

Abstract
Frataxin deficiency in Friedreich’s ataxia results from transcriptional downregulation of the FXN gene caused by expansion of the intronic trinucleotide guanine-adenine-adenine (GAA) repeats. We used multiple transcriptomic approaches to determine the molecular mechanism of transcription inhibition caused by long GAAs. We uncovered that transcription of FXN in patient cells is prematurely terminated upstream of the expanded repeats leading to the formation of a novel, truncated and stable RNA. This FXN early terminated transcript (FXN-ett) undergoes alternative, non-productive splicing and does not contribute to the synthesis of functional frataxin. The level the FXN-ett RNA directly correlates with the length of the longer of the two expanded GAA tracts. Targeting GAAs with antisense oligonucleotides or excision of the repeats eliminates the transcription impediment, diminishes expression of the aberrant FXN-ett, while increasing levels of FXN mRNA and frataxin. Non-productive transcription may represent a common phenomenon and attractive therapeutic target in diseases caused by repeat-mediated transcription aberrations.
Introduction
Friedreich’s ataxia (FRDA) is an autosomal recessive disease primarily caused by biallelic expansion of the guanine-adenine-adenine (GAA) trinucleotides located in intron 1 of the FXN gene (1,2). Although only a few GAA repeats are typically present in FXN loci of unaffected individuals, in FRDA patients this polypurine–polypyrimidine tract expands to hundreds of GAAs and can reach as many as ~1700 copies (1,3).
Since the discovery of the gene in 1996, the cause of the disease was determined, i.e. downregulation of FXN expression, making FRDA an example of a loss of function disease (1). In ~96% of patients, frataxin—the protein encoded by the FXN gene, does not carry any mutations; instead, the severe decrease of its levels leads to this progressive multisystem disorder (4,5). As determined in various tissues, frataxin levels in FRDA patients may decrease to a few percent relative to the amount detected in unaffected individuals (6–8). There is a strong correlation between mature FXN mRNA expression and the length of the expanded GAA repeats, especially the shorter of the two alleles (termed GAA1) (9,10), however, the direct mechanism of the transcriptional impediment is unknown. It is not clear whether transcription machinery is properly assembled on the promoter, how transcription progresses from initiation to elongation at FXN and what is the actual role of long GAAs in transcriptional downregulation of FXN in FRDA (11). Our prior studies, based on comprehensive RNA–seq, demonstrated an impediment of transcription elongation in intron 1 of FXN in FRDA cells (10). Contrary to numerous other repeat expansion disorders, an effect of the repeat expansions on intron retention has not been detected in FRDA samples (12), suggesting that traversing the expanded GAAs rather than later transcript maturation steps is impaired in FRDA.
Prior chromatin immunoprecipitation (ChIP) studies demonstrated a change of the chromatin environment at the FXN gene linked to expansion of the GAA repeats (13–15). Histone modifications associated with active transcription in unaffected control samples are replaced with those characteristic for transcriptionally silenced DNA in FRDA cells. These changes are localized to the regions upstream and downstream of the repeats and do not spread toward the FXN promoter/transcription start site (TSS) region (10,14–16). Furthermore, increased CpG methylation has been detected in a small differentially methylated region (DMR) in intron 1 of FXN upstream of the expanded GAAs (17). Again, the promoter and associated CpG island are not differentially methylated between control and FRDA samples. Combined, results of prior studies suggest that transcription is initiated less efficiently in FRDA cells and events downstream of the TSS are responsible for transcriptional deficiency in cells harboring expanded GAAs (10,16,18,19). Considering that the earlier described chromatin/DNA changes occur within the relatively short distance separating the TSS from GAAs (~1.5 kb), the logical question is: what is the fate of a transcript produced by RNA polymerase II (Pol II) molecules traversing an unfavorable chromatin environment, possible non-canonical DNA and DNA–RNA structures or simply homogeneous long tracts of GAA triplets that could span over 4000 bp? Answering this question is essential not only for a better understanding of the molecular events leading to FXN deficiency and FRDA but, more importantly, for the judicial design of potential therapeutic strategies based on reactivation of endogenous FXN transcription.
Herein, by applying various unbiased transcriptome analyses, in combination with genome editing and antisense oligonucleotide (ASO) treatment, we demonstrated that Pol II is recruited to the FXN promoter in FRDA cells, however, transition to productive elongation is hampered in these cells harboring expanded GAAs. Furthermore, combined RNA-seq, precision nuclear run-on sequencing (PRO-seq) and massive analysis of cDNA ends sequencing (MACE-seq) analyses showed the presence of a transcription termination zone in intron 1, upstream of the expanded GAAs indicating premature termination of RNA synthesis. We revealed that this premature termination results in forming a short, stable and aberrantly spliced transcript termed FXN early terminated transcript (FXN-ett). Targeting long GAA repeats with GAA-specific ASOs or excision of the expanded GAA repeats significantly increased FXN mRNA transcription and abrogated the formation of the aberrant, short transcript. Results of this work represent the first evidence that expanded repeats, likely via non-canonical stable DNA/DNA–RNA conformations or formation of a phase-separated domain, redirect the transcription program and stimulate the formation of an alternatively spliced and polyadenylated transcript, thus resulting in further decrease of the mature functional FXN mRNA and frataxin.
Results
Recruitment of RNA polymerase II to the FXN promoter is unaffected in FRDA cells
Our prior studies demonstrated that the promoter region required for efficient transcription of the human FXN gene extends from approximately 80 bp upstream of the TSS, through exon 1 into approximately 115 bp of intron 1 (20). The GAA tract is located 1340 bp from the exon 1—intron 1 junction, thus in proximity to the TSS. Considering that full maturation of the transcription machinery is achieved several thousand base pairs downstream of the TSS (21), expanded GAAs are encountered by an immature transcription complex. In addition, our prior analyses of RNA-seq data demonstrated that the transcription elongation rate at intron 1 of FXN is decreased in the presence of the expanded GAA repeats (10).
To determine the transcriptional status of the FXN locus, we first performed ChIP experiments using an antibody specific for total Pol II, followed by next-generation sequencing. We utilized induced pluripotent stem cells (iPSCs) derived from control and FRDA fibroblasts (FRDA 1 and CTRL 1, Supplementary Material, Table S2), with confirmed GAA expansions of ~600 repeats on both alleles and decreased FXN mRNA/protein levels compared with CTRL 1 cells (Fig. 1A–C). Frataxin is a ubiquitously expressed gene with iPSCs expressing a relatively high level of the FXN mRNA, albeit still significantly decreased by expanded GAAs (Fig. 1B). Therefore, they represent a good model to determine the transcription dynamics at the FXN locus in both control and FRDA cells. ChIP-seq analysis revealed similar occupancy of the FXN gene promoter region by Pol II in CTRL and FRDA cells, indicating that recruitment of the transcription machinery to the FXN promoter region is not affected in FRDA (Fig. 1D). Discrete bimodal distribution of Pol II reads (22) at the TSS and proximal pausing site was observed in both FRDA 1 and CTRL 1 samples (Fig. 1E). Our prior studies demonstrated that the FXN promoter sequence includes exon 1 and an adjacent fragment of intron 1 (20). This fragment, critical for transcription, was occupied by Pol II in FRDA and CTRL cells. However, in controls an additional region spanning ~70 bp and extending further into intron 1 of FXN was observed (Fig. 1E, blue bar). This region may represent Pol II transitioning into the active elongation phase in cells lacking expanded GAA repeats and indicates extended Pol II pausing in FRDA cells. As expected, Pol II ChIP-seq signal across the entire gene is low, not allowing for a detailed analysis of transcription progression at FXN.
Figure 1.
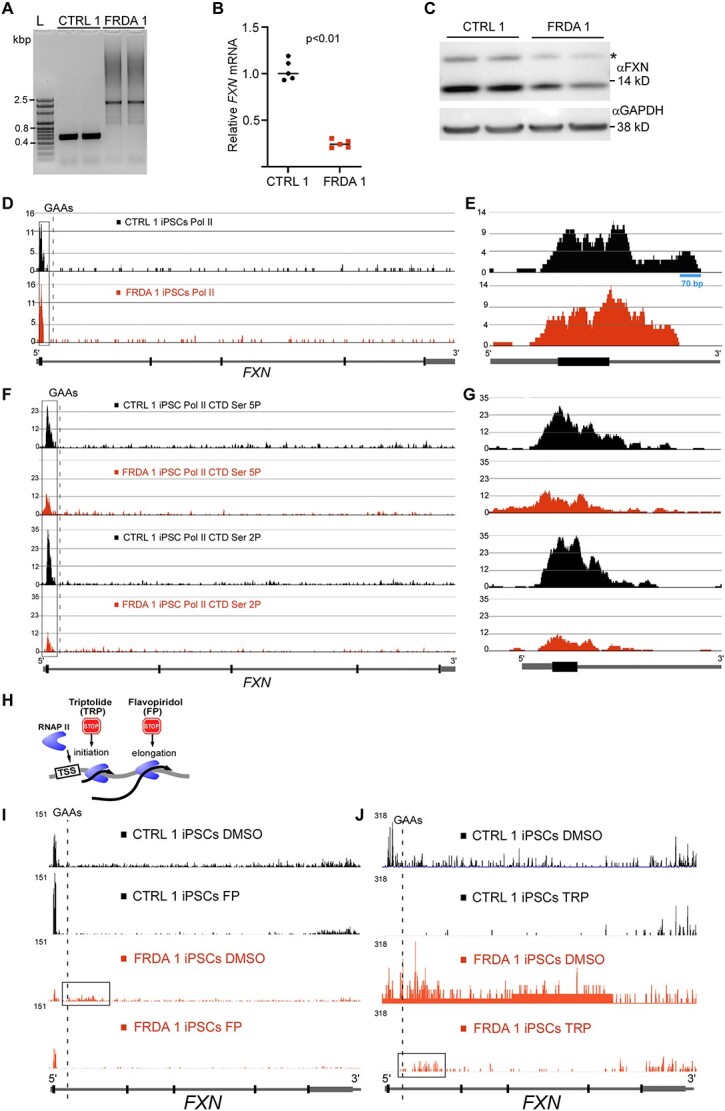
GAA repeats affect transition from the transcription initiation to productive elongation. (A) Size of the GAA repeats determined by PCR. Analysis of two control (CTRL 1) and two Friedreich’s ataxia (FRDA 1) clones of the same iPSC line are shown. Both mutated FXN alleles harbor ~600 GAAs. (B) Expression of FXN mRNA measured in FRDA 1 and CTRL 1 cells by qRT-PCR. Data from two independent qRT-PCR experiments are shown. Statistical significance was determined using the Student’s t- test. (C) Western blot analysis of frataxin protein expression in FRDA 1 and unaffected CTRL 1 iPSCs. Analysis of two independent clones is presented. GAPDH represents a loading control. Mature FXN migrates below the 14 kDa marker; asterisk denotes the intermediate form of frataxin, prior to the final maturation cleavage. (D) Total RNA Pol II ChIP-seq tracks at the FXN gene (schematic of the gene shown below tracks). Read density was normalized by MACS2. Data for control iPSCs are shown in black and for FRDA in red. Promoter region is boxed, dashed line indicates location of the GAA tract. (E) Magnification of the ChIP-seq tracks at the FXN promoter region for total Pol II reactions. (F) ChIP-seq tracks for Pol II phosphorylated at serine 5 (Ser 5P) and 2 (Ser 2P) of the CTD. Data for control iPSCs are shown in black and for FRDA in red. Independent ChIP reactions for each sample-antibody pair were performed 3–5 times, DNA was combined before NGS library preparation and sequencing. Promoter region is boxed, dashed line indicates location of the GAA tract. (G) Magnification of the ChIP-seq tracks at the FXN promoter region for Pol II CTD Ser 5P and Ser 2P. (H) Illustration of TRP and FP effects on Pol II progression. (I) Effect of elongation inhibitor FP on FXN transcription determined by PRO-seq. DMSO reactions represent vehicle controls. Black tracks represent CTRL 1 and red tracks FRDA 1 iPSCs. Schematic of the FXN gene is shown in the following. PRO-seq signal was normalized between all samples as described in Methods section. The region of intron 1 enriched in PRO-seq reads in FRDA is indicated by a box. (J) Inhibition of transcription initiation by TRP results in accumulation of PRO-seq reads in the 3’UTR as well as in intron 1 (indicated by a box). DMSO reactions represent vehicle controls. Black tracks are CTRL 1 and red tracks are FRDA 1 iPSCs. Schematic of the FXN gene is shown in the following.
Expanded GAA repeats affect the transition from initiation to productive elongation
Total Pol II occupancy does not reflect the actual engagement of the transcriptional apparatus in mRNA synthesis. Determination of the presence of Pol II phosphorylated at the serine 5 (S5P) and serine 2 (S2P) positions of the C-terminal domain (CTD) is an indicator of active transcription initiation/elongation (23,24). ChIP-seq analyses of the same FRDA 1 and CTRL 1 iPSCs demonstrated significant decreases of both Pol II S5P and S2P signals at the FXN promoter region in patient cells (Fig. 1F and G). This indicates that expanded GAA repeats located ~1 kb from the distal end of the FXN promoter affect the transition from the bound, inactive state complex to a transcriptionally active complex. To further evaluate the progression of transcription through the FXN locus in detail, we conducted precision run-on sequencing (PRO-seq) analyses in the presence of transcription initiation [triptolide (TRP)] and elongation [flavopiridol (FP)] inhibitors (Fig. 1H) (25). In agreement with ChIP-seq data, PRO-seq results revealed a lower abundance of active transcription in the promoter region of vehicle-treated FRDA cells compared with controls (Fig. 1I and J; DMSO lanes). Interestingly, in two entirely independent experiments, a noticeable increase of the PRO-seq signal was detected in intron 1 of FXN in patient iPSCs when compared with controls (Fig. 1I and J). Treatment with FP demonstrated that both FRDA and CTRL cells can actively initiate FXN transcription (increased signal at the promoter after FP treatment), although PRO-seq signal in CTRL iPSCs was significantly greater than FRDA cells. As expected, treatment with TRP resulted in an accumulation of PRO-seq reads at a transcription termination region (3’UTR) of FXN in both CTRL and FRDA iPSCs. However, similar to results obtained with vehicle treated cells, a lasting signal was detected within intron 1 of FXN, exclusively in FRDA iPSCs (Fig. 1J, boxed region). This is rather unexpected, as 50 min treatment with TRP typically allows transcribing Pol II to progress through the entire gene body to the transcription termination region as illustrated in CTRL cells. These results suggest a significant inhibition of the progression of the Pol II machinery at intron 1 containing the expanded GAAs or premature termination of transcription at this region.
RNA-seq profiling reveals RNA accumulation upstream of the expanded GAAs in different FRDA cell types
Accumulation of the actively transcribing Pol II at intron 1 should result in a detectable buildup of the RNA-seq reads at FXN in FRDA cells. Indeed, analysis of RNA-seq data obtained from FRDA 1 and CTRL 1 iPSCs (Fig. 2A and C) and primary fibroblasts (Fig. 2B and D) revealed accumulation of sequencing reads in intron 1, predominantly upstream of the expanded GAAs in FRDA cells. In addition, reexamination of previously published RNA-seq data obtained from FRDA and CTRL cardiomyocytes corroborates this finding (26). The intronic reads are also detected in control cells, although fewer than FRDA cells, especially relative to the overall decreased FXN transcription in patient cells (Fig. 2A and B).
Figure 2.

Accumulation of RNA upstream of the expanded GAAs in FRDA cells. (A) RNA-seq tracks of the FXN gene in CTRL iPSCs (black) and FRDA iPSCs (red). Dashed line indicates position of the GAA repeats. FXN gene schematic is shown in the following. (B) RNA-seq tracts displaying reads from the FXN gene in two CTRL (black) and two FRDA (red) primary fibroblast lines. (C, D) magnifications of the boxed regions of FXN upstream of the GAA repeats shown in (A) and (B), respectively. All sequencing was conducted using polyA capture, reads were mapped using STAR aligner and signals were normalized by DESeq2.
GAA expansion induces aberrant transcription termination
To further investigate the source of intronic RNA-seq and PRO-seq signals, we conducted a MACE-seq. Deep MACE-seq allows for precise differential gene expression analyses and enables the identification of novel transcripts and alternative polyadenylation (APA) signals in a strand-specific manner. MACE-seq of FRDA 1 and CTRL 1 iPSCs demonstrated, as expected, a strong PA signal at the 3’UTR (~350 nt downstream of the Stop codon) resulting from termination of canonical, full-length FXN mRNA (Fig. 3A). In addition, in FRDA cells, a signal upstream of the expanded GAA repeats was detected with an intensity comparable to the 3’UTR PA site signal (Fig. 3B). Bioinformatic analyses of the MACE-seq data indeed defined the peak upstream of the GAAs as an APA signal (Supplementary Material, Table S3). This data shows that a short, polyadenylated transcript that prematurely terminates upstream of the GAAs is preferentially accumulated in FRDA cells.
Figure 3.

MACE-seq identifies a novel, FRDA-enriched polyadenylated transcript at the FXN locus. (A) Deep MACE-seq tracks obtained from CTRL (black) and FRDA (red) iPSCs. An excess of 80 M sequencing reads was generated per sample and mapped using STAR aligner. Peaks represent putative 3’ ends of polyadenylated transcripts. Termination signal of the canonical, full length FXN mRNA is located in the 3’UTR. Bioinformatic analyses identified the presence of an alternative polyadenylated RNA terminated upstream of the expanded GAAs (see also Supplementary Material, Table S3). A schematic of the FXN gene is shown in the following. The dashed line indicates the location of the GAA repeats and the boxed region is magnified in (B). (C) Verification of directionality of the transcripts generated within the FXN locus. All forward strand reads are shown in blue and reverse in green. No antisense transcript could be detected at the FXN locus in FRDA or CTRL iPSCs. Proposed location of FAST-1 antisense RNA (27) relative to the FXN mRNA 5’UTR, exon 1 and GAA repeats is shown. Scale of the image is indicated below and examples of reads mapped to the forward and reverse strand in two RNAs, SPEN and ZBTB17, transcribed in opposite directions.
Reports of a polyadenylated antisense transcript overlapping with exon 1 and the 5’UTR of FXN exist (27,28), therefore we determined strand-specificity of the MACE-seq signal at FXN. No reads derived from the reverse strand (i.e. RNA transcribed in the opposite direction to FXN) were mapped (Fig. 3C), confirming that the short transcript terminated upstream of the GAAs is synthesized in the same direction as the full-length FXN mRNA.
Prematurely terminated FXN transcript is alternatively spliced
Next, to confirm expression of the short, prematurely terminated transcript and determine its sequence, we utilized the 3′ rapid amplification of cDNA ends (3’ RACE) approach. We identified a single, predominant ~900 bp long 3’ RACE product in CTRL cells. The length of this fragment is as expected for canonical FXN mRNA, considering the location of the FXN exon 1 primer and the known position of the major PA site in the FXN 3’UTR (Fig. 4A). Parallel analyses in FRDA iPSCs revealed a shorter ~700 bp PCR product in addition to the ~900 bp band (Fig. 4A). This 700 bp product is about half the length of the transcript predicted based on the APA identified upstream of the GAA repeats (Fig. 3; distance between the beginning of intron 1 and the first GAA is 1339 bp). The same 3’ RACE patterns were observed in multiple FRDA cell lines, derived from different patients (Fig. 6E). Next, we determined the sequence of the transcript by cloning individual gel-extracted PCR products into a TA cloning vector and performing Sanger sequencing. Bands 1 and 2 (Fig. 4A and B) represent the longer fragment (common for FRDA and CTRL cells), whereas band 3 designates a shorter, FRDA-enriched product. Sequencing of 80 individual clones revealed that the longer transcript (bands 1 and 2) represents predominantly canonical full-length FXN mRNA containing exons 1 to 5 (Fig. 4C, isoform Type I and Supplementary Material, Fig. S1). Contrarily, most transcripts (23 out of 25 clones sequenced) in FRDA cells represented by the shorter PCR product (band 3) encode RNA containing exon 1, a fragment of FXN intron 1 and terminate upstream of the expanded GAAs (Fig. 4C, isoform Type IV). Interestingly, the intronic part of this transcript lacks the first 683 nt, immediately downstream of the end of exon 1 (Fig. 4C and D and Supplementary Material, Fig. S2), thus explaining the discrepancy between the length of the expected (~1.4 kb) and experimental (~0.7 kb) PCR product obtained from the FXN transcript terminated upstream of the expanded GAA repeats. This result also indicated that the truncated FRDA-enriched transcript (FXN-ett) undergoes alternative splicing that removes a fragment of 683 nt proximal to exon 1 (Fig. 4D and Supplementary Material, Fig. S2). Additionally, examination of RNA-seq data from FRDA iPSCs revealed the presence of reads containing sequences resulting from the alternative splicing event (Supplementary Material, Fig. S3). The splicing machinery utilizes a canonical exon1/intron 1 splice donor site and recognizes the downstream alternative acceptor (SA) site. In silico alternative splicing prediction using Alternative Splice Site Predictor (29) identified the sequence at position 683 nt of intron 1 as the most likely alternative splice acceptor site in the region upstream of the GAAs (Supplementary Material, Fig. S4). Additionally, RNA structure analysis (mFold) (30) of the entire RNA fragment from the TSS to GAAs localizes this alternative SA in a single-stranded, accessible stem loop structure (Supplementary Material, Fig. S5). In addition to the predominant variants I and IV, rare FXN transcript isoforms, with modified 3′-ends downstream of exon 4 were identified in our sequencing experiments (Fig. 4C, isoforms Type II and III). All variants represented by a single clone were excluded from further analyses. In conclusion, we identified an FRDA-enriched transcript, FXN-ett, that is alternatively spliced and prematurely terminated in direct proximity to the expanded GAAs.
Figure 4.

FXN-ett is alternatively spliced and enriched in FRDA cells. (A) Results of 3’ Rapid Amplification of cDNA Ends PCR performed using FRDA and CTRL iPSCs. Results of two independent experiments are shown. NoRT represents control reactions lacking reverse transcriptase. L—1 kb Plus DNA ladder (BioLine). Bands indicated by 1, 2 and 3 were excised from the agarose gel, cloned and sequenced. (B) Isoforms of the FXN transcripts identified by DNA sequencing of 80 clones. Three independent 3’RACE/cloning experiments were conducted. Only isoforms represented by two or more independent clones are shown. Isoforms I and II were identified by sequencing the longer PCR products (bands 1 and 2), isoforms III and IV were identified only in the shorter PCR product (band 3). Isoform I represents canonical FXN mRNA (Supplementary Material, Fig. S1). The pie chart illustrates proportions of different isoforms identified in the experiment but it is not a quantitative representation of relative abundance of different isoforms transcribed at the FXN locus. (C) Schematic illustrations of isoforms I-IV. Transcript regions (exons, introns, 3’UTR) are not shown to scale. Lengths of the 3’UTR, intron 4 and intron 1 are measured from the end of the preceding exon. Spliced fragments in isoforms III and IV are depicted by a thin line. Isoform IV—the predominant 3’ RACE product identified in band 3 was termed FXN-ett. (D) DNA sequencing result of the FXN-ett transcript. A junction between the last nucleotide of exon 1 (165 nt relative to ‘A’ of the ATG initiation codon) and 848 nt of the transcript (relative +1 nt ‘A’ of the ATG) revealed splicing of a 683 nt fragment of intron 1. The full sequences of the FXN-ett transcript as well as the canonical Type I isoform are presented in Supplementary Material, Figs S1 and S2.
Figure 6.

Excision of the pathogenic expanded GAA repeats and ASO treatment increases FXN mRNA expression and alleviates formation of the early terminated transcript. (A) Schematic illustration of the exon 1—intron 1 region of the FXN gene. The exact locations of the gRNA and Cas9 cut sites (gUP—upstream of the GAA tract, gDW—downstream of the GAAs) are shown. Image not drawn to scale. Details regarding sequence and validation of the edited iPSC clones are presented in Supplementary Material, Figure S7. (B) Excision of the expanded GAAs from both alleles of FXN increases FXN mRNA expression to the level observed in unaffected controls (green symbols) and approximately 3-fold higher than in parental (P), unedited cells (gray symbols). Two independent edited ΔGAA clones are shown for each FRDA iPSC line: for FRDA 2—3B12 and 3C7; for FRDA 3—2B1 and 2D3. Expression of the FXN mRNA in CTRL 1 and FRDA 1 are shown as reference. Results of two independent experiments with two parental and 4 edited clones are shown; Student’s t-test was used to determine statistical significance. (C) Western blot analysis of frataxin expression in parental (P) and edited FRDA iPSCs. Both edited clones for FRDA 2 and FRDA 3 iPSCs are shown. Signal from mature frataxin is indicated by αFXN. Asterisk designates the 19 kDa intermediate form of frataxin. Ponceau S staining is shown as a loading control. (D) Expression of the FXN-ett RNA in parental (P, gray symbols) and edited FRDA 2 and FRDA 3 cells (green symbols). For comparison, levels of FXN-ett RNA in FRDA 1 and unaffected CTRL 1 is shown. Results of two independent experiments with two parental and four edited clones are presented; Student’s t-test was used to determine statistical significance. (E) Results of 3’ RACE reactions performed using parental (P) and isogenic edited ΔGAA FRDA 2 and ΔGAA FRDA 3 iPSC lines. Arrowheads indicate migration of FXN-ett detected only in parental, unedited cells. L designates 1 kb ladder (Bioline). (F) Effect of a GAA targeting gapmer ASO (Gap17, green symbols) on expression of FXN mRNA (left image) and FXN-ett RNA (right image). Anti MALAT 1 gapmer (39) was used as a control (gray symbols). Two different FRDA primary fibroblast lines (F4 and F7, see Supplementary Material, Table S2) were transfected with 3 nM ASO and RNA expression was determined using qRT-PCR. Significance was calculated using Student’s t-test. Sequences of the ASOs and incorporated chemical modifications are listed below and in (39). Orange designates 2’-O-methoxy-ethyl bases (2’-MOE); m—methylated C, black—phosphorothioate backbone, underlined—phosphodiester backbone.
FXN-ett level correlates with the length of the longer of the two GAA alleles
Based on the FXN-ett splicing information, we established a qRT-PCR assay to quantitatively measure its expression in FRDA and CTRL cells (Fig. 5A and B). A set of primary FRDA (14 lines) and control (12 lines) fibroblast lines from our FRDA Cell Line Repository was used to determine FXN-ett expression. As demonstrated before, FRDA fibroblasts express significantly (P < 0.0001) less FXN mRNA than controls (as measured by exon 3-exon 4 qRT-PCR assay, Fig. 5A and C), however, they exhibit significantly higher levels of the FXN-ett RNA (Fig. 5D, P = 0.0048). FRDA is an autosomal recessive disease with most known cases (~96%) attributed to the expansion of GAA repeats on both FXN alleles (1,4). A convention has been adopted terming the shorter of the two expanded alleles GAA1 and the longer GAA2 (9). To our knowledge, FRDA traits (clinical symptoms or molecular characteristics (e.g. disease onset, FXN mRNA and protein level, etc.) correlate significantly with the length of the shorter tracts (GAA1) (9,31). Likewise, the FXN mRNA expression level in the set of FRDA fibroblasts analyzed in this study correlates well with GAA1 (Fig. 5E). On the contrary, the level of FXN-ett correlates with the number of repeats in GAA2, not GAA1 (Fig. 5F and G). This is, to our knowledge, the first example of a trait in FRDA that shows a stronger correlation with the longer of the two expanded alleles. Moreover, this indicates that GAA tracts of longer lengths are likely to promote early termination of FXN transcription upstream of the GAAs and facilitate the production of FXN-ett.
Figure 5.
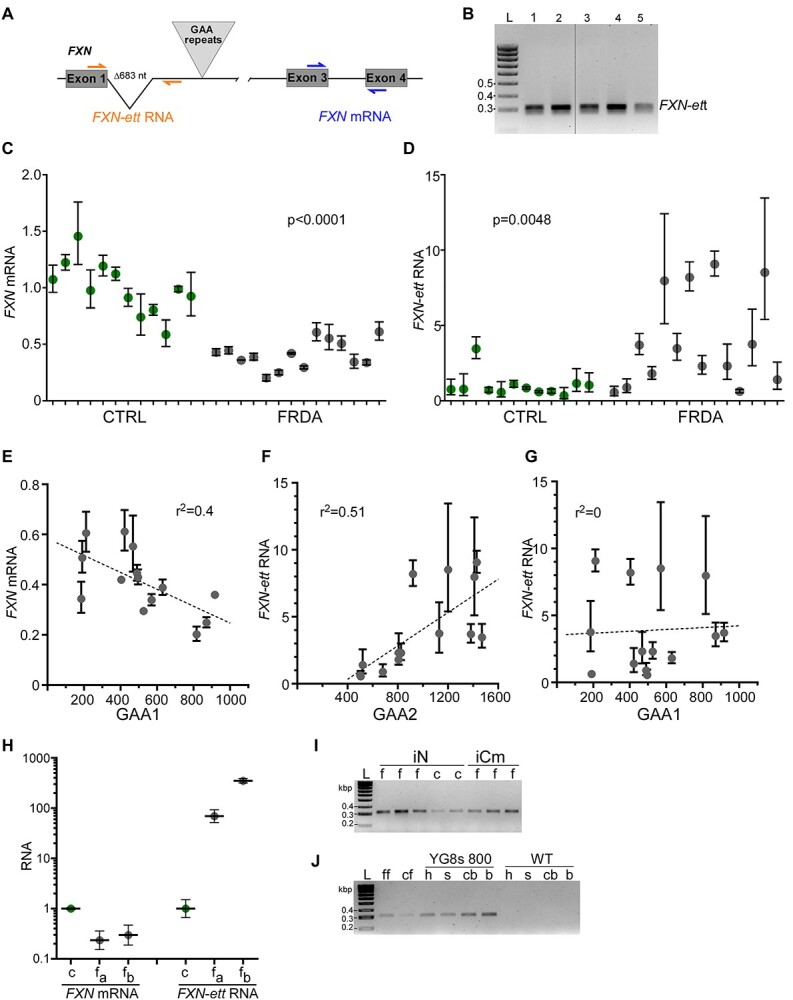
FXN-ett is expressed in different FRDA cells and correlates with GAA repeat length. (A) Location of the qRT-PCR primers used to detect FXN-ett (orange) and canonical FXN mRNA (blue). (B) Example of the FXN-ett RT-PCR product (lanes 1–5); L- represents 100 bp ladder (Bioline) (C) Expression of the FXN mRNA is reduced in FRDA fibroblasts (gray symbols) compared with CTRL lines (green symbols). All qRT-PCR reactions were performed using RNA extracted from 12 different CTRL and 14 different FRDA fibroblast lines. Statistical significance (P < 0.05) was determined by Student’s t-test. (D) FXN-ett is highly upregulated in FRDA fibroblasts. The same samples described in (C) were used for FXN-ett qRT-PCR analyses. (E) An inverse correlation between GAA1 repeat size (shorter of the two expanded GAA alleles) and FXN mRNA expression in 14 FRDA fibroblast lines analyzed in (C). The r2 value for the fit of each line is shown. (F) FXN-ett RNA expression directly correlates with the length of GAA2 (longer of the two expanded GAA alleles) in FRDA fibroblasts and does not correlate with GAA1 length (G). The r2 value for the fit of each line is shown. (H) FXN mRNA and FXN-ett expression in CTRL and FRDA autopsy heart tissues. c—unaffected control (green symbol), fa and fb—samples obtained from two different FRDA patients (gray symbols). (I) FXN-ett is detected in cortical neurons (iN) differentiated from FRDA (f) and control (c) iPSCs as well as FRDA iPSC-derived cardiomyocytes (iCm). L—100 bp ladder. (J) FXN-ett is expressed in the humanized transgenic FRDA YG8s(GAA) > 800 mouse heart (h), spinal cord (s), cerebellum (cb) and cerebrum (b). Amplification of FXN-ett RNA in FRDA (ff) and control (cf) fibroblasts is shown as positive control and wild type mouse tissues served as negative controls.
FXN-ett is stable, expressed in different cells and in FRDA humanized transgenic mice
To establish the relevance of FXN-ett expression to FRDA, we sought to determine FXN-ett expression in other cell types, including patient-derived tissues. Quantitative RT-PCR analyses showed that FXN-ett is highly upregulated in cardiac autopsy tissue obtained from FRDA patients compared with an unaffected control (Fig. 5H). In addition, FXN-ett could be readily detected in iPSC-derived neurons and cardiomyocytes (Fig. 5I). Finally, we also tested whether FXN-ett is present in FRDA humanized transgenic mice. We detected this transcript in the heart, spinal cord, cerebrum and cerebellum from the YG8s 800 mice (FRDA mouse model expressing human frataxin from a single copy YAC transgene harboring ~800 GAAs in FXN intron 1) (32), but not in wild type mice (Fig. 5J). Lastly, we established that the FXN-ett RNA is stable with ~6 h half-life as determined in two different FRDA primary fibroblast lines (Supplementary Material, Fig. S6). Taken together, these results indicate wide expression of a stable FXN-ett RNA in patient tissues and cell lines and in a FRDA animal model harboring expanded GAAs.
CRISPR-Cas9 excision of the expanded GAAs restores FXN expression and decreases FXN-ett expression
Data presented earlier indicate that a block imposed by the expanded GAAs drives premature transcription termination (PTT) and formation of the FXN-ett RNA. To confirm this, we excised both expanded GAA tracts from intron 1 of FXN using CRISPR/Cas9 editing. The gRNAs were designed downstream of the FXN-ett splice acceptor site to avoid interference with potential alternative splicing (Supplementary Material, Fig. S7). The cut sites are located 392 bp upstream and 976 bp downstream of the GAA repeats (Fig. 6A). Editing was confirmed by junction PCR, GAA repeat PCRs as well as DNA sequencing (Supplementary Material, Fig. S7). We edited two different FRDA iPSC lines (FRDA 2 and 3) and two independent ΔGAA clones were obtained for each line (Fig. 6B and C; Supplementary Material, Fig. S7). Excision of the intronic GAAs from both alleles resulted in robust increase of FXN expression both at the mRNA and protein levels in all four edited clones (Fig. 6B and C). Removal of the GAA expansion almost completely abolished formation of the early terminated FXN-ett RNA in all edited clones (Fig. 6D). Furthermore, results of the 3’ RACE experiment conducted using parental and GAA-edited lines revealed absence of the FXN-ett band, confirming that the expanded GAAs are responsible for the transcriptional impediment during early elongation and formation of the aberrantly spliced, prematurely terminated FXN-ett RNA.
Targeting expanded GAAs with ASOs decreases FXN-ett expression and elevates FXN levels
It has been demonstrated that interfering with non-canonical DNA and/or DNA–RNA structures formed by expanded GAAs via repeat-targeting ASOs increases FXN expression (33–38). We hypothesized that direct targeting of the expanded GAAs will facilitate a greater rate of Pol II passage through the expanded GAAs, resulting in increased production of FXN mRNA and decreased levels of FXN-ett RNA. Indeed, transfection of FRDA primary fibroblasts with Gap17 gapmer ASO (39) increased FXN mRNA expression relative to the control ASO, MALAT-1 (Fig. 6F). In agreement with our predictions, releasing the block imposed by the expanded repeats resulted in a significant decrease of the FXN-ett RNA level, suggesting that DNA and/or DNA/RNA conformations adopted by expanded GAAs impede progression of FXN transcription (Fig. 6F).
Discussion
A block in transcription progression caused by expanded GAA repeats has been recurrently postulated as the mechanism of FXN mRNA deficiency in FRDA. However, neither direct evidence for a repeat-induced transcription block nor the exact underlying cause of the impediment has been conclusively demonstrated in patient cells. These two outstanding issues separate us from a complete understanding of the molecular basis of FRDA pathogenesis and possibly, from successful therapies that would specifically reactivate FXN expression in patient cells.
Herein, we comprehensively addressed the first of these two challenges and, using a battery of unbiased approaches and different FRDA model systems and patient samples, we showed that transcription progression is indeed affected at the FXN locus resulting in the GAA-length dependent formation of an aberrant, early terminated and alternatively spliced transcript, FXN-ett. Interestingly, PTT in FRDA occurs immediately upstream of the expanded GAAs and correlates with the length of GAA2, the longer of the two expanded GAA alleles.
This fact is unusual as practically all disease characteristics (both molecular and clinical) defined thus far correlate with GAA1 length (5,10,40), but not unexpected. Long GAAs are likely to significantly contribute to aberrant transcription termination and formation of FXN-ett when compared with shorter GAA tracts that are, in turn, responsible for expression of residual, canonical FXN mRNA in FRDA cells.
In vitro studies on long polypurine-polypyrimidine tracts indicate that GAA-TTC repeats can adopt stable triple helical DNA structures (41–43). Furthermore, evidence exists that triple stranded DNA–RNA confirmations (R-loop like) are formed in FRDA cells (35,44,45), however, recent controversies on the antibody-based methods used to detect R-loops indicate that formation of RNA–DNA hybrids at expanded GAAs may need to be verified by additional methods (46–48). Both stable triplexes and R-loops can impede Pol II movement and stimulate PTT (49–51). The expanded GAAs in FRDA frequently exceed 1000 triplets equaling more than 3000 bp of typically uninterrupted polypurine-polypyrimidine DNA. In addition, it has been shown in different model systems that GAA expansion causes changes in the local chromatin environment, predominantly increased representation of histone marks associated with heterochromatin, such as H3K9me3 (13,14). This is accompanied by accumulation of heterochromatin proteins HP1 alpha and gamma (HP1α and HP1γ) upstream of the GAA repeats in FRDA cells when compared with controls (27), revealing a possibility that binding of these proteins to the GAAs (or non-canonical DNA/DNA–RNA conformations adopted by these sequences) may facilitate transcriptional repression. Recent studies showed that human HP1α can form phase-separated condensates (52–54). Accumulation of HP1α, its self-organization and interaction with histone H3K9me3 (overrepresented in FXN intron 1) can lead to formation of heterochromatin phase-separated condensates at the expanded GAAs that efficiently repel transcription machinery (55). These condensates are likely to be dispersed, at least to a certain extent, during DNA replication, allowing for transcription machinery to traverse expanded GAAs more efficiently during DNA synthesis compared with non-dividing, terminally differentiated cells. Therefore, contribution of the FXN-ett may be underestimated in studies conducted predominantly in dividing cells or in immature iPSC-derived neurons or cardiomyocytes. Data from patient tissues presented here demonstrate that the transcription defect maybe more severe in terminally differentiated FRDA cells, resulting in enhanced PTT, formation of the FXN-ett, and ultimately profound FXN mRNA downregulation. Limited frataxin expression data available from terminally differentiated tissues are in line with these findings (6,7). This is also consistent with observations made in animal FRDA models showing progressive, age-dependent decreases of Fxn expression in disease-relevant tissues (56).
Transcription elongation rate is linked to upstream events occurring on nascent RNA, and frequently the speed of transcription determines the ‘window of opportunity’ for processes of alternative splicing or polyadenylation (57). This model can be directly applied to the FXN gene harboring expanded GAA repeats, as our prior work indicated that transcription elongation speed is decreased in intron 1 in FRDA cells (10). Transcription deceleration creates an opportunity for the use of strong alternative splicing signals resulting in generation of aberrantly spliced FXN-ett. In addition, presence of the GAA-mediated impediment stimulates the use of alternative PA and premature termination of transcription. Greater speed of transcription through FXN in control cells, not affected by expanded GAAs, limits the opportunity for alternative splicing and polyadenylation, hence resulting in synthesis of the full length, canonical FXN mRNA (Fig. 7A and B). Targeting the expanded GAA region with ASOs or GAA-specific polyamides was demonstrated to interfere with non-canonical DNA/DNA–RNA conformations and increased expression of the FXN mRNA (35,58). More efficient progression of the transcription machinery through the GAA repeat region reduced opportunities for alternative splicing and formation of FXN-ett (Fig. 7C). Lastly, removal of the intronic GAA tracts (on both FXN alleles) using CRISPR/Cas9 excision resulted in restoration of FXN mRNA expression to the level observed in control cells and dramatically reduced FXN-ett levels, thus directly linking early transcription termination and formation of FXN-ett to the presence of a long GAA tract (Fig. 7D). Importantly, our editing strategy removed the expanded GAA region and flanking sequences, however the alternative SA site utilized to form FXN-ett was not affected (Supplementary Material, Fig. S7).
Figure 7.

Suppressing expanded GAA-induced early transcription termination by different therapeutic strategies. (A) Model of FXN transcription in control cells harboring short GAA repeats. Mainly canonical, full-length FXN mRNA is synthesized. (B) In FRDA cells with expanded GAAs, transcription of the FXN mRNA is reduced and the level of short, early terminated FXN-ett RNA is increased. (C) Gapmer ASO targeting GAAs alleviates the transcription block, decreases the amount of FXN-ett RNA while elevating the level of full-length FXN mRNA. (D) Excision of the GAAs removes the transcriptional impediment, restores FXN mRNA synthesis and eliminates FXN-ett RNA expression.
An additional possible contributor to the alternative splicing and early transcription termination at FXN in FRDA cells is DNA methylation, as the link between splicing and DNA methylation has been well established (59,60). Recently, a DMR was identified in intron 1 of FXN, upstream of the GAAs. This DMR, spanning 10 CpGs (~200 bp) immediately upstream of the SA of FXN-ett, is almost completely methylated in patients’ DNA when compared with unaffected controls (17). Mechanistically, a significant increase of DNA methylation in the FRDA-DMR may lead to enrichment of methyl-CpG binding protein 2 occupancy, recruitment of histone deacetylases and consequently alter Pol II kinetics to promote alternative splicing of the FXN-ett RNA.
What could be the consequences of FXN-ett synthesis? The most obvious direct impact of PTT on frataxin levels is unproductive utilization of the transcriptional machinery resulting in generation of a non-functional transcript, therefore decreasing the expression of full length FXN mRNA. Considering the shorter half-life of FXN-ett relative to FXN mRNA, quantitative analyses of its expression suggest that most of the FXN transcriptional activity may be directed toward synthesis of this aberrant transcript. Overall, successful transcription of an entire gene is a rather rare event. It has been determined that only ~1% of initiation events result in mRNA production and an overwhelming majority of initiations results in PTT at the promoter (61,62). At FXN, this low success rate is further exacerbated by PTT caused by expanded GAAs, resulting in profound downregulation of the FXN mRNA. The process and its consequences are strongly associated with the length of the pathogenic GAA region. Another possibility exists that FXN-ett forms stable DNA/RNA hybrid-like interactions with chromatin at the FXN gene, actively impeding progression of upstream transcription machineries. Closer examination of the FXN-ett half-life data suggests that blocking transcription by actinomycin D reduces levels of FXN-ett to ~50% of the starting amount within ~4 h, while further treatment (up to 8 h) does not reduce the level of this transcript. This is not observed for the MYC mRNA control. Such decay kinetics suggests that a fraction of FXN-ett may be resistant to degradation, perhaps owing to stable interaction with chromatin at the FXN locus.
In addition to the mechanistic insight on the FRDA FXN transcriptional defect, FXN-ett, as a novel, FRDA-enriched RNA, represents a very attractive target for therapeutic intervention. Presently, we do not have evidence for a direct pathogenic role of FXN-ett, however formation of this stable transcript may interfere with transcription of the full-length canonical FXN pre-mRNA thus reducing overall transcriptional output of the FXN locus in FRDA cells. If FXN-ett forms stable interactions with chromatin, its targeting and cleavage by specifically designed gapmer ASOs could stimulate mRNA decay pathways clearing the DNA template for subsequent Pol II complexes and enhancing the probability of productive FXN transcription. Importantly, owing to alternative splicing, FXN-ett harbors a unique RNA sequence, different from the canonical FXN pre-mRNA, thus enabling its specific targeting. An alternative, or perhaps even more attractive therapeutic strategy, owing to the current state of the ASOs used in the clinic, is the aberrant, non-productive splicing of the FXN mRNA resulting in formation of FXN-ett. Recently, a dose-dependent increase of productive transcription and protein levels was observed after targeting different types of non-productive splicing events (63). Hence, a strategy of blocking non-productive splicing of the FXN-ett RNA may lead to a natural upregulation of FXN transcription and increase of frataxin production.
In conclusion, identification of a disease-enriched, early terminated transcript at FXN not only represents a significant step forward in understanding molecular pathology of the disease but also opens completely novel and disease-specific possibilities for therapeutic intervention.
Materials and Methods
Cell lines and cell culture conditions
Protocol for derivation of human primary fibroblasts was approved by Institutional Review Board (IRB) at the University of Alabama at Birmingham (IRB N131204003). All the human FRDA and control fibroblast lines were obtained and cultured as described before (35). Briefly, biopsy material was washed 2–3 times with ~5 ml HBSS with 1X penicillin and streptomycin (Thermo Fisher Scientific), cut into small pieces and incubated in an enzyme mixture containing Collagenase IV 1 mg/ml, dispase 1 mg/ml (Stem Cell Technologies) and 0.05% trypsin EDTA (Thermo Fisher Scientific) for 30 min at 37°C with gentle shaking. Digested biopsy fragments were transferred to 15 ml conical tubes. A double volume of complete media [DMEM/F12 (Thermo Fisher Scientific), 2 mM glutamine, 20% FBS (Hyclone), 1X penicillin/streptomycin and 1X of non-essential amino acids (Thermo Fisher Scientific)] was added prior to centrifugation. Pellets were resuspended in complete media and plated onto gelatin-treated 6-well plates. Typically, outgrowths of fibroblasts were observed 7–14 days after initial plating. Established cultures were maintained in DMEM media containing 15% FBS and 1X non-essential amino acids at 37°C, 5% CO2.
Reprogramming of fibroblasts into iPSC was approved by UAB (IRB-300003993). The iPSC lines used in this study were reprogrammed using retroviral expression of reprogramming factors (FRDA 1 and CTRL 1 iPSCs) or using a Sendai virus approach (FRDA 2 and FRDA 3). Retrovirus reprogramming of fibroblasts was performed as we described before (64). In brief, fibroblasts were transduced with retroviral vectors encoding Oct3/4, Sox2, Klf-4 and c-Myc transcription factors. After 3–4 weeks of culture, iPSC colonies were manually picked and transferred to Matrigel (hESC-qualified Matrix BD)-coated 24-well plates containing mTeSR1 medium (Stem Cell Technologies) supplemented with 10 μM ROCK inhibitor Y27632 (Stemgent). Long-term culture of iPSCs was conducted under feeder-free conditions in the mTeSR1 media according to manufacturer recommendations.
Sendai virus reprogramming of FRDA iPSCs was performed using CytoTune iPS 2.0 Sendai Reprogramming Kit (Thermo Fisher Scientific) according to the manufacturers’ instructions. 70–80% confluent fibroblast cultures were transduced with CytoTune™ 2.0 KOS, CytoTune™ 2.0 c-Myc, and the CytoTune™ 2.0 Klf4 viruses at an appropriate MOI. After 24 h media was replaced with fresh fibroblast media followed by complete media changes every other day. Seven days after transduction, cells were re-plated on vitronectin-coated plates in fresh fibroblast media. Media was exchanged 24 h later for Essential 8™ medium (Thermo Fisher Scientific) followed by daily media changes. Colonies typically appeared 16–21 days after transduction. After 21 days, colonies were manually picked and transferred to Matrigel coated 48-well plates containing mTeSR1 medium supplemented with 10 μM ROCK inhibitor Y27632 (Stemgent). Long-term culture of iPSCs was conducted under feeder-free conditions in mTeSR1 media according to manufacturer recommendations.
Drosophila S2 cells (Thermo Fisher Scientific) used as spike-in control for PRO-seq experiments were grown in Schneider’s Drosophila medium supplemented with 10% FBS at room temperature in ambient CO2 as a semi-adherent monolayer in tissue culture flasks.
RNA isolation, qRT-PCR, mRNA half-life
Total RNA from cell pellets was isolated using the RNeasy Mini Kit (Qiagen). All research involving patient/patient tissues was approved by the IRB at The University of Alabama at Birmingham (IRB Protocols: N160923005 and N160922011). Autopsy specimens of heart were obtained from the FRDA tissue repository maintained at the Veterans Affairs Medical Center in Albany, NY, USA. Frozen heart tissue was homogenized by grinding thoroughly in liquid nitrogen followed by homogenization (Dounce) 50 times in RLT buffer, then the lysate was applied to RNeasy Mini Kit to extract total RNA. All extracted RNA was treated with TURBO DNA-free kit (Thermo Fisher). The qRT-PCR reactions were performed in triplicate with the Power SYBR Green RNA-to-CT 1-Step Kit (Thermo Fisher Scientific). Control reactions without reverse transcriptase (NoRT) were always included. Reactions were performed using StepOne Plus or ViiA 7 Real Time PCR instruments (Applied Biosystems). All primers used for qRT-PCR are listed in Supplementary Material, Table S4.
For mRNA half-life analyses, cells were treated with 5 μg/ml actinomycin D (Sigma-Aldrich). RNA was isolated at t = 0, 0.5, 1, 4 and 8 h and relative expression of FXN-ett RNA was analyzed by qRT-PCR. Myc-mRNA was used as a positive control. Half-life was calculated according to (65).
Western blot
Cell lysates were prepared using Passive Lysis Buffer (Promega) supplemented with Protease Inhibitor Cocktail (Millipore-Sigma). After three freeze–thaw cycles, the lysates were centrifuged at 13 000 g for 15 min at 4°C. Protein concentration was determined spectrophotometrically using Protein Assay Dye Reagent (Bio-Rad). Twenty micrograms of whole cell extract were electrophoresed on NuPAGE 4–12% Bis-Tris gels (Thermo Fisher Scientific) and then transferred onto 0.2 μm nitrocellulose membrane (Bio-Rad). Membranes were stained with Ponceau S and imaged using a Chemi Doc MP Imaging System (Bio-Rad). Frataxin was detected using anti-FXN at 1:1000 (Proteintech) for 12 h at 4°C. Actin was detected using anti-ACTIN monoclonal antibody (Santa Cruz) at 1:2000 for 12 h at 4°C. Horseradish peroxidase-conjugated rabbit anti-mouse immunoglobulin (GE Healthcare Life Science) and donkey anti-rabbit immunoglobulin (GE Healthcare Life Science) were used as secondary antibodies and incubated for 1 h at room temperature at 1:5000. Signal was quantified using Image Lab 6.0.1 software (Bio-Rad).
Chromatin immunoprecipitation sequencing
Chromatin immunoprecipitation sequencing (ChIP-seq) was performed according to a previously published protocol (15) with some modifications. 5 × 106–1 × 107 cells were cross-linked with 1% formaldehyde for 10 min and quenched in 125 mM glycine for 5 min at room temperature. Cells were washed twice with ice-cold PBS and collected by scraping from the cell culture dish. Nuclei were isolated by homogenizing (Dounce) 25 times in Farnham Lysis buffer (5 mM PIPES, pH 8.0, 85 mM KCl, 0.5% NP-40 and freshly added protease inhibitor cocktail) on ice. Chromatin shearing was performed using a Bioruptor XL sonicator (Diagenode) in RIPA Buffer (1X PBS, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS and freshly added protease inhibitor cocktail). Immunoprecipitations were carried out overnight with appropriate antibodies as indicated in Supplementary Material, Table S1 and Protein A/G-agarose beads (Life Technologies). The next day, beads were washed 5 times with LiCl IP wash buffer (100 mM Tris, pH 7.5, 500 mM LiCl, 1% NP-40, and 1% sodium deoxycholate) and once with TE buffer (10 mM Tris–HCl, pH 7.5, 0.1 mM Na2EDTA). Chromatin fragments were eluted from the beads with IP elution buffer (1% SDS, 0.1 M NaHCO3) at 65°C. Crosslinks were reversed by overnight incubation at 65°C in the presence of 200 mM NaCl followed by 3 h treatment with 0.5 mg/ml proteinase K. DNA was purified by phenol/chloroform and precipitated with 100% ethanol in the presence of GlycoBlue (Thermo Fisher Scientific). Sequencing libraries were prepared using TruSeq ChIP Library Preparation Kit (Illumina, San Diego, CA) and sequenced on an Illumina 2500 Genetic Analyzer at the UAB Heflin Center for Genomic Sciences.
FP and TRP drug treatment of FRDA and control iPSC
Induced pluripotent stem cells cultured to ~80% confluence were treated by (300 nM, FP) or (500 nM, TRP). DMSO was used as vehicle control. Cells were collected after 50 min of incubation by scraping on ice for PRO-seq analyses.
Precision nuclear run-on and sequencing
PRO-seq was performed according to the previously published protocol (25) with some modifications. Cells were harvested by scraping into ice-cold PBS, collected by brief centrifugation and then resuspended in ice-cold PBS. At this point cells were counted and supplemented with 5% drosophila S2 cells as a spike–in for normalization. After centrifugation, cell pellets were resuspended in ice-cold Dounce homogenization buffer (10 mM Tris-Cl, pH 7.4, 300 mM sucrose, 3 mM CaCl2, 2 mM MgCl2, 0.1%Triton X-100, 5 mM DTT, protease inhibitors and 4 units per ml of RNase inhibitor). Next, cells were homogenized (Dounce) 50 times in lysis buffer on ice and nuclei were collected by centrifugation. Nuclei were washed two times with Dounce homogenization buffer. After the last wash, the supernatant was discarded and nuclei were resuspended in storage buffer (10 mM Tris–HCl, pH 8.0, 25% glycerol, 5 mM MgCl2, 0.1 mM EDTA and 5 mM DTT) and snap-frozen in liquid nitrogen.
Nuclear run-on (NRO) reactions were performed by adding 100 μl of nuclei (5x106) to 100 μl of preheated (37°C) 2x NRO master mix (10 mM Tris–HCl, pH 8.0, 5 mM MgCl2, 300 mM KCl, 1 mM DTT, 250 μM ATP, 250 μM GTP, 250 μM UTP, 0.5 μM CTP, 50 μM biotin-11-CTP, and 1% Sarkosyl), and mixed by pipetting 15 times, then incubated at 37°C for 3 min. Reactions were stopped with Trizol LS, and RNA was extracted once with chloroform followed by ethanol precipitation in the presence of 1 μl GlycoBlue. Pellets were washed with 75% ethanol once and re-dissolved in DEPC-treated H2O.
RNA fragmentation was conducted using base hydrolysis. Enrichment in RNA fragments that incorporated biotin-CTP was performed as follows: fragmented RNA samples were mixed with pre-washed streptavidin beads and incubated at room temperature for 30 min. Beads were collected using a magnetic separator, washed two-times with ice-cold high-salt wash buffer (50 mM Tris–HCl pH 7.4, 2 M NaCl and 0.5%Triton X-100 in DEPC-treated H2O), two times with binding buffer (10 mM Tris–HCl pH 7.4, 300 mM NaCl and 0.1% Triton X-100), and once with low-salt wash buffer (5 mM Tris–HCl pH 7.4 and 0.1% Triton X-100). Next, RNA was recovered from beads using Trizol/chloroform extraction and precipitated with ethanol as described before. RNA pellets were dissolved in RNA adaptor ligation solution (10 μM VRA3 RNA adaptor, 1 mM ATP, 10% PEG, 4 U of RNase inhibitor, 1 U of T4 RNA ligase I and 1x T4 RNA ligase buffer) and incubated 4 h at 20°C. Subsequently, a second biotin enrichment step was performed as described earlier. After precipitation, RNA pellets were dissolved in 5 μl of DEPC-treated H2O, heat-denatured briefly at 65°C for 20 s, and placed on ice.
Next, the 5′ cap was removed from RNA fragments using 5 U RNA 5′ pyrophosphohydrolase, and 5’ OH ends were converted to 5′ phosphate by treatment with 25 U T4 polynucleotide kinase. RNA was extracted with Trizol and chloroform, precipitated and dissolved in DEPC-treated H2O. 5′ RNA adaptor ligation was conducted by incubation of RNA with RNA ligation mixture (10 μM VRA5 RNA adaptor, 1 mM ATP, 10% PEG, 4 U of RNase inhibitor, 1 U of T4 RNA ligase I and 1x T4 RNA ligase buffer) for 4 h at 20°C. This step was followed by a third biotin RNA enrichment as described before. Finally, RNA pellets were dissolved in 10 μl DEPC-treated H2O and used for reverse transcription. 10 μl of the RNA obtained after the third biotin-enrichment step was mixed with 1 μl of 12.5 mM dNTPs and 0.5 μl 100 μM RP1 reverse-transcription primer, incubated at 70°C for 2 min and immediately transferred on ice for 2 min and supplemented with 6 μl of RT buffer (5 mM DTT, 40 U of RNase inhibitor and 1 x First-strand synthesis buffer). After 5 min incubation at 37°C, 1.5 μl of Superscript III RT enzyme was added to a total volume of 20 μl and reverse transcription was carried out for 15 min at 45°C, followed by 40 min at 50°C, 10 min at 55°C and 15 min at 70°C.
Next, cDNA fragments were amplified by PCR. The optimization of PCR conditions and selection of the appropriate number of cycles was performed precisely as described in (25). After optimization, a full-scale PCR amplification was performed using entire cDNA obtained in the reverse transcription step, 250 μM dNTPs, 250 nM RP1 primer, 250 nM RPI-n primer, 2 U of Phusion DNA polymerase, 1 M betaine, 1 X HF buffer). PCR products were resolved on 8% native PAGE TBE gels. The fragment of the gel with DNA size marker was stained with ethidium bromide and based on this, PCR fragments in the range between 140 bp up to 350 bp were excised from the gel, eluted overnight in gel elution buffer (10 mM Tris–HCl, pH 8.0, 0.5 mM NH4Ac, 10 mM MgAc2, 1 mM EDTA and 0.1% SDS). Eluates were filtered through Spin-X Filters to remove gel debris, followed by phenol:chloroform extraction and ethanol precipitation. Gel-purified PCR products (PRO-seq library) were dissolved in 12 μl H2O and sequenced at the UAB Heflin Center for Genomic Sciences using Illumina TRU-seq-compatible sequencing platform.
Massive analysis of cDNA ends sequencing
MACE-seq was performed by the GenXPro GmbH company (Frankfurt, Germany) using the MACE-seq kit according to the instructions of the manufacturers. Briefly, 100 ng total RNA was reverse-transcribed to cDNA using an anchored oligo(dT) primer that preferentially hybridizes to the proximal end of poly(A) tails. Then, cDNA was randomly fragmented by sonication to an average size of 150 nt and a second adapter, carrying a unique molecular identifier (TrueQuant adapter), was ligated to the fragmented DNA according to manufacturer’s recommendations. Using primers specific for the two adapters, cDNA molecules carrying both adapters were PCR amplified in an exponential fashion (10 cycles). Fragments of 50–300 bp were sequenced using the NextSeq500 platform (Illumina).
3’ RACE, TA cloning and Sanger sequencing
3’ RACE was performed using FirstChoice® RLM-RACE Kit (Thermo Fisher Scientific) according the manufacturer’s instructions. 1 μg of DNase-treated total RNA was reverse transcribed with M-MLV Reverse Transcriptase at 42°C for 1 h using the 3’ RACE adaptor oligonucleotide. Subsequently, PCR amplification was performed with a FXN specific primer and 3’ RACE adaptor primer in the following conditions: 3 min at 94°C, 35 cycles of 30 s at 94°C, 60 s at 60°C, 90 s at 72°C followed by 7 min at 72°C. Sequences of 3’ RACE adaptor oligonucleotide and PCR primers are listed in Supplementary Material, Table S4.
PCR products were analyzed on 1% agarose gels and selected DNA fragments were excised, purified with QIAquick Gel Extraction Kit (Qiagen), cloned into sequencing vectors using TOPO TA/Sequencing PCR4 kit (Thermo Fisher Scientific) and transformed to One Shot® Mach1™-T1R competent cells (Thermo Fisher Scientific). Plasmids carrying 3’ RACE products were sequenced at the UAB Heflin Center for Genomic Sciences. Sequencing results were analyzed using SnapGene software (GSL Biotech LLC).
CRISPR/Cas9 editing of the GAA repeats
Excision of the expanded GAA tracts from both alleles of the FXN gene (homozygous excision) was achieved using CRISPR/Cas9 editing. 1 × 106 FRDA 2 or FRDA 3 iPSCs (Supplementary Material, Table S2) were transfected with 2 μg of pX330-U6-Chimeric_BB-CBhhSpCas9 (Addgene #42230) encoding SpCas9 and expressing gRNAs (gUP gRNA and gDW gRNA; Supplementary Material, Fig. S7) using a Human Stem Cell Nucleofector Kit (Lonza). Three days after nucleofection, single cells were seeded at clonal density in mTeSR medium supplemented with CloneR (STEMCELL Technologies) and cultured for 4 days. Colonies were picked manually after 10–14 days. After expansion, genomic DNA was isolated from individual clones and screened for: upstream junction (UP_jct primers); downstream junction (DW_jct primers), upstream/downstream repaired junction (UP_DW_jct primers) and for the presence of the expanded GAA repeats (GAA primers). All primers are listed in Supplementary Material, Table S4; their localization and amplicon size are schematically depicted in Supplementary Material, Figure S7. GAA PCR was performed as we described in (7). All other PCR reactions were conducted in a 25 μl reaction volume under the following conditions: initial denaturation 95°C for 2 min followed by 35 cycles of 95°C for 30 s, 60°C for 30 s and 72°C for 45 s, final extension 72°C for 7 min. Products were resolved on 1% agarose gels, DNA fragments were excised from the gel, purified with QIAquick Gel Extraction Kit (Qiagen), and then cloned and sequenced as described before (Supplementary Material, Fig. S7). Karyotype analysis was conducted by Cell Line Genetics (Madison, WI). Both heterozygous and homozygous edited clones were generated, but only homozygous were used in this study.
Design and transfection of oligonucleotides
All gapmer oligonucleotides (sequences and modifications indicated in Fig. 6F) were gifts from Ionis Pharmaceuticals. Primary fibroblast lines were seeded on 6-well plates at a density of 2 × 105 cells per well 24 h before transfection. Lipofectamine 2000 (Thermo Fisher) was used to deliver oligonucleotides at 3 nM final concentration as described in (39). Transfected cells were harvested after 48 h for FXN mRNA and FXN-ett RNA expression analyses.
Statistical analysis
When possible, a minimum of two biological replicates (different patient/control samples) or three independent experiments were conducted. For qRT-PCR experiments, three technical replicates were performed for each biological replicate and data was analyzed relative to ACT or GAPDH normalizers. Primers for all qRT-PCR reactions are listed in Supplementary Material, Table S4. Statistical significance of changes observed between samples was determined by Student’s t-test or analysis of variance utilizing GraphPad Prism v6. For western blots, signals were quantified using Image Lab 6.0.1 software (Bio-Rad) and shown relative to GAPDH or Ponceau S signal.
Analysis of next-generation sequencing data
Pro-seq
All analyses were conducted as described in (25). Briefly, sequencing reads were filtered for read quality and collapsed to remove duplicates using the fqtrim package, with the following read parameters: -C -A -q 20 -m 80 -P33. This indicates that a read must have a minimum of 80% of its reads at a qual score greater than or equal to 80. Filtered reads were then aligned using STAR aligner (—runThreadN 20, ver 2.4.2a). The .sam output was converted to bam using samtools view -b -S, and the .bam format was converted to bed (bedtools-2.17.0). Read normalization for downstream visualization and analysis was done by using bed tools to calculate the mean read density among a subset of 581 long genes, as previously described in (25). The .bed files were then converted to .bedgraph according to this normalization factor, converted to .tdf (IGVTools) and visualized in IGV.
ChIP-seq
Reads were aligned using BWA 0.7.12 and filtered to remove duplicates. Minimum mapping quality was set to 30. Peaks were identified using MACS2 2.1.1. and visualized using Partek Flow (Partek).
RNA-seq
Reads were aligned using STAR—2.5.3a and quantified with Cufflinks. Differential gene expression analysis to determine FXN mRNA levels was performed using DESeq2 3.5. Results were visualized using Partek Flow (Partek).
MACE-seq
Analyses were conducted blindly by GenXPro GmbH. Raw data was preprocessed using Cutadapt to eliminate poly-A-tails as well as bad-quality base reads. FastQC was used to assess the quality of sequencing after trimming. Cleaned reads were mapped using Bowtie2 (66). Quantification of mapped reads to each gene was performed using HT-seq (67). ENSEMBL-GTF data was used to provide genomic locations for quantification as well as additional data for annotation (such as gene name, gene description, GO-Terms etc.). APA-determination was performed according to (68). APA sites in FXN were visualized using Partek Flow (Partek).
Supplementary Material
Contributor Information
Yanjie Li, Department of Biochemistry and Molecular Genetics, University of Alabama at Birmingham, 1825 University Blvd., Birmingham, AL 35294, USA.
Jixue Li, Department of Biochemistry and Molecular Genetics, University of Alabama at Birmingham, 1825 University Blvd., Birmingham, AL 35294, USA.
Jun Wang, Department of Biochemistry and Molecular Genetics, University of Alabama at Birmingham, 1825 University Blvd., Birmingham, AL 35294, USA.
Siyuan Zhang, Department of Biochemistry and Molecular Genetics, University of Alabama at Birmingham, 1825 University Blvd., Birmingham, AL 35294, USA.
Keith Giles, Department of Biochemistry and Molecular Genetics, University of Alabama at Birmingham, 1825 University Blvd., Birmingham, AL 35294, USA.
Thazha P Prakash, Ionis Pharmaceuticals Inc., 2855 Gazelle Court, Carlsbad, CA 92010, USA.
Frank Rigo, Ionis Pharmaceuticals Inc., 2855 Gazelle Court, Carlsbad, CA 92010, USA.
Jill S Napierala, Department of Biochemistry and Molecular Genetics, University of Alabama at Birmingham, 1825 University Blvd., Birmingham, AL 35294, USA; Department of Neurology, University of Texas Southwestern Medical Center, 5323 Harry Hines Blvd., Dallas, TX 75390, USA.
Marek Napierala, Department of Biochemistry and Molecular Genetics, University of Alabama at Birmingham, 1825 University Blvd., Birmingham, AL 35294, USA; Department of Neurology, University of Texas Southwestern Medical Center, 5323 Harry Hines Blvd., Dallas, TX 75390, USA.
Conflict of Interest statement
Thazha P. Prakash and Frank Rigo are employees of Ionis Pharmaceuticals Inc. The company had no influence of performed studies, data analysis and experimental conclusions. The remaining authors declare no competing interests.
Funding
These studies were supported by research grants from the National Institutes of Health, National Institute of Neurological Disorders and Stroke R01NS081366 and R01NS121038 (to M.N.) and by Friedreich’s Ataxia Research Alliance (to M.N. and independently to J.S.N.).
Data availability
Further information and requests for resources, data and reagents should be directed to Marek Napierala (Marek.Napierala@UTSouthwestern.edu). This study’s unique/stable reagents and datasets are available with a completed Materials Transfer Agreement.
Authors’ contributions
Y.L. performed majority of the experiments and analyzed the data. J.L., S.Z. and J.S.N. performed experiments with CRISPR/Cas9 edited iPSCs. J.W. provided mouse tissues and RNAs for analyses. T.P.P. and F.R. provided ASOs. K.G. conducted bioinformatics analyses. J.S.N. and M.N. designed experiments, acquired funding and wrote the paper. All authors read the article, contributed comments and suggestions, and approved the final version of the article.
References
- 1. Campuzano, V., Montermini, L., Molto, M.D., Pianese, L., Cossee, M., Cavalcanti, F., Monros, E., Rodius, F., Duclos, F., Monticelli, A. et al. (1996) Friedreich's ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science, 271, 1423–1427. [DOI] [PubMed] [Google Scholar]
- 2. Delatycki, M.B. and Bidichandani, S.I. (2019) Friedreich ataxia- pathogenesis and implications for therapies. Neurobiol. Dis., 132, 104606. [DOI] [PubMed] [Google Scholar]
- 3. Bidichandani, S.I., Ashizawa, T. and Patel, P.I. (1998) The GAA triplet-repeat expansion in Friedreich ataxia interferes with transcription and may be associated with an unusual DNA structure. Am. J. Hum. Genet., 62, 111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cossee, M., Durr, A., Schmitt, M., Dahl, N., Trouillas, P., Allinson, P., Kostrzewa, M., Nivelon-Chevallier, A., Gustavson, K.H., Kohlschutter, A. et al. (1999) Friedreich's ataxia: point mutations and clinical presentation of compound heterozygotes. Ann. Neurol., 45, 200–206. [DOI] [PubMed] [Google Scholar]
- 5. Pandolfo, M. (2009) Friedreich ataxia: the clinical picture. J. Neurol., 256, 3–8. [DOI] [PubMed] [Google Scholar]
- 6. Koeppen, A.H., Ramirez, R.L., Becker, A.B., Bjork, S.T., Levi, S., Santambrogio, P., Parsons, P.J., Kruger, P.C., Yang, K.X., Feustel, P.J. et al. (2015) The pathogenesis of cardiomyopathy in Friedreich ataxia. PLoS One, 10, e0116396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Long, A., Napierala, J.S., Polak, U., Hauser, L., Koeppen, A.H., Lynch, D.R. and Napierala, M. (2017) Somatic instability of the expanded GAA repeats in Friedreich's ataxia. PLoS One, 12, e0189990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pianese, L., Turano, M., Lo Casale, M.S., De Biase, I., Giacchetti, M., Monticelli, A., Criscuolo, C., Filla, A. and Cocozza, S. (2004) Real time PCR quantification of frataxin mRNA in the peripheral blood leucocytes of Friedreich ataxia patients and carriers. J. Neurol. Neurosurg. Psychiatry, 75, 1061–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Filla, A., De Michele, G., Cavalcanti, F., Pianese, L., Monticelli, A., Campanella, G. and Cocozza, S. (1996) The relationship between trinucleotide (GAA) repeat length and clinical features in Friedreich ataxia. Am. J. Hum. Genet., 59, 554–560. [PMC free article] [PubMed] [Google Scholar]
- 10. Li, Y., Lu, Y., Polak, U., Lin, K., Shen, J., Farmer, J., Seyer, L., Bhalla, A.D., Rozwadowska, N., Lynch, D.R. et al. (2015) Expanded GAA repeats impede transcription elongation through the FXN gene and induce transcriptional silencing that is restricted to the FXN locus. Hum. Mol. Genet., 24, 6932–6943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Butler, J.S. and Napierala, M. (2015) Friedreich's ataxia--a case of aberrant transcription termination? Transcription, 6, 33–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sznajder, L.J., Thomas, J.D., Carrell, E.M., Reid, T., McFarland, K.N., Cleary, J.D., Oliveira, R., Nutter, C.A., Bhatt, K., Sobczak, K. et al. (2018) Intron retention induced by microsatellite expansions as a disease biomarker. Proc. Natl. Acad. Sci. U. S. A., 115, 4234–4239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Al-Mahdawi, S., Pinto, R.M., Ismail, O., Varshney, D., Lymperi, S., Sandi, C., Trabzuni, D. and Pook, M. (2008) The Friedreich ataxia GAA repeat expansion mutation induces comparable epigenetic changes in human and transgenic mouse brain and heart tissues. Hum. Mol. Genet., 17, 735–746. [DOI] [PubMed] [Google Scholar]
- 14. Herman, D., Jenssen, K., Burnett, R., Soragni, E., Perlman, S.L. and Gottesfeld, J.M. (2006) Histone deacetylase inhibitors reverse gene silencing in Friedreich's ataxia. Nat. Chem. Biol., 2, 551–558. [DOI] [PubMed] [Google Scholar]
- 15. Kim, E., Napierala, M. and Dent, S.Y. (2011) Hyperexpansion of GAA repeats affects post-initiation steps of FXN transcription in Friedreich's ataxia. Nucleic Acids Res., 39, 8366–8377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Punga, T. and Buhler, M. (2010) Long intronic GAA repeats causing Friedreich ataxia impede transcription elongation. EMBO Mol. Med., 2, 120–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rodden, L.N., Chutake, Y.K., Gilliam, K., Lam, C., Soragni, E., Hauser, L., Gilliam, M., Wiley, G., Anderson, M.P., Gottesfeld, J.M. et al. (2021) Methylated and unmethylated epialleles support variegated epigenetic silencing in Friedreich ataxia. Hum. Mol. Genet., 29, 3818–3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chutake, Y.K., Lam, C., Costello, W.N., Anderson, M. and Bidichandani, S.I. (2014) Epigenetic promoter silencing in Friedreich ataxia is dependent on repeat length. Ann. Neurol., 76, 522–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kumari, D., Biacsi, R.E. and Usdin, K. (2010) Repeat expansion affects both transcription initiation and elongation in Friedreich ataxia cells. J. Biol. Chem., 286, 4209–4215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li, J., Li, Y., Wang, J., Gonzalez, T.J., Asokan, A., Napierala, J.S. and Napierala, M. (2020) Defining transcription regulatory elements in the human Frataxin gene: implications for gene therapy. Hum. Gene Ther., 31, 839–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jonkers, I. and Lis, J.T. (2015) Getting up to speed with transcription elongation by RNA polymerase II. Nat. Rev. Mol. Cell Biol., 16, 167–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Quinodoz, M., Gobet, C., Naef, F. and Gustafson, K.B. (2014) Characteristic bimodal profiles of RNA polymerase II at thousands of active mammalian promoters. Genome Biol., 15, R85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Komarnitsky, P., Cho, E.J. and Buratowski, S. (2000) Different phosphorylated forms of RNA polymerase II and associated mRNA processing factors during transcription. Genes Dev., 14, 2452–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mayer, A., Lidschreiber, M., Siebert, M., Leike, K., Soding, J. and Cramer, P. (2010) Uniform transitions of the general RNA polymerase II transcription complex. Nat. Struct. Mol. Biol., 17, 1272–1278. [DOI] [PubMed] [Google Scholar]
- 25. Mahat, D.B., Kwak, H., Booth, G.T., Jonkers, I.H., Danko, C.G., Patel, R.K., Waters, C.T., Munson, K., Core, L.J. and Lis, J.T. (2016) Base-pair-resolution genome-wide mapping of active RNA polymerases using precision nuclear run-on (PRO-seq). Nat. Protoc., 11, 1455–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li, J., Rozwadowska, N., Clark, A., Fil, D., Napierala, J.S. and Napierala, M. (2019) Excision of the expanded GAA repeats corrects cardiomyopathy phenotypes of iPSC-derived Friedreich's ataxia cardiomyocytes. Stem Cell Res., 40, 101529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. De Biase, I., Chutake, Y.K., Rindler, P.M. and Bidichandani, S.I. (2009) Epigenetic silencing in Friedreich ataxia is associated with depletion of CTCF (CCCTC-binding factor) and antisense transcription. PLoS One, 4, e7914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mikaeili, H., Sandi, M., Bayot, A., Al-Mahdawi, S. and Pook, M.A. (2018) FAST-1 antisense RNA epigenetically alters FXN expression. Sci. Rep., 8, 17217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang, M. and Marin, A. (2006) Characterization and prediction of alternative splice sites. Gene, 366, 219–227. [DOI] [PubMed] [Google Scholar]
- 30. Zuker, M. (2003) Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res., 31, 3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lynch, D.R. and Perlman, S.L. (2015) Friedreich's ataxia: the European consortium. Lancet Neurol, 14, 130–131. [DOI] [PubMed] [Google Scholar]
- 32. Anjomani Virmouni, S., Ezzatizadeh, V., Sandi, C., Sandi, M., Al-Mahdawi, S., Chutake, Y. and Pook, M.A. (2015) A novel GAA-repeat-expansion-based mouse model of Friedreich's ataxia. Dis. Model. Mech., 8, 225–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bergquist, H., Rocha, C.S., Alvarez-Asencio, R., Nguyen, C.H., Rutland, M.W., Smith, C.I., Good, L., Nielsen, P.E. and Zain, R. (2016) Disruption of higher order DNA structures in Friedreich's ataxia (GAA)n repeats by PNA or LNA targeting. PLoS One, 11, e0165788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Grabczyk, E. and Usdin, K. (2000) Alleviating transcript insufficiency caused by Friedreich's ataxia triplet repeats. Nucleic Acids Res., 28, 4930–4937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li, L., Matsui, M. and Corey, D.R. (2016) Activating frataxin expression by repeat-targeted nucleic acids. Nat. Commun., 7, 10606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li, L., Shen, X., Liu, Z., Norrbom, M., Prakash, T.P., O'Reilly, D., Sharma, V.K., Damha, M.J., Watts, J.K., Rigo, F. et al. (2018) Activation of Frataxin protein expression by antisense oligonucleotides targeting the mutant expanded repeat. Nucleic Acid Ther, 28, 23–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shen, X., Beasley, S., Putman, J.N., Li, Y., Prakash, T.P., Rigo, F., Napierala, M. and Corey, D.R. (2019) Efficient electroporation of neuronal cells using synthetic oligonucleotides: identifying duplex RNA and antisense oligonucleotide activators of human frataxin expression. RNA, 25, 1118–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shen, X., Kilikevicius, A., O'Reilly, D., Prakash, T.P., Damha, M.J., Rigo, F. and Corey, D.R. (2018) Activating frataxin expression by single-stranded siRNAs targeting the GAA repeat expansion. Bioorg. Med. Chem. Lett., 28, 2850–2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shen, X., Wong, J., Prakash, T.P., Rigo, F., Li, Y., Napierala, M. and Corey, D.R. (2020) Progress towards drug discovery for Friedreich's ataxia: identifying synthetic oligonucleotides that more potently activate expression of human frataxin protein. Bioorg. Med. Chem., 28, 115472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Patel, M., Isaacs, C.J., Seyer, L., Brigatti, K., Gelbard, S., Strawser, C., Foerster, D., Shinnick, J., Schadt, K., Yiu, E.M. et al. (2016) Progression of Friedreich ataxia: quantitative characterization over 5 years. Ann. Clin. Transl. Neurol., 3, 684–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Khristich, A.N., Armenia, J.F., Matera, R.M., Kolchinski, A.A. and Mirkin, S.M. (2020) Large-scale contractions of Friedreich's ataxia GAA repeats in yeast occur during DNA replication due to their triplex-forming ability. Proc. Natl. Acad. Sci. U. S. A., 117, 1628–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sakamoto, N., Chastain, P.D., Parniewski, P., Ohshima, K., Pandolfo, M., Griffith, J.D. and Wells, R.D. (1999) Sticky DNA: self-association properties of long GAA.TTC repeats in R.R.Y triplex structures from Friedreich's ataxia. Mol. Cell, 3, 465–475. [DOI] [PubMed] [Google Scholar]
- 43. Zhang, J., Fakharzadeh, A., Pan, F., Roland, C. and Sagui, C. (2020) Atypical structures of GAA/TTC trinucleotide repeats underlying Friedreich's ataxia: DNA triplexes and RNA/DNA hybrids. Nucleic Acids Res., 48, 9899–9917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Grabczyk, E., Mancuso, M. and Sammarco, M.C. (2007) A persistent RNA.DNA hybrid formed by transcription of the Friedreich ataxia triplet repeat in live bacteria, and by T7 RNAP in vitro. Nucleic Acids Res., 35, 5351–5359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Groh, M., Lufino, M.M., Wade-Martins, R. and Gromak, N. (2014) R-loops associated with triplet repeat expansions promote gene silencing in Friedreich ataxia and fragile X syndrome. PLoS Genet., 10, e1004318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hartono, S.R., Malapert, A., Legros, P., Bernard, P., Chedin, F. and Vanoosthuyse, V. (2018) The affinity of the S9.6 antibody for double-stranded RNAs impacts the accurate mapping of R-loops in fission yeast. J. Mol. Biol., 430, 272–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Konig, F., Schubert, T. and Langst, G. (2017) The monoclonal S9.6 antibody exhibits highly variable binding affinities towards different R-loop sequences. PLoS One, 12, e0178875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Vanoosthuyse, V. (2018) Strengths and weaknesses of the current strategies to map and characterize R-loops. Noncoding RNA, 4, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Aguilera, A. and Garcia-Muse, T. (2012) R loops: from transcription byproducts to threats to genome stability. Mol. Cell, 46, 115–124. [DOI] [PubMed] [Google Scholar]
- 50. Bacolla, A., Wang, G. and Vasquez, K.M. (2015) New perspectives on DNA and RNA triplexes as effectors of biological activity. PLoS Genet., 11, e1005696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Crossley, M.P., Bocek, M. and Cimprich, K.A. (2019) R-loops as cellular regulators and genomic threats. Mol. Cell, 73, 398–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Keenen, M.M., Brown, D., Brennan, L.D., Renger, R., Khoo, H., Carlson, C.R., Huang, B., Grill, S.W., Narlikar, G.J. and Redding, S. (2021) HP1 proteins compact DNA into mechanically and positionally stable phase separated domains. elife, 10, e64563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Larson, A.G., Elnatan, D., Keenen, M.M., Trnka, M.J., Johnston, J.B., Burlingame, A.L., Agard, D.A., Redding, S. and Narlikar, G.J. (2017) Liquid droplet formation by HP1alpha suggests a role for phase separation in heterochromatin. Nature, 547, 236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Strom, A.R., Emelyanov, A.V., Mir, M., Fyodorov, D.V., Darzacq, X. and Karpen, G.H. (2017) Phase separation drives heterochromatin domain formation. Nature, 547, 241–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Peng, L., Li, E.M. and Xu, L.Y. (2020) From start to end: phase separation and transcriptional regulation. Biochim Biophys Acta Gene Regul Mech, 1863, 194641. [DOI] [PubMed] [Google Scholar]
- 56. Lin, H., Magrane, J., Rattelle, A., Stepanova, A., Galkin, A., Clark, E.M., Dong, Y.N., Halawani, S.M. and Lynch, D.R. (2017) Early cerebellar deficits in mitochondrial biogenesis and respiratory chain complexes in the KIKO mouse model of Friedreich ataxia. Dis. Model. Mech., 10, 1343–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bentley, D.L. (2014) Coupling mRNA processing with transcription in time and space. Nat. Rev. Genet., 15, 163–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Burnett, R., Melander, C., Puckett, J.W., Son, L.S., Wells, R.D., Dervan, P.B. and Gottesfeld, J.M. (2006) DNA sequence-specific polyamides alleviate transcription inhibition associated with long GAA.TTC repeats in Friedreich's ataxia. Proc. Natl. Acad. Sci. U. S. A., 103, 11497–11502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lev Maor, G., Yearim, A. and Ast, G. (2015) The alternative role of DNA methylation in splicing regulation. Trends Genet., 31, 274–280. [DOI] [PubMed] [Google Scholar]
- 60. Maunakea, A.K., Chepelev, I., Cui, K. and Zhao, K. (2013) Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exon recognition. Cell Res., 23, 1256–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kamieniarz-Gdula, K. and Proudfoot, N.J. (2019) Transcriptional control by premature termination: a forgotten mechanism. Trends Genet., 35, 553–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Steurer, B., Janssens, R.C., Geverts, B., Geijer, M.E., Wienholz, F., Theil, A.F., Chang, J., Dealy, S., Pothof, J., van Cappellen, W.A. et al. (2018) Live-cell analysis of endogenous GFP-RPB1 uncovers rapid turnover of initiating and promoter-paused RNA polymerase II. Proc. Natl. Acad. Sci. U. S. A., 115, E4368–E4376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lim, K.H., Han, Z., Jeon, H.Y., Kach, J., Jing, E., Weyn-Vanhentenryck, S., Downs, M., Corrionero, A., Oh, R., Scharner, J. et al. (2020) Antisense oligonucleotide modulation of non-productive alternative splicing upregulates gene expression. Nat. Commun., 11, 3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Polak, U., Hirsch, C., Ku, S., Gottesfeld, J.M., Dent, S.Y.R. and Napierala, M. (2012) Selecting and isolating colonies of human induced pluripotent stem cells reprogrammed from adult fibroblasts. J. Vis. Exp., 60, e3416. 10.3791/3416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Clement, S.L. and Lykke-Andersen, J. (2008) A tethering approach to study proteins that activate mRNA turnover in human cells. Methods Mol Biol., 419, 121–133. [DOI] [PubMed] [Google Scholar]
- 66. Langmead, B. and Salzberg, S.L. (2012) Fast gapped-read alignment with Bowtie 2. Nat. Methods, 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Anders, S., Pyl, P.T. and Huber, W. (2015) HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics, 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Muller, S., Rycak, L. Afonso-Grunz, F., Winter, P., Zawada, A.M., Damrath, E., Scheider, J., Schmah, J., Koch, I., Kahl, G. et al. (2014) APADB: a database for alternative polyadenylation and microRNA regulation events. Database (Oxford), 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Further information and requests for resources, data and reagents should be directed to Marek Napierala (Marek.Napierala@UTSouthwestern.edu). This study’s unique/stable reagents and datasets are available with a completed Materials Transfer Agreement.