

Abstract

Interactions between biomolecules and structurally disordered calcium phosphate (CaP) surfaces are crucial for the regulation of bone mineralization by noncollagenous proteins, the organization of complexes of casein and amorphous calcium phosphate (ACP) in milk, as well as for structure–function relationships of hybrid organic/inorganic interfaces in biomaterials. By a combination of advanced solid-state NMR experiments and metadynamics simulations, we examine the detailed binding of O-phospho-l-serine (Pser) and l-serine (Ser) with ACP in bone-adhesive CaP cements, whose capacity of gluing fractured bone together stems from the close integration of the organic molecules with ACP over a subnanometer scale. The proximity of each carboxy, aliphatic, and amino group of Pser/Ser to the Ca2+ and phosphate species of ACP observed from the metadynamics-derived models agreed well with results from heteronuclear solid-state NMR experiments that are sensitive to the 13C–31P and 15N–31P distances. The inorganic/organic contacts in Pser-doped cements are also contrasted with experimental and modeled data on the Pser binding at nanocrystalline HA particles grown from a Pser-bearing aqueous solution. The molecular adsorption is driven mainly by electrostatic interactions between the negatively charged carboxy/phosphate groups and Ca2+ cations of ACP, along with H bonds to either protonated or nonprotonated inorganic phosphate groups. The Pser and Ser molecules anchor at their phosphate/amino and carboxy/amino moieties, respectively, leading to an extended molecular conformation across the surface, as opposed to an “upright standing” molecule that would result from the binding of one sole functional group.
1. Introduction
Bone mineral constitutes nanoparticles of a structurally disordered and carbonate-bearing form of Ca hydroxyapatite [HA; “apatite”; Ca10(PO4)6(OH)2].1−5 Vast research efforts have been spent to understand the mechanisms of bone mineralization,5−10 which remain controversial but are believed to be governed by noncollagenous proteins (NCPs) that carry a high density of negatively charged carboxy-bearing (Asp/Glu) and phosphorylated residues.5,9−11 For decades, mainstream models for the NCP/mineral interactions involved “charge-matching” arguments assuming that the protein adopts a secondary structure with its negatively charged sidechains matching the Ca2+ positions at the apatite mineral surface to provide epitaxial crystal growth.6,7 However, doubts thereof arose from the subsequently accumulating evidence that (i) the crystal-binding domains of most NCPs lack a well-defined secondary structure10,11 and (ii) both synthetic and biogenic nanocrystalline apatite particles formed from aqueous solutions—such as body fluids—consist of a “core” of an ordered HA lattice coated by a 1–2 nm thick surface layer (“shell”) of amorphouscalcium phosphate (ACP),4,12−16 often termed the “hydrated surface layer”.2,3,17 These recent insights have led to a paradigm shift advocating simpler electrostatic models for which the net charge of the protein and its underlying distribution control the NCP binding at biogenic apatite.18−21
An accurate atomistic probing of biomolecular adsorption at structurally disordered inorganic calcium phosphate (CaP) surfaces by experimental techniques is hampered by the particle fragility, coupled with their structural disorder. The most detailed current insight is provided by magic-angle-spinning (MAS) nuclear magnetic resonance (NMR) experiments, often utilizing proteins with isotopic 13C/15N labeling at specific sites in its “crystal”-binding domain. Some MAS NMR reports targeted biomolecular binding at bone mineral,22−26 but adsorption on model systems of HA nanoparticles is most commonly encountered, encompassing the surface binding of small biomolecules27−32 and various mineralization-controlling NCPs such as statherin,33,34 osteopontin,35 osteonectin,36,37 and osteocalcin.38 Here, advanced NMR techniques relying on through-space dipolar interactions, such as 13C{31P} and 15N{31P} rotational-echo double resonance (REDOR)39 NMR experiments, have been exploited for obtaining (semi-)quantitative 13C–31P and 15N–31P interatomic distance-information along with 2D 13C–13C correlation NMR experiments that offer constraints on the surface-bound molecular conformation.
Considering the difficulties in experimental probing of the surface binding at atomic resolution, computer modeling by atomistic molecular dynamics (MD) and/or metadynamics simulations offers a rich source of structural information on the organic/inorganic interface.18−20,40−48 They may reveal the precise binding sites of the biomolecule along with its detailed surface-immobilized conformation—henceforth referred to as the binding mode of the molecule. Yet, outstanding problems in computational modeling of biomolecular adsorption at an in vitro- or in vivo-generated nanocrystalline apatite surface concern how its structural disorder and potentially pH-dependent phosphate speciation are accounted for. As discussed further in the Supporting Information, these critical aspects remain essentially ignored in all but a few very recent modeling studies.44−49
Interfaces between phosphorylated biomolecules and disordered CaP phases are not only of pivotal importance for understanding bone mineralization but also underpin casein–ACP complex-formation in milk50−53 and structure–function relationships of hybrid organic/inorganic (bio)materials, such as the bone-tissue-adhesive properties of CaP cements incorporating O-phospho-l-serine (Pser),54−59 which by their capacity to glue bone tissues together makes them promising for accelerating bone-fracture healing. As unveiled by an array of advanced MAS NMR techniques, we recently demonstrated that the “bone-gluing” strength of such a cement correlates well with its content of an amorphous “ACP/Pser” phase of Pser molecules intimately integrated with ACP across a sub-nm scale.60 An analogous “ACP/Ser” component forms in l-serine (Ser) doped cements which, however, exhibit poor bone-adhesive properties,60 as discussed and rationalized further herein.
Herein, we advance the atomic-scale
insight into the organic/inorganic
interface further by examining cements prepared from uniformly 13C/15N–enriched l-serine and O-phospho-l-serine (the latter synthesized herein
for the first time), thereby enabling direct experimental probing
of the proximity of each carboxy/aliphatic and amino group to the
inorganic phosphate moieties of ACP by 13C{31P} and 15N{31P} REDOR
NMR experimentation, respectively. These results are contrasted with
information from heteronuclear 13C{31P} and 1H{31P} correlation 2D NMR experiments that reveal
the contacts between the organic functional groups of Pser/Ser and
the inorganic 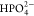 /
/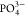 phosphate moieties of ACP. We also discuss
the similarities and differences between the inorganic/organic contacts
in the amorphous ACP/Pser and ACP/Ser biocement components with that
of nanocrystalline HA particles grown from a Pser-bearing aqueous
solution, henceforth referred to as “Pser@HA”, and constituting
a simplified model of a carboxy-bearing or/and phosphorylated protein
residue interacting with bone mineral. Moreover, the NMR-derived 13C–31P and 15N–31P interatomic-distance constraints were contrasted with results from
state-of-the-art well-tempered metadynamics MD simulations61,62 employing the INTERFACE force field and the HA-surface preparation
protocol of refs (45) and (49) to faithfully
model the adsorption of Ser and Pser molecules at the amorphous surface
of apatite nanoparticles. These structural models offer a wealth of
structural details about the Ser/Pser binding modes (encompassing
the surface-bound molecular conformation), as well as a quantification
of the relative contribution of each functional group toward the binding via a recently introduced analysis protocol.48
phosphate moieties of ACP. We also discuss
the similarities and differences between the inorganic/organic contacts
in the amorphous ACP/Pser and ACP/Ser biocement components with that
of nanocrystalline HA particles grown from a Pser-bearing aqueous
solution, henceforth referred to as “Pser@HA”, and constituting
a simplified model of a carboxy-bearing or/and phosphorylated protein
residue interacting with bone mineral. Moreover, the NMR-derived 13C–31P and 15N–31P interatomic-distance constraints were contrasted with results from
state-of-the-art well-tempered metadynamics MD simulations61,62 employing the INTERFACE force field and the HA-surface preparation
protocol of refs (45) and (49) to faithfully
model the adsorption of Ser and Pser molecules at the amorphous surface
of apatite nanoparticles. These structural models offer a wealth of
structural details about the Ser/Pser binding modes (encompassing
the surface-bound molecular conformation), as well as a quantification
of the relative contribution of each functional group toward the binding via a recently introduced analysis protocol.48
2. Materials and Methods
2.1. Preparation of 13C/15N-Enriched Samples
l-serine (Aldrich;  99.5%) and O-phospho-l-serine (Flamma SpA;
99.5%) and O-phospho-l-serine (Flamma SpA;  95%) with all isotopes at their natural
abundance were used as received. [U–13C/15N]-l-serine enriched to 98% with respect to both 13C/15N was purchased from CortecNet (France). It was used
to prepare [U–13C/15N]-O-phospho-l-serine, which was obtained as its HCl salt (Figure S1) from the corresponding BocNH-Ser-CO2-tert-Bu analogues via phosphoramidation
with tert-butyl N, N′-diisopropylcarbamimidate followed by peroxide oxidation
to yield the protected phosphate ester, which was converted into the
desired product by global deprotection under acidic conditions; see Section S3 for details. These isotopically labeled
Ser and Pser powders are for simplicity abbreviated as Ser* and Pser*,
respectively. They were used for producing a nanocrystalline HA powder
with surface-immobilized Pser* molecules, as well as cements comprising N mol % of Ser* or Pser*, which are henceforth denoted by
SerN and PserN, respectively.
95%) with all isotopes at their natural
abundance were used as received. [U–13C/15N]-l-serine enriched to 98% with respect to both 13C/15N was purchased from CortecNet (France). It was used
to prepare [U–13C/15N]-O-phospho-l-serine, which was obtained as its HCl salt (Figure S1) from the corresponding BocNH-Ser-CO2-tert-Bu analogues via phosphoramidation
with tert-butyl N, N′-diisopropylcarbamimidate followed by peroxide oxidation
to yield the protected phosphate ester, which was converted into the
desired product by global deprotection under acidic conditions; see Section S3 for details. These isotopically labeled
Ser and Pser powders are for simplicity abbreviated as Ser* and Pser*,
respectively. They were used for producing a nanocrystalline HA powder
with surface-immobilized Pser* molecules, as well as cements comprising N mol % of Ser* or Pser*, which are henceforth denoted by
SerN and PserN, respectively.
2.1.1. Pser@HA Specimen
5.0 mL of a 500
mM aqueous solution of CaCl2 in deionized water was pipetted
in a 20 mL flask placed in a water bath at 37 ± 2 °C, whereupon
Pser*·HCl was added to the solution to yield a concentration
of 10 mM. 5 mL of 300 mM (NH4)3PO4(aq) was added at 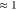 mL/min with continuous magnetic stirring.
The pH of the solution was subsequently raised to 7.5 by dropwise
addition of 1 M NaOH(aq), leading to precipitation of floccular ACP
particles. Deionized water was added to a final volume of 20 mL, and
the flask was sealed. The suspension was aged for one week at 37 ±
0.2 °C, leading to a final pH value of 5.3. The as-formed Pser@HA
particles were isolated by centrifugation, cleaned twice with deionized
water, and then dried in a desiccator at room temperature for 4 days.
mL/min with continuous magnetic stirring.
The pH of the solution was subsequently raised to 7.5 by dropwise
addition of 1 M NaOH(aq), leading to precipitation of floccular ACP
particles. Deionized water was added to a final volume of 20 mL, and
the flask was sealed. The suspension was aged for one week at 37 ±
0.2 °C, leading to a final pH value of 5.3. The as-formed Pser@HA
particles were isolated by centrifugation, cleaned twice with deionized
water, and then dried in a desiccator at room temperature for 4 days.
The Pser content was estimated as ≈ 5 wt % by contrasting the integrated 15N NMR intensity from the Pser@HA specimen with that of the Pser16 cement with known Pser content. The transmission electron microscopy images of Figure S2 reveal agglomerates of crystalline domains with variable sizes (from a few nm to 10–20 nm) that are fused together by ACP. The surface area was estimated to be 159 m2/g by the Brunauer–Emmett–Teller model63 (N2 uptake at relative pressures (P/P0) of 0.05–0.15, using a Micrometrics ASAP2020 volumetric adsorption analyzer).
The Pser@HA synthesis protocol was preceded by extensive testing of optimal preparation conditions by using Pser (Flamma SpA) with concentrations in the 5–200 mM range. As verified by Figure S3, [Pser] ≥ 20 mM yielded products with little/no surface immobilization but predominantly comprised the Ca salt of Pser, Ca[O-phospho-l-serine]·H2O,64,65 henceforth denoted CaPser.
2.1.2. Ser/Pser-Doped Cements
The SerN and PserN cements with N = {8, 16, 30} were prepared in batches of 0.25 g by first dissolving/suspending each organic Ser* and Pser* additive in 60 μL of deionized water (see Table S1), whereupon a powder of α-Ca3(PO4)2 (α-TCP) (see refs (54) and (60) for preparation details) was added at liquid-to-powder (L/P) ratio of 0.24 mL/g. The powder and liquid were mixed with a spatula for ≈30 s. The cement paste was cured at 21 °C and 30% relative humidity for 15–30 min and then transferred to a sealed plastic bag that was stored at 37 °C for 7 days at 100% humidity. All cement specimens were thereafter stored under dry conditions in a desiccator. For practical reasons, pH measurements of the cement pastes prior to their setting were performed on larger SerN/PserN batches of 3.0 g and a slightly higher L/P = 0.4 mL/g ratio. Table S1 presents the pH value associated with each cement preparation, obtained as an average over three independent preparations.
2.2. Solid-State NMR Experiments
The solid-state NMR experimentation was performed with Bruker Avance-III spectrometers operating at the magnetic fields (B0) of 9.4 T or 14.1 T, which provided {1H, 13C, 31P} Larmor frequencies of {−400.1, −100.6, −162.0} MHz and {−600.1, −150.9, −242.9} MHz, respectively, along with 60.4 MHz for 15N at 14.1 T. A fine powder of each specimen was packed in a ZrO2 rotor of outer diameters of either 2.5 mm (thin-wall), 3.2, or 4 mm, as specified along with each MAS rate (νr) in the figure captions and in Section S1, which provides all details of each NMR experiment. Full rotors were employed throughout, except for the REDOR NMR39 experiments, for which each sample was restricted to the center 1/3 volume of a 4 mm rotor. 1H/13C, 31P, and 15N chemical shifts are quoted relative to neat tetramethylsilane (TMS), 85% H3PO4(aq), and solid 15NH4Cl, respectively.
2.3. Metadynamics Simulations
The preparation
of the simulated systems and the well-tempered metadynamics simulations
were performed as described in ref (48). Here, we only recapitulate the most essential
information and refer to Section S2 and
ref (48) for details.
A box with side-lengths {3.9, 4.3, 7.8} nm (periodic boundary conditions)
contained the simulated system of three components (Figure 1a): one Ser or Pser molecule
in a water phase (2600 H2O molecules) that interfaced a
“HA slab”, whose interior consisted of an HA lattice
of the biologically relevant P63/m modification.66 The “surface”
segment was generated according to ref (45), which mimics a disordered surface at a nanocrystalline
HA-particle by introducing acidic protons at randomly selected phosphate
groups accompanied by Ca2+-cation removal until a charge-neutral
surface is obtained, whose {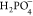 ,
,  ,
, 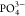 } speciation matches that of phosphate ions
in an aqueous solution at a given pH value; see Figure 1. The HA surface and the Pser/Ser protonation
states were implemented for the experimentally relevant pH values
of 4.5 for Pser and 7.4 for Ser, as explained further in Section S2.
} speciation matches that of phosphate ions
in an aqueous solution at a given pH value; see Figure 1. The HA surface and the Pser/Ser protonation
states were implemented for the experimentally relevant pH values
of 4.5 for Pser and 7.4 for Ser, as explained further in Section S2.
Figure 1.

(a) Simulated three-component system,
consisting of a “HA
slab” with a lattice of crystalline HA and a disordered surface
that interface a water phase that contains one Pser molecule (or Ser;
not shown). The surface atoms are enlarged for better visualization.
The box lengths along the z and x directions are indicated, along with the (approximate) lengths of
the water and lattice/surface domains. (b,c) Dependence of the average
number  of protons per phosphate group at the as-prepared
HA surface (red curve) along with the net number of protons of the
molecular forms of (b) Pser and (c) Ser (black curves) plotted against
the pH value of the solution. The most stable molecular form of (b)
Pser and (c) Ser at a given pH value is shown on top of each graph,
where the colored circles enclose the functional group(s) with altering
protonation states between the pH domains marked by black vertical
lines. The vertical green lines mark the simulated pH values of 4.5
and 7.4. Each number inside the graph specifies the respective pKa values of the organic functional groups, as
well as the inorganic {H3PO4, H2PO4−, and
of protons per phosphate group at the as-prepared
HA surface (red curve) along with the net number of protons of the
molecular forms of (b) Pser and (c) Ser (black curves) plotted against
the pH value of the solution. The most stable molecular form of (b)
Pser and (c) Ser at a given pH value is shown on top of each graph,
where the colored circles enclose the functional group(s) with altering
protonation states between the pH domains marked by black vertical
lines. The vertical green lines mark the simulated pH values of 4.5
and 7.4. Each number inside the graph specifies the respective pKa values of the organic functional groups, as
well as the inorganic {H3PO4, H2PO4−, and 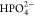 } surface species (the phosphate speciation
at the surface matches that of the surrounding solution45).
} surface species (the phosphate speciation
at the surface matches that of the surrounding solution45).
The atomistic MD simulations involved NVT ensembles
at the temperature T = 37 °C, utilizing the
GROMACS v2018.1 platform67 and the following
force-fields: INTERFACE45,49 for the HA slab, CHARMM36
(July 2017)68 for the Pser/Ser molecules,
and TIP3P69 (without Lennard-Jones terms
for H70) for water. Well-tempered metadynamics61,62 with a bias factor of γ = 5 and 32 independent “walkers”62,71 were employed to ensure an efficient sampling of the molecular conformations
and accelerate the convergence, using the variational enhanced sampling
(VES) protocol72 implemented in the PLUMED2.4
software.73 Two collective variables were
exploited for locating the most stable surface-bound molecular conformation,
involving the distance between the center of the HA slab and the COO–/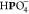 atom of the respective Ser/Pser molecule
along with an interaction-energy dependent function defined in Section S2. The reported modeled parameters and
their uncertainties are averages over 4 independent simulations.
atom of the respective Ser/Pser molecule
along with an interaction-energy dependent function defined in Section S2. The reported modeled parameters and
their uncertainties are averages over 4 independent simulations.
3. Results and Discussion
3.1. Local 31P Environments Probed by MAS NMR
Figure 2 displays 31P MAS NMR spectra recorded from the nanocrystalline Pser@HA sample and the PserN/SerN cements with N = {8, 16, 30}. The peak intensities of the single-pulse-acquired NMR spectra of the left panel quantitatively reflect the relative phase constituents of each sample, whereas those of the right panel of Figure 2 were obtained by 1H → 31P CP, thereby only revealing 31P NMR signals from phosphate groups nearby protons. The directly excited 31P MAS NMR peak shapes from the cements are complex due to the contributing signals from unreacted α-TCP (Figure S4). The absence of protons in the α-TCP structure, however, renders the 1H → 31P CPMAS spectra considerably simpler because they only comprise 31P resonances from 1H-bearing phases, which predominantly involve the amorphous ACP/Pser or ACP/Ser components.60 We henceforth focus on the 1H → 31P CPMAS NMR spectra along with the deconvolutions into their underlying NMR-peak components and the associated best-fit parameters presented in Table S6. The NMR spectra from the cements in Figure 2 overall match those presented in ref (60) from near-identical preparations with 13C/15N isotopes at natural abundance.
Figure 2.

31P MAS NMR spectra recorded at 14.00 kHz MAS either directly by single pulses (left panel) or by 1H → 31P CP (right panel) from (a,b) Pser@HA, or from the as-indicated (c–f) SerN and (g–l) PserN cements. Note that no NMR peak from remnants of the proton-free α-TCP precursor (Figure S4) appear in the CPMAS-derived spectra; the latter are shown together with the best-fit spectra (orange traces) and the deconvolutions into the as-indicated peak components given in each legend. The narrow NMR peaks at 1.4 ppm and −1.0 ppm stem from minor impurities of brushite (CaHPO4·2H2O) and CaPser (Ca[O-phospho-l-serine]·H2O), respectively, while that marked by an asterisk in (d) derives from an unknown impurity. Table S6 lists the best-fit NMR parameters.
Both the directly excited and CP-derived 31P MAS NMR
spectra of the Pser@HA specimen (Figure 2a,b) are typical for nanocrystalline HA particles,
which involve a crystalline HA “core” coated by a surface
layer (“shell”) of ACP.2−4,12−17 The “core” and “shell” components produce
a narrow and broad resonance, respectively,4,12−14 centered at the corresponding 31P chemical
shifts δP ≈ 3.0 ppm and δP ≈ 2.2 ppm (Figure 2a,b and Table S6). The lower shift
of the ACP component reflects the acidic solution (pH = 5.3) surrounding
the Pser@HA particles before their isolation, which yields a surface
layer of ACP enriched in protonated phosphate moieties4,13,14,16,74 resonating at lower 31P shifts
than nonprotonated 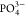 groups.75−77 Moreover, the close 1H–31P distances of
groups.75−77 Moreover, the close 1H–31P distances of 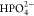 /H2PO4− groups emphasize the “ACP”
contribution in the 1H → 31P CPMAS NMR
spectrum (Figure 2b)
relative to the directly excited counterpart (Figure 2a); also see Table S6.
/H2PO4− groups emphasize the “ACP”
contribution in the 1H → 31P CPMAS NMR
spectrum (Figure 2b)
relative to the directly excited counterpart (Figure 2a); also see Table S6.
Cements prepared from α-TCP and water in the absence
of Pser
consist of disordered HA.60,78−80 Although ACP and HA particles are unstable in acidic aqueous solutions,
they become stabilized by negatively charged organic additives in weakly acidic solutions.4 The
pH value of the precursor paste before the cement setting is reduced
for increasing Pser/Ser content, while at a fixed doping level N, Pser yields a markedly lower pH value than Ser (Table S1). Hence, the highly acidic PserN cement pastes preclude HA formation, whereas the Ser8
and Ser16 samples comprise significant HA contributions (Figure 2d,f and Table S6), as proved unambiguously by the heteronuclear
correlation NMR results of Section 3.2. The progressively reduced pH values for the Pser-bearing
cements also manifest as 31P resonance displacements of
the ACP/Pser phase toward lower chemical shifts for increasing N. These acidic conditions also induced a minor brushite
(CaHPO4·2H2O) formation, as signified by
the narrow resonance at δP = 1.4 ppm60,81,82 (Figure 2h,j,l). It is instructive to contrast the 31P shifts among the ACP (≈2.2 ppm; Figure 2a,b), ACP/Ser (≈2.3
ppm; Figure 2d,f),
and ACP/Pser (0.7–1.8 ppm; Figure 2h,j,l) phases with that from amorphous tricalcium
phosphate of approximate stoichiometry Ca3(PO4)2·nH2O (δP ≈ 3 ppm)81,83,84 and the H31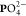 environments of amorphous CaHPO4·nH2O (δP ≈
1.5 ppm).4,77
environments of amorphous CaHPO4·nH2O (δP ≈
1.5 ppm).4,77
3.2. Organic/Inorganic Interface Probed by 1H–31P Correlation NMR
Figure 3 presents the 1H MAS NMR spectra recorded from the Pser@HA sample along with
the SerN and PserN cements. The
various 1H sites of the present specimens may be of organic
{COOH, POH, NH3, CHn} or inorganic
{OH, Hn } origin, along with H2O molecules
that may either constitute an integral component of the inorganic
ACP and HA structures (“structure-bound”), associated
with the inorganic/organic ACP/Pser and ACP/Ser components or being
mobile physisorbed species. The latter 1H2O molecules typically resonate in the 4.5–5.5 ppm spectral
region,13,14,16,74,85,86 where Figure 3 suggests
that they constitute a significant fraction of the entire proton reservoir
in the Pser@HA and PserN/SerN samples.
Moreover, the characteristic 1H NMR peak at δH ≈ 0 ppm12−14,16,74,85,86 from OH groups in the HA lattice is observed from
several specimens. We refer to Mathew et al.(60) for discussions on the various 1H
resonance assignments made from nominally identical PserN and SerN cement compositions.
} origin, along with H2O molecules
that may either constitute an integral component of the inorganic
ACP and HA structures (“structure-bound”), associated
with the inorganic/organic ACP/Pser and ACP/Ser components or being
mobile physisorbed species. The latter 1H2O molecules typically resonate in the 4.5–5.5 ppm spectral
region,13,14,16,74,85,86 where Figure 3 suggests
that they constitute a significant fraction of the entire proton reservoir
in the Pser@HA and PserN/SerN samples.
Moreover, the characteristic 1H NMR peak at δH ≈ 0 ppm12−14,16,74,85,86 from OH groups in the HA lattice is observed from
several specimens. We refer to Mathew et al.(60) for discussions on the various 1H
resonance assignments made from nominally identical PserN and SerN cement compositions.
Figure 3.

1H NMR spectra recorded at 9.4 T and 14.00 kHz MAS from the as-indicated SerN and PserN cements, as well as the Pser@HA and Pser samples. The green trace in (g) is a vertical expansion. The sharp peaks at 3.8/1.1 ppm in (f,g) stem from a minor isopropanol impurity, whereas those marked by asterisks in the NMR spectra from Pser@HA and Ser8 derive from HA-associated surface water molecules, as discussed in detail in refs (60) and (74)
The remainder of the article focuses on the probing of the organic/inorganic interface in the Pser@HA, Pser16, and Ser16 specimens, which were examined by dipolar-based MAS NMR experimentation, such as the dipolar-mediated heteronuclear multiple-quantum coherence (D-HMQC) 1H{31P} NMR spectra60,82,87,88 shown in Figure 4. They were recorded from the Pser@HA, Pser16, and Ser16 specimens using a short HMQC excitation period (τexc = 176 μs) to ensure detection predominantly of 1H and 31P sites separated by at most a few hundreds of pm. In each D-HMQC spectrum, a 2D NMR correlation peak centered at the chemical-shift pair {δ1, δ2} ≡ {δP, δH} evidences close spatial proximity between 1H and 31P sites resonating at the (average) chemical shifts δH and δP, respectively.
Figure 4.

1H{31P} D-HMQC correlation NMR spectra acquired at 34.00 kHz MAS from the (a) Ser16, (b) Pser16, and (c) Pser@HA specimens using a short HMQC excitation period of 176 μs. The 2D NMR spectra are shown together with projections along the 1H (horizontal) and 31P (vertical) dimensions at the top and to the right, respectively; the green trace in (c) is a three-fold vertical expansion of the 1H projection. Directly excited (“single-pulse”) 1H MAS NMR from crystalline Pser and Ser are displayed by red traces in (a,b). Most of the 1H resonances marked along the horizontal dimension are of organic origin, whereas the peaks assigned along the 31P dimension originate either from HA (blue color) or the ACP/Pser or ACP/Ser cement constituents (cyan color). The 2D NMR regions {δP, δH} ≈ {1.6, 5.8} ppm and {δP, δH} ≈ {4.3, 5.0} ppm marked by the respective yellow and blue rectangles in (c) are discussed in Section 3.2).
Figure 4a offers direct and unambiguous evidence for an intimate ACP/Ser integration across a sub-nm scale, whereas the Pser16 counterpart reveals HMQC correlations stemming from both intramolecular 1H–31P dipolar interactions of Pser and Pser···ACP contacts of the ACP/Pser phase (Figure 4b). The 1H{31P} D-HMQC spectral signatures observed from a PserN cement in the high-δH region reflect correlations between acidic protons and (in)organic phosphate groups. Hence, it depends strongly on the pH value before cement setting (i.e., on the Pser content; Table S1). Indeed, the higher pH = 5.3 of the solution that immersed the Pser@HA particles renders the 2D NMR spectrum in Figure 4c closer to that reported previously from a Pser4 specimen60 than that from Pser16. As expected from the results of Figures 2 and 3, the D-HMQC NMR spectra recorded from the Pser@HA powder (see Figure 4c) and the Ser16 cement (Figure 4a) manifest the HA-characteristic correlation at {δP, δH} = {2.9, 0} ppm.13,14,16,74,86
Correlations between the organic protons of
the {NH3, CHn} moieties
and 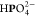 /
/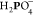 groups of ACP dominate the 1H NMR spectral range of δH ≲ 10 ppm, as becomes
evident by contrasting the projection of the 2D NMR spectra of Figure 4a,b along the 1H dimension with the single-pulse-acquired 1H MAS
NMR spectra from the crystalline Pser and Ser samples. The comparatively
more intense 2D NMR-peak intensities of the
groups of ACP dominate the 1H NMR spectral range of δH ≲ 10 ppm, as becomes
evident by contrasting the projection of the 2D NMR spectra of Figure 4a,b along the 1H dimension with the single-pulse-acquired 1H MAS
NMR spectra from the crystalline Pser and Ser samples. The comparatively
more intense 2D NMR-peak intensities of the 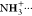 PO4 contacts with 1H shifts ≈9 ppm relative to those of CHn···PO4 (4–5 ppm) ppm of either cement suggest much shorter
distances between the inorganic phosphate species and the positively
charged
PO4 contacts with 1H shifts ≈9 ppm relative to those of CHn···PO4 (4–5 ppm) ppm of either cement suggest much shorter
distances between the inorganic phosphate species and the positively
charged  moiety than the aliphatic groups that do
not bond directly to the surface, as indeed predicted by the metadynamics
simulations (Section 3.3) and discussed further in Section 3.6.
moiety than the aliphatic groups that do
not bond directly to the surface, as indeed predicted by the metadynamics
simulations (Section 3.3) and discussed further in Section 3.6.
Moreover, the HMQC NMR spectrum
of Pser@HA reveals two broad but
resolved 2D NMR peaks (marked by blue/yellow rectangles in Figure 4c) not observed from
the cements. They are tentatively attributed to either aliphatic protons
or water molecules nearby inorganic phosphate groups, where the 2D
correlation centered at {δP, δH}
≈ {1.6, 5.8} ppm and extending toward lower 31P
and 1H chemical shifts likely reflects CHn/H2O···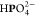 proximities. Further work is required to
reach firmer assignments, particularly for the {δP, δH} ≈ {4.3, 5.0} ppm correlation, whose
high 31P shift is atypical of
proximities. Further work is required to
reach firmer assignments, particularly for the {δP, δH} ≈ {4.3, 5.0} ppm correlation, whose
high 31P shift is atypical of  (and let alone
(and let alone 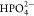 ) groups in CaP phases, although 1H–31P correlation NMR peaks at similar shifts have
been ascribed to
) groups in CaP phases, although 1H–31P correlation NMR peaks at similar shifts have
been ascribed to 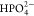 species in the HA lattice.74 As discussed further in Section S4, the signal at δH ≈ 7 ppm in the 1H{31P} D-HMQC NMR spectra from Pser and Pser@HA is characteristic
of structure-bound H2O molecules of “ACP”,
encompassing the amorphous surface of nanocrystalline HA.16,60,89
species in the HA lattice.74 As discussed further in Section S4, the signal at δH ≈ 7 ppm in the 1H{31P} D-HMQC NMR spectra from Pser and Pser@HA is characteristic
of structure-bound H2O molecules of “ACP”,
encompassing the amorphous surface of nanocrystalline HA.16,60,89
3.3. Surface Binding of Pser and Ser Modeled by Metadynamics
3.3.1. Overview and General Considerations
Metadynamics simulations were utilized to locate the energetically most favorable molecular adsorption at a structurally disordered HA surface representative for the nanocrystalline Pser@HA particles as well as the ACP component of the Pser16/Ser16 cements. The simulations were performed for a pH-dependent HA surface representative of structurally disordered forms of its crystallographic (100) and (001) faces interfacing a water phase comprising one Ser or Pser molecule with protonation states for each of pH = {4.5, 7.4}; see Figure 1 and Section S2.
Numerous experimental studies on HA nucleation/growth in the presence of both small and large biomolecules suggest a preference for molecular adsorption at the larger (100) or (101) HA surfaces relative to their smaller (001) counterpart,28,90−93 as also corroborated by previous modeling.18,40,43−45,94,95 Indeed, the consistently stronger adsorption at the “disordered” (100) surface was recently deduced by metadynamics simulations of the Pser binding at HA across a wide pH range of 4.5–14 (ref (48)): Table S3 reproduces the results for the Pser adsorption at each pH = {4.5, 7.4} value and (100)/(001) surface,48 along with those obtained herein from Ser. Hence, we focus on the results for (100) at each experimentally relevant pH = 7.4 for Ser16 and pH = 4.5 for Pser16/Pser@HA, whose most probable/representative stable binding modes are exemplified in Figure 5. No aliphatic CH/CH2 groups of either molecule are discussed because they do not bind directly at the HA surface.
Figure 5.

Representative examples of the most stable/probable metadynamics-derived molecular binding modes at a “disordered (100) HA surface” of (a) Ser at pH = 7.4 and (b) Pser at pH = 4.5. All red numbers mark selected distances (in pm) between directly bonded Ser/Pser···HA atoms, while those in black indicate C–P and N–P distances relating to the experimental constraints from NMR. The green numbers in (b) represent intramolecular C–P distances of Pser. For visualization purposes, only a few surface contacts are shown for each organic moiety. Notably, because none of the surface-binding modes of either Ser or Pser involve the sole anchoring of one functional group (Table 1), both molecules assume an extended conformation that “caps” the HA surface. We underscore that owing to the distributions of stable binding modes, no single graphical picture can capture all details of the molecular adsorption (Tables 1, S3, and S4).
There are two main groups of A···Bcontact modes (i.e.,
“bonding types”) between an organic atom site A of Ser/Pser and an inorganic surface-atom B: (i) electrostatic (“ion–ion”) interactions
among negatively charged O sites of carboxy/phosphate groups and positive
Ca2+ cations of the ACP layer at nanosized HA particles,
and (ii) H bonds between an organic (inorganic) proton and an inorganic
(organic) O site. Notably, although carboxy/phosphate groups of biomolecules
adsorb via both interaction types, each electrostatic
bond (e.g., CO···Ca2+) is around 2.5 times stronger than its H-bond
counterpart (e.g., CO··· ).48 These relative
interaction strengths underlie the current consensus that ion–ion
interactions dominate the energy landscape of biomolecular adsorption,18−21 notwithstanding that the H bonds (140–220 pm) are shorter
than the O···Ca2+ distances (240–330 pm); see Figure 5.
).48 These relative
interaction strengths underlie the current consensus that ion–ion
interactions dominate the energy landscape of biomolecular adsorption,18−21 notwithstanding that the H bonds (140–220 pm) are shorter
than the O···Ca2+ distances (240–330 pm); see Figure 5.
3.3.2. Distinct Biomolecular Binding at (100) and (001) HA Faces
Our comments made in Section S2.5 about biomolecules interacting with inorganic species at an essentially amorphous HA surface might seem to preclude any preferential adsorption at a specific HA surface type. This apparent contradiction, however, may be reconciled by noting that the consistently stronger adsorption at the “(100)” surface does not stem primarily from crystallographic/structural features—where the (hkl) notation rather specifies the crystallographic origin of the disordered (100) and (001) surfaces—but merely from its higher molar ratio nCa/nP ≈ 1.55 relative to that of nCa/nP ≈ 1.15 for (001) across the pH-range of 3.8–7.4. Consequently, the comparatively higher Ca2+ abundance (and larger nCa/nP ratio) at the disordered (100) surface relative to its (001) counterpart emphasizes the electrostatic CO/PO···Ca2+ interactions that predominantly govern the net biomolecular adsorption energy,18−21,48 as confirmed by Table S3.
The above-quoted nCa/nP values were estimated from the chemical speciations of the two outermost layers at each (100) and (001) surface at the end of the metadynamics MD simulation (≈40 ns); see Table 1 of ref (48) for the corresponding precise speciations at the outermost surface layer relevant for the pH values of the present study. Notably, due to minor ion-dissolution processes, the exact Ca2+/phosphate surface speciations vary slightly throughout each metadynamics simulation, thereby not necessarily being identical to those plotted in Figure 1b,c that represent the initially prepared outermost surface layer; see ref (48) for details.
Table 1. MD-Derived Surface-Adsorption Dataa.
| Pser | Ser | |||
|---|---|---|---|---|
| pH | 4.5 | 7.4 | 4.5 | 7.4 |
| ΔFads | –40.7 | –66.4 | –36.6 | –37.7 |
| Contributions to Binding Energyb | ||||
| phosphate | 0.72 | 0.70 | ||
| carboxy | 0.20 | 0.26 | 0.57 | 0.64 |
| amino | 0.08 | 0.04 | 0.30 | 0.16 |
| hydroxyl | 0.13 | 0.10 | ||
| Binding Mode Statisticsc | ||||
| PN | 0.51 | 0.15 | ||
| PC | 0.23 | |||
| PCN | 0.15 | 0.74 | ||
| CN | 0.41 | 0.30 | ||
| CNO | 0.55 | 0.59 | ||
Metadynamics-derived Helmholtz free energy of adsorption (ΔFads) for the Pser and Ser binding at the disordered (100) surface for pH = {4.5, 7.4}; see Section S2. A more negative ΔFads value implies a stronger surface binding. The results representative for the Pser@HA/Pser16 (pH = 4.5) and Ser16 samples (pH = 7.4) are typeset in boldface. The data for Pser are reproduced from ref (48).
Fractional contribution of each functional group to the net adsorption energy, as defined in Section S3. No aliphatic group is surface bound.
Distribution of stable binding modes, where each number reflects the probability (relative fraction/contribution) out of all stable binding modes. The capital letters P, C, N, and O represent the phosphate (Pser), carboxy, amino, and hydroxyl (Ser) group contributions, respectively. Only binding modes with probability ≥ 0.10 are listed.
3.3.3. Ser and Pser Binding Modes at HA/ACP
Figure 5a exemplifies
one of the most probable/representative Ser binding modes at ACP in
solutions at physiological pH = 7.4, relevant both for the ACP/Ser
component of the SerN cements and the amorphous surface
at HA nanoparticles, all of which involve simultaneous {COO–,  , OH} anchoring; they account for ≈60%
of all stable Ser binding modes, with the remaining constituting a
dual carboxy/amino binding (Table 1). The absence of any significant binding mode by one
functional group alone leads to a near-parallel “capping”
of the Ser molecule along the ACP surface, as illustrated in Figure 5a. Although typically
all three {COO–,
, OH} anchoring; they account for ≈60%
of all stable Ser binding modes, with the remaining constituting a
dual carboxy/amino binding (Table 1). The absence of any significant binding mode by one
functional group alone leads to a near-parallel “capping”
of the Ser molecule along the ACP surface, as illustrated in Figure 5a. Although typically
all three {COO–,  , OH} moieties bind at the HA surface, Table 1 reveals strikingly
different relative contributions of {64, 16, 10}% among the respective
groups toward stabilizing/driving the Ser adsorption. The overwhelming
carboxy-group contribution stems from its prevalent contact mode of
strong electrostatic CO···Ca2+ interactions (Table S3),
whereas the modest net contribution of ≈26% to the total adsorption
energy from the
, OH} moieties bind at the HA surface, Table 1 reveals strikingly
different relative contributions of {64, 16, 10}% among the respective
groups toward stabilizing/driving the Ser adsorption. The overwhelming
carboxy-group contribution stems from its prevalent contact mode of
strong electrostatic CO···Ca2+ interactions (Table S3),
whereas the modest net contribution of ≈26% to the total adsorption
energy from the  and OH groups together reflect their primary
(for the amino group exclusive) contact mode of weaker H bonds.
and OH groups together reflect their primary
(for the amino group exclusive) contact mode of weaker H bonds.
In neutral and alkaline solutions, Pser preferentially anchors at
the ACP surface by all three {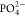 , COO–,
, COO–,  } moieties,48 leading to a significantly more negative adsorption energy than
that for Ser (Table 1). The stronger binding originates primarily from the organic phosphate
group of Pser, which is the main adsorption promoter and involved
in all stable binding modes regardless of the precise pH and (100)/(001)
surface type;48 see Table S3. However, the Pser–ACP binding weakens significantly
in the acidic solutions relevant for the Pser16 and Pser@HA sample
preparation conditions (3.9 ≤ pH ≤ 5.3). This feature
may be traced to a lower amount of Ca2+ cations at the
CaP surface and thereby fewer CO···Ca2+ and PO···Ca2+ electrostatic contacts, where the latter are
diminished further because they are superseded by weaker POH···HnPO4 bonds (Table S4) accompanying
the onset of protonation of the organic phosphate group for pH
} moieties,48 leading to a significantly more negative adsorption energy than
that for Ser (Table 1). The stronger binding originates primarily from the organic phosphate
group of Pser, which is the main adsorption promoter and involved
in all stable binding modes regardless of the precise pH and (100)/(001)
surface type;48 see Table S3. However, the Pser–ACP binding weakens significantly
in the acidic solutions relevant for the Pser16 and Pser@HA sample
preparation conditions (3.9 ≤ pH ≤ 5.3). This feature
may be traced to a lower amount of Ca2+ cations at the
CaP surface and thereby fewer CO···Ca2+ and PO···Ca2+ electrostatic contacts, where the latter are
diminished further because they are superseded by weaker POH···HnPO4 bonds (Table S4) accompanying
the onset of protonation of the organic phosphate group for pH  6.8 (Figure 1). These effects combine into a significantly weakened
Pser adsorption at pH = 4.5 (Table 1), which incidentally nearly matches that of Ser at
the comparatively Ca-richer HA surface at pH = 7.4 that promotes electrostatic
interactions.
6.8 (Figure 1). These effects combine into a significantly weakened
Pser adsorption at pH = 4.5 (Table 1), which incidentally nearly matches that of Ser at
the comparatively Ca-richer HA surface at pH = 7.4 that promotes electrostatic
interactions.
Relative to the Pser surface immobilization at
pH = 7.4, the weaker
Pser···ACP contacts in acidic solutions are reflected
in a larger distribution of distinct binding modes at pH = 4.5 (Table 1), along with an overall
more modest COO– participation: roughly half of
all surface-bound Pser molecules anchor by a dual binding of their
phosphate and amino groups, as depicted by Figure 5b, whereas all other binding constellations
occur either by the phosphate/carboxy moieties (≈23%) or by
all three groups together (≈15%); see Table 1. (We remind that all binding modes not listed
in Table 1 are insignificant,
such as the anchoring of one functional group alone of either molecule).
Yet, although the  and COO– moieties participate
in ≈90% and ≈40% of all binding modes, respectively, Table S3 reveals that their corresponding net energy contributions to stabilizing the adsorption only
amount to ≈8% (
and COO– moieties participate
in ≈90% and ≈40% of all binding modes, respectively, Table S3 reveals that their corresponding net energy contributions to stabilizing the adsorption only
amount to ≈8% ( ) and ≈20% (COO–) due to the higher CO···Ca2+ interaction-energy per bond relative to that of NH···PO and the overall dominant
PO···Ca2+ interactions.
) and ≈20% (COO–) due to the higher CO···Ca2+ interaction-energy per bond relative to that of NH···PO and the overall dominant
PO···Ca2+ interactions.
Out of all functional groups of Pser, the simulations predict the strongest binding at the ACP surface by the phosphate group. However, the broad 31P NMR peak from the ACP/Pser phase (Figures 2 and 4) cannot discriminate between the organic and inorganic phosphate contributions, which underscores the very intimate Pser···ACP contacts in the Pser/ACP phase across a sub-nm scale. Indeed, NMR experiments sensitive to the 31P–31P distance distributions revealed essentially equal average distances in synthetic/pristine ACP and the ACP/Pser components in PserN cements.60
3.4. 13C–31P Correlation NMR Reveals Ser/Pser–ACP Contacts
Figure 6 presents 1H → 13C CPMAS spectra observed from the polycrystalline Ser, Pser, and CaPser powders along with that of the nanocrystalline Pser@HA specimen and the PserN/SerN cements with N = {8, 16, 30} prepared from the Pser*/Ser* precursors. The strikingly different 13C NMR peak widths observed from the precursors relative to Pser@HA or any cement reflect the distinctly different local order of the 13C environments. Hence, even routine 13C CPMAS NMR experiments give qualitative evidence for organic/inorganic contacts. Although heteronuclear 17O–43Ca NMR experiments would enable the most direct probing of the electrostatic CO/PO···Ca2+ interactions that dominate the molecular adsorption, such experiments are precluded for NMR-signal sensitivity reasons along with further sample preparation obstacles which require isotopic 17O and 43Ca labeling.96−98 Consequently, we resorted to less challenging 13C–31P NMR experiments, which merely probe the Pser/Ser···ACP contacts via their 13C–31P distances in 13C–O···Ca2+···O–31P motifs.
Figure 6.

13C CPMAS NMR spectra obtained at 9.4 T and 9.00 kHz MAS from (a) polycrystalline Pser (black trace) and CaPser (red trace), (b) Pser@HA, and as-indicated (c–e) PserN cements, as well as (f) polycrystalline Ser along with (g,h) SerN cements. All NMR spectra were recorded from specimens prepared from 13C-enriched precursors, except for those shown in (a,f). The asterisk in (h) marks a minor peak from an unknown impurity.
Figure 7 displays 13C{31P} D-HMQC NMR87,88 spectra recorded
from the Pser16 and Ser16 cements, where each 13C–31P proximity is revealed by a 2D NMR cross peak centered at
the shift-pair {δP, δC}. Owing to
the phosphate group of Pser, however, the 13C{31P} D-HMQC spectrum observed from Pser16 primarily reflects the intramolecular 13C–31P distances,
which also account for the observed NMR-intensity increase along 13CO  13CH
13CH  13CH2 in Figure 7a; this is particularly
evident when contrasting the various peak intensities of the HMQC
projection with their counterparts of the corresponding 1H → 13C CPMAS NMR spectrum (Figure S5). The 2D NMR-peak intensities reflect semiquantitatively
the relative through-space 13C–31P distances.
The significantly emphasized intensity and the higher 13C chemical-shift dispersion of the 13COO–···31P correlations
from the Ser16 cement relative to the Pser16 counterpart (as is also
evident from the 13C CPMAS NMR spectra of Figure 6) corroborates the metadynamics
predictions (Section 3.3): the carboxy group of Ser anchors directly at the ACP surface,
in contrast with (a majority of) the Pser molecules. However, owing
to the HMQC-signal buildup across longer distances, cross-peaks associated
with all13C sites are detected for the
present τexc values (Figure 7), despite that none of the CH/CH2 groups of either Pser/Ser molecule bind directly to any species
of ACP.
13CH2 in Figure 7a; this is particularly
evident when contrasting the various peak intensities of the HMQC
projection with their counterparts of the corresponding 1H → 13C CPMAS NMR spectrum (Figure S5). The 2D NMR-peak intensities reflect semiquantitatively
the relative through-space 13C–31P distances.
The significantly emphasized intensity and the higher 13C chemical-shift dispersion of the 13COO–···31P correlations
from the Ser16 cement relative to the Pser16 counterpart (as is also
evident from the 13C CPMAS NMR spectra of Figure 6) corroborates the metadynamics
predictions (Section 3.3): the carboxy group of Ser anchors directly at the ACP surface,
in contrast with (a majority of) the Pser molecules. However, owing
to the HMQC-signal buildup across longer distances, cross-peaks associated
with all13C sites are detected for the
present τexc values (Figure 7), despite that none of the CH/CH2 groups of either Pser/Ser molecule bind directly to any species
of ACP.
Figure 7.

13C{31P} D-HMQC correlation NMR spectra obtained at B0 = 14.1 T and νr = 24 kHz from (a) Pser16 and (b) Ser16 cements by using a HMQC excitation period of (a) 1.75 ms and (b) 2.0 ms. The 2D NMR spectra are shown together with projections along the 13C (horizontal) and 31P (vertical) dimensions at the top and to the right, respectively, along with their 1H → 13C CPMAS NMR spectra acquired under the same conditions. (c) Zoom around the carboxy-resonance region of the 2D NMR spectrum in (b). The rectangles indicate 13C–31P correlations resolved at the 13C chemical shifts of 184 ppm (blue), 177/174 ppm (yellow), and 170 ppm (grey).
The close COO–···ACP
contacts
of the Ser molecules are reflected in emphasized 2D NMR-signal intensities
in the zoom around the 13COO– region
of the 2D NMR spectrum of the Ser16 cement (Figure 7c): four 2D NMR ridges are resolved at the 13C chemical shifts {184.2, 176.8, 174.0, 170.0} ppm. Notably,
none of them coincides with that of δC = 175.2 ppm
from polycrystalline Ser (Figure 6f). As is most transparent from Figure S5, the NMR peaks around δC = {184,
170} ppm are markedly emphasized (the latter peak is barely discernible
in the CPMAS NMR spectrum), whereas the comparatively reduced intensities
between 181 ppm and 172 ppm stem from Ser molecules further away from
the inorganic phosphate groups. Its significant 13C shift-dispersion
reflects a range of similar yet distinct COO–··· proximities of weakly bound Ser molecular
configurations at the ACP surface (along with some resonances from
nonbonded molecules; see Section 3.5.3 and the comments mentioned above).
proximities of weakly bound Ser molecular
configurations at the ACP surface (along with some resonances from
nonbonded molecules; see Section 3.5.3 and the comments mentioned above).
We onwards focus on the two D-HMQC NMR signals centered at the
{δP, δC} shift-pairs of {3.0, 184.2}
ppm and {2.4, 170.0} ppm, which are tentatively assigned to COO– moieties that are strongly surface bound predominantly/solely via electrostatic CO···Ca2+ interactions and CO··· H bonds, respectively. While noting that
those two contact modes were predicted by modeling (Section 3.3), the NMR-peak assignments
were based on the distinctly different 13C and 31P chemical shifts involved in each correlation: the high shifts of
the {δP, δC} = {3.0, 184.2} ppm
signal are well-aligned with COO– moieties nearby
H bonds, respectively. While noting that
those two contact modes were predicted by modeling (Section 3.3), the NMR-peak assignments
were based on the distinctly different 13C and 31P chemical shifts involved in each correlation: the high shifts of
the {δP, δC} = {3.0, 184.2} ppm
signal are well-aligned with COO– moieties nearby 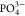 groups (see Sections 3.1 and 3.2), whereas
the lower chemical shifts of the {2.4, 170.0} ppm correlation are
commensurate with H-bond-mediated 13COO–···
groups (see Sections 3.1 and 3.2), whereas
the lower chemical shifts of the {2.4, 170.0} ppm correlation are
commensurate with H-bond-mediated 13COO–···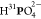 contacts, whose 13C chemical
shift incidentally matches that of the 13COOH group of polycrystalline Pser that manifests analogous inter
molecular COOH···
contacts, whose 13C chemical
shift incidentally matches that of the 13COOH group of polycrystalline Pser that manifests analogous inter
molecular COOH··· motifs.65,99,100 Notably, Figure 6 reveals a lower NMR-signal intensity at 184 ppm from
the Ser16 cement relative to its Ser8 counterpart, suggesting a concurrently
reduced number of CO···Ca2+ contacts for increasing batched Ser content, along
with earlier and more qualitative findings by Mathew et al.(60) and our inferences in Section 3.7.
motifs.65,99,100 Notably, Figure 6 reveals a lower NMR-signal intensity at 184 ppm from
the Ser16 cement relative to its Ser8 counterpart, suggesting a concurrently
reduced number of CO···Ca2+ contacts for increasing batched Ser content, along
with earlier and more qualitative findings by Mathew et al.(60) and our inferences in Section 3.7.
To our knowledge, the present study provides the first 1H/31P/13C correlation NMR signatures of surface-bound Pser molecules at nanocrystalline HA, noting that the previous 31P{1H} and 13C{1H} heteronuclear correlation spectra presented by Wang et al.(32) from similar nanocrystalline HA preparations in Pser-bearing solutions gave no convincing evidence for Pser···HA contacts. For instance, the 13C NMR features merely suggested a prevalence of Pser in crystalline environments than surface-bound ones, which likely resulted from using a very high [Pser] = 200 mM in the solution.32 Likewise, our 13C CPMAS spectra recorded from various Pser@HA syntheses with increasing Pser concentrations revealed a dominance of crystalline CaPser in all preparations with [Pser] ≥ 20 mM (Figure S3).
3.5. Quantitative Probing of Ser/Pser–ACP Contacts by 13C{31P} REDOR NMR
3.5.1. NMR-Derived Dipolar Second Moments
For a quantitative probing of the relative proximities among the {13CO, 13CH, 13CH2} sites of Ser/Pser and the inorganic phosphate groups at the HA/ACP surface, we collected the 13C{31P} REDOR NMR dephasing curves presented in Figure 8. Here, a rapid (slow) dephasing reflects short (long) distances of a given 13Cj site to the inorganic phosphate groups of ACP (with the caveat of intramolecular 13Cj–31P interactions in Pser). Table 2 collects the set of REDOR-derived dipolar second moments101−106 {M2(CO–P), M2(CH–P), M2(CH2–P)}, extracted by fitting the respective dephasing curves of each sample (Section S1.3). The precise M2(Cj–P) value depends on the underlying set of interatomic distances {r(Cj–Pk)} between a given 13C site and its nearby P atoms via a sum over [r(Cj–Pk)]−6 terms. This renders the shortest distance contributions most influential for the net M2(Cj–P) value.
Figure 8.

13C{31P} REDOR NMR dephasing curves (ΔS/S0) recorded at B0 = 14.1 T and νr = 10 kHz and plotted for increasing dipolar recoupling/dephasing periods (τrec) for the 13CO, 13CH, and 13CH2 functional groups of the (a,b) polycrystalline Pser and CaPser powders (13C at natural abundance) along with the 13C-enriched (c) Pser@HA, (d) Pser16, and (e) Ser16 specimens. Each curve in (a–e) corresponds to the best fit of the {τrec, ΔS/S0} data to eq S2, which yielded the dipolar second moments {M2(Cj–P)} listed in Table 2. The 13COO– NMR spectral region of the Ser16 sample comprises three distinct resonances at δC = {184, 177, 174.5} ppm, whose associated dephasing curves are labeled by their chemical shifts in the legend in (e). Note the different horizontal scale in (e) relative to (a–d).
Table 2. Experimental and Calculated Dipolar Second Momentsa.
|
15N–31P |
13C–31P |
|||||||
|---|---|---|---|---|---|---|---|---|
|
M2(NH3–P)/kHz2 |
M2(CO–P)/kHz2 |
M2(CH–P)/kHz2 |
M2(CH2–P)/kHz2 |
|||||
| sample | exp. | calc. | exp. | calc. | exp. | calc. | exp. | calc. |
| Ser16 | 2.7 ± 0.4 | 4.9 ± 0.7 | 11.1b | 14.0b | 8.9 | 13.0 | 7.9 | 9.6 |
| Pser16c | 2.8 ± 0.4 | 5.2 (4.0)±0.7 | 13.1 | 13.8 (8.3) | 18.2 | 25.8 (8.9) | 75.4 | 96.8 (5.7) |
| Pser@HAc | 4.0 ± 0.4 | 5.2 (4.0)±0.7 | 15.7 | 13.8 (8.3) | 20.9 | 25.8 (8.9) | 73.7 | 96.8 (5.7) |
| Pserd | 3.2 ± 0.4 | 4.5 | 18.6 | 24.5 | 18.9 | 24.8 | 79.9 | 102.5 |
| CaPser | 4.6 | 15.9 | 20.8 | 27.8 | 38.2 | 71.7 | 93.4 | |
15N–31P and 13C–31P dipolar second moments obtained for the as-indicated functional groups from 15N{31P} and 13C{31P} REDOR NMR experiments, respectively, and compared with data calculated either from metadynamics-derived structural models (for Ser16, Pser16, and Pser@HA) or from the crystal structures of Pser and CaPser.65 The M2(Cj–P) data uncertainties are ±1.7 kHz2 (NMR) and ±2.2 kHz2 (metadynamics model), with kHz2 ≡ 1000 s–2.
Net experimental/calculated values; the NMR analysis afforded the extraction of three M2(CO–P) values of {23.6, 9.3, 13.3} kHz2 for the 13CO shifts at {184, 177, 174.5} ppm, respectively.
Values within parentheses represent the contribution to the net M2(N–P) and {M2(Cj–P)} values from the Pser···HA contacts alone; the contribution from the intramolecular 15N–31P and 13Cj–31P dipolar interactions account for the difference.
The {M2(Cj–P)} and M2(N–P) values were obtained from the Pser (Flamma) and isotopically enriched Pser* samples, respectively.
The NMR-derived {M2(Cj–P)} data from the polycrystalline
Pser/CaPser powders
were validated against those calculated from their crystal structures.64,65,99 As expected103−106 and discussed further in Section S1.3, the NMR-derived dipolar second moments are consistently lower than
their theoretical counterparts (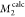 ). Yet, the
). Yet, the 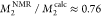 ratios are essentially constant (Table S5): a very good agreement is observed
for all relativeM2(Cj–P) values within each Pser or CaPser
structure, which remain well within the experimental uncertainties.
For Pser, the experimental and calculated M2(CO–P):M2(CH–P):M2(CH2–P) ratios relate as
1.0:1.0:4.3 and 1.0:1.0:4.2, respectively, whereas both data sets
for CaPser are 1.0:1.8:4.5.
ratios are essentially constant (Table S5): a very good agreement is observed
for all relativeM2(Cj–P) values within each Pser or CaPser
structure, which remain well within the experimental uncertainties.
For Pser, the experimental and calculated M2(CO–P):M2(CH–P):M2(CH2–P) ratios relate as
1.0:1.0:4.3 and 1.0:1.0:4.2, respectively, whereas both data sets
for CaPser are 1.0:1.8:4.5.
The markedly different relative M2(CO–P):M2(CH–P) values among the Pser and CaPser crystal structures are noteworthy: Despite the long intramolecular 13CO···31PO4 distances in both structures, the comparatively high M2(CO–P) values (particularly for Pser) originate mainly from multiple intermolecular 13COOH···31PO4 contacts via H bonds.65,99,100 Moreover, the markedly different M2(CH–P) values in Table 2 reflect distinctly different molecular conformations in the CaPser and Pser crystal structures, whose corresponding θ(P–O–Cβ–Cα) dihedral angles of 280° and 153° translate into intramolecular 13CH···P distances of 341 pm and 384 pm, respectively. Notably, such widely differing Pser conformations are readily discriminated by the 13C{31P} REDOR NMR experiments. Hence, notwithstanding that each M2(Cj–P) value is dominated by the intramolecular 13Cj–31P distance for all scenarios where intermolecular Pser···Pser contacts are negligible (thereby obscuring the herein targeted Cj···ACP contacts), the M2(CH–P) parameter offers a valuable constraint on the conformation of the surface-bound Pser molecules, such as in the Pser@HA and Pser16 systems.
3.5.2. Validation of the Metadynamics-Derived Dipolar Second Moments
The dipolar second moment M2(Cj–P) reflects all {13Cj–31Pk} distances of the adsorbed molecule to its adjacent inorganic phosphate groups (Section S1.3), but cannot unveil the precise underlying distance distribution. However, because the metadynamics simulations do offer such atomistic details (Figure 5), we first validated the modeled M2(Cj–P) data against the experimental counterparts.
Considering that all REDOR-derived M2(Cj–P) values are
underestimated by  (Table S5 and Section S1.3), a “perfect match” between the metadynamics-generated
models and the experiments should result in
(Table S5 and Section S1.3), a “perfect match” between the metadynamics-generated
models and the experiments should result in 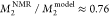 for each 13Cj–31P contact. Notwithstanding a somewhat
larger scatter in the ratios of Table S5 (which stems from higher data uncertainties of the simulated systems
than the very accurate C/P atom coordinates of the Pser/CaPser crystal
structures64,99), it is gratifying that the data
in Tables 2 and S5 confirmed our expectations for all 13Cj–31P pairs of both Ser/Pser molecules, except for the modeled M2(CO–P) values relative to the experimental
counterparts of Pser@HA and Pser16 that yielded
for each 13Cj–31P contact. Notwithstanding a somewhat
larger scatter in the ratios of Table S5 (which stems from higher data uncertainties of the simulated systems
than the very accurate C/P atom coordinates of the Pser/CaPser crystal
structures64,99), it is gratifying that the data
in Tables 2 and S5 confirmed our expectations for all 13Cj–31P pairs of both Ser/Pser molecules, except for the modeled M2(CO–P) values relative to the experimental
counterparts of Pser@HA and Pser16 that yielded 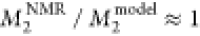 . The somewhat stronger COO–···ACP contacts in both Pser@HA and Pser16 specimens
than those predicted by the metadynamics simulations imply that the
carboxy group contributes more to the Pser binding than that suggested
by the binding mode statistics of Table 1. However, besides noting that the phosphate/amino
binding-mode population is presumably slightly overestimated at the
expense of (primarily) the phosphate/carboxy/amino counterpart, more
quantitative corrections cannot be made. We remind that the 13C{31P} D-HMQC NMR spectra also suggested weaker COO–···ACP interactions of the Pser molecules
than those of Ser (Section 3.4).
. The somewhat stronger COO–···ACP contacts in both Pser@HA and Pser16 specimens
than those predicted by the metadynamics simulations imply that the
carboxy group contributes more to the Pser binding than that suggested
by the binding mode statistics of Table 1. However, besides noting that the phosphate/amino
binding-mode population is presumably slightly overestimated at the
expense of (primarily) the phosphate/carboxy/amino counterpart, more
quantitative corrections cannot be made. We remind that the 13C{31P} D-HMQC NMR spectra also suggested weaker COO–···ACP interactions of the Pser molecules
than those of Ser (Section 3.4).
3.5.3. Discussion
The NMR-derived dipolar
second moments of the aliphatic groups of Ser (Table 2) suggest M2(Cj–P)  kHz2 as the marker of an absence of direct surface binding of any atom of a given 13Cj functional group at the ACP
surface. We remind that regardless of the (non)adsorption of Pser
at ACP, the intramolecular 13Cj–31P dipolar interactions render all M2(Cj–P) values of the
Pser@HA and Pser16 specimens markedly higher than those from Ser16,
as is evident from the dephasing curves of Figure 8. However, the simulation-derived model enables
a separation of the intra/intermolecular contributions to the net M2(Cj–P) value:
indeed, Table 2 confirms
the expectation that both CH/CH2 groups of Pser exhibit
very low dipolar second moments once their intramolecular contributions
are excluded (particularly that of M2(CH2–P), for which the organic phosphate group accounts
for ≈95% of the net value).
kHz2 as the marker of an absence of direct surface binding of any atom of a given 13Cj functional group at the ACP
surface. We remind that regardless of the (non)adsorption of Pser
at ACP, the intramolecular 13Cj–31P dipolar interactions render all M2(Cj–P) values of the
Pser@HA and Pser16 specimens markedly higher than those from Ser16,
as is evident from the dephasing curves of Figure 8. However, the simulation-derived model enables
a separation of the intra/intermolecular contributions to the net M2(Cj–P) value:
indeed, Table 2 confirms
the expectation that both CH/CH2 groups of Pser exhibit
very low dipolar second moments once their intramolecular contributions
are excluded (particularly that of M2(CH2–P), for which the organic phosphate group accounts
for ≈95% of the net value).
It is gratifying that each of the three {M2(CO–P), M2(CH–P), M2(CH2–P)} NMR-derived data agrees mutually very well among the Pser@HA and Pser16 samples, which suggests overall (very) similar Pser contacts/distances to the inorganic phosphate moieties. Hence, despite the formally distinct nature of the ACP/Pser phase of the biomedical PserN cements and the Pser adsorption at a nanocrystalline “HA” surface (Pser@HA), the local structure of their organic/inorganic interfaces must be similar in both specimens, thereby consolidating the current consensus that nanocrystalline HA is coated by a layer of “ACP”4,12−16 (although its precise chemical/structural nature remains unknown). This aspect also justifies that the single metadynamics simulation at pH = 4.5 mimics well the Pser···ACP interactions in both Pser@HA and Pser16 specimens, as well as supporting the physical relevance of the herein employed HA-surface preparation procedure.45 These important issues are discussed further in Section S2.5.
We now return to the partially overlapping 13C NMR peaks in the carboxy domain of the NMR spectrum from the Ser16 cement (Figure 6h), which, according to their distinct 13COO–···ACP contacts, were grouped into three regions in the 13C{31P} D-HMQC spectrum of Figure 7c. As expected from the δC = 184 ppm resonance attributed to CO···Ca2+ motifs (Section 3.4), its very rapid dipolar dephasing is consistent with a sizable dipolar second moment of 23.6 kHz2 (Table 2). In contrast, the spectral region marked by a yellow rectangle in Figure 7c and deconvoluted into two peak components around 177/174.5 ppm reflects more weakly surface-bound COO– groups with lower dipolar second moments of 9.3/13.3 kHz2 (Figure 8 and Table 2). Although a large M2(CO–P) value is also anticipated from COO– moieties surface bound via H bonds and giving an NMR peak at 170 ppm, its minor NMR intensity (and thereby very small population) did not permit reliable analyses of the REDOR NMR data (e.g., see Figure S6). Altogether, the varying dipolar second moments of these COO–···ACP contact modes/distances underlie the average value of M2(CO–P) ≈ 11.1 kHz2 (Table 2), which qualitatively corroborates the metadynamics predictions of direct COO–···ACP bonds.
We contrasted the D-HMQC NMR-derived and modeled relative populations of the three types of surface-bound COO– sites that produce the three spectral regions marked in Figure 7c and attributed to COO– sites bound solely by Ca2+ cations (≈184 ppm), sites weakly bound by both electrostatic and H-bond interactions (181–172 ppm), and solely by H bonds (≈170 ppm). Quantitative agreements are not expected because the 2D NMR intensities depend strongly on the precise HMQC excitation/reconversion periods. Deconvolution of the 13C projection of the D-HMQC spectrum (results not shown) yielded fractional populations of {0.27, 0.59, 0.14} for the respective {184, 181–172, 170} ppm resonance regions, whereas the corresponding metadynamics-simulation-derived fractions are {0.68, 0.27, 0.05}. While both experimental and modeled results accorded qualitatively well for the overall most sparse contact mode of solely CO···HPO42− interactions, the main discrepancy concerns the strong dominance of CO–···Ca2+ interactions predicted by the model (68% of all direct surface contacts and consistent with the results of Table S4) compared to the much lower estimate by NMR (27%). The differences presumably stem from the difficulties by NMR to accurately quantify the contributions from the 181–172 ppm resonance-region stemming from “weakly bound” carboxy groups, which may involve non-negligible 13COO– NMR signals from more distant nonbonded molecules that are not accounted for in the simulation analysis.
3.6. 15N{31P} REDOR NMR Reveals the Amino-Group Binding
Figure 9a,b displays the 1H → 15N CPMAS NMR spectra of Pser, the 15N-enriched
Ser* and Pser* precursor powders, together with those of Pser@HA,
Pser16, and Ser16. As for the 13C NMR spectra (Figure 6), the Ser/Pser adsorption
is mirrored by a significant 15N resonance broadening along
with a minor increase in the average chemical shift of ≈2 ppm
and ≈5 ppm for the Ser and Pser bearing specimens, respectively.
The amino-group binding at ACP was probed by 15N{31P} REDOR NMR experiments on the Pser@HA, Pser16, and Ser16 cements,
whose dipolar dephasing curves are displayed in Figure 9c along with that from the Pser* powder.
For the latter, Table S5 reveals a ratio
of 0.71 between the NMR-derived M2(N–P)
value and that calculated from the Pser crystal structure,99 which is close to the expected ratio of 0.76
(Section 3.5.2).
Moreover, the ratio of 0.77 between the experimental and modeled M2(P–N) data for the Pser@HA sample suggests
a very faithful metadynamics modeling of its  ACP/HA contacts (Figure 5b).
ACP/HA contacts (Figure 5b).
Figure 9.

1H → 15N CPMAS NMR spectra recorded from powders of (a) Pser (15N at natural abundance) along with the 13C/15N-enriched Ser* and Pser* precursors, and their (b) Pser@HA, Pser16, and Ser16 products. The NMR peaks at 32.8 ppm and 44.0 ppm observed from the Pser* specimen are attributed to the 15N sites of 13C/15N-enriched O-phospho-l-serine and its HCl salt, respectively. (c) 15N{31P} REDOR NMR dephasing curves recorded from the as-indicated samples. All NMR results were obtained at B0 = 14.1 T and νr = 10 kHz.
Particularly, when recalling the very good mutual
match between
the {M2(Cj–P)} sets among the Pser@HA and Pser16 specimens (Section 3.5), the significantly
lower M2(N–P) value of the Pser16
cement (as well as for Ser16) relative to Pser@HA is surprising (Table 2). It suggests much
weaker  ACP contacts in both Pser/ACP and Ser/ACP
cement components than the counterparts of the Pser@HA particles (also
see Section S1.3). Although the NMR-derived
dipolar second moments from the cements also suggest longer
ACP contacts in both Pser/ACP and Ser/ACP
cement components than the counterparts of the Pser@HA particles (also
see Section S1.3). Although the NMR-derived
dipolar second moments from the cements also suggest longer 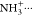 ACP distances than the metadynamics predictions,
the qualitative feature of near-equal M2(N–P) values of the models for both Ser/Pser molecules is
supported by the REDOR NMR experiments on the Pser16 and Ser16 samples.
ACP distances than the metadynamics predictions,
the qualitative feature of near-equal M2(N–P) values of the models for both Ser/Pser molecules is
supported by the REDOR NMR experiments on the Pser16 and Ser16 samples.
The amino group binds to the ACP surface exclusively via H bonds to the negatively charged O atoms of the phosphate groups. Table S4 reveals that the NH··· contact mode dominates for Pser (pH = 4.5),
whereas the amino group of Ser (pH = 7.4) forms an equal number of
bonds to
contact mode dominates for Pser (pH = 4.5),
whereas the amino group of Ser (pH = 7.4) forms an equal number of
bonds to 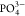 and
and 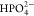 moieties. For the pH conditions of the
Pser16 and Ser16 cements, the binding-energy contribution from the
amino group is higher for Ser than Pser (Table S3), which also accords with the dipolar second moments of Table 2: although both the
experimental and modeled M2(N–P)
values for Pser16 are slightly higher than their Ser counterparts,
once excluding the intramolecular 15N–31P Pser contribution, the expectation of a stronger surface binding
of the
moieties. For the pH conditions of the
Pser16 and Ser16 cements, the binding-energy contribution from the
amino group is higher for Ser than Pser (Table S3), which also accords with the dipolar second moments of Table 2: although both the
experimental and modeled M2(N–P)
values for Pser16 are slightly higher than their Ser counterparts,
once excluding the intramolecular 15N–31P Pser contribution, the expectation of a stronger surface binding
of the  group of Ser is confirmed by its
group of Ser is confirmed by its 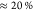 higher modeled M2(N–P) value than that of Pser.
higher modeled M2(N–P) value than that of Pser.
3.7. Implications for Bone-Adhesive Properties
We recently reported a strong correlation between the amount of the amorphous ACP/Pser component and the measured shear strength of PserN cements used for gluing two cubes of either cortical bone54 or steel60 together across a wide composition range up to N = 87 mol %.60 The shear strength reflects the bone-adhesive properties.54 Notably, both parameters exhibit a nonmonotonic trend against the batched Pser content, with an initial increase up to a plateau of near-constant shear-strengths and ACP/Pser contents in cements incorporating 40–60 mol % Pser, followed by their concurrent decrease for increasing N because the bone-adhesive-promoting ACP/Pser component becomes gradually replaced by crystalline CaPser and unreacted Pser.60 In contrast, SerN cements form an amorphous ACP/Ser phase that only develops with the batched Ser content up to N ≲ 16 mol % (which is insufficient for giving a high shear strength), whereas all remaining Ser remains unreacted.60
Consequently, the formation of an amorphous component featuring both significant organic–organic and organic–inorganic interactions appears to be a prerequisite for favorable bone-adhesive properties of an α-TCP-derived cement. Hence, the results herein combined with those of ref (60) suggest that a significant bone-adhesion cannot stem from the adsorption strength of a given biomolecule at ACP alone, because the Pser···ACP binding energy at pH = 4.5 essentially matches with that of Ser···ACP at pH = 7.4 (Table 1), implying that both Pser16/Ser16 cements feature similar Pser···ACP and Ser···ACP net interaction strengths. Nonetheless, while both Pser and Ser molecules may enter ACP/Pser and ACP/Ser phases in the respective PserN and SerN specimens with N ≲ 16 mol %, their amounts develop very differently upon increasing organic content (vide supra). Consequently, the main distinction between the PserN and SerN cements concerns their respective degrees of Pser···Pser and Ser···Ser contacts.
Due to its low Pser content ( wt %), the Pser@HA sample is expected to
involve essentially “isolated” surface-bound Pser molecules
with long Pser···Pser distances. Likewise, the metadynamics
modeling involved one sole Pser (or Ser) molecule. Hence, the good
agreement between the M2(Cj–P) NMR results of the Pser@HA and Pser16
samples, as well as with the modeled data, suggest that 16 mol % of
either Pser or Ser may be sufficiently low to be dispersed within
the ACP matrix without any significant Pser···Pser
or Ser···Ser aggregation (i.e., analogously
with a “monolayer” adsorption scenario). However, the
substantial Pser contribution to the ACP/Pser cement component in
all Pser-rich PserN cements—for which the
shear strength is maximal—cannot be reconciled with a monolayer
Pser adsorption at ACP, but must involve a significant Pser···Pser
aggregation, yet with the molecules remaining intimately integrated
also with the inorganic species of ACP (see comments in Section 3.3.3 and ref (60)). Although further work is required for a definite
proof, we propose that the high bone-adhesion/shear-strength of the
PserN cements with 40 ≲ N ≲ 60 mol % stems from a “stickiness” accompanying
the high organic content of their ACP/Pser component, in conjunction
with its dominance of the entire cement constitution. Naturally, the
“stickiness” is low in cements with batched Pser contents
≲ 20 mol %, as well as for all SerN specimens60 due to their insignificant tendency of Ser···Ser
aggregation.
wt %), the Pser@HA sample is expected to
involve essentially “isolated” surface-bound Pser molecules
with long Pser···Pser distances. Likewise, the metadynamics
modeling involved one sole Pser (or Ser) molecule. Hence, the good
agreement between the M2(Cj–P) NMR results of the Pser@HA and Pser16
samples, as well as with the modeled data, suggest that 16 mol % of
either Pser or Ser may be sufficiently low to be dispersed within
the ACP matrix without any significant Pser···Pser
or Ser···Ser aggregation (i.e., analogously
with a “monolayer” adsorption scenario). However, the
substantial Pser contribution to the ACP/Pser cement component in
all Pser-rich PserN cements—for which the
shear strength is maximal—cannot be reconciled with a monolayer
Pser adsorption at ACP, but must involve a significant Pser···Pser
aggregation, yet with the molecules remaining intimately integrated
also with the inorganic species of ACP (see comments in Section 3.3.3 and ref (60)). Although further work is required for a definite
proof, we propose that the high bone-adhesion/shear-strength of the
PserN cements with 40 ≲ N ≲ 60 mol % stems from a “stickiness” accompanying
the high organic content of their ACP/Pser component, in conjunction
with its dominance of the entire cement constitution. Naturally, the
“stickiness” is low in cements with batched Pser contents
≲ 20 mol %, as well as for all SerN specimens60 due to their insignificant tendency of Ser···Ser
aggregation.
4. Conclusions
We have presented the first atomistic probing of the Pser and Ser binding at structurally disordered CaP surfaces by a synergistic combination of advanced solid-state NMR experimentation and metadynamics MD simulations, revealing the relative proximities of each molecular functional group and their respective underlying types of bonds, as well as the conformation of the adsorbed molecules. Our study encompassed the organic/inorganic interactions in a sample of surface-bound Pser at nanocrystalline HA particles (Pser@HA) along with two biocements prepared from α-Ca3(PO4)2 doped with 16 mol % of either Pser or Ser. Notably, the very close sets of 13C–31P dipolar second moments observed from the cement and the Pser@HA sample highlight the similarities of the Pser binding at ACP and the structurally disordered surface layer of nanocrystalline HA, thereby corroborating the current consensus that it is faithfully described as “ACP”.4,13−16
The Pser and Ser adsorption is primarily mediated by electrostatic
interactions between Ca2+ cations and the negatively charged
organic COO–/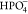 groups and, to a lesser extent, by H bonds
to the inorganic phosphate groups, which involves NH···PO4 and CO/PO···HPO4 contacts for the amino and carboxy/phosphate
groups, respectively. The dominance of electrostatic interactions
for driving the adsorption implies that the phosphate group of Pser
and the carboxy group of Ser are mainly responsible for stabilizing
their surface binding, fully consistent with earlier inferences that
biomolecules bind at bone minerals mainly via ion–ion
interactions.18−21
groups and, to a lesser extent, by H bonds
to the inorganic phosphate groups, which involves NH···PO4 and CO/PO···HPO4 contacts for the amino and carboxy/phosphate
groups, respectively. The dominance of electrostatic interactions
for driving the adsorption implies that the phosphate group of Pser
and the carboxy group of Ser are mainly responsible for stabilizing
their surface binding, fully consistent with earlier inferences that
biomolecules bind at bone minerals mainly via ion–ion
interactions.18−21
All detailed information about the number of electrostatic/H-bond
interactions and the accompanying interatomic distances were extracted
from the metadynamics models. They were validated against the NMR-derived
interatomic-distance constraints encoded by the dipolar second moments
{M2(CO–P), M2(CH–P), M2(CH2–P)} that convey the relative proximities between the 13C atoms of the respective {COO–, CH, CH2} group and the inorganic phosphate moieties of ACP, as well
as the M2(N–P) counterpart informing
about the 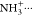 ACP contacts. The overall good agreement
confirmed the accuracy of our metadynamics simulations, notably the
validity of the herein employed HA-surface preparation protocol of
ref (45), which produces
a disordered apatite surface with pH-dependent phosphate speciation.
For our experimental conditions of 3.8 ≤ pH ≤ 5.3 (Pser@HA
and Pser16) and pH = 7.4 (Ser16), both Pser and Ser molecules anchor
at their amino groups, together with the HPO4− group of Pser and the COO– group of Ser, which leads to an extended conformation
of both surface-immobilized molecules. The OH group of Ser also participates
in the binding at ACP via both electrostatic OH···Ca2+ interactions
and H bonds to the inorganic phosphate groups, but they contribute
overall little to the net adsorption energy (≈10%). The only
discrepancy between the experiments and models concerned an underestimation
of the metadynamics-derived COO–···ACP
proximities relative to those deduced by NMR on the Pser@HA and Pser16
specimens.
ACP contacts. The overall good agreement
confirmed the accuracy of our metadynamics simulations, notably the
validity of the herein employed HA-surface preparation protocol of
ref (45), which produces
a disordered apatite surface with pH-dependent phosphate speciation.
For our experimental conditions of 3.8 ≤ pH ≤ 5.3 (Pser@HA
and Pser16) and pH = 7.4 (Ser16), both Pser and Ser molecules anchor
at their amino groups, together with the HPO4− group of Pser and the COO– group of Ser, which leads to an extended conformation
of both surface-immobilized molecules. The OH group of Ser also participates
in the binding at ACP via both electrostatic OH···Ca2+ interactions
and H bonds to the inorganic phosphate groups, but they contribute
overall little to the net adsorption energy (≈10%). The only
discrepancy between the experiments and models concerned an underestimation
of the metadynamics-derived COO–···ACP
proximities relative to those deduced by NMR on the Pser@HA and Pser16
specimens.
Besides rationalizing the distinction in bone-adhesive
properties
of Pser and Ser doped α-Ca3(PO4)2-based cements (Section 3.7), our findings settle some earlier suggestions/speculations
about which functional groups are involved in the molecular binding
of Ser and Pser at nanocrystalline HA.90,107−109 We stress that the binding modes discussed herein (Table 1 and Figure 5) are the most probable ones
from an energetic viewpoint over a distribution of several distinct
but stable modes. The nature of the comparatively weak adsorption
of small biomolecules (such as amino acids and oligopeptides) at structurally
disordered CaP surfaces must be analyzed/discussed in terms of “distributions”
and/or “effective contacts”, as those encoded by dipolar
second moments. The existence of a distribution of very similar binding
modes is indeed mirrored in the broad and typically asymmetric 13C/15N MAS NMR peak shapes observed from each functional
group which, except for the COO– moiety of Ser16,
remained unresolved. The 13C–31P correlation
NMR spectrum of the latter revealed four 13COO– resonances from groups with different proximities to ACP: the two
COO– environments closest to the inorganic phosphates
were tentatively attributed to those solely involved in electrostatic
COO–···Ca2+ interactions
(≈184 ppm) and H-bonded COO–···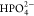 moieties (≈170 ppm). However, most
of the COO– groups bind by both interaction types,
as mirrored in a resonance-continuum across the 181–172 ppm
spectral region.
moieties (≈170 ppm). However, most
of the COO– groups bind by both interaction types,
as mirrored in a resonance-continuum across the 181–172 ppm
spectral region.
We conclude by highlighting the power of the herein-implemented combination of advanced solid-state NMR experiments with metadynamics simulations for an enhanced probing of the detailed biomolecular binding at structurally disordered CaP surfaces, which remains essentially untapped but is potentially very rewarding. Another ubiquitous tool exploited herein concerns the recently introduced Debye–Hückel-based analysis48 reviewed in the Supporting Information, which offers a straightforward decomposition of the net modeled biomolecular binding energy into its contributions from the various functional groups of the surface-immobilized molecule, as well as for quantifying each individual electrostatic/H-bond interaction energy.
Acknowledgments
This work was supported by the Swedish Foundation for Strategic Research (funder ID 501100001729; project RMA15–0110). The computations were enabled by resources provided by the Swedish National Infrastructure for Computing (SNIC) at NSC, partially funded by the Swedish Research Council through grant agreement no. 2018-05973. C.S.W. acknowledges funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement 747414. P.R.A.C. was financially supported by the Wenner-Gren Foundations. We thank Kjell Jansson and Zoltán Bacsik for TEM and surface-area measurements, respectively, and Debashis Majhi for help with processing some of the NMR spectra.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.chemmater.2c02112.
Experimental solid-state NMR conditions; metadynamics simulation parameters, data analysis, and further discussion; synthesis and characterization of Pser*; discussion on the 1H NMR shift of water in ACP; cement batch compositions; further data on the modeled surface binding and relative dipolar second moments; best-fit 31P NMR parameters; additional solid-state NMR spectra; and TEM images (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Weiner S.; Wagner H. D. THE MATERIAL BONE: Structural-Mechanical Function Relations. Annu. Rev. Mater. Sci. 1998, 28, 271–298. 10.1146/annurev.matsci.28.1.271. [DOI] [Google Scholar]
- Rey C.; Combes C.; Drouet C.; Sfihi H.; Barroug A. Physico-Chemical Properties of Nanocrystalline Apatites: Implications for Biominerals and Biomaterials. Mater. Sci. Eng. C 2007, 27, 198–205. 10.1016/j.msec.2006.05.015. [DOI] [Google Scholar]
- Combes C.; Cazalbou S.; Rey C. Apatite Biominerals. Minerals 2016, 6, 34. 10.3390/min6020034. [DOI] [Google Scholar]
- Edén M. Structure and Formation of Amorphous Calcium Phosphate and its Role as Surface Layer of Nanocrystalline Apatite: Implications for Bone Mineralization. Materialia 2021, 17, 101107. 10.1016/j.mtla.2021.101107. [DOI] [Google Scholar]
- Gower L. B. Biomimetic Model Systems for Investigating the Amorphous Precursor Pathway and Its Role in Biomineralization. Chem. Rev. 2008, 108, 4551–4627. 10.1021/cr800443h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Addadi L.; Weiner S. Interactions between Acidic Proteins and Crystals: Stereochemical Requirements in Biomineralization. Proc. Natl. Acad. Sci. U.S.A. 1985, 82, 4110–4114. 10.1073/pnas.82.12.4110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter G. K. Interfacial Aspects of Biomineralization. Curr. Opin. Solid State Mater. Sci. 1996, 1, 430–435. 10.1016/s1359-0286(96)80036-2. [DOI] [Google Scholar]
- Sharma V.; Srinivasan A.; Nikolajeff F.; Kumar S. Biomineralization Process in Hard Tissues: The Interaction Complexity within Protein and Inorganic Counterparts. Acta Biomater. 2021, 120, 20–37. 10.1016/j.actbio.2020.04.049. [DOI] [PubMed] [Google Scholar]
- Boskey A. L. Biomineralization: Conflicts, Challenges, and Opportunities. J. Cell. Biochem. 1998, 72, 83–91. . [DOI] [PubMed] [Google Scholar]
- Boskey A. L.; Villarreal-Ramirez E. Intrinsically Disordered Proteins and Biomineralization. Matrix Biol. 2016, 52–54, 43–59. 10.1016/j.matbio.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George A.; Veis A. Phosphorylated Proteins and Control over Apatite Nucleation, Crystal Growth, and Inhibition. Chem. Rev. 2008, 108, 4670–4693. 10.1021/cr0782729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isobe T.; Nakamura S.; Nemoto R.; Senna M.; Sfihi H. Solid-State Double Nuclear Magnetic Resonance Study of the Local Structure of Calcium Phosphate Nanoparticles Synthesized by a Wet-Mechanochemical Reaction. J. Phys. Chem. B 2002, 106, 5169–5176. 10.1021/jp0138936. [DOI] [Google Scholar]
- Jäger C.; Welzel T.; Meyer-Zaika W.; Epple M. A Solid-State NMR Investigation of the Structure of Nanocrystalline Hydroxyapatite. Magn. Reson. Chem. 2006, 44, 573–580. 10.1002/mrc.1774. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Von Euw S.; Fernandes F. M.; Cassaignon S.; Selmane M.; Laurent G.; Pehau-Arnaudet G.; Coelho C.; Bonhomme-Coury L.; Giraud-Guille M.-M.; et al. Water-Mediated Structuring of Bone Apatite. Nat. Mater. 2013, 12, 1144–1153. 10.1038/nmat3787. [DOI] [PubMed] [Google Scholar]
- Von Euw S.; Wang Y.; Laurent G.; Drouet C.; Babonneau F.; Nassif N.; Azaïs T. Bone Mineral: New Insights into its Chemical Composition. Sci. Rep. 2019, 9, 8456. 10.1038/s41598-019-44620-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasar O. F.; Liao W.-C.; Mathew R.; Yu Y.; Stevensson B.; Liu Y.; Shen Z.; Edén M. The Carbonate and Sodium Environments in Precipitated and Biomimetic Calcium Hydroxy-Carbonate Apatite Contrasted with Bone Mineral: Insights from Solid-State NMR. J. Phys. Chem. C 2021, 125, 10572–10592. 10.1021/acs.jpcc.0c11389. [DOI] [Google Scholar]
- Eichert D.; Sfihi H.; Combes C.; Rey C. Specific Characterisitics of Wet Nanocrystalline Apatites: Consequences on Biomaterials and Bone Tissue. Key Eng. Mater. 2004, 254–256, 927–930. 10.4028/www.scientific.net/kem.254-256.927. [DOI] [Google Scholar]
- Huq N. L.; Cross K. J.; Reynolds E. C. Molecular Modelling of a Multiphosphorylated Sequence Motif Bound to Hydroxyapatite Surfaces. J. Mol. Model. 2000, 6, 35–47. 10.1007/s0089400060035. [DOI] [Google Scholar]
- Hunter G. K.; O’Young J.; Grohe B.; Karttunen M.; Goldberg H. A. The Flexible Polyelectrolyte Hypothesis of Protein–Biomineral Interaction. Langmuir 2010, 26, 18639–18646. 10.1021/la100401r. [DOI] [PubMed] [Google Scholar]
- Azzopardi P. V.; O’Young J.; Lajoie G.; Karttunen M.; Goldberg H. A.; Hunter G. K. Roles of Electrostatics and Conformation in Protein-Crystal Interactions. PLoS One 2010, 5, e9330 10.1371/journal.pone.0009330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Addison W. N.; Miller S. J.; Ramaswamy J.; Mansouri A.; Kohn D. H.; McKee M. D. Phosphorylation-Dependent Mineral-Type Specificity for Apatite-Binding Peptide Sequences. Biomaterials 2010, 31, 9422–9430. 10.1016/j.biomaterials.2010.08.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeger C.; Groom N. S.; Bowe E. A.; Horner A.; Davies M. E.; Murray R. C.; Duer M. J. Investigation of the Nature of the Protein–Mineral Interface in Bone by Solid-State NMR. Chem. Mater. 2005, 17, 3059–3061. 10.1021/cm050492k. [DOI] [Google Scholar]
- Wise E. R.; Maltsev S.; Davies M. E.; Duer M. J.; Jaeger C.; Loveridge N.; Murray R. C.; Reid D. G. The Organic–Mineral Interface in Bone Is Predominantly Polysaccharide. Chem. Mater. 2007, 19, 5055–5057. 10.1021/cm702054c. [DOI] [Google Scholar]
- Hu Y.-Y.; Rawal A.; Schmidt-Rohr K. Strongly Bound Citrate Stabilizes the Apatite Nanocrystals in Bone. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 22425–22429. 10.1073/pnas.1009219107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikel O.; Laurencin D.; Bonhomme C.; Sroga G. E.; Besdo S.; Lorenz A.; Vashishth D. Solid State NMR Investigation of Intact Human Bone Quality: Balancing Issues and Insight into the Structure at the Organic–Mineral Interface. J. Phys. Chem. C 2012, 116, 6320–6331. 10.1021/jp2125312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikel O.; Laurencin D.; McCallum S. A.; Gundberg C. M.; Vashishth D. NMR Investigation of the Role of Osteocalcin and Osteopontin at the Organic–Inorganic Interface in Bone. Langmuir 2013, 29, 13873–13882. 10.1021/la403203w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng Y.-H.; Mou Y.; Chen P.-H.; Tsai T. W. T.; Hsieh C.-I.; Mou C.-Y.; Chan J. C. C. Solid-State P-31 NMR Study of the Formation of Hydroxyapatite in the Presence of Glutaric Acid. Magn. Reson. Chem. 2008, 46, 330–334. 10.1002/mrc.2096. [DOI] [PubMed] [Google Scholar]
- Achelhi K.; Masse S.; Laurent G.; Saoiabi A.; Laghzizil A.; Coradin T. Role of Carboxylate Chelating Agents on the Chemical, Structural and Textural Properties of Hydroxyapatite. Dalton Trans. 2010, 39, 10644–10651. 10.1039/c0dt00251h. [DOI] [PubMed] [Google Scholar]
- Hu Y.-Y.; Liu X. P.; Ma X.; Rawal A.; Prozorov T.; Akinc M.; Mallapragada S. K.; Schmidt-Rohr K. Biomimetic Self-Assembling Copolymer-Hydroxyapatite Nanocomposites with the Nanocrystal Size Controlled by Citrate. Chem. Mater. 2011, 23, 2481–2490. 10.1021/cm200355n. [DOI] [Google Scholar]
- Wu Y.-J.; Tsai W. T.; Huang S.-J.; Mou Y.; Lin C.-J.; Chan J. C. C. Hydrogen Bond Formation between Citrate and Phosphate Ions in Spherulites of Fluorapatite. Langmuir 2013, 29, 11681–11686. 10.1021/la402392b. [DOI] [PubMed] [Google Scholar]
- Davies E.; Müller K. H.; Wong W. C.; Pickard C. J.; Reid D. G.; Skepper J. N.; Duer M. J. Citrate Bridges between Mineral Platelets in Bone. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, E1354–E1363. 10.1073/pnas.1315080111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Xu Z.; Zhao W.; Sahai N. A Potential Mechanism for Amino Acid-Controlled Crystal Growth of Hydroxyapatite. J. Mater. Chem. B 2015, 3, 9157–9167. 10.1039/c5tb01036e. [DOI] [PubMed] [Google Scholar]
- Goobes G.; Stayton P. S.; Drobny G. P. Solid State NMR Studies of Molecular Recognition at Protein-Mineral Interfaces. Prog. Nucl. Magn. Reson. Spectrosc. 2007, 50, 71–85. 10.1016/j.pnmrs.2006.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P.-H.; Tseng Y.-H.; Mou Y.; Tsai Y.-L.; Guo S.-M.; Huang S.-J.; Yu S. S.-F.; Chan J. C. C. Adsorption of a Statherin Peptide Fragment on the Surface of Nanocrystallites of Hydroxyapatite. J. Am. Chem. Soc. 2008, 130, 2862–2868. 10.1021/ja076607y. [DOI] [PubMed] [Google Scholar]
- Iline-Vul T.; Nanda R.; Mateos B.; Hazan S.; Matlahov I.; Perelshtein I.; Keinan-Adamsky K.; Althoff-Ospelt G.; Konrat R.; Goobes G. Osteopontin Regulates Biomimetic Calcium Phosphate Crystallization from Disordered Mineral Layers Covering Apatite Crystallites. Sci. Rep. 2020, 10, 15722. 10.1038/s41598-020-72786-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matlahov I.; Iline-Vul T.; Abayev M.; Lee E. M. Y.; Nadav-Tsubery M.; Keinan-Adamsky K.; Gray J. J.; Goobes G. Interfacial Mineral-Peptide Properties of a Mineral Binding Peptide from Osteonectin and Bone-like Apatite. Chem. Mater. 2015, 27, 5562–5569. 10.1021/acs.chemmater.5b01696. [DOI] [Google Scholar]
- Iline-Vul T.; Matlahov I.; Grinblat J.; Keinan-Adamsky K.; Goobes G. Changes to the Disordered Phase and Apatite Crystallite Morphology during Mineralization by an Acidic Mineral Binding Peptide from Osteonectin. Biomacromolecules 2015, 16, 2656–2663. 10.1021/acs.biomac.5b00465. [DOI] [PubMed] [Google Scholar]
- Iline-Vul T.; Kulpanovich A.; Nadav-Tsubery M.; Semionov A.; Keinan-Adamsky K.; Goobes G. How Does Osteocalcin Lacking γ-Glutamic Groups Affect Biomimetic Apatite Formation and What Can we Say About its Structure in Mineral-Bound Form?. J. Struct. Biol. 2019, 207, 104–114. 10.1016/j.jsb.2019.04.014. [DOI] [PubMed] [Google Scholar]
- Gullion T.; Schaefer J. Rotational-Echo Double-Resonance NMR. J. Magn. Reson. 1989, 81, 196–200. 10.1016/0022-2364(89)90280-1. [DOI] [PubMed] [Google Scholar]
- de Leeuw N. H.; Rabone J. A. L. Molecular Dynamics Simulations of the Interaction of Citric Acid with the Hydroxyapatite (0001) and (011̅0) Surfaces in an Aqueous Environment. CrystEngComm 2007, 9, 1178–1186. 10.1039/b710974a. [DOI] [Google Scholar]
- Jiang W.; Pan H.; Cai Y.; Tao J.; Liu P.; Xu X.; Tang R. Atomic Force Microscopy Reveals Hydroxyapatite–Citrate Interfacial Structure at the Atomic Level. Langmuir 2008, 24, 12446–12451. 10.1021/la801720w. [DOI] [PubMed] [Google Scholar]
- Xu Z.; Yang Y.; Wang Z.; Mkhonto D.; Shang C.; Liu Z.-P.; Cui Q.; Sahai N. Small Molecule-Mediated Control of Hydroxyapatite Growth: Free Energy Calculations Benchmarked to Density Functional Theory. J. Comput. Chem. 2014, 35, 70–81. 10.1002/jcc.23474. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Xu Z.; Zhao W.; Chen W.; Miyoshi T.; Sahai N. Isoexergonic Conformations of Surface-Bound Citrate Regulated Bioinspired Apatite Nanocrystal Growth. ACS Appl. Mater. Interfaces 2016, 8, 28116–28123. 10.1021/acsami.6b04822. [DOI] [PubMed] [Google Scholar]
- Heinz H.; Ramezani-Dakhel H. Simulations of Inorganic–Bioorganic Interfaces to Discover New Materials: Insights, Comparisons to Experiment, Challenges, and Opportunities. Chem. Soc. Rev. 2016, 45, 412–448. 10.1039/c5cs00890e. [DOI] [PubMed] [Google Scholar]
- Lin T.-J.; Heinz H. Accurate Force Field Parameters and pH Resolved Surface Models for Hydroxyapatite to Understand Structure, Mechanics, Hydration, and Biological Interfaces. J. Phys. Chem. C 2016, 120, 4975–4992. 10.1021/acs.jpcc.5b12504. [DOI] [Google Scholar]
- Lin T.-J. Predicting Binding Affinities of Nitrogen-Containing Bisphosphonates on Hydroxyapatite Surface by Molecular Dynamics. Chem. Phys. Lett. 2019, 716, 83–92. 10.1016/j.cplett.2018.12.008. [DOI] [Google Scholar]
- Luo M.; Gao Y.; Yang S.; Quan X.; Sun D.; Liang K.; Li J.; Zhou J. Computer Simulations of the Adsorption of an N-Terminal Peptide of Statherin, SN15, and its Mutants on Hydroxyapatite Surfaces. Phys. Chem. Chem. Phys. 2019, 21, 9342–9351. 10.1039/c9cp01638d. [DOI] [PubMed] [Google Scholar]
- Stevensson B.; Edén M. Metadynamics Simulations of the pH-Dependent Adsorption of Phosphoserine and Citrate on Disordered Apatite Surfaces: What Interactions Govern the Molecular Binding?. J. Phys. Chem. B 2021, 125, 11987–12003. 10.1021/acs.jpcc.1c02325.34672586 [DOI] [Google Scholar]
- Heinz H.; Lin T.-J.; Kishore Mishra R.; Emami F. S. Thermodynamically Consistent Force Fields for the Assembly of Inorganic, Organic, and Biological Nanostructures: The INTERFACE Force Field. Langmuir 2013, 29, 1754–1765. 10.1021/la3038846. [DOI] [PubMed] [Google Scholar]
- Holt C.; Timmins P. A.; Errington N.; Leaver J. A Core-Shell Model of Calcium Phosphate Nanoclusters Stabilized by β-Casein Phosphopeptides, Derived from Sedimentation Equilibrium and Small-Angle X-Ray and Neutron-Scattering Measurements. Eur. J. Biochem. 1998, 252, 73–78. 10.1046/j.1432-1327.1998.2520073.x. [DOI] [PubMed] [Google Scholar]
- Holt C.; Sørensen E. S.; Clegg R. A. Role of Calcium Phosphate Nanoclusters in the Control of Calcification. FEBS J. 2009, 276, 2308–2323. 10.1111/j.1742-4658.2009.06958.x. [DOI] [PubMed] [Google Scholar]
- De Sa Peixoto P.; Silva J. V. C.; Laurent G.; Schmutz M.; Thomas D.; Bouchoux A.; Gésan-Guiziou G. How High Concentrations of Proteins Stabilize the Amorphous State of Calcium Orthophosphate: A Solid-State Nuclear Magnetic Resonance (NMR) Study of the Casein Case. Langmuir 2017, 33, 1256–1264. 10.1021/acs.langmuir.6b04235. [DOI] [PubMed] [Google Scholar]
- Hindmarsh J. P.; Watkinson P. Experimental Evidence for Previously Unclassified Calcium Phosphate Structures in the Casein Micelle. J. Dairy Sci. 2017, 100, 6938–6948. 10.3168/jds.2017-12623. [DOI] [PubMed] [Google Scholar]
- Pujari-Palmer M.; Guo H.; Wenner D.; Autefage H.; Spicer C. D.; Stevens M. M.; Omar O.; Thomsen P.; Edén M.; Insley G.; et al. A Novel Class of Injectable Bioceramics that Glue Tissues and Biomaterials. Materials 2018, 11, 2492. 10.3390/ma11122492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirillova A.; Kelly C.; von Windheim N.; Gall K. Bioinspired Mineral–Organic Bioresorbable Bone Adhesive. Adv. Healthcare Mater. 2018, 7, 1800467. 10.1002/adhm.201800467. [DOI] [PubMed] [Google Scholar]
- Kesseli F. P.; Lauer C. S.; Baker I.; Mirica K. A.; Van Citters D. W. Identification of a Calcium Phosphoserine Coordination Network in an Adhesive Organo–Apatitic Bone Cement System. Acta Biomater. 2020, 105, 280–289. 10.1016/j.actbio.2020.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X.; Pujari-Palmer M.; Wenner D.; Procter P.; Insley G.; Engqvist H. Adhesive Cements That Bond Soft Tissue Ex Vivo. Materials 2019, 12, 2473. 10.3390/ma12152473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pujari-Palmer M.; Giró R.; Procter P.; Bojan A.; Insley G.; Engqvist H. Factors that Determine the Adhesive Strength in a Bioinspired Bone Tissue Adhesive. Chem. Eng. 2020, 4, 19. 10.3390/chemengineering4010019. [DOI] [Google Scholar]
- Spicer C. D.; Pujari-Palmer M.; Autefage H.; Insley G.; Procter P.; Engqvist H.; Stevens M. M. Synthesis of Phospho-Amino Acid Analogues as Tissue Adhesive Cement Additives. ACS Cent. Sci. 2020, 6, 226–231. 10.1021/acscentsci.9b01149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew R.; Pujari-Palmer M.; Guo H.; Yu Y.; Stevensson B.; Engqvist H.; Edén M. Solid-State NMR Rationalizes the Bone-Adhesive Properties of Serine- and Phosphoserine-Bearing Calcium Phosphate Cements by Unveiling Their Organic/Inorganic Interface. J. Phys. Chem. C 2020, 124, 21512–21531. 10.1021/acs.jpcc.0c06224. [DOI] [Google Scholar]
- Barducci A.; Bussi G.; Parrinello M. Well-Tempered Metadynamics: A Smoothly Converging and Tunable Free-Energy Method. Phys. Rev. Lett. 2008, 100, 020603. 10.1103/PhysRevLett.100.020603. [DOI] [PubMed] [Google Scholar]
- Valsson O.; Tiwary P.; Parrinello M. Enhancing Important Fluctuations: Rare Events and Metadynamics from a Conceptual Viewpoint. Annu. Rev. Phys. Chem. 2016, 67, 159–184. 10.1146/annurev-physchem-040215-112229. [DOI] [PubMed] [Google Scholar]
- Brunauer S.; Emmett P. H.; Teller E. Adsorption of Gases in Multimolecular Layers. J. Am. Chem. Soc. 1938, 60, 309–319. 10.1021/ja01269a023. [DOI] [Google Scholar]
- Suga T.; Okabe N. Aqua(L-O-Serine Phosphato)Calcium(II). Acta Crystallogr. 1996, 52, 1894–1896. 10.1107/s0108270196002648. [DOI] [Google Scholar]
- Mathew R.; Stevensson B.; Edén M. Refined Structures of O-Phospho-L-serine and Its Calcium Salt by New Multinuclear Solid-State NMR Crystallography Methods. J. Phys. Chem. B 2021, 125, 10985–11004. 10.1021/acs.jpcb.1c05587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay M. I.; Young R. A.; Posner A. S. Crystal Structure of Hydroxyapatite. Nature 1964, 204, 1050–1052. 10.1038/2041050a0. [DOI] [PubMed] [Google Scholar]
- Abraham M. J.; Murtola T.; Schulz R.; Páll S.; Smith J. C.; Hess B.; Lindahl E. GROMACS: High Performance Molecular Simulations Through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- Bjelkmar P.; Larsson P.; Cuendet M. A.; Hess B.; Lindahl E. Implementation of the CHARMM Force Field in GROMACS: Analysis of Protein Stability Effects from Correction Maps, Virtual Interaction Sites, and Water Models. J. Chem. Theory Comput. 2010, 6, 459–466. 10.1021/ct900549r. [DOI] [PubMed] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. 10.1063/1.445869. [DOI] [Google Scholar]
- Boonstra S.; Onck P. R.; van der Giessen E. CHARMM TIP3P Water Model Suppresses Peptide Folding by Solvating the Unfolded State. J. Phys. Chem. B 2016, 120, 3692–3698. 10.1021/acs.jpcb.6b01316. [DOI] [PubMed] [Google Scholar]
- Laio A.; Gervasio F. L. Metadynamics: a Method to Simulate Rare Events and Reconstruct the Free Energy in Biophysics, Chemistry and Material Science. Rep. Prog. Phys. 2008, 71, 126601. 10.1088/0034-4885/71/12/126601. [DOI] [Google Scholar]
- Valsson O.; Parrinello M. Variational Approach to Enhanced Sampling and Free Energy Calculations. Phys. Rev. Lett. 2014, 113, 090601. 10.1103/physrevlett.113.090601. [DOI] [PubMed] [Google Scholar]
- Tribello G. A.; Bonomi M.; Branduardi D.; Camilloni C.; Bussi G. PLUMED 2: New Feathers for an Old Bird. Comput. Phys. Commun. 2014, 185, 604–613. 10.1016/j.cpc.2013.09.018. [DOI] [Google Scholar]
- Ben Osman M.; Diallo-Garcia S.; Herledan V.; Brouri D.; Yoshioka T.; Kubo J.; Millot Y.; Costentin G. Discrimination of Surface and Bulk Structure of Crystalline Hydroxyapatite Nanoparticles by NMR. J. Phys. Chem. C 2015, 119, 23008–23020. 10.1021/acs.jpcc.5b08732. [DOI] [Google Scholar]
- Turner G. L.; Smith K. A.; Kirkpatrick R. J.; Oldfieldt E. Structure and Cation Effects on Phosphorus-31 NMR Chemical Shifts and Chemical-Shift Anisotropies of Orthophosphates. J. Magn. Reson. 1986, 70, 408–415. 10.1016/0022-2364(86)90129-0. [DOI] [Google Scholar]
- Hartmann P.; Vogel J.; Schnabel B. The Influence of Short-Range Geometry on the 31P Chemical-Shift Tensor in Protonated Phosphates. J. Magn. Reson., Ser. A 1994, 111, 110–114. 10.1006/jmra.1994.1234. [DOI] [Google Scholar]
- Lu B.-Q.; Garcia N. A.; Chevrier D. M.; Zhang P.; Raiteri P.; Gale J. D.; Gebauer D. Short-Range Structure of Amorphous Calcium Hydrogen Phosphate. Cryst. Growth Des. 2019, 19, 3030–3038. 10.1021/acs.cgd.9b00274. [DOI] [Google Scholar]
- Dorozhkin S. V. Calcium Orthophosphate Cements for Biomedical Application. J. Mater. Sci. 2008, 43, 3028–3057. 10.1007/s10853-008-2527-z. [DOI] [Google Scholar]
- Reinstorf A.; Ruhnow M.; Gelinsky M.; Pompe W.; Hempel U.; Wenzel K.-W.; Simon P. Phosphoserine—a Convenient Compound for Modification of Calcium Phosphate Bone Cement Collagen Composites. J. Mater. Sci.: Mater. Med. 2004, 15, 451–455. 10.1023/b:jmsm.0000021119.14870.3d. [DOI] [PubMed] [Google Scholar]
- Brunner T. J.; Grass R. N.; Bohner M.; Stark W. J. Effect of Particle Size, Crystal Phase and Crystallinity on the Reactivity of Tricalcium Phosphate Cements for Bone Reconstruction. J. Mater. Chem. 2007, 17, 4072–4078. 10.1039/b707171j. [DOI] [Google Scholar]
- Aue W. P.; Roufosse A. H.; Glimcher M. J.; Griffin R. G. Solid-State Phosphorus-31 Nuclear Magnetic Resonance Studies of Synthetic Solid Phases of Calcium Phosphate: Potential Models of Bone Mineral. Biochemistry 1984, 23, 6110–6114. 10.1021/bi00320a032. [DOI] [PubMed] [Google Scholar]
- Yu Y.; Guo H.; Pujari-Palmer M.; Stevensson B.; Grins J.; Engqvist H.; Edén M. Advanced Solid-State 1H/31P NMR Characterization of Pyrophosphate-Doped Calcium Phosphate Cements for Biomedical Applications: The Structural Role of Pyrophosphate. Ceram. Int. 2019, 45, 20642–20655. 10.1016/j.ceramint.2019.07.047. [DOI] [Google Scholar]
- Rothwell W. P.; Waugh J. S.; Yesinowski J. P. High-Resolution Variable-Temperature 31P NMR of Solid Calcium Phosphates. J. Am. Chem. Soc. 1980, 102, 2637–2643. 10.1021/ja00528a020. [DOI] [Google Scholar]
- Tropp J.; Blumenthal N. C.; Waugh J. S. Phosphorus NMR Study of Solid Amorphous Calcium Phosphate. J. Am. Chem. Soc. 1983, 105, 22–26. 10.1021/ja00339a006. [DOI] [Google Scholar]
- Yesinowski J. P.; Eckert H. Hydrogen Environments in Calcium Phosphates: 1H MAS NMR at High Spinning Speeds. J. Am. Chem. Soc. 1987, 109, 6274–6282. 10.1021/ja00255a009. [DOI] [Google Scholar]
- Mathew R.; Turdean-Ionescu C.; Yu Y.; Stevensson B.; Izquierdo-Barba I.; García A.; Arcos D.; Vallet-Regí M.; Edén M. Proton Environments in Biomimetic Calcium Phosphates Formed from Mesoporous Bioactive CaO–SiO2–P2O5 Glasses in Vitro: Insights from Solid-State NMR. J. Phys. Chem. C 2017, 121, 13223–13238. 10.1021/acs.jpcc.7b03469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan Z. 13C/14N Heteronuclear Multiple-Quantum Correlation with Rotary Resonance and REDOR Dipolar Recoupling. J. Magn. Reson. 2007, 184, 39–43. 10.1016/j.jmr.2006.09.016. [DOI] [PubMed] [Google Scholar]
- Hu B.; Trébosc J.; Amoureux J. P. Comparison of Several Hetero-Nuclear Dipolar Recoupling NMR Methods to be Used in MAS HMQC/HSQC. J. Magn. Reson. 2008, 192, 112–122. 10.1016/j.jmr.2008.02.004. [DOI] [PubMed] [Google Scholar]
- Yasar O. F.; Liao W.-C.; Stevensson B.; Edén M. Structural Role and Spatial Distribution of Carbonate Ions in Amorphous Calcium Phosphate. J. Phys. Chem. C 2021, 125, 4675–4693. 10.1021/acs.jpcc.0c10355. [DOI] [Google Scholar]
- Moreno E. C.; Kresak M.; Hay D. I. Adsorption of Molecules of Biological Interest onto Hydroxyapatite. Calcif. Tissue Int. 1984, 36, 48–59. 10.1007/bf02405293. [DOI] [PubMed] [Google Scholar]
- Jack K. S.; Vizcarra T. G.; Trau M. Characterization and Surface Properties of Amino-Acid-Modified Carbonate-Containing Hydroxyapatite Particles. Langmuir 2007, 23, 12233–12242. 10.1021/la701848c. [DOI] [PubMed] [Google Scholar]
- Jahromi M. T.; Yao G.; Cerruti M. The Importance of Amino Acid Interactions in the Crystallization of Hydroxyapatite. J. R. Soc. Interface 2013, 10, 20120906. 10.1098/rsif.2012.0906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavafoghi M.; Cerruti M. The Role of Amino Acids in Hydroxyapatite Mineralization. J. R. Soc. Interface 2016, 13, 20160462. 10.1098/rsif.2016.0462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimola A.; Corno M.; Zicovich-Wilson C. M.; Ugliengo P. Ab Initio Modeling of Protein/Biomaterial Interactions: Glycine Adsorption at Hydroxyapatite Surfaces. J. Am. Chem. Soc. 2008, 130, 16181–16183. 10.1021/ja806520d. [DOI] [PubMed] [Google Scholar]
- Almora-Barrios N.; Austen K. F.; de Leeuw N. H. Density Functional Theory Study of the Binding of Glycine, Proline, and Hydroxyproline to the Hydroxyapatite (0001) and (011̅0) Surfaces. Langmuir 2009, 25, 5018–5025. 10.1021/la803842g. [DOI] [PubMed] [Google Scholar]
- Gerothanassis I. P. Oxygen-17 NMR Spectroscopy: Basic Principles and Applications (Part I). Prog. Nucl. Magn. Reson. Spectrosc. 2010, 56, 95–197. 10.1016/j.pnmrs.2009.09.002. [DOI] [PubMed] [Google Scholar]
- Bryce D. L. Calcium Binding Environments Probed by 43Ca NMR Spectroscopy. Dalton Trans. 2010, 39, 8593–8602. 10.1039/c0dt00416b. [DOI] [PubMed] [Google Scholar]
- Laurencin D.; Smith M. E. Development of 43Ca Solid State NMR Spectroscopy as a Probe of Local Structure in Inorganic and Molecular Materials. Prog. Nucl. Magn. Reson. Spectrosc. 2013, 68, 1–40. 10.1016/j.pnmrs.2012.05.001. [DOI] [PubMed] [Google Scholar]
- Sundaralingam M.; Putkey F. F. Molecular Structures of Amino Acids and Peptides. II. A Redetermination of the Crystal Structure of L-O-Serine Phosphate. A Very Short Phosphate-Carboxyl Hydrogen Bond. Acta Crystallogr. 1970, 26, 790–800. 10.1107/s0567740870003138. [DOI] [PubMed] [Google Scholar]
- Potrzebowski M. J.; Assfeld X.; Ganicz K.; Olejniczak S.; Cartier A.; Gardiennet C.; Tekely P. An Experimental and Theoretical Study of the 13C and 31P Chemical Shielding Tensors in Solid O-Phosphorylated Amino Acids. J. Am. Chem. Soc. 2003, 125, 4223–4232. 10.1021/ja029840z. [DOI] [PubMed] [Google Scholar]
- Van Vleck J. H. The Dipolar Broadening of Magnetic Resonance Lines in Crystals. Phys. Rev. 1948, 74, 1168–1183. 10.1103/physrev.74.1168. [DOI] [Google Scholar]
- Bertmer M.; Züchner L.; Chan J. C. C.; Eckert H. Short and Medium Range Order in Sodium Aluminoborate Glasses: 2. Site Connectivities and Cation Distributions Studied by Rotational Echo Double Resonance NMR Spectroscopy. J. Phys. Chem. B 2000, 104, 6541–6553. 10.1021/jp9941918. [DOI] [Google Scholar]
- Strojek W.; Kalwei M.; Eckert H. Dipolar NMR Strategies for Multispin Systems Involving Quadrupolar Nuclei: 31P{23Na} Rotational Echo Double Resonance (REDOR) of Crystalline Sodium Phosphates and Phosphate Glasses. J. Phys. Chem. B 2004, 108, 7061–7073. 10.1021/jp037041c. [DOI] [Google Scholar]
- Eckert H.; Elbers S.; Epping J. D.; Janssen M.; Kalwei M.; Strojek W.; Voigt U. Dipolar Solid State NMR Approaches Towards Medium-Range Structure in Oxide Glasses. Top. Curr. Chem. 2005, 246, 195–233. 10.1007/b98651. [DOI] [PubMed] [Google Scholar]
- Stevensson B.; Mathew R.; Yu Y.; Edén M. Two Heteronuclear Dipolar Results at the Price of One: Quantifying Na/P Contacts in Phosphosilicate Glasses and Biomimetic Hydroxy-Apatite. J. Magn. Reson. 2015, 251, 52–56. 10.1016/j.jmr.2014.12.002. [DOI] [PubMed] [Google Scholar]
- Edén M. Update on 27Al NMR Studies of Aluminosilicate Glasses. Annu. Rep. NMR Spectrosc. 2020, 101, 285–410. 10.1016/bs.arnmr.2020.07.002. [DOI] [Google Scholar]
- Benaziz L.; Barroug A.; Legrouri A.; Rey C.; Lebugle A. Adsorption of O-Phospho-L-Serine and L-Serine Onto Poorly Crystalline Apatite. J. Colloid Interface Sci. 2001, 238, 48–53. 10.1006/jcis.2001.7450. [DOI] [PubMed] [Google Scholar]
- Spanos N.; Klepetsanis P. G.; Koutsoukos P. G. Model Studies on the Interaction of Amino Acids With Biominerals: The Effect of L-Serine at the Hydroxyapatite–Water Interface. J. Colloid Interface Sci. 2001, 236, 260–265. 10.1006/jcis.2000.7396. [DOI] [PubMed] [Google Scholar]
- Spanos N.; Koutsoukos P. G. Model Studies of the Effect of Orthophospho-L-Serine on Biological Mineralization. Langmuir 2001, 17, 866–872. 10.1021/la0010166. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.