

Abstract

Simultaneous targeting of different antigens by bispecific antibodies (bsAbs) is permitting synergistic binding functionalities with high therapeutic potential, but is also rendering their analysis challenging. We introduce flow-induced dispersion analysis (FIDA) for the in-depth characterization of bsAbs with diverse molecular architectures and valencies under near-native conditions without potentially obstructive surface immobilization. Individual equilibrium dissociation constants are determined in solution, even in higher-order complexes with both antigens involved, hereby allowing the analysis of binding cooperativity and elucidation of a potential interference between the interactions. We further illustrate bispecific binding functionality as incremental increases in complex sizes when the bsAbs are exposed to one or two antigens. The possibility for comprehensive binding analysis with low material consumption and high matrix tolerability irrespective of molecular format and with little optimization renders FIDA a versatile tool for format selection and characterization of complex bi/multispecific protein therapeutics throughout the drug development and biomanufacturing pipeline.
Bispecific antibodies (bsAbs) represent a rapidly growing class of biotherapeutic molecules that can be engineered in numerous ways to fine-tune molecular structure and valency to fit the therapeutic purpose.1,2 Their ability to simultaneously engage two different epitopes is opening novel avenues and potencies beyond those of conventional immunoglobulins (also known as obligate mechanisms of action). Examples include, for example, T cell redirection by physically bridging tumor cells and effector T cells to induce tumor cell killing3 and dual receptor targeting for modulating receptor crosstalk.4 However, the intricate binding behavior of bsAbs is also complicating assay development. There is a need for methods capable of differentially characterizing simultaneous bsAb binding events under native conditions, irrespective of the molecular architectures of any of the binding partners. These criteria are not met by standard techniques, which are typically label-free and rely on potentially obstructive surface immobilization.5 BsAb characterizations that are unbiased by geometrical restraints are crucial considering that bsAb functionality is highly dependent on an optimal molecular format.6,7
Here, we introduce flow-induced dispersion analysis (FIDA), a diffusion-based sizing technique for in-solution characterization and binding affinity determinations of sequential binding events in higher-order bsAb complexes without any surface immobilization. Briefly, FIDA is a microfluidic technique exploiting the phenomenon of Taylor dispersion. It relies on the notion that the interaction between a molecule and a binding partner causes a change in the diffusivity of that molecule, which is quantifiable through Taylor dispersion analysis (TDA) of the sample, that is, by how much a given small plug of sample spreads in the laminar flow. If the molecule is detectable, for example, by fluorescent labeling, this apparent change in diffusivity manifests through a change in peak width and can be translated to an apparent hydrodynamic radius (Rh), that is, the size of the biomolecular complex8−10 (Figure 1A,C). This is, to the best of our knowledge, the first reporting of an assay capable of quantitatively dissecting individual binding events of bsAbs in higher-order complexes in solution.
Figure 1.

FIDA principle enables size-based detection of bispecific binding behavior. (A) Schematic illustration of the FIDA principle for bispecific antigen binding. Complexation of the fluorescent antigen with binding partners causes signal dispersion, which translates to a change in the apparent hydrodynamic radius of the antigen. The red circle indicates the fluorescently labeled (yellow stars) primary antigen used for detection, and the green circle indicates the unlabeled secondary antigen that is affecting the signal indirectly through ternary complex formation. (B) Various bsAbs were constructed through genetic fusion of anti-PD-L1 IgG1 and anti-HER2 single-domain antibodies through a flexible (GGGGS)×2 linker. Symmetric bsAb formats included sdAb C-terminal coupling on HC (CHC-IgG), sdAb C-terminal coupling on LC (CLC-IgG), and sdAb C-terminal coupling on both LC and HC (CHC-CLC-IgG). An asymmetric bsAb monovalent for each antigen (sdAb-Fc/IgG) was constructed by controlled pairing of sdAb-Fc and IgG HC by introduction of KiH mutations in the CH3 domains (illustrated as the black circle between the two CH3 domains). (C) Example of Taylor dispersion of unbound fluorescent PD-L1-DY490 (blue), monospecific CHC-IgG binding to PD-L1-DY490 (orange), and bispecific binding of CHC-IgG to PD-L1-DY490 and unlabeled HER2 (green). (D, E) FIDA complex sizing. Apparent hydrodynamic radii of antibodies in complex with fluorescently labeled (DY-490) primary antigen ± an excess of unlabeled secondary antigen. The monospecific and bispecific antibodies have been separated by stippled squares similar to (B).
Experimental Section
Design and Construction of Expression Plasmids
The expression vectors were based on a two-plasmid system, with HC and LC encoded on separate plasmids for flexibility in the HC:LC transfection ratios. All vectors used in the study originated from the pcDNA3.1 vector backbone containing antibody constant domains (IgG1 for HC and Cκ for LC) and a human serum albumin signal peptide for secretion into the culture supernatant under the control of the CMV promoter. Gene segments encoding anti-PD-L1 VH and VL,11,12 anti-HER2 sdAb,13 and CH3 heterodimerization domains were obtained from Twist BioScience and cloned using NEB HiFi Assembly (New England Biolabs) according to manufacturer instructions. The recombinant plasmids were confirmed by sequencing (Macrogen, Amsterdam, Netherlands).
Production of Antibodies
Expression plasmids were amplified in E. coli DH5α (New England Biolabs) and purified using NucleoBond Xtra Midi Plus EF (Macherey-Nagel) to ensure endotoxin removal. All antibodies were produced by transient expression in HEK293 Freestyle cells (ThermoFisher) for secretion into the culture supernatant. Plasmids encoding either HC or LC were transiently cotransfected using linear polyethylenimine (PEIPro, Polyplus-transfection) according to manufacturer instructions and transfected cells were cultured for 7 days in FreeStyle 293 Expression Medium (ThermoFisher) at 37 °C and 5% CO2. We used a 3:2 (HC:LC) transfection ratio for all symmetric antibodies.14 For asymmetric bsAbs we used a 1.5:4:1 (HC:sdAb-Fc:LC) transfection ratio optimized for the specific sdAb-Fc/IgG construct (Figures S1 and S2). After incubation, the supernatants were clarified (centrifugation at 1500×g for 10 min and 0.45 μm filtering) before purification using Protein A (HiTrap MabSelect SuRe, Cytiva, lot: 10293990) affinity chromatography connected to an ÄKTA Pure system (Cytiva). Antibodies were concentrated using Amicon Ultra-15 centrifugal filter devices (30-kDa cutoff, EMD) and the protein concentrations were measured using NanoDrop One UV–vis spectrophotometer (ThermoFisher) and theoretical extinction coefficient predicted by ProtParam (Expasy) based on the amino acid sequence of fully assembled monomeric antibodies.15
Capillary Electrophoresis
Capillary electrophoresis was performed using the High Sensitivity Protein 250 kit (#5067–1575, Agilent) with an Agilent 2100 BioAnalyzer according to manufacturer protocol without any reducing agent. The 2100 Expert software (Agilent, version B.02.11.SI824) was used for visualizing the peaks.
FIDA Equipment
Binding experiments were conducted using a FIDA One instrument employing light-emitting diode-induced fluorescence detection using an excitation wavelength of 480 nm and an emission wavelength of >515 nm (Fida Biosystems). We used standard capillaries with a 75 μm inner diameter, a 375 μm outer diameter, a 100 cm total length, and an 84 cm length to detection window. For all experiments, the capillary was coated with high sensitivity (HS) reagent (Fida Biosystems) prior to analysis. All measurements were done in triplicate unless otherwise stated.
FIDA Reagents
Human PD-L1 (ACRO Biosystem, #PD1-H5229) and HER2 (ACRO Biosystems, #HE2-H5225) were labeled with DY-490 fluorophore using a Fluorospin 490 Protein Labeling and Purification Kit (EMP Biotech, #MK-D0125) according to manufacturer instructions. PBS+0.1% BSA was used as the assay buffer for all FIDA binding experiments.
FIDA Instrument Settings
For all FIDA experiments, the assay sequence consisted of an initial capillary flush for 120 s at 3500 mbar using assay buffer followed by a 20 s loading step of assay buffer with or without an antibody analyte also at 3500 mbar. Next, indicator solution was injected at 50 mbar for 10 s, followed by a final 180–200 s loading step at 400 mbar again using assay buffer with or without antibody analyte. Data recording was done during the final loading step. The apparent Rh values were obtained by parsing Taylorgrams to FIDA Software v2.3 (Fida Biosystems) with a Taylorgram fraction of 75% and fitting for multiple species with 0.60 nm fixed Rh to account for residual free dye.
FIDA Complex Sizing
The sizing of antibody complexes was performed using assay buffer in both loading steps similarly to previous reports.16 The injected indicator solution was premixed (>15 min) 32 nM DY-490-labeled antigen and 40 nM antibody analyte with/without 80 nM unlabeled secondary antigen. To favor the formation of the ternary complex, the unlabeled secondary antigen was added 2.5-fold in excess. Control experiments without antibody analyte were also included to test for nonspecific binding between the antigens.
FIDA Affinity Determinations
Determination of equilibrium binding constants (KD) was performed in a capillary mixing mode16 using antibody analyte in varying concentrations in both of the loading steps and injecting 20 nM DY-490-labeled primary antigen with or without equimolar amounts of an unlabeled secondary antigen. The titration curves were fitted according to previously described FIDA binding models.17
In Silico Size Predictions
Structural models were generated through comparative modeling using Modeler (10.1)18 and hydrodynamic radii were estimated using HullRad19 (available through http://52.14.70.9/index.html)
Data Analysis
Data was assembled and organized using Microsoft Excel 2016 (raw data files are available through DOI: 10.11583/DTU.19801525). For analysis, we used an in-house developed Python (3.8.3) script running pandas (1.0.5) and numpy (1.18.5) for basic calculations, SciPy (1.5.0) for data fitting, and plotly (5.4.0) for data visualizations. The script is available through https://github.com/andreasvisbech/Plotly_data_analysis/releases/tag/3.1.0, and the applied commands are provided in Table S1.
Results and Discussion
Characterizing Simultaneous bsAb Binding
The versatility of bsAbs largely resides in the ability to engineer the valencies toward their binding partners and the overall structural geometry to correspond to the desired therapeutic functionality.6,7 We combined an anti-PD-L1 IgG120 with an anti-HER2 single-domain antibody (sdAb)21 to produce several symmetric IgG-like bsAbs with various molecular architectures and valencies (Figure 1B) all targeting human programmed death-ligand 1 (PD-L1) and human epidermal growth factor receptor 2 (HER2), two clinically validated cancer targets.22,23 This yielded symmetric bsAb fusion molecules with two or four potential binding sites per specificity. Fusion of sdAb onto a heavy chain (HC) or a light chain (LC) of the IgG scaffold was done through a flexible (GGGGS)×2 linker for all sdAb fusions (except sdAb-Fc molecules where the sdAbs were attached directly upstream of the natural IgG1 hinge region) since this linker typically exhibits good flexibility and stability in aqueous solutions.24 We further established an asymmetric sdAb-Fc/IgG bsAb, monovalent for both PD-L1 and HER2, using the previously described knobs-into-holes (KiH) CH3 heterodimerization platform25 (Figure 1B). The amino acid sequences of the antibody chains are available in Table S2. All antibodies were produced in HEK293F cells, followed by protein A purification, and exhibited purity, homogeneity, and stability comparable to WT anti-PD-L1 IgG1 (data not shown). The ability of the bsAbs to bind both antigens simultaneously was demonstrated as an incremental increase in apparent Rh of the biomolecular complex upon exposure to a fluorescently labeled antigen ± a second unlabeled antigen. We use the nomenclature “primary antigen” when referring to the fluorescently labeled antigen measured by FIDA and “secondary antigen” when referring to the unlabeled antigen. Since the secondary antigen does not carry any fluorescence, it can only affect the signal indirectly through complexation with the bsAb, which is in itself binding the primary antigen (Figure 1A). All bsAb constructs exhibited bispecific binding functionality when either of the antigens were used as a fluorescent probe (Figure 1D,E). Neither IgG nor sdAb-Fc showed any increase in apparent Rh upon addition of the secondary antigen, thus confirming the monospecific binding behavior of these antibodies. We observed a larger apparent Rh for unbound HER2 compared to PD-L1, which is consistent with the higher theoretical molecular weight of HER2 (70.2 kDa) compared to PD-L1 (26.0 kDa) and further reflected in the Rh change upon bsAb binding. The asymmetric sdAb-Fc/IgG showed smaller apparent Rh than the symmetric bsAbs, which is consistent with a smaller biomolecular complex size due to fewer antigens bound compared to the higher-valency symmetric bsAbs. Furthermore, the smaller apparent Rh of sdAb-Fc/IgG is also in agreement with an expected lower binding affinity, from the decreased number of binding domains compared to the symmetric bsAbs, which will effectively reduce the number of complexes formed and thus the apparent Rh. Finally, we constructed structural models of the antibody complexes (Figure S3) and found the relative differences between the computationally predicted Rh values to be in good agreement with the experimental values (Figure S4).
Selective Quantification of bsAb Interactions in Higher-Order Complexes
We generated binding curves for monospecific and bispecific binding, respectively, by titrating an antibody against a fluorescently labeled primary antigen ± equimolar amounts of the unlabeled secondary antigen to characterize interference between the two binding events. Affinity constants (KD) were determined through fitting of the binding curves using a simple 1:1 binding model (Figure 2), although similar results were obtained from the more complex excess indicator binding model17 (Table S3). We found that KD values of the engineered bsAbs were generally comparable to their monospecific counterparts, thus indicating low steric clashing and demonstrating the ability of FIDA to analyze complex molecular formats. The affinity measurements were performed in capillary mixing mode, which had previously been shown to yield comparable results to preincubated samples for a low nM affinity antibody.16 The capillary mixing analysis required only 44 nL indicator solution pr. measurement and is thus a highly efficient method for obtaining binding data.
Figure 2.

Quantitative characterization of bsAb binding. (A) Examples of antibody binding curves generated by plotting apparent hydrodynamic radius of the fluorescently labeled antigen (indicator) as a function of antibody concentration measured using FIDA. Increasing amounts of antibody increases the indicator apparent hydrodynamics radius through complexing. Inclusion of a secondary unlabeled antigen allows characterization of interference with indicator binding arising from the secondary binding event. (B) Equilibrium dissociation constants from fitting of binding curves and the related goodness of fit.
The observed KD values were in agreement with previously reported binding constants,21,26 hereby indicating that sufficient equilibrium was obtained within the analysis window. In cases where reaction kinetics are too slow or the formed complex is too big to properly diffuse within the analysis window, the analyte and indicator can be premixed prior to analysis at the expense of higher material consumption. The PD-L1 binding affinities of all symmetric bsAbs resembled the IgG binding, which was expected, as these antibodies carry the same anti-PD-L1 valency and the antigen-binding Fv domains were not changed. Examination of HER2 binding, on the other hand, indicated that the sdAb fusion site affected the binding affinity with fusion C-terminally on HC being more favorable than C-terminal fusion on LC in terms of retaining full binding affinity of the sdAb. The decrease in binding affinity is observed during both monospecific and bispecific complex formation, hereby indicating that the negative effect on binding affinity stems from the molecular engineering rather than negative cooperativity between the binding events. The example illustrates the ability of the FIDA method to dissect protein complex formation and provide insights into the individual binding events in higher order biomolecular complexes. The asymmetric sdAb-Fc/IgG likewise showed decreased HER2 binding, which might be explained by a sdAb domain that is less exposed for binding in the presence of an anti-PD-L1 Fab domain.
High-Resolution Characterization of Heterogeneous Antibody Mixtures
Lastly, we sought to challenge the accuracy of FIDA by comparing complex sizes of sdAb-Fc/IgG asymmetric bsAbs engineered with KiH CH3 domains and the same asymmetric bsAb containing WT CH3 domains. Without any HC steering, 25% of the antibody content is expected to be monospecific anti-PD-L1 IgG “contaminant” formed through random HC pairing (Figure 3A,B). Using fluorescent PD-L1 as primary antigen and unlabeled HER2 as secondary antigen, we found that the asymmetric bsAb without CH3 engineering gave rise to a slightly larger relative change in the hydrodynamic radius of the monospecific complex compared to unbound antigen than the KiH engineered bsAb (Figure 3C). The difference between the two complex sizes is expected to originate from mispaired homodimeric IgG, which is larger than the sdAb-Fc/IgG bsAb, and in itself capable of binding fluorescent PD-L1, thus increasing the apparent Rh in the sample. Correspondingly, a smaller relative increase in Rh was observed for the WT CH3 bsAb when going from the monospecific to bispecific binding because a large fraction of these antibodies have been mispaired and therefore do not contribute to the bispecific complex. Although the Rh differences observed between these biomolecular complexes are quite small, they illustrate the potential for using FIDA in high accuracy assays for unravelling protein complex formation. In theory the different complexed species in the mix should have different flow profiles, which can be fitted individually from a single Taylorgram. Practically, however, these differences are not big enough to be accurately dissected from the signal thus limiting the resolution of FIDA.
Figure 3.
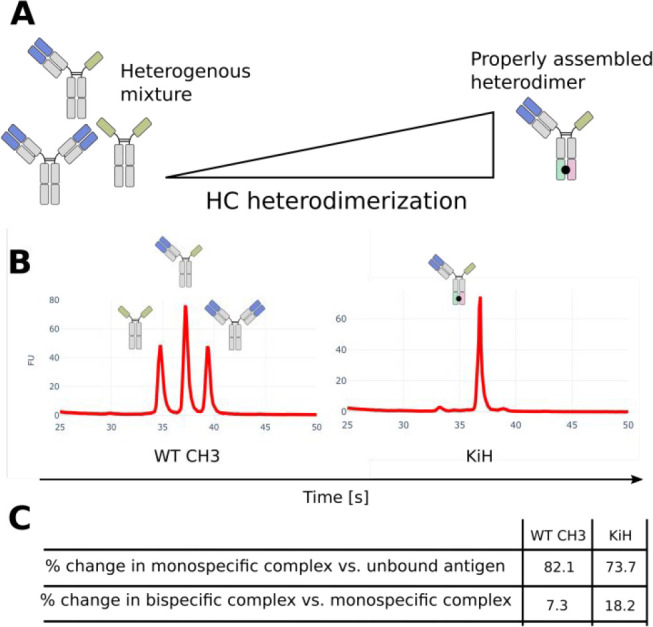
Complex Rh of asymmetric bsAbs correlate with heterodimerization. (A) CH3 steering platforms promote HC heterodimerization for assembly of asymmetric bsAbs. Without any HC heterodimerization, 50% heterodimer and 25% of each homodimer will form through random assembly. (B) Capillary electrophoresis of asymmetric sdAb-Fc/IgG bsAbs produced with (KiH) or without (WT CH3) HC steering. The electropherogram peaks represent the distribution of antibody species. (C) Overview of changes in apparent Rh of fluorescent PD-L1-DY490 upon binding of asymmetric sdAb-Fc/IgG bsAbs ± unlabeled HER2 antigen.
FIDA for Deciphering Complex bsAb Binding
In this study, we introduced FIDA, a microfluidics-based sizing technique relying on TDA, for selectively characterizing bsAb interactions events in higher order biomolecular complexes under immobilization-free binding conditions. FIDA and TDA have previously been established as valuable methods for particle sizing9,27 and analyzing biomolecular interactions in a broad size range with very low sample consumption and high tolerability to complex sample matrices.8,10,16,17,28
We demonstrate the ability of bsAbs, with different valencies and molecular architectures, to bind the antigens individually or simultaneously through incremental increases in apparent Rh of the biomolecular bsAb complexes upon stepwise exposure to one or both antigens. The incremental increase in Rh upon addition of the second antigen is not seen for either of the IgG or sdAb-Fc due to the monospecific nature of these antibodies. This allows the differentiation between binding of specific antigens as well as between different valencies.
The observed size differences for the analyzed complexes are based on changes in hydrodynamic radius, which is influenced by the tertiary structure of the analyte.9 For this reason, the apparent Rh of antibody complexes are affected by antigen size and structural format of the bsAb. This is exemplified by the Rh values of the bispecific complexes, where we did not see the same notable difference in apparent Rh when going from the tetravalent bsAb to the hexavalent bsAb as when going from bivalent IgG to any of the tetravalent bsAb (Figure 1D,E). The affinities obtained using FIDA indicate that more than two anti-HER2 binding sites in CHC-CLC-IgG are actively used, and we thus speculate that the additional CLC fusion contributes only slightly to an increase in Rh compared to the CHC fusion alone. Similarly, only a small Rh increase was observed upon secondary binding of sdAb-Fc/IgG to unlabeled PD-L1 (Figure 1E) most likely because this binding event does not markedly contribute additional diffusivity compared to the binary complex.
The examples suggest that the biomolecular complex Rh is not additive in the same way the molecular weight is. The issue also illustrates a practical limitation in complex sizing with FIDA that unlabeled binding partners must be sufficiently large so that binding events create a measurable change in diffusivity in order to do accurate complex sizing. In this study, PD-L1 (26.0 kDa) and HER2 (70.2 kDa) were generally both sufficient for eliciting measurable Rh differences when used as secondary antigen, while smaller antigens will eventually lead to limitations if used as unlabeled secondary antigens. Smaller antigens are also expected to be more prone to changes in binding behavior from the fluorescent labeling, as the tag is more likely to be situated near the epitope. Similarly, sizing analysis of low-affinity binding events is complicated by the fact that only relatively small amounts of low-affinity binding partners are captured in the complex and thus contributing to the apparent Rh. This study further sought to explore affinity binding constants under in-solution and immobilization-free binding conditions. The binding affinities of bsAbs are typically determined individually against each target,29−32 because label-free methods, which are routine for antibody KD measurements, cannot readily differentiate between simultaneous individual binding events. FIDA is relying on fluorescence detection, which allows inclusion of unlabeled binding partners that affect the sample system chemically without creating signal artifacts and thus enables discrete characterization of selected binding events during higher-order complex formation or in complex sample media. The quantitative interrogation of binding events allowed us to directly compare structurally diverse bsAbs in the same assay format without any further optimization to assess the effect of structural features on binding behavior and select the optimal candidate. Such characterization can be instrumental in the development of therapeutic bsAbs, where the molecular architecture is affecting the mechanism of action.
Conclusion
In conclusion, our work demonstrates a readily available method that, with very little optimization, can provide reproducible and accurate in-depth characterization of complex binding dynamics of bsAbs in a single-assay format. The technology allows the characterization of specific binding events in higher-order biomolecular complexes in solution under immobilization-free conditions and even in complex media such as serum, culture media, or formulations.16,17 We expect the technology to hold potential for characterizing the increasing number of complex bi/multispecific protein therapeutics that are entering drug development programs. Additionally, we believe that the technology will be relevant for biomanufacturing, quality control, and drug substance and drug product release of bsAbs in GMP manufacturing, where reliable, accurate, and easy to validate and reproduce ligand-binding assays are crucial.
Acknowledgments
This work was supported by The Novo Nordisk Foundation Grant NNF19SA0056783, NNF19SA0057794, and NNF20SA0066621. We thank Johnny Arnsdorf from The Technical University of Denmark and employees at Fida Biosystems for experimental assistance.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.2c02705.
SDS-PAGE gels, CE electropherograms, Python commands for data analysis, amino acid sequences, structural antibody models, in silico predicted molecular sizes, and fitted binding affinity constants (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Brinkmann U.; Kontermann R. E. The making of bispecific antibodies. mAbs 2017, 9 (2), 182–212. 10.1080/19420862.2016.1268307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labrijn A. F.; Janmaat M. L.; Reichert J. M.; Parren P. Bispecific antibodies: a mechanistic review of the pipeline. Nature reviews. Drug discovery 2019, 18 (8), 585–608. 10.1038/s41573-019-0028-1. [DOI] [PubMed] [Google Scholar]
- Baeuerle P. A.; Reinhardt C. Bispecific T-cell engaging antibodies for cancer therapy. Cancer research 2009, 69 (12), 4941–4944. 10.1158/0008-5472.CAN-09-0547. [DOI] [PubMed] [Google Scholar]
- Huang S.; Li C.; Armstrong E. A.; Peet C. R.; Saker J.; Amler L. C.; Sliwkowski M. X.; Harari P. M. Dual targeting of EGFR and HER3 with MEHD7945A overcomes acquired resistance to EGFR inhibitors and radiation. Cancer research 2013, 73 (2), 824–833. 10.1158/0008-5472.CAN-12-1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Register A. C.; Tarighat S. S.; Lee H. Y. Bioassay Development for Bispecific Antibodies-Challenges and Opportunities. International journal of molecular sciences 2021, 22 (10), 5350. 10.3390/ijms22105350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dengl S.; Mayer K.; Bormann F.; Duerr H.; Hoffmann E.; Nussbaum B.; Tischler M.; Wagner M.; Kuglstatter A.; Leibrock L.; Buldun C.; Georges G.; Brinkmann U. Format chain exchange (FORCE) for high-throughput generation of bispecific antibodies in combinatorial binder-format matrices. Nat. Commun. 2020, 11 (1), 4974. 10.1038/s41467-020-18477-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickopf S.; Georges G. J.; Brinkmann U. Format and geometries matter: Structure-based design defines the functionality of bispecific antibodies. Computational and structural biotechnology journal 2020, 18, 1221–1227. 10.1016/j.csbj.2020.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen H.; Østergaard J. Flow induced dispersion analysis quantifies noncovalent interactions in nanoliter samples. J. Am. Chem. Soc. 2010, 132 (12), 4070–4071. 10.1021/ja100484d. [DOI] [PubMed] [Google Scholar]
- Pedersen M. E.; Gad S. I.; Østergaard J.; Jensen H. Protein Characterization in 3D: Size, Folding, and Functional Assessment in a Unified Approach. Analytical chemistry 2019, 91 (8), 4975–4979. 10.1021/acs.analchem.9b00537. [DOI] [PubMed] [Google Scholar]
- Otzen D. E.; Buell A. K.; Jensen H. Microfluidics and the quantification of biomolecular interactions. Curr. Opin. Struct. Biol. 2021, 70, 8–15. 10.1016/j.sbi.2021.02.006. [DOI] [PubMed] [Google Scholar]
- Powles T.; Eder J. P.; Fine G. D.; Braiteh F. S.; Loriot Y.; Cruz C.; Bellmunt J.; Burris H. A.; Petrylak D. P.; Teng S. L.; Shen X.; Boyd Z.; Hegde P. S.; Chen D. S.; Vogelzang N. J. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature 2014, 515 (7528), 558–562. 10.1038/nature13904. [DOI] [PubMed] [Google Scholar]
- Irving B.; Chiu H.; Maecker H.; Mariathasan S.; Lehar S. M.; Wu Y.; Cheung J.. Anti-PD-L1 antibodies, compositions and articles of manufacture. U.S. Patent US 8,217,149B2; Genentech, Inc., 2012.
- D’Huyvetter M.; De Vos J.; Xavier C.; Pruszynski M.; Sterckx Y.; Massa S.; Raes G.; Caveliers V.; Zalutsky M. R.; Lahoutte T.; Devoogdt N. 131I-labeled Anti-HER2 Camelid sdAb as a Theranostic Tool in Cancer Treatment. Clinical cancer research 2017, 23 (21), 6616–6628. 10.1158/1078-0432.CCR-17-0310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlatter S.; Stansfield S. H.; Dinnis D. M.; Racher A. J.; Birch J. R.; James D. C. On the optimal ratio of heavy to light chain genes for efficient recombinant antibody production by CHO cells. Biotechnology progress 2005, 21 (1), 122–133. 10.1021/bp049780w. [DOI] [PubMed] [Google Scholar]
- Gasteiger E.; Hoogland C.; Gattiker A.; Duvaud S.; Wilkins M. R.; Appel R. D.; Bairoch A.. Protein Identification and Analysis Tools on the ExPASy Server. The Proteomics Protocols Handbook; Humana Press, 2005; pp 571–607. [Google Scholar]
- Pedersen M. E.; Haegebaert R.; Østergaard J.; Jensen H. Size-based characterization of adalimumab and TNF-α interactions using flow induced dispersion analysis: assessment of avidity-stabilized multiple bound species. Sci. Rep. 2021, 11 (1), 4754. 10.1038/s41598-021-84113-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen M. E.; Østergaard J.; Jensen H. In-Solution IgG Titer Determination in Fermentation Broth Using Affibodies and Flow-Induced Dispersion Analysis. ACS omega 2020, 5 (18), 10519–10524. 10.1021/acsomega.0c00791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sali A.; Blundell T. L. Comparative protein modelling by satisfaction of spatial restraints. Journal of molecular biology 1993, 234 (3), 779–815. 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Fleming P. J.; Fleming K. G. HullRad: Fast Calculations of Folded and Disordered Protein and Nucleic Acid Hydrodynamic Properties. Biophysical journal 2018, 114 (4), 856–869. 10.1016/j.bpj.2018.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goletz C.; Lischke T.; Harnack U.; Schiele P.; Danielczyk A.; Rühmann J.; Goletz S. Glyco-Engineered Anti-Human Programmed Death-Ligand 1 Antibody Mediates Stronger CD8 T Cell Activation Than Its Normal Glycosylated and Non-Glycosylated Counterparts. Frontiers in immunology 2018, 9, 1614. 10.3389/fimmu.2018.01614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaneycken I.; Devoogdt N.; Van Gassen N.; Vincke C.; Xavier C.; Wernery U.; Muyldermans S.; Lahoutte T.; Caveliers V. Preclinical screening of anti-HER2 nanobodies for molecular imaging of breast cancer. FASEB J. 2011, 25 (7), 2433–2446. 10.1096/fj.10-180331. [DOI] [PubMed] [Google Scholar]
- Oh D. Y.; Bang Y. J. HER2-targeted therapies - a role beyond breast cancer. Nature reviews. Clinical oncology 2020, 17 (1), 33–48. 10.1038/s41571-019-0268-3. [DOI] [PubMed] [Google Scholar]
- Chen L.; Han X. Anti-PD-1/PD-L1 therapy of human cancer: past, present, and future. J. Clin. Invest. 2015, 125 (9), 3384–3391. 10.1172/JCI80011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.; Zaro J. L.; Shen W. C. Fusion protein linkers: property, design and functionality. Advanced drug delivery reviews 2013, 65 (10), 1357–1369. 10.1016/j.addr.2012.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwell S.; Ridgway J. B.; Wells J. A.; Carter P. Stable heterodimers from remodeling the domain interface of a homodimer using a phage display library. Journal of molecular biology 1997, 270 (1), 26–35. 10.1006/jmbi.1997.1116. [DOI] [PubMed] [Google Scholar]
- Tan S.; Liu K.; Chai Y.; Zhang C. W.; Gao S.; Gao G. F.; Qi J. Distinct PD-L1 binding characteristics of therapeutic monoclonal antibody durvalumab. Protein & cell 2018, 9 (1), 135–139. 10.1007/s13238-017-0412-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawe A.; Hulse W. L.; Jiskoot W.; Forbes R. T. Taylor dispersion analysis compared to dynamic light scattering for the size analysis of therapeutic peptides and proteins and their aggregates. Pharm. Res. 2011, 28 (9), 2302–2310. 10.1007/s11095-011-0460-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider M. M.; Emmenegger M.; Xu C. K.; Condado Morales I.; Meisl G.; Turelli P.; Zografou C.; Zimmermann M. R.; Frey B. M.; Fiedler S.; Denninger V.; Jacquat R. P.; Madrigal L.; Ilsley A.; Kosmoliaptsis V.; Fiegler H.; Trono D.; Knowles T. P.; Aguzzi A. Microfluidic characterisation reveals broad range of SARS-CoV-2 antibody affinity in human plasma. Life science alliance 2022, 5 (2), e202101270. 10.26508/lsa.202101270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraman M.; Faroudi M.; Allen N. L.; Kmiecik K.; Gliddon D.; Seal C.; Koers A.; Wydro M. M.; Batey S.; Winnewisser J.; Young L.; Tuna M.; Doody J.; Morrow M.; Brewis N. FS118, a Bispecific Antibody Targeting LAG-3 and PD-L1, Enhances T-Cell Activation Resulting in Potent Antitumor Activity. Clinical cancer research 2020, 26 (13), 3333–3344. 10.1158/1078-0432.CCR-19-3548. [DOI] [PubMed] [Google Scholar]
- Staflin K.; et al. Target arm affinities determine preclinical efficacy and safety of anti-HER2/CD3 bispecific antibody. JCI insight 2020, 5 (7), e133757. 10.1172/jci.insight.133757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reusch U.; Duell J.; Ellwanger K.; Herbrecht C.; Knackmuss S. H.; Fucek I.; Eser M.; McAleese F.; Molkenthin V.; Gall F. L.; Topp M.; Little M.; Zhukovsky E. A. A tetravalent bispecific TandAb (CD19/CD3), AFM11, efficiently recruits T cells for the potent lysis of CD19(+) tumor cells. mAbs 2015, 7 (3), 584–604. 10.1080/19420862.2015.1029216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benschop R. J.; Chow C. K.; Tian Y.; Nelson J.; Barmettler B.; Atwell S.; Clawson D.; Chai Q.; Jones B.; Fitchett J.; Torgerson S.; Ji Y.; Bina H.; Hu N.; Ghanem M.; Manetta J.; Wroblewski V. J.; Lu J.; Allan B. W. Development of tibulizumab, a tetravalent bispecific antibody targeting BAFF and IL-17A for the treatment of autoimmune disease. mAbs 2019, 11 (6), 1175–1190. 10.1080/19420862.2019.1624463. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.