clustering based on the genes’ expression levels in single cells (Fig. 4F). Over the course of trajectory 4 pseudotime, there is decreased expression of ECM genes and increased expression of heart development and muscle contraction genes in later cell states (iCM_2, iCM_1, iCM_4). Together, these results suggest that trajectory 4 traces the pseudo-temporal timeline that ends in successful reprogramming.
Two branch points were identified along the A+M reprogramming trajectories (Fig. 4E). To understand the cell fate decisions occurring at each branch point, we determined the genes that are differentially expressed between cells immediately after each branch point (Fig. 4H; Supplemental Fig. 4G). Post branch point 1, there was sustained upregulation of genes involved in muscle tissue development and remodeling of cellular structure. There was also transient expression in trajectory 4 cells of genes involved in neuronal processes, such as synaptic transmission. Post branch point 2, trajectory 4 cells exhibited further activation of both sarcomere and transmembrane transporter genes and sustained downregulation of extracellular matrix organization genes. Together, the genes differentially expressed at these branch points suggest that cardiac fate acquisition in A+M reprogramming involves cell fate decisions that steer away from more neuron- or fibroblast-like programs.
To characterize the regulatory TF network of A+M reprogramming, we used our scATAC-seq data to determine how the motif enrichment scores of 746 mouse TFs change over trajectory 4 pseudotime (Fig. 4I–L) (Castro-Mondragon et al., 2021). Moreover, integrating our scRNA-seq data allowed us to determine which motifs were “active”; these motifs have high correlation between their accessibility in chromatin by scATAC-seq and expression of their corresponding TF by scRNA-seq. As expected, the motifs of both Ascl1 and Mef2c increased in enrichment over trajectory 4 pseudotime (Fig. 4I–K). For non-cocktail TFs, there was a gradual loss of enrichment of the motif for AP-1, while the motifs for a full spectrum of TF families critical for cardiac development and function (e.g., bHLH, Homeobox, SMAD, HMG, T-Box) became significantly enriched over pseudotime (Fig. 4I). This is in contrast to our previous observations of MGT reprogramming, in which we saw a smaller set of cardiac TFs activated (Wang et al., 2021b). Moreover, we found little overlap of either the gained or lost active motifs between our single-cell multi-omics libraries of A+M and MGT reprogramming (Fig. 4L; Supplemental Fig. 4F–H). It is worth noting that the motif of Nkx2–5 is specifically active in A+M reprogramming, as this TF is a critical regulator of cardiac cell fate (Fig. 4I, J, L). Additionally, the Tbx5 motif is also active in A+M reprogramming, even though Tbx5 has been removed as a reprogramming factor (Fig.4I, J, L). We have also identified domains of regulatory chromatin (DORCs) in both A+M and MGT reprogramming. In A+M reprogramming, we have identified 52 DORCs related to classic cardiac genes (Supplemental Fig. 4J). Consistent with our motif analysis, minimal overlapping was observed between the two sets of DORCs (Supplemental Fig. 4J). Taken together, our single-cell multi-omics analysis suggests that A+M iCM reprogramming suppresses both neuron and fibroblast programs. Furthermore, it induces a more complete, more mature cardiomyocyte-like fate – distinguishing it from MGT reprogramming at this early timepoint – by activating a distinct set of DORCs and a network of non-cocktail TFs.
Ascl1’s bi-lineage potential is restricted by Mef2c
Our single-cell multi-omics analysis suggested that Ascl1 and Mef2c both contribute to the reprogramming process; thus, we next sought to determine how Ascl1 and Mef2c interact to induce cardiac fate acquisition. First, we performed RNA-seq on fibroblasts overexpressing Ascl1 only (A-only), Mef2c only (M-only), or both Ascl1 and Mef2c (A+M) and categorized genes based on their expression changes in each condition (Fig. 5A; Supplemental Fig. 5A–D). A+M co-expression resulted in both augmented upregulation of a set of cardiac genes not activated by either factor alone (cluster 1) and augmented down-regulation of a set of immune response / vascular genes (cluster 2) (Fig. 5B, C). Conversely, gene clusters refined by A+M co-expression included a set of neuron and cardiac genes activated in A-only that was attenuated on A+M co-expression (cluster 3) (Fig. 5D, E). These data concur with our single-cell analysis, suggesting that A+M co-expression increases cardiac gene expression and decreases neuron gene expression. Next, to determine how binding of ASCL1 and MEF2C changes with A+M co-expression and how this correlates with epigenetic remodeling, we performed MEF2C ChIP-seq, ASCL1 ChIP-seq, and H3K27Ac ChIP-seq. Compared to A-only, ASCL1 binding in A+M was enriched at genes involved in cardiac muscle development, muscle contraction, and Wnt signaling (Fig. 5G). Moreover, both MEF2C binding and H3K27Ac were also enriched at these sites, implying that ASCL1 and MEF2C co-binding is important for the opening of CREs and activation of cardiac genes. Interestingly, ASCL1 depleted regions in A+M had no MEF2C binding, and H3K27Ac at these sites was reduced compared to A-only (Fig. 5H). Furthermore, ASCL1 depletion occurred at neuron-related genes. To test whether the continuous expression of Ascl1 in iCMs would restore Ascl1 induced neuron program, we profiled the expression of both cardiac and neuron genes of iCM at D58 post A+M infection. The continuous expression of Ascl1 did not change the iCM cell identity (Supplemental Fig. 5E). Together, these results suggest that ASCL1 is being redirected from its neuronal targets, becoming more restricted to cardiac genes when co-expressed with MEF2C.
Figure 5. Ascl1’s bi-lineage potential is restricted by Mef2c.
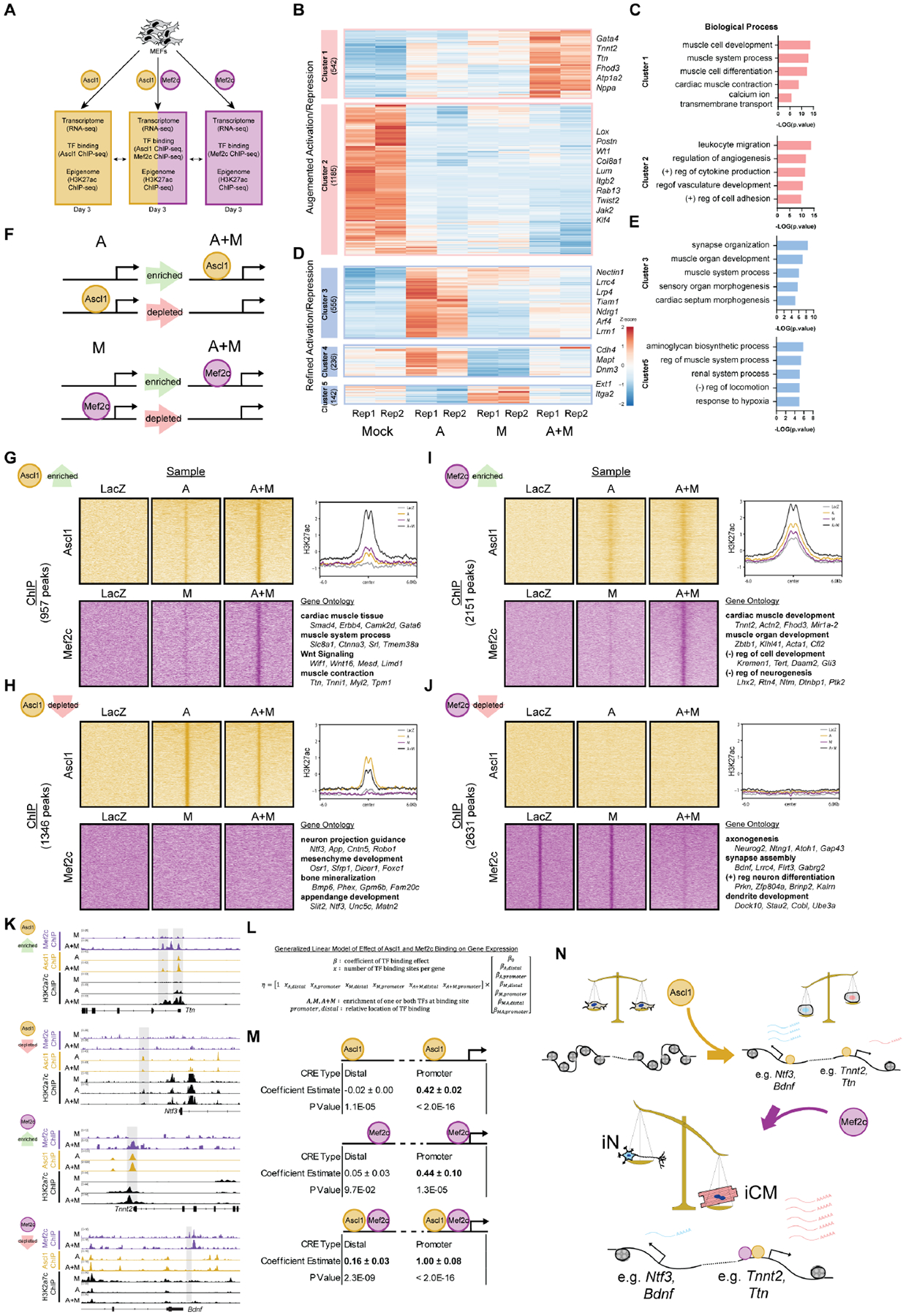
A. Schematic of the experiment design.
B-E. The augmented (B) and refined (D) activation/repression gene clusters in A+M infected MEFs compared to A, M, and mock treatment. Each column of the heat map represents a biological replicate. Bar plots showing gene ontology enrichment analysis results of augmented (C) and refined (E) activation/repression gene clusters.
F. Schematic showing categorization of A and M binding changes analyzed for the subsequent panels.
G-J. (Left) (G, H) ASCL1 ChIP-seq signal and MEF2C ChIP-seq signal at regions with enriched (G) or depleted (H) ASCL1 binding when A and M are co-expressed compared to A-only. (I, J) ASCL1 ChIP-seq signal and MEF2C ChIP-seq signal at regions with enriched (I) or depleted (J) MEF2C binding when A and M is co-expressed compared to M only. (Top right) Average enrichment plot of the H3K27Ac ChIP-seq signal for these genomic regions. (Bottom right) The significant, representative biological process of genes annotated to these genomic regions, with representative genes shown.
K. Genome tracks (generated with IGV) of representative regions and genes classified as ASCL1 enriched, ASCL1 depleted, MEF2C enriched, and MEF2C depleted when A and M are co-expressed.
L. Generalized Linear Model (GLM) of how co-operative binding of ASCL1 and MEF2C affects gene expression.
M. Effect estimates from GLM. Positive effect estimates are highlighted in bold. Effects estimates are reported ± their confidence intervals.
N. Working model of Ascl1 and Mef2c’s cooperation in driving iCM reprogramming.
See also Figure S5.
Regions of enriched MEF2C binding had pre-existing H3K27Ac signal in the LacZ negative control that was further increased in A+M (Figure 5I). GO analysis revealed that these genes were involved in muscle development processes as well as negative regulation of neurogenesis. Interestingly, ASCL1 could bind onto these regions prior to the binding of MEF2C, suggesting that ASCL1 binding could be a prerequisite for MEF2C’s binding on these regions. To test this hypothesis, we infected fibroblast with Ascl1 and Mef2c encoded retrovirus sequentially. The addition of Mef2c before Ascl1 leads to a significant drop in the reprogramming efficiency (Supplemental Fig. 5F). Regions of MEF2C depletion in A+M had MEF2C binding in our mock LacZ control as well as M-only, yet they had no ASCL1 binding nor H3K27Ac in any sample, suggesting that MEF2C’s binding at these loci did not activate CREs or lead to ASCL1 recruitment. Interestingly, GO analysis found that these genes were related to several neurogenic processes, as was seen for ASCL1 depleted regions (Fig. 5J). What’s more, we found that MEF2C binds to more muscle-related genes and fewer neuron-related genes in A+M reprogramming than in MGT reprogramming (Supplemental Fig. 5G). Taken together, these results suggest that MEF2C is binding to cardiac loci already bound – and likely made accessible by – ASCL1, promoting gene activation and reprogramming (Fig. 5K).
Because the presence of TF binding does not necessarily correlate with gene expression, we sought to determine if co-binding of ASCL1 and MEF2C has a different impact on gene expression than independent binding of either TF alone. To quantitatively assess this, we constructed a generalized linear model (GLM) (Fig. 5L). GLMs can be used to estimate the effects of a set of parameters on an outcome of interest, like gene expression. To fit the GLM, we determined the number of TF binding events of each type – binding of ASCL1, MEF2C, or both – per gene, as well as the location of the binding site relative to the promoter. Effects for binding of a single TF to a distal region were estimated to be close to zero, while binding of a single TF to a promoter had a positive effect on gene expression (Fig. 5M). In contrast, binding of both TFs to a distal region was estimated to have a positive effect, and binding of both TFs to a promoter region had the largest effect estimate overall. Thus, the combination of ASCL1 binding and MEF2C binding has a greater positive effect on gene expression than binding of either factor alone, suggesting a synergy between ASCL1 and MEF2C in activating genes.
Based on these data, we propose a A+M reprogramming model in which ASCL1 functions as a bipotent pioneer factor, opening chromatin at both neuron and cardiac gene loci, and co-expression of MEF2C shifts ASCL1’s binding away from neuron loci towards loci both distal and proximal of cardiac genes. This mode of A+M interaction leads to more pronounced activation of cardiac genes, driving the cells towards an iCM identity (Fig. 5N).
Discussion
In this study, we sought to profile regulatome dynamics in the initiation phase of direct reprogramming of diverse cell types. Generally, regulatome changes were in accordance with their respective target cell fate, with a notable exception: in iN, we observed activation of cardiac transcripts and opening of cardiac CREs, consistent with previous observation of alternative myocyte fate in neuron reprogramming. (Treutlein et al., 2016) Such ambiguous cell program activation may be caused by promiscuous binding of TFs to genes which they do not normally bind during development or the presence of alternative cell fate events. Through motif analysis, we found that not all reprogramming TFs are responsible for gene activation. Additionally, most of the non-cocktail TFs are target-cell specific. Knocking down the same TF could lead to varied outcomes in different direct reprogramming system. Such differences could be due to the extensive and dynamic epigenetic landscape shifting in each direct reprogramming despite the shared starting cell type. Nevertheless, there are non-cocktail TFs showing similar effect on direct reprogramming – specifically, Tead4 and Atf7 contribute to reprogramming efficiency in all three types of reprogramming by regulating target cell type-specific gene expression. It is known that the activity and function of TEAD family proteins in vertebrates are largely regulated by the availability and activity of TEAD co-factors. Thus, given the distinct reprogramming cocktail used for each direct reprogramming, the targets of the TEAD factors could be largely different. Such difference could lead to distinct results in the different reprogramming systems when knocking down Tead factors. As a downstream effector of the Hippo/YAP signaling pathway, Tead4 has been proposed to be involved in a metabolic switch that occurs during cell fate conversions (Di Benedetto et al., 2021; Shi et al., 2017). Phosphorylated ATF7 has been shown to interact with the histone acetyltransferases and histone demethylases, making it a potential regulator of chromatin remodeling (Maekawa et al., 2010). While further mechanistic work into the role of Tead4 and Atf7 in direct reprogramming is needed, our finding suggests that fibroblasts possess general factors/mechanisms of fate conversion that can be activated by disparate reprogramming factors.
After observing the shared cardiogenic program of iN and iCM, we demonstrated that Ascl1 can be used as a cardiac reprogramming factor with Mef2c. A+M reprogramming navigates a distinct cell fate trajectory when compared to MGT, as both factors shift and expand their binding to activate cardiac genes in a way that neither factor alone is capable of. Our use of Ascl1 as a cardiac reprogramming factor runs counter to its initial characterization as a neurodevelopmental factor (Guillemot et al., 1993). As a neuronal factor, Ascl1 has been widely used as a keystone for most of the neuron reprogramming cocktails and defined as a pioneer factor to initiate iN conversion. (Wapinski et al., 2013) To our knowledge, Ascl1 is not expressed in the heart at any developmental stage, in mammals or any other class within the animal kingdom – thus, no role for Ascl1 in cardiac fate determination has been reported. ASCL1, as a bHLH family TF, has been reported to have higher binding affinity to DNA due to its intrinsically shorter helix arm allowing more efficient and quicker clinching on DNA. (Soufi et al., 2015) Limited cardiac reprogramming capacity of other bHLH family TFs, such as Hand2 and MyoD, was detected when coupled with Mef2c (data not shown), highlighting the importance of Ascl1’s high DNA affinity to A+M reprogramming. Despite its pioneer capabilities and cross-lineage potential, Ascl1 cannot alone mediate cardiac reprogramming. Mef2c’s co-expression is necessary for the creation of iCM. The alteration of ASCL1’s binding site when MEF2C is co-expressed suggested a direct interaction of ASCL1 and MEF2C. Such synergistic relationship between MEF2C and bHLH family TFs during cardiac development has also been reported. (Wang et al., 2001) This observation also aligns with previous studies demonstrating that the binding of other TFs revolves around MEF2C. (Hashimoto et al., 2019)
The field of cellular reprogramming is a natural continuation of the insights gained through developmental biology; decades of work deducing which transcription factors are required for specification of a given cellular identity ushered in the age of “cellular alchemy” (Daley, 2012). Over the past decade, attempts at creating direct reprogramming cocktails have uniformly applied this developmental premise. Our study calls into question the assumption that transcription factors are uniquely tied to a given cellular identity – at least when used in the context of reprogramming. Like Ascl1, other TFs may possess the ability to promote the direct reprogramming of cell types unrelated to the TFs’ developmental origins. The combination – a TF that broadly remodels the epigenome, and a TF that more precisely guides gene activation – may be a strategy that can be generalized to improve the reprogramming cocktails of other cell types. Discovery of more cross-lineage TFs may force us to reconsider the ontological link between direct reprogramming and developmental biology, and, hopefully, enable us to develop more efficient, more useful reprogramming cocktails.
Limitations of the Study
This study represents one of the first endeavors to explore the fundamental differences or similarities in the epigenetic remodeling among iN, iCM and iHep. Despite our effort to achieve high quality of reprogramming, at current stage, the reprogramming platforms used are infeasible to have all possible variables adequately controlled. This limitation admittedly could render certain aspects of such comparison open to alternative explanations. For example, we found that a higher number of differentially enriched H3K27Ac peaks in iN reprogramming compared to iCM and iHep reprogramming. It is possible that iCM and iHep may exhibit increased H3K27Ac remodeling at a later time point than iN, which would suggest that epigenetic remodeling in iN occurs faster as opposed to be more widespread. Future additional study with perhaps variables being controlled may refine our conclusions drawn in this study.
For the single-cell analysis in the current study, the A+M library is made of 8682 cells and MGT library is made of 8875 cells. Although cells were harvested at day 3 from separated reprogramming experiments, we appreciate that including more cells especially from additional time points would further improve our understanding of the A+M reprogramming dynamics. We used slingshot, one of the widely used pesudotime inference programs, for our trajectory inference analysis. Although our functional data of A+M reprogrammed iCMs (Figure 3S, 3T and Supplemental Figure 3I, 3J) support the key conclusion (i.e. A+M led to a more mature iCM route) from this trajectory inference, we acknowledge that using additional trajectory inference methods could further strengthen our analysis.
STAR Methods
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Li Qian (li_qian@med.unc.edu).
Material availability
Plasmids generated in this study can be shared upon request following submission of a Material Transfer Agreement.
Data and code availability
RNA-seq, ChIP-seq, Single-cell RNA-seq datasets collected in this study have been deposited in the Gene Expression Omnibus of NCBI and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Histone H3 (acetyl K27) antibody | Abcam | ab4729, AB_2118291 |
| Recombinant Anti-MEF2C antibody | Abcam | ab211493, AB_2864417 |
| Anti-MASH1/Achaete-scute homolog 1 antibody | Abcam | ab74065, AB_1859937 |
| Anti-beta III Tubulin antibody | Abcam | ab7751, AB_306045 |
| Recombinant Anti-Albumin antibody | Abcam | ab207327, AB_2755031 |
| Recombinant Anti-Sarcomeric Alpha Actinin antibody | Abcam | ab68167, AB_11157538 |
| Cardiac Troponin T Monoclonal Antibody | ThemoFisher | MA5-12960, AB_11000742 |
| Anti-Connexin-43 antibody | Sigma | C6219, AB_476857 |
| FITC-conjugated anti-cTnT | Miltenyi | 130-119-575, AB_2751735 |
| Alexa Fluor® 488 AffiniPure Donkey Anti-Mouse IgG (H+L) | Jackson Immuno | 715-545-150, AB_2340846 |
| Alexa Fluor® 647 AffiniPure Donkey Anti-Mouse IgG (H+L) | Jackson Immuno | 715-605-150, AB_2340862 |
| Alexa Fluor® 488 AffiniPure Donkey Anti-Rabbit IgG (H+L) | Jackson Immuno | 715-545-152 |
| Alexa Fluor® 647 AffiniPure Donkey Anti-Rabbit IgG (H+L) | Jackson Immuno | 715-605-152 |
| Bacterial and virus strains | ||
| Biological samples | ||
| Chemicals, peptides, and recombinant proteins | ||
| L-Ascorbic acid | Sigma | A92902 |
| Recombinant Human VEGF 165 Protein | R&D Systems | 293-VE-010/CF |
| Recombinant Human FGF basic/FGF2/bFGF (146 aa) Protein | R&D Systems | 233-FB-025/CF |
| Recombinant Human FGF-10 Protein | R&D Systems | 345-FG-025/CF |
| StemPro™-34 SFM (1X) | ThermoFisher | 10639011 |
| N-2 Supplement (100X) | ThermoFisher | 17502048 |
| Hepatocyte Growth Factor | Sigma | H9661 |
| Recombinant Human EGF Protein, CF | R&D Systems | 236-EG-200 |
| 2-Mercaptoethanol | ThermoFisher | 21985023 |
| Nicotinamide | Sigma | N0636 |
| Dexamethasone | Sigma | D4902 |
| Insulin | Sigma | I2643 |
| Critical commercial assays | ||
| Roche KAPA Stranded mRNA-Seq kit | Roche | 07962142001 |
| truChIP® Chromatin Shearing Kit | Covaris | PN 520154 |
| chromium Single Cell Multiome ATAC + Gene expression kits | 10X Genomics | 1000285 |
| Calbryte™ 590 AM | AAT Bioquest | 20700 |
| CAMP Acquisition Systems | Multichannel Systems | S/N 102001208 |
| Deposited data | ||
| ChIP-sequencing of A+M reprogramming | This manuscript | GSE192727 |
| H3K27ac ChIP-seq of three direct reprogramming | This manuscript | GSE192770 |
| RNA-seq of A+M and MGT reprogramming | This manuscript | GSE192729 |
| RNA-seq of three direct reprogramming | This manuscript | GSE192735 |
| RNA-se1 of neonatal Cardiomyocytes | This manuscript | GSE192787 |
| RNA-seq of A+M reprogramming at late time points | This manuscript | GSE210685 |
| scATAC-seq of A+M and MGT reprogramming at Day 3 | This manuscript | GSE192738 |
| scRNA-seq of A+M and MGT reprogramming at Day 3 | This manuscript | GSE192752 |
| RNA-seq of Ascl1 Overexpressed Fibroblast | Wapinski et al., 2013 | GSE43916 |
| RNA-seq of GMT and GHMT reprogramming | Hashimoto et al., 2019 | GSE112316 |
| Experimental models: Cell lines | ||
| 293T | ATCC | CRL-3216 |
| Plat-E | Cell biolabs | RV-101 |
| Cardiac fibroblast | Isolated from neonatal mouse pups | |
| Mouse Embryonic fibroblast | Isolated from E13.5 mouse embyros | |
| Human H9F | Differentiated from H9 | |
| Experimental models: Organisms/strains | ||
| CD-1 | Charles River | 022 |
| Oligonucleotides | ||
| qRT-PCR primers | This manuscript | Supplementary Table 1 |
| Recombinant DNA | ||
| Atf3 shRNA | Sigma | TRCN0000331748 |
| Fosl2 shRNA | Sigma | TRCN0000263185 |
| Sox15 shRNA | Sigma | TRCN0000415165 |
| Sox4 shRNA | Sigma | TRCN0000234110 |
| Tead1 shRNA | Sigma | TRCN0000085865 |
| Bcl6 shRNA | Sigma | TRCN0000084653 |
| Smad2 shRNA | Sigma | TRCN0000327447 |
| Tead2 shRNA | Sigma | TRCN0000374225 |
| Tead3 shRNA | Sigma | TRCN0000301920 |
| Tead4 shRNA | Sigma | TRCN0000054774 |
| Atf7 shRNA | Sigma | TRCN0000434189 |
| pMx-Ef1a-Ascl1 | This manuscript | N/A |
| pMx-Ef1a-Mef2c | Sigma | N/A |
| pMX-Ascl1-puro | This manuscript | N/A |
| TetO-FUW-Ascl1 | Sigma | N/A |
| TetO-FUW-Brn2 | Vierbuchen et al., 2010 | N/A |
| TetO-FUW-Mytl1 | Vierbuchen et al., 2010 | N/A |
| FUW-M2rtTA | Vierbuchen et al., 2010 | Addgene_20342 |
| pGCDNsam-Hnf4α | Sekiya and Suzuki, 2011 | Addgene_33002 |
| pGCDNsam-Foxa3 | Sekiya and Suzuki, 2011 | Addgene_33005 |
| pMX-puro-MGT | Garbutt et al., 2020 | Addgene_111809 |
| TetO-FUW-EGFP | Vierbuchen et al., 2010 | Addgene_73083 |
| pGCDNsam-IRES-GFP | This manuscript | N/A |
| pMX-puro-lacZ | This manuscript | N/A |
| pLL3.7-UbC-hMEF2c | This manuscript | N/A |
| pLL3.7-UbC-hASCL1 | This manuscript | N/A |
| pGCDNsam-IRES-GFP | Sigma | N/A |
| Software and algorithms | ||
| GraphPad Prism 8 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| R 4.1.0 | R Core Team 2021 | https://www.r-proiect.ore |
| Cell Profiler | Stirling DR et al., 2021 | https://cellprofiler.org |
| FlowJo v10 | FlowJo LLC. | https://www.flowjo.com/ |
| ImageJ-Fiji | National Institutes of Health, USA | https://fiji.sc/ |
| Cell ranger version 6.0.1 | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/what-is-cell-ranger |
| Seurat 4.0.3 | Stuart et al., 2019 | https://github.com/satijalab/seurat/releases |
| MACS2 | Zhang et al., 2008 | https://github.com/macs3-project/MACS |
| Slingshot | Street et al., 2018 | https://www.bioconductor.org/packages/release/bioc/html/slingshot.html |
| Tradeseq | Van den Berge et al., 2020 | https://www.bioconductor.org/packages/release/bioc/html/tradeSeq.html |
| ChromVAR | Schep et al., 2017 | https://bioconductor.org/packages/release/bioc/html/chromVAR.html |
| FigR | Kartha et al., 2022 | https://github.com/buenrostrolab/stimATAC_analyses_code |
| HOMER | Heinz et al., 2010 | http://homer.ucsd.edu/homer/ |
| Diffbind | Stark and Brown, 2011 | https://bioconductor.org/packages/release/bioc/html/DiffBind.html |
| ChIPseeker | Yu, Wang and He, 2015 | https://bioconductor.org/packages/release/bioc/html/ChIPseeker.html |
| FastQC | Babraham Bioinformatics | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| MultiQC | Ewels et al., 2016 | https://multiqc.info/ |
| clusterProfiler | Wu et al., 2021 | https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html |
| deepTools | Ramirez et al., 2016 | https://deeptools.readthedocs.io/en/develop/ |
| pheatmap | Raivo Kolde | https://github.com/raivokolde/pheatmap |
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Salmon version 1.3.0 | Patro et al., 2017 | https://github.com/COMBINE-lab/salmon |
| Deseq2 | Love, Huber and Anders, 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| ROSE | Loven et al., 2013; Whyte et al., 2013 | http://younglab.wi.mit.edu/super_enhancer_code.html |
| Bbduk | Bbmap | https://jgi.doe.gov/data-and-tools/software-tools/bbtools/bb-tools-user-guide/bbduk-guide/ |
| Other | ||
Experimental model and subject details
Animal Handling
WT CD1 mice were purchased from Charles River (CD1 IGS). All mice were maintained in the Division of Comparative Medicine Facility at UNC-CH with standardized food, bedding, caging, and daily inspections of all cages. For all breeding, 3–6 months old one male and two female mice were set up together in one breeding cage. All experiments involving animals were performed in accordance with the University of North Carolina at Chapel Hill Institutional Animal Care and Use Committee (protocol numbers 18–204 and 21–187).
Mouse embryonic fibroblast
Breeding wildtype CD1 mice were checked for vaginal plugs daily to time insemination. The morning a plug was observed was considered embryonic day 0.5. Females were sacrificed and embryos (both male and female) were harvested on day 13.5. Following decapitation and red organ removal, embryos were mechanically and enzymatically dissociated, and the cell suspension was passed through a 40μm nylon mesh. Cells were cultured until confluence, then they were washed twice, trypsinized, filtered through a through a 40μm nylon mesh again, and frozen at P1. In all experiments, MEF were cultured in cell culture-grade incubators at 37°C 5% CO2.
Mouse cardiac fibroblast
One pup at a time, wild type P0–P3 CD1 pups (male and female) were sprayed with 70% ethanol, decapitated, and their hearts were harvested and placed in ice cold 1X DPBS. After the hearts were collected, the rest of the procedure was performed in a biosafety cabinet. Hearts were rinsed twice with ice cold DPBS then minced with a sterile razor blade until the tissue pieces were approximately 1mm in diameter. Per 20 hearts, tissue pieces were incubated in a 50mL conical tube in 10 mL pre-warmed 0.05% Trypsin-EDTA in a 37°C water bath for 10 minutes, gently mixing the tube halfway. After placing the tissue pieces back in the biosafety cabinet and allowing them to settle for 60 seconds, the trypsin-EDTA supernatant was aspirated, and the following was repeated 4–6 times: 1) 8 mL of pre-warmed 1X HBSS with 0.005% Collagenase II was added to the tissue pieces, 2) the tissue pieces and collagenase solution was vortexed at 30–40% power for 60 seconds, then incubated in a water bat at 37°C for seven minutes, 3) the tissue pieces were again vortexed at 30–40% power for 60 seconds, then returned to the biosafety cabinet and allowed to settle for 60 seconds, 4) the dissociated cells in the supernatant were transferred to a ice cold conical tube filled with IMDM 15% FBS 1X penicillin/streptomycin, and 5) 8 mL of pre-warmed 1X HBSS with 0.005% Collagenase II was reapplied to the tissue pieces. The pooled cells from the collagenase digestions were filtered through a 40 μm strainer and centrifuged at 200 × g for 5 minutes at 4 °C. The cell pellet was resuspended in a biotinylated anti-Thy1 antibody solution (20 μL antibody stock, 80 μL MACS buffer) and incubated at 4°C for 60 minutes. After diluting the cell-antibody mixture in 10mL MACS buffer, the cells were centrifuged again at 200 × g for 5 minutes at 4 °C. The cells pellet was resuspended in a Anti-Biotin MACS MicroBeads solution (20 μL MicroBeads, 80 μL MACS buffer) and incubated at 4°C for 30 minutes. After diluting the cell-beads mixture in 10mL MACS buffer, the bead-bound cells were centrifuged again at 200 × g for 5 minutes at 4 °C, then the cell-beads pellet was resuspended in 5 mL of MACS buffer. The cells were passed over a pre-equilibrated, magnet-bound MACS LS column, rinsed twice with 1 mL MACS buffer, then eluted by pressing the plunger into the column over a clean 15 mL conical tube. The eluted cells were centrifuged at 200 × g for 5 minutes at 4 °C, resuspended in a volume of IMDM 15% FBS 1X penicillin/streptomycin appropriate for the experiment, and seeded on plates or dishes pre-coated with 0.1% gelatin in 1X DPBS. In all experiments, mouse cardiac fiborblasts were cultured in cell culture-grade incubators at 37°C 5% CO2.
Method details
Virus Packaging
Pantropic lentiviruses for iN reprogramming were packaged in 293T by co-transfection with psPAX2 and pMD2.g with Lipofectamine 2000 (Thermo Fisher). Pantropic retroviruses for iHep reprogramming were packaged in 293T by co-transfection with pCMV-gagpol and pCMV-VSV-G with Lipofectamine 2000 (Thermo Fisher). Ecotropic retroviruses for iCM reprogramming were packaged in Plat-E with Lipofectamine 2000 (Thermo Fisher). For packaging of all viruses, virus harvest medium (10%FBS, 1x NEAA, and DMEM) was changed 16–18 hours after transfection. Virus-containing medium was collected 48 and 72 hours after transfection. Medium collections were combined, mixed with 40% poly-ethylene glycol (PEG) at a 4:1 volume ratio, and incubated overnight at 4°C. Virus collections were centrifuged at 3100 × g for 30 minutes at 4°C to precipitate PEG-bound virus. After aspirating the supernatant, PEG pellets were resuspended at 100 uL per 16mL of collected virus medium. Ecotropic retroviruses were used immediately after concentration by PEG; pantropic retroviruses and lentiviruses were used after freezing at −80°C.
Direct Reprogramming
P2–P3 wildtype or αMHC-GFP MEF were reprogrammed according to the previously reported protocols (Ieda et al., 2010; Sekiya and Suzuki, 2011; Vierbuchen et al., 2010) with one adjustment: for collection of H3K27Ac ChIP-seq and mRNA-seq samples, all three cell types were cultured in DMEM 10% FBS 1X NEAA until time of collection. Briefly, one day after seeding, MEF were infected with lentiviruses or retroviruses to initiate reprogramming: TetO-FUW-Brn2, TetO-FUW-Ascl1, TetO-FUW-Mytl1, and FUW-M2rtTA for iN; pGCDNsam-Hnf4α and pGCDNsam-Foxa3 for iHep; and pMX-puro-MGT for iCM. Separate vector-matched controls were used for each cell type: TetO-FUW-EGFP for iN; pGCDNsam-IRES-GFP for iHep; and pMX-puro-lacZ for iCM. Medium was changed twenty-four hours after infection, and doxycycline was added to iN samples and their control samples. Samples for mRNA-seq and H3K27ac ChIP-seq were collected 72 hours after initial infection. For other assays, medium changes occurred every 2–3 days utilizing the optimal medium for each type of reprogramming: N3 for iN, hepato-medium for iHep, and iCM medium for iCM. A+M reprogramming in MEF and neonatal mouse cardiac fibroblasts was performed with TetO-FUW-Ascl1, FUW-M2-rtTA, and pMx-Mef2c. A+M reprogramming for Figure 3T was performed with pMX-EF1a-Ascl1 and pMX-EF1a-Mef2c. Reprogramming of H9-derived fibroblasts was performed with pLL3.7-UbC-hMEF2C, pLL3.7-UbC-hASCL1, and pBabe-miR133. MGT reprogramming of mouse cardiac fibroblasts and H9-derived fibroblasts was carried out as previously described.(Garbutt et al., 2020). Fibroblast was infected with freshly concentrated MGT encoded retrovirus. From Day 2 to Day 12, cells were cultured in iCM medium (10% FBS, 20% M199 and DMEM) and the medium is changed every other day. At Day 12 post infection, cells were either harvested to examine the reprogramming efficiency or cultured in stempro medium (Stempro-34 SF medium, 1x GlutaMax, 50 μg/mL Ascorbic acid, 5 ng/mL recombinant human VEGF-165, 10ng/mL recombinant human FGF-basic, 50ng/mL recombinant human FGF-10) to be further reprogrammed to beating iCM.
RNAi
Short hairpin RNA clones in the pLKO vector were purchased from Sigma. Three clones per gene were tested, and the clone which caused greatest reduction in gene expression was used to perform RNAi in the context of direct reprogramming. For each gene, these clones are: Atf3 - TRCN0000331748, Fosl2 - TRCN0000263185, Sox15 - TRCN0000415165, Sox4 - TRCN0000234110, Tead1 - TRCN0000085865, Bcl6 - TRCN0000084653, Smad2 - TRCN0000327447, Tead2 - TRCN0000374225, Tead3 - TRCN0000301920, Tead4 - TRCN0000054774, Atf7 - TRCN0000434189. These shRNA clones were packaged into lentivirus by co-transfection with psPAX2 and pMD2.g with Lipofectamine 2000 (Thermo Fisher) in 293T. For packaging of shRNA viruses, virus harvest medium (10%FBS, 1x NEAA, and DMEM) was changed 16–18 hours after transfection. Virus-containing medium was collected 48 and 72 hours after transfection. Medium collections were combined, mixed with 40% poly-ethylene glycol (PEG) at a 4:1 volume ratio, and incubated overnight at 4°C. Virus collections were centrifuged at 3100 × g for 30 minutes at 4°C to precipitate PEG-bound virus. After aspirating the supernatant, PEG pellets were resuspended at 100 uL per 16mL of collected virus medium. One day after infecting MEF with reprogramming factors, the medium was changed, and the cells were infected with shRNA viruses.
Viral titering
Prior to their use to reprogram H9-derived fibroblasts (H9F) with varying MOIs, the approximate titer of frozen pLL3.7-UbC-hMEF2c and pLL3.7-UbC-hASCL1 lentiviruses were determined as follows. H9F were seeded in a 48-well plate at 20,000 cells / well. The next day, cells were infected with 8μg/mL polybrene in a 6-well serial dilution, starting with 8μL and diluting the virus 4-fold for each step in the series. 48-hours later, the medium was changed and puromycin selection was initiated. Selection medium was changed daily for three days, and on the final day, cell survival was assessed by phase light microscopy. Per well, cell survival was scored by the confluence of the remaining cells: 100–90%, 90–60%, 60–30%, 30–10%, 10–0%. Titer was computed for wells in which the confluence was scored in the 90–60%, 60–30%, or 30–10% ranges. Titer, defined as the volume of viral stock that will infect 100% of cells under the described titering conditions, was computed as follows:
where the “dilution factor” is the fold change in viral concentration for a given well in the dilution series (i.e. 1:1, 1:4, 1:16, etc.), and “confluence” is the percentage in the middle of the confluence range scored for a given well (i.e. 75%, 45%, or 15%). If two or more wells for a given virus were scored within the 90–10% confluence range, titer was calculated for each well then averaged.
mRNA sequencing and data analysis
RNA was extracted with TRIzol (Thermo Fisher) according to the manufacturer’s protocol. Sequencing was performed through UNC Chapel Hill’s High-Throughput Sequencing Facility. Library preparation was performed with the Roche KAPA Stranded mRNA-Seq kit. Paired-end sequencing was performed on the NovaSeq SP platform to a depth of 4 × 107 reads per sample. Data quality was checked with FastQC and MultiQC.(Ewels et al., 2016) Reads matching adapter sequences or with a mean quality score less than 20 were trimmed with Bbduk. Pseudoalignment was performed with Salmon (Patro et al., 2017) using the mm10 cDNA transcriptome as reference. Differential gene expression analysis was performed with DESeq2.(Love et al., 2014) Genes with an adjusted p value less than 0.1 and an absolute fold change greater than 1.5 compared to the relevant control. Gene ontology analysis of biological processes was performed with clusterProfiler.(Wu et al., 2021) Unsupervised hierarchical clustering and heatmaps generation were performed with pheatmap. GO terms with an adjusted p value less than 0.05 and a q value less than 0.01 were considered significant. As indicated in figure legends, supervised analyses (i.e. based on a priori comparisons between samples) were performed using gene expression changes computed by DESeq2, thus heatmaps for these analyses display Z-score-scaled DESeq2 computed log2(fold change) values. Unsupervised analyses were performed using Salmon computed TPM values, thus heatmaps for these analyses display Z-score-scaled TPM values.
ChIP sample preparation
Chromatin samples were harvested using truChIP® Chromatin Shearing Kit (Covaris, PN 520154) following the manufacturer’s protocol. Briefly, five million cells were cross-linked with Fixing Buffer A at room temperature for 10 minutes. Quenching Buffer E was added to the fixed cells, and the cells were incubated at room temperature for an additional 5 minutes. The cells were washed twice with ice-cold PBS. To extract the nuclei, Lysis Buffer B containing 1X protease inhibitors was added to the cells. The cells were incubated on ice for 10 minutes on a rocker at 4 °C. Intact nuclei were collected by centrifugation at 1,700 × g for 5 minutes at 4 °C. The nuclei were washed once with Wash Buffer C and then Shearing Buffer D3. The pelleted nuclei were resuspended in 1 mL Shearing Buffer D3 and transfered to a milliTUBE (Covaris, PN 520135). The chromatin shearing was performed with E220 Focused-ultrasonicator (Covaris) with 60sec on and 30sec off for 14 cycles. The chromatin solution was clarified by centrifugation at 20,000 g at 4 °C for 45 minutes and then pre-cleared with Dynabeads protein A (Life Technologies) for 2 hours at 4 °C. The pre-cleared chromatin sample was incubated with 50 mL of Dynabeads protein A loaded with 5 mg antibody (anti-H3K27ac, ab4729; anti-Mef2c, ab211493; anti-Ascl1, ab74065) overnight at 4 °C. The beads were washed three times with 0.1% SDS lysis buffer, once with 0.1% SDS lysis buffer/0.35 M NaCl, once with 10 mM Tris-Cl (pH 8)/1 mM EDTA/0.5% NP40/ 0.25% LiCl/0.5% NaDOC, and once with TE buffer (pH8.0). The immunoprecipitated material was eluted from the beads by heating for 45 minutes at 68 °C in 50 mM Tris-Cl (pH 7.5), 10 mM EDTA, 1% SDS. To reverse crosslinking, samples were incubated with 1.5 mg/ml Pronase at 42 °C for 2 hr followed by 67 °C for 6 hr. The samples were then processed with MinElute PCR Purification Kits (QIAGEN) and eluted in TE buffer. The ChIP DNA samples were then sent to UNC Chapel Hill’s High-Throughput Sequencing Facility libraries for ChIP-seq library preparation with Thruplex DNA Seq Library Prep kit (Takara).
ChIP sequencing and data analysis
ChIP-seq libraries were sequenced by UNC Chapel Hill’s High-Throughput Sequencing Facility on Novaseq SP at 20M PE reads/sample. Data quality was checked with FastQC and MultiQC. Reads were aligned to the mm10 reference genome using Bowtie2.(Langmead and Salzberg, 2012) MACS2 (Zhang et al., 2008) was used for peak calling. Diffbind (Stark and Brown, 2011) was used to obtain the differentially bound peaks between reprogramming and mock samples. Peaks were annotated to genes based on their distance to the nearest genes using ChIPseeker.(Yu et al., 2015) To visualize the ChIP-seq signal for peaks gained/lost during reprogramming, the computeMatrix and plotHeatmap functions from deepTools2 (Ramírez et al., 2016) were used. Gene ontology analysis of biological processes was performed with clusterProfiler. Unsupervised hierarchical clustering and heatmaps generation was performed with pheatmap. GO terms with an adjusted p value less than 0.05 and a q value less than 0.01 were considered significant.
Super enhancer classification and comparison to mouse tissues
Super enhancers were called for each type of reprogramming based on their H3K27ac ChIP-seq libraries using ROSE (Lovén et al., 2013; Whyte et al., 2013) with default parameters. To annotate the function of the super enhancer identified in our reprogramming sample, we overlapped our super enhancers to the super enhancers annotated in dbSUPER database.(Khan and Zhang, 2016)
Motif analysis
Known and de novo motif analyses of H3K27ac peaks were performed with HOMER.(Heinz et al., 2010) H3K27ac peaks were divided into enhancer and promoter peaks based on their proximity to the nearest gene’s transcription start site. Motif analysis was performed separately on the enhancer and promoter peak sets; where relevant, the combined results are reported as CRE motifs. The masked mm10 genome was preparsed into 200bp fragments for background comparison. HOMER optimized for possible de novo motifs of 8, 10, and 12bp in length. Known motifs with a p value 10−2 or lower were considered significantly enriched. Where relevant, results of analyses based on known motifs were summarized by TF family according to HOMER’s TF family classification. For a small set of known motifs categorized into multiple TF families, manual re-classification based on the experimental design of the datasets cited in HOMER was done as follows: “Paired,Homeobox” changed to “Paired”; “?,bHlH” changed to “?”; “T-box,MAD” changed to “MAD”; “RHD,bZIP” changed to “RHD”; “POU,Homeobox” changed to “POU”; “Homeobox,bHLH” changed to “Homeobox”; “POU,Homeobox,HMG” changed to “POU”; “Forkhead,bHLH” changed to “Forkhead”; “ETS,bHLH” changed to “ETS”; “CTF,Forkhead” changed to “CTS”; “IRF:bZIP” changed to “IRF”; and “ETS:IRF” changed to “IRF”.
Co-occurrence of motifs
Only known motifs that were significantly enriched in the gained CREs in each type of reprogramming were included in the analysis. The scanMotifGenomeWide.pl HOMER function was used to scan the mm10 genome for the location of the motifs. Motifs were considered to be within a gained CRE if the genomics coordinates of the H3K27ac peak were fully contained in the genomic coordinates of the peak. The presence of two known motifs within the same peak constituted co-occurrence. To determine the statistical significance of frequency of motif co-occurrences, we also generated background sequences by randomly extracting different sizes of DNA sequences from the mm10 genome which resembles the size distribution of the gained CREs. P values were calculated by the Binomial proportion test.
Analysis of motif-associated gene expression changes
For a given type of reprogramming, motifs from HOMER’s known motif database that were enriched in gained CREs were used as input for the scanMotifGenomeWide.pl HOMER function. Genomic coordinates of known motifs were matched to gained H3K27ac peaks only if they were fully contained within the peak’s genomic coordinates. Peak annotation to gene bodies was used to match known motifs to genes. The associated gene expression changes for a given motif is defined as the DESeq2-computed log2(Fold Change) values for the set of genes annotated to the peaks that contain the motif. Two-sided Wilcoxon statistical testing of motif-associated gene expression changes was performed by comparing it to the DESeq2-computed log2(Fold Change) values not matched to any motifs. After Bonferroni correction, multiplying each p value by the number of motifs tested, a motif’s associated gene expression changes were considered significant if its pBonferroni was less than 0.05.
Single-cell multi-omics library preparation and sequencing
A+M and MGT reprogramming cells from Day 3 post virus infection were harvested by trypsin and sent to UNC Chapel Hill’s Advanced Analytics Core for multi-omics library preparation. The libraries were generated using chromium Single Cell Multiome ATAC + Gene expression kits (10X Genomics) following the manufacturer’s protocol. The single-cell multi-ome libraries were sequenced by UNC Chapel Hill’s High-Throughput Sequencing Facility on a Novaseq SP flow cell aiming at 50,000 read pairs/cell for both libraries.
Single-cell multi-omics library analysis
The single-cell multi-omes libraries were first aligned to the mm10 reference genome and transcriptome. Reads were filtered, barcodes counted, peaks called and ATAC and GEX molecules counted using the cellranger-arc count. The feature barcode matrix of both RNA and ATAC-seq were loaded into R using the Read10X_h5 function from Seurat.(Hao et al., 2021) Seurat objects were created to store both the RNA and ATAC-seq using CreateSeuratObject. Per-cell quality control metrics were computed using the ATAC-seq data and cells that were outliers for these metrics were removed. Cells with unusually low or high counts for either the RNA seq or ATAC seq were also removed. ATAC peak calling was performed with the MACS2 CallPeak function. For RNA-seq data, SCTranform was used to normalize the expression data. For gene expression data, cell cycle genes were regressed out using SCTransform. For joint UMAP visualization, the weighted nearest neighbor method was used to compute a joint neighbor graph that represents both the gene expression and DNA accessibility measurements. Slingshot (Street et al., 2018) was used to construct the reprogramming progression and pseudotime inference. Tradeseq (Van den Berge et al., 2020) was used to compare the differentially expressed genes between different reprogramming trajectories. To analyze changes in motif enrichment along reprogramming trajectories, each reprogramming trajectory was separated into 5 pseudotime bins, and motif enrichment scores of 746 mouse TFs for each bin were calculated with chromVAR.(Schep et al., 2017) Motifs were clustered by enrichment score changes into different groups using unsupervised hierarchical clustering. To find active motifs, Pearson correlation co-efficient was calculated for each motif based on its motif enrichment score and expression along the reprogramming trajectory. Motifs were considered as “active motif” when its Pearson correlation co-efficient between its enrichment in accessible chromatin and its RNA expression was larger than 0.6. DORCs of A+M and MGT reprogramming were identified using FigR. (Kartha et al., 2022)
Ca2+ imaging
Calbryte 590 AM (AAT Bioquest) was reconstituted immediately prior to use at a concentration of 5mM. The dye was added to the cell culture medium to achieve a final concentration of 5uM, and the samples were incubated in the dark at 37°C 5% CO2 for 60 minutes. The samples were washed once with 1X HBSS and continued in 1X HBSS for the duration of live imaging.
Multi-electrode array recording
All instruments were purchased from Multichannel Systems. Cells were cultured on a 60-electrode glass multi-electrode array. MEAs were placed within an MEA1060-BC blanking circuit (S/N 0094), and signal was passed through a FA60SBC filter amplifier (S/N 2093). Temperature of the instrument was maintained at 37°C with a TC02 temperature regulator. Signal was recorded with a CAMP Acquisition Systems (S/N 102001208) using the MC_Rack software package.
Immunofluorescence microscopy
Cells were fixed with 4% PFA in 1X DPBS for 10 minutes, washed twice 1X PBS, permeabilized for 20 minutes in 0.1% Triton X-100 1X PBS, washed twice with 1X PBS, blocked for at least 60 minutes in 5% BSA 1X PBS, stained overnight at 4°C with primary antibody diluted in 5% BSA 1X PBS, washed three times with 1X PBS, stained for 60 minutes with secondary antibody diluted in 5% BSA 1X PBS, and lastly washed three times with 1X PBS before imaging. Primary antibodies and relevant dilutions were as follows: anti-Tuj1, 1:500 (Abcam, # ab7751); anti-Alb, 1:500 (Abcam, ab207327); anti-cTnT, 1:400 (Thermo Fisher, # MA5-12960); anti-αActinin, 1:500 (Abcam, ab68167); anti-Connexin-43, 1:500 (Sigma, C6219). Secondary antibodies and relevant dilutions were as follows: AlexaFluor 488 anti-mouse, 1:500 (Jackson Immuno, # 715-545-150), AlexaFluor 647 anti-mouse, 1:500 (Jackson Immuno, # 715-605-150), AlexaFluor 488 anti-rabbit, 1:500 (Jackson Immuno, # 715-545-152), AlexaFluor 647 anti-rabbit, 1:500 (Jackson Immuno, # 715-605-152). The quantification of the images was performed with CellProfiler(Stirling et al., 2021).
Flow cytometry
Cells were fixed, permeabilized, and immuno-stained using the Fixation/Permeabilization Kit from BD Biosciences. Cells were incubated in 1X fixation/permeabilization solution for 20 minutes, washed once 1X permeabilization/wash solution, stained with primary antibody diluted in 1X permeabilization/wash solution for 30 minutes, washed once 1X permeabilization/wash solution, stained with primary antibody diluted in 1X permeabilization/wash solution for 30 minutes, and lastly washed twice with 1X permeabilization/wash solution before flow cytometry. Samples were analyzed with an Attune NxT cytometer (Thermo Fisher). Gating in the forward and side scatter channels was performed to gate out debris and doublet cells. Primary antibodies and relevant dilutions were as follows: anti-Tuj1, 1:500 (Abcam, # ab7751); anti-Alb, 1:500 (Abcam, ab207327); anti-cTnT, 1:200 (Thermo Fisher, # MA5-12960); FITC-conjugated anti-cTnT, 1:100 (Miltenyi, # 130-119-575); anti-αActinin, 1:250 (Abcam, ab68167). Secondary antibodies and relevant dilutions were as follows: AlexaFluor 488 anti-mouse, 1:500 (Jackson Immuno, # 715-545-150), AlexaFluor 647 anti-mouse, 1:500 (Jackson Immuno, # 715-605-150), AlexaFluor 488 anti-rabbit, 1:500 (Jackson Immuno, # 715-545-152), AlexaFluor 647 anti-rabbit, 1:500 (Jackson Immuno, # 715-605-152).
Construction of generalized linear model of effect of Ascl and Mef2c binding on gene expression
To construct the model, Ascl1 and Mef2c ChIP-seq data was integrated with mRNA-seq data. For the models input, Ascl1 and Mef2c ChIP peaks enriched in the A+M samples versus the A-only or M-only samples were used. To be considered enriched, TF peaks met the cut-offs of edgeR-computed fold change > 1.5 and FDR < 0.1. For the model’s output, the gene expression changes in A+M samples versus mock control samples were used to construct the model. To avoid incorporating any inaccurate measurements of gene expression, any genes for which the standard error in the DESeq2 computed log2(fold change) was greater than log2(fold change) itself were not included in the model. Ascl1 and Mef2c peaks were annotated to genes using ChIPseeker. A peak was classified as a “Promoter” peak if it was within 3000bp upstream or downstream of the gene’s transcription start site. Otherwise, the peak was classified as a “Distal” peak. Ascl1 and Mef2c peaks were checked for overlapping genomic coordinated, allowing for no gap between them, to determine where TF co-binding occurred. Per gene, the number of peaks of each type was counted. The number of each peak of each type was used for the models input. The model was constructed using the glm() function in R.
RT-qPCR
RNA was extracted with TRIzol (Thermo Fisher) according to the manufacturers protocol. First-strand cDNA was synthesized using SuperScript IV VILO (Thermo Fisher). For comparative CT analysis, two-step qPCR followed by melt-analysis was performed with SYBR Green PCR Master Mix (Thermo Fisher) on an Applied Biosystems QuantStudio6 real-time PCR instrument (Thermo Fisher). Relative quantification of gene expression compared to control samples was calculated using the ΔΔCT method. GAPDH or a combination of GAPDH, 18S, and ACTB were used as housekeeping gene references during quantification. Primer pairs used for qPCR reactions are listed in Supplemental Table 1.
Quantification and statistical analysis
Except where otherwise stated, values are presented as the mean ± standard deviation of three or more biological replicates. The sample size for separate experiment was stated in the corresponding figure legend. Statistical significance was tested using the unpaired two-sided student’s t-test. Results for which the test returned p-value of 0.05 or less were considered significant. Computation and data manipulation was carried out with GraphPad Prism 8 and with R. (R Core Team, 2020)
Supplementary Material
Supplementary Movie 1. Spontaneous Contraction of an A+M-Reprogrammed iCM. Related to Figure 3.
Supplementary Movie 2. Spontaneous Ca2+ Oscillations in an A+M-Reprogrammed iCM. Fluorescent signal generated by CalBryte 590. Related to Figure 3Q.
Supplementary Movie 3. Example movies demonstrating the partial calcium burst and whole-cell calcium burst in the A+M iCM. Related to Supplemental Figure 3G.
Supplementary Table 1. qRT-PCR primers used in the current manuscript. Related to STAR Methods.
Highlights.
Comparison of direct reprogramming processes unveiled common regulators
Ascl1 demonstrates a cross-lineage potential to activate cardiac program
Ascl1 and Mef2c induce cardiac reprogramming with high efficiency and maturity
Ascl1 and Mef2c cooperate to induce cardiac fate and repress neuron identity
Acknowledgments:
We would like to thank all the members of the Qian and Liu labs for their helpful discussion and valuable input. We would also like to acknowledge UNC Chapel Hill’s Flow Cytometry Core (NIH/NCI P30CA016086), Advanced Analytics Core, and High-Throughput Sequencing Facility (NIH/NCI P30CA016086, NIH/NIEHS P30ES010126). This work was supported by AHA Postdoctoral Fellowship 927906 to H.F.W; NIH T32HL069768 (PI: C. Mack) and F30HL154659 to B.K.; AHA20EIA35310348 NIH/NHLBI R35HL155656 to L.Q, NIH/NHLBI R01HL139976 and R01HL139880; AHA 20EIA35320128 to J.L.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests: The authors declare that they have no competing interests.
Inclusion and Diversity: We support inclusive, diverse, and equitable conduct of research.
References
- Di Benedetto G, Parisi S, Russo T, and Passaro F (2021). YAP and TAZ Mediators at the Crossroad between Metabolic and Cellular Reprogramming. Metabolites 11. 10.3390/METABO11030154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Berge K, Roux de Bézieux H, Street K, Saelens W, Cannoodt R, Saeys Y, Dudoit S, and Clement L (2020). Trajectory-based differential expression analysis for single-cell sequencing data. Nature Communications 2020 11:1 11, 1–13. 10.1038/s41467-020-14766-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro-Mondragon JA, Riudavets-Puig R, Rauluseviciute I, Berhanu Lemma R, Turchi L, Blanc-Mathieu R, Lucas J, Boddie P, Khan A, Manosalva Pérez N, et al. (2021). JASPAR 2022: the 9th release of the open-access database of transcription factor binding profiles. Nucleic Acids Res 10.1093/NAR/GKAB1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortal A, Martignetti L, Six E, and Rausell A (2021). Gene signature extraction and cell identity recognition at the single-cell level with Cell-ID. Nat Biotechnol 39, 1095–1102. 10.1038/S41587-021-00896-6. [DOI] [PubMed] [Google Scholar]
- Daley GQ (2012). Cellular alchemy and the golden age of reprogramming. Cell 151, 1151–1154. 10.1016/j.cell.2012.11.016. [DOI] [PubMed] [Google Scholar]
- Ewels P, Magnusson M, Lundin S, and Käller M (2016). MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048. 10.1093/BIOINFORMATICS/BTW354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbutt TA, Zhou Y, Keepers B, Liu J, and Qian L (2020). An Optimized Protocol for Human Direct Cardiac Reprogramming. STAR Protocols 10.1016/j.xpro.2019.100010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillemot F, Lo LC, Johnson JE, Auerbach A, Anderson DJ, and Joyner AL (1993). Mammalian achaete-scute homolog 1 is required for the early development of olfactory and autonomic neurons. Cell 75, 463–476. 10.1016/0092-8674(93)90381-Y. [DOI] [PubMed] [Google Scholar]
- Hao Y, Hao S, Andersen-Nissen E, Mauck WM, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, et al. (2021). Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29. 10.1016/J.CELL.2021.04.048/ATTACHMENT/1990279C-028D-47D4-A5D3-C89B04E14795/MMC3.XLSX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto H, Wang Z, Garry GA, Malladi VS, Botten GA, Ye W, Zhou H, Osterwalder M, Dickel DE, Visel A, et al. (2019). Cardiac Reprogramming Factors Synergistically Activate Genome-wide Cardiogenic Stage-Specific Enhancers. Cell Stem Cell 25, 69–86.e5. 10.1016/j.stem.2019.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple Combinations of Lineage-Determining Transcription Factors Prime cis-Regulatory Elements Required for Macrophage and B Cell Identities. Molecular Cell 38, 576–589. 10.1016/J.MOLCEL.2010.05.004/ATTACHMENT/AC76CFA3-57BD-4789-B1BD-E1F2EE8F5776/MMC1.PDF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-André V, Sigova AA, Hoke HA, and Young RA (2013). Super-Enhancers in the Control of Cell Identity and Disease. Cell 155, 934– 947. 10.1016/J.CELL.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horisawa K, Udono M, Ueno K, Ohkawa Y, Nagasaki M, Sekiya S, and Suzuki A (2020). The Dynamics of Transcriptional Activation by Hepatic Reprogramming Factors. Molecular Cell 10.1016/j.molcel.2020.07.012. [DOI] [PubMed] [Google Scholar]
- Ieda M, Fu JD, Delgado-Olguin P, Vedantham V, Hayashi Y, Bruneau BG, and Srivastava D (2010). Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell 10.1016/j.cell.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kartha VK, Duarte FM, Hu Y, Ma S, Chew JG, Lareau CA, Earl A, Burkett ZD, Kohlway AS, Lebofsky R, et al. (2022). Functional inference of gene regulation using single-cell multi-omics. Cell Genomics 100166. 10.1016/J.XGEN.2022.100166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A, and Zhang X (2016). dbSUPER: a database of super-enhancers in mouse and human genome. Nucleic Acids Research 44, D164–D171. 10.1093/NAR/GKV1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nature Methods 2012 9:4 9, 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Chen O, Zheng M, Wang L, Zhou Y, Yin C, Liu J, and Qian L (2016). Re-patterning of H3K27me3, H3K4me3 and DNA methylation during fibroblast conversion into induced cardiomyocytes. Stem Cell Res 16, 507–518. 10.1016/j.scr.2016.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Wang L, Welch JD, Ma H, Zhou Y, Vaseghi HR, Yu S, Wall JB, Alimohamadi S, Zheng M, et al. (2017). Single-cell transcriptomics reconstructs fate conversion from fibroblast to cardiomyocyte. Nature 551, 100–104. 10.1038/nature24454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovén J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, and Young RA (2013). Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153, 320–334. 10.1016/J.CELL.2013.03.036/ATTACHMENT/45208297-1D89-4BD1-B65F-F995A874ADF6/MMC8.XLSX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maekawa T, Kim S, Nakai D, Makino C, Takagi T, Ogura H, Yamada K, Chatton B, and Ishii S (2010). Social isolation stress induces ATF-7 phosphorylation and impairs silencing of the 5-HT 5B receptor gene. EMBO J 29, 196–208. 10.1038/EMBOJ.2009.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris SA, Cahan P, Li H, Zhao AM, San Roman AK, Shivdasani RA, Collins JJ, and Daley GQ (2014). Dissecting engineered cell types and enhancing cell fate conversion via CellNet. Cell 158, 889–902. 10.1016/J.CELL.2014.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patro R, Duggal G, Love MI, Irizarry RA, and Kingsford C (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nature Methods 14, 417–419. 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2020). R: A language and environment for statistical computing.
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Research 44, W160–W165. 10.1093/NAR/GKW257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schep AN, Wu B, Buenrostro JD, and Greenleaf WJ (2017). chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nature Methods 2017 14:10 14, 975–978. 10.1038/nmeth.4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekiya S, and Suzuki A (2011). Direct conversion of mouse fibroblasts to hepatocyte-like cells by defined factors. Nature 475, 390–393. 10.1038/nature10263. [DOI] [PubMed] [Google Scholar]
- Shi Z, He F, Chen M, Hua L, Wang W, Jiao S, and Zhou Z (2017). DNA-binding mechanism of the Hippo pathway transcription factor TEAD4. Oncogene 36, 4362–4369. 10.1038/ONC.2017.24. [DOI] [PubMed] [Google Scholar]
- Soufi A, Garcia MF, Jaroszewicz A, Osman N, Pellegrini M, and Zaret KS (2015). Pioneer Transcription Factors Target Partial DNA Motifs on Nucleosomes to Initiate Reprogramming. Cell 161, 555. 10.1016/J.CELL.2015.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark R, and Brown G (2011). DiffBind: differential binding analysis of ChIP-Seq peak data.No Title. 10.18129/B9.bioc.DiffBind. [DOI]
- Stirling DR, Swain-Bowden MJ, Lucas AM, Carpenter AE, Cimini BA, and Goodman A (2021). CellProfiler 4: improvements in speed, utility and usability. BMC Bioinformatics 22, 1–11. 10.1186/S12859-021-04344-9/FIGURES/6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone NR, Gifford CA, Thomas R, Ivey KN, and Pollard KS (2019). Context-Specific Transcription Factor Functions Regulate Epigenomic and Transcriptional Dynamics during Cardiac Reprogramming. Cell Stem Cell 25, 87–102. 10.1016/j.stem.2019.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Street K, Risso D, Fletcher RB, Das D, Ngai J, Yosef N, Purdom E, and Dudoit S (2018). Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genomics 19. 10.1186/S12864-018-4772-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treutlein B, Yi Lee Q, Gray Camp J, Mall M, Koh W, Ali Mohammad Shariati S, Sim S, neff NF, Skotheim JM, Wernig M, et al. (2016). Dissecting direct reprogramming from fibroblast to neuron using single-cell RNA-seq. Nature 534, 391–395. 10.1038/nature18323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uosaki H, Cahan P, Lee DI, Wang S, Miyamoto M, Fernandez L, Kass DA, and Kwon C (2015). Transcriptional Landscape of Cardiomyocyte Maturation. Cell Reports 13, 1705–1716. 10.1016/J.CELREP.2015.10.032/ATTACHMENT/2DD071B5-E968-4ACF-ADAB-ECDDA203A91F/MMC6.ZIP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vierbuchen T, Ostermeier A, Pang ZP, Kokubu Y, Sudhof TC, and Wernig M (2010). Direct conversion of fibroblasts to functional neurons by defined factors. Nature 463, 1035– 1041. 10.1038/nature08797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vierbuchen T, Ling E, Cowley CJ, Couch CH, Wang X, Harmin DA, Roberts CWM, and Greenberg ME (2017). AP-1 Transcription Factors and the BAF Complex Mediate Signal-Dependent Enhancer Selection. Mol Cell 68, 1067–1082.e12. 10.1016/J.MOLCEL.2017.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang DZ, Renee Valdez M, McAnally J, Richardson J, and Olson EN (2001). The Mef2c gene is a direct transcriptional target of myogenic bHLH and MEF2 proteins during skeletal muscle development. Development 128, 4623–4633. 10.1242/DEV.128.22.4623. [DOI] [PubMed] [Google Scholar]
- Wang H, Yang Y, Liu J, and Qian L (2021a). Direct cell reprogramming: approaches, mechanisms and progress. Nature Reviews Molecular Cell Biology 1–15. 10.1038/s41580-021-00335-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Yang Y, Qian Y, Liu J, and Qian L (2021b). Delineating chromatin accessibility re-patterning at single cell level during early stage of direct cardiac reprogramming. J Mol Cell Cardiol 162, 62–71. 10.1016/J.YJMCC.2021.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wapinski OL, Vierbuchen T, Qu K, Lee QY, Chanda S, Fuentes DR, Giresi PG, Ng YH, Marro S, Neff NF, et al. (2013). Hierarchical Mechanisms for Direct Reprogramming of Fibroblasts to Neurons. Cell 155, 621–635. 10.1016/J.CELL.2013.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, and Young RA (2013). Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319. 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T, Hu E, Xu S, Chen M, Guo P, Dai Z, Feng T, Zhou L, Tang W, Zhan L, et al. (2021). clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation(China) 2, 100141. 10.1016/J.XINN.2021.100141/ATTACHMENT/04D49091-826D-4D9D-81C2-4F97B3300FCA/MMC1.PDF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang LG, and He QY (2015). ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31, 2382–2383. 10.1093/BIOINFORMATICS/BTV145. [DOI] [PubMed] [Google Scholar]
- Zaret KS, and Carroll JS (2011). Pioneer transcription factors: establishing competence for gene expression. Genes Dev 25, 2227–2241. 10.1101/GAD.176826.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nussbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biology 9, 1–9. 10.1186/GB-2008-9-9-R137/FIGURES/3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Liu Z, Welch JD, Gao X, Wang L, Garbutt T, Keepers B, Ma H, Prins JF, Shen W, et al. (2019). Single-Cell Transcriptomic Analyses of Cell Fate Transitions during Human Cardiac Reprogramming. Cell Stem Cell 0. 10.1016/j.stem.2019.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Movie 1. Spontaneous Contraction of an A+M-Reprogrammed iCM. Related to Figure 3.
Supplementary Movie 2. Spontaneous Ca2+ Oscillations in an A+M-Reprogrammed iCM. Fluorescent signal generated by CalBryte 590. Related to Figure 3Q.
Supplementary Movie 3. Example movies demonstrating the partial calcium burst and whole-cell calcium burst in the A+M iCM. Related to Supplemental Figure 3G.
Supplementary Table 1. qRT-PCR primers used in the current manuscript. Related to STAR Methods.
Data Availability Statement
RNA-seq, ChIP-seq, Single-cell RNA-seq datasets collected in this study have been deposited in the Gene Expression Omnibus of NCBI and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Histone H3 (acetyl K27) antibody | Abcam | ab4729, AB_2118291 |
| Recombinant Anti-MEF2C antibody | Abcam | ab211493, AB_2864417 |
| Anti-MASH1/Achaete-scute homolog 1 antibody | Abcam | ab74065, AB_1859937 |
| Anti-beta III Tubulin antibody | Abcam | ab7751, AB_306045 |
| Recombinant Anti-Albumin antibody | Abcam | ab207327, AB_2755031 |
| Recombinant Anti-Sarcomeric Alpha Actinin antibody | Abcam | ab68167, AB_11157538 |
| Cardiac Troponin T Monoclonal Antibody | ThemoFisher | MA5-12960, AB_11000742 |
| Anti-Connexin-43 antibody | Sigma | C6219, AB_476857 |
| FITC-conjugated anti-cTnT | Miltenyi | 130-119-575, AB_2751735 |
| Alexa Fluor® 488 AffiniPure Donkey Anti-Mouse IgG (H+L) | Jackson Immuno | 715-545-150, AB_2340846 |
| Alexa Fluor® 647 AffiniPure Donkey Anti-Mouse IgG (H+L) | Jackson Immuno | 715-605-150, AB_2340862 |
| Alexa Fluor® 488 AffiniPure Donkey Anti-Rabbit IgG (H+L) | Jackson Immuno | 715-545-152 |
| Alexa Fluor® 647 AffiniPure Donkey Anti-Rabbit IgG (H+L) | Jackson Immuno | 715-605-152 |
| Bacterial and virus strains | ||
| Biological samples | ||
| Chemicals, peptides, and recombinant proteins | ||
| L-Ascorbic acid | Sigma | A92902 |
| Recombinant Human VEGF 165 Protein | R&D Systems | 293-VE-010/CF |
| Recombinant Human FGF basic/FGF2/bFGF (146 aa) Protein | R&D Systems | 233-FB-025/CF |
| Recombinant Human FGF-10 Protein | R&D Systems | 345-FG-025/CF |
| StemPro™-34 SFM (1X) | ThermoFisher | 10639011 |
| N-2 Supplement (100X) | ThermoFisher | 17502048 |
| Hepatocyte Growth Factor | Sigma | H9661 |
| Recombinant Human EGF Protein, CF | R&D Systems | 236-EG-200 |
| 2-Mercaptoethanol | ThermoFisher | 21985023 |
| Nicotinamide | Sigma | N0636 |
| Dexamethasone | Sigma | D4902 |
| Insulin | Sigma | I2643 |
| Critical commercial assays | ||
| Roche KAPA Stranded mRNA-Seq kit | Roche | 07962142001 |
| truChIP® Chromatin Shearing Kit | Covaris | PN 520154 |
| chromium Single Cell Multiome ATAC + Gene expression kits | 10X Genomics | 1000285 |
| Calbryte™ 590 AM | AAT Bioquest | 20700 |
| CAMP Acquisition Systems | Multichannel Systems | S/N 102001208 |
| Deposited data | ||
| ChIP-sequencing of A+M reprogramming | This manuscript | GSE192727 |
| H3K27ac ChIP-seq of three direct reprogramming | This manuscript | GSE192770 |
| RNA-seq of A+M and MGT reprogramming | This manuscript | GSE192729 |
| RNA-seq of three direct reprogramming | This manuscript | GSE192735 |
| RNA-se1 of neonatal Cardiomyocytes | This manuscript | GSE192787 |
| RNA-seq of A+M reprogramming at late time points | This manuscript | GSE210685 |
| scATAC-seq of A+M and MGT reprogramming at Day 3 | This manuscript | GSE192738 |
| scRNA-seq of A+M and MGT reprogramming at Day 3 | This manuscript | GSE192752 |
| RNA-seq of Ascl1 Overexpressed Fibroblast | Wapinski et al., 2013 | GSE43916 |
| RNA-seq of GMT and GHMT reprogramming | Hashimoto et al., 2019 | GSE112316 |
| Experimental models: Cell lines | ||
| 293T | ATCC | CRL-3216 |
| Plat-E | Cell biolabs | RV-101 |
| Cardiac fibroblast | Isolated from neonatal mouse pups | |
| Mouse Embryonic fibroblast | Isolated from E13.5 mouse embyros | |
| Human H9F | Differentiated from H9 | |
| Experimental models: Organisms/strains | ||
| CD-1 | Charles River | 022 |
| Oligonucleotides | ||
| qRT-PCR primers | This manuscript | Supplementary Table 1 |
| Recombinant DNA | ||
| Atf3 shRNA | Sigma | TRCN0000331748 |
| Fosl2 shRNA | Sigma | TRCN0000263185 |
| Sox15 shRNA | Sigma | TRCN0000415165 |
| Sox4 shRNA | Sigma | TRCN0000234110 |
| Tead1 shRNA | Sigma | TRCN0000085865 |
| Bcl6 shRNA | Sigma | TRCN0000084653 |
| Smad2 shRNA | Sigma | TRCN0000327447 |
| Tead2 shRNA | Sigma | TRCN0000374225 |
| Tead3 shRNA | Sigma | TRCN0000301920 |
| Tead4 shRNA | Sigma | TRCN0000054774 |
| Atf7 shRNA | Sigma | TRCN0000434189 |
| pMx-Ef1a-Ascl1 | This manuscript | N/A |
| pMx-Ef1a-Mef2c | Sigma | N/A |
| pMX-Ascl1-puro | This manuscript | N/A |
| TetO-FUW-Ascl1 | Sigma | N/A |
| TetO-FUW-Brn2 | Vierbuchen et al., 2010 | N/A |
| TetO-FUW-Mytl1 | Vierbuchen et al., 2010 | N/A |
| FUW-M2rtTA | Vierbuchen et al., 2010 | Addgene_20342 |
| pGCDNsam-Hnf4α | Sekiya and Suzuki, 2011 | Addgene_33002 |
| pGCDNsam-Foxa3 | Sekiya and Suzuki, 2011 | Addgene_33005 |
| pMX-puro-MGT | Garbutt et al., 2020 | Addgene_111809 |
| TetO-FUW-EGFP | Vierbuchen et al., 2010 | Addgene_73083 |
| pGCDNsam-IRES-GFP | This manuscript | N/A |
| pMX-puro-lacZ | This manuscript | N/A |
| pLL3.7-UbC-hMEF2c | This manuscript | N/A |
| pLL3.7-UbC-hASCL1 | This manuscript | N/A |
| pGCDNsam-IRES-GFP | Sigma | N/A |
| Software and algorithms | ||
| GraphPad Prism 8 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| R 4.1.0 | R Core Team 2021 | https://www.r-proiect.ore |
| Cell Profiler | Stirling DR et al., 2021 | https://cellprofiler.org |
| FlowJo v10 | FlowJo LLC. | https://www.flowjo.com/ |
| ImageJ-Fiji | National Institutes of Health, USA | https://fiji.sc/ |
| Cell ranger version 6.0.1 | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/what-is-cell-ranger |
| Seurat 4.0.3 | Stuart et al., 2019 | https://github.com/satijalab/seurat/releases |
| MACS2 | Zhang et al., 2008 | https://github.com/macs3-project/MACS |
| Slingshot | Street et al., 2018 | https://www.bioconductor.org/packages/release/bioc/html/slingshot.html |
| Tradeseq | Van den Berge et al., 2020 | https://www.bioconductor.org/packages/release/bioc/html/tradeSeq.html |
| ChromVAR | Schep et al., 2017 | https://bioconductor.org/packages/release/bioc/html/chromVAR.html |
| FigR | Kartha et al., 2022 | https://github.com/buenrostrolab/stimATAC_analyses_code |
| HOMER | Heinz et al., 2010 | http://homer.ucsd.edu/homer/ |
| Diffbind | Stark and Brown, 2011 | https://bioconductor.org/packages/release/bioc/html/DiffBind.html |
| ChIPseeker | Yu, Wang and He, 2015 | https://bioconductor.org/packages/release/bioc/html/ChIPseeker.html |
| FastQC | Babraham Bioinformatics | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| MultiQC | Ewels et al., 2016 | https://multiqc.info/ |
| clusterProfiler | Wu et al., 2021 | https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html |
| deepTools | Ramirez et al., 2016 | https://deeptools.readthedocs.io/en/develop/ |
| pheatmap | Raivo Kolde | https://github.com/raivokolde/pheatmap |
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Salmon version 1.3.0 | Patro et al., 2017 | https://github.com/COMBINE-lab/salmon |
| Deseq2 | Love, Huber and Anders, 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| ROSE | Loven et al., 2013; Whyte et al., 2013 | http://younglab.wi.mit.edu/super_enhancer_code.html |
| Bbduk | Bbmap | https://jgi.doe.gov/data-and-tools/software-tools/bbtools/bb-tools-user-guide/bbduk-guide/ |
| Other | ||