

Abstract
Background
Head and neck squamous cell carcinoma (HNSCC) is one of the most frequent malignancies worldwide and is characterized by unfavorable prognosis, high lymph node metastasis and early recurrence. However, the molecular events regulating HNSCC tumorigenesis remain poorly understood. Therefore, uncovering the underlying mechanisms is urgently needed to identify novel and promising therapeutic targets for HNSCC. In this study, we aimed to explore the role of pleckstrin‐2 (PLEK2) in regulating HNSCC tumorigenesis.
Methods
The expression pattern of PLEK2 and its clinical significance in HNSCC were determined by analyzing publicly assessable datasets and our own independent HNSCC cohort. In vitro and in vivo experiments, including cell proliferation, colony formation, Matrigel invasion, tumor sphere formation, ALDEFLUOR, Western blotting assays and xenograft mouse models, were used to investigate the role of PLEK2 in regulating the malignant behaviors of HNSCC cells. The underlying molecular mechanisms for the tumor‐promoting role of PLEK2 were elucidated using co‐immunoprecipitation, cycloheximide chase analysis, ubiquitination assays, chromatin immunoprecipitation‐quantitative polymerase chain reaction, luciferase reporter assays and rescue experiments.
Results
The expression levels of PLEK2 mRNA and protein were significantly increased in HNSCC tissues, and PLEK2 overexpression was strongly associated with poor overall survival and therapeutic resistance. Additionally, PLEK2 was important for maintaining the proliferation, invasion, epithelial‐mesenchymal transition, cancer stemness and tumorigenesis of HNSCC cells and could alter the cellular metabolism of the cancer cells. Mechanistically, PLEK2 interacted with c‐Myc and reduced the association of F‐box and WD repeat domain containing 7 (FBXW7) with c‐Myc, thereby avoiding ubiquitination and subsequent proteasome‐mediated degradation of c‐Myc. Moreover, the c‐Myc signaling activated by PLEK2 was important for sustaining the aggressive malignant phenotypes and tumorigenesis of HNSCC cells. c‐Myc also directly bounded to the PLEK2 promoter and activated its transcription, forming a positive feedback loop.
Conclusions
Collectively, these findings uncover a previously unknown molecular basis of PLEK2‐enhanced c‐Myc signaling in HNSCC, suggesting that PLEK2 may represent a promising therapeutic target for treating HNSCC.
Keywords: c‐Myc, cancer stemness, FBXW7, HNSCC, PLEK2, positive feedback loop, tumorigenesis, ubiquitination‐mediated degradation
Abbreviations
- 4‐NQO
4‐Nitroquinoline 1‐oxide
- ALDH
aldehyde dehydrogenase
- ANTs
adjacent normal tissues
- Bax
Bcl‐2‐associated X protein
- Bcl‐2
B‐cell lymphoma 2
- CHIP‐qPCR
chromatin immunoprecipitation‐quantitative polymerase chain reaction
- CHX
cycloheximide
- Co‐IP
co‐immunoprecipitation
- CSCs
cancer stem cells
- DEAB
diethylaminobenzaldehyde
- DMEM
Dulbecco's Modified Eagle Medium
- EdU
5‐ethynyl‐2'‐deoxyuridine
- ELDA
extreme limiting dilution analysis
- EMT
epithelial–mesenchymal transition
- ENO1
enolase 1
- EpCAM
epithelial cell adhesion molecule
- FBS
fetal bovine serum
- FBXW7
F‐box and WD repeat domain containing 7
- GAPDH
glyceraldehyde‐3‐phosphate dehydrogenase
- HA‐FBXW7
hemagglutinin‐FBXW7
- HK
hexokinase
- HNC
head and neck cancer
- HNSCC
head and neck squamous cell carcinoma
- IHC
immunohistochemistry
- LDHA
lactate dehydrogenase A
- MCM2
minichromosome maintenance complex component 2
- MTT
3‐(4,5‐Dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide
- OCT4
octamer‐binding transcription factor 4
- OV
overexpression
- PCNA
proliferating cell nuclear antigen
- PKM2
pyruvate kinase M2
- PLEK2
pleckstrin 2
- RIPA
radioimmunoprecipitation assay
- SDS‐PAGE
sodium dodecyl sulphate‐polyacrylamide gel electrophoresis
- shRNA
short hairpin RNA
- SOX2
sex‐determining region Y‐box 2
- TCGA
The Cancer Genome Atlas
1. BACKGROUND
Head and neck cancer (HNC) is the sixth most common cancer globally, and squamous cell carcinoma (HNSCC) is the most prevalent histological subtype, constituting up to 90% of all HNC cases [1]. HNSCC is derived from the mucosal epithelium lining the oral cavity, pharynx and larynx [2, 3]. Although it is well established that HNSCC develops from different degrees of neoplasia to eventually become invasive carcinoma, most HNSCC cases are diagnosed at late phases when aggressive treatments are needed. In addition, advanced‐stage HNSCC features unfavorable clinical outcomes, a high recurrence rate and high metastatic potential. Treatments for HNSCC are multimodal, including surgery, chemoradiotherapy, targeted therapy and immunotherapy [4]. Unfortunately, the overall survival of HNSCC patients has remained stagnant over the past decade [5]. Therefore, it is imperative to elucidate the molecular events in HNSCC, which could undoubtedly provide new opportunities for improving the treatment and outcomes of this malignancy.
Pleckstrin‐2 (PLEK2) is located on chromosome 14q23.3‐q24.1 and encodes a 353‐amino acid protein widely expressed in multiple tissues. PLEK2 binds to membrane‐associated phosphatidylinositol produced by phosphatidylinositol 3‐kinase, which subsequently regulates actin cytoskeleton organization and cell spreading [6]. Although aberrant expression of PLEK2 has been shown to play a critical role in regulating many important biological processes such as inflammation, erythropoiesis and tumorigenesis [7, 8, 9], its underlying cellular and molecular mechanisms remain poorly understood. PLEK2 was reported to be highly expressed in several malignancies, including non‐small cell lung cancer [10], gallbladder cancer [11] and gastric cancer [12], while low PLEK2 mRNA levels were observed in multiple myeloma bone marrow progenitor cells [13] and prostate cancer [14]. These findings indicate that the biological functions of PLEK2 might be tumor type‐dependent and closely related to the tumor microenvironment in which cancer cells reside. Our large‐scale transcriptomic analysis of differentially expressed genes (DEGs) between HNSCC tissues and paired adjacent normal tissues (ANTs) revealed that the expression levels of PLEK2 were consistently increased in malignant specimens, indicating that PLEK2 might be involved in regulating HNSCC tumorigenesis. However, the clinical relevance of PLEK2 and its regulatory roles in HNSCC remains poorly understood.
In this study, we hypothesized that PLEK2 plays a crucial role in HNSCC tumorigenesis. Our aim was to determine the clinical significance of PLEK2 in HNSCC and to elucidate the molecular events involved in PLEK2‐mediated signaling for promoting the progression of HNSCC. First, the relationship between aberrant PLEK2 expression and unfavorable clinical outcomes in HNSCC was investigated. Additionally, the underlying molecular mechanisms for the role of PLEK2 in HNSCC tumorigenesis were systemically explored. Moreover, a regulatory signaling loop that modulates PLEK2 expression in HNSCC was identified.
2. MATERIALS AND METHODS
2.1. Patients and clinical samples
The study protocol regarding the re‐utilization of clinical samples and medical information was approved by the Ethics Committee of the First Affiliated Hospital of Sun Yat‐sen University (Guangzhou, Guangdong, China). Written informed consent was obtained from all patients. All the procedures were conducted following the Declaration of Helsinki. The following inclusion criteria were used: 1) all patients underwent primary surgery for HNSCC; 2) the diagnosis was pathologically confirmed as HNSCC. The exclusion criteria were as follows: 1) patients with more than one primary malignancy; 2) patients with incomplete clinical or prognostic data. Tumors and ANTs were obtained at the First Affiliated Hospital of Sun Yat‐sen University between January 2013 and June 2015. Fifteen paired HNSCC and ANT tissues were used for the western blotting analysis of the PLEK2 expression pattern. All tissue specimens were snap‐frozen in liquid nitrogen and stored at ‐80°C or formalin‐fixed and paraffin‐embedded. For the patients who underwent induction chemotherapy, the regimen was delivered with docetaxel 75 mg/m2 intravenously on day 1, cisplatin 75 mg/m2 intravenously on day 1 and 5‐fluorouracil 750 mg/m2 by infusion for 120 h on days 1‐5. The treatment was administered every 3 weeks for 2 cycles.
2.2. Cell lines and cell culture
The HNSCC cell lines UMSCC‐1 (SCC‐1) and UTSCC‐23 (SCC‐23) were obtained from the University of Michigan (Ann Arbor, MI, USA), and FaDu was obtained from American Type Culture Collection (Manassas, VA, USA). All cell lines were maintained in the Dulbecco's modified eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin‐streptomycin at 37°C with 5% CO2. Cisplatin‐resistant SCC‐1, SCC‐23 and FaDu cell lines were developed by chronic exposure of the parent cisplatin‐sensitive cell lines in 0.5‐25 μmol/L of cisplatin.
2.3. 3‐(4,5‐Dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT) assay
The cells were seeded in 96‐well plates at a density of 3 × 103 cells/well. In brief, MTT solution (5 mg/mL, Sigma‐Aldrich, Burlington, MA, USA) was added to each well, and the cell culture was maintained for 4 h at 37°C. Then, the supernatant was carefully removed, and the formazan product was dissolved with dimethyl sulfoxide. The absorbance was measured with a Synergy HT microplate reader (BioTek Instruments, Winooski, VT, USA) at a wavelength of 570 nm.
2.4. 5‐ethynyl‐2’‐deoxyuridine (EdU) assay
Briefly, 10 μmol/L EdU labeling solution was added to the wells, and the cells were incubated for 2 h. After fixation and permeabilization, the cells were stained with the Click‐iT® Plus reaction cocktail (Invitrogen, Carlsbad, CA, USA) for 30 min in the dark. Then the Hoechst 33342 dye was used for nuclear staining. Images were taken with an inverted fluorescence microscope (Leica Microsystems, Wetzlar, Hessen, Germany).
2.5. Colony formation assay
Cells were seeded in 6‐well plates at a density of 3000 cells/well and cultured for 2 weeks. After fixation with 4% paraformaldehyde, 0.5% crystal violet was used to visualize the colonies.
2.6. Sphere formation assay
Briefly, the cells were cultured with ultra‐low adhesion plates and maintained in DMEM/F12 medium supplemented with 1% B27 supplement (Invitrogen), 1% N2 supplement (Invitrogen), 20 ng/mL epidermal growth factor and 10 ng/mL basic fibroblast growth factor. The images of tumor spheres were captured with an inverted microscope (Leica Microsystems).
2.7. Matrigel invasion assay
Matrigel invasion assay was performed using Transwell chambers with a pore size of 8 μm (Costar, Cambridge, MA, USA). Following serum starvation, the cells in the serum‐free medium were seeded into the upper chambers, and the complete culture medium was added to the bottom chambers. After 48 h of incubation, the non‐invaded cells on the upper side of the membrane were removed with a cotton swab. Then the invaded cells were fixed with 4% paraformaldehyde and subsequently stained with 0.5% crystal violet. Six fields per chamber were imaged, and the average area of the invaded cells per field was calculated as a measure for cell invasion.
2.8. Production of lentiviral particles and cell transfection
The short hairpin RNAs (shRNAs) targeting PLEK2 were inserted into the LV3‐pGLV‐h1‐GFP‐puro vector (GenePharma, Shanghai, China) to establish recombinant lentiviral expression plasmids. The full‐length human PLEK2 was cloned to a pGCL‐GFP vector to construct the PLEK2 overexpression vector. The recombinant lentiviral vectors, packaging plasmids and envelope plasmids were co‐transfected into HEK‐293 T cells to produce lentiviruses. The c‐Myc shRNA was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). For transient transfection, siRNAs targeting PLEK2 or c‐Myc were transfected into cancer cells with Lipofectamine RNAiMAX reagent (Invitrogen) following the manufacturer's instructions. The recombinant pcDNA3‐c‐Myc (Addgene, Cambridge, MA, USA) or pcDNA3 control plasmid was transfected into cells with Lipofectamine 2000 (Invitrogen). The sequences of shPLEK2, siPLEK2 and sic‐Myc are listed in Supplementary Table S1.
2.9. Quantitative polymerase chain reaction (qPCR)
Total RNA was extracted using the Quick‐RNA™ kit (Zymo Research Corp, Irvine, CA, USA) according to the manufacturer's recommendations. The first‐strand cDNA was synthesized reversely with the SuperScript™III First‐Strand Synthesis SuperMix (Invitrogen). Briefly, the Oligo (dT)‐primers and annealing buffer were added to the RNA samples. The mixture was incubated at 65°C for 5 min, and immediately chilled on ice for 2 min. The first‐strand reaction mix and SuperScript® III/RNase Out™ enzyme mix were added to the mixture, followed by incubation at 50°C for 50 min. The reaction was terminated at 85°C for 5 min and the cDNA samples were stored at ‐20°C until use. The amplification of cDNA was performed with LightCycler 480@ SYBR Green I MasterMix (Roche, Basel, Switzerland) on a CFX96 Real‐Time PCR detection system (Bio‐Rad, Hercules, CA, USA). Thermal cycling conditions were as follows: 1 cycle for 5 min at 95°C, 40 PCR cycles of amplification (95°C for 10 s, 60°C for 20 s and 72°C for 30 s), 1 cycle for obtaining melting curve and 1 cycle for 10 s at 40°C for cooling. The relative changes in gene expression were measured with the 2−ΔΔCt method. The sequences for the primers used in qPCR are listed in Supplementary Table S1.
2.10. Measurements of glucose uptake and lactate production
To measure the glucose uptake, the cells were first cultured in media without FBS and glucose for 6 h, followed by incubation with a glucose‐free DMEM supplemented with 10 μmol/L 2‐deoxy‐2‐[(7‐nitro‐2,1,3‐benzoxadiazol‐4‐yl) amino]‐D‐glucose for 2 h. Then the samples were washed twice with phosphate‐buffered saline (PBS) and analyzed by flow cytometry (BD Biosciences, Franklin Lakes, NJ, USA). The measurement of lactate production in the samples was performed using lactate colorimetric assay kit (BioVision, Milpitas, CA, USA) according to the manufacturer's protocols. Of note, the final amount of lactate in the samples was determined by deducting the amount of lactate in the media.
2.11. ALDHEFLUOR assay
ALDEFLUOR assay was performed with the ALDEFLUOR assay kit (StemCell Technologies Inc., Vancouver, BC, Canada), following the manufacturer's instructions. For every individual sample, an aliquot was treated with diethylaminobenzaldehyde (DEAB) as a negative control. Sample analysis was conducted on a DxFLEX flow cytometer (Beckman Coulter, Inc., Brea, CA, USA).
2.12. Cell isolation and flow cytometry
The primary HNSCC tumor tissues from 6 patients were mechanically minced on ice and then digested into single‐cell suspensions using Collagenase I (Sigma‐Aldrich). Antibodies of EpCAM and CD45 were used to separate epithelial tumor cells from stromal cells and blood cells. The aldehyde dehydrogenasehigh (ALDHhigh) tumor cells from primary HNSCC cells were sorted with an ALDEFLUOR assay kit (StemCell Technologies Inc) following the manufacturer's guidelines. Then, the PLEK2 antibody was used to sort PLEK2‐high cells and PLEK2‐low cells. Similarly, the ALDHhigh cancer stem cells (CSCs) from SCC‐1 and SCC‐23 cell lines were sorted with an ALDEFLUOR assay kit (StemCell Technologies Inc). ALDHhigh and ALDHlow subpopulations were isolated by fluorescence‐activated cell sorting (FACS). For apoptosis assay, the cells with indicated treatments were collected and subsequently stained with APC‐annexin V and propidium iodide. The percentage of apoptosis cells was detected using a DxFLEX flow cytometer (Beckman Coulter, Inc.).
2.13. Western blotting
Equal amounts of prepared protein lysates were loaded into a 4%‐20% sodium dodecyl sulfate‐polyacrylamide gel electrophoresis (SDS‐PAGE) gels (Beyotime, Shanghai, China). Following electrophoretic separation, the proteins were transferred onto 0.2 μm polyvinylidene fluoride membranes using the Trans‐Blot Turbo system (Bio‐Rad). After incubating with blocking buffer (EpiZyme, Shanghai, China) for 30 min at 25°C, the membranes were incubated with primary antibodies at 4°C overnight. Then the membranes were probed with the corresponding HRP‐conjugated secondary antibodies (Proteintech, Chicago, IL, USA) for 1 h at 25°C. The Amersham ECL Prime Western Blotting Detection Reagent (Cytiva, Marlborough, MA, USA) was used to visualize the blots. The primary antibodies used in this study were as follows: anti‐PLEK2 (Proteintech), anti‐SOX2 (Proteintech), anti‐CD44 (Proteintech), anti‐CD133 (Santa Cruz Biotechnology), anti‐PCNA (Proteintech), anti‐Cyclin D1 (Proteintech), anti‐MCM2 (Cell Signaling Technology, Danvers, MA, USA), anti‐Bcl‐2 (Proteintech), anti‐Bax (Proteintech), anti‐N‐cadherin (Santa Cruz Biotechnology), anti‐E‐cadherin (Proteintech), anti‐Vimentin (Cell Signaling Technology), anti‐Snail (Santa Cruz Biotechnology), anti‐Slug (Santa Cruz Biotechnology), anti‐Twist (Abcam, Cambridge, UK), anti‐BMI‐1 (Proteintech), anti‐OCT4 (Cell Signaling Technology), anti‐Nanog (Proteintech), anti‐ENO1 (Abcam), anti‐LDHA (Cell Signaling Technology), anti‐PKM2 (Cell Signaling Technology), anti‐HK2 (Proteintech), anti‐c‐Myc (Proteintech), anti‐FBXW7 (Proteintech), and anti‐GAPDH (Proteintech).
2.14. Cycloheximide (CHX) chase assay
The cells with indicated modifications were treated with 100 μg/mL CHX. The total protein lysates were harvested at the indicated time points and subjected to western blotting to determine the half‐life of c‐Myc.
2.15. Luciferase reporter assay
The wild‐type (pGL3‐PLEK2‐wt) or mutant PLEK2 (pGL3‐PLEK2‐mut) promoter‐reporter plasmid was co‐transfected with pcDNA3.0‐c‐Myc or pcDNA‐3.0 into HNSCC cells. At 24 h post‐transfection, the relative luciferase activities were quantified with a Dual‐Luciferase Assay System (Promega, Madison, WI, USA).
2.16. Chromatin immunoprecipitation‐quantitative polymerase chain reaction (ChIP‐qPCR)
The ChIP assay was performed with the ChIP Assay Kit (Millipore, Billerica, MA, USA). The cells were incubated with 1% formaldehyde for 10 min at 25°C to cross‐link proteins and DNA. After washing with PBS twice, the cells were lysed in radioimmunoprecipitation assay (RIPA) buffer, and the DNA was broken into pieces by sonication. The chromatin fragmentation was immunoprecipitated with a primary antibody against c‐Myc or IgG overnight at 4°C. The purified DNA pellets were subjected to qPCR. The potential binding sites between c‐Myc and PLEK2 were first predicted using the R/Bioconductor packages (https://www.bioconductor.org/) including TFBSTools, JASPAR2016 and Biostrings. Then the primers for CHIP‐qPCR were designed with ApE software (https://jorgensen.biology.utah.edu/wayned/ape/) and NCBI primer blast (https://www.ncbi.nlm.nih.gov/tools/primer‐blast/). The primer sequences used for ChIP‐qPCR are available on request.
2.17. Co‐immunoprecipitation (Co‐IP) assay
The harvested cells with indicated treatments were lysed in RIPA buffer (Beyotime). Immunoprecipitation was performed using the primary antibodies and IgG control antibody in the presence of protein A/G magnetic beads for 6 h at 4°C. After washing with the elution buffer thrice, the protein complexes were heated at 100°C for 10 min in the SDS sample buffer. Immunoblotting was performed as described above. To suppress the proteasome activity, the cells with indicated modifications were treated with MG132 (20 μmol/L, 8 h). The total protein lysates were harvested and subjected to immunoblotting.
2.18. Immunohistochemistry (IHC) analysis
After deparaffinization and rehydration in a descending series of ethanol, the formalin‐fixed paraffin‐embedded tissue specimens were blocked with goat serum for 2 h at 25°C, then probed with primary antibodies in a cold room overnight. After washing with PBS thrice, the sections were incubated in the horseradish peroxidase‐labeled secondary antibody solution for 1 h at 25°C. A 3,3'‐diaminobenzidine substrate kit was used to detect the staining signal. For the quantitative IHC analysis, the IHC score of interested proteins equals to the staining intensity (on a scale of 0‐3: negative = 0, weak = 1, moderate = 2, and strong = 3) × the percentage of cells stained (on a scale of 0‐4: 0 = 0%, 1 = 1%‐25%, 2 = 26%–50%, 3 = 51%‐75%, and 4 = 76%‐100%). The percentage of Ki67 positive cells was used to evaluate Ki67 staining intensity in xenograft tumor tissues.
2.19. Tumor models
All the mice used in this study were obtained from Guangdong Medical Laboratory Animal Center (Foshan, Guangdong, China). For the 4‐nitroquinoline 1‐oxide (4‐NQO) induced HNSCC mouse model, six‐week‐old C57BL/6 male mice were treated with 4‐NQO in drinking water (50 μg/mL) for 16 weeks and then received regular drinking water for another 8‐10 weeks. For the xenograft mouse model, 6‐week‐old immune‐deficient BALB/c nude mice were injected subcutaneously with 2 × 106 HNSCC cells in the dorsal flank region. Four weeks after the injection, mice were sacrificed and tumor tissues were excised by surgery. The tumor size and weight were measured and recorded. The collected tumor samples were fixed with 4% paraformaldehyde for immunohistochemical analysis. For limit‐dilution transplantation in nude mice, different numbers of FACS‐sorted cells (EpCAM+/ALDH+/PLEK2+ and EpCAM+/ALDH+/PLEK2‐) from primary HNSCC tumors were mixed with Matrigel and subcutaneously injected into nude mice for 4 weeks. Similarly, the ALDHhigh CSCs from SCC‐1 or SCC‐23 cells were infected lentiviruses expressing PLEK2 or control lentiviruses. Then, a different number of cells with indicated modifications were subcutaneously into the nude mice for 4 weeks. The tumor‐initiating capacity of cancer cells was analyzed using the Extreme Limiting Dilution Analysis (ELDA) software (http://bioinf.wehi.edu.au/software/elda/).
2.20. Determine the clinical relevance of PLEK2 in cancer with publicly accessible databases
The public datasets comparing the genome‐wide gene expression profiling between tumor tissues and normal tissues/precancerous lesions were downloaded from the Gene Expression Omnibus database (https://www.ncbi.nlm.nih.gov/geo/). The accession numbers were GSE127165, GSE25099, GSE31056, GSE30784, GSE85195 and GSE41613. For The Cancer Genome Atlas (TCGA) data analysis, the RNA‐seq expression profile data and the corresponding clinical information of all TCGA tumor types were retrieved from The National Cancer Institute Genomic Data Commons (https://gdc.cancer.gov/). The X tile software (https://medicine.yale.edu/lab/rimm/research/‐software/) was used to determine the best cutoff point for dividing the cancer patients into high and low PLEK2 expression groups.
2.21. RNA‐Seq analysis
Fourteen paired HNSCC and ANT tissues were used for RNA‐seq analysis. Sequencing was performed with the BGISEQ‐500 platform at the Beijing Genomics Institute (Beijing, China). The hierarchical indexing for spliced alignment of transcripts pipeline (http://www.ccb.jhu.edu/software/hisat/) was applied to align reads against the reference genome (human assembly GRCh37/hg19). The clean reads were mapped to reference genes with the Bowtie2 software, and the expression values of candidate genes were calculated with the RNA‐Seq by Expectation‐Maximization software. The DEGs were identified using the DEGseq. The cutoff of DEGs was absolute fold change ≥ 2 and adjusted P value ≤ 0.001.
2.22. Statistical analysis
All data were presented as the mean ± standard deviation, and the statistical analyses were performed using the Graphpad Prism version 9.0 (GraphPad Software, San Diego, CA, USA). Differences between the two groups were analyzed by independent sample t‐test or paired t‐test. One‐way analysis of variance followed by post hoc Tukey's test was performed to compare the differences among multiple groups. Overall survival was defined as the date of diagnosis to final follow‐up or to the date of death from any cause. The survival differences between different groups were calculated using the Kaplan‐Meier method and analyzed by log‐rank test. The Pearson correlation analysis was used to determine statistical correlations. P < 0.05 was considered statistically significant.
3. RESULTS
3.1. PLEK2 overexpression was an unfavorable prognostic factor for HNSCC
In total, the RNA‐Seq analysis identified 10814 significantly altered genes (6655 upregulated and 4159 downregulated) between 14 paired HNSCC tissues and ANTs (Figure 1A), and the expression of PLEK2 was significantly higher in tumor tissues than in the ANTs (Figure 1B). In addition, the top five upregulated genes in HNSCC tissues compared to ANTs were listed in Supplementary Table S2. We then evaluated the expression pattern of PLEK2 in HNSCC using publicly accessible datasets. The results showed that PLEK2 expression was significantly higher in HNSCC tissues than in paired ANTs in both TCGA HNSCC and GSE127165 cohorts (Figure 1C,D). Similarly, higher PLEK2 levels were found in malignant specimens than in normal tissues in the GSE25099 and GSE31056 datasets (Figure 1E,F). Interestingly, similar observations were made between tumor tissues and precancerous lesions in the GSE30784 and GSE85195 datasets (Figure 1G,H). Survival analysis further revealed that upregulation of PLEK2 in HNSCC tissues was significantly associated with poor overall survival in different independent study cohorts (Figure 1I,J).
FIGURE 1.
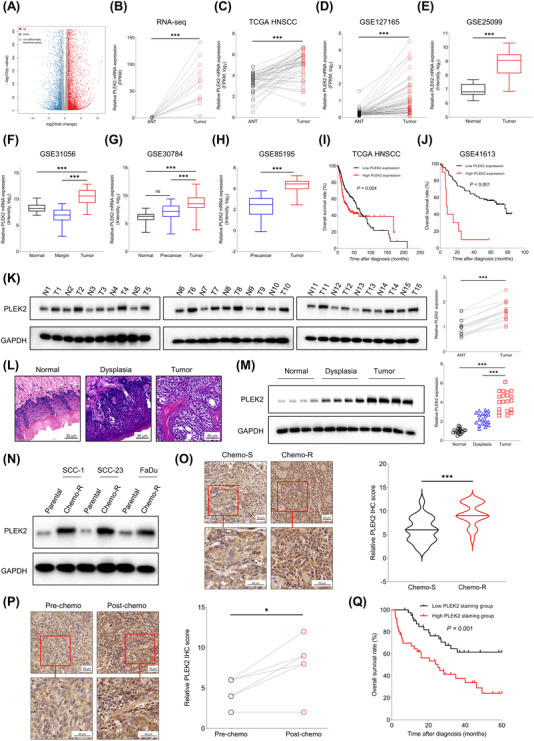
High PLEK2 expression predicts unfavorable clinical outcome in HNSCC patients. (A) RNA‐seq analysis of the DEGs between 14 paired HNSCC tissues and ANTs. (B) PLEK2 was among the top dysregulated genes based on transcriptomics analysis. (C‐D) Expression of PLEK2 in tumor tissues and paired ANTs from TCGA HNSCC and GSE127165 cohorts. (E‐F) Expression of PLEK2 in tumor tissues and normal tissues in GSE25099 and GSE31056. (G‐H) Expression of PLEK2 in normal tissues, precancerous tissues and tumor tissues in GSE30784 and GSE85195 cohorts. (I‐J) Correlation between PLEK2 expression and overall survival of HNSCC patients in TCGA HNSCC and GSE41613 cohorts. (K) Expression levels of PLEK2 in 15 paired tumor tissues and normal tissues detected by Western blotting. PLEK2 was significantly increased in tumor tissues compared to ANTs. (L‐M) PLEK2 is progressively upregulated from normal, dysplasia to cancer tissues in the 4‐NQO‐induced HNSCC mouse model. (N) Expression of PLEK2 protein in parental HNSCC cell lines and corresponding chemoresistant cell lines. (O) Quantitative IHC staining score of PLEK2 in specimens of HNSCC patients with chemosensitive and chemoresistant characteristics. (P) IHC analysis of PLEK2 expression in specimens of HNSCC patients before and after chemotherapy. (Q) HNSCC patients in the high PLEK2 staining group had worse overall survival than those in the low PLEK2 staining group. *P < 0.05, ***P < 0.001. Abbreviations: PLEK2, pleckstrin 2; ANTs, adjacent normal tissues; TCGA, The Cancer Genome Atlas; HNSCC, head and neck squamous cell carcinoma; 4‐NQO, 4‐Nitroquinoline 1‐oxide; IHC, immunohistochemistry; Chemo‐S, chemosensitive; Chemo‐R, chemoresistant
We then assessed the expression levels of PLEK2 using HNSCC tissues and ANTs from patients who received surgical treatments in our hospital. The results showed that PLEK2 was consistently upregulated in 15 paired tumor specimens compared to ANTs (Figure 1K). A 4‐NQO‐induced HNSCC mouse model was further constructed to validate the dynamic changes in PLEK2 observed in the publicly accessible datasets. Our results showed that PLEK2 was gradually increased from normal and dysplasia to cancer tissues (Figure 1L,M), indicating that PLEK2 might play an important role in modulating tumorigenesis in HNSCC. To further determine the clinical significance of PLEK2 in HNSCC, the correlation between PLEK2 expression and chemotherapy was evaluated. The cisplatin‐resistant HNSCC cell lines were established and then verified. As expected, the cisplatin‐resistant cell lines were more resistant to cisplatin than their respective parental cell lines (Supplementary Figure S1A). Our results demonstrated that PLEK2 was significantly upregulated in chemoresistant cancer cell lines compared to chemosensitive controls at both the mRNA and protein levels (Figure 1N, Supplementary Figure S1B). In addition, an HNSCC cohort including 42 patients receiving chemotherapy was stratified into a chemosensitive group (n = 21) and a chemoresistant group (n = 21). The IHC results showed that PLEK2 was overexpressed in the chemoresistant group compared to the chemosensitive group (Figure 1O). Moreover, chemotherapy increased the expression levels of PLEK2 in treatment‐resistant individuals (Figure 1P). The association between PLEK2 expression and overall survival of HNSCC was also evaluated using our own HNSCC cohort. The survival analysis revealed that the HNSCC patients in the high PLEK2 staining group had worse overall survival than those in the low PLEK2 staining group (Figure 1Q). High PLEK2 expression was also associated with unfavorable overall survival in many cancer types from the TCGA cohort, including low‐grade glioma (LGG), liver hepatocellular carcinoma (LIHC), lung adenocarcinoma (LUAD), ovarian cancer (OV), pancreatic adenocarcinoma (PAAD), sarcoma (SARC), skin cutaneous melanoma (SKCM) and uveal melanoma (UVM) (Supplementary Figure S2).
3.2. PLEK2 promoted the malignant characteristics in HNSCC
To study the unknown functions of PLEK2 in HNSCC, we first generated stable PLEK2 knockdown HNSCC cell lines (Figure 2A, Supplementary Figure S3). MTT, colony formation and EdU assays showed that downregulation of PLEK2 significantly restrained the growth and clonogenicity ability of HNSCC cells (Figure 2B‐D). In addition, the invasive capacity of HNSCC cells was dramatically reduced following PLEK2 knockdown (Figure 2E). The apoptosis rate was higher in the PLEK2 knockdown cells compared to the control cells (Supplementary Figure S4). The xenograft mouse model revealed that shPLEK2 markedly decreased the weight and volume of SCC‐1 or SCC‐23 cell‐derived tumors grown in nude mice compared to cells treated with shCTRL (Figure 2F,G, Supplementary Figure S5A‐B). PLEK2 was then stably overexpressed in HNSCC cells to further determine its functional roles (Figure 2H, Supplementary Figure S5C‐D). Our results demonstrated that ectopic expression of PLEK2 significantly enhanced the proliferation, clonogenicity and invasion of HNSCC cells (Figure 2I‐L). More importantly, the in vivo results further confirmed that PLEK2 overexpression remarkably promoted tumor growth in both SCC‐1 and SCC‐23 cells (Figure 2M,N, Supplementary Figure S5E‐F).
FIGURE 2.
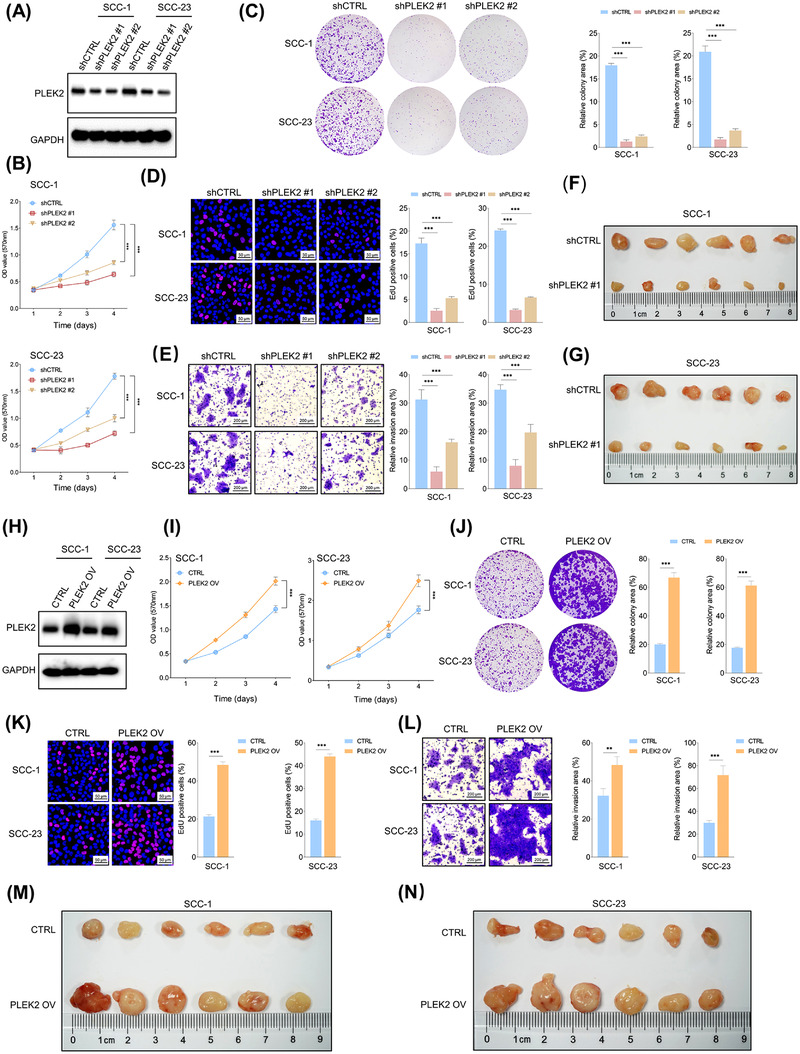
Overexpression of PLEK2 enhances the malignant characteristics of HNSCC cells. (A) Validation of the extent of endogenous PLEK2 depletion in response to shPLEK2 in cancer cells. (B) An MTT assay of the indicated cells with or without PLEK2 depletion was performed. (C) Colony formation assay of HNSCC cells with the indicated modifications. (D) The percentage of EdU positive cells in the PLEK2‐ depleted and control groups. (E) Representative images of invaded cells with the indicated modifications. (F‐G) Xenograft tumors formed by cancer cells with the indicated treatments in the SCC‐1 and SCC‐23 cell lines. (H) Validation of the extent of PLEK2 upregulation by PLEK2 OV in the indicated cells. (I‐K) MTT, colony formation and EdU assays of cancer cells with the indicated treatments. (L) Matrigel invasion assay of cancer cells with the indicated modifications. (M‐N) Xenograft tumors formed by the indicated cells with or without PLEK2 overexpression. **P < 0.01, ***P < 0.001. Abbreviations: PLEK2, pleckstrin 2; HNSCC, head and neck squamous cell carcinoma; MTT, 3‐(4,5‐Dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide; EdU, 5‐ethynyl‐2'‐deoxyuridine; OV, overexpression; CTRL, control
3.3. PLEK2 controlled cancer stemness in HNSCC
As PLEK2 is strongly correlated with chemoresistance and tumors acquire drug resistance due to the presence of CSCs [15], the correlation between PLEK2 and cancer stemness was further explored. Our results revealed that tumor formation capacity was significantly suppressed in PLEK2 knockdown cells (Figure 3A), and the opposite findings were observed when PLEK2 was overexpressed (Figure 3B), indicating that PLEK2 might play an important role in regulating HNSCC stemness. The ALDEFLUOR assay revealed that the percentage of cells with high ALDH activity was markedly lower in the shPLEK2 group than in the shCTRL group (Figure 3C, Supplementary Figure S6A), and overexpression of PLEK2 significantly increased the proportion of ALDH‐positive cells (Figure 3D, Supplementary Figure S6B). Notably, the expression levels of PLEK2 were remarkably higher in ALDHhigh CSCs than in ALDHlow tumor cells (Supplementary Figure S6C‐D).
FIGURE 3.
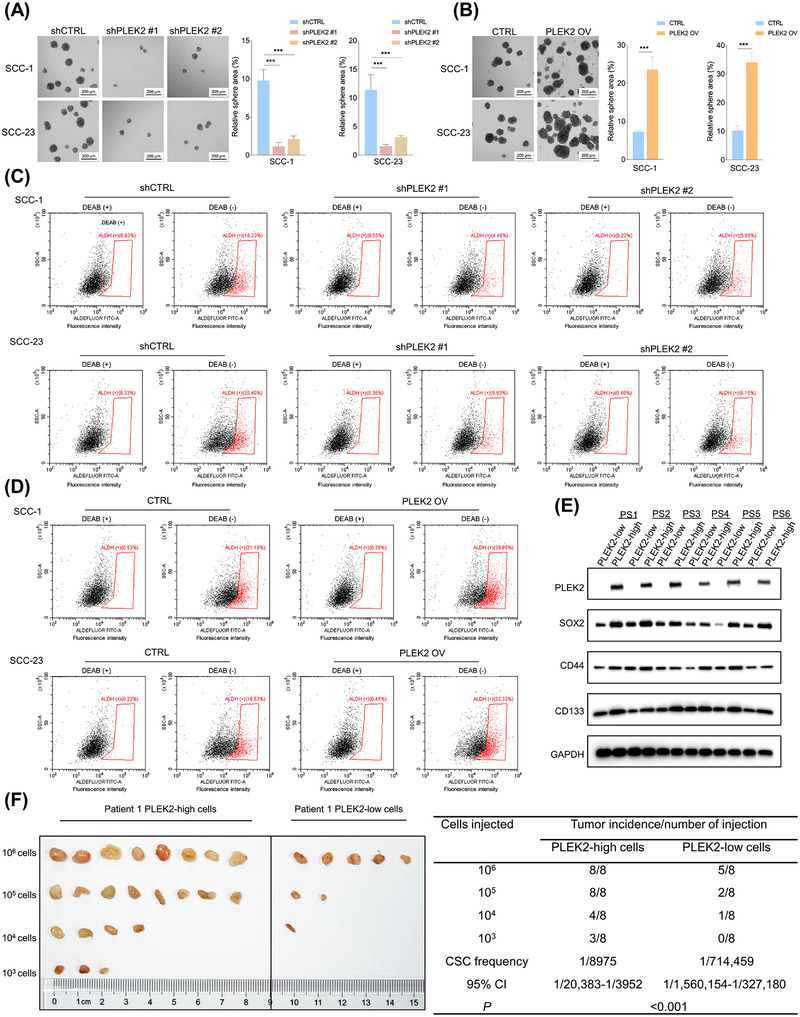
PLEK2 controls HNSCC cancer stemness. (A) Tumor spheres of the indicated cells with or without PLEK2 depletion were detected. (B) The tumor sphere formation capacity of HNSCC cells with the indicated modifications. (C) ALDEFLUOR assay of the percentage of ALDH positive cells in PLEK2 depleted groups and the control group. (D) ALDEFLUOR assay of the percentage of ALDH positive cells in the PLEK2 overexpression and control groups. (E) Immunoblot analyses of PLEK2, SOX2, CD44, and CD133 expression in PLEK2‐high cells and PLEK2‐low cells from six patients are shown. (F) In vivo limiting dilution analysis of PLEK2‐high cells and PLEK2‐low cells from patient 1. These cells were subcutaneously injected into nude mice (n = 8) at the indicated cell dose. Tumor formation ability and stem cell frequency were analyzed using ELDA software. ***P < 0.001. Abbreviations: PLEK2, pleckstrin 2; HNSCC, head and neck squamous cell carcinoma; ALDH, aldehyde dehydrogenase; SOX2, sex‐determining region Y‐box 2; ELDA, extreme limiting dilution analysis; DEAB, diethylaminobenzaldehyde; PS: patients; CSC, cancer stem cells; CI: confidence interval
To further determine whether PLEK2 controls cancer stemness, flow cytometry was used to isolate PLEK2‐high‐expressing cells from PLEK2‐low‐expressing cells in primary HNSCC cancer cells derived from six independent patients. Our results showed that the expression of SOX2, CD44 and CD133, which are well‐established markers for maintaining cancer stemness in HNSCC cells, was consistently upregulated in PLEK2‐high cells compared to PLEK2‐low cells (Figure 3E, Supplementary Figure S6E). To further determine the role of PLEK2 in controlling HNSCC tumorigenesis, we injected 1 × 103‐1 × 106 PLEK2‐high or PLEK2‐low primary HNSCC cells separated from tumor specimens into the subcutaneous space on the back of nude mice to perform an in vivo limiting dilution tumorigenicity assay. We found that PLEK2‐high cells exhibited a marked increase in stem cell frequency (1/8975) compared with PLEK2‐low cells (1/714,459) (Figure 3F), indicating that PLEK2 could be crucial for maintaining the tumorigenic potential of CSCs in nude mice. Similarly, SCC‐1 or SCC‐23 cells with PLEK2 overexpression exhibited markedly higher tumor‐forming ability and stem cell frequency than the control cells (Supplementary Table S3‐S4).
3.4. The effects of PLEK2 depletion or overexpression on the expression of important molecular markers and glycolytic metabolism in HNSCC
As our results demonstrated that PLEK2 was crucial for controlling the malignant phenotypes of HNSCC cells, potentially important molecular markers affected by PLEK2 were further investigated. As shown in Figure 4A, not surprisingly, the expression of proliferation markers, including PCNA, cyclin D1 and MCM2, was significantly decreased following PLEK2 downregulation and vice versa. Interestingly, we also observed that the anti‐apoptotic protein Bcl‐2 was reduced, while the pro‐apoptotic protein Bax was increased in PLEK2 knockdown cells. The opposing results were observed in PLEK2‐overexpressing cells, further supporting that PLEK2 upregulation protects cells against chemotherapy by conferring increased resistance to apoptosis‐induced signaling.
FIGURE 4.
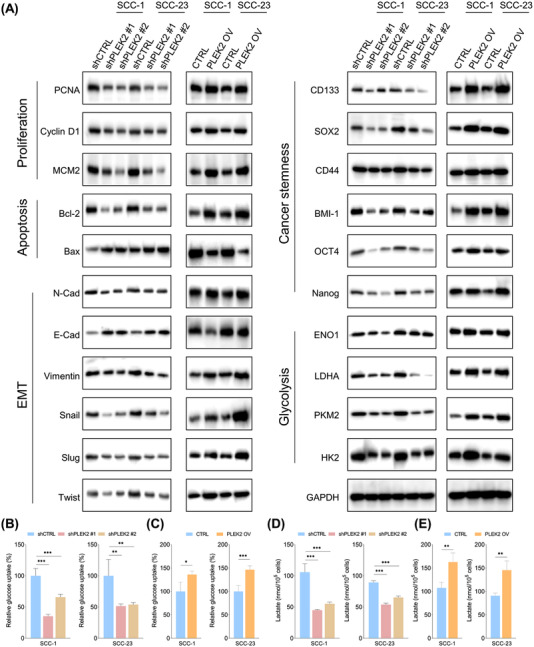
The effects of PLEK2 on important signaling pathways and metabolism in HNSCC. (A) Expression of proliferation markers (PCNA, Cyclin D1 and MCM2), apoptosis‐related markers (Bcl‐2, Bax), EMT related markers (N‐cadherin, E‐cadherin, Vimentin, Snail, Slug and Twist), cancer stemness markers (CD133, SOX2, CD44, BMI‐1, OCT4 and Nanog), and glycolysis related metabolic enzymes (ENO1, LDHA, PKM2 and HK2) in indicated cells with or without PLEK2 depletion as well as in the indicated cells with or without PLEK2 overexpression. (B) Relative glucose uptake in cancer cells with the indicated modifications. (C) Relative glucose uptake in the indicated cells with or without PLEK2 overexpression. (D‐E) Relative lactate production in the indicated cells with or without PLEK2 depletion and in cells with or without PLEK2 overexpression. *P < 0.05, **P < 0.01, ***P < 0.001. Abbreviations: PLEK2, pleckstrin 2; HNSCC, head and neck squamous cell carcinoma; PCNA, proliferating cell nuclear antigen; MCM2, minichromosome maintenance complex component 2; Bcl‐2, B‐cell lymphoma 2; Bax, Bcl‐2‐associated X protein; EMT, epithelial‐mesenchymal transition; SOX2, sex‐determining region Y‐box 2; OCT4, octamer‐binding transcription factor 4; ENO1, enolase 1; LDHA, lactate dehydrogenase A, PKM2, pyruvate kinase M2; HK, hexokinase
As epithelial‐to‐mesenchymal transition (EMT) is associated with invasion and the generation and maintenance of CSCs [16], we assessed the changes in the EMT markers, N‐cadherin, E‐cadherin, vimentin, snail, slug and twist in PLEK2‐downregulated or PLEK2‐upregulated cells. Our results demonstrated a significant decrease in E‐cadherin levels and a dramatic increase in N‐cadherin, vimentin, snail, slug and twist in PLEK2‐overexpressing HNSCC cells. In contrast, opposite findings were observed in PLEK2 knockdown cells. As expected, PLEK2 depletion significantly reduced the expression of stemness‐related markers, including CD133, SOX2, CD44, BMI‐1, OCT4 and Nanog, while PLEK2 overexpression markedly induced their expression (Figure 4A).
The effects of PLEK2 on metabolic changes in HNSCC cells were also investigated. The immunoblotting results showed that downregulation of PLEK2 inhibited the expression of ENO1, LDHA, PKM2 and HK2, which are critical metabolic enzymes controlling glycolysis (Figure 4A). In addition, PLEK2 overexpression led to the opposite findings. To further validate the effects of PLEK2 on cellular metabolism, we measured glucose uptake and lactate production in HNSCC cells with PLEK2 depletion and overexpression. Our results showed that knockdown of PLEK2 reduced glucose uptake and lactate production in HNSCC cells, while overexpression of PLEK2 enhanced glucose uptake and lactate production (Figure 4B‐E).
3.5. PLEK2 increased the expression of c‐Myc by enhancing its stability
To explore the underlying mechanisms by which PLEK2 promotes HNSCC, we first searched for the potential proteins that might interact with PLEK2. Interestingly, based on the BioGRID (https://thebiogrid.org/) predicted results and the high throughput results reported by Heidelberger et al. [17], PLEK2 was one of the highest‐ranked proteins that might interact with c‐Myc. The MYC oncogene is a master regulator that controls proliferation, apoptosis, EMT, stemness and glycolysis in cancer cells. Therefore, we speculated that PLEK2 might exert its functions by interacting with c‐Myc. To test this hypothesis, endogenous and exogenous co‐immunoprecipitation (Co‐IP) assays were performed to detect the potential interaction between PLEK2 and c‐Myc. The results consistently demonstrated that PLEK2 interacted with c‐Myc (Figure 5A,B).
FIGURE 5.
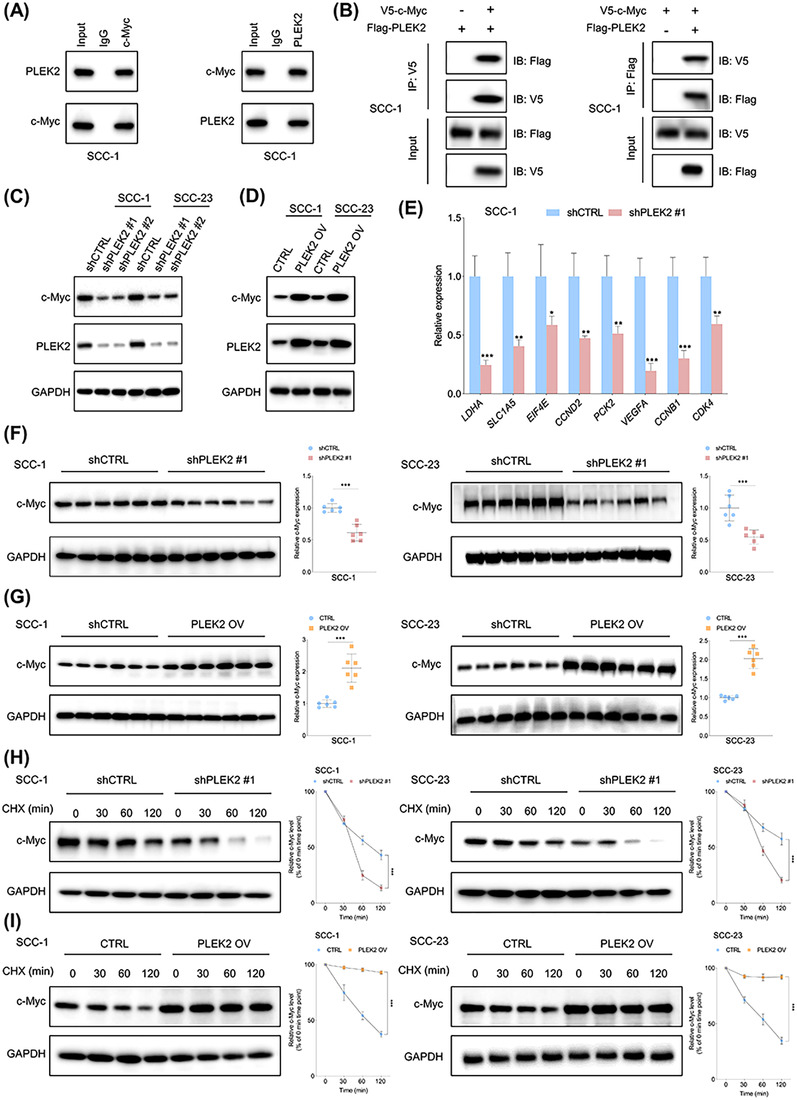
PLEK2 regulates c‐Myc expression by modulating its stability. (A) Endogenous Co‐IP revealed that PLEK2 interacts with c‐Myc. (B) The interaction between PLEK2 and c‐Myc was further evaluated by exogenous Co‐IP. (C‐D) Expression of c‐Myc in the indicated cells with or without PLEK2 depletion and in cells with or without PLEK2 overexpression. (E) The effects of PLEK2 depletion on the expression of c‐Myc targets in SCC‐1 cells. (F) Expression of c‐Myc in xenograft tumor tissues formed by the indicated cells with or without PLEK2 depletion. (G) Expression of c‐Myc in xenograft tumor tissues from the PLEK2 overexpression and the control groups. (H) HNSCC cells with the indicated modifications were treated with CHX for the indicated time periods. (I) HNSCC cells with or without PLEK2 overexpression were treated with CHX for various periods of time. *P < 0.05, **P < 0.01, ***P < 0.001. Abbreviations: PLEK2, pleckstrin 2; Co‐IP, co‐immunoprecipitation, HNSCC, head and neck squamous cell carcinoma; CHX, cycloheximide
Based on the TCGA HNSCC cohort, we found that PLEK2 was positively correlated with c‐Myc downstream targets such as LDHA, ENO1, FOSL1, MTHFD1 and CTSC (Supplementary Figure S7A), indicating that PLEK2 might modulate the expression levels of c‐Myc. Immunoblotting results showed that PLEK2 depletion reduced the expression of c‐Myc, while overexpression of PLEK2 increased c‐Myc levels in HNSCC cells (Figure 5C,D). In addition, downregulation of PLEK2 significantly inhibited mRNA expression of the c‐Myc targets LDHA, SLC1A5, EIF4E, CCND2, PCK2, VEGFA, CCNB1 and CDK4 in both SCC‐1 and SCC‐23 cells (Figure 5E, Supplementary Figure S7B). Moreover, we found that expression of c‐Myc was significantly lower in the xenograft tumor tissues from the shPLEK2 group compared to the control group (Figure 5F), whereas c‐Myc levels were higher in xenograft tumor tissues derived from the PLEK2 overexpression group (Figure 5G). Next, we explored whether PLEK2 modulated c‐Myc expression by regulating the stability of c‐Myc. Treatment of HNSCC cells with the protein synthesis inhibitor CHX demonstrated that PLEK2 depletion reduced c‐Myc expression by shortening its half‐life (Figure 5H). In contrast, PLEK2 overexpression stabilized the expression of c‐Myc by prolonging its half‐life (Figure 5I).
3.6. PLEK2 stabilized c‐Myc by hindering its interaction with FBXW7
c‐Myc stability is regulated by ubiquitination‐dependent proteasomal degradation [18]. PLEK2 depletion on c‐Myc expression was abrogated by adding the proteasome inhibitor MG132 (Figure 6A), indicating that PLEK2 enhances c‐Myc stability by preventing its proteasome‐dependent degradation. As expected, PLEK2 depletion increased c‐Myc polyubiquitylation, while PLEK2 overexpression decreased c‐Myc polyubiquitylation in both SCC‐1 and SCC‐23 cells (Figure 6B). The most extensively characterized pathway regulating c‐Myc protein stability involves FBXW7 [19]. Interestingly, we found that FBXW7 reduced c‐Myc protein levels as expected, while co‐expression of PLEK2 with FBXW7 enhanced c‐Myc levels, suggesting an opposing effect of PLEK2 on FBXW7 function (Figure 6C). More importantly, the Co‐IP results showed that PLEK2 overexpression reduced the binding of FBXW7 to c‐Myc in HNSCC cells, whereas PLEK2 depletion enhanced their interaction (Figure 6D‐E). These results indicated that PLEK2 could hinder the binding of FBXW7 to c‐Myc, decreasing the degradation of c‐Myc. We next examined the combined effects of PLEK2 and FBXW7 on c‐Myc ubiquitination. Our results demonstrated that overexpression of FBXW7 increased c‐Myc polyubiquitination, which was reduced by co‐expression of PLEK2 (Figure 6F). In contrast, downregulation of FBXW7 reduced c‐Myc polyubiquitylation, while co‐expression of PLEK2 further enhanced the suppressive effect of FBXW7 inhibition on c‐Myc polyubiquitylation (Figure 6G).
FIGURE 6.
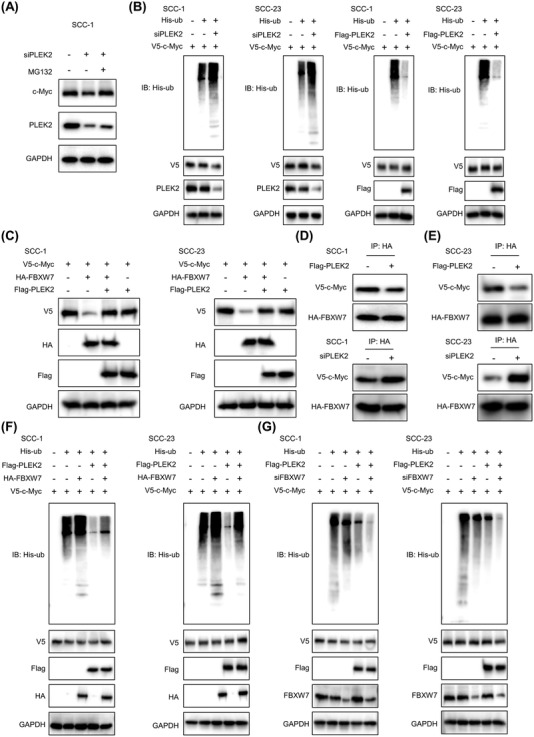
PLEK2 stabilizes c‐Myc by hindering its interaction with FBXW7. (A) SCC‐1 cells with the indicated modification were treated with or without MG132. Western blotting analyses were performed using the indicated antibodies. (B) Ubiquitination levels of c‐Myc in the indicated cells with or without PLEK2 depletion and those with or without PLEK2 overexpression were detected by Co‐IP. (C) Immunoblot analyses of c‐Myc levels in the indicated cells transfected with flag‐PLEK2 and HA‐FBXW7, flag‐PLEK2 alone or HA‐FBXW7 alone. (D‐E) A vector expressing HA‐FBXW7 was transfected into HNSCC cells with the indicated treatments. Immunoprecipitation and immunoblotting analyses were performed using the indicated antibodies. (F‐G) Ubiquitination levels of c‐Myc in HNSCC cells with the indicated modifications were detected by Co‐IP. Abbreviations: PLEK2, pleckstrin 2; FBXW7, F‐box and WD repeat domain containing 7; Co‐IP, co‐immunoprecipitation, HA‐FBXW7, hemagglutinin‐FBXW7; HNSCC, head and neck squamous cell carcinoma; His‐ub, His‐ubiquitin
3.7. Depletion of c‐Myc abrogated the tumor‐promoting effects of PLEK2 in HNSCC
Here, we investigated whether PLEK2 promoted HNSCC tumorigenesis by modulating c‐Myc. The expression of c‐Myc in PLEK2‐overexpressing cells was significantly reduced following the transfection of shc‐Myc (Supplementary Figure S8A). Our results showed that the proliferation, colony formation and invasion capacities of cancer cells promoted by PLEK2 overexpression were significantly reduced following c‐Myc depletion (Figure 7A‐C). Similarly, knockdown of c‐Myc also inhibited the tumor sphere formation capability of HNSCC cells, which was enhanced by PLEK2 upregulation (Figure 7D). More importantly, PLEK2 overexpression significantly increased the weight and volume of xenograft tumors, and these enhancing effects were abrogated by c‐Myc depletion in both SCC‐1 and SCC‐23 cells (Figure 7E,F). IHC results revealed that PLEK2 overexpression significantly enhanced the staining intensities of Ki‐67 and CD44 in xenograft tumor tissues, and these promoting effects were reduced by c‐Myc downregulation (Figure 7G, Supplementary Figure S8B‐C). The effects of FBXW7 depletion on the malignant activities of HNSCC cells subjected to PLEK2 downregulation were also investigated. Our results showed that FBXW7 depletion partially rescued the tumor‐suppressive effects of PLEK2 inhibition (Supplementary Figure S9).
FIGURE 7.
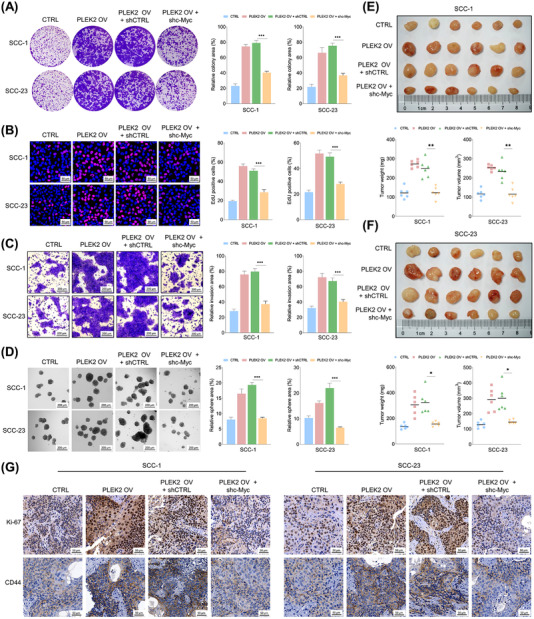
Downregulation of c‐Myc counteracts the tumor‐promoting effects of PLEK2 in HNSCC. (A‐B) Colony formation and EdU assays of HNSCC cells with the indicated modifications. (C) The invasive capacity of HNSCC cells with the indicated treatments was examined by Matrigel invasion assay. (D) The tumor sphere formation capability of HNSCC cells with the indicated modifications. (E‐F) The size, weight and volume of xenograft tumors formed by SCC‐1 or SCC‐23 cells with the indicated modifications. (G) Representative IHC staining intensities of Ki‐67 and CD44 in xenograft tumor tissues formed by SCC‐1 or SCC‐23 cells with the indicated treatments. *P < 0.05, **P < 0.01, ***P < 0.001. Abbreviations: PLEK2, pleckstrin 2; HNSCC, head and neck squamous cell carcinoma; IHC, immunohistochemistry
3.8. c‐Myc directly regulated PLEK2 expression in HNSCC cells
Based on bioinformatics analysis of potential transcription factors binding to PLEK2, we found that c‐Myc has one potential binding site on the promoter region of PLEK2 (Supplementary Figure S10A). c‐Myc depletion significantly reduced PLEK2 expression at both the mRNA and protein levels in HNSCC cells (Figure 8A,B, Supplementary Figure S10B‐C). Likewise, PLEK2 expression was enhanced by c‐Myc upregulation, which was restored by c‐Myc knockdown (Figure 8C). Luciferase assays revealed that c‐Myc enhanced the luciferase activity of the wild‐type PLEK2 promoter, whereas c‐Myc did not affect the activity of the mutant‐type PLEK2 promoter (Figure 8D‐E). ChIP‐qPCR showed that c‐Myc was enriched at the binding site in the promoter region of PLEK2 (Figure 8F). These findings indicate that c‐Myc directly regulates PLEK2 expression in HNSCC cells.
FIGURE 8.
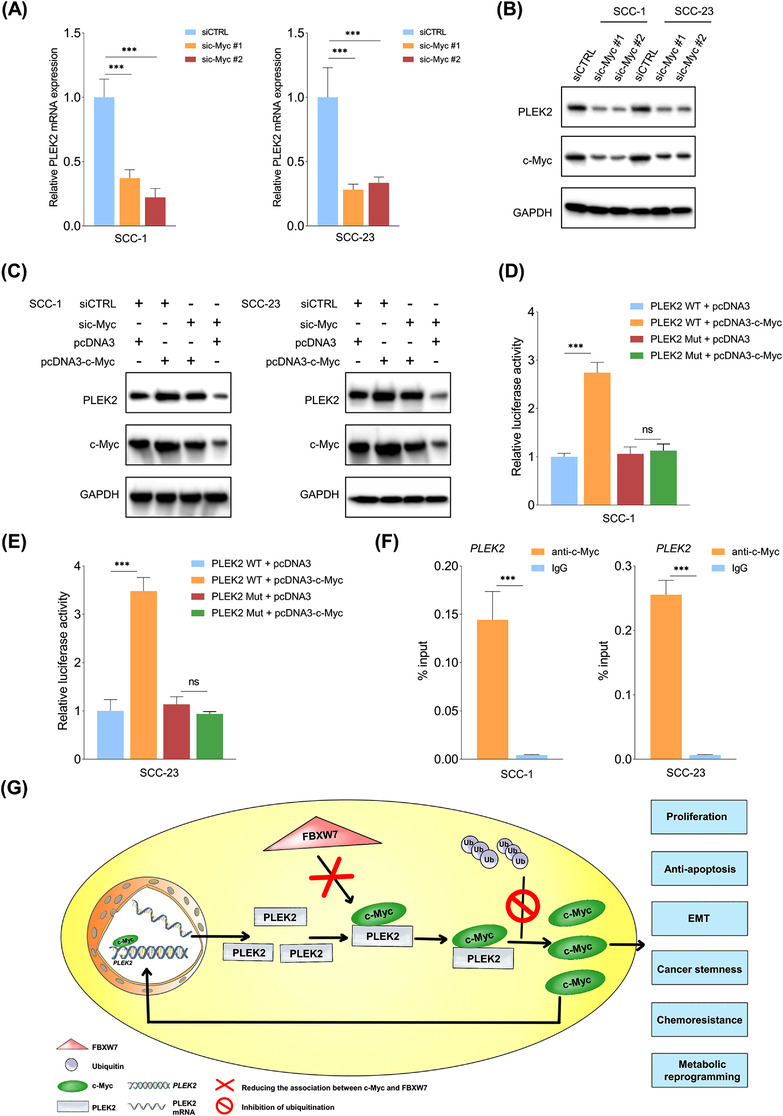
c‐Myc directly regulates PLEK2 expression in HNSCC cells. (A‐B) Expression of PLEK2 mRNA and protein in the indicated cells subjected to c‐Myc depletion. (C) Expression of PLEK2 in SCC‐1 or SCC‐23 cells transfected with siCTRL, sic‐Myc, pcDNA3 and pcDNA3‐c‐Myc alone or in combination. (D‐E) Relative luciferase activities of HNSCC cells with the indicated modifications. (F) ChIP analysis of c‐Myc occupancy at the PLEK2 promoter in SCC‐1 and SCC‐23 cells. (G) Proposed model for the tumor‐promoting role of PLEK2 in HNSCC tumorigenesis. ***P < 0.001, ns: not significant. Abbreviations: PLEK2, pleckstrin 2; HNSCC, head and neck squamous cell carcinoma; CHIP‐qPCR, chromatin immunoprecipitation‐quantitative polymerase chain reaction; PLEK2 WT, PLEK2 wild type; PLEK2 Mut, PLEK2 mutant
4. DISCUSSION
In this study, we first revealed that PLEK2 was highly expressed in HNSCC and that its upregulation was associated with chemoresistance and unfavorable clinical outcomes in HNSCC. Mechanistically, PLEK2 exerted strong tumor‐promoting effects on HNSCC cells by hindering the interaction between c‐Myc and FBXW7, which subsequently enhanced c‐Myc stability. Additionally, c‐Myc directly regulated PLEK2 expression, forming a positive feedback loop driving HNSCC progression. The proposed model for the PLEK2‐c‐Myc feedback circuit in regulating HNSCC tumorigenesis is illustrated in Figure 8G. Although previous studies have revealed that abnormal expression of PLEK2 is associated with poor clinical outcomes in some types of cancer [9, 12], its expression pattern and clinical significance in HNSCC remain unknown. A recent bioinformatics analysis of public datasets demonstrated that PLEK2 might be a potential molecular target for HNSCC [20]. We experimentally validated the prognostic significance of PLEK2 overexpression in HNSCC. PLEK2 has been linked to tumor growth and metastasis [9, 10, 12]. However, most of these findings have not been further explored to determine the exact function of PLEK2 in regulating cellular activities and the biological processes of cancer cells. In addition, the role of PLEK2 in HNSCC tumorigenesis is completely unknown. Here, we uncovered a novel PLEK2‐c‐Myc positive feedback loop driving HNSCC tumorigenesis, which might represent a potential therapeutic target for treating HNSCC.
The reasons we chose PLEK2 as the molecular target were as follows: first, although PLEK2 was not among the most upregulated genes (Supplementary Table S2), our transcriptomic data revealed that it was one of the most consistently increased genes in the tumor tissues compared to the paired ANTs (data not shown). In addition, based on the analysis of publicly available datasets, it was consistently upregulated in tumor tissues in different independent HNSCC cohorts. These findings indicated that PLEK2 might play an important role in regulating HNSCC tumorigenesis. Interestingly, it should be pointed out that the PLEK2 level was found to be higher in normal tissues than in the margins of tumor tissues in GSE31056. As described in the dataset, all the specimens were obtained from patients with histologically normal surgical resection margins, indicating that these margins were probably free of tumor cells. In addition, to achieve adequate surgical margins, the muscular tissue components might be removed. Thus, a higher percentage of muscle tissues might be in surgical margins than in normal tissues. Further, the Human Protein Atlas (https://www.atlasantibodies.com/resources/human‐protein‐atlas/) showed that PLEK2 is enriched in epithelial cells but barely detectable in myocytes. Therefore, it is possible to observe that PLEK2 was lower in the surgical margins than in the normal tissues. Further experiments are warranted to determine the expression patterns of PLEK2 mRNA and proteins between surgical margins and normal tissues.
Chemoresistance is one of the biggest challenges in conquering HNSCC. Consistent with our findings, Wang et al. [9] reported that downregulation of PLEK2 increased the chemosensitivity of esophageal squamous cell carcinoma cells. However, the underlying molecular mechanism is unknown. Cancer stem cells are believed to play an important role in acquiring drug resistance [21, 22]. Our results showed that PLEK2‐positive cells from primary HNSCC tissues had a significantly stronger tumor‐initiating capacity than PLEK2 negative cells. In addition, PLEK2 overexpression promoted cancer cell stemness properties, including enhancing the tumor sphere formation capacity, increasing the ALDH‐positive subpopulation, and improving the tumor‐forming capability. These data consistently indicate that PLEK2 modulates the cancer stemness of HNSCC. The EMT process is critical for generating and maintaining CSCs [16]. In addition, recent findings revealed that glycolysis was more active in CSCs than in the bulk of cancer cells [23]. Therefore, it was reasonable to expect that PLEK2 also significantly affected the expression of EMT markers and glycolytic metabolism in HNSCC, providing direct evidence for PLEK2 being a driver of cancer stemness in HNSCC. Therefore, targeting PLEK2 might be a promising strategy for overcoming chemoresistance in HNSCC.
Mechanistically, we provided compelling evidence showing that PLEK2 interacted with c‐Myc and regulated its expression by modulating its stability. It is well established that c‐Myc dictates many different cancer cellular functions, including uncontrolled proliferation, immortalization, EMT, cancer stemness, metastasis and metabolic reprogramming [24, 25]. As c‐Myc plays a central regulatory role in tumorigenesis and PLEK2 modulates the expression of c‐Myc, it is no wonder that PLEK2 regulates many important biological processes critical for HNSCC initiation and progression. Although targeting the c‐Myc oncoprotein directly is a good strategy for treating cancer, it has been a major challenge for decades due to its “undruggable” protein structure [26]. Therefore, exploring other alternatives for blocking the c‐Myc signaling pathway is important for improving anticancer outcomes. Because PLEK2 functions as an upstream regulator of c‐Myc, developing efficient and selective agents against PLEK2 might provide a practical and effective new approach for suppressing c‐Myc activity.
As a protein with a short half‐life, c‐Myc's levels are tightly regulated, and it is degraded by the ubiquitin‐proteasome pathway. One well‐established mechanism whereby this occurs is through the binding of FBXW7 and c‐Myc, which directly results in c‐Myc degradation [19]. Interestingly, we observed that PLEK2 hindered the interaction between c‐Myc and FBXW7, consequently reducing the ubiquitination levels of c‐Myc and increasing its accumulation. As a powerful transcription factor, c‐Myc exerts its biological functions by regulating its target genes. Interestingly, we demonstrated that c‐Myc directly bounded to the promoter region of PLEK2 and regulated its expression. Collectively, these results indicated that PLEK2 promoted the expression of c‐Myc by increasing its stability by hindering the interaction between c‐Myc and FBXW7. Accumulating c‐Myc enters the nucleus and promotes the expression of PLEK2, forming a positive feedback loop to promote the tumorigenesis of HNSCC.
There were some limitations in our study. First, the global mRNA and protein alterations in HNSCC cells in response to PLEK2 depletion or overexpression were not sufficiently clear and warrant further investigation. Second, the identification of selective inhibitors of PLEK2 by high‐throughput screening is urgently needed to test their potential for treating patient‐derived xenograft (PDX) models of HNSCC, especially those with chemoresistance.
In summary, our findings demonstrate a previously unknown mechanism underlying the stabilization of the c‐Myc protein and activation of the c‐Myc signaling pathway. PLEK2 plays a critical role in promoting chemoresistance, proliferation, anti‐apoptosis, EMT, cancer stemness and metabolic reprogramming, suggesting PLEK2 as a novel and promising molecular target for combating HNSCC.
DECLARATIONS
COMPETING INTERESTS
The authors declare that they have no competing interests.
AUTHOR CONTRIBUTIONS
ZXY, SDL, SWJ, SSS and CL performed the experiments. ZXY, RW, GB and CL analyzed, interpreted the data and wrote the paper. All authors have read and approved the final manuscript.
AVAILABILITY OF DATA AND MATERIALS
The data that support the findings of this study are available from the corresponding authors upon reasonable request. The public datasets comparing the genome‐wide gene expression profiling between tumor tissues and normal tissues/precancerous lesions were downloaded from the Gene Expression Omnibus database (https://www.ncbi.nlm.nih.gov/geo/). The accession numbers were GSE127165, GSE25099, GSE31056, GSE30784, GSE85195 and GSE41613.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
The study protocol regarding the re‐utilization of clinical samples and medical information was approved by the Ethics Committee of the First Affiliated Hospital of Sun Yat‐sen University (Guangzhou, China). Written informed consent was obtained from all patients for the use of tissue samples and clinical data.
CONSENT FOR PUBLICATION
Not applicable.
Supporting information
Supporting Information
ACKNOWLEDGEMENTS
This work was supported by the National Natural Science Foundation of China (81901006), Guangdong Basic and Applied Basic Research Foundation (2020A1515110051), Scientific Research Talent Cultivation Project of Stomatological Hospital, Southern Medical University (RC202005) and Science Research Cultivation Program of Stomatological Hospital, Southern Medical University (PY2020002).
Zhao X, Shu D, Sun W, Si S, Ran W, Guo B, et al. PLEK2 promotes cancer stemness and tumorigenesis of head and neck squamous cell carcinoma via the c‐Myc‐mediated positive feedback loop. Cancer Commun. 2022;42:987–1007. 10.1002/cac2.12349
Contributor Information
Wei Ran, Email: ranweigd@163.com.
Bing Guo, Email: guobing@mail.sysu.edu.cn.
Li Cui, Email: licui@smu.edu.cn.
REFERENCES
- 1. Johnson DE, Burtness B, Leemans CR, Lui VWY, Bauman JE, Grandis JR. Head and neck squamous cell carcinoma. Nat Rev Dis Primers. 2020;6(1):92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cui L, Zhao X, Jin Z, Wang H, Yang SF, Hu S. Melatonin modulates metabolic remodeling in HNSCC by suppressing MTHFD1L‐formate axis. J Pineal Res. 2021;71(4):e12767. [DOI] [PubMed] [Google Scholar]
- 3. Shin YY, Seo Y, Oh SJ, Ahn JS, Song MH, Kang MJ, et al. Melatonin and verteporfin synergistically suppress the growth and stemness of head and neck squamous cell carcinoma through the regulation of mitochondrial dynamics. J Pineal Res. 2022;72(1):e12779. [DOI] [PubMed] [Google Scholar]
- 4. Oosting SF, Haddad RI. Best Practice in Systemic Therapy for Head and Neck Squamous Cell Carcinoma. Front Oncol. 2019;9:815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ju H, Hu Z, Wei D, Huang J, Zhang X, Rui M, et al. A novel intronic circular RNA, circGNG7, inhibits head and neck squamous cell carcinoma progression by blocking the phosphorylation of heat shock protein 27 at Ser78 and Ser82. Cancer Commun (Lond). 2021;41(11):1152‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bach TL, Kerr WT, Wang Y, Bauman EM, Kine P, Whiteman EL, et al. PI3K regulates pleckstrin‐2 in T‐cell cytoskeletal reorganization. Blood. 2007;109(3):1147‐55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang G, Zhou Q, Xu Y, Zhao B. Emerging Roles of Pleckstrin‐2 Beyond Cell Spreading. Front Cell Dev Biol. 2021;9:768238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Feola M, Zamperone A, Moskop D, Chen H, Casu C, Lama D, et al. Pleckstrin‐2 is essential for erythropoiesis in beta‐thalassemic mice, reducing apoptosis and enhancing enucleation. Commun Biol. 2021;4(1):517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang F, Zhang C, Cheng H, Liu C, Lu Z, Zheng S, et al. TGF‐beta‐induced PLEK2 promotes metastasis and chemoresistance in oesophageal squamous cell carcinoma by regulating LCN2. Cell Death Dis. 2021;12(10):901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wu DM, Deng SH, Zhou J, Han R, Liu T, Zhang T, et al. PLEK2 mediates metastasis and vascular invasion via the ubiquitin‐dependent degradation of SHIP2 in non‐small cell lung cancer. Int J Cancer. 2020;146(9):2563‐75. [DOI] [PubMed] [Google Scholar]
- 11. Shen H, He M, Lin R, Zhan M, Xu S, Huang X, et al. PLEK2 promotes gallbladder cancer invasion and metastasis through EGFR/CCL2 pathway. J Exp Clin Cancer Res. 2019;38(1):247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang J, He Z, Sun B, Huang W, Xiang J, Chen Z, et al. Pleckstrin‐2 as a Prognostic Factor and Mediator of Gastric Cancer Progression. Gastroenterol Res Pract. 2021;2021:5527387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yang Q, Li K, Li X, Liu J. Identification of Key Genes and Pathways in Myeloma side population cells by Bioinformatics Analysis. Int J Med Sci. 2020;17(14):2063‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang Y, Lin J, Yan K, Wang J. Identification of a Robust Five‐Gene Risk Model in Prostate Cancer: A Robust Likelihood‐Based Survival Analysis. Int J Genomics. 2020;2020:1097602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhou HM, Zhang JG, Zhang X, Li Q. Targeting cancer stem cells for reversing therapy resistance: mechanism, signaling, and prospective agents. Signal Transduct Target Ther. 2021;6(1):62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Celia‐Terrassa T, Jolly MK. Cancer Stem Cells and Epithelial‐to‐Mesenchymal Transition in Cancer Metastasis. Cold Spring Harb Perspect Med. 2020;10(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Heidelberger JB, Voigt A, Borisova ME, Petrosino G, Ruf S, Wagner SA, et al. Proteomic profiling of VCP substrates links VCP to K6‐linked ubiquitylation and c‐Myc function. EMBO Rep. 2018;19(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gregory MA, Hann SR. c‐Myc proteolysis by the ubiquitin‐proteasome pathway: stabilization of c‐Myc in Burkitt's lymphoma cells. Mol Cell Biol. 2000;20(7):2423‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. King B, Trimarchi T, Reavie L, Xu L, Mullenders J, Ntziachristos P, et al. The ubiquitin ligase FBXW7 modulates leukemia‐initiating cell activity by regulating MYC stability. Cell. 2013;153(7):1552‐66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang J, Sun Z, Wang J, Tian Q, Huang R, Wang H, et al. Expression and prognostic potential of PLEK2 in head and neck squamous cell carcinoma based on bioinformatics analysis. Cancer Med. 2021;10(18):6515‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Picon H, Guddati AK. Mechanisms of resistance in head and neck cancer. Am J Cancer Res. 2020;10(9):2742‐51. [PMC free article] [PubMed] [Google Scholar]
- 22. Zhao J. Cancer stem cells and chemoresistance: The smartest survives the raid. Pharmacol Ther. 2016;160:145‐58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yasuda T, Ishimoto T, Baba H. Conflicting metabolic alterations in cancer stem cells and regulation by the stromal niche. Regen Ther. 2021;17:8‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yoshida GJ. Emerging roles of Myc in stem cell biology and novel tumor therapies. J Exp Clin Cancer Res. 2018;37(1):173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Miller DM, Thomas SD, Islam A, Muench D, Sedoris K. c‐Myc and cancer metabolism. Clin Cancer Res. 2012;18(20):5546‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen H, Liu H, Qing G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct Target Ther. 2018;3:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding authors upon reasonable request. The public datasets comparing the genome‐wide gene expression profiling between tumor tissues and normal tissues/precancerous lesions were downloaded from the Gene Expression Omnibus database (https://www.ncbi.nlm.nih.gov/geo/). The accession numbers were GSE127165, GSE25099, GSE31056, GSE30784, GSE85195 and GSE41613.