

Abstract
Africa, the ancestral home of all modern humans, is the most informative continent for understanding the human genome and its contribution to complex disease. To better understand the genetics of schizophrenia, we studied the illness in the Xhosa population of South Africa, recruiting 909 cases and 917 age-, gender-, and residence-matched controls. Individuals with schizophrenia were significantly more likely than controls to harbor private, severely damaging mutations in genes that are critical to synaptic function, including neural circuitry mediated by the neurotransmitters glutamine, γ-aminobutyric acid, and dopamine. Schizophrenia is genetically highly heterogeneous, involving severe ultrarare mutations in genes that are critical to synaptic plasticity. The depth of genetic variation in Africa revealed this relationship with a moderate sample size and informed our understanding of the genetics of schizophrenia worldwide.
Schizophrenia is a disabling neurodevelopmental disorder characterized by aberrant perceptions, thought, and social connectivity. An evolutionary perspective is particularly valuable for understanding the genetic origins of the disorder (1). Because fewer children are born to persons with schizophrenia, mutations underlying the illness are under negative selection. Therefore, the genetic architecture of schizophrenia is characterized by damaging mutations that are very recent or de novo and thus individually extremely rare (2–5). Common variants with individually small effects on schizophrenia have also been reported (6). Genes implicated by both common and rare alleles operate in pathways that are essential to brain development, including histone modification, neuronal migration, transcriptional regulation, immune function, and synaptic integrity (3–6).
Until now, nearly all genetic studies of schizophrenia have been based in populations of European or Asian ancestries. The goal of the present study was to identify and characterize genetic influences on schizophrenia in the Xhosa population of South Africa. The study was undertaken not because the Xhosa have an unusual prevalence of schizophrenia, but because African populations harbor the greatest wealth of human genetic diversity (7). Nearly 99% of human evolution after the chimpanzee-human divergence 5 to 6 million years ago took place before human migrations from Africa to Eurasia 50,000 to 100,000 years ago. Because relatively small numbers of individuals migrated (8), a very large number of alleles remained on the African continent, creating an African-specific tranche of human genetic variation. Alleles of the African tranche are more rare than the common single-nucleotide polymorphisms (SNPs) shared by all populations and more common than recent mutations that appeared subsequently in all populations. In the absence of studies of ancestral African populations, alleles of the African tranche are missing from our understanding of human disease.
The Xhosa trace their history to the migration of Bantu people from the Great Lakes region of eastern Africa to southern Africa centuries ago. Until the arrival of these migrants, southern Africa was occupied exclusively by San peoples, who diverged from other modern humans at least 100,000 years ago (9). Archaeological, linguistic, and DNA evidence indicate that the Xhosa people are descended from the admixture of these Bantu and San populations (10–12). The Xhosa now live throughout South Africa and are the largest population of the Eastern Cape region.
For this project, participants with schizophrenia (cases) were recruited from psychiatric inpatient units and outpatient health clinics in the Eastern Cape Province and Western Cape Province (Fig. 1A). Controls were recruited from the same locales, including patients presenting with conditions not related to mental health. Cases and controls were matched for age, gender, education, and region of recruitment (13).
Fig. 1. The Xhosa of South Africa.
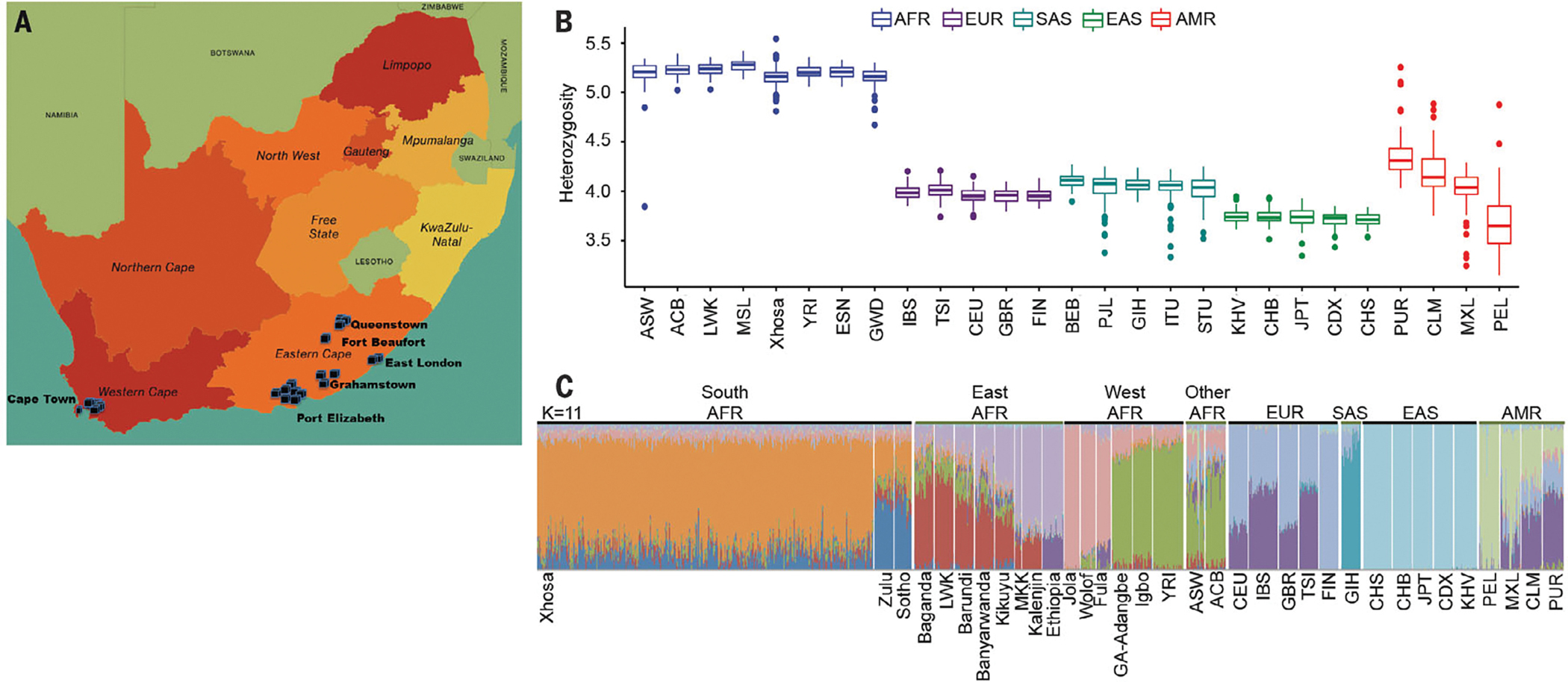
(A) Sites of the Western Cape and Eastern Cape provinces of South Africa, where cases and controls were recruited. (B) Heterozygosity at coding sequence 31.7 Mb of the human genome, calculated in 10-kb intervals, in the Xhosa and other populations from all continents. (C) Population structure of the Xhosa with respect to other world populations, evaluated by using ADMIXTURE version 1.3.0 with K = 6. Genotypes from populations other than the Xhosa are from the African Genome Variation Project and the 1000 Genomes Project. Sources and abbreviations for all populations are listed in the materials and methods.
A total of 2092 individuals, all self-identifying as Xhosa, enrolled in the study. The final cohort for genetic analysis was 1826 individuals, comprising 909 cases and 917 controls. More detailed information regarding sampling strategy, consent, and clinical and demographic features of the cohort is provided in tables S1 and S2 (13). DNA from participants was evaluated by whole-exome sequencing, with quality control and variant interpretation being based on established experimental and bioinformatics methods (figs. S1 to S5) (13).
As expected, genetic variation among the Xhosa (regardless of case-control status) was far greater than among non-Africans (Fig. 1B). Analysis of the Xhosa vis-a-vis other African populations suggested the closest genetic relationship to the Zulu and Sotho populations, their geographic neighbors (Fig. 1C and figs. S6 to S10).
To characterize the genetic architecture of schizophrenia in the Xhosa population, we evaluated contributions from both rare alleles and common alleles. We first compared the numbers of cases versus controls carrying at least one private damaging variant in a gene that was intolerant to such mutations (table S3) (13). A “private variant” was defined as a variant that appeared in only one case or only one control among our participants and that was absent from other population databases (13). “Damaging variants” were defined as nonsense mutations, frameshift mutations, and splice-disrupting and missense mutations that were predicted to be damaging by multiple criteria (13).
Definition of private damaging variants in mutation-intolerant genes yielded “case genes,” which harbored such mutations only in cases, and “control genes,” which harbored such mutations only in controls. For example, CNTNAP1 was a case gene, harboring four different private damaging variants in cases and none in controls, whereas MUC5B was a control gene, harboring seven different private damaging variants in controls and none in cases. Analyses were carried out for minimum thresholds of one, two, or three different private damaging mutations per case gene or per control gene.
Comparisons of cases and controls yielded four significant results. First, cases were significantly more likely than controls to harbor private damaging mutations in case genes or control genes that were intolerant to such mutations (Fig. 2A). Results were similar for thresholds of one, two, and three private damaging mutations per gene and were robust to definitions of intolerance (table S4). These results paralleled those of our prior study of exclusively de novo events in schizophrenia cases and controls (4). As a control for this approach, we compared the numbers of cases versus controls harboring at least one private benign mutation in mutation-intolerant genes (13). There were no significant differences between cases and controls for these benign events (Fig. 2A).
Fig. 2. Distributions of damaging mutations in cases and controls.
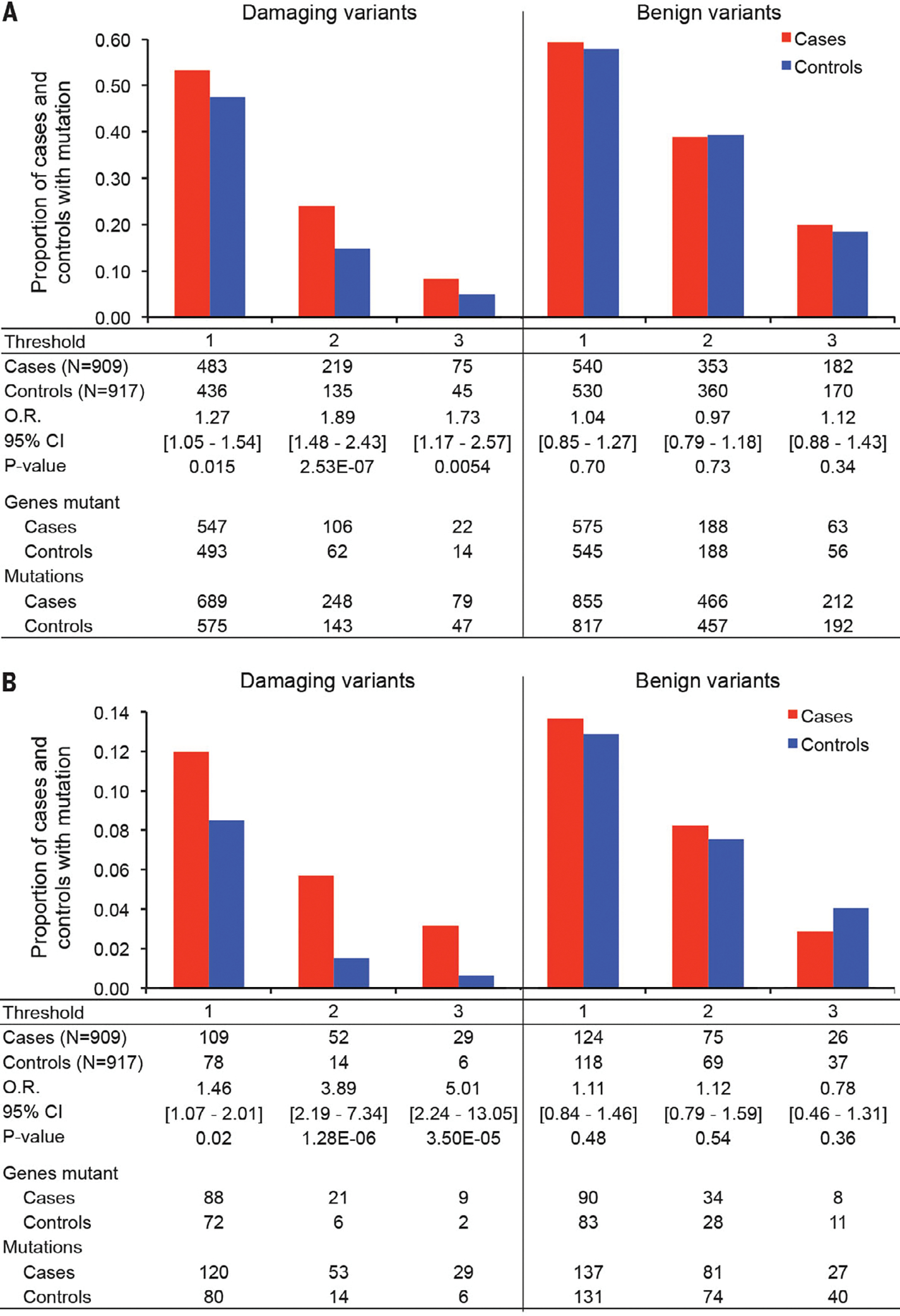
Histograms indicate the proportions of cases and controls with private damaging mutations in genes with such mutations only in cases (case genes) or only in controls (control genes). Thresholds indicate the minimum number of private damaging mutations that define a case gene or a control gene. (A) Proportions of cases and controls with either private damaging or benign mutations in all case genes or control genes. O.R., odds ratio. (B) Proportions of cases and controls with either private damaging or benign mutations in synaptic genes (15).
Differences in mutation distributions in cases versus controls were consistent with an oligogenic model; that is, the hypothesis that the illness may be caused by one or a few severe damaging variants. Case status was significantly associated with more private damaging mutations per individual [Firth logistic regression, P = 0.0002 (13)]. The odds ratio representing the increased risk of being a case associated with each additional mutation was 1.25 [95% confidence interval (CI), 1.11–1.41] (table S5).
Second, cases were significantly more likely than controls to carry private damaging mutations, specifically in genes that are highly expressed in brain (14) (table S6) and in genes that are involved in synaptic functioning (15) (Fig. 2B). Increasingly strong effects were found for subsets of synaptic genes harboring two or three private damaging mutations (Fig. 2B). Again, as a control for the approach, we compared the numbers of cases versus controls harboring at least one private benign mutation in mutation-intolerant synaptic genes. There were no significant differences between cases and controls.
Proportions of HIV-positive individuals differed by case-control status and by gender (table S1). To determine whether HIV status was a confounder for the genomics analyses, we recalculated all comparisons in two ways: by using HIV status as a covariate in regression and excluding all HIV-positive individuals. Results did not differ from those reported above (table S7).
Third, integration of gene dysregulation profiles from analysis of postmortem brain tissues from individuals with schizophrenia, autism, or bipolar disorder (16) with mutational profiles of Xhosa cases and controls revealed that Xhosa cases were significantly more likely than controls to carry private damaging mutations in genes that are downregulated in schizophrenia or autism, or up-regulated in bipolar disorder (Fig. 3). By contrast, controls were not enriched for damaging mutations in any set of genes that are dysregulated in these illnesses.
Fig. 3. Proportions of cases and controls with mutations in genes that are dysregulated in severe mental illness.
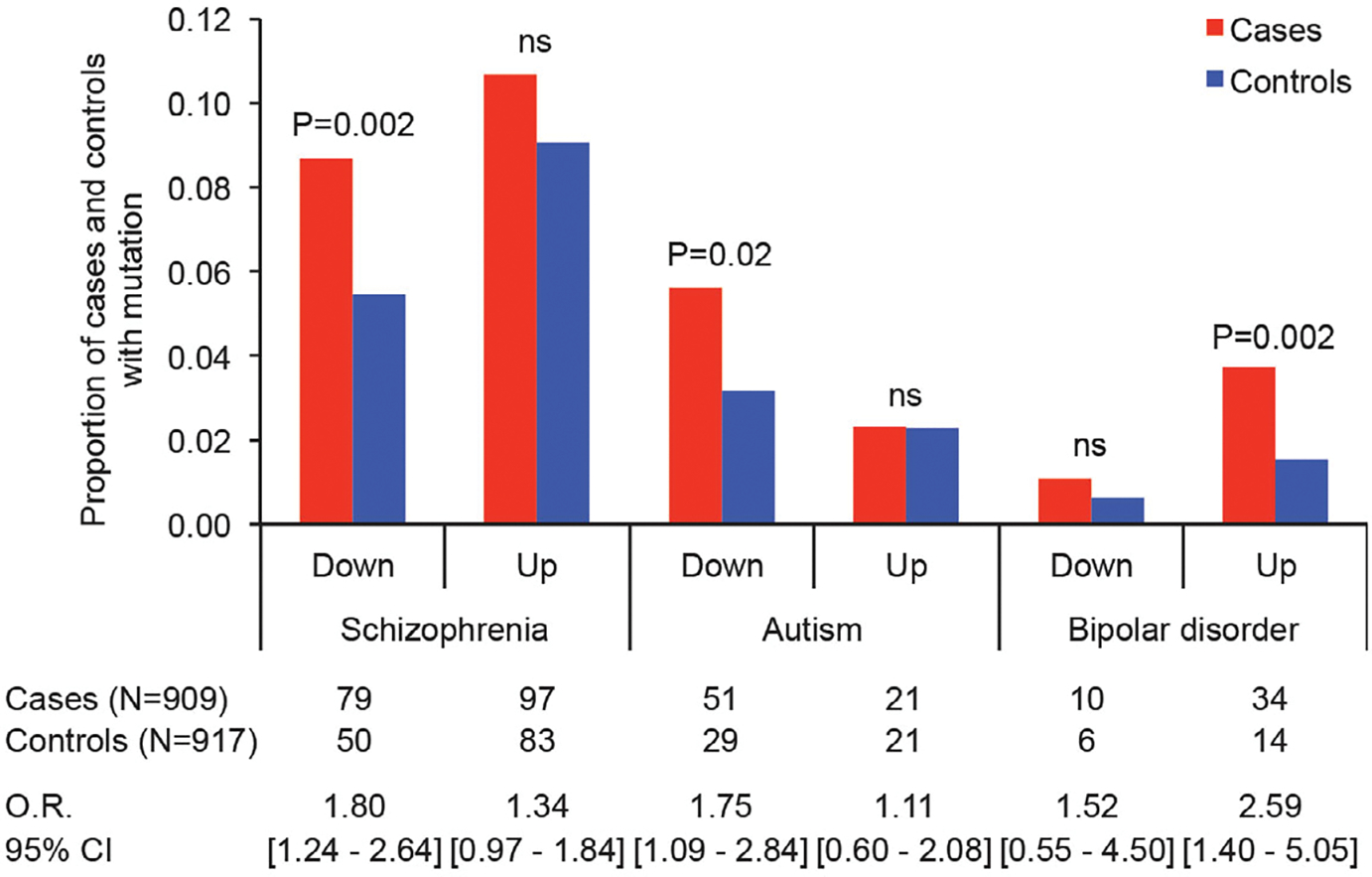
Previous studies evaluated differential gene expression in the postmortem brain of genes from individuals with schizophrenia, autism, and bipolar disorder (16). Integrating these data with mutational profiles of cases and controls reveals that cases were significantly more likely than controls to carry private damaging mutations in genes that are downregulated in schizophrenia or autism or upregulated in bipolar disorder. ns, not significant.
The possibility that individual genes were enriched for rare variants in cases versus controls was evaluated by using SKAT (sequence kernel association test) (17). No single gene was significant after multiple-comparison adjustment, although marginal results may prove significant in future studies with larger sample sizes (13) (table S8 and fig. S11).
To evaluate the African tranche of variation, we compared distributions of African-specific exonic variants of frequency 0.01 to 0.10 in cases versus controls by logistic regression, adjusting for covariates (13). In addition, common exonic variants (defined as minor allele frequency MAF ≥ 0.01 in the Xhosa, regardless of frequency in other populations) were evaluated by using the same methods. Q-Q plots and Manhattan plots for these analyses are shown in fig. S12. With the caveat that sample size was low for an exome-wide association design, one SNP was significant after exome-wide multiple-comparison adjustment (table S9): rs12600437 in the 3′ untranslated region of zinc finger protein ZFP3 had a higher minor allele frequency in controls than in cases.
Cases and controls were assessed for copy number variants (CNVs) in 15 genomic regions enriched for CNVs in schizophrenia and other psychiatric disorders (18). Of the 904 cases and 912 controls with exome data that could be evaluated for CNVs (19), three cases and one control carried a deletion at chromosome 15q11.2, three cases carried a duplication at chromosome 16p13.11, and one control carried a duplication at chromosome 16p11.2 (fig. S13). In addition, one control had karyotype XXY, and one control had karyotype XYY.
Fourth, to determine whether results for the Xhosa generalized to non-African populations, we evaluated data from a case-control study of schizophrenia in the Swedish population (3) using the same methods. We applied our quality control criteria to the Swedish dataset, yielding data from 4436 cases and 5713 controls, then selected random subsets of 909 Swedish cases and 909 Swedish controls 10,000 times, matching for critical covariates (13). For each Swedish subset, we counted the numbers of cases and controls harboring qualifying private mutations, using the same criteria as for the Xhosa cases and controls, then calculated P values from the median numbers of cases and controls for each threshold (table S10). As with the Xhosa, Swedish cases were significantly more likely than controls to harbor private damaging mutations in case genes or control genes that are intolerant to such mutations.
Results for the Xhosa yielded generally larger effect sizes than did the results for the Swedes, reflecting the greater depth of genetic variation of the Xhosa population and hence more rigorous definitions of case-only and control-only genes. For the same number of cases and controls, greater genetic variation in Africa provides more power to detect relationships of genes to phenotypes. The concordance of analyses of the Xhosa and Swedish populations is important, because many findings in genetic studies of schizophrenia do not replicate across populations (20). Results from both populations support disruptions in synaptic signaling and plasticity as being critical to the development of schizophrenia.
We can make some inferences about the causes of schizophrenia given these results. In the Xhosa, private damaging mutations in genes that are critical to synaptic plasticity and neural circuitry were enriched in participants with schizophrenia, with one or a few severe mutations in each affected individual. Human brain circuitry comprises some 100 million neurons interconnected by synapses, which are the communication hubs for transmitting and processing information. Dynamic changes in dendritic structure and synaptic organization are choreographed by complex signaling cascades involving thousands of proteins, including scaffolds, channels, receptors, kinases, adhesion molecules, signaling enzymes, and cytoskeleton components. These changes are ultimately responsible for learning, memory, and brain function (21).
Genes encoding synaptic proteins are well conserved, intolerant of mutations, and enriched for de novo mutations associated with neurodevelopmental disorders, including intellectual disability, autism, and schizophrenia (15). Among Xhosa cases, synaptic genes harboring private damaging mutations included glutamate (GRIA2), γ-aminobutyric acid (GABRB1, GABRB2, GABRA5, and GABRD), dopamine (DRD2), and glycine (GLRA2) receptors; voltage-gated calcium channels (CACNA1A and CACNA1C); scaffold proteins (DLG1, DLG3, and DLGAP1); cell adhesion molecules (CNTNAP1, CTNNB1, CTNNA2, and CTNND2); and multiple postsynaptic density signaling proteins, kinases, and phosphatases (Fig. 4). Synaptic genes that are disrupted by private damaging mutations in multiple cases include CACNA1C, DLGAP1, and huntingtin (HTT) and its associated kinase kalirin (KALRN), each of which was mutant in three cases, and CNTNAP1, which was mutant in four cases.
Fig. 4. Synaptic genes harboring damaging mutations in Xhosa persons with schizophrenia.
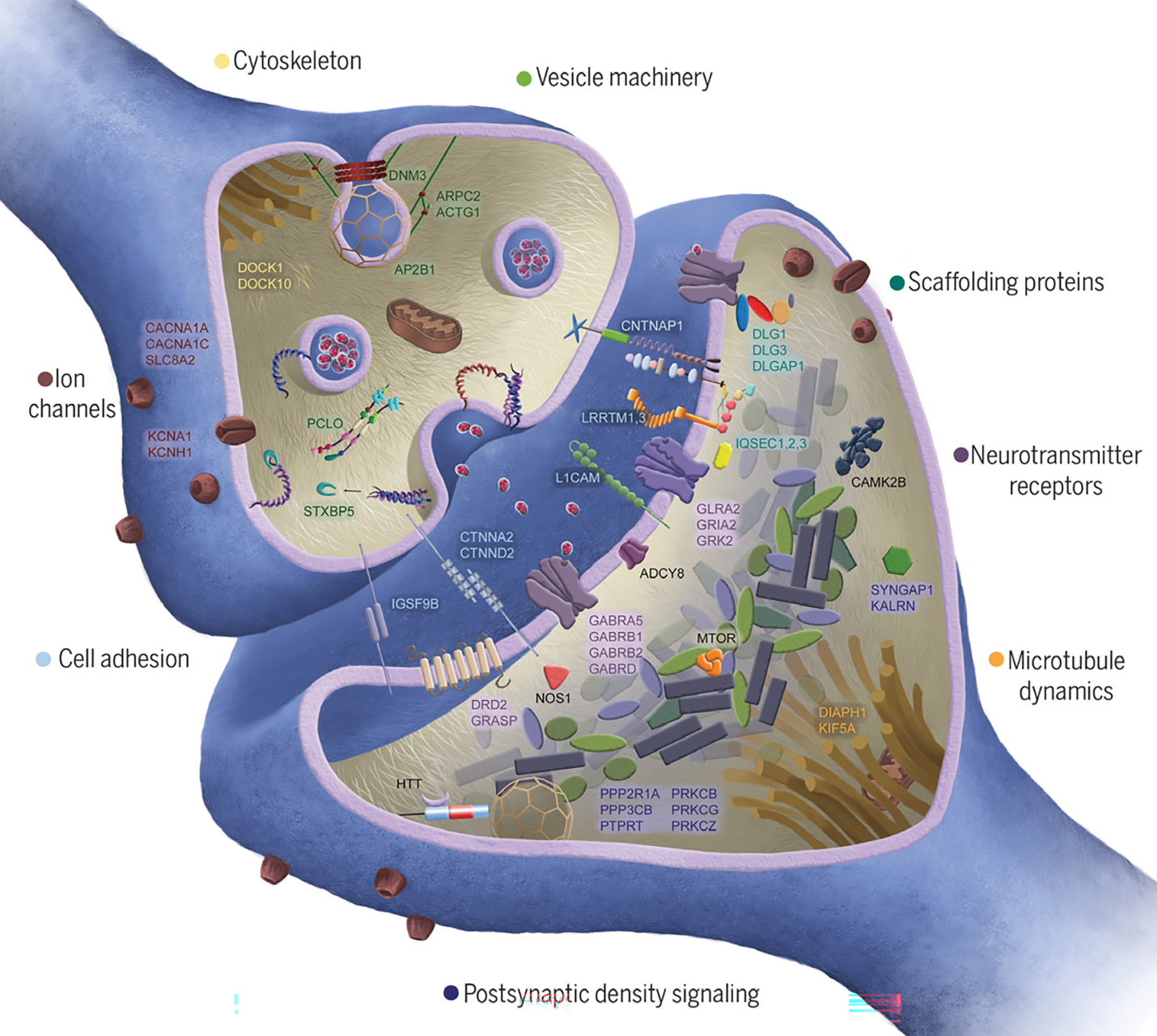
Proteins shown in the figure are encoded by genes that are mutant in at least one individual with schizophrenia. Synaptic genes collectively harbor a significantly greater burden of private damaging mutations in cases compared with controls. No single gene is significantly enriched for private damaging events in cases; most genes have a private damaging variant in one case and no controls.
In both African and non-African populations, a causal role for private damaging mutations in genes that are important to brain development is consistent with the nature of schizophrenia and with selection against it. Although schizophrenia is highly heritable, most cases are sporadic, and affected individuals have significantly fewer children, all of which are consistent with a critical role for de novo and recent ultrarare mutations (1). As a result, schizophrenia is characterized by extreme genetic heterogeneity, with no single gene explaining schizophrenia in more than a small number of patients (22).
Advances in treatments for schizophrenia depend on characterizing shared mechanisms underlying the illness. Results from African and European cohorts converge, both implicating disruptions in synaptic architecture and plasticity. Current antipsychotic medications generally act as antagonists and/or agonists at neurotransmitter receptors, mostly targeting dopamine, serotonin, and adrenergic receptors (23). These agents broadly affect neurotransmitter firing in neuronal circuits throughout the central nervous system, rather than narrowly targeting specific molecular pathways. Although helpful for reducing psychotic symptoms, these agents are not curative and generally do not address the neurocognitive and social difficulties inherent to the disorder (23). The synaptic genes that are disrupted in our cases encode structural components of neurotransmitter and ion channel receptors, cell adhesion proteins, scaffolding proteins, and postsynaptic density signaling molecules (21). Interventions designed to remediate disruptions in synaptic structural organization and intracellular signaling pathways potentially offer more specific therapeutic benefits.
Private damaging mutations in genes that are critical to brain function also appear in controls. For this study, controls were recruited from health clinics, which is a conservative bias because damaging mutations in controls may confer susceptibility to conditions that share genetic influences with schizophrenia. Whether a person harboring a severe mutation in a critical gene develops schizophrenia depends on the biology of the gene, the consequences of the mutation for gene function, and secondary events. For many patients, schizophrenia may be oligogenic, involving a few severe germline mutations and/or brain-specific severe somatic events, either genetic or epigenetic (1, 24). Secondary events may also include nongenetic brain injury during development. To the extent that these secondary events are stochastic, a damaging mutation in a critical gene may not lead to schizophrenia in the person in whom it first appears, but only in subsequent generations.
Finally, the infrastructure, capacity building, and research cohorts currently being established by H3Africa offer enormous promise for gene discovery for complex human phenotypes (25). Human biology is universal, and the study of human genomics in Africa provides an invaluable scientific opportunity to better detect and define genes that are critical for health worldwide.
Supplementary Material
ACKNOWLEDGMENTS
This study owes its success to African scientists and medical personnel and the grace of the Xhosa community. Institutional review committees at the University of Cape Town, Walter Sisulu University, Rhodes University, the University of Washington, and Columbia University approved the study. B. Malagas (University of Cape Town data verification officer) audited and reconciled the database.
Funding:
This work was supported by NIH grants U01MH096754 (D.J.S.), U01MH096756 (E.S.S.), U01MH096844 (M.-C.K., J.M.M., and T.W.), and UM1HG008898 with supplementary funding from the National Institute of Mental Health (R.A.G.), the South African Medical Research Council (D.J.S. and R.A.G.), and a NARSAD grant (to S.G).
Footnotes
Competing interests: D.J.S. receives research grants and honoraria from Lundbeck Pharmaceuticals and from Sun Pharmaceuticals IndustriesLtd. T.W. is a consultant to Color Genomics. R.A.G. receives payment for genetic testing from Baylor Genetics. All other authors declare no competing interests.
SUPPLEMENTARY MATERIALS
Data and materials availability:
Exome data from consenting participants have been deposited to dbGaP (accession no. phs000959.v1).
REFERENCES AND NOTES
- 1.McClellan J, King MC, Cell 141, 210–217 (2010). [DOI] [PubMed] [Google Scholar]
- 2.Ganna A et al. , Am. J. Hum. Genet. 102, 1204–1211 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Genovese G et al. , Nat. Neurosci. 19, 1433–1441 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gulsuner S et al. , Cell 154, 518–529 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fromer M et al. , Nature 506, 179–184 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schizophrenia Working Group of the Psychiatric Genomics Consortium, Nature 511, 421–427 (2014).25056061 [Google Scholar]
- 7.McClellan JM, Lehner T, King MC, Cell 171, 261–264 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Cavalli-Sforza LL, Menozzi P, Piazza A, The History and Geography of Human Genes (Princeton Univ. Press, 1994). [Google Scholar]
- 9.Chimusa ER et al. , PLOS Genet. 11, e1005052 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peires JB, The House of Phalo: A History of the Xhosa People in the Days of Their Independence (Univ. of California Press, 1982). [Google Scholar]
- 11.Newman JL, The Peopling of Africa: A Geographic Interpretation (Yale Univ. Press, 1995). [Google Scholar]
- 12.Phillipson DW, African Archaeology (Cambridge Univ. Press, ed. 3, 2005). [Google Scholar]
- 13.Materials and methods are available as supplementary materials.
- 14.Wells A et al. , Nucleic Acids Res. 43, 10804–10820 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koopmans F et al. , Neuron 103, 217–234.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gandal MJ et al. , Science 362, eaat8127 (2018).30545856 [Google Scholar]
- 17.Ionita-Laza I, Lee S, Makarov V, Buxbaum JD, Lin X, Am. J. Hum. Genet. 92, 841–853 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marshall CR et al. , Nat. Genet. 49, 27–35 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krumm N et al. , Genome Res. 22, 1525–1532 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Curtis D, Psychiatr. Genet. 28, 85–89 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Li J et al. , Sci. Signal. 9, rs8 (2016). [DOI] [PubMed] [Google Scholar]
- 22.Singh T et al. , Nat. Neurosci. 19, 571–577 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaar SJ, Natesan S, McCutcheon R, Howes OD, Neuropharmacology, 107704 (2019). [DOI] [PubMed] [Google Scholar]
- 24.Huang WC, Bennett K, Gregg C, Trends Neurosci. 41, 925–937 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rotimi C et al. , Science 344, 1346–1348 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Exome data from consenting participants have been deposited to dbGaP (accession no. phs000959.v1).