

Graphical Abstract
The use of asymmetric catalysis in the enantioselective construction of all-carbon quaternary stereocenters is a contemporary challenge in organic synthesis.2–7 Conjugate addition reactions of carbon nucleophiles to β,β-disubstituted α,β-unsaturated carbonyl compounds has emerged as an effective strategy to address this challenge.8 Copper-catalyzed asymmetric conjugate addition reactions generally employ highly reactive nucleophiles such as dialkylzinc,9–17 triorganoaluminum,18–27 organozirconium,28–30 and organomagnesium reagents.31–34 Organoaluminum nucleophiles have also been reported to participate in asymmetric conjugate addition reactions to β,β-disubstituted α,β-unsaturated carbonyl compounds under rhodium catalysis.35 Unfortunately, use of such highly reactive organometallic species require rigorous exclusion of moisture and oxygen, and can limit the functional group tolerance of these transformations.
Efforts by Hayashi and coworkers have led to the development of rhodium complexes to catalyze the addition of organoboron nucleophiles to α,β-unsaturated carbonyl compounds with excellent levels of enantioselectivity.36 In contrast to the previously mentioned organometallic nucleophiles, organoboron nucleophiles are easily handled and stored on the benchtop, and the mild nature of these reagents allow for rhodium-catalyzed conjugate addition reactions to tolerate a wide variety of functional groups. While reports of rhodium-catalyzed conjugate addition with β,β-disubstituted α,β-unsaturated carbonyl compounds are rare, chiral diene ligated rhodium complexes have proven to be effective catalysts for the formation of stereogenic all-carbon quaternary centers via this reaction manifold. Unfortunately, commercially available boronic acids are not suitable for this process, which often requires the use of tetraaryl borates37 or boroxines.38 Despite the associated advantages of rhodium-catalyzed conjugate addition, the high cost of rhodium, and oxygen sensitivity of these processes are undesirable.
Palladium catalysis has recently emerged as a more robust and cost-effective alternative to rhodium-catalyzed conjugate addition processes. The asymmetric construction of tertiary β-substituted ketones utilizing a cationic palladium(II)-DuPHOS complex was described by Minnaard and coworkers in 2011, and displays a remarkable tolerance to air and moisture.39 Unfortunately, the conditions outlined in this report are not suitable for the asymmetric construction of quaternary stereocenters. A subsequent report from Lu and colleagues disclosed that a cationic palladium(II)-bipyridine complex is a competent catalyst for the reaction of β,β-disubstituted α,β-unsaturated ketones, although it is worth noting that these conditions do not allow for the construction of enantioenriched all-carbon quaternary centers.40 Given our interests in the asymmetric construction of all-carbon quaternary stereocenters, we focused our attention toward an enantioselective variant of this transformation. Described herein are recent developments in the field of palladium-catalyzed conjugate addition toward the asymmetric construction of all-carbon quaternary centers.
Reaction Development and Substrate Scope
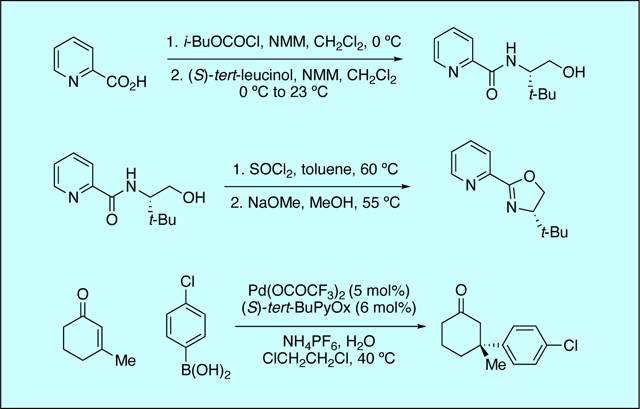
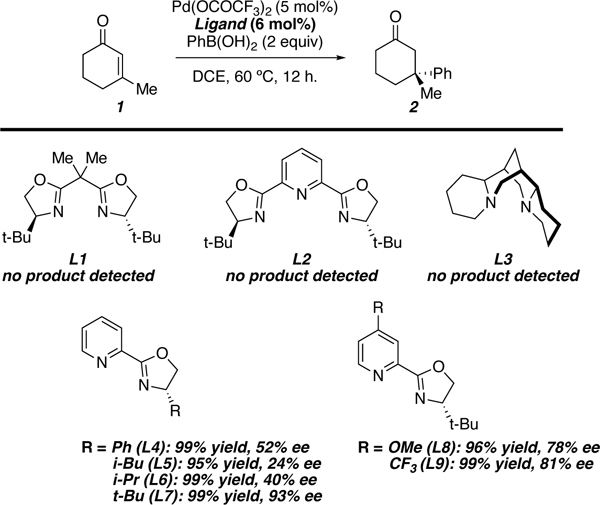
In 2011 our laboratory disclosed the first report of an asymmetric palladium-catalyzed conjugate addition to generate all-carbon quaternary stereocenters.41 Our studies revealed that chiral pyridineoxazoline (PyOx) ligands (L4-L9) complexed to Pd(OCOCF3)2 were particularly adept in providing the conjugate addition products in high yield. Other chiral ligands such as bisoxazoline (L1), pyridinebisoxazoline (L2), and (−)-spartine (L3) did not give rise to catalytically active palladium complexes. The palladium catalyst derived from t-BuPyOx (L7) provided the highest levels of enantioselectivity, whereas ligands derived from phenylalanine (L4), leucine (L5), or valine (L6) provided significantly lower levels of enantioselectivity. Finally, electronic rich (L8), and electron poor (L9) t-BuPyOx derivatives were both inferior to the unsubstituted variant (Table 1). While t-BuPyOx can be synthesized in one step from methyl picolinimidate and tert-leucinol, the two-step procedure outlined in the original Organic Syntheses article was more efficient to produce larger quantities of t-BuPyOx in our hands.
Table 1.
Investigation of Chiral Ligands
|
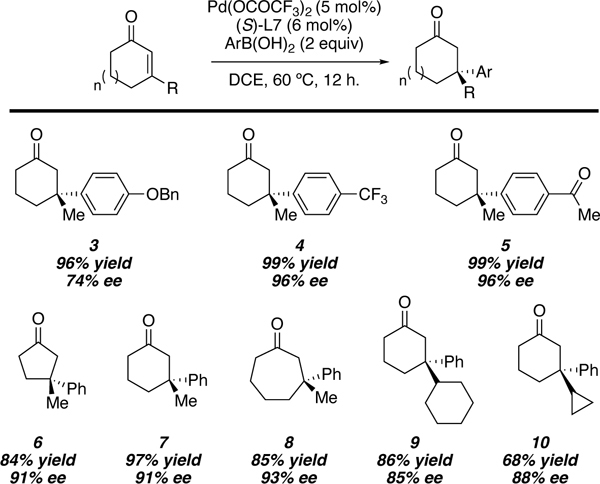
Our initial report details the addition of a wide variety of boronic acids including electron rich (product 3), electron poor (products 4 and 5), and boronic acids containing potentially reactive functional groups such as ketones (product 5). Notably, electron rich boronic acids tend to exhibit lower levels of enantioselectivity. With respect to the enone component, the reaction of five-, six-, and seven-membered rings were well tolerated (products 6, 7, and 8 respectively). Moreover, substrates with increased steric hindrance about the enone react smoothly under the standard reaction conditions to generate highly congested all-carbon quaternary centers (products 9 and 10). After our initial report, Stanley and coworkers disclosed a similar study that details the conjugate addition of arylboronic acids to β-aryl α,β-unsaturated ketones to yield enantioenriched bis-benzylic quaternary stereocenters.42
Despite the broad substrate scope and efficiency of the process, attempts to scale up the reaction with gram quantities of 1 were initially unsuccessful (Scheme 1A, Eq. 1). To this end, we set out to study the mechanism computationally in collaboration with Ken Houk’s group at UCLA and devise an improved set of reaction conditions to address the problem of scalability.43 An important feature of our proposed mechanism is the enantiodetermining carbopalladation to provide the palladium(II) enolate (IV), which must then be protonated to close the catalytic cycle (Scheme 1B). We initially hypothesized that the boronic acid could act as a proton source to turn over the catalytic cycle; however, the lack of scalability of the reaction caused us to reconsider this hypothesis. We posited that addition of an external proton source would be beneficial for the reaction. To our delight, addition of 5 equivalents of water to the previously employed reaction conditions allowed for facile scale-up of this process with no change in yield or enantioselectivity (Scheme 1A, Eq. 2).
Scheme 1.

Gram-scale reaction and role of water
We then turned our attention to the addition of metal salts containing weakly coordinating counter ions (PF6−, SbF6−, BF4−, etc) in an effort to improve the activity of the palladium catalyst. This would allow for lower catalyst loadings and lower reaction temperatures. We hypothesize that weakly coordinating anions could undergo a salt metathesis with the palladium(II) trifluoroacetate, resulting in a more reactive, cationic palladium(II) species. In line with our hypothesis, addition of NaCl (Table 3, entry 1) inhibited reactivity, presumably due to the strongly coordinating nature of chloride anion. Conversely, when sodium salts bearing weakly coordinating counterions were employed, enhanced reactivity was observed (Table 3, entries 2–4); however, the enantioselectivity of the process was diminished. Enantioselectivity could be restored at the expense of the reaction rate when tetrabutylammonium salts were employed (Table 3, entries 5–6). Sodium tetraphenylborate (Table 3, entry 7) failed to promote the desired reactivity due to concomitant formation of biphenyl, presumably via an oxidative coupling of the tetraphenylborate nucleophile. Finally, ammonium salts were effective in providing an optimal balance of reactivity and enantioselectivity (Table 3, entries 8–9), with the hexafluorophosphate anion yielding the best result (Table 3, entry 9).
Table 3.
Effect of Salt Additives
| ||||
|---|---|---|---|---|
| entry | additive | time (h) | yield | ee |
| 1 | NaCl | 24 | trace | − |
| 2 | NaBF4 | 8 | 81 | 88 |
| 3 | NaPF6 | 6 | 97 | 87 |
| 4 | NaSbF6 | 5 | 99 | 81 |
| 5 | n-Bu4NPF6 | 24 | 98 | 90 |
| 6 | n-Bu4NBF4 | 24 | 95 | 88 |
| 7 | NaBPh4 | 24 | trace | − |
| 8 | NH4BF4 | 15 | 93 | 89 |
| 9 | NH4PF6 | 12 | 96 | 91 |
The combination of 5 equivalents of water, and NH4PF6 allowed for the reaction to be conducted at lower temperatures, and with lower catalyst loadings to provide product 2 in essentially the same yield and enantioselectivity as the standard reaction conditions (Scheme 2A). Moreover, the addition of water, and NH4PF6 allows for more efficient reaction of substrates that proved difficult under the initial set of conditions (Scheme 2B).
Scheme 2.

Low Catalyst Load and Reactivity of Difficult Substrates
Subsequent efforts from our group have led to the expansion of this method to include chromenones as electrophiles, providing access to tertiary stereocenter containing flavanones (Scheme 3A).44 Unfortunately, even the improved conditions outlined above were unable to promote the reaction of C(2)-substituted chromenones. Interestingly, Stanley and coworkers reported the reaction of C(2)-substituted chromenones with a palladium(II)-phenanthroline complex in the presence of aqueous Na(OCOCF3); however, these reaction conditions generate a racemic mixture of the products bearing tetrasubstituted stereocenters.45 Subsequent investigation of different ligand scaffolds in collaboration with Sukwon Hong’s group of GIST, revealed that pyridine-dihydroisoquinoline (PyDHIQ) ligands, and an oxygen atmosphere allowed for smooth reaction of C(2)-substituted chromenone electrophiles with excellent levels of enantioselectivity (Scheme 3B).46 Finally, we became interested in the reaction of 3-acyl cyclohexenones, a challenging substrate class due to the possibility of the formation of constitutional isomers. Employing palladium(II) trifluoroacetate and (S)-t-BuPyOx allows for selective formation of compound 20, which bears a quaternary stereocenter at the exclusion of forming a tertiary center via addition to the other end of the olefin (Scheme 3C).47
Scheme 3.

Selected Electrophile Examples
Applications in Synthesis
The robust nature and scalability of this reaction has led to its use in the synthesis natural products and active pharmaceutical ingredients. In 2019, AbbVie reported the synthesis of ABBV-2222 (24), which is a preclinical candidate for the treatment of cystic fibrosis.48 Synthesis of ABBV-2222 was accomplished via a key conjugate addition of boronic acid 22 to chromenone 21 to produce 13 grams of conjugate addition adduct 23 with excellent enantioselectivity, in a single pass. Enantioenriched flavanone 23 could then be carried forward through 5 more steps to generate >130 g of the active pharmaceutical ingredient 24 (Scheme 4).49
Scheme 4.

Synthesis of ABBV-2222
Additionally, our group has utilized a palladium-catalyzed conjugate addition to accomplish a formal synthesis of (+)-dichroanone and (+)-taiwaniaquinone H.50 Plotting the enantiomeric ratio versus the Hammett (σP) value for a variety of arylboronic acids revealed a strong positive linear correlation. This suggests that the difference in the diastereomeric transition states in the enantiodetermining step is larger with more electron withdrawing para substituents on the arylboronic acid. To this end, para-halogenated boronic acid 25 was selected to undergo conjugate addition with enone 1 to provide enantioenriched β-quaternary ketone 26 in excellent yield and enantioselectivity. This material was then advanced to intermediate 27, which is a known intermediate in the synthesis of (+)-dichroanone and (+)-taiwaniaquinone H.51
More recently, Wood and coworkers utilized an enantioselective palladium-catalyzed conjugate addition toward the total synthesis of (−)-caesalpinflavans A and B in 2019.52 Addition of phenyl boronic acid to chromenone 30 allowed for enantioselective synthesis of flavanone 31. A divergent approach from this key intermediate can then grant access to (−)-caesalpinflavans A and B (Scheme 6). This strategy highlights the mild nature of boronic acid nucleophiles, which allowed for the use of 30 in its unprotected form.
Scheme 6.

Synthesis of (−)-Caesalpinflavens A and B
Conclusion
In summary, the asymmetric palladium-catalyzed conjugate addition of arylboronic acids to β,β-disubstituted α,β-unsaturated carbonyl compounds has emerged as a useful strategy for the construction of enantioenriched quaternary centers. The t-BuPyOx ligand required for the catalytically active cationic palladium(II) complex is easily synthesized in two steps from commercially available starting materials, as outlined in the original article. Furthermore, this process is advantageous due to the use of commercially available boronic acid nucleophiles, which are mild and easily handled on the benchtop. Finally, the reaction is easily scaled and extremely tolerant to air and water. The aforementioned attributes make this strategy attractive for the facile construction of enantioenriched building blocks toward the synthesis of complex molecules.
Scheme 5.

Formal Synthesis of (+)-Dichroanone and (+)-Taiwaniaquinone H
Table 2.
Selected substrate scope
|
Biographies

Stephen R. Sardini was born in Fairfax, VA in 1990 and earned his B.S. in chemistry from The Ohio State University in 2012. He then earned his Ph.D. in 2020 under the direction of Prof. M. Kevin Brown at Indiana University. Stephen is currently an NIH Ruth L. Kirschstein NRSA postdoctoral fellow conducting research in the laboratory of Prof. Brian Stoltz at the California Institute of Technology.

Brian M. Stoltz was born in Philadelphia, PA in 1970 and obtained his B.S. degree from the Indiana University of Pennsylvania in Indiana, PA. After acquiring his Ph.D. at Yale University in the laboratory of John L. Wood, he commenced an NIH postdoctoral fellowship at Harvard under E. J. Corey. In 2000, he accepted a position at Caltech, where he is currently a Professor of Chemistry. His research interests lie in the development of new methodologies with general applications in synthetic chemistry.
References
- 1.Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, California, 91125, United States. Email: stoltz@caltech.edu. We thank the NIH (R01GM080269) and Caltech for financial support. S.R.S thanks the NIH for a Ruth L. Kirschstein NRSA Postdoctoral fellowship (F32GM139300–02). [Google Scholar]
- 2.Denissova I; Barriault L. Tetrahedron 2003, 59, 10105–10146. [Google Scholar]
- 3.Douglas CJ; Overman LE. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 5363–5367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Christoffers J; Baro A. Adv. Synth. Catal. 2005, 347, 1473–1482. [Google Scholar]
- 5.Trost BM; Jiang C. Synthesis 2006, 369–396. [Google Scholar]
- 6.Mohr JT Stoltz BM. Chem. –Asian J. 2007, 2, 1476–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cozzi PG; Hilgraf R; Zimmermann N. Eur. J. Org. Chem. 2007, 36, 5969–5994. [Google Scholar]
- 8.Hawner C; Alexakis A. Chem. Commun. 2010, 46, 7295–7306. [DOI] [PubMed] [Google Scholar]
- 9.Feringa BL. Acc. Chem. Res. 2000, 33, 346–353. [DOI] [PubMed] [Google Scholar]
- 10.Wu J; Mampreian DM; Hoveyda AH. J. Am. Chem. Soc. 2005, 127, 4584–4585. [DOI] [PubMed] [Google Scholar]
- 11.Hird AW; Hoveyda AH. J. Am. Chem. Soc. 2005, 127, 14988–14989. [DOI] [PubMed] [Google Scholar]
- 12.Wilsily A; Fillion EJ. Am. Chem. Soc. 2006, 128, 2774–2775. [DOI] [PubMed] [Google Scholar]
- 13.Lee K-S; Brown MK; Hird AW; Hoveyda AH. J. Am. Chem. Soc. 2006, 128, 7182–7184. [DOI] [PubMed] [Google Scholar]
- 14.Brown MK; May TL; Baxter CA; Hoveyda AH. Angew. Chem., Int. Ed. 2007, 46, 1097–1100. [DOI] [PubMed] [Google Scholar]
- 15.Wilsey A; Fillion E. Org. Lett. 2008, 10, 2801–2804. [DOI] [PubMed] [Google Scholar]
- 16.Wilsey A; Fillion EJ. Org. Chem. 2009, 74, 8583–8594. [DOI] [PubMed] [Google Scholar]
- 17.Dumas AM; Fillion E. Acc. Chem. Res. 2010, 43, 440–454. [DOI] [PubMed] [Google Scholar]
- 18.D’Augustin M; Palais L; Alexakis A. Angew. Chem., Int. Ed. 2005, 44, 1376–1378. [DOI] [PubMed] [Google Scholar]
- 19.Fuchs N; d’Augustin M; Humam M; Alexakis A; Taras R; Gladiali S. Tetrahedron: Assym. 2005, 16, 3143–3146. [Google Scholar]
- 20.Vuagnoux-d’Augustin M; Alexakis A. Chem. –Eur. J. 2007, 13, 9647–9662. [DOI] [PubMed] [Google Scholar]
- 21.Vuagnoux-d’Augustin M; Kehrli M; Alexakis A. Synlett 2007, 2057–2060. [Google Scholar]
- 22.Palais L; Mikhel IS; Bournaud C; Micouin L; Falciola CA; Vuagnoux-d’Augustin M; Rosset S; Bernardinelli G; Alexakis A. Angew. Chem., Int. Ed. 2007, 46, 7462–7465. [DOI] [PubMed] [Google Scholar]
- 23.May TL; Brown MK; Hoveyda AH. Angew. Chem., Int. Ed. 2008, 47, 7358–7362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hawner C; Li K; Cirriez V; Alexakis A. Angew. Chem., Int. Ed. 2008, 47, 8211–8214. [DOI] [PubMed] [Google Scholar]
- 25.Ladjel C; Fuchs N; Zhao J; Bernardinelli G Alexakis A. Eur. J. Org. Chem. 2009. 4949–4955. [Google Scholar]
- 26.Palais L; Alexakis A. Chem. –Eur. J. 2009, 15, 10473–10485. [DOI] [PubMed] [Google Scholar]
- 27.Müller D; Hawner C; Tissot M; Palais L; Alexakis A. Synlett 2010, 1694–1698. [Google Scholar]
- 28.Roth PMC; Sidera M; Maksymowicz RM; Fletcher SP. Nat. Protoc. 2014, 9, 104–111. [DOI] [PubMed] [Google Scholar]
- 29.Gao Z; Fletcher SP. Chem. Sci. 2016, 8, 641–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ardkhean R; Mortimore M; Paton RS; Fletcher SP. Chem Sci. 2018, 9, 2628–2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martin D; Kehrli S; Vuagnoux-d’Augustin M; Clavier H; Mauduit M; Alexakis AJ. Am. Chem. Soc. 2006, 128, 8416–8417. [DOI] [PubMed] [Google Scholar]
- 32.Hénon H; Mauduit M; Alexakis A. Angew. Chem., Int. Ed. 2008, 47, 9122–9124. [DOI] [PubMed] [Google Scholar]
- 33.Matsumoto Y; Yamada K-I; Tomioka KJ. Org. Chem. 2008, 73, 4578–4581. [DOI] [PubMed] [Google Scholar]
- 34.Kehrli S; Martin D; Rix D; Mauduit M; Alexakis A. Chem. –Eur. J. 2010. 16, 9890–9904. [DOI] [PubMed] [Google Scholar]
- 35.Hawner C; Müller D; Gremaud L; Felouat A; Woodward S; Alexakis A. Angew. Chem., Int. Ed. 2010, 49, 7769–7772. [DOI] [PubMed] [Google Scholar]
- 36.Hayashi T; Yamasaki K. Chem. Rev. 2003, 103, 2829–2844. [DOI] [PubMed] [Google Scholar]
- 37.Shintani R; Tsutsumi Y; Nagaosa M; Nishimura T; Hayashi TJ. Am. Chem. Soc. 2009, 131, 13588–13589. [DOI] [PubMed] [Google Scholar]
- 38.Shintani R; Takeda M; Nishimura T; Hayashi T. Angew. Chem., Int. Ed. 2010, 49, 3969–3971. [DOI] [PubMed] [Google Scholar]
- 39.Gini F; Hessen B; Minnaard AJ. Org. Lett, 2005, 7, 5309–5312. [DOI] [PubMed] [Google Scholar]
- 40.Lin S; Lu X. Org. Lett. 2010, 12, 2536–2539. [DOI] [PubMed] [Google Scholar]
- 41.Kikushima K; Holder J; Gattie M; Stoltz BMJ .Am. Chem. Soc. 2011, 133, 6902–6905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kadam A; Ellern A; Stanley L. Org. Lett. 2017, 19, 4062–4065. [DOI] [PubMed] [Google Scholar]
- 43.Holder J; Zou L; Marziale A; Liu P; Lan Y; Gatti M; Kikushima K; Houk KN; Stoltz BM. J. Am. Chem. Soc. 2013, 135, 14996–15007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Holder J; Marziale A; Gatti M; Mao B; Stoltz BM. Chem. Eur. J. 2013, 19, 74–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gerten A; Stanley L. Tetrahedron Lett. 2016, 57, 5460–5463. [Google Scholar]
- 46.Baek D; Ryu H; Ryu J Lee J; Stoltz BM; Hong S. Chem. Sci. 2020, 11, 4602–4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Holder J; Goodman E; Kikushima K; Gatti M; Marziale, Stoltz BM. Tetrahedron 2015, 71, 5781–5792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang X; Liu B; Searle X; Yeung C; Bogdan A; Greszler S; Singh A; Fan Y; Swensen A; Vortherms T; Balut C; Jia Y; Desino K; Gao W; Yong H; Tse C; Kym PJ. Med. Chem. 2018, 61, 1436–1449 [DOI] [PubMed] [Google Scholar]
- 49.Greszler S; Shelat B; Voight E. Org. Lett. 2019, 21, 5725–5727. [DOI] [PubMed] [Google Scholar]
- 50.Shockley S; Holder J; Stoltz BM. Org. Lett. 2014, 16, 6362–6365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li L-Q; Li M-M; Chen D; Liu H-M; Geng H-C; Jin J; Qin H-B. Tetrahedron Lett. 2014, 55, 5960–5962. [Google Scholar]
- 52.Timmerman J; Sims N; Wood JL. J. Am. Chem. Soc. 2019, 141, 10082–10090. [DOI] [PubMed] [Google Scholar]