

Abstract
Metabolic alterations shared between the nervous system and skin fibroblasts have emerged in amyotrophic lateral sclerosis (ALS). Recently, we found that a subgroup of sporadic ALS (sALS) fibroblasts (sALS1) is characterized by metabolic profiles distinct from other sALS cases (sALS2) and controls, suggesting that metabolic therapies could be effective in sALS. The metabolic modulators nicotinamide riboside and pterostilbene (EH301) are under clinical development for the treatment of ALS. Here, we studied the transcriptome and metabolome of sALS cells to understand the molecular bases of sALS metabotypes and the impact of EH301. Metabolomics and transcriptomics were investigated at baseline and after EH301 treatment. Moreover, weighted gene coexpression network analysis (WGCNA) was used to investigate the association of the metabolic and clinical features. We found that the sALS1 transcriptome is distinct from sALS2 and that EH301 modifies gene expression differently in sALS1, sALS2 and the controls. Furthermore, EH301 had strong protective effects against metabolic stress, an effect linked to the antiinflammatory and antioxidant pathways. WGCNA revealed that the ALS functional rating scale and metabotypes are associated with gene modules enriched for the cell cycle, immunity, autophagy and metabolic genes, which are modified by EH301. The meta-analysis of publicly available transcriptomic data from induced motor neurons by Answer ALS confirmed the functional associations of genes correlated with disease traits. A subset of genes differentially expressed in sALS fibroblasts was used in a machine learning model to predict disease progression. In conclusion, multiomic analyses highlighted the differential metabolic and transcriptomic profiles in patient-derived fibroblast sALS, which translate into differential responses to the investigational drug EH301.
Introduction
Amyotrophic lateral sclerosis (ALS) is a rapidly progressive neurodegenerative disease involving the upper and lower motor neurons. Although only 10–15% of the total cases are associated with known genetic mutations, most ALS patients have no familial history and no clear genetic alterations (sALS) (1). While familial ALS-associated mutations are found in genes involved in several key cellular mechanisms, such as RNA binding, proteostasis, mitochondrial function and cytoskeletal organization (2), the pathological mechanisms leading to sALS are still largely unknown. Therefore, there remains a lack of sALS biomarkers and specific therapeutic targets. Although the clinical course in sporadic and familial cases reaches a generally predictable outcome, variability between patients has been recognized as an important factor in ALS, whose onset and progression are probably the result of complex interactions between genome, epigenome and environment. Moreover, it is plausible that the disease is triggered by a variety of molecular abnormalities, ultimately converging onto common pathogenic pathways that lead to motor neuron death. Thus, the complexity and heterogeneity of sALS pathogenesis are probable causes for the failure of numerous clinical trials, so that currently there are only two approved drugs, Riluzole and Edaravone, which have modest clinical effects. As sALS heterogeneity has become evident, precision medicine approaches are gaining increasing attention. The ability to stratify ALS patients could inform the design of more precisely targeted therapeutic approaches and increase the probability of finding effective treatments for specific groups of patients. Understanding the differences and similarities among sALS patients at the molecular level could also contribute to the discovery of biomarkers that would improve the reliability of trial endpoints and potentially provide a foundation for patient stratification and early intervention.
A recurring observation in ALS patients is the dysregulation of energy metabolism, with increased energy consumption and loss of fat mass, which sometimes even precedes disease onset (3). In addition, increased glucose metabolism has been described in the central nervous system of ALS patients (4). Importantly, hypermetabolism in ALS is associated with greater functional decline and accelerated mortality (5). Indeed, there is mounting evidence that metabolic alterations in ALS patients are common between the affected cells of the nervous system and other cell types. Specifically, we reported aberrantly increased energy metabolism in sALS patient-derived primary skin fibroblasts (6,7). Furthermore, our recent work showed that a subset of sALS fibroblasts is characterized by a distinct metabolic profile, which we defined as the ‘sALS1’ metabotype, associated with acceleration through the transsulfuration pathway for glutathione synthesis and glucose hypermetabolism (8). The sALS1 metabotype was also identified based on targeted metabolomics in human plasma, indicating a direct relationship between skin fibroblasts and systemic metabolism (8). These observations suggest that sALS patients could be stratified based on metabotypes, which may correspond to different pathogenic mechanisms and susceptibility to therapeutic interventions targeting specific metabolic alterations.
Energy metabolism alterations are often accompanied by oxidative stress and a modification of the redox state of the cell, which is largely controlled by the levels of nicotinamide adenine dinucleotide (NAD). NAD functions as an electron carrier in many redox reactions and as a cosubstrate for poly (adenosine diphosphate (ADP)-ribose) polymerases, ADP-ribosyl cyclases and sirtuins. Therefore, NAD is a crucial element in the coordinated signaling between DNA, proteins and metabolism (9). NAD declines with age (10) and there is evidence of NAD metabolism imbalance in ALS patients and animal models (11). Boosting NAD availability is considered a viable approach to restore or sustain cell metabolism, and it has been shown to be protective in in vitro (12) and in vivo ALS models (11). In particular, the NAD precursor nicotinamide riboside (NR) has been deemed safe and effective in increasing the circulating NAD levels in humans (13).
In addition to boosting NAD levels, another approach to modulate metabolism is through the administration of polyphenols, a class of molecules with known antioxidant properties and protective effects against diseases associated with aging (14). Pterostilbene (PT) is a polyphenol analog of resveratrol, but with improved bioavailability (15), which has been shown to be protective in animal models of metabolic diseases (16) and neurodegeneration (17).
Recently, it was shown that the combination of NR and PT increases survival and delays motor neuron degeneration in the SOD1G93A mouse model of familial ALS (18). Further, the therapeutic potential of a NR and PT combination (EH301) was suggested by a pilot clinical study, showing slower decline and improved muscle function in a small cohort of ALS patients (19). Based on these promising early results, a larger two-dose, randomized, double-blind one-year trial has been initiated [The NO-ALS Study: a Trial of NR/PT Supplement in ALS (20)].
In this study, to better understand the molecular bases of sALS metabotypes and the impact of EH301 on sALS metabolism, we analyzed the metabolome and transcriptome of fibroblasts from patients with different sALS metabotypes and control individuals, before and after the treatment with EH301. Furthermore, we performed weighted gene coexpression network analysis (WGCNA) in transcriptomic data from sALS fibroblasts. These analyses identified specific gene modules that correlated with clinical features and were modified by EH301 treatment. To confirm the fibroblast results in cell types affected by the disease, we performed WGCNA on a publicly available transcriptomic dataset from induced motor neurons (iMNs) from Answer ALS (21). Finally, we performed a proof-of-concept experiment to test the ability of a machine learning model to predict disease progression based on the expression of a few genes differentially expressed in sALS fibroblasts.
Results
Transcriptomic analysis reveals more differentially expressed genes in sALS1 than sALS2 fibroblasts and different transcriptional responses to EH301
To characterize the gene expression profiles of sALS subgroups, we performed 3'RNAseq analysis on fibroblast lines from control, sALS1 and sALS2 subjects (n = 6 per group). In sALS1, there were 281 differentially expressed genes (DEGs) (Padj. <0.05) relative to the controls (Supplementary Material, Table S1) whereas only one gene reached statistical significance in the comparison between sALS2 and controls (Fig. 1A, Supplementary Material, Table S2). Interestingly, several genes relevant to neuronal function and development were differentially expressed in sALS1. For example, stathmin 2 (STMN2) was downregulated in sALS1 by ~80%. STMN2 has been linked to transactive response DNA-binding protein (TDP)-43 dysfunction (22) and a novel STMN2 genetic variant has been associated with ALS risk, onset and progression (23). Furthermore, the most upregulated gene in sALS1 was an antisense RNA for the kinesin family member 5C (KIF5C-AS1). KIF5C is highly expressed in the brain and enriched in motor neurons (24), where it regulates axonal transport (25), and alterations of KIF5C are associated with intellectual disabilities and cortical development malformations (26,27). In addition, Yip1 interacting factor homolog A (YIF1A), which was upregulated in sALS1, interacts with the ALS8 related protein vesicle associated membrane protein associated protein B (VAPB) (28) involved in neuronal ER–Golgi interactions (29). Histone cluster 1 H4 family member C (HIST1H4C), a replication-dependent component of the nucleosome, was among the top downregulated genes in sALS1. Mutations affecting lysine 91 in HIST1H4C have been associated with a syndrome characterized by developmental anomalies and intellectual disabilities, indicating the importance of chromatin organization for the correct development and function of the nervous system (30). Sex determining region Y-box transcription factor 9 (SOX9), a transcription factor (TF) that controls several aspects of neurodevelopment (31) and is highly expressed in astrocytes and neural progenitor cells (32), was downregulated in sALS1. Spectrin repeat containing nuclear envelope protein 2 (SYNE2) (nesprin), involved in the organellar subcellular organization (33) and associated with muscular dystrophy (34), was also downregulated in sALS1. The only gene significantly downregulated in sALS2 compared with the controls was meiotic recombination 11 homolog 1 (MRE11), which encodes a double-strand break repair protein implicated in DNA damage response (35).
Figure 1.

Different sALS metabotypes show distinct gene expression profiles and transcriptional responses to EH301. (A) Volcano plot of DEGs in sALS1 (left) versus control and sALS2 (right) versus control fibroblasts (n = 6 per group). Red dots represent DEGs with P-value < 0.05 after FDR adjustment. Known ALS-related disease genes are labeled. (B) Gene ontology pathways with significantly enriched BPs and MFs of upregulated and downregulated DEGs in sALS1compared to control. (C) Venn diagram of DEGs with P-value < 0.05 after FDR adjustment modified by EH301 in control (green), sALS1 (blue) and sALS2 (red) fibroblasts (n = 6 per group). (D) Gene ontology pathways significantly enriched in EH301 treated controls (green) and sALS1 (blue) fibroblasts, (n = 6 per group), all DEGs with P-value < 0.05 after FDR adjustment were included in the analysis.
In addition to examining individual gene expression profiles, we performed pathway analysis of biological processes (BPs) and molecular function (MF) of sALS1 DEGs by Webgestalt (36), which revealed that upregulated genes are involved in vesicular and protein transport and in extracellular matrix organization (Fig. 1B, Supplementary Material, Table S3). Among the downregulated genes, the most enriched pathways in sALS1 were linked to cell cycle progression and cytoskeletal function (Fig. 1B).
Next, we evaluated the effects of EH301 on fibroblast transcriptomic profiles. Cells were exposed to EH301 (NR 1 mM, PT 10 μM) for 48 h before RNA extraction. Interestingly, we found that EH301 affected a larger number of genes in sALS1 and sALS2 fibroblasts than in the controls (233 genes in sALS1, 202 genes in sALS2 and 77 genes in controls), with a little overlap between groups (Fig. 1C, Supplementary Material, Tables S4–S6). In control fibroblasts, pathway analysis of the DEGs between vehicle- and EH301-treated cells indicated that EH301 modified the expression of genes involved in mRNA processing (Fig. 1D, Supplementary Material, Tables S7–S9). For example, synaptotagmin binding cytoplasmic RNA interacting protein (SYNCRIP), a ribonucleoprotein involved in RNA stabilization and editing, which has been associated with intellectual disabilities (37–39), was downregulated by EH301. CWC 22 spliceosome associated protein homolog (CWC22) and CWC 27 spliceosome associated cyclophilin (CWC27), which cooperate during spliceosome assembly and are linked to developmental defects (40), were also downregulated in control fibroblasts by EH301. In sALS1 fibroblasts, EH301 influenced downstream steps of protein biosynthesis by modifying the expression of genes involved in ribosome organization, translation initiation and protein localization (Fig. 1D). Several genes encoding ribosomal proteins, components of the 60S and the 40S subunits, were upregulated. Furthermore, two elements of the eukaryotic initiation factor 3 complex were differentially expressed after treatment. Eukaryotic translation initiation factor 3 subunit F (EIF3F), a positive regulator of NOTCH signaling (41), was upregulated. Conversely, eukaryotic translation initiation factor 3 subunit J (EIF3J), involved in the recognition of starting codons (42) and in ribosome recycling (43), was downregulated. Signal peptidase complex catalytic subunit SEC11A (SEC11A), which mediates the import of nascent proteins into the ER (44), was upregulated whereas lysine, aspartate, glutamate, and leucine endoplasmic reticulum protein retention receptor 3 (KDELR3), which mediates protein trafficking from Golgi to ER and involved in the stress response (45), was downregulated by EH301. B cell receptor associated protein 31 (BCAP31), a chaperone abundant in the ER and involved in transmembrane protein export (46,47) and in the assembly of mitochondrial complex I (48), was upregulated. Both KDELR3 and BCAP31 have been associated with pathologies of the nervous system (49,50). Of note, treatment with EH301 normalized YIF1A expression in sALS1 whereas STMN2 and KIF5C-AS1 expression remained altered. Surprisingly, no pathway was found to be significantly enriched in sALS2 fibroblasts, even though the expression of 187 genes was altered by EH301 in this group. Interestingly, the expression of MRE11, the only gene significantly downregulated in sALS2 versus control (CTL), was increased in sALS2 only after EH301 treatment (trending toward statistical significance, Padj = 0.073, log2 fold change = 0.53), therefore moving its expression to levels closer to that of the controls.
In summary, RNAseq in human primary fibroblasts confirmed that based on the number of DEGs the sALS1 samples are more distinct from the controls than sALS2 and that genes involved in neurodevelopment and neuronal function are differentially regulated in sALS1 fibroblasts. Moreover, pathway analysis indicates that EH301 affects sALS1, sALS2 and control fibroblast gene expression differently, mostly affecting mRNA splicing and stability in controls and protein biosynthesis and localization in sALS1 whereas no specific pathways were identified in EH301-treated sALS2.
The metabolite profiles of sALS and control fibroblasts are modified by EH301
Next, we investigated how treatment with EH301 affects the metabolite profiles of sALS1, sALS2 and control fibroblasts. We performed targeted metabolomics in the same cell lines (n = 6 per group) as used for transcriptomics under the same cell culture conditions. After excluding low-abundance hits, 166 metabolites were used for analysis.
The metabolomic profiles showed that sALS1 had reduced cystathionine and increased betaine compared with the controls (Supplementary Material, Tables S10–S12), corroborating previously reported differences in the transsulfuration pathway (8). Cystathionine levels were unchanged in EH301-treated sALS1 but decreased in both control and sALS2 fibroblasts (Fig. 2A). Oxidized glutathione was increased after treatment in the controls but unchanged in sALS1 and sALS2 fibroblasts (Fig. 2A). Together, these results indicate that EH301 modulates the transsulfuration pathway but does not correct the alterations observed in sALS1. As expected, the metabolic pathway analysis showed that EH301 modifies metabolites of the nicotinate and nicotinamide pathway in all groups, increasing the availability of NAD precursors and NAD (Fig. 2A and B). EH301-treated sALS2 also showed decreased fumarate and malate compared with the vehicle-treated cells, suggesting that NAD derived from NR accelerates the tricarboxylic acid cycle (TCA) cycle (Fig. 2A). D-glyceraldehyde-3-phosphate was significantly increased at baseline in sALS2 compared with that in the controls and was normalized by EH301 (Fig. 2A), pointing to the accelerated flux of NAD-dependent reactions in the EH301-treated sALS2 cells. Riboflavin, the precursor of flavin mononucleotide and flavin adenine dinucleotide, was decreased at baseline in both sALS1 and sALS2 fibroblasts and returned to control levels after treatment with EH301 (Fig. 2A). The pentose phosphate pathway was also affected by EH3101 in the control, sALS1 and sALS2 fibroblasts, with an increased concentration of ribose and phosphorylated ribose in the treated cells (Fig. 2A and B). Pyrimidine metabolism was modified by EH301 only in sALS1 and sALS2 fibroblasts whereas the controls were not affected (Fig. 2A and B). Purine metabolism, which was reported to be altered in sALS1 at baseline (8), was affected by EH301 in control and sALS1 cells (Fig. 2A and B). Interestingly, EH301 increased 1-methyladenosine in all groups. 1-Methyladenosine is found in the S-adenosylmethionine (SAM)-dependent modification of RNA regulating mRNA localization, stability, translation and splicing (51). 1-Methyladenosine also responds to stress, decreasing upon glucose or amino acid starvation and increasing after heat shock (51). 1-Methyladenosine can also modify tRNA, regulating its stability and folding (52) and is found in mtDNA-encoded transcripts (53). Thus, some of the effects of EH301 on transcription and metabolism could be mediated by the regulation of SAM-dependent epigenetic marks on RNA.
Figure 2.

EH301 affects nucleotide and amino acid metabolism and protects fibroblasts from thiol depletion stress. (A) Z score heatmap of metabolites significantly modified by EH301 in control, sALS1 and sALS2 fibroblasts (n = 6 per group), P-value < 0.05 after FDR adjustment. (B) Scatter plot of metabolic pathways enriched in control (square), sALS1 (circle) and sALS2 (triangle) fibroblasts after treatment with EH301, shape size reflects number of metabolites. (C) Percentage cell death in control, sALS1 and sALS2 fibroblasts grown in methionine/cystine depleted medium for 72 h and treated with EH301 or its single components (NR, PT). Bars represent average +/− standard error of mean of at least three independent experiments. One-way ANOVA followed by Fisher least significant difference (LSD). *P < 0.05, **P < 0.01, ****P < 0.0001. (D) Volcano plot of transcripts modified by EH301 in control fibroblasts (n = 3 per condition) grown for 48 h in methionine/cystine depleted medium compared to vehicle treatment. Red dots represent DEGs with log2 fold change greater than 1 with P-value < 0.05 after FDR adjustment. Some representative genes belonging to enriched GO pathways are labeled. (E) Significantly enriched pathways of upregulated and downregulated genes in control fibroblasts grown in methionine/cystine depleted medium for 48 h and treated with EH301 (n = 3 per condition).
The alanine, aspartate and glutamate metabolic pathways were globally altered by EH301 in sALS1 and sALS2 fibroblasts (Fig. 2A and B). Specifically, glutamate was decreased at baseline in sALS1, but not in sALS2, compared with that in controls (Fig. 2A and B). This selective glutamate decrease in sALS1 could be due to the increased extrusion of glutamate in exchange for cystine by the solute carrier family 7 member 11 (SLC7A11) transporter, which was shown to be upregulated in sALS1 fibroblasts (8). EH301 decreased glutamate levels in all groups, a potentially disease-relevant effect, as extracellular glutamate homeostasis is known to be dysregulated in ALS (54).
EH301 strongly protects fibroblasts from cell death induced by thiol group depletion
Although EH301 did not correct the characteristic imbalances of transsulfuration metabolites in sALS1, we investigated whether the increased NAD availability and modification of amino acid metabolism by EH301 could improve cell viability under metabolic stress induced by methionine and cystine deprivation. To this end, we cultured cells for 72 h in methionine and cystine-depleted medium, in the presence or absence of EH301 or its individual components (NR or PT). The depletion of methionine and cystine caused similar levels of cell death in all groups (Fig. 2C). Interestingly, the addition of NR alone was effective in protecting sALS1 fibroblasts from cell death, whereas the viability of sALS2 and the controls was not improved (Fig. 2C). Treatment with PT alone was sufficient to prevent cell death in all groups (Fig. 2C). The combination of NR and PT (EH301) had comparable effects to PT alone. These results further indicate that the metabolic alterations of sALS1 are different than of sALS2 and potentially more responsive to nicotinamide derivatives. They also indicate that PT is the most potent compound in protecting cells from the profound redox stress derived from thiol group depletion.
Transcriptomic analysis performed on the control cells exposed to medium depleted of methionine and cystine showed that treatment with EH301 reduces expression of genes involved in inflammation and apoptosis (Fig. 2D and E, Supplementary Material, Tables S13–S14). Of note, the expression of the stress response factor activating transcription factor 3 (ATF3) was downregulated by EH301, while the level of activating transcription factor 5 (ATF5), which promotes the expression of chaperones and prosurvival factors (55), was increased (Fig. 2D). Members of the kinesin family and tumor protein p53 (TP53) were among the genes upregulated by EH301 in fibroblasts grown in the absence of methionine and cystine. Protein arginine methyltransferase 1 (PRMT1) and protein arginine methyltransferase 2 (PRMT2), which regulate the DNA damage response and other signaling pathways through SAM-dependent arginine methylation, were also increased by EH301. On the other hand, interleukins (C-X-C motif chemokine ligand 1 (CXCL1), C-X-C motif chemokine ligand 2 (CXCL2), C-X-C motif chemokine ligand 3 (CXCL3), C-X-C motif chemokine ligand 5 (CXCL5), C-X-C motif chemokine ligand 6 (CXCL6), C-X-C motif chemokine ligand 8 (CXCL8)) were downregulated by the treatment. The transcripts of superoxide dismutase 2 (SOD2) and different metallothionein isoforms were reduced in fibroblasts treated with EH301 compared with the vehicle, further indicating that EH301 acts through antioxidant and antiinflammatory mechanisms, which prevent the need for upregulation of free radical scavengers and stress response genes under thiol-depleted conditions.
Weighted gene co-expression network analysis highlights associations between fibroblast transcriptional profiles and ALS clinical traits, which are altered by EH301 treatment
WGCNA is a powerful unbiased method for the analysis of transcriptome-wide changes due to disease state (56,57). WGCNA differs from more traditional differential gene expression analysis methods because it considers groups of genes with highly similar expression patterns across samples as part of a set of interconnected modules, rather than considering genes as single entities. This type of analysis increases the statistical power available to identify significant associations with phenotypic traits by minimizing noise. It may also provide more comprehensive information on complex BPs (58). The method works by using expression data from every gene in every sample to create a network that represents overall patterns of gene expression. Several matrices are constructed stepwise to encode the correlations of each gene with every other gene in all samples, the relative connection strength of these correlations, and the proportional interconnectedness of each gene to all other genes across all samples. These networks of values are then used as input to hierarchical clustering algorithms to identify groups of highly coexpressed genes called modules (56,57).
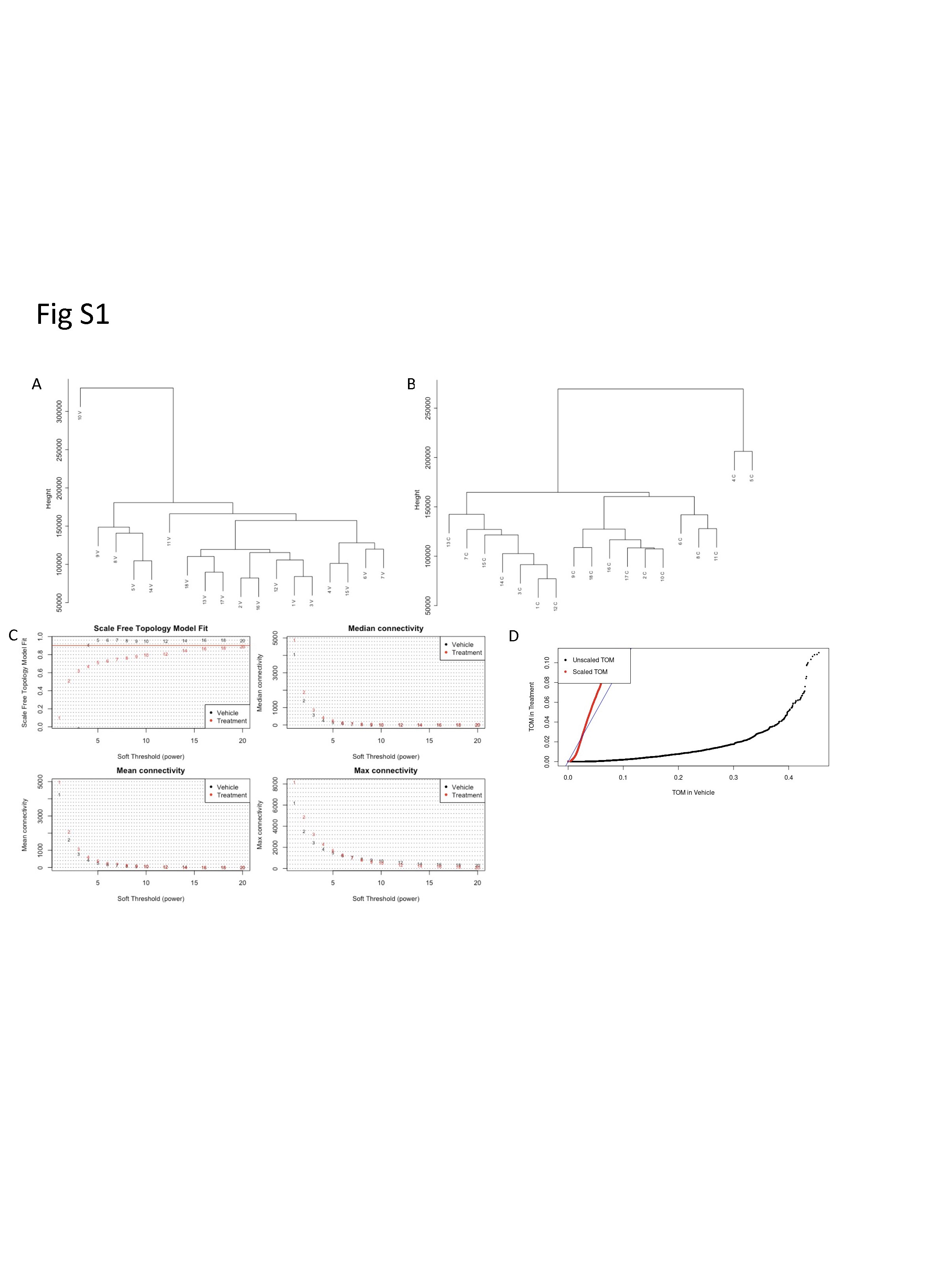
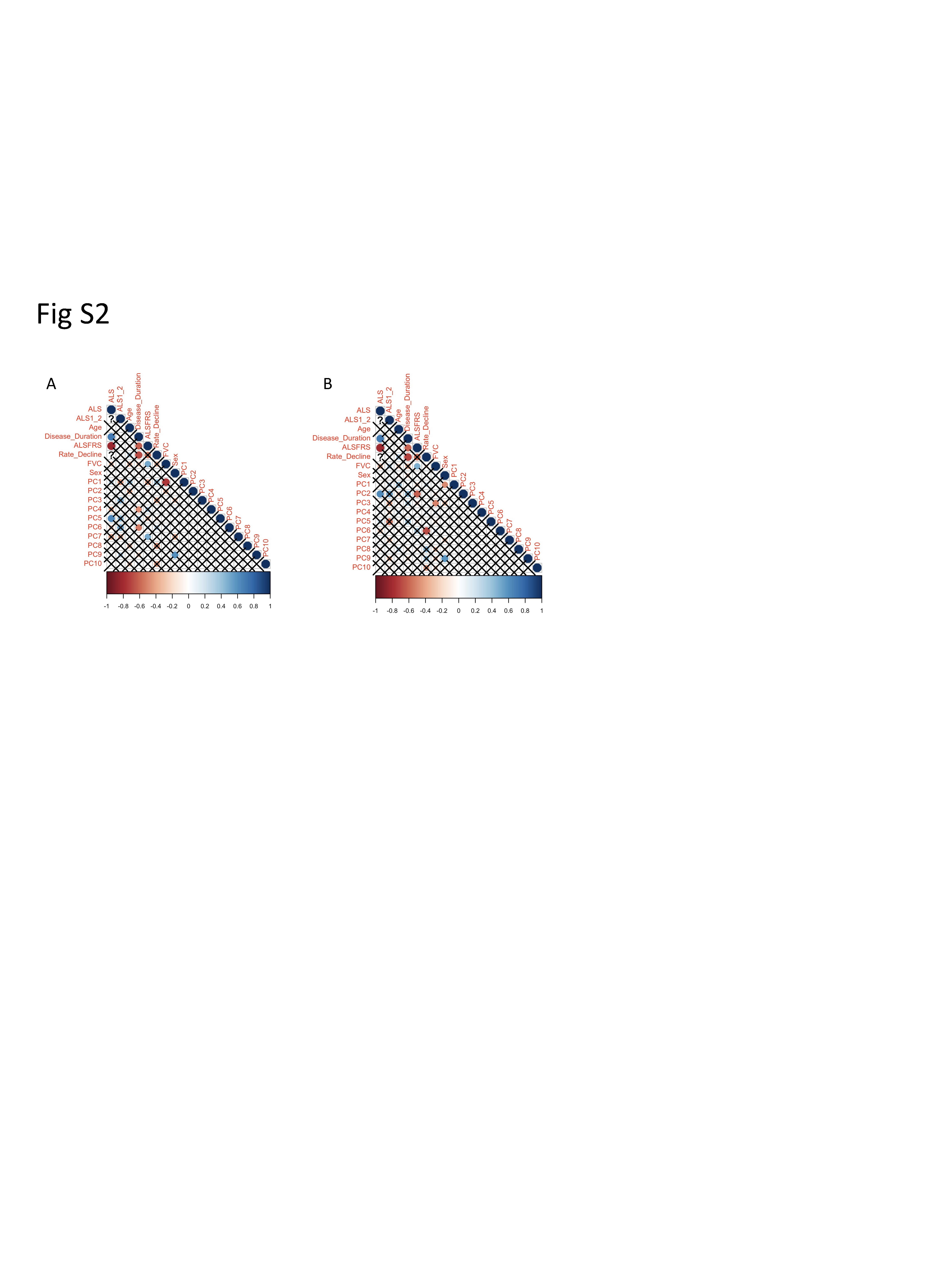
To apply the WGCNA framework to our fibroblast gene expression dataset, we first constructed a coexpression matrix using normalized expression data for 17 662 genes. For this analysis, we included all the vehicle-treated lines except one sALS1 (10 V) that was identified as an outlier based on its extreme distance from all other samples in hierarchical clustering (Supplementary Material, Fig. S1). Note that 10 V was only excluded from WGCNA, due to this method’s higher sensitivity to outliers (59), and not the other analyses described above. We applied the same method to construct a matrix using all the EH301-treated samples. After clustering, we identified 25 modules in the vehicle network and 90 in the EH301 network (Fig. 3A and B). We next correlated module gene expression with six disease traits (disease status—ALS or control, disease subgroup—sALS1 or sALS2 -, disease duration, amyotrophic lateral sclerosis functional rating scale (ALSFRS)-R, rate of ALSFRS-R decline and forced vital capacity (FVC%)), as well as sex and age, to identify modules that are strongly associated with important disease parameters. Disease duration, ALSFRS-R, rate of decline and FVC% are all relevant markers of ALS severity, which were significantly correlated with each other, as expected (Supplementary Material, Fig. S2). We found no significant correlations between age and any of the disease traits or the first 10 principal components, which cumulatively explain over half of the total variance in the dataset, derived from gene expression in either vehicle or EH301 samples, suggesting that age does not significantly contribute to the variance in gene expression in our data. Sex significantly correlated with vehicle PC9 and EH301 PC1 and PC9 but did not correlate with any of the disease traits (Supplementary Material, Fig. S2). Nevertheless, we opted to include sex and age in our analysis as they are potential biologically relevant variables in ALS. In the vehicle network, 10 modules (40%) were significantly associated with one or more traits (Fig. 3C) whereas in the EH301 network, 38 modules (44%) had significant associations with one or more traits (Fig. 3D).
Figure 3.

EH301 modifies network structure and associations of modules with disease traits. (A, B) Dendogram of hierarchical clustering of gene co-expression dissimilarity values, constructed from the TOM, for vehicle-treated samples (A) and EH301-treated samples (B). Colors correspond to module assignments (see Supplementary Material Fig. S1 for network construction parameters). (C, D) Heatmaps showing correlations between module eigengene expression values and clinical traits for vehicle-treated samples (C) and EH301-treated samples (D). The numbers in each box are P-values, while box colors correspond to the correlation coefficient. For clarity, only modules with at least 1 significant trait association are shown.
We then performed Gene Ontology (GO) pathway analyses using the GO: BP, GO: MF and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases on the significantly associated modules and found that in the vehicle network 6 out of 10 modules (Table 1) had a significant enrichment for one or more pathways whereas in the EH301 network 23 out of 38 modules (Table 2) had a significant enrichment for one or more pathways. The module significantly associated with the largest number of traits in the vehicle network was the Greenyellow, which was associated with disease status, disease duration, ALSFRS-R score and nearly reached significant association (P = 0.06) for FVC%. GO analysis showed that the set of genes comprising the Greenyellow module were functionally enriched for genes involved in cell cycle, chromatin modifications and DNA damage repair (Fig. 4A, Supplementary Material, Table S15). The Turquoise module significantly associated with FVC% and neared significance for association with disease metabotype, ALSFRS-R score and rate of decline. Genes belonging to the Turquoise module were functionally enriched for pathways related to DNA damage repair, autophagy and protein catabolism, cell cycle, innate immunity and mitochondrial function (Fig. 4B, Supplementary Material, Table S15). Interestingly, the Turquoise module was also significantly enriched for genes annotated by the KEGG database as important for ALS pathogenesis (KEGG hsa05014). Finally, the Salmon module, which significantly associated with disease metabotype, contained genes belonging to pathways related to autophagy and protein catabolism (Fig. 4C, Supplementary Material, Table S15). Clustering of module eigengene expression revealed that the Salmon, Turquoise and Greenyellow modules had highly dissimilar expression patterns from each other (Supplementary Material, Fig. S3). Furthermore, when comparing the GO terms enriched in these three modules, we found minimal overlap in terms enriched in the Greenyellow and Salmon modules (Fig. 4D), suggesting that the genes comprising them have mostly distinct functions. Although the Turquoise module has GO terms showing an over 50% overlap with those found in the Salmon and Greenyellow modules, it also has over 1000 unique GO terms, indicating that it contains genes that have functional annotations not represented in either of the other two modules. Thus, based on their eigengene expression and GO enrichment, the three modules identified in the vehicle network are non-redundant.
Table 1.
Vehicle network modules with a significant trait association and GO annotation
| ALS versus CTL | sALS1 versus sALS2 | Disease duration | ALSFRS-R | Rate of decline | FVC% | Age | Sex |
|---|---|---|---|---|---|---|---|
| Greenyellow | Salmon | Greenyellow | Greenyellow | Turquoise | Brown | ||
| Darkred | |||||||
| Orange |
Table 2.
EH301 network modules with a significant trait association and GO annotation
| ALS versus CTL | sALS1 versus sALS2 | Disease duration | ALSFRS-R | Rate of decline | FVC% | Age | Sex |
|---|---|---|---|---|---|---|---|
| Brown | White | Lightpink2 | Darkviolet | Darkviolet | Red | Tan3 | Salmon1 |
| Indianred1 | Skyblue | Red | Red | Salmon1 | Lavenderblush3 | Navajowhite2 | |
| Brown | Sienna4 | Magenta4 | Slateblue | Darkred | Slateblue | ||
| Firebrick | Salmon1 | Mediumpurple1 | Magenta4 | ||||
| Slateblue | Lavenderblush3 | Mediumpurple1 | |||||
| Brown | Lightskyblue4 | Darkorange | |||||
| Magenta4 | Mediumorchid4 | ||||||
| Magenta3 |
Figure 4.

Both vehicle and EH301 disease-associated modules have overlapping and unique GO functional annotations. (A–C) Top 20 most significant non-redundant GO: BP terms (identified using the simplify function from the clusterProfiler package to merge terms with more than 40% overlapping annotated genes) enriched in the Greenyellow (A), Turquoise (B) and Salmon (C) modules from the vehicle network. (D) Venn diagram showing overlap of all significantly enriched GO terms in the Greenyellow, Turquoise and Salmon vehicle modules. (E, F) Top 20 most significant GO terms enriched in the Brown (E) and Red (F) modules from the EH301 network. (G) Venn Diagram showing overlap of all significantly enriched GO terms in the Brown, Red, Slateblue and Magenta4 EH301 modules.
In the EH301 network, four modules were associated with at least three traits. The Brown module was associated with disease status, disease metabotype and ALSFRS-R and included genes enriched for cell cycle, chromatin modifications, DNA damage repair, nucleic acid metabolism and transcriptional activity GO terms (Fig. 4E, Supplementary Material, Table S15). The Red module associated with ALSFRS-R, rate of decline and FVC% and included genes enriched for chemotaxis, antigen processing and immunity (Fig. 4F, Supplementary Material, Table S15). The Slateblue module was associated with ALSFRS-R, FVC% and sex and included genes enriched for RNA stem-loop and scaffold protein binding (Supplementary Material, Table S15). Finally, the Magenta4 module was associated with ALSFRS-R, rate of decline and sex and included genes enriched for Fanconi anemia pathway, a pathway activated by DNA damage (Supplementary Material, Table S15). Clustering of module eigengene expression revealed that Brown, Slateblue and Magenta4 cluster together whereas Red does not (Supplementary Material, Fig. S3). There was minimal overlap in GO enrichment terms among the four modules in the EH301 network that associate with traits (Fig. 4G). This indicates that although the gene expression signatures of three of the four trait-associated modules in the EH301 network are similar, all four modules are functionally distinct.
Comparing the vehicle and EH301 networks revealed a striking difference as the EH301 network included nearly four times the number of modules observed in the vehicle-treated cells. However, the intramodular connectivity was comparable between the two networks (Table 3), indicating that modules are clustered with a similar robustness in both networks. On the other hand, the total average connectivity and extramodular connectivity were significantly higher in the EH301 network (Supplementary Material, Table S15). This indicates that individual modules are more highly connected to each other in the EH301 than in the vehicle network. Modules from the two networks were compared based on their components using Fisher’s exact test, and modules were paired if P < 0.05. This analysis revealed that 22 out of 25 vehicle modules have a corresponding module in the EH301 network (Supplementary Material, Fig. S4). Of these 22 pairs, four significantly associated with only one common trait whereas the Greenyellow and Brown pair associated with two traits common to both (disease status and ALSFRS-R). When the GO terms from all modules that significantly associated with a trait in both networks were compared, most terms associated with disease status in the Greenyellow vehicle module were also found in the Brown EH301 module (Fig. 5A) and were related to cell cycle and DNA replication (Fig. 4). This suggests that the genes that correlate with disease status likely share similar functions, regardless of treatment with EH301, but the latter introduces new associations with genes annotated with different functions, including nucleic acid metabolism and transcriptional regulation (Figs 5A and4E). However, for other traits including disease metabotype (Fig. 5B), disease duration (Fig. 5C) and FVC% (Fig. 5E), the overlap between GO terms from the vehicle and EH301 networks was small or absent. GO terms differing between sALS1 and sALS2 in vehicle conditions were related to autophagy and protein catabolism (Fig. 4C) whereas GO terms after EH301 treatment were related to glycolysis, extracellular matrix organization, cell cycle and transcription (Supplementary Material, Table S15). Vehicle modules associated with disease duration were enriched for terms related to cell cycle and DNA damage repair whereas EH301 modules were enriched for terms related to cell adhesion, membrane polarization and cation homeostasis (Fig. 4A and Supplementary Material, Table S15). The Turquoise vehicle module associated with FVC% was enriched for terms related to mitochondrial function, innate immunity, cell cycle and autophagy (Fig. 4B) whereas the EH301 modules associated with FVC% were enriched for terms including major histocompatibility complex (MHC) complex assembly and antigen presentation (Fig. 4F) and cell cycle (Fig. 4E, and Supplementary Material, Table S15). This suggests that EH301 modifies gene sets associated with disease traits and that these genes regulate more diverse functions than those from the vehicle network. Similar to disease status (sALS or control), there was a substantial overlap in GO terms between the Greenyellow vehicle module and Brown EH301 module associated with ALSFRS-R (Fig. 5D), but there were also several modules from the EH301 network with no GO overlap with vehicle modules. These were enriched for terms including sugar alcohol metabolism, RNA and protein binding, MHC complex assembly and cell adhesion and motility (Supplementary Material, Table S15). Overall, comparison of the two networks revealed conservation of most of the vehicle modules after EH301 treatment, including the functionally similar Greenyellow/Brown pair that associated with disease status and ALSFRS-R. However, a large disparity was found in the functional annotation of the other modules associated with clinical traits in the two networks, indicating that EH301 modulates the expression of gene sets that are significantly correlated with various clinical traits.
Table 3.
Vehicle and EH301 network structure comparison parameters
| Parameter | Vehicle network average | EH301 network average | P-value |
|---|---|---|---|
| Total connectivity | 184.45 | 243.75 | 2.04 e-118 |
| Intramodular connectivity | 140.0 | 144.0 | 0.18 |
| Extramodular connectivity | 44.0 | 100.0 | 0.00 |
Figure 5.

Minimal overlap exists between GO terms from vehicle and EH301 modules associated with specific disease traits. (A–E) Venn Diagrams comparing overlap between all significantly enriched GO terms from all modules significantly associated with the listed clinical traits, from vehicle and EH301 networks. Due to the number of EH301 modules associated with ALSFRS-R score, only the 4 modules containing common terms are shown.
To investigate the transcriptional regulation underlying the effects of disease and EH301 treatment on gene expression, we performed a TF binding site enrichment analysis on all genes in modules significantly associated with disease traits in both networks. The TF binding sites most enriched in genes associated with disease in both vehicle and EH301 modules were those of the E2F and the Sp families of TFs (Supplementary Material, Table S16). Importantly, enrichment of binding sites of several TFs was found only in disease-associated modules of the EH301 network, indicating that these transcriptional effects are EH301-specific (Supplementary Material, Table S16). These TFs have also been associated with ALS in patients or model systems and include activating enhancer binding protein 2 (AP-2), forkhead box protein O1A (FOXO1A) (60), sterol regulatory element binding transcription factor 1 (SREBP1) (61), metal regulatory transcription factor (MTF)-1 (62,63) and retinoic acid receptor beta (RARB) (64).
Finally, we aimed to identify ‘hub’ genes from important modules, or genes that drive the expression profile of each module, while also correlating significantly with disease traits. Hub genes may be useful as biomarkers for potential classification of patients based on their clinical characteristics and disease severity. For each gene, we calculated a significance score, denoting how strongly that gene associates with a trait, and a module membership score, denoting how closely that gene’s expression pattern matches the average module eigengene expression, or how strongly that gene ‘belongs’ to that module (56). We identified the most interconnected genes in each module using connection strengths calculated from the topological overlap matrix (TOM) and visualized with VisANT (65). For each of the relevant modules in the vehicle network (Supplementary Material, Fig. S5) and the EH301 network (Supplementary Material, Fig. S6), we selected the 50 most significant genes for each associated trait as well as the top 50 most strongly connected genes within the associated module. To illustrate the potential application of this analysis to discover disease biomarkers, we chose five genes from the Greenyellow vehicle module that were significantly associated with ALSFRS-R. To this end, genes were ranked in the order of significance for correlation with ALSFRS-R and chosen if they (1) were part of the network hub identified by VisANT and (2) were identified as differentially expressed in the sALS1 versus CTL comparison in vehicle samples. Expression patterns of these five genes (distinct subgroup of the rat sarcoma virus family member 3 (DIRAS3), G2 and S-phase expressed 1 (GTSE1), ribonucleotide reductase regulatory subunit M2 (RRM2), cell division cycle associated 5 (CDCA5) and holliday junction recognition protein (HJURP)) showed clear differences between control and sALS1 samples, and to a lesser extent between control and sALS2 (Fig. 6A). We next examined if the expression of these genes could be used to group samples based on their ALSFRS-R score. Dendograms were constructed from hierarchical clustering first of ALSFRS-R score and then of average expression of the five genes for vehicle-treated samples (Fig. 6B, left) and EH301-treated samples (Fig. 6B, right). In both vehicle- and EH301-treated fibroblasts, clustering based on ALSFRS-R significantly matched the clustering based on gene expression, indicating that expression of these five selected genes can be used to group samples based on their ALSFRS-R scores.
Figure 6.

A set of five hub genes that correlate strongly with ALSFRS-R can be used to cluster samples into groups that correspond to their ALSFRS-R scores. (A) Heatmap showing Z-scores of normalized expression values of the five genes chosen as ‘markers’ of ALSFRS-R score, along with aggregated average expression values of all five and ALSFRS-R score for each sample. (B) Dendograms created by hierarchical clustering of ALSFRS-R score on the left, and average expression of the five marker genes on the right, for vehicle samples (left) and EH301 samples (right). Central gray lines show matching of each sample over the two trees, and colored lines in the tree denote matching clusters. P-value for Baker’s gamma index calculated using 100-fold permutation.
WGCNA of transcriptomic data from ALS induced motor neurons supports and extends fibroblast results
To extend the WGCNA analysis to a cell type affected by the disease, we utilized transcriptomic and clinical data from 124 (99 ALS and 25 control) iMN lines obtained by the Answer ALS project (21). We constructed a new network (Fig. 7A) using expression data from the 22 653 genes that passed quality control filters and identified 38 modules. We next calculated associations between modules and six traits (disease status, baseline ALSFRS-R, most recent ALSFRS-R, ALSFRS-R progression slope, age and sex) and found 23 modules (60%) with a significant association with one or more traits (Fig. 7B). GO analysis revealed that 20/23 modules had significant enrichment for one or more GO: MF, GO: BP and KEGG pathways (Table 4). Of these, Blue, Magenta and Tan associated with both disease status and one or more measures of ALSFRS-R. Genes in both the Blue and Tan modules were functionally enriched for terms associated with nucleotide metabolism and several types of protein modifications, and Tan module genes were also enriched for terms involved in the mitochondrial electron transport chain (Fig. 7C and D). Genes in the Magenta module were enriched for pathways related to signal transduction through G-protein coupled receptors (Fig. 7E).
Figure 7.

WGCNA of transcriptomic data from iMNs confirms the association of several GO pathways with disease traits found in fibroblasts. (A) Dendogram of hierarchical clustering of gene co-expression dissimilarity values, constructed from the TOM, for all iMN samples (n = 124). Colors correspond to module assignments. (B) Heatmap showing correlations between module eigengene expression values and clinical traits. The numbers in each box are P-values, while box colors correspond to the correlation coefficient. For clarity, only modules with at least 1 significant trait association are shown. (C–G) Top 20 most significant non-redundant GO: BP terms (identified using the simplify function from the clusterProfiler package to merge terms with more than 40% overlapping annotated genes) terms enriched in the Blue (C), Tan (D), Magenta (E), Turquoise (F) and Purple (G) modules. (H, I) Venn diagram showing overlap of all significantly enriched GO terms from all modules associated with disease status (H) or ALSFRS-R (I) in the iMN and vehicle fibroblast networks. (J) ROC curves for each of the six cross-validation runs of the classification model (trained and tested on the 62 iMN samples with ALFRS-R progression data) with computable ROC. AUC values for each curve indicated on bottom right.
Table 4.
iMN network modules with a significant trait association and GO annotation
| ALS | ALSFRS-R baseline | ALSFRS-R latest | ALSFRS-R progression | Age | Sex |
|---|---|---|---|---|---|
| Blue | Cyan | Magenta | Blue | Honeydew1 | Bisque4 |
| Magenta | Tan | Magenta | Darkgrey | ||
| Tan | Lightgreen | Tan | Bisque4 | ||
| Maroon | Yellowgreen | ||||
| Yellowgreen | Lightpink4 | ||||
| Cyan | Coral1 | ||||
| Lightpink4 | Darkred | ||||
| Darkgreen | Sienna3 | ||||
| Orangered4 | Black | ||||
| Lightyellow | Purple | ||||
| Turquoise |
sALS, sporadic amyotrophic lateral sclerosis; ALSFRS-R, revised ALS functional rating scale; DEGs, differentially expressed genes; FVC, forced vital capacity; GO, gene ontology; GSH, glutathione; iMN, induced motor neurons; NAD, nicotinamide adenine dinucleotide; NR, nicotinamide riboside; PT, pterostilbene; TF, transcription factor; WGCNA, weighted gene co-expression network analysis.
Next, we evaluated which modules in the iMN network contained genes from the Greenyellow module in the vehicle-treated fibroblast network. We found that the majority of the Greenyellow genes were found in the Purple and Turquoise modules in the iMN network, both of which significantly associate with ALSFRS-R progression slope. Accordingly, GO analysis revealed that both Purple and Turquoise genes were enriched for pathways related to cell cycle and development, similar to the pathways identified in the fibroblast Greenyellow module (Fig. 7F and G, Supplementary Material, Table S15). We then evaluated whether the genes associated with traits in the vehicle fibroblast network had similar functional GO annotations to the genes associated with the same traits in the iMN network. There was a little overlap in GO enrichment between modules associated with disease status in the fibroblast and iMN networks (Fig. 7H). However, for all traits related to ALSFRS-R, there was a large overlap in the GO terms identified in each network, with approximately ~66% of the GO terms associated with ALSFRS-R in the fibroblast network also associating with one or more of the ALSFRS-R measures in the iMN network (Fig. 7I). Therefore, genes related to ALSFRS-R, a measure of disease severity, share similar functions, including cell cycle and nucleic acid metabolism, in fibroblasts and iMNs.
Finally, to provide an initial proof of concept of the potential predictive value of the five genes related to ALSFRS-R progression in fibroblasts (DIRAS3, GTSE1, RRM2, CDCA5 and HJURP), we used multinomial logistic regression to classify ALS iMN samples based on the expression of these genes. We used a 10-fold cross validation approach, in which we randomly divided the 62 ALS samples with available ALSFRS-R progression slopes into 10 sets. In each iteration, one set was used as the training data (n = 6) and the model was tested on the remaining 9 sets (n = 56). We arbitrarily defined cases as ‘fast progressors’ if their ALSFRS-R progression slope values were one standard deviation or more below the mean. Using this method, we obtained an average accuracy of 78.9% (95% CI: 66.0–91.6%), precision of 80.1% (95% CI: 67.6–92.6%), false positive rate of 1.4% (95% CI: −1.4–4.2%) and false negative rate of 17.0% (95% CI: 6.3–28.0%), indicating good specificity and fair sensitivity. We then computed receiver-operating characteristic (ROC) curves to evaluate model performance for six of the ten iterations (four iterations were unusable due to the absence of any fast progressors in the test set) and obtained an average area under the curve (AUC) value of 0.767 (95% CI: 0.651–0.883). ROC curves showing the performance of the model for the 6 iterations are shown in Figure 7J. This approach represents an example of how genes associated with ALSFRS-R in fibroblasts could be utilized to discriminate patients with fast disease progression relative to all other ALS cases in disease-relevant iMNs.
Discussion
Numerous lines of evidence suggest that metabolic alterations in ALS patients are related to both pathogenesis and prognosis (3–5). Therefore, metabolism could be a viable therapeutic target for the disease. However, the biochemical and molecular underpinnings of metabolic dysregulation remain largely unknown, especially in sALS, hindering the development of effective targeted approaches to correct them. The mechanisms leading to metabolic dysregulation can differ in sALS metabotypes resulting in different sets of biomarkers and susceptibility to the effects of treatments. Efforts have been made to use biofluids to identify metabolic biomarkers for stratification of sALS patients (6,7,66), but high variability associated with environmental factors can be challenging. An alternative approach to using biofluids for unbiased-omic studies is to obtain cells from patients and investigate their metabolism under homogeneous conditions to identify specific markers indicative of sALS metabotypes that can then be validated and in patients and used in clinical settings. Our studies have utilized primary skin fibroblasts from sALS patients to first identify a hypermetabolic phenotype relative to healthy controls (6,7) and, more recently, to define a subtype of sALS (sALS1) metabolically characterized by enhanced transsulfuration metabolism, which was then confirmed in patient plasma (8). This finding raised the possibility that sALS1 cases are differentially responsive to therapies that modulate cellular metabolism. The NR and PT combination drug EH301 (19) is one of these therapies, as it is designed to elevate NAD levels and activate sirtuins. EH301 was successful in a pilot ALS clinical trial (19) and is currently being investigated in a larger phase 3 clinical study. However, the metabolic effects of EH301 in different subtypes of sALS patients are still unknown.
In this study, we took advantage of a set of sALS patient-derived fibroblasts with defined metabotype (i.e. sALS1 and sALS2) and controls based on previous published work (8). In these groups of cell lines, we performed in-depth transcriptomic and targeted metabolomic analyses to further elucidate profiles that characterize the subtypes and to assess how EH301 treatment affects gene expression and metabolism in the different metabotypes. We identified striking differences in the number of DEGs, relative to the control cells, between untreated sALS1 and sALS2 lines (281 versus 1), and in the response to EH301, whereby only approximately 5% of the genes modified by the drug were in common between sALS1 and sALS2. Notably, EH301 affected a much smaller number of genes in control lines compared to both sALS groups (77 versus >200), further indicating group-specific responses to the drug. In addition to the expected increase of NAD and its precursors, targeted metabolomics revealed that EH301 affected nucleotide metabolism in all groups as well as amino acid metabolism in both sALS groups. Cysteine and methionine metabolism was only affected in sALS2 suggesting that the intrinsic transsulfuration alterations of sALS1 lines are not corrected, possibly because the effect of EH301 drives metabolism in a similar direction as the adaptive mechanisms that naturally take place in sALS1. EH301 had a potent protective effect against metabolic stress induced by thiol group deprivation in all groups, and this effect was largely mediated by PT because NR alone had a moderate effect only in sALS1. This result supports efficacy of EH301 in antagonizing the deleterious effects of cellular stress, but do not indicate metabotype specificity of PT. However, it needs to be noted that the stress paradigm we used is very severe and using cell death as readout may not allow for detection of more subtle phenotypic differences.
To complement and extend the differential gene expression analysis we used WGCNA, which allows association of gene expression modules with several phenotypic variables, such as clinical parameters or other indices of pathology. This approach may provide more sensitivity to detect differences in disease characteristics among patients than traditional grouped pairwise-comparison approaches (58). WGCNA has been recently used to identify new risk genes and putative drug targets associated with major neurodegenerative diseases (67–69). We used WGCNA to construct networks for vehicle- and EH301-treated samples and identified several modules in each network that significantly associated with disease and measures of disease severity. In both networks, genes belonging to modules associated with disease and ALSFRS-R were highly enriched for pathways related to the cell cycle, DNA damage repair and nucleic acid metabolism. TF binding site analysis of disease-associated genes in both networks revealed enrichment of E2F and Sp family TF binding sites. TFs in the E2F family are essential regulators of the cell cycle and apoptosis and have been involved in the response to DNA damage in cultured neurons (70,71). E2F transcription factor 1 (E2F1) has been shown to have upregulated expression correlated with markers of aberrant cell cycle re-entry in postmortem spinal cord and motor cortex samples from ALS patients (72,73). Interestingly, in addition to its canonical role regulating the cell cycle, E2F1 is also important for controlling several aspects of global metabolic homeostasis (74). Sp family TFs have been demonstrated to be key for modulating apoptosis in cultured neurons undergoing oxidative stress caused by glutathione depletion, and Sp1 knockdown is protective in G93A superoxide dismutase 1 (SOD1) mutant mice (75,76). Sp1 and specificity protein transcription factor 3 (Sp3) can also interact with E2F1 and cooperatively regulate transcription as a complex (77). Although the cell cycle has not been traditionally considered relevant in post-mitotic cells, such as motor neurons, increasing evidence suggests that cell cycle genes can be dysregulated in neurodegenerative disease. This dysregulation may promote aberrant re-entry into the cell cycle, leading to neuronal death (71,73,78). Genes associated with cell cycle also have important functions in the maintenance of the cytoskeleton. Notably, mutations in kinesin genes, such as KIF5A, have been linked to ALS and other motor neuron diseases (79). Our results confirm that cell cycle genes may be involved in ALS pathogenesis.
Despite the common effects of ALS on gene expression identified in both vehicle and EH301 networks, we also identified EH301-specific alterations. We found that EH301 markedly modifies network structure relative to vehicle, suggesting global alterations in patterns of gene expression. Evidence that EH301 produces widespread changes in transcriptional regulation is also provided by the enrichment of several TF binding sites in genes belonging to modules associated with disease, which were not found in the vehicle network. Furthermore, we identified substantial differences in the functions of genes associated with disease traits between EH301 and vehicle networks, such as cell adhesion and immune response which are unique to EH301. Therefore, EH301 may modulate genes and pathways relevant to mechanisms of ALS pathogenesis through transcriptional regulation. These findings will be valuable in interpreting the results of the ongoing clinical trial of EH301 in ALS patients.
In this study, we utilized fibroblasts as a model system, as these cells have been extensively used by us and many other groups for the investigation of molecular, biochemical and metabolic changes in ALS. Nevertheless, we deemed it important to compare the gene expression modules obtained from ALS fibroblasts with modules from an independent, larger dataset from iMNs publicly available from the Answer ALS database. We observed a small overlap in GO terms enriched in genes belonging to modules associated with disease status in fibroblasts and iMNs, including nucleic acid metabolism terms. On the other hand, there was a substantial overlap in the functional annotations of genes associated with ALSFRS-R in both datasets, indicating disease severity-related gene expression changes common to both cell types. Previous studies have used genomic and transcriptomic data from human motor cortex to characterize sALS metabotypes, and have identified immune response, cell adhesion, cytoskeletal organization and cell cycle among the most dysregulated pathways in specific sALS subgroups relative to control (66,80). These findings coincide with our results on pathways affected in sALS fibroblasts and those identified in sALS iMNs, further supporting the notion that different subsets of sALS cases are characterized by distinct gene expression profiles that are common among multiple cell types and tissues.
Purine and pyrimidine metabolism alterations were consistently identified in metabolomic and transcriptomic analyses in ALS fibroblasts and transcriptomics analyses in ALS iMNs. Alterations in nucleotide metabolism have also been shown in spinal cord from ALS patients (81). In post-mitotic cells, nucleotide metabolism is required for proper DNA damage repair (82), and many of the mechanisms thought to be involved in ALS pathogenesis, such as oxidative stress and energy imbalance can lead to a loss of DNA integrity and high burden of DNA damage (83–85). Therefore, nucleotide metabolism may be integral to the mechanisms leading to motor neuron degeneration in ALS, and nucleotide metabolism may provide new targets for therapeutic intervention.
Finally, we confirmed in iMNs the association of the five biomarker genes with ALSFRS-R that we found in fibroblasts. In agreement with the GO terms associated with disease severity identified in both fibroblasts and iMNs, four of these genes (HJURP, DIRAS3, CDCA5 and GTSE1) are associated with cell cycle and DNA damage repair, while RRM2 is involved in purine and pyrimidine metabolism. To establish an initial proof of concept that the expression of a small subset of genes in patient-derived cells could be used to predict disease progression, we employed a logistic regression model based on the expression of the five biomarker genes in iMNs to identify fast progressing sALS cases. Cross-validation of ROC results showed an average accuracy of 78.9% and precision of 80.1%, which indicates that the combination of the expression of these genes could potentially identify fast progressing sALS cases. This proof-of-concept assessment was performed on a relatively small dataset available from Answer ALS (n = 62), but we think that the encouraging results warrant the extension of these studies to larger datasets from patient-derived cells, when they become available.
In conclusion, multiomic analyses of patient-derived fibroblasts highlight differential metabolic and transcriptomic profiles in sALS metabotypes, which translate into differential responses to the investigational drug EH301. In the future, it will be important to apply similar multi-omics and machine learning strategies to readily available patient materials, such as white blood cells, to develop viable predictive biomarkers of disease progression and response to EH301 and other therapeutics.
Materials and Methods
Cell culture
A total of 18 primary fibroblasts lines derived from healthy donors or ALS patients (Table 5) were maintained in culture as previously reported (7). Sporadic ALS subclasses (sALS1 and sALS2) were defined based on previously published metabolic profiles (8). For experiments, cells were plated in Dulbecco’s modified Eagle’s medium (DMEM) medium containing 5 mM glucose and 2 mM glutamine, 10% fetal bovine serum (FBS), 1% of 100× antibiotic/antimycotic (sterile filtered 10 000 units of penicillin, 10 mg of streptomycin and 25 μg of amphotericin B per ml, and 2.5 μg/ml Plasmocin). Methionine and cystine depleted DMEM contained 10 mM glucose and 2 mM glutamine, 10% FBS, 1% of 100× antibiotic/antimycotic (sterile filtered 10 000 units of penicillin, 10 mg of streptomycin and 25 μg of amphotericin B per ml, and 2.5 μg/ml Plasmocin). Cells were assessed at the comparable passage number. EH301 was defined as the combination of 1 mM NR and 10 μM PT for the time specified in the text and figure legends.
Table 5.
Characteristics of sALS and control subjects at time of biopsy
| ID | Age at biopsy | Sex | Disease duration at time of skin biopsy (months) | ALSFRS-R total at biopsy | Rate of ALSFRS-R decline* | FVC% | |
|---|---|---|---|---|---|---|---|
| Control | 1 | 62 | F | ||||
| 2 | 63 | M | |||||
| 3 | 53 | F | |||||
| 4 | 79 | M | |||||
| 5 | 60 | F | |||||
| 6 | 73 | M | |||||
| sALS1 | 7 | 67 | F | 24 | 34 | 0.58 | 97 |
| 8 | 73 | M | 10 | 24 | 2.4 | 38 | |
| 9 | 67 | F | 14 | 35 | 0.93 | 78 | |
| 10 | 64 | M | 24 | 38 | 0.42 | 55 | |
| 11 | 66 | F | 5 | 40 | 1.6 | 118 | |
| 12 | 56 | M | 34 | 39 | 0.26 | 81 | |
| sALS2 | 13 | 72 | M | 12 | 39 | 0.75 | 105 |
| 14 | 73 | F | 6 | 39 | 1.5 | 59 | |
| 15 | 66 | F | 11 | 43 | 0.45 | 102 | |
| 16 | 68 | M | 32 | 32 | 0.5 | 78 | |
| 17 | 69 | F | 15 | 29 | 1.27 | 96 | |
| 18 | 55 | M | 10 | 33 | 1.5 | 116 |
a% Calculated as ((48−ALSFRS at skin BX)/disease duration at skin BX).
RNAseq and quantitative expression analysis
About 500 ng of total RNA extracted with TRIzol (Invitrogen) were used by the Weill Cornell Genomics Resources Core Facility to prepare 3’RNAseq libraries using the Lexogen QuantSeq 3’ mRNA-Seq Library Prep Kit forward (FWD) for Illumina. The libraries were quantified on a Molecular Devices Spectra Max M2 plate reader (with the intercalating dye QuantiFluor) and pooled accordingly for maximum evenness. The pool was quantified by digital PCR and sequenced on 1 lane of an Illumina NextSeq500 sequencer, single end 1 × 86 bp, and demultiplexed based upon six base i7 indices using Illumina bcl2fastq2 software (version 2.18; Illumina, Inc., San Diego, CA).
Illumina adapters were removed from the demultiplexed fastq files using Trimmomatic version 0.36 (86). The trimmed reads were aligned to the human genome assembly GRCh38.p13 using the STAR aligner version 2.7.0f (87). The output SAM files were converted to BAM using SAMtools version 1.8 (88), and the number of reads overlapping each gene in the gff3 file on the forward strand were counted using HTSeq-count version 0.6.1 (89). The R package DESeq2 version 1.24.0 (90) was used to obtain both normalized and variance stabilized counts and to identify genes that were differentially expressed between controls and sALS lines. Genes with less than 10 total raw reads were filtered out before running the DESeq2 model, and all other filtering parameters were kept as DESeq2’s defaults. Pathway analysis was performed with the free web tool WebGestalt (36), and the gprofiler2 (91) and clusterProfiler packages (92), and the cutoff for significance was a false discovery rate (FDR) corrected P-value <0.05.
Metabolomics
Targeted metabolomics was performed by the Proteomics and Metabolomics Core Facility at Weill Cornell Medicine in New York. Metabolites were rapidly extracted in 80% ice-cold methanol; samples were cleared by centrifugation at 14 000×g for 20 min at 4°C and stored at −80°C until analysis. The Weill Cornell Medicine Meyer Cancer Center Proteomics & Metabolomics Core Facility performed hydrophilic interaction liquid chromatography-mass spectrometry (MS) for relative quantification of polar metabolite profiles. Metabolites were measured on a Q Exactive Orbitrap mass spectrometer (Thermo Scientific), which was coupled to a Vanquish ultra-performance liquid chromatography (UPLC) system (Thermo Scientific) via an Ion Max ion source with a heated electrospray ionization (HESI) II probe (Thermo Scientific). A Sequant ZIC-pHILIC column (2.1 mm i.d. ×150 mm, particle size of 5 μm, Millipore Sigma) was used for separation. Total protein, determined by BCA Assay, was used for normalization, and MS peak data was processed using XCalibur 4.1 (Thermo Scientific) to obtain metabolite signal intensity for relative quantification. Relative abundance data were normalized with a log transformation and analyzed with the free online tool MetaboAnalyst 5.0 (93). Identification was done using an in-house library established with known chemical standards, and required exact mass (within 5 ppm) and standard retention times.
Cell viability
To measure viability, cells were plated in methionine and cystine depleted medium on 96 well glass bottom plates (Cellvis). After 72 h, cells were stained with HOECHST 33342 (Invitrogen), Calcein AM (Invitrogen), Propidium Iodide (Invitrogen) for 30 min at 37°C. Fluorescence was acquired with the ImageXpress Pico Automated Imaging System. Viability threshold was determined by a combination of calcein and propidium iodide signals as previously reported (94).
Weighted gene co-expression network analysis
WGCNA has been previously described (56,59). In brief, we used normalized gene expression data from DESEq2 as input for the functions included in the WGCNA package available from Comprehensive R Archive Network (CRAN). Induced motor neurons data was additionally normalized to remove batch effects using the limma: removeBatchEffect() function (95). We generated a TOM based on expression values from 17 662 genes in the fibroblast dataset and 22 663 genes in the iMN dataset and identified modules using a dynamic tree cutting algorithm based on hierarchical clustering of TOM dissimilarity values. After optimization to maintain scale-free topology, we set the ideal soft power threshold value at 4 for the vehicle network, 20 for the EH301 network, and 8 for the iMN network. To allow for direct comparisons between the vehicle and EH301 networks, quantile-quantile (Q-Q) scaling was performed such that the 95% quantiles of both matrices matched. For all networks the module merging parameter was kept consistent at 80%. Pairwise Pearson’s correlations were used to calculate associations between modules and disease traits. Pathway analysis was done in the same way as described for RNAseq. Hierarchical clustering of ALSFRS-R and gene expression was done using the dendextend package (96). Correlation between dendograms was calculated using Baker’s gamma index, and significance was determined using a permutation test (97). Logistic regression using the glm() function was used to calculate a model predicting ALSFRS-R progression. To evaluate model performance, we used a ten-fold cross validation approach, in which we randomly divided samples into ten sets. In each iteration, one set was used as the test data and the model was trained on the remaining 9 sets. Model performance was evaluated by calculating precision, accuracy, false positive rate and false negative rate with the caret package (98), and with receiver operating characteristic analysis done with the receiver operating characteristic R package (ROCR) package (99).
Statistical analyses
A Wald test was used to determine statistical significance of differential gene expression, with the cutoff being a False Discovery Rate of <5% after Benjamini-Hochberg correction. Significance for differential metabolite abundance was determined with one-way analysis of variance (ANOVA) with post-hoc t-tests, with the cutoff being a False Discovery Rate of <5% after Benjamini-Hochberg correction. All data visualization was done in R using the ggplot2, pheatmap, corrplot and venndiagram packages available from CRAN. Z scores were calculated from normalized counts for each gene using the standard formula (x−μ)/σ, where x is the sample value, μ is the population mean and σ is the population standard deviation.
Availability of data and materials
All R packages are available from CRAN. Full differential expression and gene ontology analysis results are included in Supplementary Material, Tables. Raw transcriptomic data will be made available from Gene Expression Omnibus. Transcriptomic data from induced motor neurons are available from Answer ALS.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank Elysium Health for providing N.R. and P.T. for this study. We thank Dr Hiroshi Mitsumoto (Columbia University) and the COSMOS initiative for providing the fibroblast and plasma samples utilized in this work. Fibroblast samples and clinical data were obtained from the NINDS Repository. We acknowledge the contribution of the Weill Cornell Genomics Resource Core Facility, and the Medicine Meyer Cancer Center Proteomics & Metabolomics Core Facility. Muscular Dystrophy Association (grant 602762 to C.K.).
Conflict of Interest statement. Drs Holly E. Holmes and Ryan W. Dellinger are employed by Elysium Health. The other authors do not have conflicts of interest to declare.
Contributor Information
Jasmine A Fels, Feil Family Brain and Mind Research Institute, Weill Cornell Medicine, 407 East 61st Street, New York, NY 10065, USA; Neuroscience Graduate Program, Weill Cornell Graduate School of Medical Sciences, 1300 York Ave, New York, NY 10065, USA.
Gabriella Casalena, Feil Family Brain and Mind Research Institute, Weill Cornell Medicine, 407 East 61st Street, New York, NY 10065, USA.
Csaba Konrad, Feil Family Brain and Mind Research Institute, Weill Cornell Medicine, 407 East 61st Street, New York, NY 10065, USA.
Holly E Holmes, Elysium Health New York, New York, NY 10013, USA.
Ryan W Dellinger, Elysium Health New York, New York, NY 10013, USA.
Giovanni Manfredi, Feil Family Brain and Mind Research Institute, Weill Cornell Medicine, 407 East 61st Street, New York, NY 10065, USA.
Funding
This work was supported by funds from ALS Association grant ALS-90452 (to G.M.), National Institute of Health/National Institute of Neurological Disorders and Stroke grant R35 NS122209-01 (to G.M.), and Muscular Dystrophy Association grant 602762 (to C.K.).
Declarations
Ethics approval and consent to participate
All human samples were obtained from repositories of de-identified cell lines. The academic centers that provided cell lines had approved IRB protocols in place.
References
- 1. Turner, M.R., Al-Chalabi, A., Chio, A., Hardiman, O., Kiernan, M.C., Rohrer, J.D., Rowe, J., Seeley, W. and Talbot, K. (2017) Genetic screening in sporadic ALS and FTD. J. Neurol. Neurosurg. Psychiatry, 88, 1042–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kiernan, M.C., Vucic, S., Talbot, K., McDermott, C.J., Hardiman, O., Shefner, J.M., Al-Chalabi, A., Huynh, W., Cudkowicz, M., Talman, P. et al. (2021) Improving clinical trial outcomes in amyotrophic lateral sclerosis. Nat. Rev. Neurol., 17, 104–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Huisman, M.H., Seelen, M., van Doormaal, P.T., de Jong, S.W., de Vries, J.H., van der Kooi, A.J., de Visser, M., Schelhaas, H.J., van den Berg, L.H. and Veldink, J.H. (2015) Effect of presymptomatic body mass index and consumption of fat and alcohol on amyotrophic lateral sclerosis. JAMA Neurol, 72, 1155–1162. [DOI] [PubMed] [Google Scholar]
- 4. Cistaro, A., Valentini, M.C., Chio, A., Nobili, F., Calvo, A., Moglia, C., Montuschi, A., Morbelli, S., Salmaso, D., Fania, P. et al. (2012) Brain hypermetabolism in amyotrophic lateral sclerosis: a FDG PET study in ALS of spinal and bulbar onset. Eur. J. Nucl. Med. Mol. Imaging, 39, 251–259. [DOI] [PubMed] [Google Scholar]
- 5. Steyn, F.J., Ioannides, Z.A., van Eijk, R.P.A., Heggie, S., Thorpe, K.A., Ceslis, A., Heshmat, S., Henders, A.K., Wray, N.R., van den Berg, L.H. et al. (2018) Hypermetabolism in ALS is associated with greater functional decline and shorter survival. J. Neurol. Neurosurg. Psychiatry, 89, 1016–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kirk, K., Gennings, C., Hupf, J.C., Tadesse, S., D'Aurelio, M., Kawamata, H., Valsecchi, F., Mitsumoto, H., Groups, A.P.C.S. and Manfredi, G. (2014) Bioenergetic markers in skin fibroblasts of sporadic amyotrophic lateral sclerosis and progressive lateral sclerosis patients. Ann. Neurol., 76, 620–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Konrad, C., Kawamata, H., Bredvik, K.G., Arreguin, A.J., Cajamarca, S.A., Hupf, J.C., Ravits, J.M., Miller, T.M., Maragakis, N.J., Hales, C.M. et al. (2017) Fibroblast bioenergetics to classify amyotrophic lateral sclerosis patients. Mol. Neurodegener., 12, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen, Q., Konrad, C., Sandhu, D., Roychoudhury, D., Schwartz, B.I., Cheng, R.R., Bredvik, K., Kawamata, H., Calder, E.L., Studer, L. et al. (2020) Accelerated transsulfuration metabolically defines a discrete subclass of amyotrophic lateral sclerosis patients. Neurobiol. Dis., 144, 105025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Belenky, P., Bogan, K.L. and Brenner, C. (2007) NAD+ metabolism in health and disease. Trends Biochem. Sci., 32, 12–19. [DOI] [PubMed] [Google Scholar]
- 10. Massudi, H., Grant, R., Braidy, N., Guest, J., Farnsworth, B. and Guillemin, G.J. (2012) Age-associated changes in oxidative stress and NAD+ metabolism in human tissue. PLoS One, 7, e42357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Harlan, B.A., Killoy, K.M., Pehar, M., Liu, L., Auwerx, J. and Vargas, M.R. (2020) Evaluation of the NAD(+) biosynthetic pathway in ALS patients and effect of modulating NAD(+) levels in hSOD1-linked ALS mouse models. Exp. Neurol., 327, 113219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Harlan, B.A., Pehar, M., Sharma, D.R., Beeson, G., Beeson, C.C. and Vargas, M.R. (2016) Enhancing NAD+ salvage pathway reverts the toxicity of primary astrocytes expressing amyotrophic lateral sclerosis-linked mutant superoxide dismutase 1 (SOD1). J. Biol. Chem., 291, 10836–10846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dellinger, R.W., Santos, S.R., Morris, M., Evans, M., Alminana, D., Guarente, L. and Marcotulli, E. (2017) Repeat dose NRPT (nicotinamide riboside and pterostilbene) increases NAD(+) levels in humans safely and sustainably: a randomized, double-blind, placebo-controlled study. NPJ Aging Mech. Dis., 3, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li, Y.R., Li, S. and Lin, C.C. (2018) Effect of resveratrol and pterostilbene on aging and longevity. Biofactors, 44, 69–82. [DOI] [PubMed] [Google Scholar]
- 15. Wang, P. and Sang, S. (2018) Metabolism and pharmacokinetics of resveratrol and pterostilbene. Biofactors, 44, 16–25. [DOI] [PubMed] [Google Scholar]
- 16. Gomez-Zorita, S., Milton-Laskibar, I., Aguirre, L., Fernandez-Quintela, A., Xiao, J. and Portillo, M.P. (2021) Effects of pterostilbene on diabetes, liver steatosis and serum lipids. Curr. Med. Chem., 28, 238–252. [DOI] [PubMed] [Google Scholar]
- 17. Freyssin, A., Page, G., Fauconneau, B. and Rioux Bilan, A. (2020) Natural stilbenes effects in animal models of Alzheimer's disease. Neural Regen. Res., 15, 843–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Obrador, E., Salvador, R., Marchio, P., Lopez-Blanch, R., Jihad-Jebbar, A., Rivera, P., Valles, S.L., Banacloche, S., Alcacer, J., Colomer, N. et al. (2020) Nicotinamide riboside and pterostilbene cooperatively delay motor neuron failure in ALS SOD1(G93A) mice. Mol. Neurobiol., 58, 1345–1371. [DOI] [PubMed] [Google Scholar]
- 19. de la Rubia, J.E., Drehmer, E., Platero, J.L., Benlloch, M., Caplliure-Llopis, J., Villaron-Casales, C., de Bernardo, N., AlarcOn, J., Fuente, C., Carrera, S. et al. (2019) Efficacy and tolerability of EH301 for amyotrophic lateral sclerosis: a randomized, double-blind, placebo-controlled human pilot study. Amyotroph Lateral Scler. Frontotemporal Degener., 20, 115–122. [DOI] [PubMed] [Google Scholar]
- 20. NO-ALS . https://clinicaltrials.gov/ct2/show/NCT04562831?term=eh301&cond=ALS&draw=2&rank=2.
- 21. Baxi, E.G., Thompson, T., Li, J., Kaye, J.A., Lim, R.G., Wu, J., Ramamoorthy, D., Lima, L., Vaibhav, V., Matlock, A. et al. (2022) Answer ALS, a large-scale resource for sporadic and familial ALS combining clinical and multi-omics data from induced pluripotent cell lines. Nat. Neurosci., 25, 226–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Klim, J.R., Williams, L.A., Limone, F., Guerra San Juan, I., Davis-Dusenbery, B.N., Mordes, D.A., Burberry, A., Steinbaugh, M.J., Gamage, K.K., Kirchner, R. et al. (2019) ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nat. Neurosci., 22, 167–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Theunissen, F., Anderton, R.S., Mastaglia, F.L., Flynn, L.L., Winter, S.J., James, I., Bedlack, R., Hodgetts, S., Fletcher, S., Wilton, S.D. et al. (2021) Novel STMN2 variant linked to amyotrophic lateral sclerosis risk and clinical phenotype. Front. Aging Neurosci., 13, 658226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kanai, Y., Okada, Y., Tanaka, Y., Harada, A., Terada, S. and Hirokawa, N. (2000) KIF5C, a novel neuronal kinesin enriched in motor neurons. J. Neurosci., 20, 6374–6384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Padzik, A., Deshpande, P., Hollos, P., Franker, M., Rannikko, E.H., Cai, D., Prus, P., Magard, M., Westerlund, N., Verhey, K.J. et al. (2016) KIF5C S176 Phosphorylation regulates microtubule binding and transport efficiency in mammalian neurons. Front. Cell. Neurosci., 10, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Poirier, K., Lebrun, N., Broix, L., Tian, G., Saillour, Y., Boscheron, C., Parrini, E., Valence, S., Pierre, B.S., Oger, M. et al. (2013) Mutations in TUBG1, DYNC1H1, KIF5C and KIF2A cause malformations of cortical development and microcephaly. Nat. Genet., 45, 639–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Michels, S., Foss, K., Park, K., Golden-Grant, K., Saneto, R., Lopez, J. and Mirzaa, G.M. (2017) Mutations of KIF5C cause a neurodevelopmental disorder of infantile-onset epilepsy, absent language, and distinctive malformations of cortical development. Am. J. Med. Genet. A, 173, 3127–3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nishimura, A.L., Mitne-Neto, M., Silva, H.C., Richieri-Costa, A., Middleton, S., Cascio, D., Kok, F., Oliveira, J.R., Gillingwater, T., Webb, J. et al. (2004) A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am. J. Hum. Genet., 75, 822–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kuijpers, M., Yu, K.L., Teuling, E., Akhmanova, A., Jaarsma, D. and Hoogenraad, C.C. (2013) The ALS8 protein VAPB interacts with the ER-Golgi recycling protein YIF1A and regulates membrane delivery into dendrites. EMBO J., 32, 2056–2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tessadori, F., Giltay, J.C., Hurst, J.A., Massink, M.P., Duran, K., Vos, H.R., van Es, R.M., Deciphering Developmental Disorders, S, Scott, R.H., van Gassen, K.L.I. et al. (2017) Germline mutations affecting the histone H4 core cause a developmental syndrome by altering DNA damage response and cell cycle control. Nat. Genet., 49, 1642–1646. [DOI] [PubMed] [Google Scholar]
- 31. Byun, J.S., Oh, M., Lee, S., Gil, J.E., Mo, Y., Ku, B., Kim, W.K., Oh, K.J., Lee, E.W., Bae, K.H. et al. (2020) The transcription factor PITX1 drives astrocyte differentiation by regulating the SOX9 gene. J. Biol. Chem., 295, 13677–13690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sun, W., Cornwell, A., Li, J., Peng, S., Osorio, M.J., Aalling, N., Wang, S., Benraiss, A., Lou, N., Goldman, S.A. et al. (2017) SOX9 Is an astrocyte-specific nuclear marker in the adult brain outside the neurogenic regions. J. Neurosci., 37, 4493–4507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang, Q., Ragnauth, C.D., Skepper, J.N., Worth, N.F., Warren, D.T., Roberts, R.G., Weissberg, P.L., Ellis, J.A. and Shanahan, C.M. (2005) Nesprin-2 is a multi-isomeric protein that binds lamin and emerin at the nuclear envelope and forms a subcellular network in skeletal muscle. J. Cell Sci., 118, 673–687. [DOI] [PubMed] [Google Scholar]
- 34. Zhang, X., Xu, R., Zhu, B., Yang, X., Ding, X., Duan, S., Xu, T., Zhuang, Y. and Han, M. (2007) Syne-1 and Syne-2 play crucial roles in myonuclear anchorage and motor neuron innervation. Development, 134, 901–908. [DOI] [PubMed] [Google Scholar]
- 35. Rahman, S., Canny, M.D., Buschmann, T.A. and Latham, M.P. (2020) A survey of reported disease-related mutations in the MRE11-RAD50-NBS1 complex. Cell, 9, 1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liao, Y., Wang, J., Jaehnig, E.J., Shi, Z. and Zhang, B. (2019) WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res., 47, W199–W205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rizzo, F., Nizzardo, M., Vashisht, S., Molteni, E., Melzi, V., Taiana, M., Salani, S., Santonicola, P., Di Schiavi, E., Bucchia, M. et al. (2019) Key role of SMN/SYNCRIP and RNA-Motif 7 in spinal muscular atrophy: RNA-Seq and motif analysis of human motor neurons. Brain, 142, 276–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lelieveld, S.H., Reijnders, M.R., Pfundt, R., Yntema, H.G., Kamsteeg, E.J., de Vries, P., de Vries, B.B., Willemsen, M.H., Kleefstra, T., Lohner, K. et al. (2016) Meta-analysis of 2,104 trios provides support for 10 new genes for intellectual disability. Nat. Neurosci., 19, 1194–1196. [DOI] [PubMed] [Google Scholar]
- 39. Gillentine, M.A., Wang, T., Hoekzema, K., Rosenfeld, J., Liu, P., Guo, H., Kim, C.N., De Vries, B.B.A., Vissers, L., Nordenskjold, M. et al. (2021) Rare deleterious mutations of HNRNP genes result in shared neurodevelopmental disorders. Genome. Med., 13, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Busetto, V., Barbosa, I., Basquin, J., Marquenet, E., Hocq, R., Hennion, M., Paternina, J.A., Namane, A., Conti, E., Bensaude, O. et al. (2020) Structural and functional insights into CWC27/CWC22 heterodimer linking the exon junction complex to spliceosomes. Nucleic Acids Res., 48, 5670–5683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Moretti, J., Chastagner, P., Gastaldello, S., Heuss, S.F., Dirac, A.M., Bernards, R., Masucci, M.G., Israel, A. and Brou, C. (2010) The translation initiation factor 3f (eIF3f) exhibits a deubiquitinase activity regulating Notch activation. PLoS Biol., 8, e1000545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Elantak, L., Wagner, S., Herrmannova, A., Karaskova, M., Rutkai, E., Lukavsky, P.J. and Valasek, L. (2010) The indispensable N-terminal half of eIF3j/HCR1 cooperates with its structurally conserved binding partner eIF3b/PRT1-RRM and with eIF1A in stringent AUG selection. J. Mol. Biol., 396, 1097–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Young, D.J. and Guydosh, N.R. (2019) Hcr1/eIF3j Is a 60S ribosomal subunit recycling accessory factor in vivo. Cell Rep., 28, 39, e34–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nickel, W. and Rabouille, C. (2009) Mechanisms of regulated unconventional protein secretion. Nat. Rev. Mol. Cell Biol, 10, 148–155. [DOI] [PubMed] [Google Scholar]
- 45. Trychta, K.A., Back, S., Henderson, M.J. and Harvey, B.K. (2018) KDEL receptors are differentially regulated to maintain the ER proteome under calcium deficiency. Cell Rep., 25, 1829, e1826–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Annaert, W.G., Becker, B., Kistner, U., Reth, M. and Jahn, R. (1997) Export of cellubrevin from the endoplasmic reticulum is controlled by BAP31. J. Cell Biol., 139, 1397–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wakana, Y., Takai, S., Nakajima, K., Tani, K., Yamamoto, A., Watson, P., Stephens, D.J., Hauri, H.P. and Tagaya, M. (2008) Bap31 is an itinerant protein that moves between the peripheral endoplasmic reticulum (ER) and a juxtanuclear compartment related to ER-associated Degradation. Mol. Biol. Cell, 19, 1825–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Namba, T. (2019) BAP31 regulates mitochondrial function via interaction with Tom40 within ER-mitochondria contact sites. Sci. Adv., 5, eaaw1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jin, H., Komita, M. and Aoe, T. (2017) The role of BiP retrieval by the KDEL receptor in the early secretory pathway and its effect on protein quality control and neurodegeneration. Front. Mol. Neurosci., 10, 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Whalen, S., Shaw, M., Mignot, C., Heron, D., Bastaraud, S.C., Walti, C.C., Liebelt, J., Elmslie, F., Yap, P., Hurst, J. et al. (2021) Further delineation of BCAP31-linked intellectual disability: description of 17 new families with LoF and missense variants. Eur. J. Hum. Genet., 29, 1405–1417 in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dominissini, D., Nachtergaele, S., Moshitch-Moshkovitz, S., Peer, E., Kol, N., Ben-Haim, M.S., Dai, Q., Di Segni, A., Salmon-Divon, M., Clark, W.C. et al. (2016) The dynamic N(1)-methyladenosine methylome in eukaryotic messenger RNA. Nature, 530, 441–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhang, C. and Jia, G. (2018) Reversible RNA modification N(1)-methyladenosine (m(1)A) in mRNA and tRNA. Genomics Proteomics Bioinformatics, 16, 155–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Li, X., Xiong, X., Zhang, M., Wang, K., Chen, Y., Zhou, J., Mao, Y., Lv, J., Yi, D., Chen, X.W. et al. (2017) Base-resolution mapping reveals distinct m(1)A methylome in nuclear- and mitochondrial-encoded transcripts. Mol. Cell, 68, 993, e1009–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sattler, R. and Rothstein, J.D. (2006) Regulation and dysregulation of glutamate transporters. Handb. Exp. Pharmacol, 175, 277–303. [DOI] [PubMed] [Google Scholar]
- 55. Sears, T.K. and Angelastro, J.M. (2017) The transcription factor ATF5: role in cellular differentiation, stress responses, and cancer. Oncotarget, 8, 84595–84609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Langfelder, P. and Horvath, S. (2008) WGCNA: an R package for weighted correlation network analysis. BMC Bioinform., 9, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhang, B. and Horvath, S. (2005) A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol., 4, Article17. [DOI] [PubMed] [Google Scholar]
- 58. Gaiteri, C., Ding, Y., French, B., Tseng, G.C. and Sibille, E. (2014) Beyond modules and hubs: the potential of gene coexpression networks for investigating molecular mechanisms of complex brain disorders. Genes Brain Behav., 13, 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Langfelder, P., Luo, R., Oldham, M.C. and Horvath, S. (2011) Is my network module preserved and reproducible? PLoS Comput. Biol., 7, e1001057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bernardini, C., Censi, F., Lattanzi, W., Barba, M., Calcagnini, G., Giuliani, A., Tasca, G., Sabatelli, M., Ricci, E. and Michetti, F. (2013) Mitochondrial network genes in the skeletal muscle of amyotrophic lateral sclerosis patients. PLoS One, 8, e57739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Taghibiglou, C., Lu, J., Mackenzie, I.R., Wang, Y.T. and Cashman, N.R. (2011) Sterol regulatory element binding protein-1 (SREBP1) activation in motor neurons in excitotoxicity and amyotrophic lateral sclerosis (ALS): Indip, a potential therapeutic peptide. Biochem. Biophys. Res. Commun., 413, 159–163. [DOI] [PubMed] [Google Scholar]
- 62. Dangoumau, A., Marouillat, S., Coelho, R., Wurmser, F., Brulard, C., Haouari, S., Laumonnier, F., Corcia, P., Andres, C.R., Blasco, H. et al. (2021) Dysregulations of expression of genes of the Ubiquitin/SUMO pathways in an in vitro model of amyotrophic lateral sclerosis combining oxidative stress and SOD1 gene mutation. Int. J. Mol. Sci., 22, 1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Morahan, J.M., Yu, B., Trent, R.J. and Pamphlett, R. (2007) Genetic susceptibility to environmental toxicants in ALS. Am. J. Med. Genet. B Neuropsychiatr. Genet., 144B, 885–890. [DOI] [PubMed] [Google Scholar]
- 64. Kim, K.Y., Kim, Y.R., Choi, K.W., Lee, M., Lee, S., Im, W., Shin, J.Y., Kim, J.Y., Hong, Y.H., Kim, M. et al. (2020) Downregulated miR-18b-5p triggers apoptosis by inhibition of calcium signaling and neuronal cell differentiation in transgenic SOD1 (G93A) mice and SOD1 (G17S and G86S) ALS patients. Transl. Neurodegener., 9, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hu, Z., Snitkin, E.S. and DeLisi, C. (2008) VisANT: an integrative framework for networks in systems biology. Brief. Bioinform., 9, 317–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Aronica, E., Baas, F., Iyer, A., ten Asbroek, A.L., Morello, G. and Cavallaro, S. (2015) Molecular classification of amyotrophic lateral sclerosis by unsupervised clustering of gene expression in motor cortex. Neurobiol. Dis., 74, 359–376. [DOI] [PubMed] [Google Scholar]
- 67. Wang, J.C., Ramaswami, G. and Geschwind, D.H. (2021) Gene co-expression network analysis in human spinal cord highlights mechanisms underlying amyotrophic lateral sclerosis susceptibility. Sci. Rep., 11, 5748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Mostafavi, S., Gaiteri, C., Sullivan, S.E., White, C.C., Tasaki, S., Xu, J., Taga, M., Klein, H.U., Patrick, E., Komashko, V. et al. (2018) A molecular network of the aging human brain provides insights into the pathology and cognitive decline of Alzheimer's disease. Nat. Neurosci., 21, 811–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sun, Y., Lin, J. and Zhang, L. (2019) The application of weighted gene co-expression network analysis in identifying key modules and hub genes associated with disease status in Alzheimer's disease. Ann. Transl. Med, 7, 800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Park, D.S., Morris, E.J., Padmanabhan, J., Shelanski, M.L., Geller, H.M. and Greene, L.A. (1998) Cyclin-dependent kinases participate in death of neurons evoked by DNA-damaging agents. J. Cell Biol., 143, 457–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Zhang, Y., Song, X. and Herrup, K. (2020) Context-dependent functions of E2F1: cell cycle, cell death, and DNA damage repair in cortical neurons. Mol. Neurobiol., 57, 2377–2390. [DOI] [PubMed] [Google Scholar]
- 72. Ranganathan, S. and Bowser, R. (2003) Alterations in G(1) to S phase cell-cycle regulators during amyotrophic lateral sclerosis. Am. J. Pathol., 162, 823–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Joseph, C., Mangani, A.S., Gupta, V., Chitranshi, N., Shen, T., Dheer, Y., Kb, D., Mirzaei, M., You, Y., Graham, S.L. et al. (2020) Cell cycle deficits in neurodegenerative disorders: uncovering molecular mechanisms to drive innovative therapeutic development. Aging Dis., 11, 946–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Denechaud, P.D., Fajas, L. and Giralt, A. (2017) E2F1, a novel regulator of metabolism. Front Endocrinol. (Lausanne), 8, 311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ryu, H., Lee, J., Zaman, K., Kubilis, J., Ferrante, R.J., Ross, B.D., Neve, R. and Ratan, R.R. (2003) Sp1 and Sp3 are oxidative stress-inducible, antideath transcription factors in cortical neurons. J. Neurosci., 23, 3597–3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Garcia-Morales, V., Rodriguez-Bey, G., Gomez-Perez, L., Dominguez-Vias, G., Gonzalez-Forero, D., Portillo, F., Campos-Caro, A., Gento-Caro, A., Issaoui, N., Soler, R.M. et al. (2019) Sp1-regulated expression of p11 contributes to motor neuron degeneration by membrane insertion of TASK1. Nat. Commun., 10, 3784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Rotheneder, H., Geymayer, S. and Haidweger, E. (1999) Transcription factors of the Sp1 family: interaction with E2F and regulation of the murine thymidine kinase promoter. J. Mol. Biol., 293, 1005–1015. [DOI] [PubMed] [Google Scholar]
- 78. Zhang, X., Song, S. and Peng, W. (2021) Cell cycle deregulation in neurodegenerative diseases. Int. J. Neurosci., in press., 1–15. [DOI] [PubMed] [Google Scholar]
- 79. Nicolas, A., Kenna, K.P., Renton, A.E., Ticozzi, N., Faghri, F., Chia, R., Dominov, J.A., Kenna, B.J., Nalls, M.A., Keagle, P. et al. (2018) genome-wide analyses identify KIF5A as a novel ALS gene. Neuron, 97, 1268, e1266–e1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Morello, G., Guarnaccia, M., Spampinato, A.G., Salomone, S., D'Agata, V., Conforti, F.L., Aronica, E. and Cavallaro, S. (2019) Integrative multi-omic analysis identifies new drivers and pathways in molecularly distinct subtypes of ALS. Sci. Rep., 9, 9968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Veyrat-Durebex, C., Bris, C., Codron, P., Bocca, C., Chupin, S., Corcia, P., Vourc'h, P., Hergesheimer, R., Cassereau, J., Funalot, B. et al. (2019) Metabo-lipidomics of fibroblasts and mitochondrial-endoplasmic reticulum extracts from ALS patients shows alterations in purine, pyrimidine, energetic, and phospholipid metabolisms. Mol. Neurobiol., 56, 5780–5791. [DOI] [PubMed] [Google Scholar]
- 82. Fishel, M.L., Vasko, M.R. and Kelley, M.R. (2007) DNA repair in neurons: so if they don't divide what's to repair? Mutat. Res., 614, 24–36. [DOI] [PubMed] [Google Scholar]
- 83. Penndorf, D., Witte, O.W. and Kretz, A. (2018) DNA plasticity and damage in amyotrophic lateral sclerosis. Neural Regen. Res., 13, 173–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Schwartz, E.I., Smilenov, L.B., Price, M.A., Osredkar, T., Baker, R.A., Ghosh, S., Shi, F.D., Vollmer, T.L., Lencinas, A., Stearns, D.M. et al. (2007) Cell cycle activation in postmitotic neurons is essential for DNA repair. Cell Cycle, 6, 318–329. [DOI] [PubMed] [Google Scholar]
- 85. Coppede, F. (2011) An overview of DNA repair in amyotrophic lateral sclerosis. Sci. World J., 11, 1679–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Bolger, A.M., Lohse, M. and Usadel, B. (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics, 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Dobin, A., Davis, C.A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., Batut, P., Chaisson, M. and Gingeras, T.R. (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics, 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., Marth, G., Abecasis, G., Durbin, R. and Genome Project Data Processing, S (2009) The sequence alignment/map format and SAMtools. Bioinformatics, 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Anders, S., Pyl, P.T. and Huber, W. (2015) HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics, 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Love, M.I., Huber, W. and Anders, S. (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol., 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Xia, J., Psychogios, N., Young, N. and Wishart, D.S. (2009) MetaboAnalyst: a web server for metabolomic data analysis and interpretation. Nucleic Acids Res., 37, W652–W660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Wu, T., Hu, E., Xu, S., Chen, M., Guo, P., Dai, Z., Feng, T., Zhou, L., Tang, W., Zhan, L. et al. (2021) clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation (N Y), 2, 100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Raudvere, U., Kolberg, L., Kuzmin, I., Arak, T., Adler, P., Peterson, H. and Vilo, J. (2019) g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res., 47, W191–W198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Granatiero, V., Sayles, N.M., Savino, A.M., Konrad, C., Kharas, M.G., Kawamata, H. and Manfredi, G. (2021) Modulation of the IGF1R-MTOR pathway attenuates motor neuron toxicity of human ALS SOD1(G93A) astrocytes. Autophagy, 17, 4029–4042, in press, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Ritchie, M.E., Phipson, B., Wu, D., Hu, Y., Law, C.W., Shi, W. and Smyth, G.K. (2015) limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res., 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Galili, T. (2015) dendextend: an R package for visualizing, adjusting and comparing trees of hierarchical clustering. Bioinformatics, 31, 3718–3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Baker, F.B. and Hubert, L.J. (1975) Measuring the power of hierarchical cluster analysis. J. Am. Calif. Assoc., 70, 31–38. [Google Scholar]
- 98. Kuhn, M. (2012) The caret Package. J. Stat. Softw., 28, 1–26. [Google Scholar]
- 99. Sing, T., Sander, O., Beerenwinkel, N. and Lengauer, T. (2005) ROCR: visualizing classifier performance in R. Bioinformatics, 21, 3940–3941. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All R packages are available from CRAN. Full differential expression and gene ontology analysis results are included in Supplementary Material, Tables. Raw transcriptomic data will be made available from Gene Expression Omnibus. Transcriptomic data from induced motor neurons are available from Answer ALS.