

Abstract

Functionalized chiral indole derivatives are privileged and versatile organic frameworks encountered in numerous pharmaceutically active agents and biologically active natural products. The catalytic asymmetric Friedel–Crafts reaction of indoles, catalyzed by chiral metal complexes or chiral organocatalysts, is one of the most powerful and atom-economical approaches to access optically active indole derivatives. Consequently, a wide range of electrophilic partners including α,β-unsaturated ketones, esters, amides, imines, β,γ-unsaturated α-keto- and α-ketiminoesters, ketimines, nitroalkenes, and many others have been successfully employed to achieve a plethora of functionalized chiral indole moieties. In particular, strategies for C–H functionalization in the phenyl of indoles require incorporation of a directing or blocking group in the phenyl or azole ring of indole. The discovery of chiral catalysts which can control enantiodiscrimination has gained a great deal of attention in recent years. This review will provide an updated account on the application of the asymmetric Friedel–Crafts reaction of indoles in the synthesis of diverse chiral indole derivatives, covering the timeframe from 2011 to today.
1. Introduction
As the most widely distributed heterocyclic compounds in nature, indole scaffolds are privileged and versatile organic frameworks encountered in numerous pharmaceuticals, agrochemicals, material science, and bioactive compounds in nature.1 Among the more than 10000 pharmacologically active indole derivatives that have been discovered, more than 200 have been advanced either as drugs or as clinical drug candidates (Figure 1).2
Figure 1.

Pharmaceutically relevant chiral indole derivatives.
Moreover, they also serve as versatile building blocks in the construction of valuable natural products.3 Although direct and selective synthesis of indole derivatives is quite challenging, owing to their structural diversity and complexity, enormous efforts have been devoted to develop direct alkylation of indoles in recent years.4 In particular, strategies for the synthesis of optically active indolyl derivatives have attracted a great deal of attention due to the ubiquity of their biological relevance.5 In this regard, the catalytic asymmetric Friedel–Crafts (F–C) reaction of indoles, catalyzed by chiral metal complexes or chiral organocatalysts, is one of the most powerful and atom-economical approaches that provides direct access to optically active indole derivatives in high enantioselectivities.6 A wide range of electrophilic partners including α,β-unsaturated ketones, esters, amides, imines, β,γ-unsaturated α-keto- and α-ketiminoesters, ketimines, nitroalkenes, and many others have been successfully employed to achieve a plethora of functionalized chiral indole moieties (Scheme 1).7
Scheme 1. Representative Enantioselective F–C Reactions of Indoles.

Indole is an electron-rich heteroaromatic system that exhibits high nucleophilic reactivities for the electrophilic alkylation at the C3- and C2-positions of the azole ring.8 Nevertheless, C–H functionalization in the phenyl ring of indoles often requires incorporating either directing or blocking groups in the phenyl or azole ring or necessitating harsher reaction conditions or the use of transition metals as catalysts.9 In the past two decades, the discovery of chiral catalysts that can control enantiodiscrimination of aromatic electrophilic substitution has attracted a great deal of attention in the asymmetric F–C reactions of indoles.10 After the pioneering report of Johannsen et al. on enantioselective F–C alkylation of indoles catalyzed by a chiral (tol-binap)–Cu(I) complex11 followed by an elegant discovery by Austin and MacMillan that reports enantioselective Michael addition of indoles to enals, catalyzed by chiral imidazolidinones,12 the field of catalytic asymmetric F–C reaction of indoles has been progressing at an astonishing pace. A tutorial review on the topic was published in 2009, which mainly covers metal or Brønsted acid-catalyzed F–C alkylation in the azole ring of indole.13 Moreover, Beletskaya et al. have reported asymmetric F–C alkylation of indoles with activated alkenes, employing chiral Lewis acids complexes and Brønsted acid catalysts.14 Likewise, two short reviews including one on the enantioselective arylation reactions15 and another on F–C reactions of naphthols and phenols that touch upon the topic have appeared in the literature.16 Recently, organocatalytic asymmetric dearomatization of indole to indolines and indolenines and their applications in the total synthesis of natural products has also been reviewed.17 Nevertheless, the flourishing achievements in the field have been ever growing in the past few years. In this review, we provide an updated account on the catalytic asymmetric F–C reactions of indoles catalyzed by chiral metal complexes or chiral organocatalysts. We aim to catalogue recent developments and strategies of this vibrant research field, covering literature from 2011 to today. This review will cover reactions involving the classical mechanism of aromatic electrophilic substitution, whereas redox reactions including oxidative couplings or oxidative cyclizations are not considered due to the length limitations. Moreover, the review is organized mainly based on the nature of electrophilic partners employed in catalytic asymmetric F–C reactions with indoles. In addition, examples of catalytic asymmetric functionalization of indoles are also included.
2. Conjugate Addition to Electrophilic C=C Bonds
2.1. Reactions with α,β-Unsaturated Ketones
The catalytic asymmetric F–C reaction of indoles with α,β-unsaturated ketones is considerably challenging. The inherent difficulty in the stereodifferentiation of the two faces exists due to steric similarity of the two carbonyl substituents. Nevertheless, enantiomerically enriched indole derivatives derived from such reactions are promising scaffolds, implicated in the synthesis of natural products and bioactive compounds. The asymmetric transition metal catalysis, employing transition metal in the presence of chiral ligands, is widely known in the construction of chiral-functionalized indoles. Kim et al. disclosed the first utilization of chiral dicationic palladium complex L1 as an efficient catalyst in the asymmetric F–C reaction of indoles 1 with γ,δ-unsaturated β-keto phosphonates 2.18 The synthetic strategy produced the corresponding alkylation adducts in fair to high yields and excellent enantioselectivities (up to 99% ee; Scheme 2a). The salient feature of the synthetic strategy includes low catalyst loading and the use of an air- and moisture-stable catalyst system at room temperature. In another report, the indole nucleus was functionalized at the C3-position with a functionality having a 1,4-difunctionalized moiety and benzylic stereogenic center. The synthetic approach utilized enantioselective F–C alkylation of indoles 4 with functionalized electrophilic partners, (E)-1-aryl-4-benzyloxybut-2-en-1-ones 5, catalyzed by chiral [Hf{(R)-3,3′-Br2-BINOL}(OtBu)2]2 complex L2.19 The reaction afforded the corresponding optically active alkylated indoles in good yields and low to high enantiomeric excess (up to 97% ee; Scheme 2b). The simple experimental procedure coupled with the commercial availability of ligands, in both enantiomeric forms, employed in the catalyst system makes this synthetic strategy quite attractive. Chatterjee et al. reported the first application of Fiaud’s acid (trans-1-hydroxy-2,5-diphenylphospholane-1-oxide) L3 as a chiral Brønsted acid catalyst in the enantioselective F–C alkylation between indoles 7 and 2-butene-1,4-diones 8.20 The reaction provided the corresponding alkylated indoles in appreciable yields and high enantioselectivities (up to 91% ee; Scheme 2c). The superiority of the method lies in the sufficiently milder reaction conditions, which offer a broader substrate scope. Moreover, the use of Fiaud’s acid as a catalyst could offer advantages of fine-tuning of steric and electronic properties of the 2,5-diarylphospholane backbone in comparison to widely employed BINOL-derived phosphoric acids, which offer limited opportunities for structural modifications. In 2019, Zhou et al. developed an elegant strategy for the synthesis of alkylated indoles 12, bearing a stereogenic center at the α-position relative to the carbonyl function of the electrophile.21 The production of such alkylated indoles in asymmetric catalysis is quite challenging since it requires enantioselective proton transfer of highly active enol intermediates. The synthetic strategy was based on chiral spiro-phosphoric acid (SPA) L4-catalyzed F–C conjugate addition of indoles 10 with 4-chromanone-derived enones 11 to afford versatile indoles, bearing cyclic ketones in excellent yields and high enantiomeric excess (up to 98% ee; Scheme 2d). The scope of the synthetic approach was quite broad since diverse indoles bearing both steric and electronically demanding substituents at the C5–C7-positions could furnish the corresponding products in good to high yields and excellent enantioselectivities. Moreover, C3- and C4-substituted indoles were also quite compatible. The C3-substituted indole delivered the corresponding product in moderate yield (80%), whereas C4-substituted indole yielded the desired product in high yield (87–90%) and high to excellent enantioselectivities (87–94% ee), although requiring slightly modified reaction conditions. Likewise, the scope of electrophilic partner was also quite diverse since electronically demanding substituents at the C6- and C7-positions of the phenyl ring proceeded smoothly to afford the desired products in high yields and excellent enantioselectivities (up to 98% ee). The practical utility of the synthetic strategy was demonstrated by transforming products into valuable polycyclic compounds. To gain a deeper insight into the reaction mechanism, density functional theory (DFT) studies suggested that SPA initially played an important role with Brønsted acid to initiate the addition reaction, followed by serving as a chiral proton-transfer shuttle to accelerate the proton-transfer reaction of the enol intermediate.
Scheme 2. Enantioselective F–C Alkylation of Indoles with (a) γ-δ-Unsaturated β-Keto Phosphonates, (b) (E)-1-Aryl-4-benzyloxybut-2-en-1-ones, (c) 2-Alkene-1,4-diones, and (d) Exocyclic Enones.

In view of the pharmacological importance of C7-functionalized indoles and their prevalence in naturally occurring bioactive alkaloids, Zhao et al. devised a synthetic strategy for the catalytic regio- and enantioselective F–C alkylation of indoles at the C7-position.7b In order to achieve regioselective alkylation at the C7-position, the challenge of competing alkylation at the more nucleophilic C3-position was overcome by introducing an amino function at the C4-position, which turned the C7-position more nucleophilic. The synthetic methodology utilizes chiral phosphoric acid (CPA) L5 as a catalyst in the enantioselective F–C alkylation of aminoindoles 13 and 16 with electrophilic acceptors diaryl 2-butene-1,4-diones 14 and 3-aroyl acrylates 17, respectively, to afford the corresponding substituted chiral indole derivatives in appreciable yields and low to high enantiomeric excess (up to 94% ee; Scheme 3). The generality of the method was diverse since a wide range of electrophilic acceptors, bearing electron-donating and electron-withdrawing groups in the aryl ring of both 14 and 17, were well-tolerated, providing the alkylated indoles in fair to high yields (up to 98%) and high enantiomeric excess (up to 96% ee). However, the presence of a substituent in the azole ring of indole 13 was detrimental for both reaction yield and stereoselectivity. Moreover, the practical utility of the reaction was demonstrated by a gram-scale preparation of the C7-functionalized indole.
Scheme 3. F–C Reaction of Aminoindoles with Diaryl 2-Butene-1,4-Diones and 3-Aroylacrylates.

To delineate the absolute configuration of the C7-alkylated product, a plausible transition state was proposed. Activation of both substrates by the CPA catalyst through cooperative hydrogen-bonding (H-bonding) interactions in a chiral pocket facilitates the attack of indole from the Re-face of activated electrophilic acceptors 14 or 17, affording the corresponding product in S-configuration (Scheme 3).
2.2. Reactions with α,β-Unsaturated Esters
The α,β-unsaturated esters are intriguing electrophilic partners in numerous catalytic asymmetric F–C alkylation of indoles. Chen et al. developed chiral bis(oxazoline) (BOX) ligand L6 for the highly enantioselective F–C alkylation of indoles 19 with alkylidene malonate 20.22 The ligand-designed strategy was based on connecting oxazoline rings to an sp2-hybridized bridge carbon in order to create a larger bridge angle. Moreover, substituents attached to the end of the double bond as well as on the oxazoline rings were also fine-tuned to achieve favorable steric and electronic effects. Under the catalysis of 10 mol % of the Cu(II)/L6 complex, the reaction provided the desired alkylated products 21 in excellent enantioselectivities (up to 99% ee) and high yields (Scheme 4).
Scheme 4. Asymmetric F–C Reaction of Indoles with Alkylidene Malonate.

In 2015, Oyama and Nakada disclosed a synthetic protocol for the enantioselective F–C reaction of indoles 23 with cyclic α-alkylidene β-oxo imides 22 as electrophilic partners.23 Using 10 mol % of the Cu(II)/L7 complex catalyst system and low reaction temperature (−60 °C), the method afforded the corresponding alkylated indoles 24 in high yields (up to 89%) and excellent enantioselectivities (up to 97% ee; Scheme 5a). The origin of higher enantioselectivity was linked to the formation of a complex between the catalyst and the rigid conformation of the substrate, which, in turn, originated due to the intramolecular H-bonding by the acidic imide hydrogen. Consequently, formation of chelate complex between the two imide carbonyls with the metal cation facilitated differentiating enantiotopic faces of the reacting double bond, resulting in enhanced enantioselectivity (Scheme 5a). In another study, Mocarska et al. reported a series of thiourea-based organocatalysts for the preparation of chiral 5-((1H-indol-3-yl)(aryl)methyl)-2,2-dimethyl-1,3-dioxane-4,6-diones 28 through asymmetric F–C reaction of indole 27 with 5-arylidene-2,2-dimethyl-1–3-dioxane-4–6-diones 26.24 The developed synthetic protocol utilizes the effective catalyst L8, affording the corresponding products in quantitative yield but with moderate enantiomeric ratios (up to 78:22 er; Scheme 5b). Moreover, the bulkier side chain in the catalyst, such as the tert-butyl group, appeared to increase the effectiveness of the catalyst. In general, the enantioselection of the reaction was strongly dependent upon the nature of substrate.
Scheme 5. Asymmetric F–C Reaction of Indoles with (a) Cyclic α-Alkylidene β-Oxo Imides and (b) 5-Arylidene-2,2-dimethyl-1-3-dioxane-4-6-diones.

In search of more sustainable organic processes, Bolm et al. developed an asymmetric F–C reaction of indoles 29 with arylidene malonates 30, using ball milling techniques under solvent-free conditions.7c The chiral complex of the copper/BOX catalyst, derived from the combination of CuCl/AgNTf2/L9, provided the desired alkylated indoles 31 in excellent yields and good enantioselectivities (up to 91:9 er; Scheme 6a). The scope of the method was diverse as a broad variety of indoles and arylidene malonates bearing functionalities with steric and electronic properties were tolerated under the optimized conditions. Nevertheless, C2-substitued indole and indoles having electron-donating groups at the C4- and C5-positions markedly reduced either the enantioselectivity or yield of the reactions. Moreover, the enantioselectivity of the reaction could be enhanced by the use a pentafluorophenol additive. Finally, the practicality of mechanochemical approach was validated by a 4-fold scale-up experiment to afford the scaled-up product in high yield (90%) and excellent enantioselectivity (91:9 er). In the same year, Arai et al. reported the enantioselective construction of bisindolylmethane derivatives 34 via F–C reaction between indoles 32 and methylene indolinones 33 as α,β-unsaturated amide electrophilic partners.25 The synthetic strategy employed a tosylated bis(imidazolidine)pyridine (Ts-PyBidine)-Ni(OTf)2 complex as a catalyst system to provide the desired chiral bisindolylmethanes in high yields and enantioselectivities (up to 95% ee; Scheme 6b). The generality of the synthetic approach was found to be quite diverse as introduction of both electron-donating and electron-withdrawing functions at the C5-position of N(H)-indoles reacted smoothly, affording the corresponding bisindolylmethanes in excellent enantioselectivities (up to 95% ee). Nevertheless, the use of N-methylindole as a reaction partner reduced both yield (30%) and enantioselectivity (42% ee) of the reaction. Similarly, reactions between C5- and C6-substituted indolinones with N(H)-indoles also proceeded efficiently, albeit with lower enantioselectivities in case of the N(H)-indole substrate bearing electron-withdrawing substituents. Notably, reactions of N(H)-methylene indolinone or electrophilic substrates having a methoxycarbonyl function replaced with a phenyl group with N(H)-indole did not proceed well, and the desired products could not be detected. Moreover, the scope of the strategy was further extended by transforming the product into important synthetic derivatives, containing an asymmetric quaternary carbon center in a highly diastereoselective fashion.
Scheme 6. Asymmetric F–C Reaction of Indoles with (a) Benzylidene Malonates and (b) Methylene Indolinones.

The catalytic cycle for the reaction was proposed, as depicted in Figure 2. The reaction started off by the coordination of methylene indolinone with the Ts-PyBidine-Ni(OTf)2 complex to generate intermediate A. The intermediacy of A was confirmed by electrospray ionization mass spectrometry. It is noteworthy that the tosyl group on the imidazoline ligand played a significant role in increasing the acidity of the Ni complex, which, in turn, enhanced the reactivity of methylene indolinone. The asymmetric F–C reaction then proceeded to deliver complex B, which, in turn, underwent diastereoselective protonation to furnish the kinetic product 34b. Nevertheless, the kinetic product tends to epimerize easily over the silica column, producing the diastereomixture.
Figure 2.

Plausible reaction mechanism for the asymmetric synthesis of bisindolylmethanes.
Due to the importance of β-amino acids bearing indoles in bioactive compounds and their involvement as an important structural unit in many bioactive natural compounds, Beletskaya et al. reported an elegant method for the synthesis of versatile β3-tryptophan derivatives.26 The synthetic design was based on the F–C reaction between indoles 35 and phthaloyl-protected aminoethylenemalonate 36, catalyzed by chiral complex of the copper/iPrBox catalyst system, derived from Cu(II) triflate and ligand iPrBox L7. Under mild reaction conditions, the synthetic method furnishes the desired β3-tryptophan derivatives in excellent yields and high enantiomeric excess (up to 99% ee; Scheme 7). The phthaloyl group as the N-protection of aminomethylenemalonates’ electrophilic partner was crucial for the success of the reaction because other protecting groups such as BocNH, CbzNH, and AcNH either failed to proceed or rendered lower conversions. Installation of these protecting groups resulted in intramolecular H-bonding between NH and the carbonyl oxygen of ester group, making aminomethylenemalonate substrates less reactive. The generality of the approach was also tested, which revealed that diverse indoles bearing electron-donating and electron-withdrawing groups at C1–C6-positions participated smoothly to yield the desired products in high yields and excellent enantioselectivities. However, reaction of 2-phenylindole failed to proceed, presumably due to steric demand. Likewise, the less bulky methyl substituent at the C2-position delivered the target product in excellent yield (99%), albeit with moderate enantioselectivity (76% ee). Similarly, the N-methylindole delivered the target product in good yield (83%) but in low enantioselectivity (74% ee). This observation suggested that the NH proton of indole plays a significant role in the enhancement of enantioinduction, which was in line with previously published reports.27
Scheme 7. Asymmetric F–C Reaction of Indoles with Phthalimidomethylenemalonate.

Based on X-ray analysis, the absolute configuration of chiral center in one of the alkylated products was found to be R. The configurations of other products were tentatively assigned accordingly. A stereochemical model for the stereochemical outcome of the reaction was proposed (Scheme 7, bottom). The intermediate, complex of phthalimidomethylenemalonate 36/Cu(OTf)2/L7, was initially formed, and the indole nucleophile then attacked the activated phthalimide function from the Si-face.
2.3. Reaction with β,γ-Unsaturated α-Keto- and α-Ketiminoesters
The β,γ-unsaturated α-keto- and α-ketiminoesters are diverse electrophilic reactants that are successfully employed in many enantioselective F–C alkylations of indoles.28 In 2014, Zhang et al. disclosed the reversal of enantioselective F–C of indoles by slightly tuning the amide units of chiral ligands L-RaPr2L11 and L-RamBu2L12, derived from reacting L-ramipril with 2,6-diisopropylaniline and 3,5-ditertbutylaniline, respectively.29 The F–C alkylation between indoles 38 and β,γ-unsaturated α-ketoesters 39, catalyzed by catalyst systems based on the Ni(II) complexes of chiral ligands (L-RaPr2-Ni(OTf)2) L11 and (L-RamBu2-Ni(OTf)2) L12, afforded the corresponding alkylated products (40) in moderate to high reversed or unreversed enantioselectivities (up to 95% ee) and high yields (Scheme 8a). The reaction of 2-methylindole proceeded smoothly with both catalyst systems, rendering the corresponding alkylated products in high yields and reversed enantioselectivities. Similarly, reactions of indole or 2-phenyl indole as substrates catalyzed by the L-RaPr2-Ni(OTf)2 complex catalytic system occurred favorably, delivering the corresponding products in high yields and enantioselectivities. Nevertheless, moderate reversed or unreversed enantioselectivity was observed when L-RamBu2-Ni(OTf)2 complex L12 was used as a catalyst. Recently, Antilla et al. reported a F–C alkylation/N-hemiacetalization cascade process of aminoindoles to produce functionalized 1,7-annulated indole scaffolds, which are frequently found in numerous pharmaceuticals and natural products.30 The chiral magnesium bis(phosphate) complex L13 catalyzed reactions between indoles 41 and β,γ-unsaturated α-ketoesters 42, which served as a dielectrophile, and underwent a F–C alkylation/N-hemiacetalization cascade reaction to afford the desired annulated indoles 43 in high yield (up to 98%) and enantiomeric excess (up to 99% ee). The magnesium phosphate was found to be a better promoter compared to calcium phosphate. Moreover, 10 mol % of catalyst loading was needed for the improved enantioselectivity. The generality of the approach was evaluated under optimized reaction conditions. The method was quite diverse as the reaction between 4-aminoindole and diverse β,γ-unsaturated α-ketoesters, bearing a wide range of electron-donating or electron-withdrawing functions at the ortho-, meta-, and para-positions on the aryl ring, proceeded smoothly, affording the corresponding chiral alkylated products with similar yields and stereoselectivities. Likewise, reaction of different C2-, C3-, and N-substituted 4-aminoindoles with phenyl β,γ-unsaturated α-ketoester also occurred smoothly, providing the desired product in high yield and good to excellent enantio- and diastereoselectivities (Scheme 8b).
Scheme 8. Asymmetric F–C Reaction of (a) Indoles with β,γ-Unsaturated α-Ketoesters and (b) Aminoindoles with β,γ-Unsaturated α-Ketoesters.

The study of mechanistic insights suggested that the NH proton of indole played a significant role in the production of desired alkylated product since N-methylated indole failed to produce the desired C7-functionalized product. Moreover, on the basis of previous reports and experimental outcomes,31 a dual activation mode in the active transition state was proposed (Scheme 8b, bottom). Activation of the dicarbonyl function of β,γ-unsaturated α-ketoester by Lewis acidic Mg2+ and concomitant activation of 4-aminoindole by P=O through H-bonding in a cooperative manner, followed by the attack of indole on the activated C=C bond from Re-face in a tight chiral pocket, produces the 1,4-adduct, which spontaneously undergoes N-hemiacetalization to deliver the corresponding annulated indole. The 3,3-disubstituted 2-oxindole skeleton is an important structural motif frequently found in numerous natural products and several drug candidates. Wang et al. reported the synthesis of chiral 3,3-disubstituted oxindoles 45 by the F–C reaction between indole 27 and isatin-derived β,γ-unsaturated α-ketoester 44.32 Under the catalysis of the chiral Cu(II)/L14 complex, the synthetic approach provided chiral 2-oxindoles 45, with an all-carbon quaternary chiral center, in high yields and remarkable enantiocontrol (up to >99%; Scheme 9). Under the optimized reaction conditions, a wide range of N-protected α-ketoesters or substituted 2-oxindoles bearing either electron-donating or electron-withdrawing functions at the C5- and C7-positions in the aromatic ring efficiently provided the corresponding products in high yields and excellent enantioselectivities.
Scheme 9. Enantioselective F–C Reaction of Indole with β,γ-Unsaturated α-Ketoester.

Given the importance of 2-substituted indoles as a structural framework present in many alkaloids and bioactive compounds, Zhou et al. developed a mild synthetic protocol for the preparation of C2-functionalized indole derivatives 48 by F–C reaction between 3-substituted indoles 47 and β,γ-unsaturated α-ketiminoesters 46, catalyzed by CPA L15 (Scheme 10).33 In view of the highly efficient activation of imines by CPA catalysts, β,γ-unsaturated α-ketiminoesters 46 were envisaged as electrophilic coupling partners to deliver the corresponding C2-functionalized indole scaffolds 48, possessing α-ketiminoester motifs in fair to good yields and excellent enantiomeric excess. The generality of the methodology was investigated, which revealed that either electron-donating or electron-withdrawing groups in the aromatic ring of β,γ-unsaturated α-ketiminoesters 46 were well-tolerated, producing the corresponding alkylated products in high enantioselectivities (up to 99% ee). Similarly, screening the scope of 3-substituted indole suggested that the electron-donating groups in the phenyl ring afforded the corresponding products in high yields and enantioselectivities. Nevertheless, the presence of electron-withdrawing substituents in the indole substrate failed to produce the desired alkylated products. In addition, F–C alkylation between β,γ-unsaturated α-ketiminoesters 46 and simple indole produced the corresponding C3-alkylated product in a high yield but with poor stereoselectivity. The disparity in the stereochemical outcomes observed in two different reactions of 3-substituted indoles and indole with the same electrophilic partner, β,γ-unsaturated α-ketiminoester, hinted at the involvement of two plausible transition states in the reaction (Scheme 10, bottom).34 As illustrated, in both cases, the CPA catalyst synergistically activates both the reaction partners. However, in case of 3-substituted indoles, the NH activation through H-bonding by the catalyst generates a H-bonding closer to the C2-position of indole, which, in turn, results in spatial arrangement that favors a nucleophilic attack from one face of the activated electrophilic partner, resulting in higher stereoselectivity. On the other hand, activation of a simple indole substrate through H-bonding between the indole NH and oxygen of the catalyst turned out to be relatively distant from the C3-position. This, in turn, causes poor facial discrimination by the unsubstituted indole nucleophile (Scheme 10, bottom).
Scheme 10. Enantioselective F–C Reaction of C3-Substituted Indoles with β,γ-Unsaturated α-Ketiminoesters.

On the basis of the above hypothesis, the same research group devised a synthetic methodology based on CPA-catalyzed C3 functionalization of indoles with β,γ-unsaturated α-ketiminoesters 46 to achieve the corresponding indole derivatives 50 with improved stereoselectivity.34 In their approach, an NH function was introduced at the C4-position of indole in anticipation that the activation of the indole substrate by a catalyst would produce H-bonding closer to the C3-position, which, in turn, may create a spatial arrangement that would favor enhanced facial discrimination by the indole nucleophile (Scheme 11a, bottom). The proposed hypothesis was then verified by a CPA-catalyzed F–C reaction of tert-butyl 1H-indol-4-ylcarbamate 49 with β,γ-unsaturated α-ketiminoesters 46. The method improved the stereochemical outcome of the desired product. A wide range of β,γ-unsaturated α-ketiminoesters 46 that contain electron-donating and electron-withdrawing groups at different positions of the aromatic ring reacted smoothly to provide the corresponding functionalized indoles 50 in moderate to high yields (up to 86%) and high enantioselectivities (up to 94% ee; Scheme 11a). The mechanistic insights of the reaction were further explored by DFT studies, which was in line with the observed experimental stereochemical outcomes. The computed relative energy difference of transition states of reactants in the presence of the CPA catalyst revealed that the triple hydrogen-bonded complex, A-TS-Re, was found to be 2.7 kcal/mol lower in energy than that of the A-TS-Si counterpart, thus favoring the nucleophilic attack from the Re-face of the activated electrophilic partner (Scheme 11a, bottom). Due to the improved stereoselectivity in F–C reactions of 4-aminoindoles reported previously, Zhao et al. envisaged a highly regio- and enantioselective F–C alkylation of indole at the C7-position.35 The synthetic method employs incorporating para-directing function at C4 of the indole, which enabled regioselective alkylation at the C7-position in the presence of a C3 nucleophilic site. The CPA-catalyzed reaction between sterically hindered N-benzyl-1H-indol-4-amine 52 and β,γ-unsaturated α-ketiminoesters 51 provided the corresponding C7-functionalized chiral indoles in high yields (up to 97%) and moderate to excellent enantioselectivities (up to 99% ee; Scheme 11b). With respect to the substrate scope of β,γ-unsaturated α-ketiminoesters, aromatic substrates with diverse substitution patterns of different electronic nature in the aromatic ring reacted smoothly to afford the corresponding alkylated products in high yields and excellent stereoselectivities.
Scheme 11. Enantioselective F–C Reaction of Aminoindoles with β,γ-Unsaturated α-Ketiminoesters.

2.4. Reactions with Nitroalkenes
The enantioselective F–C alkylation of indoles with nitroalkenes is regarded as a fundamental transformation in the construction of functionalized indoles. The atom-economical process increases molecular complexity through the construction of carbon–carbon bonds, providing access to important pharmaceutical and biologically relevant intermediates.36 Moreover, the process offers a wide application scope and synthetic versatility since the nitro function in the alkylated product can be easily manipulated into important functionalities.37 Consequently, numerous protocols have been designed to realize catalytic F–C reaction of indoles with nitroalkenes. Among them, organocatalysis has particularly emerged as a powerful synthetic strategy.38 Hirata and Yamanaka performed DFT calculations to investigate the reaction mechanism and the origin of high stereoselectivity of the 3,3′-substituted (R)-BINOL-derived CPA L15-catalyzed F–C reaction between nitroalkenes 55 and indoles 54 (Scheme 12).39 The bifunctional activation of both 54 and 55 by the catalyst through H-bonding furnishes a two-point binding cyclic transition structure (TS-1), which results in placing both substrates closer to the C2-symmetric reaction space which ultimately leads to asymmetric induction. The sterically demanding bulky SiPh3 group at the 3,3′-position of CPA L15 was crucial for achieving higher enantioselectivity. For example, CPA L15 rendered higher enantioselectivity (91% ee) compared to that when the substituents at 3,3′-positions were replaced with less bulky 9-anthryl groups (91% ee). The bulkier SiPh3 group reduces the C2-symmetric reaction space, resulting in an increase of the energy difference between the transition states, which possibly could have yielded the S and R products, and consequently enhances the stereoselectivity (TS-2, Scheme 12, bottom).
Scheme 12. Enantioselective F–C Reaction of Indoles with Nitrostyrenes.

Trifluoromethyl function is known to modify the reactivity, bioactivity, and stability of organic compounds. As a result, the synthesis of trifluoromethyl compounds has received a great deal of attention in pharmaceutical, agricultural, and materials science. Jia et al. developed a synthetic method for the production of functionalized indoles 59 with a trifluoromethylated all-carbon quaternary stereocenter.40 The synthetic strategy was based on employing β-CF3-β-substituted nitrostyrenes 58 as electrophilic partners in the F–C reaction with indoles 57, catalyzed by the Ni(ClO4)2-chiral BOX L17 complex catalyst system. The incorporation of a CF3 moiety at the β-position in nitrostyrene was expected to increase the reactivity of the electrophilic partner due to the electron-withdrawing ability of the CF3 function. The reaction provided access to corresponding functionalized indoles 59 in good yields (up to 95%) and high to excellent enantioselectivities (up to 96% ee; Scheme 13a). The generality of the reaction was quite diverse since either electron-donating or electron-withdrawing moieties in the phenyl ring of indoles or aryl rings of the nitrostyrenes reacted smoothly to provide the desired products in high yields and good to excellent enantioselectivities. Nevertheless, a nitrostyrene substrate bearing ortho-substituent in the aryl ring failed to produce any desired product. Likewise, 1-Me- and 2-Me-indoles turned out to be inferior substrates, yielding the corresponding products in either poor yields or low enantioselectivities. In view of the remarkable reactivities, the synthetic utility of β-CF3-β-substituted nitrostyrenes 61 was extended to F–C alkylation with 4,7-dihydroindoles 60 to produce C2-substituted indoles 62, bearing a trifluoromethylated all-carbon quaternary stereocenter.41 The Ni(ClO4)2-chiral BOX L17 complex-catalyzed reaction initially furnished the corresponding dihydroindoles in good to high yields (up to 95%) and high enantioselectivities (up to 91% ee; Scheme 13b). The oxidation of the resulting alkylated products with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) then afforded the corresponding C2-alkylated indoles 62 without a significant loss in enantiomeric purity. Recently, Akiyama et al. employed a BINOL-derived calcium CPA complex as a Lewis acid catalyst for an efficient enantioselective F–C alkylation between indoles 63 and α-CF3-substituted β-nitrostyrenes 64 to generate the corresponding indoles 65, bearing an all-carbon quaternary stereocenter.42 Under optimized reaction conditions, a wide range of substituted nitrostyrenes having electron-donating, electron-withdrawing, or heteroaromatic functions reacted smoothly with C6- and C7-substituted indoles, bearing diverse substituents, irrespective of their electron nature, to afford the corresponding alkylated products in high yields (up to 99%) and excellent enantioselectivities (up to 98% ee; Scheme 13c). The calcium CPA complex-based catalyst system was required for the progress of the transformation since the CPA catalyst alone could not promote the reaction. The lack of reactivity of the CPA catalyst was linked to the lower basicity of the electrophilic partner due to the presence of the CF3 group, which, in turn, impeded its effective coordination ability with the catalyst. On the other hand, metal phosphates were thought to create a better chiral environment. Based on DFT calculations, the origin of high stereocontrol was proposed. The calcium CPA complex acts as a bifunctional catalyst, activating both nitrostyrene and indole through the bidentate coordination with the calcium (Lewis acidic site) and H-bonding with phosphoryl oxygen, respectively. The attack of an incoming indole nucleophile in a chiral pocket occurs from the Re-face, affording the corresponding product in S-configuration.
Scheme 13. Enantioselective F–C Reaction of (a) Indoles with β-CF3-β-Aryl Nitrostyrenes, (b) 4,7-Dihydroindoles with β-CF3-β-Aryl Nitrostyrenes, and (c) Indoles with β-CF3-β-Alkyl Nitrostyrenes.

The same research group previously reported a synthetic approach based on CPA L5-catalyzed enantioselective F–C reaction of indoles 66 with β-alkoxycarbonyl-β-disubstituted nitroalkenes 67 in the creation of functionalized indoles 68 that contain all-carbon quaternary centers.43 The key feature of the synthetic strategy was the use of nitroalkene, containing an ester group at the β-position, which not only made the electrophilic partner more reactive but also allowed further chemical manipulations of the resulting F–C alkylated product. The scope of the reaction was quite diverse as a wide range of nitrostyrenes bearing substituents in the aryl ring, irrespective of their electronic nature, and indoles with diverse functions including neutral, electron-donating, or electron-withdrawing groups reacted smoothly to afford the corresponding alkylated products in good to high yields (up to 88%) and high to excellent enantioselectivities (up to 94% ee; Scheme 14a). The synthetic utility of the synthetic method was validated by transforming the corresponding alkylated products into synthetically important structural frameworks. Inspired by the remarkable performance of bifunctional thiourea catalysts, Fan and Kass designed a charge-containing derivative of privileged Schreiner’s thiourea catalyst L19 that contains a cationic N-methylpyridinium ion center with a noncoordinating anionic counterion and chiral 2-indanol as a substituent.36 The design of catalyst L19 was based on the fact that, compared to Schreiner’s thiourea, charge-containing derivatives of the thiourea catalyst are known to significantly increase the rate of reactions of several organic transformations.44 The catalytic performance of synthesized catalyst L19 was then evaluated in a F–C reaction between a series of indoles 69 and trans-β-nitrostyrene 70. The reaction provided the corresponding alkylated products in a good yield and a high enantioselective ratio (up to 95:5 er; Scheme 14b). Although substituents on the aryl ring of nitrostyrene had almost little or no effect on the rate of the reaction, electron-donating and electron-withdrawing substituents on the indole substrate led to an increase and decrease of the reaction’s reactivity, respectively. Under the reaction conditions, the mechanistic studies suggested a second-order transformation, hinting that the dimer of thiourea was the active catalyst species. In another study, Arai et al. reported a novel synthetic method for the preparation of highly substituted chiral indoles 74 based on the chiral complex of Ni(OTf)2/bis(imidazolidine)pyridine L20-catalyzed F–C reaction between 2-vinylindoles 72 and nitroalkenes 73.45 Under the optimized reaction conditions, a wide range of nitrostyrenes bearing different substituents in the aryl ring, irrespective of their electronic nature, reacted smoothly to provide the corresponding chiral 2-vinylindoles in good to high yields (up to 90%) and good enantioselectivities (up to 85% ee; Scheme 14c). The synthetic utility of the reported protocol was demonstrated through conversion of the obtained chiral indoles into useful chiral scaffolds for diverse possibilities in synthetic chemistry.
Scheme 14. Asymmetric F–C Alkylation of Indoles with (a) Disubstituted Nitroalkanes and (b,c) Monosubstituted Nitroalkanes.

Squaramide-based catalysts, bearing a conformationally rigid cyclobutene ring, contain a stronger H-bonding unit and a tertiary amine function in its structure. The remarkable activation potential of these catalysts lies in their stronger hydrogen bond formation ability through NH H-bonding donors. Their stronger H-bonding ability arises due to the concomitant enhancement in the aromatic character of a four-membered ring that exists in their structural architecture. As a result, the use of squaramide-derived bifunctional catalyst has been on the rise in diverse asymmetric organocatalytic transformations. Xu et al. reported an elegant strategy for the asymmetric functionalization of indole in the phenyl ring by reacting indoles 75 with (E)-2-nitroallylic acetate 76 under the catalysis of bifunctional squaramide catalyst L21.46 In general, enantioselective functionalization in the phenyl ring of indoles is relatively more challenging compared to that in the azole ring. Therefore, functionalization of indoles in the carbocyclic ring is less frequently studied. The switchable, regiodivergent synthetic methodology was based on installing a hydroxy function at the C4-position of the indole, which, in turn, served as a directing/activating group to achieve functionalization at the C5-position in preference over the C3-position. The synthetic approach provided access to a wide range of enantiopure tetrahydropyranoindoles 75 through a cascade process, involving asymmetric F–C alkylation at the C5-position of the indole followed by oxa-Michael cyclization (Scheme 15a). The preferential functionalization at the C5-position over the C3-position was believed to be due to the less Gibbs free energy requirement suggested by control experiments and thermodynamic calculations. The scope of the reaction was quite diverse as the reaction was not affected by the presence of either a bulkier substituent or electron-withdrawing and electron-donating groups in the allylic acetate substrates, generating the corresponding products in good yields (up to 98%) and high to excellent diastereoselectivities (up to >20:1 dr) and enantioselectivities (up to >99% ee). Moreover, the reaction also occurred smoothly with heterocyclic-substituted 2-nitroallyl acetates. Later, Pedro et al. also exploited activating/directing effects of the hydroxy function in the phenyl ring of the indole and reported the enantioselective F–C alkylation in the phenyl ring to produce regioisomeric indoles.47 The bifunctional squaramide L22-catalyzed F–C alkylation between hydroxyindoles 78 and nitrostyrenes 79 provided the corresponding chiral nitroalkylated indoles 80 in high regio- and enantioselectivities (up to 99% ee; Scheme 15b). In the case of 4-hydroxyindoles, in addition to the generation of desired C5-alkylated indole product, formation of minor C5–C7-dialkylated and/or C7-alkylated side products was also observed. Nevertheless, appropriate choice of solvent (CHCl3) and catalyst loading (2 mol %) led to enhanced regioselectivity, affording the desired C5-alkylated product in good yields (up to 80%) and good to high enantioselectivities (up to 92% ee). Moreover, functionalization of 5-hydroxyindoles in the organocatalytic F–C reaction with β-nitrostyrenes was also examined. The reaction with a wide range of β-aryl- and β-heteroarylnitroalkenes occurred smoothly to afford the corresponding C4-alkylated products in high yields (up to 98%) and better enantioselectivities (up to 95% ee) than those of the 4-hydroxyindole counterpart. The synthetic protocol was also extended to the F–C alkylation of 6-hydroxyindoles, providing the corresponding C7-alkylated products in good yields (up to 92%) and excellent enantioselectivities (up to 94% ee). Nevertheless, 6-hydroxyindoles were proven to be less reactive than their C4- and C5-hydroxyindole counterparts. Finally, the F–C alkylation of 7-hydroxyindole unfortunately led to the formation of a mixture of C6- and C4-alkylated products, in addition to C4–C6-dialkylated product, in poor regio- and enantioselectivity. The interference between the NH of indole and the C7 hydroxyl group was thought to be responsible for such poor regio- and stereochemical outcomes. A tentative transition state was proposed in order to rationalize the observed regio- and enantioselectivity (Scheme 15b, bottom). The cooperative activation of nitro and hydroxyl groups through H-bonding by squaramide and the tertiary amine group of the catalyst, respectively, favors the attack of the indole nucleophile from the Si-face of activated nitrostyrene to produce S-configured products. Taking advantage of the activating/directing effects of the hydroxy function in the phenyl ring of indole, Pedro et al. developed a synthetic protocol for F–C alkylation of 4-hydroxyindoles 81 with nitroenyne 82, catalyzed by Rawal’s chiral squaramide L23 (Scheme 15c).7f After careful optimization of the reaction conditions, the desired C5-alkylated indole was obtained, together with C5–C7-dialkylated and C7-alkylated side products in a 13:3:1 ratio in good yield (78%) and excellent enantioselectivity (up to 96% ee). However, the use of 5-hydroxyindole as an alkylating partner resulted in the exclusive formation of C4-alkylated indole in high efficiency (94% yield, 96% ee). Unfortunately, C7-hydroxyindole under the given conditions yielded a complex mixture of products. Finally, the substrate scope of nitroenyne suggested that both phenyl and para-chlorophenyl substituents were compatible with the catalyst system to efficiently afford the corresponding C4- and C5-alkylated indoles.
Scheme 15. Asymmetric F–C Alkylation in the Phenyl Ring of (a) Hydroxyindoles and (b,c) Substituted Indoles.

Li et al. recently developed an elegant protocol for the synthesis of optically active dihydrofuranoindoles 86 due to its appealing bioactive properties and high pharmaceutical value.48 The bifunctional squaramide L24-catalyzed F–C alkylation/annulation cascade process between 1,3-dinucleophilic hydroxyindoles 85 and dielectrophilic β,β-bromonitrostyrenes 84 provided the corresponding fused indole products 86 in good to high yields (up to 95%) and high to excellent enantioselectivities (Scheme 16). The use of K2CO3 as a basic additive was crucial for the higher conversion rate and enantioselectivity of the reaction. The experimental evidence suggested that hydrobromic acid produced in the aftermath of the alkylation step in the domino process protonated the basic site of the catalyst, which deteriorated its catalytic activity. The generality of the reaction suggested that the position and nature of substitutions in the aryl ring of nitrostyrenes, irrespective of their electronic nature, had no or very little effect on the progress of the cascade process. The synthetic approach provides dihydrofuranoindoles exclusively as trans-diastereomers (up to 99% ee). The synthetic utility of the protocol was demonstrated by performing the domino reaction on a gram scale, followed by further synthetic manipulation of the functionalities in the dihydrofuranoindole product.
Scheme 16. Asymmetric Synthesis of Dihydrofuranoindole via Domino F–C Alkylation/Annulation.

A plausible mechanism for the domino process was proposed, as illustrated in Figure 3. The cooperative double H-bonding activation of the nitrostyrene substrate through H-bonding between the squaramide moiety of the catalyst and nitro group of nitroalkene 84 and concurrent deprotonation of phenolic hydroxyl group of indole by quinuclidine nitrogen of the catalyst from the bottom face permitted the attack of incoming indole nucleophile on the activated nitrostyrene from the Si-face. The subsequent annulation through the displacement of bromine by a phenoxide anion under a thermodynamically controlled process then finally furnished the desired dihydrofuranoindoles exclusively as trans-diastereomers (Figure 3).
Figure 3.

Proposed mechanism for the product formation and stereoselectivity.
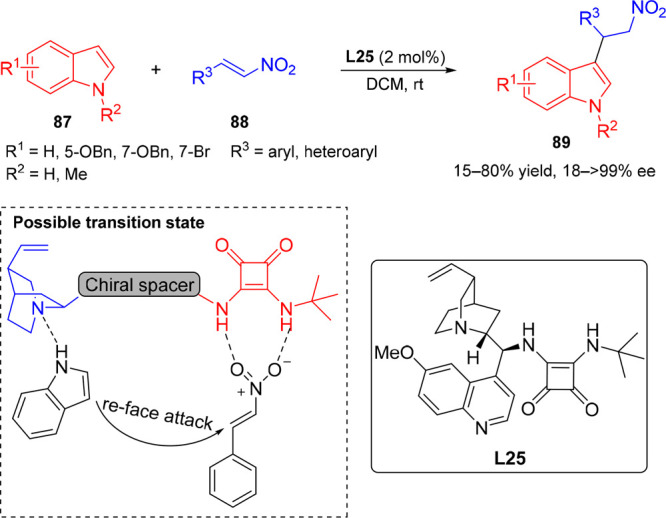
Recently, Tanyeli et al. reported bifunctional squaramide L25-catalyzed F–C alkylation between indoles 87 and nitrostyrenes 88 in the construction 3-substituted indole derivatives 89 under milder reaction conditions.49 Under optimized conditions, the developed protocol could tolerate a wide range of both electron-donating and electron-withdrawing groups in the aryl rings of both indole and nitrostyrene, affording the corresponding products in good yields and low to excellent enantioselectivities (up to 92% ee) (Scheme 17). Based on the experimental studies, a plausible transition state model for the stereochemical outcome was proposed (Scheme 17, bottom). The reaction started from the activation of indole by the L25 catalyst through H-bonding between the NH group and quinuclidine nitrogen of the catalyst and concomitant cooperative double H-bonding activation of the nitro function of nitrostyrene, orienting the activated nitrostyrene in a chiral pocket in such a way that it facilitates the attack of the indole nucleophile from the Re-face to deliver the desired product.
Scheme 17. Enantioselective F–C Reaction of Indoles with trans-β-Nitrostyrene.
In another study, Rachwalski et al. synthesized chiral aziridine phosphines for the asymmetric F–C alkylation of indoles.50 The catalyst system based on the (CuOTf)2·benzene complex/L26 catalyzes the reaction between indoles (90) and β-nitrostyrene (91) in the presence of triethylamine to afford the corresponding products in moderate yields and good enantioselectivities (up to 92% ee; Scheme 18). The proposed mechanism suggests formation of an orthogonal system between the catalyst and substrates. Creation of an orthogonal complex reduces the steric hindrance between the isopropyl-containing ring of catalyst and the phenyl ring of indole. Moreover, the quasi-trans orientation of the phenyl substituent of styrene to indole with respect to the new bond being formed favors the attack of the indole nucleophile from the Si-face of activated nitrostyrene (Scheme 18, bottom).
Scheme 18. Enantioselective F–C Reaction of Indoles with Nitroolefins.

Recently, Shi et al. developed a spiro-fused BOX chiral ligand L27-based catalyst system, containing Ni(ClO4)2·6H2O/L27 complex, for the asymmetric Michael-type F–C reaction of indoles 93 with β-CF3-β-disubstituted nitroalkenes 94.51 The design of ligand L27 was based on incorporating rigid spirocyclic units, which limited its flexibility and the related complex (Ni(ClO4)2·6H2O/L27, which, in turn, allowed a better stereocontrol. Moreover, the C2 symmetry of ligand L27 limited the number of possible transition states, and their chiral binaphthyl side arms can create a deep chiral pocket, which, in turn, increases confinement of β-disubstituted nitroalkene 94 for enhanced enantioselectivity. The Ni(ClO4)2·6H2O/L27-catalyzed protocol offers easy access to functionalized indoles 95, bearing a trifluoromethylated all-carbon quaternary center in moderate to high yields and high enantiomeric excess (up to >99.9% ee; Scheme 19a). The generality of the reaction was quite general since C5- and C6-substituted indoles bearing electron-donating or electron-withdrawing groups occurred smoothly to afford the corresponding indole derivatives in high yields and enantioselectivities (up to >99% ee; Scheme 19a). Nevertheless, C5-substituted indoles bearing electron-donating groups were proven to be superior in both reactivity and enantioselectivity compared to indoles containing electron-withdrawing functions. Moreover, C7-substituted indoles, bearing 7-OMe or 7-Me groups, were also compatible with the catalytic system, delivering the desired products in high enantioselectivities albeit in low yields, presumably due to the steric hindrance. Similarly, substituted nitroalkenes containing electron-donating or electron-withdrawing groups at ortho- or meta-positions of the phenyl ring were also compatible, although substituted nitroalkenes containing electron-withdrawing groups were superior in delivering the corresponding products in higher enantioselectivities than were the substituted nitroalkenes bearing electron-donating functions. A plausible reaction pathway was suggested, indicating that the deep chiral pocket serving as a confined reaction space stabilizes β-disubstituted nitroalkene 94 through noncovalent interactions and favoring the attack of an incoming indole nucleophile from the Re-face of the β-disubstituted nitroalkene to afford the corresponding alkylated product in R-configuration. In 2021, Al-Majid et al. reported the synthesis of chiral 2,5-bis(oxazolinyl)thiophene ligands and studied their utility in the asymmetric F–C reaction of indoles 96 with β-nitroolefins 97.52 The methodology employs 15 mol % of the Cu(OTf)2/L28 complex catalyst system to provide access to the corresponding functionalized indoles 98 in low to moderate yields and poor to good enantiomeric excess (up to 81% ee; Scheme 19b). A wide range of nitroolefins containing diverse substituents on the aryl ring, irrespective of electronic nature, were well-tolerated. Nevertheless, bulkier substituents such as 4-CF3, 2,4-dichloro, 2-NO2, and 4-MeO in the aryl ring or 2-thienyl function impeded the rate of the reaction, resulting in comparatively lower yields (37–48%) of the corresponding products.
Scheme 19. Michael-Type F–C Reaction of Indoles with (a) β-CF3-β-Disubstituted Nitroalkene and (b) β-Nitroolefins.

A plausible reaction pathway for the transformation was proposed, as illustrated in Figure 4. Based on the experimental evidence, the attack of the incoming indole nucleophile on the activated nitrostyrene (intermediates II) favorably occurs from the sterically less hindered Si-face to deliver S-configured product.
Figure 4.

Plausible mechanism for the reaction of indoles with β-nitroolefins.
3. 1,2-Nucleophilic Addition to C=X Bond
3.1. Addition to C=N Bonds
Imines are versatile electrophiles and have been employed extensively in the enantioselective F–C reaction of indoles in the construction of functionalized chiral indole derivatives, which are potential intermediates in pharmaceutically active agents and biologically active natural products.13,53 Moreover, the synthesis of 3-indolyl-3-aminooxindoles has attracted a great deal of attention from the synthetic community due to their potential antimalarial and antitumor activities. Duan et al. recently disclosed an efficient strategy for the highly enantioselective synthesis of 3-indolyl-3-aminooxindoles 101 based on the aza-F–C reaction of isatin-derived ketimines 100 with indoles 99, catalyzed by a quinine-derived bifunctional phase-transfer catalyst.7d The synthetic methodology has a broader substrate scope since a wide range of N-Cbz-ketimines bearing diverse substituents at C5- and C6-positions, regardless of their electronic and steric nature, reacted smoothly with indole to provide the corresponding 3-indolyl-3-aminooxindoles bearing a tetrasubstituted stereocenter in high yields and moderate to high enantioselectivities (up to 94% ee; Scheme 20). Nevertheless, electron-donating substituents, such as a Me group, at the C7-position of N-Cbz-ketimines delivered the corresponding product in high enantioselectivity compared to that with substrates bearing electron-withdrawing substituents (7-Cl and 7-Br). Likewise, indoles bearing electron-donating groups at C4- and C5-positions provided the desired products in enantioselectivities higher than those of substituted indoles bearing electron-withdrawing group at the same positions. Based on control experiments, the quaternary ammonium center in the catalyst was important for the catalytic activity. The N-Cbz-ketimine activation through H-bonding by the catalyst and the electrostatic interaction of the nucleophilic indole anion with the quaternary ammonium center kept these substrates in a tight chiral pocket, resulting in high enantioselectivities.
Scheme 20. Enantioselective Aza-F–C Reaction of Substituted Indoles with Ketimines.

Due to the involvement of 2-substituted indoles in several alkaloids and bioactive compounds, Chen et al. disclosed a synthetic approach for the direct F–C C2-alkylation of 3-substituted indoles 102 with aldimines 103, employing highly acidic BINOL-derived chiral disulfonimide (DSI) L30 as a catalyst.10b The optimized protocol works efficiently for a broad scope of 3-substituted indoles and diverse aldimines to afford a variety chiral 2-indolyl methanamine derivatives 104 with excellent yield (up to 91%) and enantioselectivity (up to 98%; Scheme 21). Moreover, increasing the reaction temperature led to an increased enantioselectivity although with a low yield of the product. The synthetic utility of the method was demonstrated through gram-scale synthesis of various 2-indolyl methanamines. Based on control experiments, a plausible transition state for the reaction was proposed (Scheme 21). The cooperative activations of both substrates, indole and aldimine, by DSI L30 through H-bonding orient the attack of the indole from the Si-face of the active imine to afford the corresponding R-configured product.
Scheme 21. Asymmetric F–C C2-Alkylation of Indoles with Aldimines.

In another study, Fu et al. reported a catalyst system based on the Cu(OTf)2-BOX/L31 complex for the catalytic enantioselective F–C reaction of indoles 105 with N-sulfonyl aldimines 106 to produce chiral 3-indolylmethanamines 107.54 The structure of the ligand was crucial for achieving higher stereochemical outcomes. Moreover, the (S)-Bn-BOX was found to be superior to the (S)-i-Pr-BOX. The scope of the protocol allowed employing both electron-donating and electron-withdrawing groups at the C5- and C6-positions of indole. Likewise, N-sulfonyl aldimines bearing electron-withdrawing moieties in the aryl group were also compatible with the catalyst system, affording the corresponding alkylated products in high yields (up to 90%) and high excellent enantioselectivities (up to >99%; Scheme 22a). However, the reaction failed to produce the desired product when aldimine, containing a p-OMe group in the phenyl ring, was used as an electrophilic substrate. With a view to improve the catalytic performance of the CPA catalyst, Jiang et al. developed BINOL-derived double axial bisphosphorylimide L32 to improve the catalytic performance of CPA and employed in the F–C alkylation of indole 108 with aryl/alkyl N-tosyl imines 109.55 In the initial screening, reaction of equimolar amounts of indole with imine produced the corresponding chiral 3-indolylmethanamines 110 in 15 min, which was rapidly converted to the undesired bis(indolyl)methane product. The poor chemoselectivity was linked to the strong acidity of catalyst L32. Therefore, in order to improve the chemoselectivity and to suppress the formation of unwanted side product, the acidity of the catalyst was decreased by adding 0.2 mol % of 4-(dimethylamino)pyridine (DMAP) as an efficient additive. This, in turn, suppressed the reactivity of the catalyst and gratifyingly improved the selective formation of the desired chiral 3-indolylmethanamines and eliminated the formation of the undesired bis(indolyl)methane product, although slowing the rate of the reaction to 120 min. Under optimized reaction conditions, a wide range of diverse aryl imines, bearing various substituents in the phenyl ring, irrespective of their electronic nature, as well as alkyl imines reacted smoothly to generate the corresponding desired 3-indolylmethanamines 110 in high yields (up to 99%) and excellent enantioselectivities (up to >99%; Scheme 22b). The synthetic utility of the protocol was demonstrated by a gram-scale synthesis of 3-indolylmethanamines in enantiopure form and almost quantitative yields by employing aryl and alkyl imines as electrophilic reacting partners. In another report, Ishihara et al. disclosed the first report on the utility of low-reactive ketimines 112, lacking electron-withdrawing group functionalization, as electrophilic acceptors in the aza-F–C reaction with indoles 111 in the production of chiral 3-indolylmethanamines 113.56 The simple ketimines are less reactive due to their strong basicity, which can potentially neutralize the catalyst and subsequently deactivate it. To overcome this problem, a stronger Brønsted acid, chiral monopotassium binaphthyldisulfonate L33, was employed as a catalyst in the reaction. It is pertinent to mention that the initial screening studies with the corresponding chiral disulfonic acid of monopotassium L33 suggested that it was too strongly Brønsted acidic, which was thought to be due to the activation of one acid function by the other acid moiety that coexists within the catalyst. The stronger acidity of the chiral disulfonic acid catalyst thus led to the conversion of the desired chiral 3-indolylmethanamines into the undesired bis(indolyl)methane product. Therefore, monopotassium binaphthyldisulfonate L33, which had acidity substantially weaker than that of the corresponding chiral disulfonic acid counterpart, was employed in the reaction which consequently suppressed the generation of undesired bis(indolyl)methane side products. Under optimized conditions, various substituted N-Bn-indoles, bearing an electron-donating MeO group at the C5–C7-positions, reacted smoothly to provide the corresponding alkylated indoles in high enantioselectivity (up to 97% ee), although 4-MeO-substituted indole produced the desired product in low yield, presumably due to steric reasons. Examining the scope of low-reactive ketimines revealed that p-Me, p-OMe, p-F, p-Br, and p-I substituents in the aryl ring of ketimines as well as aliphatic ketimines were very compatible with catalyst system, affording the corresponding products in high yields (up to 99%) and excellent enantioselectivities (up to 97% ee; Scheme 22c).
Scheme 22. Enantioselective F–C Alkylation of Indoles with N-Tosylimines.

Due to the remarkable versatility of BINOL-derived CPA catalysts, Bolm et al. disclosed CPA L5-catalyzed F–C alkylation of indoles 114 with trifluoropyruvate-derived imines 115 to produce quaternary α-amino acids 116.57 The N-Boc-protected 3,3,3-trifluoropyruvate imine was proven to be the most efficient substrate, which reacted smoothly with a wide range of indoles, bearing numerous substituents at the C5-position with different electronic nature. Similarly, C6-substituted indoles bearing electron-withdrawing groups were also compatible with the catalyst system, generating the corresponding quaternary α-amino acids in excellent yields (up to 99%) and high to excellent enantiomeric ratios (up to 98:2; Scheme 23a). Akiyama et al. exploited the utility of CPA L5 toward the synthesis of optically pure 2-indolylmethylamines 118, bearing a trifluoromethyl function.58 The F–C reaction between 4,7-dihydroindole 60 with N-unprotected aryl trifluoromethyl ketimines 117, followed by oxidation of the resulting adducts with DDQ, produced the desired 2-indolylmethylamines 118. Under optimized reaction conditions, CPA L5-catalyzed reaction of 4,7-dihydroindole with a wide range of N-unprotected aryl trifluoromethyl ketimines, bearing substituents at C3- and C4-positions of the phenyl ring, regardless of their electronic nature, were well-tolerated to afford the corresponding products in good yields (up to >99%) and high enantioselectivities (up to 95% ee; Scheme 23b). Nevertheless, trifluoromethyl ketimine, bearing a 2-OMe substituent in the phenyl ring, rendered the product in very low enantioselectivity, probably due to the background reaction arising due to the activation of the imine function through H-bonding between the NH with methoxy group.
Scheme 23. Enantioselective F–C Alkylation of (a) Indoles with Trifluoropyruvate-Derived Imines and (b) 4,7-Dihydroindole with N-Unprotected Aryl Trifluoromethyl Ketimines.

The promising bioactivity of trifluoromethyldihydroquinazoline scaffolds has spurred significant interest in the synthesis of CF3-substituted tertiary carbinamines. Ma et al. disclosed the BINOL-derived CPA L5-catalyzed aza-F–C reaction of indoles 119 with cyclic N-acylketimines 120 to construct chiral trifluoromethyldihydroquinazolines 121 that contain tetrasubstituted carbon stereocenters bearing the nitrogen atom and CF3 group.59 Due to the role of 2,4,6-triisopropylphenyl as a substituent at 3,3′-positions of the catalyst, appropriate solvent selection (CH2ClCH2Cl) and optimum temperature (−35 °C) were crucial for the enhanced enantioselectivity of the products. Moreover, cyclic ketimines, bearing diverse substituents in the aryl ring, irrespective of their electronic nature, and substituted indoles having either electron-donating or electron-withdrawing groups at C5–C7-positions were well-tolerated to provide the corresponding products in high yields (up to 98%) and excellent enantioselectivities (up to >99% ee; Scheme 24a). In another report, Kim et al. employed cyclic N-sulfimines 123 as electrophilic acceptors in the CPA L34-catalyzed F–C reaction with indoles 122 to furnish the corresponding 3-indolyl sulfamidate scaffolds 124 in moderate to good yields and excellent enantioselectivities (up to 97% ee).60 Lowering the reaction temperature was crucial for the enhanced enantioselectivity of the products. Under optimized conditions, substituted indoles bearing substituents at C5–C7-positions, irrespective of their electronic or steric nature, could efficiently afford the corresponding products (Scheme 24b).
Scheme 24. Enantioselective F–C Alkylation of Indoles with (a) Cyclic N-Acylketimines and (b) Cyclic N-Sulfimines.

In another study, Nakamura et al. employed cyclic 4-aryl-3-oxo-1,2,5-thiadiazol-1,1-oxides 126, as cyclic ketimines, for the enantioselective aza-F–C reaction with indoles 125, catalyzed by imidazoline-CPA catalyst L35.61 Under the optimized conditions, reaction of diverse cyclic ketamines containing either electron-donating or electron-withdrawing groups in the aryl ring of imine reacted smoothly with substituted indoles, bearing either electron-donating or electron-withdrawing groups at C5–C7-positions, providing the corresponding functionalized indole derivative 127 in high yields (up to 99%) and excellent enantioselectivities (up to 99% ee; Scheme 25a). Moreover, cyclic ketimines having 2-naphthyl or 3-thienyl as an aryl function were also compatible with the synthetic protocol. Based on experimental evidence, a plausible transition state was proposed (Scheme 25a, bottom). The cooperative activation of both cyclic ketamine and indole by the bifunctional CPA catalyst through H-bonding in a chiral pocket force the incoming indole nucleophile to approach from the sterically less demanding Re-face of C=N to produce the R-configured stereoisomer in high enantioselectivity. In another report, Jia et al. disclosed a synthetic protocol for a direct enantioselective F–C C2-alkylation of indoles 128 with cyclic N-sulfonyl α-ketiminoesters 129 as electrophilic acceptors.7g For higher conversion and enhanced enantioselectivity, reaction conditions were optimized by considering the appropriate selection of Lewis acid, solvent, and chiral ligand. Moreover, the type of substituents on the oxazoline ring or the nature of the linker of two oxazoline units was important for improved enantioselectivity and a high product yield. Under the catalysis of the Zn(OTf)2/BOX L36 complex, a wide range of 3-methylindoles, bearing substituents at the C4–C6-positions, irrespective of their electronic nature, reacted smoothly with diverse cyclic N-sulfonyl α-ketiminoesters, bearing various substituents regardless of their electronic nature, at the C5-position to afford access to the corresponding products in good yields (up to 99%) and high enantioselectivities (up to 99% ee; Scheme 25b).
Scheme 25. Enantioselective F–C Alkylation of Indoles with (a) Cyclic Ketimines and (b) Cyclic N-Sulfonyl α-Ketiminoesters.

3.2. Addition to C=O Bonds
The asymmetric F–C alkylation of indoles with a carbonyl that functions as an electrophilic partner can lead to the generation of diverse functionalized indoles having a quaternary chiral carbon, which are privileged scaffolds frequently found in natural products and biologically active compounds. The increased utility and importance of chiral fluorinated compounds in pharmaceutical and agrochemicals prompted great interest in the catalytic F–C alkylation of indoles with trifluoromethyl ketones. Jia et al. disclosed a synthetic strategy for a direct enantioselective F–C alkylation of indoles 131 with trifluoropyruvates 132 to produce trifluoromethylated α-hydroxyesters 133.7g The screening of chiral ligands suggested that the role of substituents on the oxazoline ring or the nature of the linker of two oxazoline units was crucial for achieving enhanced enantioselectivity and a high yield of product. Under optimized conditions, the Cu(OTf)2/BOX L37 complex-catalyzed reaction of 3-methyl- or 3-phenyl indoles, bearing electron-donating or electron-withdrawing groups on the C5- or C6-position, with trifluoropyruvates having a methyl or ethyl ester function were well-tolerated in the catalyst system to generate the corresponding C2-alkylated products 133, bearing quaternary stereogenic centers, in good yields (up to 97%) and excellent enantioselectivities (up to 97% ee; Scheme 26a). Previously, Dong et al. utilized chiral squaramide catalyst L38 as an efficient catalyst for the enantioselective F–C reaction between trifluoropyruvates 135 and indoles 134.62 The use of a C3-symmetric chiral catalyst was advantageous due its high efficiency and stability. Moreover, the catalyst can be recycled due its poor solubility in organic solvents which, in turn, permitted its utilization in five repeated cycles, without losing efficiency and stereoselectivity. The scope of the protocol was diverse as numerous substituted indoles 134 bearing various substituents in the C4-, C5-, and C7-positions, irrespective of their electronic nature, reacted smoothly to provide access to the corresponding functionalized indole derivatives 136 in good yields (up to >99%) and high enantioselectivities (up to 99% ee; Scheme 26b). Recently, Wang et al. reported the enantioselective preparation of chiral trifluoromethylated indoles 139 by F–C reaction of indoles 137 with trifluoromethyl pyruvates 138, catalyzed by Trost’s dinuclear zinc complex L39 catalyst system.7a The binuclear zinc catalyst, derived from the reaction of chiral (S,S)-Trost’s ligand and 2 equiv of ZnEt2, was able to catalyze the reaction between an array of substituted indoles 137, possessing either electron-donating or electron-withdrawing groups at the C4–C7-positions, with trifluoromethyl pyruvates 138 as electrophilic partners to afford the corresponding trifluoromethylated indoles 139 in good yields (up to 95%) and enantiomeric excess (up to 88% ee; Scheme 26c). In order to develop a general synthetic method with broader substrate scope, Wolf and Zheng exploited asymmetric catalysis with chiral BOX ligands, employing the Cu(OTf)2-BOX L40 complex of as an efficient catalyst system for the F–C reaction between indoles 140 with alkyl trifluoropyruvates 141.63 Under the optimized conditions, the synthetic protocol could tolerate a wide range of indoles bearing either electron-donating or electron-withdrawing groups at the C5-position and a methyl substituent at the C1,C6–C7-positions to afford the corresponding alkylated products in high yields (up to 99%) and good enantioselectivities (up to 94% ee; Scheme 26d). However, the temperature of the reaction proved to be a critical factor and required optimization for each substrate individually.
Scheme 26. Asymmetric F–C Reaction of Indoles with (a–c) Trifluoropyruvates and (d) Alkyl Trifluoropyruvates.

In another study, Ma and Kass developed BINOL-derived chiral CPA catalyst L41, containing 3,3′-phosphonium ion substituents, and employed the F–C alkylation of indoles 143 with 2,2,2-trifluoromethyl aryl ketones 144 in the synthesis of chiral trifluoromethylated indoles 145.64 The synthetic protocol offers high tolerance to both substrates, i.e., indoles, bearing either electron-donating or electron-withdrawing substituents at the C5–C7-positions, as well as 2,2,2-trifluoroacetophenone possessing an electron-withdrawing halogen or CF3 group at the para-position of the aryl ring, providing the corresponding functionalized indoles 145 in high yields (up to 94%) and good enantioselectivities (up to 91% ee; Scheme 27a). The comparative enantioselectivity and reactivity of the charged CPA catalyst with the noncharged counterpart was also examined. The designed charged CPA catalyst was proven to be orders-of-magnitude more reactive than their noncharged analogues. Moreover, the positively charged ion centers were believed to be responsible for the higher reactivity and stereoselectivity of the charged catalyst. Recently, Zhao et al. exploited the activating/directing effects of the amino function at the C4-position of the phenyl ring of indoles 146 in the regioselective and enantioselective F–C alkylation with trifluoromethyl ketones 147 to construct C7-functionalized indoles.65 The SPINOL-derived spirocyclic phosphoric acid L42-catalyzed reaction provided access to diverse C7-functionalized indoles 148. The scope of the protocol was quite diverse since a wide range of trifluoromethyl ketones, including aromatic, heteroaromatic, and aliphatic trifluoromethyl ketones reacted smoothly under the optimized conditions, providing access to the corresponding C7-indolyl trifluoromethyl alcohols in high yields (up to 98%) and excellent enantioselectivities (up to >99% ee). Moreover, substituted aromatic trifluoromethyl ketones bearing electron-donating and electron-withdrawing groups at the para-position of the phenyl ring were also compatible. Nevertheless, relatively lower yields were observed in the case of substituted aromatic ketones, possessing electron-donating substituents as well as aliphatic trifluoromethyl ketones. The low yields were attributed to the decreased electrophilic character of these electrophilic partners. Similarly, substituted 4-aminoindoles bearing bulkier phenyl and isopropyl groups at the C2- and C3-positions also led to the generation of the corresponding products in lower yields (21 and 49%), although in high enantioselectivities (91 and 89% ee; Scheme 27b). However, reaction of unsubstituted 4-aminoindole retarded the progress of the reaction, due to the condensation of the amino function with the carbonyl group with an electrophilic partner, affording the desired product in trace amounts.
Scheme 27. Enantioselective F–C Reaction of Indoles with Trifluoromethyl Ketones at the (a) C3-Position and (b) C7-Position.

Based on experimental evidence and previous reports, a plausible transition state for the observed regioselectivity was proposed, as depicted in Figure 5. Activation of both substrates, indole and trifluoromethyl ketone, by the bifunctional L42 catalyst through cooperative H-bonding can lead to two possible pathways, TS-I and TS-II. However, steric repulsion in TS-I, due to the existence of a C4-amino moiety in closer proximity to the aryl function of trifluoromethyl ketone, makes it unfavorable. Therefore, the reaction proceeds through the more favorable TS-II pathway, affording C7-alkylated product.
Figure 5.

Plausible transition states for enantioselective F–C reaction of 4-aminoindoles with trifluoromethyl ketones.
Aldehydes are useful electrophiles commonly employed in F–C reactions. However, participation of aldehydes in the F–C reaction with indoles is quite challenging, due to the successive formation of bisindole byproduct, which originates from the expected hydroxyl product. Thus, hydroxyl alkylation of indoles via the F–C reaction was restricted only to ketones. In 2020, Tang et al. disclosed the first report of employing phenylglyoxal as an electrophilic partner for the enantioselective hydroxyl alkylation of indoles, catalyzed by CPA L43 (Scheme 28a).66 Previous attempts to use phenylglyoxal as an electrophilic substrate in the F–C reaction with indole led to the exclusive formation of bisindole product only.67 The generation of undesired bisindole byproduct was suppressed by installing a bulkier tert-butyl substituent at the C2-position of indole. Moreover, a low catalyst loading (0.1 mol %) was also employed in the reaction. The bulkier substituent at the C2-position of indole seemingly developed steric hindrance, and a low catalyst loading presumably inhibited the protonation of the hydroxyl function, thus suppressing subsequent elimination, which, in turn, impeded the production of a bisindole side reaction. Under optimized reaction conditions, phenylglyoxal bearing distinct substituents at the para- and meta-positions, irrespective of their electronic nature, and aromatic formyl aldehydes having thiophene, 1-naphthalenyl, and 2-naphthalenyl functions reacted smoothly to provide the corresponding α-hydroxyl ketones in high yields (up to 99%) and good enantioselectivities (up to 97% ee; (Scheme 28a). Nevertheless, a less reactive alkyl formyl aldehyde rendered the product in low yield and enantioselectivity. Screening the scope of the indole substrate suggested that a bulky substituent (tert-Bu) at the C2-position was critical for the higher enantioselectivity of the desired product since the less bulky substituent led a decrease in enantioselectivity. Based on experimental evidence, a plausible transition state (TS-I) for the observed stereoselectivity was proposed (Scheme 28, bottom). The cooperative activation of both substrates by bifunctional CPA catalyst through H-bonding in a chiral pocket force the incoming indole nucleophile to approach from Re-face of the activated arylglyoxal to produce S-configured stereoisomer in high enantioselectivity. In another report, Enders et al. exploited the catalytic potential of cinchona alkaloid-derived organocatalyst L44 for the enantioselective F–C reaction of indoles 152 with pyrazole-4,5-dione derivatives 153 as electrophilic partners.68 Under optimized reaction conditions, a wide range of indoles bearing substituents at N1-, C5-, and C7-positions, irrespective of their electronic nature, reacted smoothly with diverse pyrazolone substrates having different substituents on the N1- (R1 group) and C3-positions (R2 group), generating the corresponding adducts in high yields (up to 99%) and enantiomeric ratios (up to 94:6 er; Scheme 28b). Nevertheless, a sterically bulkier group at the C3-position (R2 group) marginally reduced the yield of the product.
Scheme 28. Enantioselective F–C Reaction of Indoles with (a) Aryl- and Alkylglyoxals and (b) Pyrazole-4,5-diones.

4. Reactions with Other Electrophilic Partners
4.1. Synthesis of Fused Cyclic Indole Derivatives
The synthesis of fused indole polycyclic skeletons has become increasingly important due to their widespread presence in many alkaloids and important organic compounds. Therefore, development of a straightforward strategy for the construction of indole polycyclic systems with skeletal and stereochemical diversity is of great significance.10c Although asymmetric F–C alkylation of indoles with alkenes has evolved as a useful technique for the asymmetric functionalization of indoles in recent years, the creation of quaternary stereogenic centers through hydroarylation of unactivated olefins remains elusive and a formidable task. Recently, List et al. developed an elegant atom-economical strategy for the organocatalytic enantioselective intramolecular hydroarylations of unactivated, electronically neutral olefins, with indoles 155.10a The unprecedented transformation of diverse olefins, catalyzed by strong and confined imidodiphosphorimidate (IDPi) Brønsted acid catalyst L45, delivered the corresponding tetrahydrocarbazoles 156 bearing quaternary stereogenic centers in high yields and excellent enantioselectivities (up to 94% ee; Scheme 29). The synthetic protocol has a broad substrate scope since alkyl boronate, an aliphatic iodide and azide, is well-tolerated and compatible, affording the corresponding hydroarylation products efficiently. Moreover, substituted indoles bearing both electron-donating and electron-withdrawing groups were well-tolerated and compatible with the catalyst system. The synthetic utility of the method was demonstrated by further transforming the hydroarylation products into bioactive molecules, without any decrease in enantiopurity.
Scheme 29. Asymmetric Intramolecular F–C Reaction of Indoles with Unactivated Olefins.
Based on controlled experiments, a plausible reaction mechanism was suggested, as illustrated in Figure 6. The reaction commenced by the protonation of olefin 155 to generate ion pair I. The attack of the indole nucleophile, governed by the chiral counteranion coordinated with an indole substrate through H-bonding, then leads to spirocyclization to generate a spiroindolenine cation that is associated with the IDPi anion (ion pair II). The subsequent stereoretentive rapid migration of the most electron-rich alkyl function then generates the corresponding tetrahydrocarbazole 156 and restoration of catalyst L45 upon proton transfer.
Figure 6.

Proposed reaction mechanism for the intramolecular F–C reaction of unactivated olefins with indoles.
Recently, You et al. developed an atom-economical approach based on the intramolecular asymmetric allylic alkylation of indoles 157 in the construction of tetrahydrocarbazoles 158, which are ubiquitous in natural products.69 The metal-free organocatalytic strategy was envisioned utilizing allylic primary alcohols as the electrophilic precursors. A highly acidic chiral Brønsted acid, imidodiphosphorimidate (IDPi) L46, was used as a catalyst, which helped to circumvent the issue of low reactivity of allylic primary alcohols. The generality of the reaction was quite diverse, as different malonate esters were well-tolerated, although an understandably sterically bulkier group decreases the enantioselectivity of the product. Likewise, under optimized conditions, indoles bearing diverse functionalities including electron-donating (e.g., Me, OMe, OBn) and electron-withdrawing groups (e.g., F, Cl, Br, I, CF3, CO2Me, and NO2) at C5- and C6-positions reacted smoothly to afford the corresponding desired products in good to excellent yields and enantioselectivities (up to 93% ee; Scheme 30). Nevertheless, the nucleophilicity of the indole bearing a strong electron-withdrawing group, for instance NO2, was greatly diminished, rendering the corresponding product in low yield (31%). Similarly, substituents at the C4- or C7-position were proven to be detrimental to the asymmetric induction, affording the desired products in low to moderate enantioselectivities.
Scheme 30. Enantioselective Synthesis of Tetrahydrocarbazoles via Intramolecular Asymmetric Allylic Alkylation of Indoles.

In another recent study, Wang et al. disclosed an elegant synthetic strategy for the highly enantioselective synthesis of azepino[3,4,5-cd]indole derivatives 161, which are important structural frameworks frequently found in pharmaceuticals and bioactive compounds.70 The synthetic method was based on the cooperative Cu/Ir catalysis-catalyzed asymmetric 1,3-dipolar (3 + 4) cycloaddition between azomethine ylides and ambiphilic π-allyl iridium species, which were generated in situ from aldimine esters 160 and 4-indolyl allylic carbonates 159, respectively. For improved stereochemical outcomes, the deployment of chiral Phosferrox ligand L47 and chiral phosphoramidite ligand L48 in generating a combined Cu(I)/(S,Sp)-L47/Ir(I)/(Sa,S,S)-L48 dual catalyst system was proven to be an optimum catalytic system to realize the desired transformation. Likewise, the addition of Zn(OTf)2 (0.5 equiv) as a promoter and p-chlorobenzaldehyde (two equivalents) for suppressing the undesired imine decomposition was needed for improved yield of the reaction. Through the operations of asymmetric allylic alkylation was followed by intramolecular F–C alkylation, the synthetic method provides access to an array of 3,4-fused tricyclic indoles bearing three stereogenic centers in high diastereo- and enantioselectivities (Scheme 31). The superiority of the stereodivergent method lies in the late-stage epimerization, which permitted precise creation of all eight stereoisomers of the azepino[3,4,5-cd]indole derivatives. In addition, screening of substrates scope suggested that a variety aromatic aldehyde-derived imine esters, containing diverse electron-donating or electron-withdrawing groups at the ortho-, para-, or meta-position of the phenyl ring, were well-tolerated in the catalytic system, affording the corresponding 3,4-fused tricyclic indoles in high diastereoselective and excellent enantioselectivities (up to 99% ee). Moreover, the 2-amino-γ-butyrolactone-derived aldimine ester also participated smoothly under the synergistic catalysis system.
Scheme 31. Stereoselective Synthesis of Chiral Azepino[3,4,5-cd]indoles.

Zhao et al. utilized activating/directing effects of the hydroxy group in the phenyl ring and disclosed an enantioselective F–C alkylation/lactonization cascade process by reacting methyleneoxindoles 162 with hydroxyindoles 163, catalyzed by bifunctional squaramide catalyst L21.6b The methodology was applicable to diverse indoles having a hydroxy group at the C4–C6-positions of the phenyl ring of indole, affording the corresponding pyrrolodihydrocoumarins in moderate yields (up to 99%) and high enantio- and diastereoselectivities (up to 99% ee and >20:1 dr; Scheme 32). Nevertheless, the reaction between 7-hydroxyindole and 3-trifluoroethyleneoxindole was shown to be sluggish, affording the corresponding product in low enantioselectivity (25% ee). This observation suggested that H-bonding activation by the quinuclidine nitrogen of the catalyst and the hydroxy group of indole was crucial for high enantioselectivity. The competitive H-bonding between the NH of indole and the C7 hydroxyl group was thought to be responsible for such a poor stereochemical outcome.
Scheme 32. Enantioselective F–C Reaction of Hydroxyindoles.

Recently, Feng et al. disclosed an elegant strategy for the enantioselective synthesis of dihydrocarbazoles by domino F–C alkylation/annulation of indoles 165 with diazoacetoacetate enones 166, catalyzed by dual metallic system of Rh(II)/chiral N,N′-dioxide-Sc(III) complex L49.71 The generality of the reaction was quite diverse since a wide range of substituted indoles bearing either electron-donating or electron-withdrawing groups at different positions of the phenyl ring reacted smoothly with a variety of diazoacetoacetate enones, having different ester groups, to afford the corresponding chiral dihydrocarbazoles in moderate yields and excellent enantioselectivities (up to 99% ee; Scheme 33). The controlled experiments suggested that NaBArF4 played the role of accelerating the initial addition step rather than the insertion step and acted as an acid to recycle the chiral scandium catalyst. Moreover, the dual metallic system Sc(III)/L49 and Rh(II) was needed for the annulation step in the cascade process. Moreover, the presence of Lewis acid Sc(OTf)3 facilitated increasing the electrophilicity of the Rh-carbenoid, generated from the F–C adduct through the activation of the β-ketoester function. The observed stereochemical outcomes were due to the attack of indole on the activated diazoacetoacetate enone, coordinated with the N,N′-dioxide-Sc(III) complex, from the sterically less hindered β-Re-face to generate S-configured F–C adduct. The latter then undergoes annulation to generate the spirocyclic intermediate, which, in turn, undergoes 1,2-migraton/isomerization to afford the corresponding dihydrocarbazoles.
Scheme 33. F–C Alkylation of Indoles with Diazoacetoacetate Enones.

Li et al. recently reported the enantioselective F–C alkylation of indole 27 with α-(3-isoindolinonyl) propargylic alcohols 168 to produce the corresponding C3-alkylated indoles 169.72 The key feature of the CPA L50-catalyzed synthetic approach was based on employing the 3-isoindolinonyl function as an auxiliary group in the propargylic alcohol substrate in order to transform it into a reactive intermediate, hence enabling its participation in the subsequent catalytic F–C alkylation. The synthetic strategy provided access to a variety of C3-functionalized indoles in high regio- and enantioselectivities (Scheme 34a). However, under the optimized conditions, reactions of 3-substituted indoles 170 with α-(3-isoindolinonyl) propargylic alcohols 168 proceeded with completely different regioselectivity, generating diverse spirocyclic heterocycles 171 in high enantioselectivities (up to >99% ee) (Scheme 34b). The sterically hindered 3-substituted indoles result in the 1,4-addition to the propargylic N-acylimine intermediate, leading to the allene intermediate, which undergoes protonation followed by cyclization to produce the spirocyclic heterocycles 171. The isolation of a key covalently bonded CPA adduct in the reaction suggested a covalent activation mode for the transformation. Acting as a precatalyst, the covalent phosphate ester is believed to produce propargylic N-acylimine with the regeneration of CPA L50 (Scheme 34c). The scope of the reaction was quite diverse since a variety of α-(3-isoindolinonyl) propargylic alcohols bearing various alkyne substituents reacted smoothly with either indole or 3-methylindole to afford the corresponding products in high yields and enantioselectivities.
Scheme 34. Enantioselective F–C Reaction of Indole with (a,b) α-(3-Isoindolinonyl) Propargylic Alcohols and (c) Mechanism of the Reaction between Indole and α-(3-Isoindolinonyl) Propargylic Alcohols.

Zhou et al. recently disclosed the Pictet–Spengler reaction on the benzene ring of indoles in the creation of polycyclic indole derivatives 174. The reaction between 2-(1H-indol-7-yl)anilines 172 with isatins 173 was catalyzed by the (R)-CPA L51 catalyst.10c Design of the synthetic strategy implies generation of ketimine from the condensation of 2-(1H-indol-7-yl)anilines 172 and isatins 173 followed by C6-selective enantioselective aza-F–C alkylation. A wide range of mono- or disubstituted indoles containing different functionalities at the C2- and C3-positions of the azole ring were compatible with the catalytic system, affording the corresponding polycyclic indole derivatives 174 in good to high yields and moderate to high enantioselectivities (up to 93% ee; Scheme 35). Based on control experiments, a plausible transition state was proposed to rationalize the observed stereochemistry of the products. The activation of both indole and ketimine substrates by the (R)-CPA L51 catalyst through cooperative H-bonding activations in a confined chiral cavity facilitated the attack of the indole from the Re-face of the C=N bond to deliver the S-configured product (Scheme 35, bottom).
Scheme 35. Synthesis of Polycyclic Indole Derivatives Bearing Spiro Quaternary Stereocenters.

4.2. Functionalization in the Azole Ring of Indole
Elegant synthetic methodologies have been recently developed for the enantioselective F–C reaction of indoles to construct C2- and C3-substituted indole motifs, which are potential intermediates in the synthesis of numerous alkaloids and medicinally relevant compounds.73 Masson et al. developed a synthetic protocol for the enantioselective aza-F–C reaction of indoles 175 with γ-hydroxy-γ-lactams 176, employing the CPA L52 ion pair catalyst system.74 Under optimized conditions, the scope of the reaction was quite diverse since a wide range of substituted indoles bearing either electron-donating or electron-withdrawing groups reacted smoothly with diverse hydroxylactams, containing Br or I at the para-position on the N-aryl group, providing the corresponding 5-indolylpyrrolidinones in good yields and high enantioselectivities (up to 98% ee; Scheme 36a). Based on experimental evidence, CPA L52 was believed to play the dual role of catalyzing the dehydration of hydroxylactam to produce a chiral ion pair and then controlling the face of attack through H-bonding with NH of the indole and phosphoryl oxygen. Li et al. disclosed an efficient strategy for the organocatalytic asymmetric F–C reaction between indoles 178 and indole-derived hydroxylactams 179 to produce chiral isoindolo-β-carboline derivatives 180.75 The CPA L53-catalyzed synthetic strategy could tolerate a wide range of substituted indoles containing electron-donating or electron-withdrawing groups at the C4–C6-positions and reacted smoothly with a series of substituted hydroxylactams that bore electron-donating or electron-withdrawing groups at different positions of the indole function, affording the corresponding products in low to excellent yields and moderate to very high enantiomeric excess (up to 99% ee; Scheme 36b). In the same year, Jia et al. disclosed a dual catalyst system based on Au/CPA L51 for the catalytic redox annulation of nitroalkynes 182 with indoles derivatives 181 to construct the corresponding indolin-3-ones 183.76 The dual catalyst system could efficiently tolerate diverse nitroalkynes bearing Cl, F, or Ph at the C4- or C5-position and substituted indoles possessing either electron-donating or electron-withdrawing groups at the C5- or C6-position to deliver the corresponding products, bearing quaternary stereocenters at the C2-position in good yields and excellent enantioselectivities (up to 96% ee; Scheme 36c). The AuCl3 (7 mol %)-catalyzed control experiment on 1-nitro-2-(phenylethynyl)benzene, in the absence of indole, resulted in the production of isatogen (nitrone). The subsequent addition of indole and then a catalytic amount of CPA L51 (10 mol %) along with molecular sieves led to the production of the corresponding indolin-3-one. These observations hinted that the process was based on a dual catalysis system of Au/CPA L51.
Scheme 36. Enantioselective F–C C3-Alkylation of Indoles with (a) γ-Hydroxy-γ-lactams, (b) Indole-Derived Hydroxylactams, and (c) Nitroalkynes.

In another study, Song et al. reported a synthetic strategy based on a cooperative cation-binding catalysis approach, employing a chiral oligoethylene glycol L54/KF catalyst system for a highly enantioselective F–C reaction between indoles 184 and α-amidosulfones 185, which was used as a synthetic equivalent for the in situ production of sensitive imine.77 Under the optimized conditions, a wide range of α-amidosulfones, containing aromatic functions with diverse substituents, regardless of their electron and steric nature, were well-tolerated. Likewise, α-amidosulfones bearing heteroaromatic and aliphatic substituents also reacted smoothly to afford the corresponding indolyl-1-alkylamine derivatives 186 in high yields and excellent enantioselectivities (up to 99% ee; Scheme 37a). Elaboration on the mechanistic insights suggested that complexation of KF with chiral oligoethylene glycol L54 was a key step for the in situ generation of a cation-binding catalyst system that contained a densely confined chiral space. Thereafter, the simultaneous activation of both imine and indole substrates in a close proximity, within the confined chiral space, delivers the corresponding product in a highly stereoselective fashion. Recently, Wang et al. developed a catalyst system based on visible light photoredox and chiral phosphate catalysts [Ir(dF(CF3)ppy)2(dtbbpy)](PF6)/L55 and demonstrated its utility in the F–C reaction of indoles 187 toward the synthesis of indolyl-1-alkylamine derivatives 189.78 The combined catalyst system was designed to facilitate in situ conversion of α-amino acid-derived redox-active esters (RAEs) to the corresponding N-acyl imines, which, in turn, participated as electrophilic partners in the subsequent F–C reaction with indoles. The reaction protocol tolerated a wide range of substituted indoles, bearing either electron-donating or electron-withdrawing groups at the C4–C7-positions, as well as nonaromatic amino acid and N-acarylglycine-derived RAEs to afford the corresponding products in high enantioselectivities (up to 97% ee; Scheme 37b). Based on control experiments, a plausible reaction mechanism was proposed, illustrated in Figure 7. The irradiated Ir*(III) was initially quenched by indole to produce the reductive Ir(II), which, in turn, provided single electron transfer (SET) to N-(acyloxy)phthalimide (188) to generate the corresponding Ir(III) and α-aminoalkyl radical, respectively. The conversion of Ir(III) to oxidative Ir*(III) progressed by blue light irradiation, which, in turn, oxidized the α-aminoalkyl radical to produce the corresponding protonated N-acyl imine. On the other hand, the cooperative activations of both N-acyl imine and indole substrates by a chiral bifunctional phosphate catalyst in a confined chiral environment led to the attack of the indole nucleophile from the Si-face of the activated imine to furnish the corresponding R-configured product.
Scheme 37. Enantioselective F–C Reaction of Indole with (a) α-Amidosulfones and (b) α-Amino Acid-Derived Redox-Active Esters.

Figure 7.

Proposed reaction mechanism for the asymmetric F–C reaction of indole with α-amino acid-derived RAEs.
Functionalization of enamides are attractive transformations in accessing chiral amine derivatives, which are valuable building blocks in pharmaceutical chemistry and for the construction of important organic molecules.79 Combining the visible light photoredox and chiral lithium phosphate L56 catalysis approach, the same research group disclosed the multicomponent dicarbofunctionalization of enamides 190 with RAEs 191 and indoles 192.80 The methodology features broad substrate scopes and mild reaction conditions. Under the optimum condition, a variety of substituted indoles bearing either electron-donating or electron-withdrawing groups in the C5–C7-positions were well-tolerated to afford highly functionalized chiral amine derivatives in moderate yields and good enantioselectivities (up to 96% ee; Scheme 38). Likewise, a wide range of RAEs such as N-(acyloxy)phthalimides, phenylacetyl acid-derived RAEs bearing different substituents in the phenyl ring, isobutyric acid and pivalic acid-derived RAEs, naphthyl, and hetereoaryl acetyl acid-derived RAEs were all compatible with the catalyst system, delivering the corresponding functionalized chiral amine derivatives 193 in good yields and moderate enantioselectivities. The mechanistic studies suggested that the reaction may proceed in two plausible pathways (A and B) (Figure 8). In path A, the dynamic assembly of enamide and RAE within the pocket of chiral lithium phosphate, which helps them aggregate through H-bonding, leads to the formation of charge-transfer complex (CTC) I, which, in turn, could be excited by either direct irradiation or Ru(II)-mediated energy transfer, resulting in an electron transfer from enamide to RAE and subsequent generation of intermediate II. In path B, enamide is first excited by either direct irradiation or energy transfer (ET) to the corresponding triplet excited state, which, in turn, undergoes dynamic assembly with RAE and chiral lithium phosphate to produce CTC II. The latter could generate intermediate II through ET, which, in turn, undergoes homolytic cleavage of the N–O bond followed by decarboxylation and radical recombination to generate intermediate III. The subsequent proton-transfer release of phthalimide and the leftover chiral iminium intermediate are then attacked by the indole through TS-1 to produce the functionalized chiral amine derivative and the release of the catalyst.
Scheme 38. Asymmetric Synthesis of Chiral C3-Alkylated Indole Scaffolds.

Figure 8.

Proposed reaction pathway for the formation of chiral indole derivatives.
Recently, Kim et al. developed a synthetic protocol based on the CPA L57-catalyzed F–C alkylation of indoles 195 with 3-indolyl sulfamidates 194 for the creation of bisindolylarylmethane derivatives 196 bearing a phenylsulfamate group.81 The existence of bisindolylarylmethane derivatives is widespread in numerous natural products and pharmaceutical agents. Under optimized conditions, the generality of the synthetic method could tolerate a wide range of 3-indolyl sulfamidates with diverse substituted indoles bearing electron-donating or electron-withdrawing groups at the C5–C7-positions, affording the corresponding of bisindolylarylmethane derivatives in good yields (up to 89%) and moderate to high enantioselectivities (88% ee; Scheme 39). Nevertheless, electron-withdrawing moieties in the phenyl ring of 3-indolyl sulfamidates afforded enantioselectivities better than those of their electron-donating counterparts. On the contrary, indoles with electron-donating substituents provided reaction yields and enantioselectivities slightly better than those of indoles containing electron-withdrawing groups. Moreover, the utility of synthesized compounds was examined through biological screening, which revealed their effectiveness in controlling the degeneration of peripheral nerve.
Scheme 39. Asymmetric F–C Alkylation of Indoles with 3-Indolyl Sulfamidates.

Based on experimental evidence and previous reports,82 a plausible mechanism for the reaction was proposed (Figure 9). The CPA catalyst L57 induces ring opening of 3-indolyl sulfamidates to produce benzylideneindolenine intermediate II, which, in turn, coordinates with a catalyst to transform into a stable ion pair III. The nucleophilic attack of N-methylindole on the ion pair III then generates sulfamate intermediate IV, which subsequently rearomatizes to yield the desired bisindolylmethane sulfamate product.
Figure 9.

Proposed reaction mechanism for the synthesis of bisindolylmethane sulfamate.
Chiral α-trifluoromethyl tertiary alcohols are important molecular frameworks frequently employed in the preparation of agrochemicals and pharmaceuticals.83 Yang et al. recently reported an efficient strategy based on the CPA L42-catalyzed F–C reaction of indoles 197 with benzothiazole-bearing trifluoromethyl ketone hydrates 198. The reaction produces α-trifluoromethyl tertiary alcohols 199, bearing benzothiazole and indole rings (Scheme 40).84 Under optimized conditions, a variety of substituted indoles bearing either electron-donating or electron-withdrawing groups were well-tolerated, providing the corresponding alkylated products with predominately R-configuration in high yields (up to 99%) and enantioselectivities (up to >99%). Control experiments were performed to determine the mechanism for stereochemical outcome of the reaction. First, CPA L42 induces dehydration of trifluoromethyl ketone hydrate to generate the corresponding trifluoromethyl ketone. The cooperative activation of both ketone and indole by a chiral bifunctional catalyst through H-bonding poses both substrates in close proximity in the chiral pocket in such a way that it avoid steric repulsion between the benzothiazole moiety of the ketone and catalyst substituent and hence facilitates π–π interactions between the substrates. The attack of the indole nucleophile is favored from the Si-face of activated ketone to generate R-configured product (Figure 10).
Scheme 40. Asymmetric F–C Alkylation of Indoles with Trifluoromethyl Ketone Hydrates.

Figure 10.

Plausible transition states for F–C reaction of indole with benzothiazole trifluoromethyl ketone.
Recently, Liao et al. disclosed a unified protocol for the asymmetric construction of versatile C2-substituted indole derivatives by the reaction of indoles 201 with azadienes 200 using CPA catalyst L43.85 The operationally facile strategy provides direct access to the synthesis of C2-substituted triarylmethane scaffolds 202 in fair to high yields (41–98%) and enantioselectivity (up to 99% ee; Scheme 41). Under optimized conditions, a series of azadienes bearing diverse substituents in the aromatic rings with both steric and electronic nature were tolerated and compatible with the catalyst system, delivering the corresponding triarylmethane moieties in high yields and excellent enantioselectivities. However, relatively lower yields (44–77%) were obtained when azadienes having electron-donating functions were used as electrophilic coupling partners. Likewise, substituted azadienes having alkyl substituents at the para- and meta-positions of the benzylidene ring were also well-tolerated, furnishing the corresponding products in high yields. Nevertheless, alkyl substituents bearing electron-withdrawing groups (F, Cl, Br) rendered the corresponding products in low yields but in excellent enantioselectivities (up to 99% ee). Similarly, diverse indoles containing substituents at the C5- and C6-positions reacted smoothly to render the corresponding products in low to appreciable yields and high enantiomeric excess (80–97% ee). Contrary, the use of 7-methyl indole greatly diminished the yield of the reaction, presumably due to the high steric demand.
Scheme 41. Asymmetric F–C Reaction of Indoles with Azadienes.

Based on experimental evidence, a plausible transition state for the observed stereochemical outcomes of the reaction was proposed (Figure 11). The cooperative activation of both indole and azadiene substrates by bifunctional catalyst L43 through H-bonding assembled both substrates in a chiral pocket such that 1,4-addition of indole on azadiene to produce S-configured facilitated intermediate (S)-202a′, which, in turn, rapidly isomerizes to afford the corresponding product in the S-configuration.
Figure 11.

Proposed reaction mechanism for the synthesis of C2-substituted indole derivatives.
The axially chiral styrenes find widespread applications as stereochemical relay agents as well as chiral ligands in diverse asymmetric organic transformations, including in the synthesis of natural products, optically pure materials, and asymmetric catalysis. Due to their synthetic utility, Lv et al. recently disclosed a synthetic strategy for the construction of axially chiral acyclic styrenes based on theF–C reaction between indoles 204 and ortho-alkynylnaphthols 203, catalyzed by SPINOL-derived CPA L58.10d Based on the nature of C2- or C3-subtituted indoles used as nucleophilic partners, a wide range of axially chiral acyclic styrenes linked through C3- and C2-positions of indoles were prepared in high yields and enantioselectivities (up to 98% ee; Scheme 42). The scope of the reaction was quite general since a wide range substituted indoles, bearing either electron-donating or electron-withdrawing groups at the C4–C7-positions, were quite compatible with the catalytic system. In particular, sterically demanding indole substrates bearing C3-substituents such as Ph, 4-methyl phenyl, and 4-chloro phenyl reacted smoothly to provide the corresponding C2-alkylated products in good to high yields and excellent enantioselectivities (up to 98% ee). Likewise, C2-substituted indoles containing bulkier groups such as Me and Ph also smoothly provided the corresponding C3-alkylated products in high yields (81 and 87%) and enantioselectivities (95 and 96% ee). Nevertheless, insertion of an electron-withdrawing group (e.g., Br) in the naphthalene ring of ortho-alkynylnaphthols slightly diminished both the yield and the enantioselectivity. Based on control experiments, the intermediacy of vinylidene quinone methide (VQM) was proposed. Activation of ortho-alkynylnaphthols 203 by CPA L58 through H-bonding interactions generates the chiral (aS)-VQM complex, followed by two H-bonding interactions that facilitate the attack of indole from the (aR)-face to afford the product with observed stereochemistry.
Scheme 42. C2- and C3-Selective F–C Alkylation of Indoles with ortho-Alkynylnaphthols.

The catalytic enantioselective F–C reaction of indoles with enal electrophilic partners can provide access to functionalized chiral indole derivatives. However, higher electrophilicity of the aldehyde function often leads to overalkylation, making selective 1,4-addition problematic.86 This problem was circumvented by aminocatalytic activation of conjugated aldehyde to produce a conjugated iminium ion, which underwent facile F–C alkylation of indoles.12 Sarotti et al. adopted a rational design approach by employing fast DFT calculations to re-engineer a new levoglucosenone-derived organocatalyst L59 for the asymmetric F–C alkylation of indoles 207.87 The initially designed organocatalyst based on the ONIOM-derived in silico screening, although proven to be a highly efficient catalyst in the asymmetric Diels–Alder reaction between (E)-cinnamaldehyde and cyclopentadiene, failed to provide enantiodiscrimination in the F–C alkylation of indole, rendering null selectivity. Based on modeling, the competing transition structures from the ONIOM method and after an intense survey of the potential energy surface, prediction of the most efficient catalytic system for the asymmetric F–C alkylation becomes possible. The computationally developed catalyst was synthesized and then successfully evaluated experimentally in the F–C alkylation of indoles with (E)-cinnamaldehyde to afford the corresponding alkylated indoles 209 in excellent enantioselectivity (enantiomeric ratios reaching up to 92:8 er; Scheme 43).
Scheme 43. Organocatalyzed Enantioselective F–C Reaction of Indole with Cinnamaldehydes.

Recently, Yang et al. disclosed a synthetic protocol based on the F–C reaction between indoles 210 and β-substituted cyclopentenimines 211 to produce C3-functionalized indoles, bearing chiral all-carbon quaternary stereocenters, under CPA L60 catalysis.88 The hydrolysis of the resultant chiral imine products with basic alumina or reduction with l-Selectride then provided facile access to the corresponding β-indolyl-β-methyl cyclopentanones 212 and cyclopentylamides 213, respectively, with high diastereoselectivities and enantiomeric ratios (Scheme 44a). The generality of the method was quite diverse as neutral, electron-donating, and electron-withdrawing groups at the C5-position of indole were well-tolerated. Similarly, C6-substituted indoles also reacted smoothly to provide the corresponding alkylated products in high yields (up to 99%) and good to excellent enantiomeric ratios (up to 99.5:0.5 er). The synthetic application of methodology was demonstrated by the stereoselective reduction of chiral cyclopentanone with l-Selectride to produce the corresponding chiral cyclopentanol in high yield and diastereoselectivity (99% yield, 85:15 dr). Likewise, the deprotection of cyclopentylamide with SmI2 efficiently produced the corresponding cyclopentylamine (93% yield, 81:19 dr). Zhou et al. employed aurone-derived azadienes 214 as electrophilic coupling partners in the F–C reaction with indoles 215 to produce optically pure heterotriarylmethanes 216.89 The CPA-catalyzed reaction tolerated very well the electron-donating or electron-withdrawing groups in the indole substrate, generating the corresponding heterotriarylmethanes in high yields (up to 95%) and good to high enantioselectivities (up to 98% ee; Scheme 44b). Based on the experimental results, a plausible transition-state model for the reaction was proposed. The synergetic H-bonding activation of both substrates by the catalyst followed by shielding of the Si-face of azadiene by triisopropyl phenyl groups at the 3,3′-positions of CPA favored the Re-face nucleophilic attack by indole to afford S-configured heterotriarylmethanes.
Scheme 44. Asymmetric F–C Reaction of Indoles with (a) Azadienes and (b) β-Cyclopentenimines.

5. Conclusion and Outlook
In conclusion, this review provides an updated account on the application of asymmetric F–C reactions of indoles in the construction of diverse chiral indole derivatives bearing stereogenic centers. Given the simplicity and atom economy, the catalytic asymmetric F–C reaction of indole, catalyzed by chiral metal complexes or chiral organocatalysts, represents one of the most powerful approaches to access optically active indole derivatives, with increased molecular complexity through the construction of a carbon–carbon bond. A wide range of electrophilic partners including activated ketones and alkenes, ketoesters, imines, nitrones, allylic alcohols, and many others have been successfully employed to achieve a plethora of functionalized chiral indole moieties. In general, the indole exhibits high nucleophilic reactivities for the electrophilic alkylation at C3- and C2-positions of the azole ring, whereas F–C alkylation in the benzene ring often requires incorporation of a directing/activation group, blocking groups in the azole ring, or necessitating harsher reaction conditions or the use of transition metals as catalysts. The development of enantioselective alkylation of less active C2-position is quite challenging, in particular, for indoles having no substituents on the N1- and C3-positions. The catalytic alkylation of 4,7-dihydroindole with a suitable electrophilic partner followed by the oxidation of the alkylation product offers an alternative for the synthesis of C2-alkylated chiral indoles. Nevertheless, direct F–C C2-alkylation of 3-substituted indoles is far less explored. Chiral metal complexes derived from chiral ligands work efficiently in catalyzing enantioselective F–C reaction of diverse indole derivatives in a stereodefined manner. However, the development of easily tunable and stable metal-free chiral organocatalysts such as BINOL-derived CPA, thiourea, urea, squaramides, and chincona alkaloid derivatives have made asymmetric F–C reactions operationally simple and very productive. Nevertheless, despite significant advances, liabilities associated with organocatalytic enantioselective F–C reactions using Brønsted acids including high catalyst loading and longer reaction time hamper their practical utility. In the case of CPA, introducing different functionalities at 3,3′-positions of the binaphthyl backbone can generate steric bulk around the active site and may also influence the electron density of the entire catalyst, depending on the electronic nature of the installed substituents. Nevertheless, optimization of a selected substituent is often required for an optimized individual enantioselective conversion. Moreover, the lower acidity of BINOL-derived CPA catalysts generally requires more basic substrates for sufficient activation. In addition, due to relatively less confined chiral environment around the acidic functionality in the active site often needs a structurally biased substrate for high stereoselectivity. Therefore, the design of more acidic chiral catalysts such as N-triflyl phosphoramide that contain a more confined chiral environment around the active site is necessary for promoting the reaction of less reactive substrates with higher stereoinduction. Likewise, bifunctional organocatalysts can enable activation of substrates demanding greater stereochemical control and regiocontrol. Similarly, deployment of electrophilic partners with great versatility and abundant substrates scopes will further enhance the practical applications of chiral indole building blocks for synthetic and medicinal chemistry. Similarly, the incorporation of electrophilic partners with great versatility and abundant substrates scopes will further enhance the practical applications of chiral indole building blocks for synthetic and medicinal chemistry. Therefore, we believe that this study will not only offer a comprehensive overview of recent advancements in enantioselective F–C reaction of indoles but also provide a new window and motivate synthetic chemists to confront the upcoming daunting challenges in this field.
Acknowledgments
The authors extend their appreciation to the Deputyship for Research & Innovation, Ministry of Education in Saudi Arabia, for funding this research work through the project number DRI 74.
Glossary
Abbreviations
- F–C
Friedel–Crafts
- CPA
chiral phosphoric acid
- SPA
spiro phosphoric acid
- BINOL
1,1′-bi-2-naphthol
- ee
enantioselectivity
- er
enantiomeric ratio
- DCM
dichloromethane
- THF
tetrahydrofuran
- DFT
density functional theory
- ONIOM
our own n-layered integrated molecular orbital and molecular mechanics method
- RAEs
redox-active ester
- CTC
charge-transfer complex
- TS
transition state
- VQM
vinylidene quinone methide
- Hal
halogen
- PMB
para-methoxybenzyl
- TMB
2,4,6-trimethylbenzene
The authors declare no competing financial interest.
References
- a Kochanowska-Karamyan A. J.; Hamann M. T. Marine indole alkaloids: Potential new drug leads for the control of depression and anxiety. Chem. Rev. 2010, 110 (8), 4489–4497. 10.1021/cr900211p. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Schmidt A. W.; Reddy K. R.; Knölker H.-J. Occurrence, biogenesis, and synthesis of biologically active carbazole alkaloids. Chem. Rev. 2012, 112 (6), 3193–3328. 10.1021/cr200447s. [DOI] [PubMed] [Google Scholar]; c Zhang M.-Z.; Chen Q.; Yang G.-F. A review on recent developments of indole-containing antiviral agents. Eur. J. Med. Chem. 2015, 89, 421–441. 10.1016/j.ejmech.2014.10.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Bronner S. M.; Im G. Y. J.; Garg N. K.. Indoles and Indolizidines; Wiley-VCH: Weinheim, Germany, 2011; pp 221–265. [Google Scholar]; b Kaushik N. K.; Kaushik N.; Attri P.; Kumar N.; Kim C. H.; Verma A. K.; Choi E. H. Biomedical importance of indoles. Molecules 2013, 18 (6), 6620–6662. 10.3390/molecules18066620. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Singh T. P.; Singh O. M. Recent progress in biological activities of indole and indole alkaloids. Mini-Rev. Med. Chem. 2018, 18 (1), 9–25. 10.2174/1389557517666170807123201. [DOI] [PubMed] [Google Scholar]; d Zhai Y.-J.; Huo G.-M.; Zhang Q.; Li D.; Wang D.-C.; Qi J.-Z.; Han W.-B.; Gao J.-M. Phaeosphaones: tyrosinase inhibitory thiodiketopiperazines from an endophytic Phaeosphaeria fuckelii. J. Nat. Prod. 2020, 83 (5), 1592–1597. 10.1021/acs.jnatprod.0c00046. [DOI] [PubMed] [Google Scholar]
- a Zheng C.; You S.-L. Exploring the chemistry of spiroindolenines by mechanistically-driven reaction development: asymmetric Pictet–Spengler-type reactions and beyond. Acc. Chem. Res. 2020, 53 (4), 974–987. 10.1021/acs.accounts.0c00074. [DOI] [PubMed] [Google Scholar]; b Khan S.; Ahmad T.; Rasheed T.; Ullah N. Recent applications of vinylethylene carbonates in Pd-catalyzed allylic substitution and annulation reactions: Synthesis of multifunctional allylic and cyclic structural motifs. Coord. Chem. Rev. 2022, 462, 214526. 10.1016/j.ccr.2022.214526. [DOI] [Google Scholar]; c Ahmad T.; Ullah N. The oxa-Michael reaction in the synthesis of 5-and 6-membered oxygen-containing heterocycles. Org. Chem. Front. 2021, 8 (6), 1329–1344. 10.1039/D0QO01312A. [DOI] [Google Scholar]
- Shiri M. Indoles in multicomponent processes (MCPs). Chem. Rev. 2012, 112 (6), 3508–3549. 10.1021/cr2003954. [DOI] [PubMed] [Google Scholar]
- a D’yakonov V. A.; Trapeznikova O. A.; de Meijere A.; Dzhemilev U. M. Metal Complex Catalysis in the Synthesis of Spirocarbocycles. Chem. Rev. 2014, 114, 5775. 10.1021/cr400291c. [DOI] [PubMed] [Google Scholar]; b Xie X.; Huang W.; Peng C.; Han B. Organocatalytic Asymmetric Synthesis of Six-Membered Carbocycle-Based Spiro Compounds. Adv. Synth. Catal. 2018, 360 (2), 194–228. 10.1002/adsc.201700927. [DOI] [Google Scholar]; c Xu P.-W.; Yu J.-S.; Chen C.; Cao Z.-Y.; Zhou F.; Zhou J. Catalytic enantioselective construction of spiro quaternary carbon stereocenters. ACS Catal. 2019, 9 (3), 1820–1882. 10.1021/acscatal.8b03694. [DOI] [Google Scholar]
- a Arai T.; Kakino J. Catalytic Asymmetric Synthesis of 3-Indolyl Methanamines Using Unprotected Indoles and N-Boc Imines under Basic Conditions. Angew. Chem., Int. Ed. 2016, 55 (49), 15263–15267. 10.1002/anie.201607679. [DOI] [PubMed] [Google Scholar]; b Xiao M.; Xu D.; Liang W.; Wu W.; Chan A. S.; Zhao J. Organocatalytic enantioselective Friedel–Crafts alkylation/lactonization reaction of hydroxyindoles with methyleneoxindoles. Adv. Synth. Catal. 2018, 360 (5), 917–924. 10.1002/adsc.201701089. [DOI] [Google Scholar]
- a Hua Y.-Z.; Chen J.-W.; Yang H.; Wang M.-C. Asymmetric Friedel–Crafts Alkylation of Indoles with Trifluoromethyl Pyruvate Catalyzed by a Dinuclear Zinc Catalyst. J. Org. Chem. 2018, 83 (3), 1160–1166. 10.1021/acs.joc.7b02599. [DOI] [PubMed] [Google Scholar]; b Huang T.; Zhao Y.; Meng S.; Chan A. S.; Zhao J. C7-Functionalization of Indoles via Organocatalytic Enantioselective Friedel-Crafts Alkylation of 4-Amino-indoles with 2-Butene-1, 4-diones and 3-Aroylacrylates. Adv. Synth. Catal. 2019, 361 (15), 3632–3638. 10.1002/adsc.201900377. [DOI] [Google Scholar]; c Staleva P.; Hernández J. G.; Bolm C. Mechanochemical Copper-Catalyzed Asymmetric Michael-Type Friedel–Crafts Alkylation of Indoles with Arylidene Malonates. Chem.—Eur. J. 2019, 25 (39), 9202–9205. 10.1002/chem.201901826. [DOI] [PubMed] [Google Scholar]; d Li J.; Wei Z.; Cao J.; Liang D.; Lin Y.; Duan H. Aymmetric Aza-Friedel–Crafts Reaction of Isatin-Derived Ketimines with Indoles Catalyzed by a Chiral Phase-Transfer Catalyst. J. Org. Chem. 2022, 87 (5), 2532–2542. 10.1021/acs.joc.1c02477. [DOI] [PubMed] [Google Scholar]; e He H.; Cao Y.; Xu J.; Antilla J. C. Catalytic Asymmetric C-7 Friedel–Crafts Alkylation/N-Hemiacetalization of 4-Aminoindoles. Org. Lett. 2021, 23 (8), 3010–3014. 10.1021/acs.orglett.1c00699. [DOI] [PubMed] [Google Scholar]; f Montesinos-Magraner M.; Lluna-Galán C.; Cernicharo-Toledo F.; Vila C.; Blay G.; Pedro J. R. Enantioselective Friedel–Crafts reaction of hydroxyarenes with nitroenynes to access chiral heterocycles via sequential catalysis. Org. Biomol. Chem. 2021, 19 (32), 6990–6994. 10.1039/D1OB01238J. [DOI] [PubMed] [Google Scholar]; g Huang B.-B.; Wu L.; Liu R.-R.; Xing L.-L.; Liang R.-X.; Jia Y.-X. Enantioselective Friedel–Crafts C2-alkylation of 3-substituted indoles with trifluoropyruvates and cyclic N-sulfonyl α-ketiminoesters. Org. Chem. Front. 2018, 5 (6), 929–932. 10.1039/C7QO01014A. [DOI] [Google Scholar]
- a Dalpozzo R. Strategies for the asymmetric functionalization of indoles: an update. Chem. Soc. Rev. 2015, 44 (3), 742–778. 10.1039/C4CS00209A. [DOI] [PubMed] [Google Scholar]; b Bera K.; Schneider C. Brønsted Acid Catalyzed [3+ 2]-Cycloaddition of Cyclic Enamides with in Situ Generated 2-Methide-2 H-indoles: Enantioselective Synthesis of Indolo [1, 2-a] indoles. Org. Lett. 2016, 18 (21), 5660–5663. 10.1021/acs.orglett.6b02898. [DOI] [PubMed] [Google Scholar]
- a Shah T. A.; De P. B.; Pradhan S.; Punniyamurthy T. Transition-metal-catalyzed site-selective C7-functionalization of indoles: advancement and future prospects. Chem. Commun. 2019, 55 (5), 572–587. 10.1039/C8CC04116D. [DOI] [PubMed] [Google Scholar]; b Chen S.; Zhang M.; Su R.; Chen X.; Feng B.; Yang Y.; You J. C2/C4 Regioselective heteroarylation of indoles by tuning C–H metalation modes. ACS Catal. 2019, 9 (7), 6372–6379. 10.1021/acscatal.9b01273. [DOI] [Google Scholar]
- a Zhang P.; Tsuji N.; Ouyang J.; List B. Strong and confined acids catalyze asymmetric intramolecular hydroarylations of unactivated olefins with indoles. J. Am. Chem. Soc. 2021, 143 (2), 675–680. 10.1021/jacs.0c12042. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Sun P.; Jia Z.-H.; Tang L.; Zheng H.; Li Z.-R.; Chen L.-Y.; Li Y. Enantioselective synthesis of 2-indolyl methanamine derivatives through disulfonimide-catalyzed Friedel–Crafts C2-alkylation of 3-substituted indoles with imines. Org. Biomol. Chem. 2022, 20 (9), 1916–1925. 10.1039/D1OB02281D. [DOI] [PubMed] [Google Scholar]; c Wang X.-W.; Huang W.-J.; Wang H.; Wu B.; Zhou Y.-G. Chiral-Phosphoric-Acid-Catalyzed C6-Selective Pictet–Spengler Reactions for Construction of Polycyclic Indoles Containing Spiro Quaternary Stereocenters. Org. Lett. 2022, 24 (8), 1727–1731. 10.1021/acs.orglett.2c00368. [DOI] [PubMed] [Google Scholar]; d Zhang W.; Song R.; Yang D.; Lv J. Construction of Axially Chiral Styrenes Linking an Indole Moiety by Chiral Phosphoric Acid. J. Org. Chem. 2022, 87 (5), 2853–2863. 10.1021/acs.joc.1c02750. [DOI] [PubMed] [Google Scholar]
- Johannsen M. An enantioselective synthesis of heteroaromatic N-tosyl α-amino acids. Chem. Commun. 1999, (21), 2233–2234. 10.1039/a906758b. [DOI] [Google Scholar]
- Austin J. F.; MacMillan D. W. Enantioselective organocatalytic indole alkylations. Design of a new and highly effective chiral amine for iminium catalysis. J. Am. Chem. Soc. 2002, 124 (7), 1172–1173. 10.1021/ja017255c. [DOI] [PubMed] [Google Scholar]
- Bandini M.; Eichholzer A. Catalytic functionalization of indoles in a new dimension. Angew. Chem., Int. Ed. 2009, 48 (51), 9608–9644. 10.1002/anie.200901843. [DOI] [PubMed] [Google Scholar]
- Beletskaya I. P.; Averin A. D. Asymmetric friedel-crafts reactions of indole and its derivatives. Curr. Organocatalysis 2015, 3 (1), 60–83. 10.2174/2213337202666150505230013. [DOI] [Google Scholar]
- Liu R.-R.; Liang R.-X.; Jia Y.-X. Construction of Benzylic Stereogenic Carbon Centers through Enantioselective Arylation Reactions. Synlett 2018, 29 (02), 157–168. 10.1055/s-0036-1590923. [DOI] [Google Scholar]
- Montesinos-Magraner M.; Vila C.; Blay G.; Pedro J. R. Catalytic enantioselective Friedel–Crafts reactions of naphthols and electron-rich phenols. Synthesis 2016, 48 (14), 2151–2164. 10.1055/s-0035-1561434. [DOI] [Google Scholar]
- Sheng F.-T.; Wang J.-Y.; Tan W.; Zhang Y.-C.; Shi F. Progresses in organocatalytic asymmetric dearomatization reactions of indole derivatives. Org. Chem. Front. 2020, 7 (23), 3967–3998. 10.1039/D0QO01124J. [DOI] [Google Scholar]
- Kang Y. K.; Kwon B. K.; Mang J. Y.; Kim D. Y. Chiral Pd-catalyzed enantioselective Friedel–Crafts reaction of indoles with γ, δ-unsaturated β-keto phosphonates. Tetrahedron Lett. 2011, 52 (25), 3247–3249. 10.1016/j.tetlet.2011.04.084. [DOI] [Google Scholar]
- Blay G.; Fernandez I.; Muñoz M. C.; Pedro J. R.; Vila C. Enantioselective Friedel–Crafts Alkylation of Indoles with (E)-1-Aryl-4-benzyloxybut-2-en-1-ones Catalyzed by an (R)-3, 3′-Br2BINOLate–Hafnium (IV) Complex. Eur. J. Org. Chem. 2013, 2013 (10), 1902–1907. 10.1002/ejoc.201201636. [DOI] [Google Scholar]
- Chatterjee S.; Hintermann L.; Mandal M.; Achari A.; Gupta S.; Jaisankar P. Fiaud’s acid: a Brønsted acid catalyst for enantioselective Friedel–Crafts Alkylation of indoles with 2-alkene-1, 4-diones. Org. Lett. 2017, 19 (13), 3426–3429. 10.1021/acs.orglett.7b01383. [DOI] [PubMed] [Google Scholar]
- Li Y.-P.; Li Z.-Q.; Zhou B.; Li M.-L.; Xue X.-S.; Zhu S.-F.; Zhou Q.-L. Chiral Spiro Phosphoric Acid-Catalyzed Friedel–Crafts Conjugate Addition/Enantioselective Protonation Reactions. ACS Catal. 2019, 9 (7), 6522–6529. 10.1021/acscatal.9b01502. [DOI] [Google Scholar]
- Chen H.; Du F.; Liu L.; Li J.; Zhao Q.; Fu B. Malonate-type bis (oxazoline) ligands with sp2 hybridized bridge carbon: Synthesis and application in Friedel–Crafts alkylation and allylic alkylation. Tetrahedron 2011, 67 (49), 9602–9608. 10.1016/j.tet.2011.09.106. [DOI] [Google Scholar]
- Oyama H.; Nakada M. Highly enantioselective catalytic Friedel–Crafts reactions of cyclic α-alkylidene β-oxo imides. Tetrahedron Asymmetry 2015, 26 (4), 195–202. 10.1016/j.tetasy.2015.01.004. [DOI] [Google Scholar]
- Najda-Mocarska E.; Zakaszewska A.; Janikowska K.; Makowiec S. New thiourea organocatalysts and their application for the synthesis of 5-(1 H-indol-3-yl) methyl-2, 2-dimethyl-1, 3-dioxane-4, 6-diones a source of chiral 3-indoylmethyl ketenes. Synth. Commun. 2018, 48 (1), 14–25. 10.1080/00397911.2017.1383432. [DOI] [Google Scholar]
- Sato R.; Tosaka T.; Masu H.; Arai T. Catalytic Asymmetric Synthesis of Chiral Bis (Indolyl) Methanes Using a Ts-PyBidine–Nickel Complex. J. Org. Chem. 2019, 84 (21), 14248–14257. 10.1021/acs.joc.9b02006. [DOI] [PubMed] [Google Scholar]
- Tarasenko E. A.; Shestakov I. V.; Rybakov V. B.; Beletskaya I. P. Enantioselective Copper (II)/Box-Catalyzed Synthesis of Chiral β3-Tryptophan Derivatives. ChemCatChem. 2019, 11 (16), 3913–3918. 10.1002/cctc.201900575. [DOI] [Google Scholar]
- a Wu J.; Wang D.; Wu F.; Wan B. Cu (OTf) 2-Catalyzed Asymmetric Friedel–Crafts Alkylation Reaction of Indoles with Arylidene Malonates Using Bis (sulfonamide)-Diamine Ligands. J. Org. Chem. 2013, 78 (11), 5611–5617. 10.1021/jo400747d. [DOI] [PubMed] [Google Scholar]; b Liu L.; Li J.; Wang M.; Du F.; Qin Z.; Fu B. Synthesis of heteroarylidene malonate derived bis (thiazolines) and their application in the catalyzed Friedel–Crafts reaction. Tetrahedron Asymmetry 2011, 22 (5), 550–557. 10.1016/j.tetasy.2011.03.003. [DOI] [Google Scholar]
- a Rueping M.; Nachtsheim B. J.; Moreth S. A.; Bolte M. Asymmetric Brønsted Acid Catalysis: Enantioselective Nucleophilic Substitutions and 1, 4-Additions. Angew. Chem., Int. Ed. 2008, 47 (3), 593–596. 10.1002/anie.200703668. [DOI] [PubMed] [Google Scholar]; b Spangler J. E.; Davies H. M. Catalytic asymmetric synthesis of pyrroloindolines via a rhodium (II)-catalyzed annulation of indoles. J. Am. Chem. Soc. 2013, 135 (18), 6802–6805. 10.1021/ja4025337. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Yang N.; Liu X.; Guo J.; Zhang X.; Lin L.; Hu C.; Feng X. Reversal of enantioselective Friedel–Crafts C3-alkylation of pyrrole by slightly tuning the amide units of N, N′-dioxide ligands. Chem. Commun. 2015, 51 (40), 8432–8435. 10.1039/C4CC10055G. [DOI] [PubMed] [Google Scholar]
- He H.; Cao Y.; Xu J.; Antilla J. C. Catalytic Asymmetric C-7 Friedel–Crafts Alkylation/N-Hemiacetalization of 4-Aminoindoles. Org. Lett. 2021, 23 (8), 3010–3014. 10.1021/acs.orglett.1c00699. [DOI] [PubMed] [Google Scholar]
- Zhong X.; Lv J.; Luo S. Enantio-and diastereoselective cyclopropanation of β, γ-unsaturated α-ketoester by a chiral phosphate/indium (III) complex. Org. Lett. 2017, 19 (12), 3331–3334. 10.1021/acs.orglett.7b01577. [DOI] [PubMed] [Google Scholar]
- Li J.; Lu W.; Lu Y.; Zha Z.; Wang Z. Construction of Chiral All-Carbon Quaternary Stereocenters by Asymmetric Friedel– Crafts Reaction of Isatin Derivatives. Chin. J. Chem. 2022, 40 (2), 195–200. 10.1002/cjoc.202100696. [DOI] [Google Scholar]
- Bi B.; Lou Q.-X.; Ding Y.-Y.; Chen S.-W.; Zhang S.-S.; Hu W.-H.; Zhao J.-L. Chiral Phosphoric Acid Catalyzed Highly Enantioselective Friedel–Crafts Alkylation Reaction of C3-Substituted Indoles to β, γ-Unsaturated α-Ketimino Esters. Org. Lett. 2015, 17 (3), 540–543. 10.1021/ol5035222. [DOI] [PubMed] [Google Scholar]
- Ding Y. Y.; Xu D. F.; Meng S. S.; Li Y.; Zhao J. L. Chiral Phosphoric Acid Catalyzed Enantioselective Friedel-Crafts Reaction of N-Protected 4-Aminoindoles with β, γ-Unsaturated α-Ketimino Esters. Adv. Synth. Catal. 2018, 360 (4), 664–669. 10.1002/adsc.201701354. [DOI] [Google Scholar]
- Xun W.; Xu B.; Chen B.; Meng S.; Chan A. S.; Qiu F. G.; Zhao J. Regio and enantioselective organocatalytic Friedel–Crafts alkylation of 4-aminoindoles at the C7-position. Org. Lett. 2018, 20 (3), 590–593. 10.1021/acs.orglett.7b03703. [DOI] [PubMed] [Google Scholar]
- Fan Y.; Kass S. R. Enantioselective Friedel–Crafts alkylation between nitroalkenes and indoles catalyzed by charge activated thiourea organocatalysts. J. Org. Chem. 2017, 82 (24), 13288–13296. 10.1021/acs.joc.7b02411. [DOI] [PubMed] [Google Scholar]
- a Ballini R.; Bosica G.; Fiorini D.; Palmieri A.; Petrini M. Conjugate additions of nitroalkanes to electron-poor alkenes: Recent results. Chem. Rev. 2005, 105 (3), 933–972. 10.1021/cr040602r. [DOI] [PubMed] [Google Scholar]; b Aitken L. S.; Arezki N. R.; Dell’Isola A.; Cobb A. J. Asymmetric organocatalysis and the nitro group functionality. Synthesis 2013, 45 (19), 2627–2648. 10.1055/s-0033-1338522. [DOI] [Google Scholar]
- a Ganesh M.; Seidel D. Catalytic enantioselective additions of indoles to nitroalkenes. J. Am. Chem. Soc. 2008, 130 (49), 16464–16465. 10.1021/ja8063292. [DOI] [PubMed] [Google Scholar]; b Arai T.; Yokoyama N. Tandem Catalytic Asymmetric Friedel–Crafts/Henry Reaction: Control of Three Contiguous Acyclic Stereocenters. Angew. Chem., Int. Ed. 2008, 47 (27), 4989–4992. 10.1002/anie.200801373. [DOI] [PubMed] [Google Scholar]
- Hirata T.; Yamanaka M. DFT Study of Chiral-Phosphoric-Acid-Catalyzed Enantioselective Friedel–Crafts Reaction of Indole with Nitroalkene: Bifunctionality and Substituent Effect of Phosphoric Acid. Chem.—Asian J. 2011, 6 (2), 510–516. 10.1002/asia.201000596. [DOI] [PubMed] [Google Scholar]
- Wu H.; Liu R.-R.; Jia Y.-X. Asymmetric Friedel–Crafts Alkylation Reaction in the Construction of Trifluoromethylated All-Carbon Quaternary Stereocenters. Synlett 2014, 25 (04), 457–460. [Google Scholar]
- Wu H.; Liu R.-R.; Shen C.; Zhang M.-D.; Gao J.; Jia Y.-X. Enantioselective Friedel–Crafts reaction of 4, 7-dihydroindoles with β-CF 3-β-disubstituted nitroalkenes. Org. Chem. Front. 2015, 2 (2), 124–126. 10.1039/C4QO00265B. [DOI] [Google Scholar]
- Ibáñez I.; Kaneko M.; Kamei Y.; Tsutsumi R.; Yamanaka M.; Akiyama T. Enantioselective Friedel–Crafts alkylation reaction of indoles with α-trifluoromethylated β-nitrostyrenes catalyzed by chiral binol metal phosphate. ACS Catal. 2019, 9 (8), 6903–6909. 10.1021/acscatal.9b01811. [DOI] [Google Scholar]
- Mori K.; Wakazawa M.; Akiyama T. Stereoselective construction of all-carbon quaternary center by means of chiral phosphoric acid: highly enantioselective Friedel–Crafts reaction of indoles with β, β-disubstituted nitroalkenes. Chem. Sci. 2014, 5 (5), 1799–1803. 10.1039/C3SC53542H. [DOI] [Google Scholar]
- a Ma J.; Kass S. R. Electrostatically Enhanced Phosphoric Acids: A Tool in Brønsted Acid Catalysis. Org. Lett. 2016, 18 (22), 5812–5815. 10.1021/acs.orglett.6b02750. [DOI] [PubMed] [Google Scholar]; b Samet M.; Buhle J.; Zhou Y.; Kass S. R. Charge-enhanced acidity and catalyst activation. J. Am. Chem. Soc. 2015, 137 (14), 4678–4680. 10.1021/jacs.5b01805. [DOI] [PubMed] [Google Scholar]
- Arai T.; Tsuchida A.; Miyazaki T.; Awata A. Catalytic Asymmetric Synthesis of Chiral 2-Vinylindole Scaffolds by Friedel–Crafts Reaction. Org. Lett. 2017, 19 (4), 758–761. 10.1021/acs.orglett.6b03584. [DOI] [PubMed] [Google Scholar]
- Liu J.-Y.; Yang X.-C.; Lu H.; Gu Y.-C.; Xu P.-F. Organocatalytic, Enantioselective Friedel–Crafts Reaction of Indoles in the Carbocyclic Ring and Electron-Rich Phenols. Org. Lett. 2018, 20 (8), 2190–2194. 10.1021/acs.orglett.8b00503. [DOI] [PubMed] [Google Scholar]
- Vila C.; Rostoll-Berenguer J.; Sánchez-García R.; Blay G.; Fernández I.; Muñoz M. C.; Pedro J. R. Enantioselective synthesis of 2-amino-1, 1-diarylalkanes bearing a carbocyclic ring substituted indole through asymmetric catalytic reaction of hydroxyindoles with nitroalkenes. J. Org. Chem. 2018, 83 (12), 6397–6407. 10.1021/acs.joc.8b00612. [DOI] [PubMed] [Google Scholar]
- Dong X.; Tang Z.; Ye L.; Shi Z.; Zhao Z.; Li X. Stereoselective Synthesis of Dihydrofuranoindoles via the Friedel–Crafts Alkylation/Annulation Cascade Process. J. Org. Chem. 2020, 85 (18), 11607–11617. 10.1021/acs.joc.0c00444. [DOI] [PubMed] [Google Scholar]
- Dündar E.; Tanyeli C. Enantioselective Friedel-Crafts alkylation of indole with nitroalkenes in the presence of bifunctional squaramide organocatalysts. Tetrahedron Lett. 2021, 73, 153153. 10.1016/j.tetlet.2021.153153. [DOI] [Google Scholar]
- Buchcic A.; Zawisza A.; Leśniak S.; Rachwalski M. Asymmetric Friedel–Crafts alkylation of indoles catalyzed by chiral aziridine-phosphines. Catalysts 2020, 10 (9), 971. 10.3390/catal10090971. [DOI] [Google Scholar]
- Bao R. L.-Y.; Shi L.; Fu K. Highly enantioselective construction of CF3-bearing all-carbon quaternary stereocenters: Chiral spiro-fused bisoxazoline ligands with 1, 1′-binaphthyl sidearm for asymmetric Michael-type Friedel-Crafts reaction. Chin. Chem. Lett. 2022, 33 (5), 2415–2419. 10.1016/j.cclet.2021.10.062. [DOI] [Google Scholar]
- Islam M. S.; Alammari A. S.; Barakat A.; Alshahrani S.; Haukka M.; Al-Majid A. M. Exploiting the Chiral Ligands of Bis (imidazolinyl)-and Bis (oxazolinyl) thiophenes—Synthesis and Application in Cu-Catalyzed Friedel–Crafts Asymmetric Alkylation. Molecules 2021, 26 (23), 7408. 10.3390/molecules26237408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor S. E.; Maresh J. J. Chemistry and biology of monoterpene indole alkaloid biosynthesis. Nat. Prod. Rep. 2006, 23 (4), 532–547. 10.1039/b512615k. [DOI] [PubMed] [Google Scholar]
- Liu L.; Zhao Q.; Du F.; Chen H.; Qin Z.; Fu B. Highly enantioselective Friedel–Crafts reaction of indoles with N-sulfonyl aldimines catalyzed by heteroarylidene malonate-type bis (oxazoline) copper (II) complexes. Tetrahedron Asymmetry 2011, 22 (20–22), 1874–1878. 10.1016/j.tetasy.2011.10.017. [DOI] [Google Scholar]
- Wu K.; Jiang Y. J.; Fan Y. S.; Sha D.; Zhang S. Double Axially Chiral Bisphosphorylimides Catalyzed Highly Enantioselective and Efficient Friedel–Crafts Reaction of Indoles with Imines. Chem.—Eur. J. 2013, 19 (2), 474–478. 10.1002/chem.201202900. [DOI] [PubMed] [Google Scholar]
- Hatano M.; Mochizuki T.; Nishikawa K.; Ishihara K. Enantioselective Aza-Friedel–Crafts Reaction of Indoles with Ketimines Catalyzed by Chiral Potassium Binaphthyldisulfonates. ACS Catal. 2018, 8 (1), 349–353. 10.1021/acscatal.7b03708. [DOI] [Google Scholar]
- Husmann R.; Sugiono E.; Mersmann S.; Raabe G.; Rueping M.; Bolm C. Enantioselective organocatalytic synthesis of quaternary α-amino acids bearing a CF3 moiety. Org. Lett. 2011, 13 (5), 1044–1047. 10.1021/ol103093r. [DOI] [PubMed] [Google Scholar]
- Uchikura T.; Suzuki R.; Suda Y.; Akiyama T. Enantioselective Synthesis of 2-Substituted Indoles Bearing Trifluoromethyl Moiety by the Friedel-Crafts Alkylation Reaction of 4, 7-Dihydroindole with N– H Trifluoromethyl Ketimines. ChemCatChem. 2020, 12 (19), 4784–4787. 10.1002/cctc.202000920. [DOI] [Google Scholar]
- Zhang K. F.; Nie J.; Guo R.; Zheng Y.; Ma J. A. Chiral Phosphoric Acid-Catalyzed Asymmetric Aza-Friedel–Crafts Reaction of Indoles with Cyclic N-Acylketimines: Enantioselective Synthesis of Trifluoromethyldihydroquinazolines. Adv. Synth. Catal. 2013, 355 (17), 3497–3502. 10.1002/adsc.201300534. [DOI] [Google Scholar]
- Lee S. G.; Kim S.-G. An asymmetric Brønsted acid-catalyzed Friedel–Crafts reaction of indoles with cyclic N-sulfimines. RSC Adv. 2017, 7 (54), 34283–34286. 10.1039/C7RA06244C. [DOI] [Google Scholar]
- Nakamura S.; Furukawa T.; Hatanaka T.; Funahashi Y. Enantioselective aza-Friedel–Crafts reaction of cyclic ketimines with indoles using chiral imidazoline–phosphoric acid catalysts. Chem. Commun. 2018, 54 (31), 3811–3814. 10.1039/C8CC00594J. [DOI] [PubMed] [Google Scholar]
- Han X.; Liu B.; Zhou H.-B.; Dong C. Enhanced efficiency of recyclable C3-symmetric cinchonine-squaramides in the asymmetric Friedel–Crafts reaction of indoles with alkyl trifluoropyruvate. Tetrahedron Asymmetry 2012, 23 (18–19), 1332–1337. 10.1016/j.tetasy.2012.08.015. [DOI] [Google Scholar]
- Wolf C.; Zhang P. Asymmetric Friedel–Crafts reaction of indoles with ethyl trifluoropyruvate using a copper (I)-bisoxazolidine catalyst. Adv. Synth. Catal. 2011, 353 (5), 760–766. 10.1002/adsc.201000918. [DOI] [Google Scholar]
- Ma J.; Kass S. R. Asymmetric arylation of 2, 2, 2-trifluoroacetophenones catalyzed by chiral electrostatically-enhanced phosphoric acids. Org. Lett. 2018, 20 (9), 2689–2692. 10.1021/acs.orglett.8b00900. [DOI] [PubMed] [Google Scholar]
- Cai L.; Zhao Y.; Huang T.; Meng S.; Jia X.; Chan A. S.; Zhao J. Chiral phosphoric-acid-catalyzed regioselective and enantioselective C7-friedel–crafts alkylation of 4-aminoindoles with trifluoromethyl ketones. Org. Lett. 2019, 21 (10), 3538–3542. 10.1021/acs.orglett.9b00821. [DOI] [PubMed] [Google Scholar]
- Deng X.-f.; Wang Y.-w.; Zhang S.-q.; Li L.; Li G.-x.; Zhao G.; Tang Z. An organocatalytic asymmetric Friedel–Crafts reaction of 2-substituted indoles with aldehydes: enantioselective synthesis of α-hydroxyl ketones by low loading of chiral phosphoric acid. Chem. Commun. 2020, 56 (16), 2499–2502. 10.1039/C9CC09637J. [DOI] [PubMed] [Google Scholar]
- Dong H. M.; Lu H. H.; Lu L. Q.; Chen C. B.; Xiao W. J. Asymmetric Friedel–Crafts Alkylations of Indoles with Ethyl Glyoxylate Catalyzed by (S)-BINOL-Titanium (IV) Complex: Direct Access to Enantiomerically Enriched 3-Indolyl (hydroxy) acetates. Adv. Synth. Catal. 2007, 349 (10), 1597–1603. 10.1002/adsc.200600495. [DOI] [Google Scholar]
- Vetica F.; Chauhan P.; Mahajan S.; Raabe G.; Enders D. Asymmetric Organocatalytic Friedel–Crafts Hydroxyalkylation of Indoles Using Electrophilic Pyrazole-4, 5-diones. Synthesis 2018, 50 (05), 1039–1046. 10.1055/s-0036-1591860. [DOI] [Google Scholar]
- Zou L.-M.; Huang X.-Y.; Zheng C.; Cheng Y.-Z.; You S.-L. Chiral Brønsted Acid-Catalyzed Intramolecular Asymmetric Allylic Alkylation of Indoles with Primary Alcohols. Org. Lett. 2022, 24 (19), 3544–3548. 10.1021/acs.orglett.2c01253. [DOI] [PubMed] [Google Scholar]
- Xiao L.; Li B.; Xiao F.; Fu C.; Wei L.; Dang Y.; Dong X.-Q.; Wang C.-J. Stereodivergent synthesis of enantioenriched azepino [3, 4, 5-cd]-indoles via cooperative Cu/Ir-catalyzed asymmetric allylic alkylation and intramolecular Friedel–Crafts reaction. Chem. Sci. 2022, 13 (17), 4801–4812. 10.1039/D1SC07271D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu L.; Cao W.; Wang K.; Liu X.; Feng X. Asymmetric synthesis of dihydrocarbazoles through a Friedel–Crafts alkylation/annulation sequential reaction of indoles. Chem. Commun. 2021, 57 (97), 13138–13141. 10.1039/D1CC05099K. [DOI] [PubMed] [Google Scholar]
- Qian C.; Liu M.; Sun J.; Li P. Chiral phosphoric acid-catalyzed regio- and enantioselective reactions of functionalized propargylic alcohols. Org. Chem. Front. 2022, 9 (5), 1234–1240. 10.1039/D1QO01864G. [DOI] [Google Scholar]
- a Ishikura M.; Abe T.; Choshi T.; Hibino S. Simple indole alkaloids and those with a nonrearranged monoterpenoid unit. Nat. Prod. Rep. 2015, 32 (10), 1389–1471. 10.1039/C5NP00032G. [DOI] [PubMed] [Google Scholar]; b Hatano M.; Toh K.; Ishihara K. Enantioselective Aza-Friedel–Crafts Reaction of Indoles and Pyrroles Catalyzed by Chiral C 1-Symmetric Bis (phosphoric Acid). Org. Lett. 2020, 22 (24), 9614–9620. 10.1021/acs.orglett.0c03662. [DOI] [PubMed] [Google Scholar]; c Tanaka H.; Ukegawa N.; Uyanik M.; Ishihara K. Hypoiodite-Catalyzed Oxidative Umpolung of Indoles for Enantioselective Dearomatization. J. Am. Chem. Soc. 2022, 144 (13), 5756–5761. 10.1021/jacs.2c01852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courant T.; Kumarn S.; He L.; Retailleau P.; Masson G. Chiral Phosphoric Acid-Catalyzed Enantioselective Aza-Friedel–Crafts Alkylation of Indoles with γ-Hydroxy-γ-lactams. Adv. Synth. Catal. 2013, 355 (5), 836–840. 10.1002/adsc.201201008. [DOI] [Google Scholar]
- Fang F.; Hua G.; Shi F.; Li P. Organocatalytic enantioselective Friedel–Crafts reaction: an efficient access to chiral isoindolo-β-carboline derivatives. Org. Biomol. Chem. 2015, 13 (15), 4395–4398. 10.1039/C5OB00175G. [DOI] [PubMed] [Google Scholar]
- Liu R. R.; Ye S. C.; Lu C. J.; Zhuang G. L.; Gao J. R.; Jia Y. X. Dual Catalysis for the Redox Annulation of Nitroalkynes with Indoles: Enantioselective Construction of Indolin-3-ones Bearing Quaternary Stereocenters. Angew. Chem., Int. Ed. 2015, 54 (38), 11205–11208. 10.1002/anie.201504697. [DOI] [PubMed] [Google Scholar]
- Kim M. J.; Xue L.; Liu Y.; Paladhi S.; Park S. J.; Yan H.; Song C. E. Cooperative Cation-Binding Catalysis as an Efficient Approach for Enantioselective Friedel–Crafts Reaction of Indoles and Pyrrole. Adv. Synth. Catal. 2017, 359 (5), 811–823. 10.1002/adsc.201601198. [DOI] [Google Scholar]
- Shen M.-L.; Shen Y.; Wang P.-S. Merging Visible-Light Photoredox and Chiral Phosphate Catalysis for Asymmetric Friedel–Crafts Reaction with in Situ Generation of N-Acyl Imines. Org. Lett. 2019, 21 (9), 2993–2997. 10.1021/acs.orglett.9b00442. [DOI] [PubMed] [Google Scholar]
- Ohfune Y.; Shinada T. Enantio-and diastereoselective construction of α, α-disubstituted α-amino acids for the synthesis of biologically active compounds. Eur. J. Org. Chem. 2005, 2005 (24), 5127–5143. 10.1002/ejoc.200500434. [DOI] [Google Scholar]
- Shen Y.; Shen M.-L.; Wang P.-S. Light-Mediated Chiral Phosphate Catalysis for Asymmetric Dicarbofunctionalization of Enamides. ACS Catal. 2020, 10 (15), 8247–8253. 10.1021/acscatal.0c02660. [DOI] [Google Scholar]
- Kim Y.; Lee J.; Jung J.; Kim S.-G. Chiral Brønsted acid-catalyzed Friedel–Crafts reaction of 3-indolylsulfamidates with indoles: Synthesis of enantioenriched bisindolylmethane sulfamates. Tetrahedron Lett. 2019, 60 (25), 1625–1630. 10.1016/j.tetlet.2019.05.003. [DOI] [Google Scholar]
- Ma C.; Jiang F.; Sheng F. T.; Jiao Y.; Mei G. J.; Shi F. Design and catalytic asymmetric construction of axially chiral 3, 3′-bisindole skeletons. Angew. Chem., Int. Ed. 2019, 58 (10), 3014–3020. 10.1002/anie.201811177. [DOI] [PubMed] [Google Scholar]
- Jiang H.-X.; Zhuang D.-M.; Huang Y.; Cao X.-X.; Yao J.-H.; Li J.-Y.; Wang J.-Y.; Zhang C.; Jiang B. Design, synthesis, and biological evaluation of novel trifluoromethyl indoles as potent HIV-1 NNRTIs with an improved drug resistance profile. Org. Biomol. Chem. 2014, 12 (21), 3446–3458. 10.1039/C3OB42186D. [DOI] [PubMed] [Google Scholar]
- Wang W.; Xiong W.; Wang J.; Wang Q.-A.; Yang W. Brønsted Acid-Catalyzed Asymmetric Friedel–Crafts Alkylation of Indoles with Benzothiazole-Bearing Trifluoromethyl Ketone Hydrates. J. Org. Chem. 2020, 85 (6), 4398–4407. 10.1021/acs.joc.0c00116. [DOI] [PubMed] [Google Scholar]
- Lu Z.-y.; Hu J.-t.; Lan W.-q.; Mo X.-q.; Zhou S.; Tang Y.-f.; Yuan W.-c.; Zhang X.-m.; Liao L.-h. Enantioselective synthesis of hetero-triarylmethanes by chiral phosphoric acid-catalyzed 1, 4-addition of 3-substituted indoles with azadienes. Tetrahedron Lett. 2021, 67, 152862. 10.1016/j.tetlet.2021.152862. [DOI] [Google Scholar]
- Ko S.; Lin C.; Tu Z.; Wang Y.-F.; Wang C.-C.; Yao C.-F. CAN and iodine-catalyzed reaction of indole or 1-methylindole with α, β-unsaturated ketone or aldehyde. Tetrahedron Lett. 2006, 47 (4), 487–492. 10.1016/j.tetlet.2005.11.058. [DOI] [Google Scholar]
- Gerosa G. G.; Marcarino M. O.; Spanevello R. A.; Suárez A. G.; Sarotti A. M. Re-engineering organocatalysts for asymmetric Friedel–Crafts alkylation of indoles through computational studies. J. Org. Chem. 2020, 85 (15), 9969–9978. 10.1021/acs.joc.0c01256. [DOI] [PubMed] [Google Scholar]
- Liu W.; Rajkumar S.; Wu W.; Huang Z.; Yang X. Asymmetric Synthesis of β-Indolyl Cyclopentanones and Cyclopentylamides with an All-Carbon Quaternary Stereocenter via Chiral Phosphoric Acid Catalyzed Friedel–Crafts Alkylation Reactions. Org. Lett. 2019, 21 (10), 3563–3567. 10.1021/acs.orglett.9b00963. [DOI] [PubMed] [Google Scholar]
- Xie H.-P.; Wu B.; Wang X.-W.; Zhou Y.-G. Chiral Brønsted acid-catalyzed conjugate addition of indoles to azadienes: Enantioselective synthesis of hetero-triarylmethanes. Chin. J. Catal. 2019, 40 (10), 1566–1575. 10.1016/S1872-2067(19)63396-6. [DOI] [Google Scholar]