

Abstract
Although autism is typically characterized by differences in language, social interaction and restrictive, repetitive behaviors, it is becoming more well known in the field that alterations in energy metabolism and mitochondrial function are comorbid disorders in autism. The synaptic cell adhesion molecule, neurexin-1 (NRXN1), has previously been implicated in autism, and here we show that in Drosophila melanogaster, the homologue of NRXN1, called Nrx-1, regulates energy metabolism and nutrient homeostasis. First, we show that Nrx-1-null flies exhibit decreased resistance to nutrient deprivation and heat stress compared to controls. Additionally, Nrx-1 mutants exhibit a significantly altered metabolic profile characterized by decreased lipid and carbohydrate stores. Nrx-1-null Drosophila also exhibit diminished levels of nicotinamide adenine dinucleotide (NAD+), an important coenzyme in major energy metabolism pathways. Moreover, loss of Nrx-1 resulted in striking abnormalities in mitochondrial morphology in the flight muscle of Nrx-1-null Drosophila and impaired flight ability in these flies. Further, following a mechanical shock Nrx-1-null flies exhibited seizure-like activity, a phenotype previously linked to defects in mitochondrial metabolism and a common symptom of patients with NRXN1 deletions. The current studies indicate a novel role for NRXN1 in the regulation of energy metabolism and uncover a clinically relevant seizure phenotype in Drosophila lacking Nrx-1.
Introduction
Autism spectrum disorder, henceforth ‘autism,’ is a neurodevelopmental condition that affects 1 in 44 children in the United States, and is mainly characterized by differences in language, socialization and sensory perception. In addition to these core symptoms, co-morbid disorders such as sleep disruptions, seizures and increased stress and anxiety are also common in autistic individuals (1–10). Furthermore, alterations in energy metabolism and mitochondrial functioning have been identified in autistic individuals in several biochemical, anatomical and neuroimaging studies. For example, neuroimaging studies using positron emission tomography and nuclear magnetic resonance spectroscopy have shown decreased glucose utilization and reduced adenosine triphosphate (ATP) levels in the cerebral cortices of autistic individuals (11,12). A more recent meta-analysis found the prevalence of mitochondrial disease in autistic children to be 5.0%, which is much higher than the ~ 0.01% prevalence in the general population (13). Additionally, levels of biochemical markers of mitochondrial dysfunction, such as lactate, pyruvate, ubiquinone and carnitine, are significantly altered in autistic individuals compared to a control population (13–21). Further, there is evidence to suggest enhanced oxidative stress in autistic patients (22–25), which can lead to impairments in electron transport chain activity. In some autistic patients, these abnormalities in energy metabolism can be explained by mutations in mitochondrial or nuclear DNA; however, in most cases the cause for abnormal energy metabolism in autistic patients is unclear.
There is increasing evidence to show that disruptions in the gene, neurexin-1 (NRXN1), are associated with autism (26–34). NRXN1 deletions have also been associated with other neuropsychiatric conditions such as schizophrenia (35,36). Interestingly, there is evidence of altered brain energy metabolism in the pathophysiology of schizophrenia as well (37–39), suggesting a potential shared mechanism for metabolic deficits between autism and schizophrenia.
NRXN1 is a synaptic cell-adhesion molecule that is primarily expressed in the central and peripheral nervous systems and is involved in synaptic function and transmission (26,40). In vertebrates, there are three NRXN genes, NRXN1, NRXN2 and NRXN3, each of which have promoters for α, β and γ isoforms. NRXNs are also subject to extensive alternative splicing, resulting in hundreds of differentially spliced isoforms (41). Thus, understanding of how NRXN1 may be involved in disruptions in energy metabolism in autism and schizophrenia presents a challenge in vertebrates, with so many different isoforms of NRXN that may be functioning at the synapse.
Invertebrates such as Drosophila melanogaster only contain a single gene encoding, α-neurexin, Nrx-1, which is not subject to alternative splicing (41), lending a simplified system in which we can study how Nrx-1 may regulate energy metabolism. Additionally, the Nrx-1 protein has an identical domain structural organization to vertebrate α-neurexins (41). Researchers studying Nrx-1 in flies have identified that loss of Nrx-1 expression results in defects at the synapse, including abnormal pre- and postsynaptic structure, altered bouton number and impaired synaptic transmission (42–44). Nrx-1-null Drosophila also exhibit behavioral phenotypes that are pertinent to autism, such as disruptions in sleep and associative learning (45–47). Together, these findings indicate that the Nrx-1 mutant fly model exhibits physiological and behavioral phenotypes relevant to autism and is a valuable tool to further investigate how metabolic alterations may contribute to autism.
In this study, we show that Nrx-1-null mutants display decreased resistance to starvation stress and heat stress, which led us to question the metabolic status of Nrx-1-null mutants. We demonstrate through metabolomics and complimentary colorimetric assays that Nrx-1 mutant flies display a shift in metabolic signature, in addition to decreased lipid and carbohydrate energy stores. Additionally, we show that Nrx-1-null Drosophila exhibit increased levels of tryptophan and kynurenine and decreased levels of nicotinamide adenine dinucleotide (NAD+) and a diminished NAD+/NADH ratio. Further, Nrx-1 mutants show severe morphological disruptions in flight muscle mitochondria and impaired flight ability. Finally, Nrx-1 also exhibits seizure-like activity following an acute mechanical stimulation, which is reminiscent of clinical data showing seizures in human patients with NRXN1 deletions (48–54). Together, these findings illustrate a novel role for Nrx-1 in the regulation of energy metabolism in Drosophila and identify a clinically relevant seizure phenotype.
Results
Nrx-1 mutants show decreased resistance to starvation and heat stress
We first set out to investigate the role of Nrx-1 in regulating or responding to environmental stress, as it relates to stress and anxiety in autism (8). Specifically, we queried whether Nrx-1-null flies could withstand environmental stressors, in this case starvation stress and HS, compared to iso31Bw- (wild-type) conspecifics. For these studies, we utilized two null mutant alleles, Nrx-1273 and Nrx-1241 (42), and after six generation out-crosses with the iso31Bw- strain all homozygous Nrx-1 mutants were viable. To avoid potential confounds due to age or sex, we limited our studies to 3- to 5-day-old male flies.
For our starvation stress experiments, we recorded the survival time of flies of each genotype on a 2% agar medium. The 2% agar medium, while providing zero nutritional value, allowed for a source of water and eliminated desiccation as a cause of mortality. These experiments were performed using the Drosophila Activity Monitoring (DAM) system, and thus we determined the time of death for each fly by identifying the last recorded activity count. We found that the Nrx-1 mutants died significantly faster than their wild-type counterparts when deprived of nutrients (Fig. 1A). As an added control, we replicated these experiments using transheterozygous Nrx-1273/241 mutant flies to ensure that the decreased starvation resistance that we observed in the homozygous Nrx-1 mutants was not due to any extraneous background mutation in either allele. The Nrx-1273/241 transheterozygotes also exhibited significantly decreased resistance to starvation stress compared to control flies (Supplementary Material, Fig. S1A), indicating that this increased sensitivity to stressors is a result of loss of Nrx-1. This finding led us to wonder whether Nrx-1 mutants are hypersensitive to stressors other than starvation stress, so we also assessed survival of Nrx-1 mutants after heat stress. To measure the response of the Nrx-1 mutants and controls flies to heat stress, we exposed flies of each genotype to 37°C for 1 h, returned all flies to 25°C after heat exposure, and then assessed for survival 24 h later (55). We found that the Nrx-1 mutants had significantly decreased survival rates following acute high heat exposure (Fig. 1B). This finding, combined with our starvation stress experiments, suggests that Nrx-1 mutants are hypersensitive to environmental stressors, such as nutrient deprivation and heat stress.
Figure 1.

Nrx-1 mutants have decreased resistance to environmental stressors, such as starvation stress and heat stress. (A) Survival curve of Nrx-1 mutants (red and blue lines) and wild-type iso31Bw- controls (black line) during starvation. Log-rank (Mantel–Cox) test with Bonferroni-corrected significance threshold for multiple comparisons indicated that Nrx-1273/273 and Nrx-1241/241 mutant flies die significantly faster than wild-type flies when on media containing 0% sucrose and 2% agar. Nrx-1273/273 and Nrx-1241/241 were not significantly different from each other in starvation resistance. Sample number (N) per genotype: iso31Bw- N = 64, Nrx-1273/273 N = 61, Nrx-1241/241 N = 58. (B) Survival curve of each genotype 24 h after an acute HS of exposure to 37°C for 1 h. Log-rank (Mantel-Cox) test with Bonferroni-corrected significance threshold for multiple comparisons results indicated that Nrx-1273/273 and Nrx-1241/241 mutant flies have significantly reduced resistance to HS compared to wild-type flies. Additionally, Nrx-1241/241 flies were more sensitive to HS than Nrx-1273/273 flies. Sample number (N) per genotype: iso31Bw- N = 280, Nrx-1273/273 N = 270, Nrx-1241/241 N = 270. Values represent mean ± SEM. ** = P < 0.01. **** = P < 0.0001.
Together, these data show that Nrx-1 mutants have decreased resistance to nutrient deprivation and heat stress, which indicates potential alterations in energy metabolism in the Nrx-1 mutants. Survival during starvation has shown to be influenced by lipid and glycogen content (56,57). Additionally, survival following heat exposure also has metabolic implications as it has been shown that HS causes significant depletion of metabolites involved in energy metabolism, such as glycogen and triglycerides (58–60). Thus, it follows that an organism with an altered metabolic profile characterized by decreased energy stores would have decreased survival rates when undergoing nutrient deprivation or HS. Therefore, our findings led us to hypothesize that the Nrx-1 mutants have an altered metabolic profile.
Nrx-1 mutants exhibit metabolic alterations, characterized by decreased lipids and carbohydrates
To understand whether altered metabolic homeostasis underlies the decreased resistance to nutrient deprivation and heat stress in the Nrx-1 mutants, we began with a broad approach by performing a metabolomics screen for primary metabolite and complex lipid levels. Six independent samples of 40 whole flies per genotype were analyzed for primary metabolites and complex lipids by mass spectrometry. From the resultant datasets, we used principle component analysis to determine whether we could segregate the Nrx-1 mutants from their wild-type conspecifics based solely on their primary metabolite and complex lipid metabolite signature. We found that the PCA plots for both primary metabolism and complex lipids showed a clear separation of iso31Bw- from both Nrx-1273/273 and Nrx-1241/241 mutants, whereas the Nrx-1 mutants still overlapped with each other (Fig. 2A and B). These data indicate a significant shift in primary metabolites and complex lipid metabolites in the Nrx-1 mutants relative to control animals.
Figure 2.

Nrx-1 mutants exhibit an altered biochemical signature, with decreased lipids and carbohydrates. Principal component analysis of (A) primary metabolites and (B) complex lipids of 3- to 5-day-old iso31Bw- control (red), Nrx-1241/241 (green), and Nrx-1273/273 (blue) flies reveals a clear shift in (A) primary metabolite concentrations and (B) lipid levels between the Nrx-1 mutant flies and iso31Bw- flies. Shown are the biplot of the first two components extracted from a principal component analysis, with point colors indicating group membership (red = iso31Bw-, green = Nrx-1241/241, blue = Nrx-1273/273) and normal data ellipses for each group. (C) Quantification of triglycerides in 3- to 5-day-old adult male flies. Each data point represents an independent sample of 3 fly bodies. One-way ANOVA with Tukey’s multiple comparisons post-hoc test indicated that Nrx-1-null mutant flies have decreased triglycerides (P < 0.0001) compared to iso31Bw- control flies. Sample number (N) per genotype: iso31Bw- N = 12, Nrx-1273/273 N = 14, Nrx-1241/241 N = 14. (D) Quantification of glucose levels using GC/TOF MS reveals that the Nrx-1 mutant flies have reduced glucose levels relative to the iso31w- mutant flies. The box plot depicts the autoscaled peak height of glucose on the y-axis. Mann–Whitney U test revealed that both Nrx-1273/273 (P = 0.021421) and Nrx-1241/241 (P = 0.019413) flies have decreased glucose levels compared to iso31Bw- flies. Six samples of 40 flies for each genotype were used for the analysis. (E) Quantification of glycogen in 3- to 5-day-old adult male flies. Each data point represents an independent sample of 4 fly bodies. One-way ANOVA with Tukey’s multiple comparisons post-hoc test indicated that Nrx-1-null mutant flies have decreased glycogen stores (P < 0.0001) compared to wild-type conspecifics. Sample number (N) per genotype: iso31Bw- N = 15, Nrx-1273/273 N = 15, Nrx-1241/241 N = 14. (F) Average amount of food consumed by flies of each genotype after an 18-h overnight fasting period. Each data point represents an independent sample of 5 whole flies. One-way ANOVA with Tukey’s multiple comparisons post-hoc test showed that Nrx-1-null mutant flies eat the same amount of food as iso31Bw- control flies (P = 0.6406). Sample number (N) per genotype: iso31Bw- N = 14, Nrx-1273/273 N = 14, Nrx-1241/241 N = 14. (G) Average activity during wake is shown for individual flies of each genotype. Kruskal–Wallis test with Dunn’s multiple comparisons test indicated that both Nrx-1273/273 flies (P = 0.0001) and Nrx-1241/241 flies (P < 0.0001) show decreased activity during wake compared to iso31Bw- control flies. Sample number (N) per genotype: iso31Bw- N = 51, Nrx-1273/273 N = 52, Nrx-1241/241 N = 56. (H) Average activity counts per 24-h day are shown for individual flies of each genotype, values shown are the average of 3 days of activity data combined. One-way ANOVA with Tukey’s multiple comparisons post-hoc test revealed that Nrx-1273/273 mutant flies show increased daily activity (P = 0.0003), whereas Nrx-1241/241 flies show no difference in daily activity (P > 0.9999) compared to iso31Bw- control flies. Sample number (N) per genotype: iso31Bw- N = 53, Nrx-1273/273 N = 53, Nrx-1241/241 N = 58. Values represent mean ± SEM.
We next performed a more detailed review of the known metabolites that were significantly altered in both the Nrx-1273/273 and Nrx-1241/241 mutants compared to wild-type flies according to a Mann–Whitney U test with false discovery rate corrected P-value. Within this analysis, we were intrigued to find that all but one of the glycerolipids that were included in the analysis was significantly decreased in the Nrx-1 mutants relative to iso31Bw- controls (Table 1). In contrast, other lipid classes had a mix of lipids that were increased or decreased in the Nrx-1 mutants. Glycerolipids are a large group of molecules that includes triglycerides, which represent over 90% of stored lipids in Drosophila. Importantly, triglycerides have previously been crucial for survival during starvation and heat stress (56–60). We sought to validate this finding of decreased glycerolipids by performing an additional colorimetric assay to measure triglyceride levels in the bodies of Nrx-1 mutants and iso31Bw- controls. We chose to conduct these experiments using decapitated bodies only, as opposed to whole bodies, for a few reasons. First, eye pigment has the potential to interfere with accurate absorbance measures in these colorimetric assays (61). Second, the primary tissue for the storage of fuel molecules, such as triglycerides and glycogen, is the fat body in Drosophila. Additionally, there is important crosstalk between central and peripheral tissues in the regulation of energy metabolism (62–65). Thus, we were interested in whether loss of Nrx-1 leads to systemic alterations in nutrient homeostasis. Consistent with the observed decrease in glycerolipids in our metabolomics experiments, we found that Nrx-1273/273 and Nrx-1241/241 homozygous mutants, as well as Nrx-1273/241 transheterozygous mutants, exhibit significantly diminished triglyceride levels compared to control animals (Fig. 2C and Supplementary Material, Fig. S1B).
Table 1.
Significantly altered complex lipids in Nrx-1 mutants. We analyzed 3- to 5-day-old adult male flies of each genotype for complex lipids by the CSH-QTOF MS/MS. Six samples of 40 flies for each genotype were used for the analysis. Table only includes known lipids that were significantly different (P < 0.05) in both the Nrx-1273/273 and Nrx-1241/241 groups when compared to iso31Bw- controls by a Mann–Whitney U test with false discovery rate corrected P-value. Fold change, calculated as FC = median (metabolite in group 1)/median (metabolite in group 2), is listed for Nrx-1273/273 versus iso31Bw-, and Nrx-1241/241 versus iso31Bw-. Boxes shaded in green indicate a significant decrease in lipid levels in Nrx-1 mutants, and boxes shaded in red indicate a significant increase in lipid levels in Nrx-1 mutants. A more detailed version of Table 1 can be viewed in the Supplemental Material, Table S1
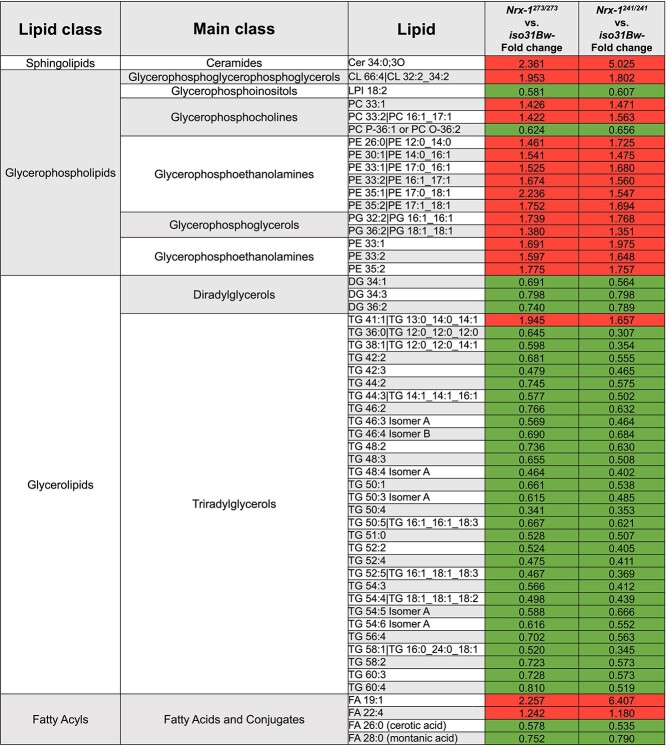
|
In addition to diminished glycerolipids, our metabolomics experiments also revealed that Nrx-1 mutants have decreased glucose levels compared to wild-type conspecifics (Fig. 2D). Although glucose is not the primary insect blood sugar, it is the precursor for glycogen, which is the primary form of carbohydrate storage in Drosophila (66). As mentioned previously, glycogen content is important for survival during nutrient deprivation and heat stress. Given that glucose was decreased in the Nrx-1 mutants, we sought to uncover whether glycogen was also diminished using a colorimetric assay with Nrx-1 mutant and wild-type bodies. Consistent with our data showing decreased glucose levels, these colorimetric experiments revealed that Nrx-1 mutants have significantly diminished glycogen levels compared to wild-type controls (Fig. 2E).
Taken together, these findings suggest that Nrx-1 plays a role in the regulation of lipid and carbohydrate metabolism. Although there are several possible hypotheses that could explain how Nrx-1 may regulate energy metabolism, we first began to gather clues to these questions by assessing feeding and activity levels in the Nrx-1 mutant flies. To assess feeding levels, we placed fasted flies on cornmeal-molasses medium that was spiked with Brilliant Blue FCF, and then performed spectrophotometry on samples from whole flies to determine the amount of labeled food ingested. For these experiments, we chose to starve the flies for 18 h before the feeding period as it has been shown that after prolonged periods of starvation, feeding is driven by taste-independent mechanisms (67); thus, we can avoid potential confounds due to taste perception. We ultimately found no significant difference in feeding levels between the Nrx-1 mutants and their wild-type controls (Fig. 2F), suggesting that reduced energy stores cannot be attributed to decreased food intake. Next, we considered the hypothesis that Nrx-1 mutants are hyperactive and thereby burn through their lipid and carbohydrate stores faster than control flies. To determine daily activity levels of the Nrx-1 mutants, we used the DAM System. We analyzed our data in terms of waking activity and average daily activity counts. These measures differ in that the waking activity is a measure of how active a fly is during wake times, whereas average daily activity count is a 3-day average of the total activity throughout the course of an entire 24-h day. These experiments revealed that both Nrx-1273/273 and Nrx-1241/241 mutants actually display decreased waking activity compared to iso31Bw- controls (Fig. 2G). Conversely, in terms of average total daily activity the Nrx-1273/273 mutant flies exhibit increased total activity counts, and the Nrx-1241/241 flies show no difference in activity compared to control flies (Fig. 2H). Although the Nrx-1273/273 mutants showed increased average daily activity (Fig. 2H), we believe this can be attributed to decreased and fragmented sleep in these flies lending to more time awake and therefore an increase in daily activity counts (Supplementary Material, Fig. S2). Additionally, the fact that we only see an increase in average daily activity counts in the Nrx-1273/273 mutants and not the Nrx-1241/241 mutants suggests that increased activity alone cannot explain the decreased lipid and carbohydrates seen in both Nrx-1 mutant alleles. Thus, it appears that decreased lipid and carbohydrate stores cannot be fully explained by decreased feeding or hyperactivity, as the Nrx-1 mutants exhibit normal feeding levels and show decreased waking activity compared to controls. Additionally, these alterations in energy stores cannot be attributed to body size as the Nrx-1 mutants have similar body weight compared to controls (Supplementary Material, Fig.S3).
Nrx-1 mutants show altered tryptophan, kynurenine and NAD + levels
Our metabolomics studies indicated significant alterations in lipid metabolism and carbohydrate metabolism in the Nrx-1 mutants, and thus we decided to further examine this dataset to assess for any additional alterations of components of energy metabolism (Supplementary Material, Table S2). Interestingly, we found that Nrx-1 mutants exhibited significantly elevated levels of tryptophan and elevated levels of kynurenine according to a Mann–Whitney U test (Fig. 3A and B, Supplementary Material, Table S2). Tryptophan is converted to kynurenine in the de novo synthesis pathway of NAD+ (Supplementary Material, Fig. S4). Notably, nicotinamide (NAM), the precursor for NAD+ in the salvage pathway was unchanged in the Nrx-1 mutants compared to controls (Supplementary Material, Table S3). Moreover, nicotinic acid, the precursor for NAD+ in the Preiss-Handler pathway, was significantly increased only in the Nrx-1241/241 mutants and not in the Nrx-1273/273 mutants (Supplementary Material, Table S3). These findings suggest that there may be dysregulation mainly in the de novo synthesis pathway of NAD+. NAD+ is a crucial regulator of energy metabolism by functioning as a coenzyme in redox reactions within the major energy production pathways, including glycolysis, the tricarboxylic acid (TCA) cycle, fatty acid oxidation (β-oxidation), and oxidative phosphorylation (OXPHOS) (68). Based on our findings of dysregulated NAD+ precursors, we decided to assess NAD+ levels in the Nrx-1 mutants. We performed these experiments using a colorimetric assay with bodies of Nrx-1 mutants and wild-type controls. Interestingly, we found that the Nrx-1 mutants showed significantly diminished NAD+ levels (Fig. 3C), although there were no significant changes in NADH levels (Fig. 3D). Additionally, the NAD+/NADH ratio was significantly decreased in the Nrx-1 mutants compared to controls, which was likely driven by the decrease in NAD+ levels (Fig. 3E). The observed decrease in NAD+ and the NAD+/NADH ratio could potentially contribute to the metabolic impairments seen in the Nrx-1 mutants, as diminished NAD+ levels have been shown to be involved in the development of behavioral, mitochondrial and metabolic disorders (69–75).
Figure 3.

Nrx-1 mutants exhibit altered tryptophan, kynurenine and NAD+ levels. Quantification of (A) tryptophan and (B) kynurenine by GC/TOF-MS in Nrx-1 mutants and iso31Bw- controls. Six samples of 40 flies for each genotype were used for the analysis. Box plots depict the autoscaled peak height of metabolites on the y-axis. Mann–Whitney U test revealed that both Nrx-1273/273 (P = 0.021421) and Nrx-1241/241 (P = 0.019413) flies exhibit elevated tryptophan levels. Similarly for kynurenine, Nrx-1273/273 (P = 0.021421) and Nrx-1241/241 (P = 0.019413) flies showed increased kynurenine levels compared to wild-type flies. Additionally, Mann–Whitney U test indicated no significant difference in tryptophan or kynurenine levels between Nrx-1273/273 and Nrx-1241/241 flies. Quantification of (C) NAD+, (D) NADH and the (E) NAD+/NADH ratio in Nrx-1 mutants and wild-type flies. The levels of NAD+ and NADH were normalized to protein content. Each data point represents an independent sample of 10 fly bodies. One-way ANOVA followed by Tukey–HSD post-hoc test indicated that compared to iso31Bw- controls, NAD+ levels were significantly decreased in both Nrx-1273/273 (P < 0.0001) and Nrx-1241/241 (P = 0.0001) flies. NADH levels were not significantly different than controls. The NAD+/NADH ratio was significantly diminished in both Nrx-1273/273 and Nrx-1241/241 flies (P < 0.0001). These experiments were performed three independent times. The NAD+ and NADH values shown (C and D) are representative figures from one experiment. The NAD+/NADH ratio is internally normalized, allowing data to be pooled from all three independent experiments. Sample number (N) per genotype for NAD+ and NADH data = 3. Sample number (N) per genotype for NAD+/NADH ratio data: iso31Bw- N = 7, Nrx-1273/273 N = 8, Nrx-1241/241 N = 8. Values represent mean ± SEM.
To assess the hypothesis of whether decreased NAD+ levels underlie the diminished stress resistance in the Nrx-1 mutants, we attempted treating the Nrx-1 mutants with NAD+ precursors, nicotinamide riboside (NR) and NAM. Treatment with NR or NAM was not sufficient to improve starvation resistance to wild-type levels in Nrx-1273/273 and Nrx-1241/241 flies (Supplementary Material, Figs S5 and S6). In fact, Nrx-1273/273 flies treated with NAM showed significantly worsened starvation resistance compared to vehicle treated counterparts (Supplementary Material, Fig. S5). There are several reasons as to why treatment with NAD+ precursors may have been insufficient to improve starvation resistance in the Nrx-1 mutants, for example the drug dosage or timing of drug delivery may not have been optimal for phenotypic rescue. Additionally, it could be possible that the starvation resistance phenotype in the Nrx-1 mutants is caused by a combination of factors whereby treatment with NAD+ precursors alone is insufficient for rescue.
Loss of Nrx-1 leads to abnormal mitochondrial morphology and impaired flight
Our findings thus far closely mimic those seen in a previous study from our lab, which showed diminished lipid and carbohydrate stores as well as decreased NAD+ levels in a Drosophila model of Fragile X Syndrome, which is the leading monogenic cause of autism (76). This previous study also observed striking morphological abnormalities within flight muscle mitochondria. Thus, we pondered whether Nrx-1 mutants would exhibit similar morphological alterations in their mitochondria as well. For these experiments, longitudinal sections of indirect flight muscle in the thorax of Nrx-1 mutants and controls were prepared for transmission electron microscopy. We chose this section of tissue specifically because the flight muscle requires robust mitochondrial functioning for its high energy demands. Additionally, this tissue has a structured organization of mitochondria lining the space in between individual myofibrils, making it optimal for comparison among conspecifics. In the wild-type animals, we observed mitochondria that appeared intact, as well as densely packed and regularly distributed along the muscle fibers (Fig. 4A and D). Conversely, the mitochondria in the Nrx-1273/273 and Nrx-1241/241 mutants appeared irregularly spaced between myofibrils (Fig. 4B, C, E and F). Upon closer inspection using higher magnification, it was clear that mitochondria in the Nrx-1 mutants also appeared severely damaged with disruptions in the cristae (Fig. 4E and F).
Figure 4.

Loss of Nrx-1 leads to abnormal mitochondrial morphology and impaired flight. Longitudinal sections of indirect flight muscle of iso31Bw- flies (A and D), Nrx-1273/273 flies (B and E) and Nrx-1241/241 flies (C and F) were prepared from isolated thoraces for transmission electron microscopy experiments. (A–C) Electron micrographs of Drosophila flight muscle at 7500X magnification. Scale bar indicates 2 microns. (D–F) Electron micrographs of Drosophila flight muscle at 20 000X magnification. Scale bar indicates 800 nm. Images show mitochondria (M) aligned between rows of myofibrils (F), where Nrx-1 mutants have irregularly spaced mitochondria with marked disruptions in the cristae. Arrowheads denote disruptions in cristae. Asterisks signify areas devoid of cristae. (G) Quantification of the average landing height for flies of each genotype in a glycerol flight assay. Each data point represents an individual fly. Kruskal–Wallis test with Dunn’s multiple comparisons post-hoc test showed that Nrx-1273/273 and Nrx-1241/241 flies performed significantly worse on a glycerol flight assay compared to control flies. Sample number (N) per genotype: iso31Bw- N = 66, Nrx-1273/273 N = 67, Nrx-1241/241 N = 63. Values represent mean ± SEM. **** = P < 0.0001.
Given that mitochondrial ultrastructure and function are closely linked, and that the observed pathological lesions were within the mitochondria of indirect flight muscle, we decided to assess flight ability in the Nrx-1 mutants. Consistent with the severely disrupted ultrastructure found in flight muscle mitochondria in the absence of Nrx-1, we found that Nrx-1273/273 and Nrx-1241/241 homozygous mutants, as well as Nrx-1273/241 transheterozygous mutants, had significantly weakened flight ability compared to wild-type conspecifics (Fig. 4G and Supplementary Material, Fig. S1C). It is likely that the morphological abnormalities in flight muscle mitochondria contribute to this decreased flight fitness in the Nrx-1 mutants. Additionally, glycogen is the energy reserve that is used during flight in Drosophila (77), and therefore it is possible that decreased glycogen levels in the Nrx-1 mutants (Fig. 2E) also contribute to reduced flight ability in the Nrx-1 mutants. Overall, these findings demonstrate that loss of Nrx-1 gives rise to pathological disruptions in mitochondria in indirect flight muscle, and importantly that the functional output of this tissue, flight ability, is disrupted in Nrx-1 mutants.
Nrx-1 mutant flies exhibit seizure-like behavior after mechanical stimulation
The striking mitochondrial defects that we identified, coupled with multiple reports in the clinical literature that patients with deletions in NRXN1 exhibit seizures (48–54), prompted us to assess seizure susceptibility in the Nrx-1 mutants. Mitochondrial dysfunction has emerged as a primary cause of epilepsy given the critical role of mitochondrial OXPHOS in the generation of the ATP required to maintain the proper balance between excitatory and inhibitory neural transmission (78–81). Given that our findings thus far are suggestive of potential mitochondrial dysfunction in the Nrx-1 mutants, and that NRXN1 plays an important role in the regulation of synaptic function and transmission (40), we hypothesized that Nrx-1 mutants would be hypersensitive to mechanical shock. To assess for seizure susceptibility, Nrx-1 mutants and controls were placed in vials and vortexed for 10 s (82). These experiments revealed that following an acute mechanical shock, Nrx-1273/273, Nrx-1241/241 and Nrx-1273/241 mutants exhibited a significantly higher percentage of flies showing seizure-like activity, consisting of hyperactivity, leg twitching and paralysis, as well as increased time to recovery after mechanical stimulation compared to control flies (Fig. 5A and B, Supplementary Material, Fig. S1D and E). These findings indicate that loss of Nrx-1 contributes to seizure-like activity in Drosophila and are consistent with the clinical data wherein patients with NRXN1 deletions exhibit seizures.
Figure 5.
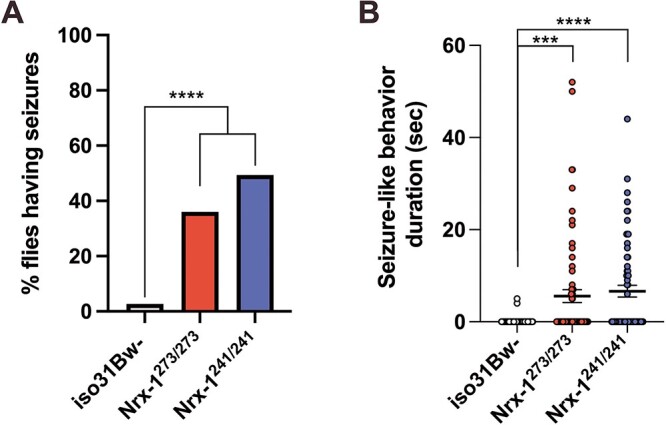
Nrx-1 mutant flies exhibit seizure-like activity after mechanical stimulation. (A) Quantification of the percentage of flies seizing per genotype. Flies were collected into groups of 5 flies per vial prior to a 10-s mechanical shock. Chi-square test was used to compare the penetrance of seizures. Sample number (N) per genotype: iso31Bw- N = 75, Nrx-1273/273 N = 75, Nrx-1241/241 N = 75. (B) Quantification of the duration of seizure-like activity after mechanical shock for flies of each genotype. Each data point represents an individual fly. Kruskal-Wallis test with Dunn’s multiple comparisons post-hoc test showed that Nrx-1273/273 (P = 0.0003) and Nrx-1241/241 (P < 0.0001) flies show a significant increase in the time taken to regain their posture following a mechanical shock. This quantification period was capped at a maximum of 60 s, and thus flies that continued seizure-like behavior beyond that period were excluded from this analysis. Sample number (N) per genotype: iso31Bw- N = 75, Nrx-1273/273 N = 68, Nrx-1241/241 N = 62. Values represent mean ± SEM. *** = P < 0.001, **** = P < 0.0001.
Discussion
In our pursuit to better understand how NRXN1 contributes to the pathophysiology of autism, we uncovered striking alterations in stress resistance, energy metabolism, mitochondrial morphology and seizure-like activity following the loss of Nrx-1 in Drosophila melanogaster. Although neurexins are conventionally considered regulators of synaptic properties in the brain (40), our findings suggest that the loss of NRXN1 also has systemic implications for energy metabolism, which could have a potential impact on behavior or contribute to the increased prevalence of metabolic disorders seen in neurodevelopmental disorders, such as autism and schizophrenia. Here, we show that Drosophila lacking Nrx-1 exhibit decreased resistance to environmental stress, specifically starvation stress and heat stress. These findings lead us to investigate the metabolic status of the Nrx-1 mutants using metabolomics and complementary colorimetric assays, which resulted in the discovery that Nrx-1 mutant flies have a distinct metabolic profile as well as decreased lipid and carbohydrate stores. It is likely that these deficient energy stores are underlying the observed decrease in survival under starvation and heat stress in the Nrx-1 mutants, as reduced energy stores have been linked to starvation sensitivity and heat stress exposure (56–59). Interestingly, similar alterations in nutrient homeostasis and energy metabolism have been revealed in Nrxn1 mutant mice by the International Mouse Phenotyping Consortium (https://www.mousephenotype.org/data/genes/MGI:1096391) (83), which is an international effort in phenotyping the 20 000 protein-coding genes in the mouse genome. Specifically, Nrxn1 mutant male mice were found to have decreased total body fat, increased lean mass, as well as decreased total cholesterol and high-density lipoprotein-cholesterol compared to wild-type controls. These findings are consistent with decreased lipid stores seen in the Nrx-1 mutant flies. Additionally, female Nrxn1 mutant mice had decreased fasting circulating glucose levels compared to wild-type animals, which closely mimics our findings of altered carbohydrate metabolism in Nrx-1 mutant flies. Together, these findings indicate that both Drosophila and mice lacking NRXN1 show deficits in lipid and carbohydrate metabolism, which suggests a potential conserved effect of loss of NRXN1 between invertebrates and vertebrates.
Furthermore, the findings presented here are consistent with those of other animal models of neurodevelopmental disorders that have identified metabolic differences. Our lab previously demonstrated a significantly altered metabolome, reduced triglyceride and glycogen stores and reduced starvation resistance in a Drosophila model of Fragile X Syndrome (76). Additionally, mice mutant for the Fragile X proteins, Fragile X mental retardation 1 (FMR1) and Fragile X mental retardation syndrome-related protein 2 have reduced fat deposits, hypoglycemia and increased sensitivity to insulin (84). Metabolic alterations are also seen in animal models of neurofibromatosis type 1 (NF1), which is a monogenic cancer predisposition syndrome resulting from loss of function in the neurofibromin protein (Nf1) that is also characterized by increased susceptibility to neurocognitive impairments, such as attention-deficit/hyperactivity disorder and autism. For example, Nf1 haploinsufficient mice have reduced fat mass, increased glucose clearance, and increased insulin sensitivity (85). Additionally, loss of Nf1 in Drosophila leads to increased metabolic rate, increased feeding, decreased triglycerides and altered lipid turnover kinetics (86). Moreover, a recent study of mouse models of Down syndrome, 16p11.2 deletion syndrome, and Fragile X Syndrome demonstrated sex-specific alterations in basal energy metabolism as well as differences in key plasma metabolites related to the TCA cycle (87). Our study contributes additional evidence to this growing body of literature, illustrating dysfunctional energy metabolism as a co-morbid disorder in autism and suggesting a role for NRXN1 within this narrative.
The current study provides compelling evidence that Nrx-1 has a role in the regulation of energy metabolism in Drosophila. However, there are still several remaining questions as to how this cell adhesion molecule that is primarily thought to function at the synapse may contribute to broad systemic alterations in nutrient homeostasis. In Drosophila, Nrx-1 is expressed broadly across the central and peripheral nervous system, as well as in the muscle, gut and neuromuscular junction (42,47,88). Thus, it is possible that Nrx-1 could regulate energy metabolism by regulating synaptic transmission within a neuronal population involved in metabolic regulation. In Drosophila, the insulin-producing neurons have been shown to regulate metabolism and are located within the pars intercerebralis region of the brain (89–93). In vertebrates, NRXN1α regulates insulin granule docking and insulin secretion via actions in the insulin-producing pancreatic β-cells (94,95). Therefore, misregulation of the release of Drosophila insulin-like peptides may lead to the observed metabolic alterations in the Nrx-1 flies. It is well known that there is important communication between the brain and peripheral tissues in the regulation of energy metabolism and nutrient homeostasis (62–65), and thus Nrx-1 could be acting cell non-autonomously to regulate the storage of lipids and carbohydrates within peripheral tissues. Future studies will be imperative to elucidate the mechanisms by which Nrx-1 regulates the processes presented here.
Beyond the potential routes that loss of Nrx-1 function could alter metabolism discussed above, the metabolic defects seen in Nrx-1-null flies could also be an effect of their diminished NAD+ levels or potential altered mitochondrial function. Given the importance of an optimal NAD+/NADH ratio in several redox reactions within energy metabolism pathways such as glycolysis, the TCA cycle, β-oxidation and OXPHOS, the observed decrease in NAD+ levels and the NAD+/NADH ratio could certainly impact energy metabolism in the Nrx-1 mutants. Although we found that NAD+ precursor treatment was not sufficient to rescue starvation resistance, the diminished NAD+ levels we observed in the Nrx-1 mutants may not be solely explained by alterations in NAD+ biosynthesis, as the total pool size of NAD+ depends on the relative rates of synthesis and degradation. In addition to possible alterations in NAD+ biosynthesis, an imbalance in NAD+ consumption due to the aberrations in other metabolic pathways in which NAD+ is a cofactor, which is another possible area of future investigation. Thus, the failure of NAD+ precursor treatment to rescue starvation resistance does not provide conclusive evidence that the observed decrease in NAD+ and the NAD+/NADH ratio are not linked to the behavioral phenotypes we see in the Nrx-1 mutants, as there are several remaining experiments that are yet to be performed. Additionally, it would be worthwhile to assess whether NAD+ precursors are able to improve other metabolic, mitochondrial or behavioral phenotypes in the Nrx-1 mutants.
Additionally, we also discovered noticeable disruptions and atrophy in the cristae in mitochondria within the flight muscle of Nrx-1-null flies. In support of the notion that the observed mitochondrial abnormalities are detrimental to the Nrx-1 mutants, we also uncovered significantly weakened flight ability in these flies. Again, these findings are in line with what has been seen previously in a Drosophila model of Fragile X Syndrome, wherein dfmr1 mutant flies had decreased NAD+ levels and altered mitochondrial morphology and indications of mitochondrial dysfunction (76). There is increasing evidence in mouse models as well supporting the hypothesis that mitochondrial defects may be central to the etiology of autism and other neurodevelopmental disorders (96–102). Mice with a missense mutation in the mitochondrial DNA complex I ND6 gene exhibit impaired social behavior, increased repetitive behaviors and anxiety, decreased seizure threshold, as well as alterations in mitochondrial respiratory function and reactive oxygen species (102). These findings illustrate that altered mitochondrial function can lead to ‘autistic-like’ phenotypes.
In addition to findings in animal models, there is considerable research showing evidence of increased prevalence of mitochondrial disease in autistic individuals compared to the general population, wherein autistic individuals have decreased cortical glucose utilization and ATP levels, altered levels of biochemical markers of mitochondrial dysfunction, altered tryptophan levels, as well as increased oxidative stress (11–25,103–107). It will be highly informative for future studies to determine whether these alterations in NAD+ levels and mitochondrial morphology in the Nrx-1 mutants contribute to potential mitochondrial dysfunction or additional behavioral phenotypes related to autism.
Furthermore, these studies show that loss of Nrx-1 results in mechanically-induced seizure-like activity. These findings are clinically relevant, as patients with deletions in NRXN1 also exhibit seizures (48–54) and autistic individuals are at an increased risk for epilepsy, as the average prevalence of epilepsy in autistic children is 12% and reaches 26% by adolescence (9), compared to the <1% of children with epilepsy in the general population. The identification of seizures in the Nrx-1-null flies illuminates a behavioral paradigm that can be used to assess the efficacy of potential therapeutic options for autistic individuals who experience seizures. Future studies will be crucial to further investigate how loss of Nrx-1 contributes to seizure-like activity in the Nrx-1 mutants, as these seizures could be the result of altered synaptic transmission, altered mitochondrial function and bioenergetics, or a combination of these factors.
Although there has been a greater focus on investigating the role of NRXN1 deletions in neurodevelopmental disorders, there is evidence that indicates that mutations in the paralogous NRXN2 and NRXN3 are also associated with autism and schizophrenia (108–113). Thus, future studies that examine the extent to which our metabolic and mitochondrial findings apply to the other members of the NRXN protein family in vertebrate models would expand our understanding of shared and divergent features of NRXN1–3.
In conclusion, our work depicts a novel role for neurexins in the regulation of energy metabolism by using a combination of metabolomic and physiological methodologies. Moreover, we have identified behavioral alterations in stress resistance, flight ability and seizure-like activity resulting from Nrx-1 deficiency. These studies establish a foundation for future inquiries into the function of neurexins and the role of metabolic dysfunction in the etiology of autism.
Materials and Methods
Fly genetics and husbandry
Fly strains that contain the Nrx-1273 and Nrx-1241 alleles are described in Li et al. (42). These flies were outcrossed in the w1118(iso31Bw-) background for six generations and maintained over a third chromosome balancer to avoid recombination events. The fly strains were cultured on a standard cornmeal molasses medium and maintained in the presence of 12:12 h light:dark (LD) cycle at 25°C.
Starvation stress
Male flies were collected 0–1 days post eclosion and entrained to a stringent LD cycle for 3–4 days at 25°C. Flies were then placed in individual glass tubes containing 0% sucrose, 2% agar and loaded into activity monitors (Trikinetics, DAM2 system, Waltham, MA) that were then returned to a LD light cycle at 25°C. The experiment was concluded when all flies were confirmed dead. Time of death after the onset of starvation was determined by the final activity count recorded by the activity monitors.
Heat stress
The HS protocol was modified from Folk et al. (55). We placed 3- to 5-day-old adult male flies in groups of 10 into empty plastic vials without CO2. The empty vials were then submerged in a 37°C water bath for 1 h. After heat exposure, flies were flipped to new plastic vials containing standard cornmeal molasses medium and then returned to 25°C. After 24 h, the number of dead flies per vial was recorded.
Metabolomics
We collected 3- to 5-day-old adult male flies on dry ice at approximately ZT0-ZT1. Flies of each genotype were then pooled into six independent groups comprising 40 whole flies each. These samples were then sent for metabolomics analysis of primary metabolites and complex lipids at the West Coast Metabolomics Center (https://metabolomics.ucdavis.edu/). Primary metabolites were probed using ALEX-CIS gas chromatography/time-of-flight mass spectrometry (GC/TOF-MS) and complex lipids were analyzed using the charged surface hybrid-electrospray ionization quadrupole time of flight mass spectrometer tandem mass spectrometry (CSH-ESI QTOF MS/MS). A more detailed description of the data acquisition procedures and chromatographic parameters can be found in Fiehn et al. (114) and Matyash et al. (115).
Triglyceride measurement
Measurement of triglycerides was performed as described in Weisz et al. (76). Briefly, 3- to 5-day-old adult male flies were collected on dry ice at approximately ZT0-ZT1. Samples for triglyceride measurement contained 3 flies each, and fly heads were removed prior to homogenization. Fly bodies were homogenized in lysis buffer that contained 140 mM NaCl, 50 mM Tris–HCl (pH 7.4), 20% Triton X-100 and 1X protease inhibitors (Roche, Indianapolis, IN). The homogenate was then centrifuged at 15871×g for 10 min at 4°C. Triglyceride concentration for each sample was determined in triplicate using the Triglyceride LiquiColor kit (Stanbio Laboratory, Boerne, TX), and was normalized to protein content using the Pierce BCA Protein Assay Kit (Thermo Scientific, Rockford, IL).
Glycogen measurement
Measurement of glycogen was performed as described in Weisz et al. (76). Briefly, 3- to 5-day-old adult male flies were collected on dry ice at approximately ZT0-ZT1. Samples each had 4 decapitated fly bodies, which were homogenized in 200 μl of 0.1 M NaOH. The homogenate was centrifuged at 15871×g for 10 min at 4°C. The supernatant was extracted from each sample and 20 μl of the lysate was treated with 5 mg/ml Amyloglucosidase (Sigma-Aldrich, Saint Louis, MO) in 0.2 M acetate, pH 4.8, as this enzyme catabolizes glycogen to yield free glucose molecules. Concurrently, another 20 μl aliquot of the lysate was treated with 0.2 M acetate, pH 4.8 alone. Both reactions incubated for 2 h at 37°C. Subsequently, the free glucose content in each reaction was measured in triplicate with the Amplex Red Glucose/Glucose Oxidase Assay kit (Molecular Probes, Eugene, OR). The glycogen concentration of each sample was determined by subtracting the values of free glucose in the untreated samples. The protein concentration of these reactions was then determined with the Pierce BCA Protein Assay Kit (Thermo Scientific, Rockford, IL) for normalization.
Feeding
This feeding assay was modified from Edgecomb et al. (116). Briefly, 3- to 5-day-old adult male flies of each genotype were fasted for an 18-h period in plastic vials with 0% sucrose, 2% agar medium. After the fasting period, flies were flipped onto standard cornmeal molasses medium containing 2.5 mg/ml Brilliant Blue FCF (Sigma Aldrich). After 1 h of feeding, flies were anesthetized using CO2 and sorted into groups of 5 flies each into 1.5 ml Safelock Eppendorf tubes. The tubes were placed onto dry ice for 10 min, transferred onto wet ice, and then 130 μl of chilled PBST was added to each tube. Samples were homogenized and centrifuged at max speed for 15 min at 4°C. After centrifuging, 50 μl of supernatant for each sample was loaded into duplicate wells on a 96-well plate and absorbance was measured at 620 nm. The concentration of Brilliant Blue FCF consumed in each sample was determined using a standard curve.
Activity/sleep levels
Male flies of the appropriate genotype were collected 0–1 days post eclosion and entrained to a stringent LD cycle for 3–4 days at 25°C. Flies were then placed in individual tubes containing 5% sucrose, 2% agar, and loaded into monitors (Trikinetics, DAM2 system, Waltham, MA) that were returned to an LD cycle at 25°C. The activity of these flies, as indicated by beam breaks, was measured from days 2 to 4. The daily activity of each fly was averaged over the 3 days used for analysis, and the average daily activity of each genotype was determined. Sleep was also measured during this 3-day period; sleep was defined as a period of 5 min of inactivity. Flies that died at any time during the assay were excluded from analysis.
Weight measurement
1.5 ml microcentrifuge tubes were first pre-weighed on a Mettler AE 100 Analytical Balance. Then, 3- to 5-day-old adult male flies of the appropriate genotype were placed into the tubes in groups of 10–20. Each sample was then weighed and the weight/fly was calculated.
NAD +/NADH Quantification
Measurement of NAD+/NADH was performed as described in Weisz et al. (76). The concentrations of nicotinamide nucleotides were measured using the NAD+/NADH Quantification Colorimetric Kit (BioVision, Milpitas, CA). Briefly, adult male flies aged 3 to 5 days were collected on dry ice. Fly heads were removed prior to homogenization and the decapitated fly bodies were pooled in groups of 10. The samples were homogenized for 30 pulses in 400 μl of the NAD+/NADH Extraction Buffer supplied in the kit, and the homogenate was centrifuged at top speed for 5 min at 4°C to remove debris. The cycling reaction was carried out as per the manufacturer’s instructions for 2 h and the nicotinamide nucleotide concentrations of each standard and sample were determined in duplicate. The protein concentration of each sample was measured with the Pierce BCA Protein Assay Kit (Thermo Scientific, Rockford, IL) and the nicotinamide nucleotide concentration of each sample was then normalized to its respective protein content.
NAD + precursor food preparation
Standard cornmeal molasses fly food was melted down to a liquid and subsequently cooled to 60°C. NR was obtained from Sigma (SMB00907) and added to the medium at a final concentration of 500 μM. NAM was obtained from Sigma (481907) and was added to the medium at a final concentration of 30 mg NAM/100 g medium. Both NR and NAM were solubilized in water prior to adding them to the medium. A commensurate amount of water without NR or NAM was added to a separate batch of medium as a vehicle control medium.
Transmission electron microscopy
Tissues for electron microscopic examination were fixed with 2.5% glutaraldehyde, 2.0% paraformaldehyde in 0.1 M sodium cacodylate buffer, pH 7.4, overnight at 4°C. After subsequent buffer washes, the samples were post-fixed in 2.0% osmium tetroxide for 1 h at room temperature, and then washed again in buffer followed by dH2O. After dehydration through a graded ethanol series, the tissue was infiltrated and embedded in EMbed-812 (Electron Microscopy Sciences, Fort Washington, PA). Thin sections were stained with lead citrate and examined with a JEOL 1010 electron microscope fitted with a Hamamatsu digital camera and AMT Advantage image capture software.
Glycerol flight assay
The flight assay was modified from Elkins et al. (117). The inside of a 1000 ml glass graduated cylinder was coated with a thin layer of glycerol, and a group of 5–10 adult male flies of the appropriate genotype was quickly flipped into the cylinder using a funnel. Immediately after flipping the flies into the cylinder, a dry erase marker was used to indicate where each fly landed within the cylinder. The landing height was recorded as the corresponding measurement in ml on the cylinder. The glycerol was then rinsed out and renewed for each group of flies.
Seizure assay
Male flies of each genotype were collected 0–1 days post eclosion and entrained to a stringent LD cycle for 3–4 days at 25°C. Flies of each genotype were placed into empty plastic vials without CO2 in groups of 5 flies per vial. To induce seizure-like activity, the vials were vortexed at maximum speed for 10 s (82). The flies were video recorded for quantification of the number of flies seizing per vial and the time taken to regain their posture following the mechanical shock. Analysis of the time to recovery was capped at a maximum of 60 s. Genotypes were blinded to the scorer during the analysis.
Statistics
For the metabolomics data, univariate between-group comparisons were conducted by Christopher Brydges at the West Coast Metabolomics Center. Mann–Whitney U tests (non-parametric) were conducted and uncorrected and false discovery rate corrected P-values were calculated. Bayes factors were calculated for the same comparisons using a Bayesian Mann–Whitney U test, using a default prior distribution (118). The PCA plots are the biplots of the first two components extracted from a principal components analysis, with normal data ellipses for each group. For all other experiments, the Prism software package (GraphPad Software, v9.2.0) was used to generate graphs and perform statistical analyses. Parametric data was analyzed using either t-tests or one-way analysis of variance (ANOVA) with post hoc Tukey’s tests, as appropriate. Non-parametric data was analyzed using Mann–Whitney tests or Kruskal–Wallis tests with post hoc Dunn’s tests, as appropriate. Chi-square test or Fisher’s exact test were used to compare penetrance of seizure-like behavior. The log-rank (Mantel–Cox) test with a Bonferroni-corrected significance threshold for multiple comparisons was used to examine differences in stress resistance between genotypes.
Supplementary Material
Acknowledgements
We thank Meridith Toth at the University of Pennsylvania for assistance with teaching the colorimetric assays; Kelly Paglia, Christopher Brydges, and the team at the West Coast Metabolomics Center at the University of California, Davis for performing and analyzing our metabolomics study; Biao Zuo and Inna Martynyuk at the Electron Microscopy Resource Laboratory at the University of Pennsylvania for assistance with our electron microscopy experiments; William Haury at the University of Pennsylvania for help with Drosophila husbandry; and Vishnu Cuddapah at the University of Pennsylvania for feedback and advice on the seizure assay.
Conflicts of Interest statement. None declared.
Contributor Information
Kyra A Levy, Department of Genetics, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA 19104, USA; Neuroscience Graduate Group, University of Pennsylvania, Philadelphia, PA 19104, USA; Autism Spectrum Program of Excellence, University of Pennsylvania, Philadelphia, PA 19104, USA.
Eliana D Weisz, Department of Genetics, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA 19104, USA; Autism Spectrum Program of Excellence, University of Pennsylvania, Philadelphia, PA 19104, USA.
Thomas A Jongens, Department of Genetics, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA 19104, USA; Autism Spectrum Program of Excellence, University of Pennsylvania, Philadelphia, PA 19104, USA.
Funding
Autism Spectrum Program of Excellence at the University of Pennsylvania (https://aspe.med.upenn.edu); United States Department of Defense Autism (Grant AR1101189); National Institutes of Health (Grant MH126257).
References
- 1. Malow, B.A. (2004) Sleep disorders, epilepsy, and autism. Ment. Retard. Dev. Disabil. Res. Rev., 10, 122–125. [DOI] [PubMed] [Google Scholar]
- 2. Devnani, P.A. and Hegde, A.U. (2015) Autism and sleep disorders. J. Pediatr. Neurosci., 10, 304–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Herrmann, S. (2016) Counting sheep: sleep disorders in children with autism spectrum disorders. J. Pediatr. Health Care, 30, 143–154. [DOI] [PubMed] [Google Scholar]
- 4. Volkmar, F.R. and Nelson, D.S. (1990) Seizure disorders in autism. J. Am. Acad. Child Adolesc. Psychiatry, 29, 127–129. [DOI] [PubMed] [Google Scholar]
- 5. Matson, J.L. and Neal, D. (2009) Seizures and epilepsy and their relationship to autism spectrum disorders. Res. Autism Spectr. Disord., 3, 999–1005. [Google Scholar]
- 6. Gillott, A., Furniss, F. and Walter, A. (2001) Anxiety in high-functioning children with autism. Autism, 5, 277–286. [DOI] [PubMed] [Google Scholar]
- 7. Gillott, A. and Standen, P.J. (2007) Levels of anxiety and sources of stress in adults with autism. J. Intellect. Disabil., 11, 359–370. [DOI] [PubMed] [Google Scholar]
- 8. White, S.W., Oswald, D., Ollendick, T. and Scahill, L. (2009) Anxiety in children and adolescents with autism spectrum disorders. Clin. Psychol. Rev., 29, 216–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Viscidi, E.W., Triche, E.W., Pescosolido, M.F., McLean, R.L., Joseph, R.M., Spence, S.J. and Morrow, E.M. (2013) Clinical characteristics of children with autism spectrum disorder and co-occurring epilepsy. PLoS One, 8, e67797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Frye, R.E., Casanova, M.F., Fatemi, S.H., Folsom, T.D., Reutiman, T.J., Brown, G.L., Edelson, S.M., Slattery, J.C. and Adams, J.B. (2016) Neuropathological mechanisms of seizures in autism spectrum disorder. Front. Neurosci., 10, 192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schifter, T., Hoffman, J.M., Hatten, H.P., Hanson, M.W., Coleman, R.E. and DeLong, G.R. (1994) Neuroimaging in infantile autism. J. Child Neurol., 9, 155–161. [DOI] [PubMed] [Google Scholar]
- 12. Minshew, N.J., Goldstein, G., Dombrowski, S.M., Panchalingam, K. and Pettegrew, J.W. (1993) A preliminary 31P MRS study of autism: evidence for undersynthesis and increased degradation of brain membranes. Biol. Psychiatry, 33, 762–773. [DOI] [PubMed] [Google Scholar]
- 13. Rossignol, D.A. and Frye, R.E. (2012) Mitochondrial dysfunction in autism spectrum disorders: a systematic review and meta-analysis. Mol. Psychiatry, 17, 290–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Moreno, H., Borjas, L., Arrieta, A., Sáez, L., Prassad, A., Estévez, J. and Bonilla, E. (1992) Clinical heterogeneity of the autistic syndrome: a study of 60 families. Investig. Clin., 33, 13–31. [PubMed] [Google Scholar]
- 15. Kurup, R.K. and Kurup, P.A. (2003) A hypothalamic digoxin-mediated model for autism. Int. J. Neurosci., 113, 1537–1559. [DOI] [PubMed] [Google Scholar]
- 16. Filipek, P.A., Juranek, J., Nguyen, M.T., Cummings, C. and Gargus, J.J. (2004) Relative carnitine deficiency in autism. J. Autism Dev. Disord., 34, 615–623. [DOI] [PubMed] [Google Scholar]
- 17. Mostafa, G.A., El-Gamal, H.A., El-Wakkad, A.S., El-Shorbagy, O.E. and Hamza, M.M. (2005) Polyunsaturated fatty acids, carnitine and lactate as biological markers of brain energy in autistic children. Int J Child Neuropsychiatry, 2, 179–188. [Google Scholar]
- 18. Al-Mosalem, O.A., El-Ansary, A., Attas, O. and Al-Ayadhi, L. (2009) Metabolic biomarkers related to energy metabolism in Saudi autistic children. Clin. Biochem., 42, 949–957. [DOI] [PubMed] [Google Scholar]
- 19. Giulivi, C., Zhang, Y.-F., Omanska-Klusek, A., Ross-Inta, C., Wong, S., Hertz-Picciotto, I., Tassone, F. and Pessah, I.N. (2010) Mitochondrial dysfunction in autism. JAMA, 304, 2389–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. El-Ansary, A., Al-Daihan, S., Al-Dbass, A., and Al-Ayadhi, L. Measurement of selected ions related to oxidative stress and energy metabolism in Saudi autistic children. Clinical Biochemistry, 43, 63–70. [DOI] [PubMed] [Google Scholar]
- 21. Chugani, D.C., Sundram, B.S., Behen, M., Lee, M.L. and Moore, G.J. (1999) Evidence of altered energy metabolism in autistic children. Prog. Neuro-Psychopharmacol. Biol. Psychiatry, 23, 635–641. [DOI] [PubMed] [Google Scholar]
- 22. Chauhan, A. and Chauhan, V. (2006) Oxidative stress in autism. Pathophysiology, 13, 171–181. [DOI] [PubMed] [Google Scholar]
- 23. Chauhan, A., Chauhan, V., Brown, W.T. and Cohen, I. (2004) Oxidative stress in autism: Increased lipid peroxidation and reduced serum levels of ceruloplasmin and transferrin - the antioxidant proteins. Life Sci., 75, 2539–2549. [DOI] [PubMed] [Google Scholar]
- 24. Al-Gadani, Y., El-Ansary, A., Attas, O. and Al-Ayadhi, L. (2009) Metabolic biomarkers related to oxidative stress and antioxidant status in Saudi autistic children. Clin. Biochem., 42, 1032–1040. [DOI] [PubMed] [Google Scholar]
- 25. Zoroglu, S.S., Armutcu, F., Ozen, S., Gurel, A., Sivasli, E., Yetkin, O. and Meram, I. (2004) Increased oxidative stress and altered activities of erythrocyte free radical scavenging enzymes in autism. Eur. Arch. Psychiatry Clin. Neurosci., 254, 143–147. [DOI] [PubMed] [Google Scholar]
- 26. Südhof, T.C. (2008) Neuroligins and neurexins link synaptic function to cognitive disease. Nature, 455, 903–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Feng, J., Schroer, R., Yan, J., Song, W., Yang, C., Bockholt, A., Cook, E.H., Skinner, C., Schwartz, C.E. and Sommer, S.S. (2006) High frequency of neurexin 1β signal peptide structural variants in patients with autism. Neurosci. Lett., 409, 10–13. [DOI] [PubMed] [Google Scholar]
- 28. Yan, J., Noltner, K., Feng, J., Li, W., Schroer, R., Skinner, C., Zeng, W., Schwartz, C.E. and Sommer, S.S. (2008) Neurexin 1α structural variants associated with autism. Neurosci. Lett., 438, 368–370. [DOI] [PubMed] [Google Scholar]
- 29. Glessner, J.T., Wang, K., Cai, G., Korvatska, O., Kim, C.E., Wood, S., Zhang, H., Estes, A., Brune, C.W., Bradfield, J.P. et al. (2009) Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature, 459, 569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bucan, M., Abrahams, B.S., Wang, K., Glessner, J.T., Herman, E.I., Sonnenblick, L.I., Retuerto, A.I.A., Imielinski, M., Hadley, D., Bradfield, J.P. et al. (2009) Genome-wide analyses of exonic copy number variants in a family-based study point to novel autism susceptibility genes. PLoS Genet., 5, e1000536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Marshall, C.R., Noor, A., Vincent, J.B., Lionel, A.C., Feuk, L., Skaug, J., Shago, M., Moessner, R., Pinto, D., Ren, Y. et al. (2008) Structural variation of chromosomes in autism spectrum disorder. Am. J. Hum. Genet., 82, 477–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Morrow, E.M., Yoo, S.-Y., Flavell, S.W., Kim, T.-K., Lin, Y., Hill, R.S., Mukaddes, N.M., Balkhy, S., Gascon, G., Hashmi, A. et al. (2008) Identifying autism loci and genes by tracing recent shared ancestry. Science, 321, 218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Szatmari, P., Paterson, A., Zwaigenbaum, L., Roberts, W., Brian, J., Liu, X.-Q., Vincent, J., Skaug, J., Thompson, A., Senman, L. et al. (2007) Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat. Genet., 39, 319–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kim, H.-G., Kishikawa, S., Higgins, A.W., Seong, I.-S., Donovan, D.J., Shen, Y., Lally, E., Weiss, L.A., Najm, J., Kutsche, K. et al. (2008) Disruption of Neurexin 1 associated with autism spectrum disorder. Am. J. Hum. Genet., 82, 199–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kirov, G., Gumus, D., Chen, W., Norton, N., Georgieva, L., Sari, M., O’Donovan, M.C., Erdogan, F., Owen, M.J., Ropers, H.-H. et al. (2008) Comparative genome hybridization suggests a role for NRXN1 and APBA2 in schizophrenia. Hum. Mol. Genet., 17, 458–465. [DOI] [PubMed] [Google Scholar]
- 36.(2008) Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature, 455, 237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pruett, B.S. and Meador-Woodruff, J.H. (2020) Evidence for altered energy metabolism, increased lactate, and decreased pH in schizophrenia brain: a focused review and meta-analysis of human postmortem and magnetic resonance spectroscopy studies. Schizophr. Res., 223, 29–42. [DOI] [PubMed] [Google Scholar]
- 38. Martins-de-Souza, D., Harris, L.W., Guest, P.C. and Bahn, S. (2011) The role of energy metabolism dysfunction and oxidative stress in schizophrenia revealed by proteomics. Antioxid. Redox Signal., 15, 2067–2079. [DOI] [PubMed] [Google Scholar]
- 39. Prabakaran, S., Swatton, J.E., Ryan, M.M., Huffaker, S.J., Huang, J.-J., Griffin, J.L., Wayland, M., Freeman, T., Dudbridge, F., Lilley, K.S. et al. (2004) Mitochondrial dysfunction in schizophrenia: evidence for compromised brain metabolism and oxidative stress. Mol. Psychiatry, 9, 684–697. [DOI] [PubMed] [Google Scholar]
- 40. Südhof, T.C. (2017) Synaptic neurexin complexes: a molecular code for the logic of neural circuits. Cell, 171, 745–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tabuchi, K. and Südhof, T.C. (2002) Structure and evolution of neurexin genes: insight into the mechanism of alternative splicing. Genomics, 79, 849–859. [DOI] [PubMed] [Google Scholar]
- 42. Li, J., Ashley, J., Budnik, V. and Bhat, M.A. (2007) Crucial ole of Drosophila Neurexin in proper active zone apposition to postsynaptic densities, synaptic growth, and synaptic transmission. Neuron, 55, 741–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chen, K., Gracheva, E.O., Yu, S.-C., Sheng, Q., Richmond, J. and Featherstone, D.E. (2010) Neurexin in embryonic drosophila neuromuscular junctions. PLoS One, 5, e11115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Xing, G., Li, M., Sun, Y., Rui, M., Zhuang, Y., Lv, H., Han, J., Jia, Z. and Xie, W. (2018) Neurexin–Neuroligin 1 regulates synaptic morphology and functions via the WAVE regulatory complex in Drosophila neuromuscular junction. elife, 7, e30457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Larkin, A., Chen, M.-Y., Kirszenblat, L., Reinhard, J., van Swinderen, B. and Claudianos, C. (2015) Neurexin-1 regulates sleep and synaptic plasticity in Drosophila melanogaster. Eur. J. Neurosci., 42, 2455–2466. [DOI] [PubMed] [Google Scholar]
- 46. Tong, H., Li, Q., Zhang, Z.C., Li, Y. and Han, J. (2016) Neurexin regulates nighttime sleep by modulating synaptic transmission. Sci. Rep., 6, 38246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zeng, X., Sun, M., Liu, L., Chen, F., Wei, L. and Xie, W. (2007) Neurexin-1 is required for synapse formation and larvae associative learning in Drosophila. FEBS Lett., 581, 2509–2516. [DOI] [PubMed] [Google Scholar]
- 48. Al Shehhi, M., Forman, E.B., Fitzgerald, J.E., McInerney, V., Krawczyk, J., Shen, S., Betts, D.R., Ardle, L.M., Gorman, K.M., King, M.D. et al. (2019) NRXN1 deletion syndrome; phenotypic and penetrance data from 34 families. Eur. J. Med. Genet., 62, 204–209. [DOI] [PubMed] [Google Scholar]
- 49. Béna, F., Bruno, D.L., Eriksson, M., van Ravenswaaij-Arts, C., Stark, Z., Dijkhuizen, T., Gerkes, E., Gimelli, S., Ganesamoorthy, D., Thuresson, A.C. et al. (2013) Molecular and clinical characterization of 25 individuals with exonic deletions of NRXN1 and comprehensive review of the literature. Am. J. Med. Genet. B Neuropsychiatr. Genet., 162, 388–403. [DOI] [PubMed] [Google Scholar]
- 50. Schaaf, C.P., Boone, P.M., Sampath, S., Williams, C., Bader, P.I., Mueller, J.M., Shchelochkov, O.A., Brown, C.W., Crawford, H.P., Phalen, J.A. et al. (2012) Phenotypic spectrum and genotype–phenotype correlations of NRXN1 exon deletions. Eur. J. Hum. Genet., 20, 1240–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ching, M.S.L., Shen, Y., Tan, W.-H., Jeste, S.S., Morrow, E.M., Chen, X., Mukaddes, N.M., Yoo, S.-Y., Hanson, E., Hundley, R. et al. (2010) Deletions of NRXN1 (neurexin-1) predispose to a wide spectrum of developmental disorders. Am. J. Med. Genet. B Neuropsychiatr. Genet., 153B, 937–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Viñas-Jornet, M., Esteba-Castillo, S., Gabau, E., Ribas-Vidal, N., Baena, N., San, J., Ruiz, A., Coll, M.D., Novell, R. and Guitart, M. (2014) A common cognitive, psychiatric, and dysmorphic phenotype in carriers of NRXN1 deletion. Mol. Genet. Genomic Med., 2, 512–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Angione, K., Eschbach, K., Smith, G., Joshi, C. and Demarest, S. (2019) Genetic testing in a cohort of patients with potential epilepsy with myoclonic-atonic seizures. Epilepsy Res., 150, 70–77. [DOI] [PubMed] [Google Scholar]
- 54. Harrison, V., Connell, L., Hayesmoore, J., McParland, J., Pike, M.G. and Blair, E. (2011) Compound heterozygous deletion of NRXN1 causing severe developmental delay with early onset epilepsy in two sisters. Am. J. Med. Genet. A, 155, 2826–2831. [DOI] [PubMed] [Google Scholar]
- 55. Folk, D.G., Zwollo, P., Rand, D.M. and Gilchrist, G.W. (2006) Selection on knockdown performance in Drosophila melanogaster impacts thermotolerance and heat-shock response differently in females and males. J. Exp. Biol., 209, 3964–3973. [DOI] [PubMed] [Google Scholar]
- 56. Slocumb, M.E., Regalado, J.M., Yoshizawa, M., Neely, G.G., Masek, P., Gibbs, A.G. and Keene, A.C. (2015) Enhanced sleep is an evolutionarily adaptive response to starvation stress in drosophila. PLoS One, 10, e0131275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Masek, P., Reynolds, L.A., Bollinger, W.L., Moody, C., Mehta, A., Murakami, K., Yoshizawa, M., Gibbs, A.G. and Keene, A.C. (2014) Altered regulation of sleep and feeding contribute to starvation resistance in Drosophila. J. Exp. Biol., 217, 3122–3132, jeb.103309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Malmendal, A., Overgaard, J., Bundy, J.G., Sørensen, J.G., Nielsen, N.C., Loeschcke, V. and Holmstrup, M. (2006) Metabolomic profiling of heat stress: hardening and recovery of homeostasis in Drosophila. Am. J. Physiol.-Regul. Integr. Comp. Physiol., 291, R205–R212. [DOI] [PubMed] [Google Scholar]
- 59. Klepsatel, P., Gáliková, M., Xu, Y. and Kühnlein, R.P. (2016) Thermal stress depletes energy reserves in Drosophila. Sci. Rep., 6, 33667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Klepsatel, P., Wildridge, D. and Gáliková, M. (2019) Temperature induces changes in Drosophila energy stores. Sci. Rep., 9, 5239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Tennessen, J.M., Barry, W.E., Cox, J. and Thummel, C.S. (2014) Methods for studying metabolism in Drosophila. Methods, 68, 105–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Xu, K., Zheng, X. and Sehgal, A. (2008) Regulation of feeding and metabolism by neuronal and peripheral clocks in Drosophila. Cell Metab., 8, 289–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Iijima, K., Zhao, L., Shenton, C. and Iijima-Ando, K. (2009) Regulation of energy stores and feeding by neuronal and peripheral CREB activity in Drosophila. PLoS One, 4, e8498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Pool, A.-H. and Scott, K. (2014) Feeding regulation in Drosophila. Curr. Opin. Neurobiol., 29, 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Géminard, C., Rulifson, E.J. and Léopold, P. (2009) Remote control of Insul7in secretion by fat cells in Drosophila. Cell Metab., 10, 199–207. [DOI] [PubMed] [Google Scholar]
- 66. Arrese, E.L. and Soulages, J.L. (2010) Insect fat body: energy, metabolism, and regulation. Annu. Rev. Entomol., 55, 207–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Dus, M., Min, S., Keene, A.C., Lee, G.Y. and Suh, G.S.B. (2011) Taste-independent detection of the caloric content of sugar in Drosophila. Proc. Natl. Acad. Sci., 108, 11644–11649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Okabe, K., Yaku, K., Tobe, K. and Nakagawa, T. (2019) Implications of altered NAD metabolism in metabolic disorders. J. Biomed. Sci., 26, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Yoshino, J., Mills, K.F., Yoon, M.J. and Imai, S. (2011) Nicotinamide Mononucleotide, a key NAD+ intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab., 14, 528–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Zhang, H., Ryu, D., Wu, Y., Gariani, K., Wang, X., Luan, P., D’Amico, D., Ropelle, E.R., Lutolf, M.P., Aebersold, R. et al. (2016) NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science, 352, 1436–1443. [DOI] [PubMed] [Google Scholar]
- 71. Yoshino, J., Baur, J.A. and Imai, S. (2018) NAD+ intermediates: the biology and therapeutic potential of NMN and NR. Cell Metab., 27, 513–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Mills, K.F., Yoshida, S., Stein, L.R., Grozio, A., Kubota, S., Sasaki, Y., Redpath, P., Migaud, M.E., Apte, R.S., Uchida, K. et al. (2016) Long-term administration of Nicotinamide mononucleotide mitigates age-associated physiological decline in mice. Cell Metab., 24, 795–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Cantó, C., Houtkooper, R.H., Pirinen, E., Youn, D.Y., Oosterveer, M.H., Cen, Y., Fernandez-Marcos, P.J., Yamamoto, H., Andreux, P.A., Cettour-Rose, P. et al. (2012) The NAD+ precursor Nicotinamide Riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab., 15, 838–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Mitchell, S.J., Bernier, M., Aon, M.A., Cortassa, S., Kim, E.Y., Fang, E.F., Palacios, H.H., Ali, A., Navas-Enamorado, I., Di Francesco, A. et al. (2018) Nicotinamide improves aspects of Healthspan, but not lifespan, in mice. Cell Metab., 27, 667–676.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Julius, U. (2015) Niacin as antidyslipidemic drug. Can. J. Physiol. Pharmacol., 93, 1043–1054. [DOI] [PubMed] [Google Scholar]
- 76. Weisz, E.D., Towheed, A., Monyak, R.E., Toth, M.S., Wallace, D.C. and Jongens, T.A. (2018) Loss of Drosophila FMRP leads to alterations in energy metabolism and mitochondrial function. Hum. Mol. Genet., 27, 95–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Williams, C.M., Barness, L.A. and Sawyer, W.H. (1943) The utilization of glycogen by flies during flight and some aspects of the physiological ageing of drosophila. Biol. Bull., 84, 263–272. [Google Scholar]
- 78. Patel, M. (2004) Mitochondrial dysfunction and oxidative stress: cause and consequence of epileptic seizures. Free Radic. Biol. Med., 37, 1951–1962. [DOI] [PubMed] [Google Scholar]
- 79. Zsurka, G. and Kunz, W.S. (2015) Mitochondrial dysfunction and seizures: the neuronal energy crisis. Lancet Neurol., 14, 956–966. [DOI] [PubMed] [Google Scholar]
- 80. Kovac, S., Dinkova Kostova, A.T., Herrmann, A.M., Melzer, N., Meuth, S.G. and Gorji, A. (2017) Metabolic and homeostatic changes in seizures and acquired epilepsy—mitochondria, calcium dynamics and reactive oxygen species. Int. J. Mol. Sci., 18, 1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Cock, H.R. (2002) The role of mitochondria and oxidative stress in neuronal damage after brief and prolonged seizures. Progress in Brain Research, Do seizures damage the brain, Elsevier, 135, 187–196. [DOI] [PubMed] [Google Scholar]
- 82. Ganetzky, B. and Wu, C.-F. (1982) Indirect suppression involving behavioral mutants with altered nerve excitability in Drosophila Melanogaster. Genetics, 100, 597–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Dickinson, M.E., Flenniken, A.M., Ji, X., Teboul, L., Wong, M.D., White, J.K., Meehan, T.F., Weninger, W.J., Westerberg, H., Adissu, H. et al. (2016) High-throughput discovery of novel developmental phenotypes. Nature, 537, 508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Lumaban, J.G. and Nelson, D.L. (2015) The Fragile X proteins Fmrp and Fxr2p cooperate to regulate glucose metabolism in mice. Hum. Mol. Genet., 24, 2175–2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Tritz, R., Benson, T., Harris, V., Hudson, F.Z., Mintz, J., Zhang, H., Kennard, S., Chen, W., Stepp, D.W., Csanyi, G. et al. (2021) Nf1 heterozygous mice recapitulate the anthropometric and metabolic features of human neurofibromatosis type 1. Transl. Res., 228, 52–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Botero, V., Stanhope, B.A., Brown, E.B., Grenci, E.C., Boto, T., Park, S.J., King, L.B., Murphy, K.R., Colodner, K.J., Walker, J.A. et al. (2021) Neurofibromin regulates metabolic rate via neuronal mechanisms in Drosophila. Nat. Commun., 12, 4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Menzies, C., Naz, S., Patten, D., Alquier, T., Bennett, B.M. and Lacoste, B. (2021) Distinct basal metabolism in three mouse models of neurodevelopmental disorders. eNeuro, 8, ENEURO.0292-20.2021. Published 2021 Apr 16. doi: 10.1523/ENEURO.0292-20.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Sun, M., Zeng, X. and Xie, W. (2016) Temporal and spatial expression of Drosophila Neurexin during the life cycle visualized using a DNRX-Gal4/UAS-reporter. Sci. China Life Sci., 59, 68–77. [DOI] [PubMed] [Google Scholar]
- 89. Rulifson, E.J., Kim, S.K. and Nusse, R. (2002) Ablation of insulin-producing neurons in flies: growth and diabetic phenotypes. Science, 296, 1118–1120. [DOI] [PubMed] [Google Scholar]
- 90. Ikeya, T., Galic, M., Belawat, P., Nairz, K. and Hafen, E. (2002) Nutrient-dependent expression of insulin-like peptides from Neuroendocrine cells in the CNS contributes to growth regulation in Drosophila. Curr. Biol., 12, 1293–1300. [DOI] [PubMed] [Google Scholar]
- 91. Broughton, S.J., Piper, M.D.W., Ikeya, T., Bass, T.M., Jacobson, J., Driege, Y., Martinez, P., Hafen, E., Withers, D.J., Leevers, S.J. et al. (2005) Longer lifespan, altered metabolism, and stress resistance in Drosophila from ablation of cells making insulin-like ligands. Proc. Natl. Acad. Sci., 102, 3105–3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Géminard, C., Arquier, N., Layalle, S., Bourouis, M., Slaidina, M., Delanoue, R., Bjordal, M., Ohanna, M., Ma, M., Colombani, J. et al. (2006) Control of metabolism and growth through insulin-like peptides in Drosophila. Diabetes, 55, S5–S8. [Google Scholar]
- 93. Haselton, A., Sharmin, E., Schrader, J., Sah, M., Poon, P. and Fridell, Y.-W.C. (2010) Partial ablation of adult Drosophilainsulin-producing neurons modulates glucose homeostasis and extends life span without insulin resistance. Cell Cycle, 9, 3135–3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Suckow, A.T., Comoletti, D., Waldrop, M.A., Mosedale, M., Egodage, S., Taylor, P. and Chessler, S.D. (2008) Expression of neurexin, neuroligin, and their cytoplasmic binding partners in the pancreatic β-cells and the involvement of neuroligin in insulin secretion. Endocrinology, 149, 6006–6017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Mosedale, M., Egodage, S., Calma, R.C., Chi, N.-W. and Chessler, S.D. (2012) Neurexin-1α contributes to insulin-containing secretory granule docking *. J. Biol. Chem., 287, 6350–6361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Napoli, E., Ross-Inta, C., Wong, S., Hung, C., Fujisawa, Y., Sakaguchi, D., Angelastro, J., Omanska-Klusek, A., Schoenfeld, R. and Giulivi, C. (2012) Mitochondrial dysfunction in Pten Haplo-insufficient mice with social deficits and repetitive behavior: interplay between Pten and p53. PLoS One, 7, e42504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Großer, E., Hirt, U., Janc, O.A., Menzfeld, C., Fischer, M., Kempkes, B., Vogelgesang, S., Manzke, T.U., Opitz, L., Salinas-Riester, G. et al. (2012) Oxidative burden and mitochondrial dysfunction in a mouse model of Rett syndrome. Neurobiol. Dis., 48, 102–114. [DOI] [PubMed] [Google Scholar]
- 98. Yui, K., Sato, A. and Imataka, G. (2015) Mitochondrial dysfunction and its relationship with mTOR signaling and oxidative damage in autism spectrum disorders. Mini Rev. Med. Chem., 15, 373–389. [DOI] [PubMed] [Google Scholar]
- 99. Pei, L. and Wallace, D.C. (2018) Mitochondrial etiology of neuropsychiatric disorders. Biol. Psychiatry, 83, 722–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Yang, J., Fu, X., Liao, X. and Li, Y. (2020) Nrf2 activators as dietary phytochemicals against oxidative stress, inflammation, and mitochondrial dysfunction in autism spectrum disorders: a systematic review. Front. Psychiatry, 11, 1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Ahn, Y., Sabouny, R., Villa, B.R., Yee, N.C., Mychasiuk, R., Uddin, G.M., Rho, J.M. and Shutt, T.E. (2020) Aberrant mitochondrial morphology and function in the BTBR mouse model of autism is improved by two weeks of Ketogenic diet. Int. J. Mol. Sci., 21, 3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Yardeni, T., Cristancho, A.G., McCoy, A.J., Schaefer, P.M., McManus, M.J., Marsh, E.D. and Wallace, D.C. (2021) An mtDNA mutant mouse demonstrates that mitochondrial deficiency can result in autism endophenotypes. Proc. Natl. Acad. Sci., 118, e2021429118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Lombard, J. (1998) Autism: a mitochondrial disorder? Med. Hypotheses, 50, 497–500. [DOI] [PubMed] [Google Scholar]
- 104. Rossignol, D.A. and Bradstreet, J.J. (2008) Evidence of mitochondrial dysfunction in autism and implications for treatment. Am. J. Biochem. Biotechnol., 4, 208–217. [Google Scholar]
- 105. El-Ansary, A., Bjørklund, G., Chirumbolo, S. and Alnakhli, O.M. (2017) Predictive value of selected biomarkers related to metabolism and oxidative stress in children with autism spectrum disorder. Metab. Brain Dis., 32, 1209–1221. [DOI] [PubMed] [Google Scholar]
- 106. Palmieri, L. and Persico, A.M. (2010) Mitochondrial dysfunction in autism spectrum disorders: cause or effect? Biochim. Biophys. Acta BBA-Bioenerg., 1797, 1130–1137. [DOI] [PubMed] [Google Scholar]
- 107. Boccuto, L., Chen, C.-F., Pittman, A.R., Skinner, C.D., McCartney, H.J., Jones, K., Bochner, B.R., Stevenson, R.E. and Schwartz, C.E. (2013) Decreased tryptophan metabolism in patients with autism spectrum disorders. Mol. Autism, 4, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Tromp, A., Mowry, B. and Giacomotto, J. (2021) Neurexins in autism and schizophrenia—a review of patient mutations, mouse models and potential future directions. Mol. Psychiatry, 26, 747–760. [DOI] [PubMed] [Google Scholar]
- 109. Mohrmann, I., Gillessen-Kaesbach, G., Siebert, R., Caliebe, A. and Hellenbroich, Y. (2011) A de novo 0.57 Mb microdeletion in chromosome 11q13.1 in a patient with speech problems, autistic traits, dysmorphic features and multiple endocrine neoplasia type 1. Eur. J. Med. Genet., 54, e461–e464. [DOI] [PubMed] [Google Scholar]
- 110. Hu, X., Zhang, J., Jin, C., Mi, W., Wang, F., Ma, W., Ma, C., Yang, Y., Li, W., Zhang, H. et al. (2013) Association study of NRXN3 polymorphisms with schizophrenia and risperidone-induced bodyweight gain in Chinese Han population. Prog. Neuro-Psychopharmacol. Biol. Psychiatry, 43, 197–202. [DOI] [PubMed] [Google Scholar]
- 111. Gauthier, J., Siddiqui, T.J., Huashan, P., Yokomaku, D., Hamdan, F.F., Champagne, N., Lapointe, M., Spiegelman, D., Noreau, A., Lafrenière, R.G. et al. (2011) Truncating mutations in NRXN2 and NRXN1 in autism spectrum disorders and schizophrenia. Hum. Genet., 130, 563–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Boyle, M.I., Jespersgaard, C., Nazaryan, L., Ravn, K., Brøndum-Nielsen, K., Bisgaard, A.-M. and Tümer, Z. (2015) Deletion of 11q12.3–11q13.1 in a patient with intellectual disability and childhood facial features resembling Cornelia de Lange syndrome. Gene, 572, 130–134. [DOI] [PubMed] [Google Scholar]
- 113. Yuan, H., Wang, Q., Liu, Y., Yang, W., He, Y., Gusella, J.F., Song, J. and Shen, Y. (2018) A rare exonic NRXN3 deletion segregating with neurodevelopmental and neuropsychiatric conditions in a three-generation Chinese family. Am. J. Med. Genet. B Neuropsychiatr. Genet., 177, 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Fiehn, O., Wohlgemuth, G., Scholz, M., Kind, T., Lee, D.Y., Lu, Y., Moon, S. and Nikolau, B. (2008) Quality control for plant metabolomics: reporting MSI-compliant studies. Plant J., 53, 691–704. [DOI] [PubMed] [Google Scholar]
- 115. Matyash, V., Liebisch, G., Kurzchalia, T.V., Shevchenko, A. and Schwudke, D. (2008) Lipid extraction by methyl-tert-butyl ether for high-throughput lipidomics. J. Lipid Res., 49, 1137–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Edgecomb, R.S., Harth, C.E. and Schneiderman, A.M. (1994) Regulation of feeding behavior in adult Drosophila melanogaster varies with feeding regime and nutritional state. J. Exp. Biol., 197, 215–235, 21. [DOI] [PubMed] [Google Scholar]
- 117. Elkins, T., Ganetzky, B. and Wu, C.F. (1986) A Drosophila mutation that eliminates a calcium-dependent potassium current. Proc. Natl. Acad. Sci., 83, 8415–8419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. van Doorn, J., Ly, A., Marsman, M. and Wagenmakers, E.-J. (2020) Bayesian rank-based hypothesis testing for the rank sum test, the signed rank test, and Spearman’s. J. Appl. Stat., 47, 2984–3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.