

Abstract
Background
Maintenance of cancer stem‐like cell (CSC) stemness supported by aberrantly regulated cancer cell metabolism is critical for CSC self‐renewal and tumor progression. As a key glycolytic enzyme, hexokinase 2 (HK2) plays an instrumental role in aerobic glycolysis and tumor progression. However, whether HK2 directly contribute to CSC stemness maintenance in small cell lung cancer (SCLC) is largely unclear. In this study, we aimed to investgate whether HK2 independent of its glycolytic activity is directly involved in stemness maintenance of CSC in SCLC.
Methods
Immunoblotting analyses were conducted to determine the expression of HK2 in SCLC CSCs and their differentiated counterparts. CSC‐like properties and tumorigenesis of SCLC cells with or without HK2 depletion or overexpression were examined by sphere formation assay and xenograft mouse model. Immunoprecipitation and mass spectrometry analyses were performed to identify the binding proteins of CD133. The expression levels of CD133‐associated and CSC‐relevant proteins were evaluated by immunoblotting, immunoprecipitation, immunofluorescence, and immunohistochemistry assay. RNA expression levels of Nanog, POU5F1, Lin28, HK2, Prominin‐1 were analyzed through quantitative reverse transcription PCR. Polyubiquitination of CD133 was examined by in vitro or in vivo ubiquitination assay. CD133+ cells were sorted by flow cytometry using an anti‐CD133 antibody.
Results
We demonstrated that HK2 expression was much higher in CSCs of SCLC than in their differentiated counterparts. HK2 depletion inhibited CSC stemness and promoted CSC differentiation. Mechanistically, non‐mitochondrial HK2 directly interacted with CD133 and enhanced CD133 expression without affecting CD133 mRNA levels. The interaction of HK2 and CD133 promoted the binding of the deubiquitinase ubiquitin‐specific protease 11 (USP11) to CD133, thereby inhibiting CD133 polyubiquitylation and degradation. HK2‐mediated upregulation of CD133 expression enhanced the expression of cell renewal regulators, SCLC cell stemness, and tumor growth in mice. In addition, HK2 expression was positively correlated with CD133 expression in human SCLC specimens, and their expression levels were associated with poor prognosis of SCLC patients.
Conclusions
These results revealed a critical non‐metabolic function of HK2 in promotion of cancer cell stemness. Our findings provided new insights into the multifaceted roles of HK2 in tumor development.
Keywords: HK2, metabolic enzyme, non‐metabolic function, cancer stem‐like cell, CD133, USP11, ubiquitylation, SCLC
Abbreviations
- ACN
acetonitrile
- ALDH
aldehyde dehydrogenase
- ATCC
American Type Culture Collection
- α‐KGDH
α‐ketoglutarate dehydrogenase
- CHX
cycloheximide
- CSC
cancer stem cell
- DAB
diaminobenzidine
- DAPI
4’,6‐diamidino‐2‐phenylindole
- DEAB
diethylaminobenzaldehyde
- DMEM
Dulbecco's modified Eagle's medium
- DTT
dithiothreitol
- EDTA
ethylene diamine tetraacetic acid
- EGFR
epidermal growth factor receptor
- FBS
fetal bovine serum
- FDR
false discovery rate
- G6P
glucose‐6‐phosphate
- GBM
glioblastoma
- GST
glutathione S‐transferase
- HCC
hepatocellular carcinoma
- HE
hematoxylin and eosin stain
- HRP
horseradish peroxidase
- HK2
Hexokinase 2
- IP
immunoprecipitation
- IACUC
Institutional Animal Care and Use Committee
- IPTG
isopropyl β‐D‐1‐thiogalactopyranoside
- IgG
immunoglobin G
- IF
immunofluorescence
- IHC
immunohistochemistry
- KLF4
Kruppel‐like factor 4
- KHKA
ketohexokinase isoform A
- LB
Luria‐Bertani
- MS
mass spectrometry
- MBD
mitochondrial binding domain
- Oct4
octamer‐binding transcription factor 4
- OS
overall survival
- PCK1
phosphoenolpyruvate carboxykinase 1
- PCR
polymerase chain reaction
- PGK1
phosphoglycerate kinase 1
- PVDF
polyvinylidene fluoride
- PI3K
phosphoinositide 3‐kinase
- qRT‐PCR
quantitative reverse transcription PCR
- SCLC
small cell lung cancer
- shRNA
short hairpin RNA
- SDS‐PAGE
sodium dodecyl sulfate‐polyacrylamide gel electrophoresis
- SDS
sodium dodecyl sulfate
- siRNA
small interfering RNA
- SD
standard deviation
- TFA
trifluoroacetic acid
- TMA
tissue microarray
- ub
ubiquitin
- USP11
ubiquitin‐specific protease 11
- VDAC
voltage dependent anion channel
- WT
wild‐type
1. BACKGROUND
Small cell lung cancer (SCLC), which is a neuroendocrine cancer that accounts for ∼15% of all lung cancers, is an exceptionally lethal malignancy [1, 2]. SCLC is characterized by rapid growth, early metastasis, and high relapse rates, with a 5‐year survival rate of less than 5% [3, 4, 5, 6]. SCLC often quickly develops resistance to chemotherapy and radiotherapy [7, 8] and exhibits only moderate responses to immune checkpoint blockade therapy [9]. Critical understanding of the mechanism underlying SCLC progression is essential for developing more effective therapeutic strategies against SCLC.
Cancer stem‐like cells (CSCs) represent a proportion of cells in solid and hematopoietic tumors and possess special characteristics, such as self‐renewal, tumorigenic ability, heterogeneity, metastasis, drug resistance, and recurrence [10, 11]. These cells often express the pluripotency transcription factors octamer‐binding transcription factor 4 (Oct4), Nanog, c‐Myc, and Kruppel‐like factor 4 (KLF4) and the translation reprogramming factor Lin28 along with selected CSC markers, such as aldehyde dehydrogenase (ALDH) isozymes and CD133 (also known as prominin‐1) [12]. CD133 is a pentaspan transmembrane glycoprotein and the most commonly used marker for isolating CSC populations from different tumors [13]. CD133+ CSCs of SCLC are highly tumorigenic and chemoresistant [14]. In hepatocellular carcinoma (HCC) cells or glioblastoma (GBM) cells, CD133 activates AKT through regulation of epidermal growth factor receptor (EGFR) internalization or interaction with phosphoinositide 3‐kinase (PI3K) 85 kDa regulatory subunit (p85) [15, 16, 17, 18]. In colon and ovarian cancer cells, CD133 promotes β‐catenin signaling to enhance the proliferation of tumor cells and tumorigenesis [19]. In addition, CD133 depletion reduced Oct4 expression and the self‐renewal ability of head and neck squamous cell carcinoma cells and HCC cells [20, 21]. However, whether CD133 expression is post‐translationally regulated in SCLC cells, thereby promoting tumor cell stemness remains largely unknown.
Aberrantly regulated metabolic enzymes harness regulation of the balance between anabolic and catabolic processes, which are critical for cancer cell proliferation and stemness [22, 23, 24, 25]. CSCs are more glycolysis‐dependent than their corresponding differentiated cells [26]. Recent studies have shown that metabolic enzymes possess noncanonical functions, which are critical for many cellular activities, such as gene expression and DNA damage repair [27, 28, 29]. However, whether metabolic enzymes directly contribute to cancer stemness through their nonmetabolic functions remains unclear.
Hexokinases (HKs) catalyze the first step of glycolysis to produce metabolic intermediate glucose‐6‐phosphate (G6P). Four HK isozymes, denoted as HK1, HK2, HK3, and HK4, have been identified in mammals. Among them, HK1 and HK2, which bind to mitochondrial outer membrane via mitochondrial voltage dependent anion channel (VDAC), have high affinity for glucose. Unlike HK1, which is highly expressed in all adult tissues, HK2 is expressed only in adult heart and muscle tissues and the cells derived from embryos. In tumor cells, HK2 is often overexpressed and the mitochondria‐localized HK2 promoted tumor development through its glycolytic function [30, 31, 32]. However, whether HK2 independent of its glycolytic activity plays a role in tumor cell stemness is unclear.
In this study, we explored the involvement of HK2 in regulating the CSC properties of SCLC and determined its underlying mechanisms in vivo and in vitro.
2. MATERIALS AND METHODS
2.1. Cell lines and cell culture
The human SCLC cell lines NCI‐H446, NCI‐H1048, and NCI‐H69 and the human embryonic kidney cell line HEK‐293T were purchased from American Type Culture Collection (ATCC; Manassas, VA, USA). NCI‐H446 and NCI‐H69 cells were cultured in RPMI 1640 medium (Corning, NY, USA) supplemented with 10% fetal bovine serum (FBS; Corning, NY, USA), penicillin (100 U/mL; Sigma, St Louis, MO, USA) and streptomycin (100mg/ml, Sigma) in a humidified incubator at 37°C with 5% CO2. NCI‐H1048 cells were cultured in DMEM:F12 (Gibco, Carlsbad, CA, USA) supplemented with 10% FBS, 0.005 mg/mL insulin (Sigma), 0.01 mg/mL transferrin (Gibco), 30 nmol/L sodium selenite (Sigma), 10 nmol/L hydrocortisone (Sigma), 10 nmol/L beta‐estradiol (Sigma), 4.5 mmol/L L‐glutamine (Gibco), penicillin (100 U/mL) and streptomycin (100 mg/mL) in a humid incubator at 37°C with 5% CO2. HEK‐293T cells were cultured in DMEM (Gibco) supplemented with 10% FBS, penicillin (100 U/mL) and streptomycin (100 mg/mL) in a humidified incubator at 37°C with 5% CO2.
2.2. Lentivirus production and stable cell‐line construction
Short hairpin RNA (shRNA) and wild‐type plasmids were constructed by SyngenTech company (Beijing, China). Briefly, polymerase chain reaction (PCR)‐amplified human HK2, USP11, and CD133 were cloned into pLV‐CMV‐MCS‐puro‐3XFlag vector or pzDonor‐CMV‐MCS‐neo‐HA vector. shRNA target sequences were cloned into PLKO.1‐puro vector. Then, HEK‐293T cells were cotransfected with the recombinant plasmids and packaging plasmids (pLP1, pLP2 and pLP/VSVG; Thermo Fisher Scientific, Waltham, MA, USA) using Lipofectamine 3000 according to the instructions (Thermo Fisher Scientific). Forty‐eight hours later, lentivirus particles were collected and stored at ‐80°C. shRNA sequences are listed in Supplementary Table S1.
For stable cell line construction, cells were infected with lentivirus in culture medium supplemented with 5 μg/mL polybrene (Sigma) for 24 hours. The cells were treated with puromycin (Gibco) or geneticin (Gibco) for positive clones selection. The gene expression efficiency was determined by quantitative reverse transcription PCR (qRT‐PCR) or immunoblotting analyses.
2.3. Sphere formation assay
NCI‐H446 and NCI‐H1048 cells (1 × 103) were seeded in ultralow attachment 6‐well plates (Corning) in DMEM/F12 (Gibco) supplemented with 20 μL/mL B27 (Gibco), 20 ng/mL epidermal growth factor (Gibco), and 20 ng/mL basic fibroblast growth factor (PeproTech, Hamada, Rehovot, Israel). After culture for one week, the number and diameters of spheres were determined under an inverted fluorescence microscope (Olympus, Tokyo, Japan).
For the second‐passage sphere formation, the first‐passage spheroids were trypsinized by trypsin (Gibco) and plated in 6‐well plates at a density of 1 × 103 cell/well in the media described above. After culture for one week, the numbers and diameters of spheres were determined under an inverted fluorescence microscope (Olympus).
2.4. Measurement of glucose consumption and lactate production
Monolayer and sphere‐forming cells were seeded into 6‐well plates. Twenty‐four hours later, the culture medium was collected and analyzed as previously described [33]. Lactate production and glucose uptake were determined using a lactate colorimetric assay kit (#K627, BioVision, Milpitas, CA, USA) and glucose colorimetric assay kit (#K606, BioVision), respectively. Briefly, a standard curve was generated by using the standards provided from the kit. The OD values of glucose or lactate in the medium before and after cell culture was detected, and the corresponding concentration was calculated by using the standard curve. The cells were collected for total protein content measurement. Glucose consumption and lactate production values were calculated with relative protein content.
2.5. Immunoblotting and immunoprecipitation assays
Cells were harvested, and proteins were extracted using lysis buffer (Thermo Fisher Scientific) supplemented with protease and phosphatase inhibitor cocktail (Invitrogen, Waltham, MA, USA). Then, the protein concentration was quantified with a BCA protein assay kit (Thermo Fisher Scientific) according to the manufacturer's protocol. Immunoblotting and immunoprecipitation were performed as previously described [34]. Briefly, protein lysates were electrophoresed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS‐PAGE) gel, followed by transferring proteins to polyvinylidene fluoride (PVDF) membranes (Merck, Burlington, MA, USA). Antibodies against HK2 (1:1000; ab209847, Abcam, Cambridge, MA, USA), Lin28 (1:1000; ab109751, Abcam), Nanog (1:1000; ab109250, Abcam), PDK1 (1:1000; ab202468, Abcam), PGK1 (1:1000; ab199438, Abcam), PFKP (1:1000; ab204131, Abcam), COX IV (1:1000; ab202554, Abcam), PKM2 (1:1000; #4053, Cell Signaling Technology, Danvers, MA, USA), Oct4 (1:1000; #2840, Cell Signaling Technology), HA (1:1000; #3726, Cell Signaling Technology), GST (1:1000; #2625, Cell Signaling Technology), Ubiquitin (1:1000; #3933, Cell Signaling Technology), CD133 (1:1000; #64326, Cell Signaling Technology), GLUT1 (1:1000; A11170, Abclonal, Wuhan, China), LDHA (1:500; sc‐137243, Santa Cruz Biotechnology, Dallas, TX, USA), USP11 (1:1000; sc‐365528, Santa Cruz Biotechnology), Flag (1:3000; F1804, Sigma), Tubulin (1:10000; T9026, Sigma), and GAPDH (1:10000; HRP‐60004, Proteintech, Wuhan, China) were used as primary antibodies and incubated with the membranes overnight at 4°C. After washing, the membranes were incubated with the secondary anti‐rabbit IgG HRP‐linked antibody (1:5000; #7074, Cell Signaling Technology) or anti‐mouse IgG HRP‐linked antibody (1:5000; #7076, Cell Signaling Technology) at room temperature for 1 h. Then proteins were visualized by HRP substrates reactions with GE Amersham Imager (GE Healthcare, Boston, MA, USA)
For immunoprecipitation assay, cell lysates were incubated with primary antibodies for 3 h at 4°C. The antigen‐antibody mixture was then mixed with Protein A/G PLUS‐Agarose (sc‐2003, Santa Cruz Biotechnology) and incubated for another 12 h at 4°C After washing, the agarose beads were boiled with 2 × SDS‐loading buffer (B1007, Applygen, Beijing, China) and the associated proteins were detected by immunoblotting. The following primary antibodies were used: anti‐HK2 antibody (1:100; ab209847, Abcam),.anti‐CD133 antibody (1:100; #64326, Cell Signaling Technology), anti‐USP11 antibody (1:50; sc‐365528, Santa Cruz Biotechnology) and normal rabbit IgG (1:5000; #2729, Cell Signaling Technology).
2.6. Protein half‐life detection
For the CD133 protein half‐life determination, NCI‐H1048 cells expressing specific shRNAs or the recombinant plasmids were treated with 100 μg/mL Cycloheximide (CHX; HY‐12320, MedChemExpress, Monmouth Junction, NJ, USA) for different periods of time. The cells were collected, and immunoblotting analyses were performed with an anti‐CD133 antibody (1:1000; #64326, Cell Signaling Technology).
2.7. In vitro deubiquitylation assay
HEK‐293T cells were transfected with both HA‐tagged ubiquitin plasmid (HA‐ub) and Flag‐tagged CD133 expressing plasmid (Flag‐CD133) HA‐ub and Flag‐CD133. After 48 hours, the cells were treated with MG132 (50 μM; S2619, Selleckchem, Houston, TX, USA) for an additional 8 hours. The cells were then lysed by IP lysis buffer (87787, Thermo Fisher Scientific), and ubiquitinated Flag‐CD133 was purified from the cell extracts with anti‐Flag magnetic beads (M8823, Sigma). Following extensive washing by lysis buffer for three times, Flag‐CD133 was eluted with 3 × Flag‐peptides (F4799, Sigma).
For the in vitro deubiquitylation assay, ubiquitinated CD133 protein was incubated with recombinant GST‐USP11 (ab269114, Abcam) in deubiquitylation buffer (50 mmol/L Tris‐HCl pH 8.0, 50 mmol/L NaCl, 1 mmol/L ethylene diamine tetraacetic acid [EDTA], 10 mmol/L dithiothreitol [DTT], and 5% glycerol) at 37°C for 2 hours. CD133 ubiquitylation was examined via immunoblotting analyses.
2.8. Immunofluorescence analysis
Immunofluorescence analyses were performed as described previously [35]. Briefly, cells were fixed and incubated with primary antibodies at 4°C overnight. After washing, Alexa Fluor 488‐conjugated (1:1000; #4408, Cell Signaling Technology) or Alexa Fluor 555‐conjugated (1:1000; #4413, Cell Signaling Technology) secondary antibodies were added and incubated for 1 h at room temperature. DAPI (C0065, Solarbio, Beijing, China) was used to stain the nucleus. Cells were examined using microscope (Olympus, Tokyo, Japan).
For paraffin sections, tissue sections were dewaxed and hydrated, followed by heat induced epitope retrieval step for permeabilization and blocking. After washing, immunofluorescence analysis was performed as mentioned above.
The following primary antibodies were used: anti‐HK2 antibody (1:100; ab104836, Abcam), anti‐Lin28 antibody (1:100; ab109751, Abcam), anti‐Nanog antibody (1:100; ab109250, Abcam), anti‐Oct4 antibody (1:200; #2840, Cell Signaling Technology), anti‐CD133 antibody (1:200; #64326, Cell Signaling Technology) and anti‐USP11 antibody (1:100; PA5‐82061, Thermo Fisher Scientific).
2.9. Liquid chromatography‐mass spectrometry/mass spectrometry (LC‐MS/MS) analysis
As described previously [36], CD133 was immunoprecipitated from the designated cells and the sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS‐PAGE) was performed. The SDS‐PAGE gel was stained with Coomassie brilliant blue using Protein Stains H (C510041; SanGon Biotech, Shanghai, China). The In‐gel CD133 proteins were digested overnight in 12.5 ng/μL trypsin (650279, Sigma) in 25 mmol/L NH4HCO3. The peptides were extracted three times with 60% acetonitrile (ACN) / 0.1% trifluoroacetic acid (TFA) and dried completely in a vacuum centrifuge. LC‐MS/MS analysis was performed on a Q Exactive mass spectrometer (Thermo Fisher Scientific) that was coupled to an Easy nLC instrument (Thermo Fisher Scientific) for 60 min.
MS analysis was performed using the MASCOT engine (Matrix Science, version 2.2; London, UK) embedded in Proteome Discoverer 1.4 (Thermo Electron, San Jose, CA.) against the UniProt Human database (https://www.uniprot.org/proteomes/UP000005640) and the decoy database (https://www.matrixscience.com/help/decoy_help.html). MS data were searched against the UniProt database (https://www.uniprot.org/). The cutoff of the global false discovery rate (FDR) for peptide and protein identification was set to 0.01. LC‐MS/MS was performed in Shanghai Applied Protein Technology Co., Ltd (Shanghai, China).
2.10. Glutathione S‐transferase (GST) pulldown assay
Briefly, equal amounts of Flag‐CD133 protein, which was purified from HEK‐293T cells, were incubated with or without purified His‐HK2 protein (NBC1‐18389, Novus Biologicals, Minneapolis, MN, USA) in a modified reaction buffer (25 mmol/L Tris‐HCl [pH 7.5], 5 mmol/L beta‐glycerophosphate, 2 mmol/L dithiothreitol [DTT], 0.1 mmol/L Na3VO4, and 10 mmol/L MgCl2, 0.5 mmol/L ATP) at 25°C for 1 hour. The reaction mixture was then incubated with purified GST‐USP11 protein (ab269114, Abcam) in GST pull‐down reaction buffer together with glutathione‐sepharose 4B beads (17075601, Cytiva, Uppsala, Sweden). The glutathione‐sepharose 4B beads were then washed three times with reaction buffer, eluted by elution buffer (C600325, Sangon Biotech) and then subjected to immunoblotting analysis.
Purified His‐HK2 protein was incubated with purified GST or GST‐CD133 (ab160218, Abcam) in GST pull‐down reaction buffer together with glutathione‐sepharose 4B beads. The glutathione agarose beads were then washed with elution buffer and then subjected to immunoblotting analysis. The recombinant proteins were purified as described previously [35]. Briefly, BL21 (DE3) expressing pGEX‐6P‐GST were cultured in 250 mL of Luria‐Bertani (LB) broth medium and treated with isopropyl β‐D‐1‐thiogalactopyranoside (IPTG) for 16 h at 30°C before lysis via sonication. Cell lysates were incubated with glutathione‐sepharose 4B beads (17075601, Cytiva), then washed with a modified reaction buffer (25 mmol/L Tris‐HCl [pH 7.5], 5 mmol/L beta‐glycerophosphate, 2 mmol/L dithiothreitol [DTT], 0.1 mmol/L Na3VO4, and 10 mmol/L MgCl2, 0.5 mmol/L ATP) three times and eluted by elution buffer (C600325, Sangon Biotech).
2.11. RNA extraction and quantitative qRT‐PCR analysis
Total RNA was extracted using TRIzol reagent (Thermo Fisher Scientific) according to the manufacturer's protocol. cDNA was synthesized with a RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific), and qRT‐PCR was performed with SYBR Green Select Master Mix (Invitrogen) on an ABI 7900HT Real‐Time PCR Thermocycler (Life Technologies, Houston, TX, USA). Relative mRNA expression was determined using the 2–ΔΔCt method, and actin was used as an endogenous reference. The primers are listed in Supplementary Table S2.
2.12. ALDH activity analysis
For ALDH activity measurement, an ALDEFLUOR™ reagent system (01700, STEMCELL, Cambridge, MA, USA) was used. The fluorescence intensity of labeled NCI‐H69 and NCI‐H1048 cells (1×106/mL) was analyzed via flow cytometry (BD LSRII, San Jose, CA, USA) after the addition of ALDH substrate buffer (01700, STEMCELL). A specific inhibitor of ALDH, diethylaminobenzaldehyde (DEAB; 01700, STEMCELL), was used as a control for background fluorescence.
2.13. CD133+ cells sorting
NCI‐H1048 or NCI‐H69 cells were digested and separated into a single‐cell suspension with PBS, then adjusted to the density of 1×107/mL, which were subsequently incubated with APC‐conjugated anti‐CD133 antibody (1:100 dilution; 397906, Biolegend, San Diego, CA, USA) on ice for 30 min in the dark. Following two washes with PBS, cells were resuspended with 500 μL PBS and subjected to isolation by flow cytometry (LSRII, BD). Negative control was determined by using equal amounts of APC‐conjugated immunoglobulin G (IgG) (M1310G05, Biolegend) stained cell.
2.14. Mitochondrial fractionation isolation
Mitochondrial fractionation of NCI‐H1048 or NCI‐H69 monolayer and spheroid cells were extracted by using Mitochondria/Cytosol Fractionation Kit (ab65320, Abcam, Cambridge, MA, USA) according to the manufacturer's protocol. Briefly, SCLC monolayer cells and spheroid cells were collected and washed with PBS. Cells were then resuspended in cytosol extraction buffer and incubated on ice for 10 minutes, followed by homogenization for non‐mitochondrial fractionation extraction. After centrifugation, mitochondrial components were collected and resuspended in mitochondrial extraction buffer. Both mitochondrial fractionations and non‐mitochondrial fractionations were used for immunoprecipitation analyses. Mitochondrial COX IV (1:1000; ab202554, Abcam) and GAPDH (1:10000; HRP‐60004, Proteintech, Wuhan, China) were used as loading control [37].
2.15. Hexokinase (HK) activity analysis
The HK activity of tumor cells was determined using hexokinase activity assay kit (ab136957, Abcam) following the manufacturer's instructions. Briefly, tumor cells were rapidly homogenized in HK assay buffer for 10 minutes on ice. After centrifugation, the supernatant was collected for colorimetric analysis at OD450nm. NADH standard curve was made to calculate the hexokinase activity according to OD values. The cellular NADH levels were normalized to the protein amounts of the samples.
2.16. Mice
Athymic BALB/c nude mice (4‐5 weeks old) or NOD/SCID mice (4‐5 weeks old) were purchased from the Huafukang Bioscience (Beijing, China). Tumor cells were subcutaneously injected into the flanks of mice. The mice were euthanized by carbon dioxide narcosis after five or six weeks, and tumours were dissected, fixed in 4% formaldehyde and embedded in paraffin for further study.
All mice were housed under specific pathogen‐free and controlled conditions (25‐27°C, 45%‐55% humidity, 12 h day/night cycle). All animal experiments were conducted in accordance with guidelines approved by the Institutional Animal Care and Use Committee (IACUC) of the National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College.
2.17. Animal studies
To evaluate tumor growth in mouse models, tumor cells (2×106) were subcutaneously injected into the flanks of 4∼5‐week‐old female athymic BALB/c nude mice, as described previously [38]. Tumor length (a) and minor diameter (b) were monitored once a week, and tumor size was calculated using the following formula: volume = a×b2/2.
For the limiting dilution assay, cells were diluted and prepared at 3×106, 1×106, 5×105 and 1×105 cells per 100 μL. Then, equal amounts of cells were subcutaneously injected into the flanks of 4∼5‐week‐old female NOD/SCID mice. Tumor volume was monitored and calculated as described above. Limiting dilution analysis was calculated using extreme limiting dilution analysis software (https://bioinf.wehi.edu.au/software/elda/) as previously described [39].
2.18. Patients and tissue samples
Fresh frozen SCLC tissues and matched normal tissues for tissue arrays were obtained from 90 patients who underwent radical resections from January 2011 to January 2015 at the National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College (Beijing, China). Specimens without clear pathological, clinical and survival information were excluded. A paraffin‐embedded tissue microarray (TMA) was prepared by Superbiotek, Inc (Shanghai, China). Ethics approval was granted by the Committee for the Ethics Review of Research Involving Human Subjects of the National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College. The basic information and clinical status of SCLC patient cohort were shown in Supplementary Table S3.
2.19. Hematoxylin and eosin (H&E) staining and immunohistochemical (IHC) analysis
H&E staining was performed and evaluated by two pathologists independently, and high tumor/stroma ratio areas were marked in the paraffin‐embedded specimens, which were used for tissue arrays.
IHC analyses of paraffin sections were performed as previously described [40]. Briefly, paraffin‐embedded samples were deparaffinized by xylene and rehydrated with different concentrations of ethanol. After heat induced antigen retrieval, the tissue sections were permeabilizated and blocked with animal serum. The specimens were then incubated with primary antibody overnight at 4°C. After washing, HRP conjugated secondary antibody was added for incubation, followed by diaminobenzidine (DAB) staining (DA1010, Solarbio, Beijing, China). The nuclear counterstaining was performed with hematoxylin (G1080, Solarbio). Sections were then dehydrated and sealed with mounting medium. Images were visualized by using Leica Aperio ScanScope XT Slide Scanner (Wetzlar, Germany) and processed with Leica Aperio ImageScope. The following primary antibodies were used: anti‐HK2 antibody (1:100; ab104836, Abcam), anti‐Lin28 antibody (1:100; ab109751, Abcam), anti‐Nanog antibody (1:100; ab109250, Abcam), anti‐Oct4 antibody (1:100; #2840, Cell Signaling Technology) and anti‐CD133 antibody (1:100; #64326, Cell Signaling Technology). The IHC scores were assessed by two independent authors blinded to the section treatment. IHC scoring was based on the percentage of positive cells and the staining intensity as previously described [41].
2.20. Statistical analysis
Statistical analyses were conducted with a two‐tailed unpaired Student's t test, and all data are expressed as the mean ± standard deviation (SD) obtained in at least three independent experiments. Overall survival was analyzed with Kaplan‐Meier curves (from the date of surgery to death); differences in survival among patient subgroups were analyzed using a log‐rank test [42]. The correlation between HK2 and CD133 levels was analyzed using a Pearson correlation coefficient. All the data were analyzed with GraphPad Prism 8 (GraphPad Software, San Diego, CA, USA). P values of less than 0.05 were considered significant. All statistical tests were two‐tailed.
3. RESULTS
3.1. HK2 expression is elevated in SCLC CSCs and correlates with poor prognosis of SCLC patients
Glycolysis is elevated in CSCs of non‐small cell lung cancer and gastric cancer compared to their differentiated counterparts [43, 44]. In line with these findings, stem‐like spheroids of H446 and H1048 cells exhibited higher glucose uptake and lactate production than their monolayer cells (Figure 1A,B). Immunoblotting analyses of the glycolytic enzyme HK2, PKM2, PFKP, PDK1, GLUT1, and LDHA showed that only HK2 expression was upregulated in SCLC CSCs, which also exhibited increased expression of Nanog, Oct4 and Lin28 (Figure 1C,D and Supplementary Figure S1A‐B). Consistent results were also observed by immunofluorescence staining (Figure 1E). In addition, qRT‐PCR analyses showed that the mRNA levels of HK2, Nanog, POU5F1 (encoding Oct4) and Lin28 were enhanced in CSCs compared to the levels in their counterpart monolayer cells (Supplementary Figure S1C‐D). These results suggested that SCLC CSCs enhance aerobic glycolysis, with greatly increased HK2 expression.
FIGURE 1.
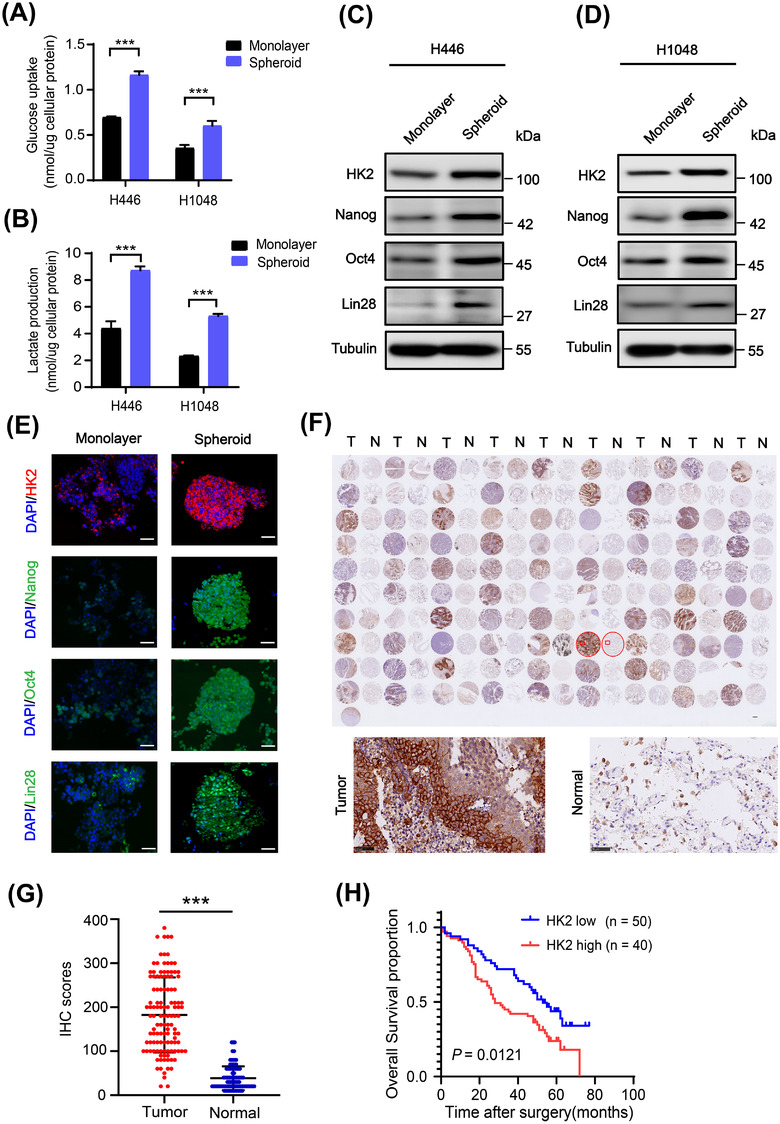
HK2 expression is elevated in SCLC CSCs and correlates with poor prognosis of SCLC patients. (A‐B) Glucose consumption (A) and lactate production (B) of H446 and H1048 cell monolayers or spheroids were determined. Data shown are the mean ± SD (n = 3). ***, P < 0.001. (C‐D) Immunoblotting analyses of the lysis of H446 (C) and H1048 (D) cell monolayers or spheroids were performed with the indicated antibodies. (E) Immunofluorescence analyses were performed with the indicated antibodies in monolayer H1048 cells or spheroid cells. DAPI was used for nuclear staining. Scale bars: 50 μm. (F‐G) Ninety human SCLC specimens with their adjacent tissues were analyzed by IHC with an anti‐HK2 antibody (F, top). Representative and magnified IHC images imagines of SCLC tissues and normal tissues were shown (F, bottom). IHC scores of HK2 expression were calculated (G). N, adjacent normal tissue; T, tumor tissue. Scale bars: 50 μm. (H) Kaplan‐Meier plots of the overall survival time of 90 SCLC patients with low (score ≤ 4) or high (score >4) expression levels of HK2. P values are calculated using a log‐rank test (two‐tailed). Abbreviations: HK2, hexokinase 2; SCLC, small cell lung cancer; CSC, cancer stem cell; SD, standard deviation; DAPI, 4',6‐diamidino‐2‐phenylindole; IHC, immunohistochemistry
To determine the expression levels of HK2 in human SCLC tissues, we performed IHC analyses of SCLC tissues and their adjacent normal tissues, which were also stained with H&E (Supplementary Figure S1E). The results showed that HK2 expression levels were substantially higher in the tumor tissues than in the adjacent normal tissues (Figure 1F,G). Notably, Kaplan‐Meier survival analysis demonstrated that high HK2 levels were correlated with poor overall survival of SCLC patients (Figure 1H). These results strongly suggested that HK2 is highly expressed in SCLC and a biomarker for poor prognosis of SCLC patients.
3.2. HK2 promotes CSC stemness and tumor growth
To determine the role of HK2 in SCLC cell stemness, we stably depleted HK2 using two different HK2 short hairpin RNAs (shRNAs) in H1048, H69, and H446 SCLC cells. HK2 depletion reduced the protein (Figure 2A,B and Supplementary Figure S2A) and mRNA (Supplementary Figure S2B) expression levels of Nanog, POU5F1, and Lin28 and inhibited tumor cell spheroid formation by reducing spheroid diameters and numbers either in the first‐generation spheres or the second‐generation spheres (Figure 2C and Supplementary Figure S2C‐D). Notably, HK2 depletion also reduced the ALDH+ (stem cell marker) population of H1048 cells (Supplementary Figure S2E), suggesting that HK2 specifically affects CSC stemness.
FIGURE 2.

HK2 promotes CSC stemness and tumor growth. (A‐B) Immunoblotting analyses of H1048 (A) and H69 (B) cells with or without HK2 depletion were performed with the indicated antibodies. (C) The sphere formation ability of H1048 cells or H1048 cells with or without HK2 depletion in their first and the second passage was examined. Representative images were displayed (top), and the diameters and number of spheres were determined (bottom). Data shown are the mean ± SD (n = 3). ***, P < 0.001. Scale bars: 100 μm. (D‐E) Immunoblotting analyses of H1048 (D) and H69 (E) cells with or without HK2 overexpression were performed with the indicated antibodies. (F) The sphere formation ability of H1048 cells with or without HK2 overexpression in their first and the second passage was examined. Representative images were displayed (top), and the diameters and number of spheres were determined (bottom). Data shown are the mean ± SD (n = 3). **, P < 0.01, ***, P < 0.001. Scale bars: 100 μm. (G) Immunodeficient mice (n = 10) were subcutaneously inoculated with an equal number (2 × 106) of H1048 cells with or without HK2 shRNA expression. Tumor sizes (top) and volumes (bottom) were measured and calculated. Data represent the means ± SD of ten mice per group. ***, P < 0.001. (H) IHC staining of mouse tumor tissues derived from H1048 cells with or without HK2 shRNA expression was performed with the indicated antibodies. Representative images were displayed (left), and the IHC scores were calculated (right). Data shown are the mean ± SD (n = 5). ***, P < 0.001. Scale bars: 100 μm. (I) Different numbers of H1048 cells with or without HK2 overexpression were subcutaneously injected into NOD/SCID mice (n = 10). Images of the tumors are shown on the left. Scale bar (right): 1 cm. Stem cell frequencies were estimated as the ratio 1/x with the upper and lower 95% confidence intervals (middle). ELDA analysis was performed (right). Abbreviations: HK2, hexokinase 2; CSC, cancer stem cell; SD, standard deviation; IHC, immunohistochemistry; ELDA, extreme limiting dilution analysis
In contrast to HK2 depletion, HK2 overexpression in H1048, H69 and H446 cells increased the expression levels of Nanog, Oct4, and Lin28 (Figure 2D,E and Supplementary Figure S2F); spheroid formation ability (Figure 2F and Supplementary Figure S2G‐H); and the proportion of ALDH+ cells (Supplementary Figure S2I‐J).
We next determined the role of HK2 in tumor growth by subcutaneously injecting H1048 and H1048 cells with or without HK2 depletion or HK2 overexpression into mice. We showed that HK2 depletion inhibited tumor growth in mice (Figure 2G), with a corresponding reduction in Nanog and Lin28 expression in tumor tissues (Figure 2H). In contrast, HK2 overexpression significantly promoted tumorigenic ability, even when a small number of tumor cells were injected (Figure 2I). These results strongly suggested that HK2 promotes SCLC stemness and tumor growth.
3.3. HK2 interacts with CD133 and inhibits polyubiquitylation‐dependent CD133 degradation
CD133+ CSCs in SCLC are highly tumorigenic [14]. Cell sorting and immunoblotting analyses of CD133– and CD133+ subpopulations from H1048 cells or H69 cells showed that the expression levels of HK2 and Nanog were dramatically higher in the CD133+ cells than in the CD133– cells (Supplementary Figure S3A). Notably, HK2 depletion reduced CD133 expression (Figure 3A), and HK2 overexpression enhanced CD133 expression in SCLC cells (Figure 3B) without affecting the mRNA levels of prominin‐1 (encoding CD133) (Supplementary Figure S3B). Treatment of the cells with the proteasome inhibitor MG132 blocked HK2 depletion‐induced CD133 downregulation (Supplementary Figure S3C), suggesting that HK2 stabilized CD133 by inhibiting proteasome‐dependent CD133 degradation. In line with these findings, HK2 depletion (Figure 3C and Supplementary Figure S3D) or HK2 overexpression (Figure 3D and Supplementary Figure S3E) in both H1048 and H69 cells decreased or increased CD133 half‐life, respectively, in the presence of cycloheximide (CHX), a protein translation inhibitor to eliminate the transcriptional effect. Correspondingly, HK2 depletion (Figure 3E) or overexpression (Figure 3F) increased or reduced CD133 polyubiquitylation, respectively. These results indicated that HK2 expression inhibits CD133 polyubiquitination and proteasomal degradation.
FIGURE 3.

HK2 interacts with CD133 and inhibits polyubiquitylation‐dependent CD133 degradation. (A‐B) HK2 was depleted (A) or overexpressed (B) in H1048 or H69 cells. Immunoblotting analyses were performed with an anti‐CD133 antibody. (C‐D) H1048 cells with or without HK2 depletion (C) or overexpression (D) were treated with CHX (100 μg/mL) for the indicated periods of time. Immunoblotting analyses were performed with the indicated antibodies (left). Quantification of CD133 expression levels relative to tubulin expression levels is shown (right). (E‐F) H1048 cells with or without HK2 depletion (E) or overexpression (F) were treated with MG132 (50 μmol/L) for 8 hours. Immunoprecipitation with an anti‐CD133 antibody and immunoblotting analyses were performed with the indicated antibodies. (G) Purified His‐HK2 was incubated with purified GST or GST‐CD133 together with glutathione‐sepharose 4B beads. GST pulldown assay and immunoblotting analyses were performed with the indicated antibodies. (H) Mitochondrial and non‐mitochondrial fractions of H1048 monolayers or spheroids were prepared and subjected to immunoprecipitation with an anti‐HK2 antibody. Immunoblotting analyses were performed with the indicated antibodies. (I) RNAi‐resistant WT HK2, inactive HK2 D209/657A, or HK2 △N16 was expressed in H1048 and H69 cells with depletion of endogenous HK2. Immunoblotting analyses were performed with the indicated antibodies. (J) H1048 cells with or without WT HK2 or inactive HK2 D209/657A overexpression were treated with MG132 (50 μmol/L) for 8 hours. Immunoprecipitation and immunoblotting analyses were performed with the indicated antibodies. (K) H1048 cells with or without expression of inactive HK2 D209/657A were treated with CHX (100 μg/mL) for the indicated periods of time. Immunoblotting analyses were performed with the indicated antibodies (upper). Quantification of CD133 expression levels relative to tubulin expression levels is shown (lower). Abbreviations: HK2, hexokinase 2; CHX, cycloheximide; IP, immunoprecipitation; WCL, whole cell lysate; WT, wide type
To further determine the relationship between HK2 and CD133, we performed coimmunoprecipitation analyses with limited amounts of either an anti‐HK2 (Supplementary Figure S3F) or an anti‐CD133 (Supplementary Figure S3G) antibody so that these antibodies were saturated and could precipitate comparable amounts of the proteins in SCLC cells in spheroids and monolayers. We found that HK2 could bind to CD133. Notably, the interaction between HK2 with CD133 was largely enhanced in CSCs compared to their monolayer counterparts (Supplementary Figure S3F‐G). Consistently, immunofluorescence analyses showed that the colocalization of HK2 with CD133 was amplified in spheroid cells than in monolayer cells (Supplementary Figure S3H) and detected in human SCLC tissues (Supplementary Figure S3I). Moreover, in vitro GST pulldown assay showed that purified GST‐CD133 directly bound to purified His‐HK2 (Figure 3G). These results suggested that HK2 directly interacts with CD133 and inhibits polyubiquitylation‐dependent CD133 degradation.
Mitochondrial outer membrane‐localized HK2 catalyzes glycolysis [45, 46]. Immunoprecipitation analyses of mitochondrial and non‐mitochondrial fractions of SCLC monolayer cells or spheroid cells showed that the interaction of HK2 and CD133 occurred in non‐mitochondrial fractions rather than in the mitochondrial fractions (Figure 3H and Supplementary Figure S3J), revealing a noncanonical function of non‐mitochondrial HK2 in the stabilization of CD133. Consistently, reconstituted expression of RNAi‐resistant HK2 △N16 with depletion of mitochondrial binding domain (MBD), the first N‐terminal 16 amino acids (MIASHLLAYFFTELNH) mitochondrial binding domain (MBD) [47], increased CD133 expression compared to reconstituted expression of its wild‐type (WT) counterpart (Figure 3I). Of note, reconstituted expression of Flag‐HK2 D209/657A, a glucose‐binding inactive mutant that lost mitochondria‐cytosol translocation [47, 48] (Supplementary Figure S3K), was unable to increase CD133 expression (Figure 3I) and failed to suppress CD133 polyubiquitination (Figure 3J) or alter the half‐life of CD133 (Figure 3K). These results indicated that cytosolic HK2 stabilizes CD133 in a manner dependent on the metabolic activity of HK2.
3.4. The interaction between HK2 and CD133 induces binding of USP11 to CD133 and inhibits CD133 polyubiquitylation and degradation
To determine the mechanism underlying HK2‐enhanced CD133 stability, we immunoprecipitated CD133 from H1048 cells with or without HK2 overexpression and performed mass spectrometry (MS) analyses. We showed that the deubiquitinase, USP11, is a CD133‐associated protein (Supplementary Figure S4A), the expression of which was not obviously altered by CD133 expression levels (Supplementary Figure S4B).
Coimmunoprecipitation analyses showed that the SCLC spheroid cells exhibited an enhanced association of CD133 with USP11 compared to their monolayer counterparts (Figure 4A). Consistently, immunofluorescence analyses showed increased colocalization of USP11 with CD133 in spheroid cells compared to monolayer cells (Supplementary Figure S4C). These results indicated that the interaction between CD133 and USP11 is distinctly regulated between CSCs and their differentiated cells.
FIGURE 4.

The interaction between HK2 and CD133 induces binding of USP11 to CD133 and inhibits CD133 polyubiquitylation and degradation. (A) SCLC monolayer or spheroid cells were lysed. Immunoprecipitation and immunoblotting analyses were performed with the indicated antibodies. (B) 293T cells expressing HA‐Ub were transfected with or without a Flag‐CD133 plasmid, followed by treatment with MG132 (50 μmol/L) for 8 hours. CD133 purified by immunoprecipitation was incubated with or without bacterially purified GST‐USP11. Immunoblotting analyses were performed with the indicated antibodies (upper). The purification efficiency was determined using SDS‐PAGE and Coomassie brilliant blue staining (lower). (C) shRNA resistant WT Flag‐USP11 or inactive Flag‐USP11(C318S) mutant was expressed in H1048 USP11 depletion cells. Immunoblotting analyses were performed with the indicated antibodies. (D) H1048 cells expressing two different USP11 shRNAs or a control shRNA were treated with MG132 (50 μmol/L) for 8 hours. Immunoprecipitation and immunoblotting analyses were performed with the indicated antibodies. (E) Two different USP11 shRNAs or a control shRNA were expressed in H1048 cells. Immunoblotting analyses were performed with the indicated antibodies. (F) H1048 cells expressing two different USP11 shRNAs or a control shRNA were examined using a first and the second passage sphere formation assay. Representative images were displayed (upper). The diameters and number of spheres were determined (lower). Data shown are the mean ± SD (n = 3). ***, P < 0.001. Scale bars: 100 μm. (G) 293T cells were transfected with or without Flag‐CD133. Purified CD133 was incubated with or without His‐HK2, followed by incubation with purified GST‐USP11. GST pulldown assay and immunoblotting analyses were performed (upper). The purification efficiency was determined using SDS‐PAGE and coomassie brilliant blue staining (lower). (H) H1048 CSCs with endogenous HK2 depletion and with or without reconstituted expression of RNAi‐resistant WT Flag‐rHK2 or an inactive Flag‐rHK2 D209/657A mutant were analyzed by immunoprecipitation and immunoblotting with the indicated antibodies. (I) RNAi‐resistant WT HK2, inactive HK2 D209/657A, or HK2 △N16 was expressed in H1048 cells with depletion of endogenous HK2. Immunoprecipitation and immunoblotting analyses were performed with the indicated antibodies. Abbreviations: HK2, hexokinase 2; USP11, ubiquitin‐specific protease 11; SCLC, small cell lung cancer; SDS‐PAGE; sodium dodecyl sulfate polyacrylamide gel electrophoresis; shRNA, short hairpin RNA; SD, standard deviation; WT, wide type; IP, immunoprecipitation; WCL, whole cell lysate
In vitro deubiquitylation analyses showed that purified USP11 decreased polyubiquitylation of purified CD133 (Figure 4B). Overexpression of shRNA resistant wild‐type (WT) USP11, but not the catalytically inactive USP11(C318S) mutant, in SCLC cells increased CD133 expression (Figure 4C and Supplementary Figure S4D) and cancer stemness, reflected by increased expression levels of Oct4 and Lin28 (Figure 4C and Supplementary Figure S4D) and sphere formation ability (Supplementary Figure S4E). In contrast, USP11 depletion in SCLC cells increased CD133 polyubiquitylation (Figure 4D), decreased the expression levels of CD133, Oct4, and Lin28 (Figure 4D,E and Supplementary Figure S4F), and reduced the sphere formation ability of the cells (Figure 4F). The effect of USP11 depletion on CD133 polyubiquitylation and expression was abrogated by reconstituted expression of shRNA resistant WT USP11 but not inactive USP11 (C318S; Supplementary Figure S4G). In addition, MG132 treatment blocked USP11 depletion‐reduced CD133 expression (Supplementary Figure S4H). These results indicated that USP11 enhances CD133 expression by inhibiting CD133 polyubiquitination‐ and proteasome‐dependent degradation.
To determine the role of HK2 in USP11‐regulated CD133 expression, we performed an in vitro GST pulldown assay and showed that purified His‐CD133 bound to GST‐USP11. Notably, the binding of USP11 to CD133 was largely increased under preincubation CD133 with purified HK2 (Figure 4G). In addition, HK2 depletion in H1048 CSCs decreased the association between USP11 and CD133 without altering USP11 expression, and this decrease was rescued by reconstituted expression of WT HK2 but not the inactive HK2 D209/657A mutant (Figure 4H). Notably, HK2 △N16 expression increased the association between CD133 to USP11 compared to wild‐type HK2 expression (Figure 4I). These results indicated that non‐mitochondrial HK2 increases the binding of USP11 to CD133, thereby reducing CD133 polyubiquitylation and enhancing its expression.
3.5. HK2 promotes cancer stem‐like properties via CD133
CD133 expression promotes cancer stemness [14]. HK2 overexpression enhanced the expression of Oct4 and Lin28 (Figure 5A) and CSC sphere formation (Supplementary Figure S5A), and this increase was largely abrogated by depletion of USP11. Of note, USP11 depletion‐induced inhibitory effect was eliminated by ectopic expression of CD133. In addition, HK2 depletion‐reduced Oct4 and Lin28 expression and CSC spheroid formation were also greatly rescued by overexpression of CD133 (Figure 5B and Supplementary Figure S5B).
FIGURE 5.
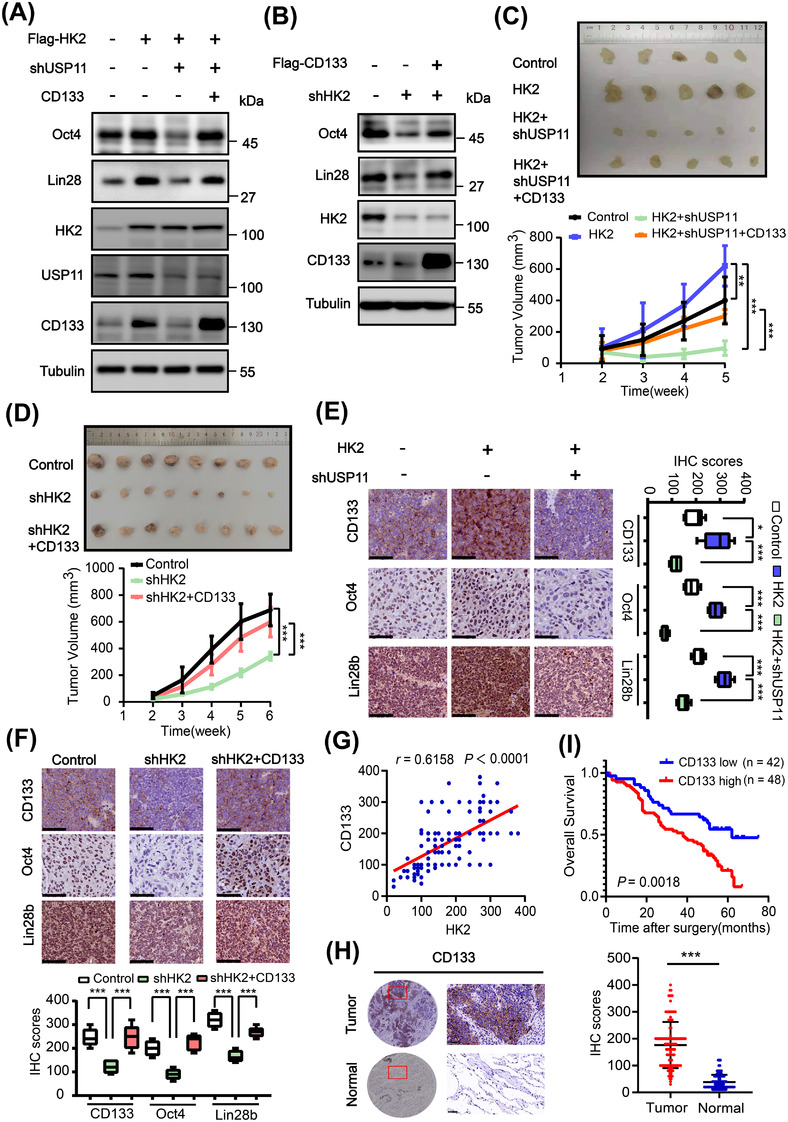
HK2 promotes cancer stem‐like properties via CD133. (A) H1048 cells with or without the indicated expression of Flag‐HK2, USP11 shRNA, and CD133 were analyzed via immunoblotting with the indicated antibodies. (B) H1048 cells with or without expression of HK2 shRNA and Flag‐CD133 were analyzed via immunoblotting with the indicated antibodies. (C) H1048 cells with or without the indicated combined expression of Flag‐HK2, USP11 shRNA, and CD133 were subcutaneously injected into the flank regions of nude mice. Tumor sizes (upper) and volumes (lower) were measured and calculated. Data represent the means ± SD of five mice per group. **, P < 0.01, ***; P < 0.001. (D) H1048 cells with or without HK2 shRNA expression or combined expression of HK2 shRNA and Flag‐CD133 were subcutaneously injected into the flank regions of nude mice. Tumor sizes (top) and volumes (bottom) were measured and calculated. Data represent the means ± SD of eight mice per group. ***, P < 0.001. (E‐F) IHC staining of tumor tissues was performed with the indicated antibodies. Representative images were shown (left or upper). IHC scores (right or lower) were calculated. Data represent the means ± SD (n = 5). *, P < 0.05; ***, P < 0.001. Scale bars: 100 μm. (G) Correlation between HK2 and CD133 expression in an SCLC tissue microarray was analyzed using a two‐tailed Pearson correlation coefficient. (H) Representative and magnified IHC images of CD133 protein expression (left) in paired SCLC tumor and adjacent normal tissues from a tissue array were shown. Analysis of the IHC scores of CD133 expression was performed (right). N, adjacent normal tissue; T, tumor tissue. Scale bars: 50 μm. (I) Kaplan–Meier survival curves were compared using a log‐rank test in 90 patients. The expression of CD133 was classified as low (score ≤ 4) or high (score >4). Abbreviations: HK2, hexokinase 2; USP11, ubiquitin‐specific protease 11; shRNA, short hairpin RNA; SCLC, small cell lung cancer; SD, standard deviation; IHC, immunohistochemistry
We next subcutaneously injected H1048 cells with or without HK2 overexpression or depletion in the presence or absence of CD133 overexpression or USP11 depletion. We found that HK2 overexpression enhanced tumor growth (Figure 5C). This enhancement was abrogated by USP11 depletion, but the abrogation was rescued by ectopic expression of CD133 (Figure 5C). In contrast, HK2 depletion inhibited tumor growth, and this inhibition was alleviated by CD133 overexpression (Figure 5D). IHC analysis of tumor tissues showed that HK2 overexpression increased expression of CD133, nuclear Oct4, and Lin28, and this increase was inhibited by USP11 depletion (Figure 5E). Conversely, HK2 depletion decreased expression of CD133, Oct4, and Lin28, and this decrease was rescued by ectopic CD133 expression (Figure 5F). These results indicated that HK2 promotes USP11‐mediated CD133 expression and tumor growth.
To determine the clinical significance of HK2‐regulated CD133 expression, we performed IHC analyses of 90 SCLC tissue specimens. We found that HK2 protein expression levels were positively correlated with CD133 expression (Figure 5G). In addition, CD133 expression levels in SCLC tissues were much higher than those in adjacent normal tissues (Figure 5H) and inversely correlated with the survival time of SCLC patients (Figure 5I). These results strongly suggested that HK2‐stabilized CD133 expression promotes clinical aggressiveness of SCLC.
4. DISCUSSION
Metabolic enzymes possess multifaceted roles in promoting tumorigenesis [22, 24]. We demonstrated that HK2 expression is much higher in SCLC specimens than in their normal adjacent tissues and correlates with poor prognosis. Importantly, HK2 is highly expressed in SCLC CSCs compared to differentiated cells, and depletion of HK2 reduced CSC spheroid formation ability and promoted CSC differentiation, with corresponding reductions in the protein and mRNA levels of Nanog, Oct4, and Lin28. Notably, HK2 interacted with CD133 and regulated CD133 expression without affecting the CD133 mRNA level. Mechanistically, non‐mitochondrial HK2 bound to CD133, thereby enhancing the binding of USP11 to CD133 and reducing CD133 polyubiquitylation and degradation. In addition, HK2 enhanced the expression of transcription factors and the translation reprogramming factor Lin28 which are critical for cell renewal, SCLC cell stemness, and tumor growth in mice in a CD133 expression‐dependent manner, and these effects were mediated by USP11‐dependent CD133 deubiquitylation (Figure 6).
FIGURE 6.

A schematic displays the mechanism underlying HK2‐regulated cancer cell stemness. In cancer stem cells, HK2 expression is increased. Non‐mitochondrial HK2 binds to CD133 and subsequently promotes the interaction between USP11 and CD133, leading to inhibition of CD133 polyubiquitlytion and increase of CD133 stability and CD133 maintenance (left). In differentiated cancer cells, HK2 binds to outer membrane of mitochondria, promoting ubiquitin‐proteasome system‐mediated CD133 degradation. The light green parts represent the cytoplasm. The red solid arrows represent positive regulation or increased expression of the indicated factors. Black solid arrows represent decrease of gene expression. The black dotted line indicates that the regulation will not occur
It has been shown that multiple metabolic enzymes exert a noncanonical function in diverse cellular activities in tumor cells. Several metabolic enzymes, including phosphoglycerate kinase 1 (PGK1), pyruvate kinase M2 (PKM2), ketohexokinase isoform A (KHKA), phosphoenolpyruvate carboxykinase 1 (PCK1), and choline kinase α, can function as protein kinases and phosphorylate a variety of protein substrates to promote tumor development [27, 28, 33, 36, 49–60]. Mitochondrial α‐ketoglutarate dehydrogenase (α‐KGDH) translocates into the nucleus and associates with KAT2A, which is known as a histone acetyltransferase. α‐KGDH‐associated KAT2A gains a new function and acts as a histone H3 succinyltransferase to regulate gene expression by locally catalyzing succinyl‐CoA generated by α‐KGDH [29, 61, 62]. It was previously shown that the mitochondria‐localized HK2 promoted tumor development through its glycolytic function [30, 31, 32]. K63‐linked ubiquitination by HectH9 ubiquitin E3 ligase enhanced the mitochondrial localization and glycolytic function of HK2, thereby promoting prostate CSC expansion and prostate CSC‐associated chemoresistance [63]. In esophageal CSCs, HK2 expression was upregulated by Hsp27‐dependent AKT activation and required for maintenance of glycolysis and CSC phenotypes [64], further supporting the role of glycolic function of HK2 in tumor cell stemness. In contrast, the current report showed that HK2 promoted SCLC cell stemness dependent on CD133 upregulation but independent of the mitochondrial localization of HK2, highlighting a critical noncanonical role of HK2 in regulating CSC stemness. Our findings suggested that enhanced glycolysis promotes the disassociation of HK2 from mitochondria and subsequent binding of HK2 to CD133, during which the conformation of HK2 regulated by its enzymatic activity is essential. Importantly, the clinical significance of this regulation was evidenced by the positive correlation between HK2 and CD133 expression in human SCLC specimens and their association with SCLC patient overall survival. These findings underscore the potential to treat human SCLC by disrupting HK2‐mediated CD133 expression.
Our study revealed that the binding of HK2 to CD133 promotes the binding of USP11 to CD133. The detailed mechanism underlying the HK2‐mediated CD133‐USP11 interaction remains to be elucidated. Given that glycolytic enzymes often possess multifaced roles in tumor development, it would be of importance to determine whether HK2 regulates critical cellular activities with yet unidentified noncanonical functions in future studies.
5. CONCLUSIONS
This study revealed that HK2 possessed a moonlighting function to promote cancer cell stemness through enhanced interaction between USP11 and CD133 and subsequent deubiquitylation and stabilization of CD133 and then upregulated expression of Oct4 and Lin28. The delineated noncanonical function of HK2 promoted SCLC cell proliferation and tumor growth.
DECLARATIONS
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
The use of human SCLC specimens and the study protocol were approved by the Institute Research Medical Ethics Committee at the National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College (permit number: NCC2021C‐052). All tissue samples were collected in compliance with an informed consent policy. All animal experiments were conducted in accordance with guidelines approved by the Institutional Animal Care and Use Committee (IACUC) of the National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College (permit number: A019).
CONSENT FOR PUBLICATION
Not applicable.
AVAILABILITY OF DATA AND MATERIALS
The data that support the findings of this study are available from the corresponding author upon reasonable request.
COMPETING INTERESTS
The authors declare that they have no conflicts of interest.
AUTHORS’ CONTRIBUTIONS
Zhimin Lu conceived and designed the study. Jie He provided critical scientific input. Juhong Wang, Yannan Yang, Fei Shao, Wei Wang, Xueying Yang, and Sijin Sun performed the experiments. Jie He, Yibo Gao, and Xiaoli Feng established the patient cohort and biobanking of specimen for tissue microarray. Renda Li and Hong Cheng collected clinical features of the patients. Juhong Wang wrote the draft manuscript. Zhimin Lu and Yibo Gao revised the manuscript.
Supporting information
Supporting Information
ACKNOWLEDGEMENTS
Not applicable.
Wang J, Shao F, Yang Y, Wang W, Yang X, Li R, et al. A non‐metabolic function of hexokinase 2 in small cell lung cancer: promotes cancer cell stemness by increasing USP11‐mediated CD133 stability. Cancer Commun. 2022;42:1008–1027. 10.1002/cac2.12351
Contributor Information
Yibo Gao, Email: gaoyibo@cicams.ac.cn.
Jie He, Email: hejie@cicams.ac.cn.
Zhimin Lu, Email: zhiminlu@zju.edu.cn.
REFERENCES
- 1. Yang S, Zhang Z, Wang Q. Emerging therapies for small cell lung cancer. J Hematol Oncol. 2019;12(1):47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. van Meerbeeck JP, Fennell DA, De Ruysscher DK. Small‐cell lung cancer. Lancet. 2011;378(9804):1741–55. [DOI] [PubMed] [Google Scholar]
- 3. Bunn PA, Jr. , Minna JD, Augustyn A, Gazdar AF, Ouadah Y, Krasnow MA, et al. Small Cell Lung Cancer: Can Recent Advances in Biology and Molecular Biology Be Translated into Improved Outcomes? J Thorac Oncol. 2016;11(4):453–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Semenova EA, Nagel R, Berns A. Origins, genetic landscape, and emerging therapies of small cell lung cancer. Genes Dev. 2015;29(14):1447–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Farago AF, Keane FK. Current standards for clinical management of small cell lung cancer. Transl Lung Cancer Res. 2018;7(1):69–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yu J, Wang S, Zhao W, Duan J, Wang Z, Chen H, et al. Mechanistic Exploration of Cancer Stem Cell Marker Voltage‐Dependent Calcium Channel α2δ1 Subunit‐mediated Chemotherapy Resistance in Small‐Cell Lung Cancer. Clin Cancer Res. 2018;24(9):2148–58. [DOI] [PubMed] [Google Scholar]
- 7. Kalemkerian GP, Schneider BJ. Advances in Small Cell Lung Cancer. Hematol Oncol Clin North Am. 2017;31(1):143–56. [DOI] [PubMed] [Google Scholar]
- 8. Zimmerman S, Das A, Wang S, Julian R, Gandhi L, Wolf J. 2017‐2018 Scientific Advances in Thoracic Oncology: Small Cell Lung Cancer. J Thorac Oncol. 2019;14(5):768–83. [DOI] [PubMed] [Google Scholar]
- 9. Dómine M, Moran T, Isla D, Martí JL, Sullivan I, Provencio M, et al. SEOM clinical guidelines for the treatment of small‐cell lung cancer (SCLC) (2019). Clin Transl Oncol. 2020;22(2):245–55. [DOI] [PubMed] [Google Scholar]
- 10. Peng F, Wang JH, Fan WJ, Meng YT, Li MM, Li TT, et al. Glycolysis gatekeeper PDK1 reprograms breast cancer stem cells under hypoxia. Oncogene. 2018;37(8):1062–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sheng Y, Yu C, Liu Y, Hu C, Ma R, Lu X, et al. FOXM1 regulates leukemia stem cell quiescence and survival in MLL‐rearranged AML. Nat Commun. 2020;11(1):928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Testa U, Castelli G, Pelosi E. Lung Cancers: Molecular Characterization, Clonal Heterogeneity and Evolution, and Cancer Stem Cells. Cancers (Basel). 2018;10(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Najafi M, Farhood B, Mortezaee K. Cancer stem cells (CSCs) in cancer progression and therapy. J Cell Physiol. 2019;234(6):8381–95. [DOI] [PubMed] [Google Scholar]
- 14. Sarvi S, Mackinnon AC, Avlonitis N, Bradley M, Rintoul RC, Rassl DM, et al. CD133+ cancer stem‐like cells in small cell lung cancer are highly tumorigenic and chemoresistant but sensitive to a novel neuropeptide antagonist. Cancer Res. 2014;74(5):1554–65. [DOI] [PubMed] [Google Scholar]
- 15. Jang JW, Song Y, Kim SH, Kim JS, Kim KM, Choi EK, et al. CD133 confers cancer stem‐like cell properties by stabilizing EGFR‐AKT signaling in hepatocellular carcinoma. Cancer Lett. 2017;389:1–10. [DOI] [PubMed] [Google Scholar]
- 16. Ma S, Lee TK, Zheng BJ, Chan KW, Guan XY. CD133+ HCC cancer stem cells confer chemoresistance by preferential expression of the Akt/PKB survival pathway. Oncogene. 2008;27(12):1749–58. [DOI] [PubMed] [Google Scholar]
- 17. Wei Y, Jiang Y, Zou F, Liu Y, Wang S, Xu N, et al. Activation of PI3K/Akt pathway by CD133‐p85 interaction promotes tumorigenic capacity of glioma stem cells. Proc Natl Acad Sci USA. 2013;110(17):6829–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Boivin D, Labbe D, Fontaine N, Lamy S, Beaulieu E, Gingras D, et al. The stem cell marker CD133 (prominin‐1) is phosphorylated on cytoplasmic tyrosine‐828 and tyrosine‐852 by Src and Fyn tyrosine kinases. Biochemistry. 2009;48(18):3998–4007. [DOI] [PubMed] [Google Scholar]
- 19. Mak AB, Nixon AM, Kittanakom S, Stewart JM, Chen GI, Curak J, et al. Regulation of CD133 by HDAC6 promotes β‐catenin signaling to suppress cancer cell differentiation. Cell Rep. 2012;2(4):951–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu YM, Li XF, Liu H, Wu XL. Ultrasound‐targeted microbubble destruction‐mediated downregulation of CD133 inhibits epithelial‐mesenchymal transition, stemness and migratory ability of liver cancer stem cells. Oncol Rep. 2015;34(6):2977–86. [DOI] [PubMed] [Google Scholar]
- 21. Chen YS, Wu MJ, Huang CY, Lin SC, Chuang TH, Yu CC, et al. CD133/Src axis mediates tumor initiating property and epithelial‐mesenchymal transition of head and neck cancer. PLoS One. 2011;6(11):e28053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xu D, Shao F, Bian X, Meng Y, Liang T, Lu Z. The evolving landscape of noncanonical functions of metabolic enzymes in cancer and other pathologies. Cell Metab. 2021;33(1):33–50. [DOI] [PubMed] [Google Scholar]
- 23. Li X, Egervari G, Wang Y, Berger SL, Lu Z. Regulation of chromatin and gene expression by metabolic enzymes and metabolites. Nat Rev Mol Cell Biol. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang Y, Xia Y, Lu Z. Metabolic features of cancer cells. Cancer Commun (Lond). 2018;38(1):65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Folmes CD, Dzeja PP, Nelson TJ, Terzic A. Metabolic plasticity in stem cell homeostasis and differentiation. Cell Stem Cell. 2012;11(5):596–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Feng W, Gentles A, Nair RV, Huang M, Lin Y, Lee CY, et al. Targeting unique metabolic properties of breast tumor initiating cells. Stem Cells. 2014;32(7):1734–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xu D, Wang Z, Xia Y, Shao F, Xia W, Wei Y, et al. The gluconeogenic enzyme PCK1 phosphorylates INSIG1/2 for lipogenesis. Nature. 2020;580(7804):530–5. [DOI] [PubMed] [Google Scholar]
- 28. Xu D, Li X, Shao F, Lv G, Lv H, Lee JH, et al. The protein kinase activity of fructokinase A specifies the antioxidant responses of tumor cells by phosphorylating p62. Sci Adv. 2019;5(4):eaav4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang Y, Guo YR, Liu K, Yin Z, Liu R, Xia Y, et al. KAT2A coupled with the alpha‐KGDH complex acts as a histone H3 succinyltransferase. Nature. 2017;552(7684):273–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Roberts DJ, Tan‐Sah VP, Ding EY, Smith JM, Miyamoto S. Hexokinase‐II positively regulates glucose starvation‐induced autophagy through TORC1 inhibition. Mol Cell. 2014;53(4):521–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang L, Xiong H, Wu F, Zhang Y, Wang J, Zhao L, et al. Hexokinase 2‐mediated Warburg effect is required for PTEN‐ and p53‐deficiency‐driven prostate cancer growth. Cell Rep. 2014;8(5):1461–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jiao L, Zhang HL, Li DD, Yang KL, Tang J, Li X, et al. Regulation of glycolytic metabolism by autophagy in liver cancer involves selective autophagic degradation of HK2 (hexokinase 2). Autophagy. 2018;14(4):671–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Qian X, Li X, Shi Z, Xia Y, Cai Q, Xu D, et al. PTEN Suppresses glycolysis by dephosphorylating and inhibiting autophosphorylated PGK1. Mol Cell. 2019;76(3):516–27 e7. [DOI] [PubMed] [Google Scholar]
- 34. Ji H, Ding Z, Hawke D, Xing D, Jiang BH, Mills GB, et al. AKT‐dependent phosphorylation of Niban regulates nucleophosmin‐ and MDM2‐mediated p53 stability and cell apoptosis. EMBO Rep. 2012;13(6):554–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zheng Y, Li X, Qian X, Wang Y, Lee JH, Xia Y, et al. Secreted and O‐GlcNAcylated MIF binds to the human EGF receptor and inhibits its activation. Nat Cell Biol. 2015;17(10):1348–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liu R, Lee JH, Li J, Yu R, Tan L, Xia Y, et al. Choline kinase alpha 2 acts as a protein kinase to promote lipolysis of lipid droplets. Mol Cell. 2021;81(13):2722–35 e9. [DOI] [PubMed] [Google Scholar]
- 37. Zhong M, Lu Z, Foster DA. Downregulating PKC delta provides a PI3K/Akt‐independent survival signal that overcomes apoptotic signals generated by c‐Src overexpression. Oncogene. 2002;21(7):1071–8. [DOI] [PubMed] [Google Scholar]
- 38. Lee JH, Liu R, Li J, Wang Y, Tan L, Li XJ, et al. EGFR‐Phosphorylated Platelet Isoform of Phosphofructokinase 1 Promotes PI3K Activation. Mol Cell. 2018;70(2):197–210 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hu Y, Smyth GK. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods. 2009;347(1‐2):70–8. [DOI] [PubMed] [Google Scholar]
- 40. Shao F, Yang X, Wang W, Wang J, Guo W, Feng X, et al. Associations of PGK1 promoter hypomethylation and PGK1‐mediated PDHK1 phosphorylation with cancer stage and prognosis: a TCGA pan‐cancer analysis. Cancer Commun (Lond). 2019;39(1):54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yang X, Shao F, Shi S, Feng X, Wang W, Wang Y, et al. Prognostic impact of metabolism reprogramming markers Acetyl‐CoA synthetase 2 Phosphorylation and Ketohexokinase‐A expression in non‐small‐cell lung carcinoma. Front Oncol. 2019;9:1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lee JH, Liu R, Li J, Zhang C, Wang Y, Cai Q, et al. Stabilization of phosphofructokinase 1 platelet isoform by AKT promotes tumorigenesis. Nat Commun. 2017;8(1):949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Xie H, Hanai J, Ren JG, Kats L, Burgess K, Bhargava P, et al. Targeting lactate dehydrogenase–a inhibits tumorigenesis and tumor progression in mouse models of lung cancer and impacts tumor‐initiating cells. Cell Metab. 2014;19(5):795–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yang T, Shu X, Zhang HW, Sun LX, Yu L, Liu J, et al. Enolase 1 regulates stem cell‐like properties in gastric cancer cells by stimulating glycolysis. Cell Death Dis. 2020;11(10):870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bock FJ, Tait SWG. Mitochondria as multifaceted regulators of cell death. Nat Rev Mol Cell Biol. 2020;21(2):85–100. [DOI] [PubMed] [Google Scholar]
- 46. John S, Weiss JN, Ribalet B. Subcellular localization of hexokinases I and II directs the metabolic fate of glucose. PLoS One. 2011;6(3):e17674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nawaz MH, Ferreira JC, Nedyalkova L, Zhu H, Carrasco‐López C, Kirmizialtin S, et al. The catalytic inactivation of the N‐half of human hexokinase 2 and structural and biochemical characterization of its mitochondrial conformation. Biosci Rep. 2018;38(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Robey RB, Hay N. Mitochondrial hexokinases, novel mediators of the antiapoptotic effects of growth factors and Akt. Oncogene. 2006;25(34):4683–96. [DOI] [PubMed] [Google Scholar]
- 49. Yang W, Xia Y, Hawke D, Li X, Liang J, Xing D, et al. PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell. 2012;150(4):685–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Li X, Jiang Y, Meisenhelder J, Yang W, Hawke DH, Zheng Y, et al. Mitochondria‐translocated PGK1 functions as a protein kinase to coordinate glycolysis and the TCA cycle in tumorigenesis. Mol Cell. 2016;61(5):705–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Li X, Qian X, Peng LX, Jiang Y, Hawke DH, Zheng Y, et al. A splicing switch from ketohexokinase‐C to ketohexokinase‐A drives hepatocellular carcinoma formation. Nat Cell Biol. 2016;18(5):561–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jiang Y, Wang Y, Wang T, Hawke DH, Zheng Y, Li X, et al. PKM2 phosphorylates MLC2 and regulates cytokinesis of tumour cells. Nat Commun. 2014;5:5566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jiang Y, Li X, Yang W, Hawke DH, Zheng Y, Xia Y, et al. PKM2 regulates chromosome segregation and mitosis progression of tumor cells. Mol Cell. 2014;53(1):75–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ma Q, Meng Z, Meng Y, Liu R, Lu Z. A moonlighting function of choline kinase alpha 2 in the initiation of lipid droplet lipolysis in cancer cells. Cancer Commun (Lond). 2021;41(10):933‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jiang H, Zhu L, Xu D, Lu Z. A newly discovered role of metabolic enzyme PCK1 as a protein kinase to promote cancer lipogenesis. Cancer Commun (Lond). 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Jiang H, Lin Q, Ma L, Luo S, Jiang X, Fang J, et al. Fructose and fructose kinase in cancer and other pathologies. J Genet Genomics. 2021;48(7):531–9. [DOI] [PubMed] [Google Scholar]
- 57. Li X, Qian X, Lu Z. Fructokinase A acts as a protein kinase to promote nucleotide synthesis. Cell Cycle. 2016;15(20):2689–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lu Z. PKM2 functions as a histone kinase. Cell Cycle. 2012;11(22):4101–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Li X, Zheng Y, Lu Z. PGK1 is a new member of the protein kinome. Cell Cycle. 2016;15(14):1803–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Qian X, Li X, Lu Z. Protein kinase activity of the glycolytic enzyme PGK1 regulates autophagy to promote tumorigenesis. Autophagy. 2017;13(7):1246–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wang Y, Guo YR, Xing D, Tao YJ, Lu Z. Supramolecular assembly of KAT2A with succinyl‐CoA for histone succinylation. Cell Discov. 2018;4:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tong Y, Guo D, Yan D, Ma C, Shao F, Wang Y, et al. KAT2A succinyltransferase activity‐mediated 14‐3‐3zeta upregulation promotes beta‐catenin stabilization‐dependent glycolysis and proliferation of pancreatic carcinoma cells. Cancer Lett. 2020;469:1–10. [DOI] [PubMed] [Google Scholar]
- 63. Lee HJ, Li CF, Ruan D, He J, Montal ED, Lorenz S, et al. Non‐proteolytic ubiquitination of Hexokinase 2 by HectH9 controls tumor metabolism and cancer stem cell expansion. Nat Commun. 2019;10(1):2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Liu CC, Chou KT, Hsu JW, Lin JH, Hsu TW, Yen DH, et al. High metabolic rate and stem cell characteristics of esophageal cancer stem‐like cells depend on the Hsp27‐AKT‐HK2 pathway. Int J Cancer. 2019;145(8):2144–56. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.