

Summary
The immune checkpoint inhibitor (ICI) pembrolizumab is FDA-approved for treatment of solid tumors with high tumor mutational burden (TMB-high,≥10 variants/Mb). However, the extent to which TMB-high generalizes as an accurate biomarker in diverse patient populations is largely unknown. Using two clinical cohorts, we investigated the interplay between genetic ancestry, TMB, and tumor-only versus tumor-normal paired sequencing in solid tumors. TMB estimates from tumor-only panels substantially overclassified individuals into the clinically important TMB-high group due to germline contamination, and this bias was particularly pronounced in patients with Asian/African ancestry. Among patients with non-small cell lung cancer treated with ICIs, those misclassified as TMB-high from tumor-only panels did not associate with improved outcomes. TMB-high was significantly associated with improved outcomes only in European ancestries and merits validation in non-European ancestry populations. Ancestry-aware tumor-only TMB calibration and ancestry-diverse biomarker studies are critical to ensure that existing disparities are not exacerbated in precision medicine.
Keywords: Tumor mutational burden, genetic ancestry, immunotherapy, biomarker, genomics, cancer disparities
Graphical Abstract
eTOC Blurb:
Analyzing tumor-only genetic sequencing data, Nassar et al. highlight overestimation of mutation counts in non-Europeans. Recognizing the utility of TMB as a biomarker of response to immune checkpoint inhibitors, they propose an ancestry-informed calibration of mutational burden to mitigate biases in immunotherapy treatment allocation whenever matched-normal sequencing data is unavailable.
Introduction
Immune checkpoint inhibitors (ICIs) have introduced a new paradigm of cancer management in the past decade(Robert, 2020). However, racial and ethnic minorities are underrepresented in clinical trials of ICIs(Nazha et al., 2019), which limits the generalizability of emerging biomarkers. Moreover, analyses of patients from trials still center around race and ethnicity, which capture a complicated mix of social constructs and genetic ancestry(Nazha et al., 2019).
Recently, pembrolizumab received FDA approval for the treatment of patients with unresectable or metastatic tumors classified as tumor mutational burden (TMB)-high (≥10 variants/Mb) and have progressed on prior therapy(Marcus et al., 2021; Subbiah et al., 2020). More recent work(Cristescu et al., 2022) builds on the phase II KEYNOTE-158 study, and shows that among 1772 patients receiving pembrolizumab monotherapy, overall response rate is 31.4% in 433 patients with TMB ≥175 mutations/exome versus 9.5% in 1339 patients with TMB<175 mutations/exome. Importantly, this association is independent of PD-L1 expression, tumor type, or MSI status. As TMB is adopted in clinical decision-making, generalizability of TMB to diverse real-world settings is a prerequisite to its validity and utility as a biomarker. The use of tumor-only next generation sequencing panels(Chalmers et al., 2017) poses an additional challenge for TMB estimation, where in-silico methods for germline variant exclusion that rely on reference data, may lead to higher rates of false positive somatic variants in populations that are underrepresented in the reference(Fancello et al., 2019) (Figure 1). Parikh et al.(Parikh et al., 2020) show that among 50 tumor samples from 10 different tumor types, TMB-only estimates, based on filtering germline variants from population databases, are significantly inflated compared to estimates from paired tumor/normal TMB.
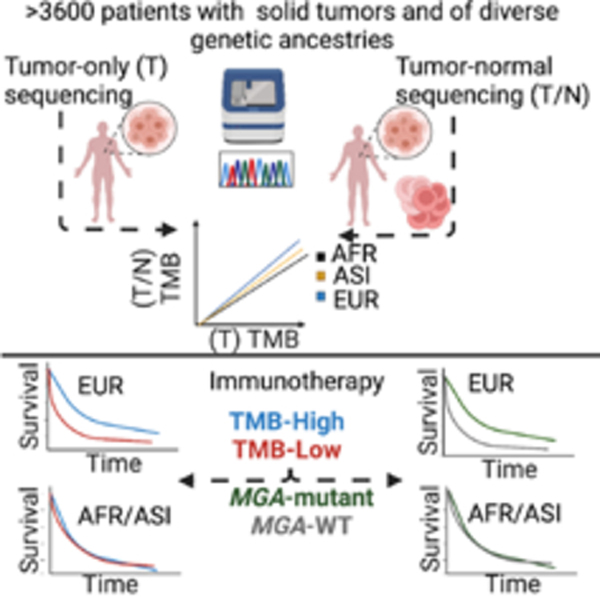
Figure 1: See also Figures S1-S4 and Tables S1-S2 TMB overestimation in tumor-only sequencing panels driven by false positive germline variants in non-Europeans.
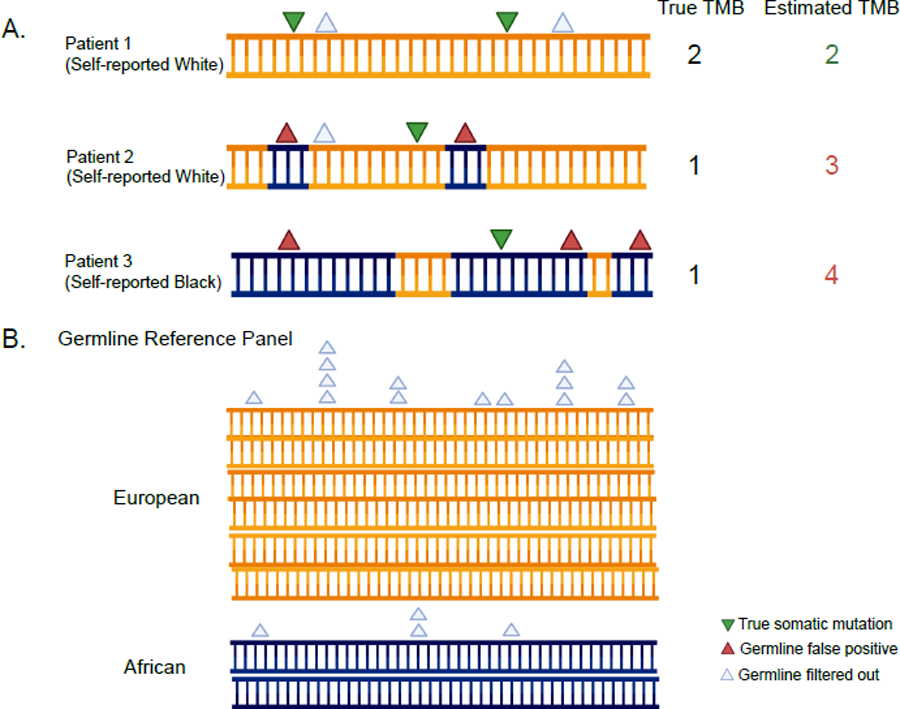
A. Annotating somatic and germline variants across self-reported race and effect on TMB calculations. The orange color in the DNA molecule refers to the European component of the genome and the navy-blue color refers to the African component. B. Germline reference panels where non-Europeans are underrepresented. This is driven by genetic ancestry and not by race as an individual with high non-European ancestry will have the same bias in their tumor-only TMB estimate whether they self-report as white or non-white. See also Figures S1-S4 and Tables S1 and S2.
Separate from the question of accurate TMB estimation, it is critical to understand the generalizability of TMB as a biomarker in diverse patient cohorts, as the TMB-high cutoff is established in studies of primarily white patients with European ancestry(Marabelle et al., 2020a; Marabelle et al., 2020b). The emergence of large-scale patient sequencing, detailed multi-ethnic reference panels, and computational tools enables robust inference of genetic ancestry(Jorde and Bamshad, 2020; Kumar et al., 2010; Martin et al., 2019; Price et al., 2006; Sakaue et al., 2020), defined as the contribution of genetic material from an ancestral population into a contemporary individual. Several studies leverage genetic ancestry to identify relationships with somatic drivers in cancer(Carrot-Zhang et al., 2020; Carrot-Zhang et al., 2021; Yuan et al., 2018). However, clinical outcomes of patients with non-European ancestry, particularly in the ICI setting, are scarce.
Herein, we analyze genetic ancestry for >2000 patients with common solid tumor types treated with ICIs and determine genomic and clinical correlatives. We confirm ancestry-biomarker associations in an independent cohort and specifically highlight ancestry-specific biases in TMB estimates from tumor-only sequencing panels. We point to a potential influence of these biases on patient outcomes and propose a strategy for TMB recalibration.
Results
Genetic Ancestry Improves Population Classification of Self-reported non-Whites
To assess the value of genetic ancestry in population studies, we utilized 8,193 patients from the entire DFCI/PROFILE cohort for cancers of interest with genetic data available. We inferred genetic ancestry using principal component analysis in a large multi-ancestry population including our target data, focusing on two "continental" components capturing European/African/Asian ancestry (see STAR Methods). Compared to self-reported race (which can be arbitrarily defined or missing), genetic analysis identified an additional 42% and 38% of patients with African and Asian ancestry, respectively: 219/312 patients with African ancestry were self-reported Black/African American and 53/312 were self-reported White; 248/343 patients with East Asian ancestry were self-reported Asian and 57/343 were self-reported White (Figure S1A).
TMB Inflation in Tumor-Only Sequencing Panels by Ancestry and Race
Calling somatic variation from tumor-only sequencing is prone to miscalling germline variants as somatic (Garofalo et al., 2016; Parikh et al., 2020) and necessitates filtering out germline false-positive variants using population reference panels. As the available reference panels for European populations are larger than those for non-Europeans (for example, gnomAD v2.1 reference panel contains 56,885 European exomes but only 8,128 African exomes), we hypothesized that the degree of inflation would be particularly pronounced for tumor-only TMB estimates in individuals with non-European ancestry (Figure 1 A-B). Using the TCGA cohort of 3,618 samples (STAR Methods; Figure S1B) and genomic variant calls from Oncopanel-restricted genes (Table S1), we compared the correlation between TMB from WES paired tumor-normal samples (hereby referred to as TMB paired) versus TMB tumor-only from the same samples estimated by blinding the germline variant calls, restricting to Oncopanel baitsets (v1-v3), and applying the Oncopanel germline filtering pipeline including filtering variants occurring in >0.1% of the TOPMED freeze 8 database (see STAR Methods). As expected, TMB tumor-only was significantly higher than TMB-paired (median TMB tumor-only=5.3 vs. median TMB-paired=3.3, p<0.0001), although the two estimates were generally correlated (Pearson correlation coefficient for Oncopanel v3=0.92) (Table S1, Figure S2A-C). Importantly, TMB tumor-only inflation was significantly higher in non-Europeans compared to Europeans (Figure S2A-C): On average, TMB was inflated 2.2-fold in non-Europeans (computed as the median of the ratio TMBtumor-only / TMBpaired) versus 1.5-fold in Europeans.
To investigate the phenomenon of differential inflation by continental ancestry in tumor-only sequencing panels in contemporary, real-world sequencing, we turned to data from >120,000 tumors across multiple institutions collected by AACR Project GENIE. We used MSK-IMPACT and Yale as the gold standard panel for estimating TMB (paired tumor/normal sequencing) and data from seven other sequencing centers as the representative tumor-only panels. Because the panels differ in the types and numbers of genes assayed, we did not attempt to compare TMB directly but instead compared per-variant variant allele frequency (VAF) distributions, which should be insensitive to the number of variants in the platform. Since rare germline variants generally have a VAF of 50%, we expect germline contamination to lead towards more high-VAF variants, which was clearly observed in the tumor-only cohorts compared to the tumor/normal paired panels (MSK-IMPACT and Yale) (Figure S3). Moreover, more high-VAF variants were generally observed for tumors from non-white patients (race serving as a crude proxy for ancestry, which was not available) in the tumor-only cohorts but not the tumor/normal paired cohorts.
Confirmation of ancestry-specific tumor-only TMB bias with normal-matched data
To confirm the relationship between ancestry and tumor-only TMB, we turned to data from a real-world cohort of 456 patients sequenced using MSKCC IMPACT with tumor-normal as ground truth. Genetic ancestry for each patient was computed in prior work (see STAR Methods). In the MSKCC IMPACT cohort, patients with Asian and African ancestry (excluding Admixed populations, see STAR Methods) were oversampled for two groups: 327 Non-Small Cell Lung Cancer (NSCLC) patients on ICIs, and 139 randomly selected other cancers (Table S2). For each sample, the matched normal sample was blinded and tumor-only TMB was estimated using the conventional reference-based filtering approach, with the difference between tumor-only TMB and tumor/normal matched TMB being the outcome of interest (see STAR Methods). To allow a fair comparison, tumor/normal TMB was estimated using the same workflow and treated as the ground truth (see STAR Methods). As expected, TMB inflation was more pronounced in tumor-only TMB compared to tumor/normal TMB, especially among non-Europeans (Figure S4). Non-European populations had a significantly larger log-transformed TMB difference in both the NSCLC cohort (1.4-fold; p=1.1x10−09) and the pan-cancer cohort (1.4-fold; p=1.1x10−06) (Table S2). These findings confirmed the excess tumor-only TMB estimates in non-European patients with direct matched-normal sequencing.
TMB Calibration Eliminates Ancestry Differences in Most Cancer Types
We used TCGA data to estimate ancestry-specific calibration coefficients for panel TMB tumor-only using the TMB paired WES as a benchmark, accounting for panel version and cancer type (see STAR Methods). We then applied the calibration coefficients trained in TCGA to the full DFCI/PROFILE (n=8193 patients) to compute recalibrated and ancestry corrected TMB, which we call TMB-c. Prior to recalibration in the DFCI/PROFILE cohort, non-Europeans had significantly higher TMB compared to Europeans across four of seven cancer types (CRC, EGC, HNSCC, NSCLC) (Table S3, Figure 2A). After TMB calibration, only TMB-c among patients with CRC and HNSCC remained significantly higher in non-Europeans compared to Europeans (the difference remained significant for CRC after adjusting for microsatellite instability status); EGC and NSCLC no longer exhibited significant ancestry differences in TMB-c (Figure 2B).
Figure 2: See also Figure S5 and Tables S3-S4. Differential TMB estimates across continental ancestral populations in the DFCI cohorts prior and after calibration and effects of TMB calibration on clinical outcomes.
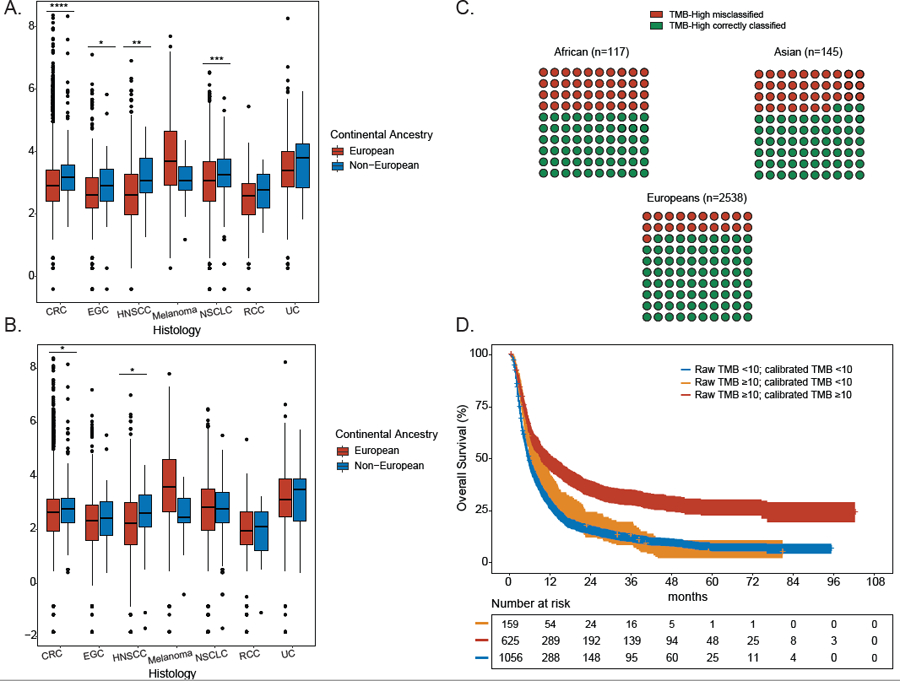
A. Distribution of uncalibrated TMB estimates for each of 7 cancer types shown by genetic ancestry. A two-sided binomial test was used to establish P values (* <0.05, ** <0.01, *** <0.001, **** <0.0001). Data are represented as boxplots. The horizontal lines reflect the median, the lower and upper whiskers indicate 1.5 x the interquartile ranges. Circles are outliers. B. Distribution of calibrated TMB estimates for each of 7 cancer types shown by genetic ancestry. A two-sided binomial test was used to establish P values (* <0.05, ** <0.01, *** <0.001, **** <0.0001). Data are represented as boxplots. The horizontal lines reflect the median, the lower and upper whiskers indicate 1.5 x the interquartile ranges. Circles are outliers. C. 10x10 dot plot showing TMB misclassification rates for TMB-high tumors in each ancestral population in the entire DFCI cohort (n=2800 TMB-high patients). D. Impact of TMB calibration on overall survival in ICI-treated patients at DFCI (n=1840 patients). Patients were stratified into: (a) true TMB-low (raw TMB<10; calibrated TMB<10), true TMB-high (raw TMB≥10; calibrated TMB ≥10), and false TMB-high (raw TMB ≥10, calibrated TMB<10). CRC: colorectal cancer, EGC: esophagogastric cancer (EGC), HNSCC: head and neck squamous cell carcinoma, melanoma, NSCLC: non-small cell lung cancer, UC: urothelial carcinoma, RCC: renal cell carcinoma. See also Figure S5 and Tables S3-S4.
We investigated whether additional granularity in ancestry beyond the continental populations would substantially alter our TMB calibration method. We inferred fine-scale genetic ancestry based on 11 principal components (PCs) that corresponded to sub-continental populations in the 1000 Genomes reference (see STAR Methods). In the TCGA data, we then evaluated the impact of an increasing number of ancestry PCs on the TMB difference, defined as (TMBTumor-only – TMBTumor-Normal). TMB difference was only significantly associated with PCs 1–3 (PC3 correlating with admixed American populations in the 1000 Genomes reference), consistent with our assumption that TMB miscalibration is primarily driven by continental ancestries underrepresented in germline reference panels. The addition of PC3 to our model for computing TMB-c in the DFCI/PROFILE samples led to reclassification of only 1.2% individuals, compared to the model with continental (African/Asian) ancestry alone (see STAR Methods, Table S4). We thus used TMB-c from the continental model for all subsequent analyses.
Potential Impact of TMB calibration in tumor-only panels on clinical decision-making
Having established the presence of differential inflation by ancestry in tumor-only panels, we hypothesized that 1) differential inflation of TMB in non-European populations sequenced with tumor-only panels would likely lead to increasing rates of TMB-high misclassification in non-Europeans relative to Europeans, 2) TMB-c would better predict clinical outcomes to ICI. We computed TMB and TMB-c for each of 8,193 patients in the DFCI cohort (Figure S1B) and concentrated on patients that had TMB-high tumors prior to calibration (n=2800). Individuals with non-European ancestry and TMB-high tumors had significantly higher rates of false TMB-high (i.e. raw TMB-high corrected to TMB-c low) compared to Europeans (African: 51/117, 43.6%; Asian: 54/145, 37%; European: 528/2538, 21%, p<0.0001, Figure 2C). For example, out of 100 patients of European ancestry with mixed tumor types, 21 would be expected to have false TMB-H. In a similar-sized cohort of patients of Asian or African descent, an estimated 37 and 44, respectively, would be expected to have false TMB-H, suggesting that an additional 17% of Asian and 23% of Africans would have their tumors erroneously called as false TMB-H and thus be eligible for ICIs solely due to tumor-only TMB inflation.
Next, to evaluate the clinical impact of TMB misclassification, we investigated the association between TMB-c reclassification and outcomes in two cohorts: 1) 1840 patients treated with ICI at DFCI with one of seven cancer types (Table 1, Table S3) and 2) 234 patients with NSCLC treated with ICI at MSKCC and with available tumor-only and tumor/normal data. For the MSKCC cohort (Table 2), we applied the TMB calibration coefficients trained in TCGA as a proof-of-concept that this methodology applies to non-Oncopanel targeted panels as well. For the PROFILE/DFCI cohort, we stratified the population into: (a) true TMB-high (TMB high and TMB-c high) (b) false TMB-high (TMB high but TMB-c low), and (c) true TMB-low (TMB low and TMB-c low); no patients were observed with TMB-low but TMB-c high. For the MSKCC cohort, paired tumor/normal was available and represented ground truth to which we compared our calibration method (see STAR Methods).
Table 1:
Baseline clinical characteristics of the DFCI population. See also Table S3.
| DFCI (n=1840) | ||
|---|---|---|
|
| ||
| N (median) | % (range) | |
| Age at ICI start | 65 | 22–93 |
|
| ||
| Tumor Type | ||
| CRC | 78 | 4.2 |
| EGC | 144 | 7.8 |
| HNSCC | 100 | 5.4 |
| Melanoma | 314 | 17.1 |
| Non-small cell lung carcinoma | 879 | 47.8 |
| RCC | 155 | 8.4 |
| Urothelial Carcinoma | 170 | 9.2 |
|
| ||
| Sex | ||
| Male | 1071 | 58.2 |
| Female | 769 | 41.8 |
|
| ||
| Site of specimen sequenced | ||
| Primary | 871 | 47.3 |
| Metastatic | 950 | 51.6 |
| Unspecified | 19 | 1.0 |
|
| ||
| Genetic Ancestry | ||
| African | 83 | 4.5 |
| Asian | 90 | 4.9 |
| Europeans | 1667 | 90.6 |
|
| ||
| Self-Reported Race | ||
| African Americans | 60 | 3.3 |
| Asians | 70 | 3.8 |
| Whites | 1646 | 89.5 |
| Other | 64 | 3.5 |
|
| ||
| ICI type | ||
| Single | 1300 | 70.7 |
| Combination | 540 | 29.3 |
|
| ||
| ICI class | ||
| Anti-PD-1/PD-L1 | 1577 | 85.7 |
| Anti-CTLA-4 | 33 | 1.8 |
| Anti-PD-1/PD-L1 + anti-CTLA-4 | 230 | 12.5 |
|
| ||
| Number of prior lines | ||
| 0 | 919 | 49.9 |
| 1 | 595 | 32.3 |
| ≥2 | 326 | 17.7 |
Table 2.
Clinical and demographic information on 1898 ICI-treated patients of different solid tumor types sequenced with MSK-IMPACT
|
| ||
|---|---|---|
| N (median) | % (range) | |
| Age at ICI start | 67 | 22–88 |
|
| ||
| Histology | ||
| Adenocarcinoma | 1388 | 73.1 |
| Squamous Cell Carcinoma | 236 | 12.4 |
| Non-Small Cell Lung Cancer, NOS | 132 | 7 |
| Large Cell Neuroendocrine Carcinoma | 42 | 2.2 |
| Other | 100 | 5.3 |
|
| ||
| Sex | ||
| Male | 1003 | 52.8 |
| Female | 895 | 47.2 |
|
| ||
| Site of specimen sequenced | ||
| Primary | 977 | 51.5 |
| Metastatic | 914 | 48.2 |
| Local Recurrence | 2 | 0.1 |
| Unspecified | 5 | 0.3 |
|
| ||
| Genetic Ancestry | ||
| Ashkenazi European | 325 | 17.1 |
| Non-Ashkenazi European | 1209 | 63.7 |
| East Asian | 113 | 6 |
| South Asian | 19 | 1 |
| African | 78 | 4.1 |
| Admixed/Other | 154 | 8.1 |
|
| ||
| Self-Reported Race | ||
| African Americans | 109 | 5.7 |
| Asian | 132 | 7 |
| Native American/Pacific Islander | 4 | 0.2 |
| Whites | 1556 | 82 |
| Other/Unknown | 97 | 5.1 |
|
| ||
| ICI type | ||
| Atezolizumab | 231 | 12.2 |
| Durvalumab | 146 | 7.7 |
| Ipilimumab | 58 | 3.1 |
| Nivolumab | 576 | 30.3 |
| Pembrolizumab | 887 | 46.7 |
|
| ||
In the DFCI/PROFILE cohort, after accounting for tumor type and genetic ancestry as covariates in a cox proportional hazards model, true TMB-high tumors had overall significantly superior outcomes relative to false TMB-high and true TMB-low tumors (median OS 27.7 months; 95% CI [22.6–36.2] vs. 16.3 months, 95% CI [13.7–18.7] vs. 14.4 months, 95% CI [13.1–16.2], respectively; p<0.0001, Figure 2D). Importantly, no significant difference in OS was observed between false TMB-high and true TMB-low tumors in the DFCI ICI cohort (p=0.66; Figure S5) even after accounting for tumor type and genetic ancestry.
For validation, we turned to the MSKCC cohort of patients with NSCLC treated with ICIs and with available TMB and gene-level mutation status (n=234 patients). Raw tumor-only, calibrated tumor-only TMB (TMB-c), and tumor/normal paired TMB were computed for 234 patients with NSCLC treated with ICIs. True TMB-high tumors were defined as either 1) TMB-high in both the tumor/normal and raw tumor-only calculations or 2) TMB-high in both raw tumor-only TMB and tumor-only TMB-c. True TMB-low tumors were either 1) both TMB-low by tumor/normal and raw tumor-only calculations or 2) TMB-low in both raw tumor-only TMB low and tumor-only TMB-c. False TMB-high tumors had high TMB by raw tumor-only calculations that corrected to low TMB by tumor/normal calculations or TMB-c. Similar to the DFCI cohort, using paired tumor/normal TMB or tumor-only TMB-c as a gold standard for TMB status classification, patients with true TMB-high tumors had significantly improved outcomes relative to patients with true TMB-low tumors and patients with false TMB-high tumors (Figure 3A,B). Reassuringly, associations between TMB assignment and OS were similar between paired tumor/normal TMB (Figure 3A) and tumor-only TMB-c (Figure 3B), which meant that our calibration method was accurate in reassigning TMB in an independent sequencing platform of similar bait-set size.
Figure 3. Effects of TMB calibration on clinical outcomes in the MSKCC cohort of patients with NSCLC treated with ICI (n=234 patients).
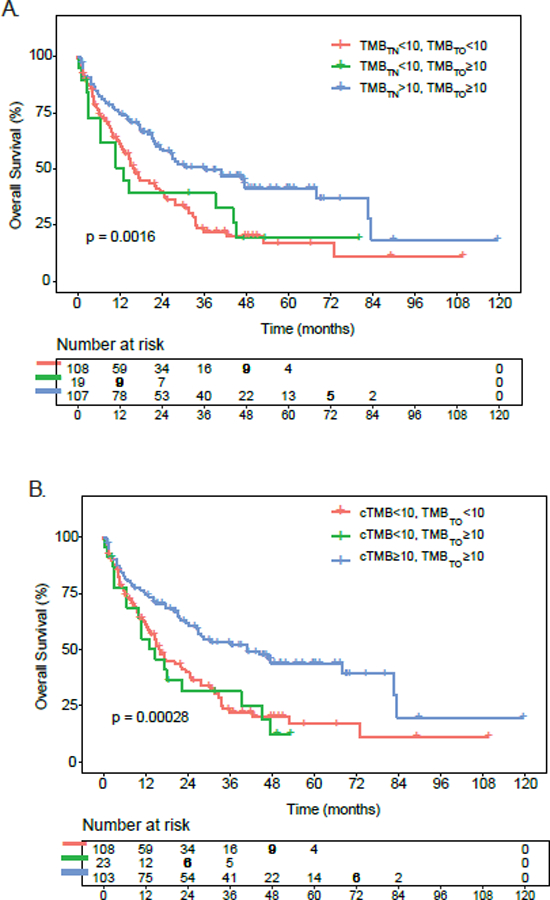
A. Impact of tumor-only versus paired tumor/normal TMB on overall survival in ICI-treated patients with NSCLC at MSKCC. Patients were stratified into: (a) true TMB-low (raw TMB<10; tumor/normal TMB<10), true TMB-high (raw TMB≥10; tumor/normal TMB ≥10), and false TMB-high (raw TMB ≥10, tumor/normal TMB<10). B. Impact of TMB calibration on overall survival in ICI-treated patients with NSCLC at MSKCC. Patients were stratified into: (a) true TMB-low (raw TMB<10; calibrated TMB<10), true TMB-high (raw TMB≥10; calibrated TMB ≥10), and false TMB-high (raw TMB ≥10, calibrated TMB<10). cTMB= calibrated TMB; TMBTO= raw tumor-only TMB; TMBTN= paired tumor/normal TMB.
Evaluating the trans-ethnic portability of TMB as an ICI biomarker
Previous clinical trials and observational studies were either depleted for non-Europeans or relied on self-reported race(Gandhi et al., 2018; Hellmann et al., 2019; Nazha et al., 2019; Paz-Ares et al., 2018), motivating us to investigate the prognostic effect of TMB across ancestries in our ICI cohort (Figure 4A,B). In the PROFILE/DFCI cohort, no effect of ancestry (taken as continuous indices or as categories) on OS was observed for any cancer type, after controlling for prior lines of therapy, ICI type, TMB-c, treatment prior to sequencing and histologic subtype; although statistical power was insufficient to rule out moderate effect-size differences. Asian ancestry was nominally associated with worse OS in RCC (p=0.036, Figure 4B) and worse TTF in NSCLC (p=0.012; Figure 4C, Table S5).
Figure 4: See also Table S5. Adjusted hazard ratios (HR) of clinical outcomes of different ancestral populations treated with ICI therapy in different cancer types.
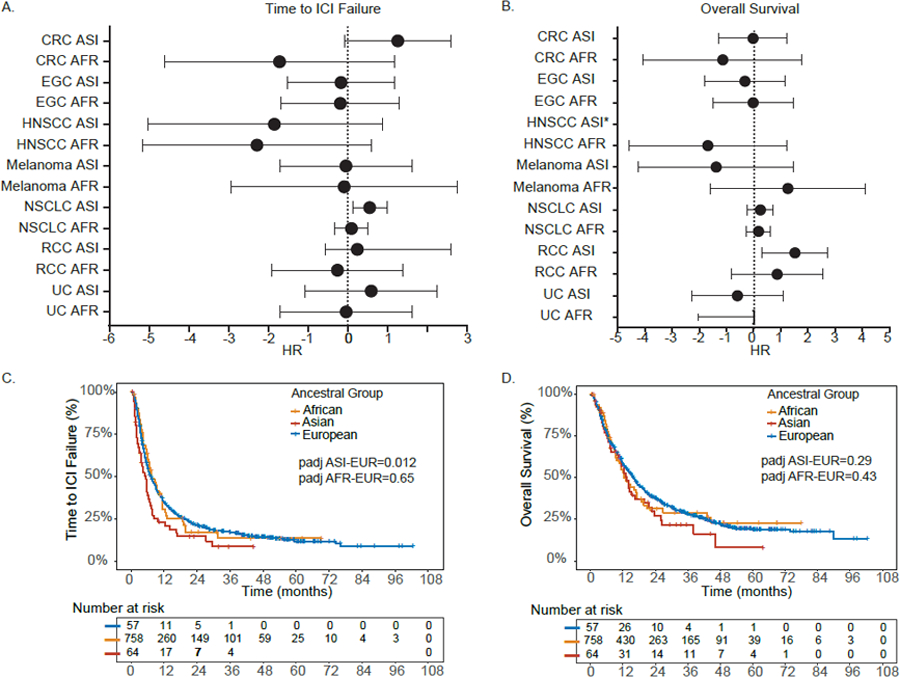
A. Adjusted HR ratios for time to ICI failure in the DFCI ICI cohort (n=1840). Data are presented as adjusted HR with 95% CI (reference group EUR). B. Adjusted HR ratios for overall survival. All p-values and hazard ratios in A and B are of the Wald x2 test from the Cox regression analysis, adjusted as detailed in the STAR Methods section. Data are presented as adjusted HR with 95% CI (reference group EUR). * A horizontal line is not shown for HNSCC given only 1 patient had an event and confidence interval ranged from 0 to infinity. C. Time to ICI failure and genetic ancestry in DFCI patients with NSCLC treated with ICI. D. Overall survival and genetic ancestry in DFCI patients with NSCLC treated with ICI. See also Table S5.
We investigated the trans-ethnic portability of TMB-c in DFCI/PROFILE and MSKCC patients with NSCLC treated with ICI. In the DFCI/PROFILE cohort, multivariable Cox regression showed significantly longer OS in patients of European ancestry with high TMB-c (adjusted hazard ratio (HR)=0.64; 95% CI=0.53–0.77, p<0.0001), after adjusting for number of prior lines of therapy, type of ICI, treatment relative to sequencing date, and histologic subtype (Figure 5A). In contrast, higher TMB-c was not associated with OS in patients of Asian (HR=1.00, 95% CI=0.95–1.05, Figure 5B) or African ancestry (HR=0.99, 95% CI=0.94–1.06, Figure 5C Table S6), although this sample size (n=57 for Asians and n=64 for Africans) had limited power to identify an association in down-sampling analyses (see STAR Methods). Higher TMB-c was associated with improved (p<0.0001) TTF in patients of European ancestry and no significant difference in TTF in patients of Asian, and African ancestry (Table S6). Accounting for histology subtype in the MSKCC NSCLC cohort, higher TMB (paired tumor/normal) was associated with significantly improved survival among Europeans (n=1,534, adjusted hazard ratio (HR)=0.7; 95% CI=0.62–0.81, p<0.0001, Figure 5D) treated with ICIs while no association was noted among Asians (n=132; Figure 5E) and Africans (n=78, Figure 5F). Given the relatively small number of non-European individuals even in these large cohorts across two major institutions, our findings motivate further study of TMB as a biomarker in ancestrally diverse individuals.
Figure 5: See also Table S6. Ancestry-specific associations between TMB status and overall survival (OS) among ICI-treated patients with NSCLC.
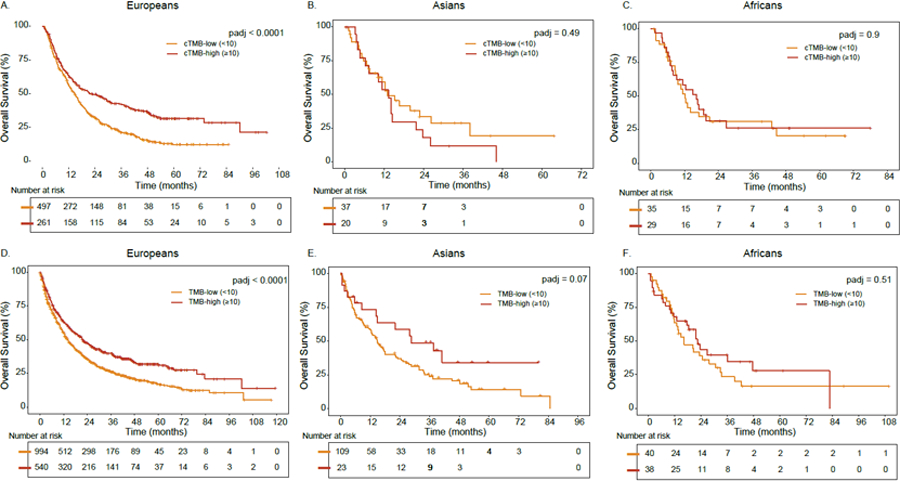
A. Association between TMB and OS in European ancestry in the DFCI cohort. B. Association between TMB and OS in Asian ancestry in the DFCI cohort. C. Association between TMB and OS in African ancestry in the DFCI cohort. D. Association between TMB and OS in European ancestry in the MSKCC cohort. E. Association between TMB and OS in Asian ancestry in the MSKCC cohort. F. Association between TMB and OS in African ancestry in the MSKCC cohort. For the DFCI and MSKCC cohorts, calibrated TMB and tumor/normal TMB were analyzed, respectively. P-values were adjusted for prior lines of therapy, ICI type, TMB-c, treatment prior to sequencing and histologic subtype. See also Table S6.
Ancestry-specific Effects of Gene Alterations (GA) on ICI Outcomes in NSCLC
As an exploratory analysis, we sought to identify individual GA biomarkers that may lack transancestry portability. We analyzed individual GAs with significant ancestry differences in their prognostic effect using an ancestry-interaction scan across 35 genes with >5% GA frequency (see STAR Methods). After accounting for type of ICI, treatment relative to sequencing, histologic subtype, prior lines of therapy and using an FDR<10%, MGA gene alterations were associated with longer OS and TTF in Europeans (Figure 6A, Table S7), were not associated with outcomes in Africans (Figure 6B), and were associated with shorter OS and TTF in Asians (Figure 6C, Table S7), with the difference significant in a formal ancestry interaction test that accounts for power (OS: pinteraction=0.029; TTF: pinteraction=0.03, q-value<0.1; Table S7). In line with prior work by (Sun et al., 2021), the HR for associations between MGA alterations and OS among Europeans was similar (HRDFCI=0.48; HRSun et al.=0.39).
Figure 6: See also Tables S7-S8. Impact of MGA genomic alterations on overall survival (OS) in ICI-treated patients with NSCLC.
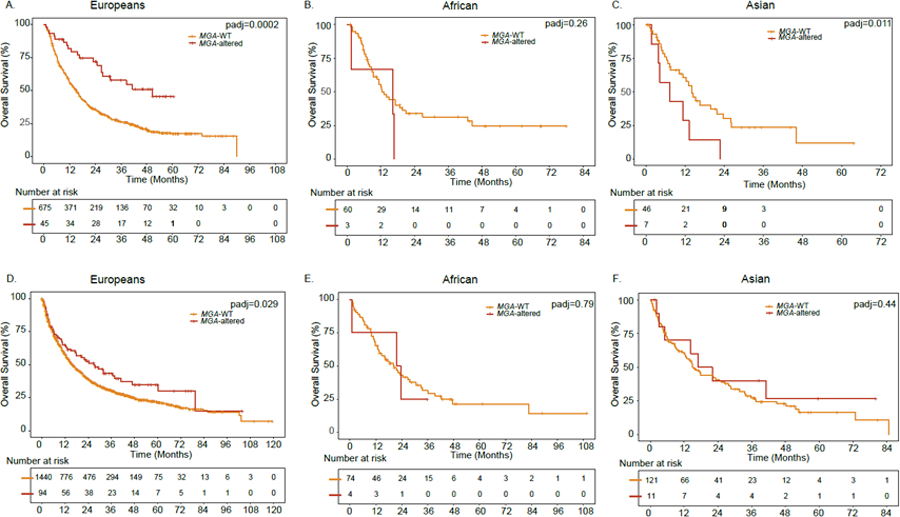
A. OS in the DFCI NSCLC cohort of European ancestry. B. OS in the DFCI NSCLC cohort of African ancestry. C. OS in the DFCI NSCLC cohort of Asian ancestry. D. OS in the MSKCC NSCLC cohort of European ancestry. E. OS in the MSKCC NSCLC cohort of African ancestry. F. OS in the MSKCC NSCLC cohort of Asian ancestry. WT: Wild type. P-values were adjusted for prior lines of therapy, ICI type, TMB-c, treatment prior to sequencing and histologic subtype. See also Tables S7-S8.
To test whether these qualitative differences in the effect direction may be driven by local tumor-only inference biases, we turned to tumor/normal data from the MSKCC NSCLC cohort treated with ICIs (n=1898). After accounting for histologic subtype, Europeans with tumors harboring MGA gene alterations (Table S8) had significantly improved OS compared to Europeans with WT tumors (median OS for MGA-mutant tumors=26.9 months versus median OS for MGA-WT tumors=15.6 months, p=0.029; Figure 6D). However, no association between MGA gene alterations and OS was observed in the African or Asian ancestry individuals (Figure 6E, F). We again caution that the sample sizes for non-European individuals were small for both the DFCI/PROFILE and MSKCC cohorts and thus no definitive conclusions can be made regarding MGA as a biomarker in non-Europeans.
Discussion
In this work, using genetic ancestry, we investigated the generalizability of predictive biomarkers in the ICI setting. In two independent cohorts (DFCI, MSKCC), we showed that TMB estimates from tumor-only panels substantially overclassify individuals into the clinically important TMB-high group, particularly in non-Europeans. We then identified two examples of ancestry-specific biomarkers in patients with NSCLC treated with ICI. We found no evidence that TMB generalizes to non-European populations as a biomarker for improved ICI response although we acknowledge that this may be limited by the small sample size of non-Europeans in both DFCI and MSKCC cohorts. Next, among individual genomic alterations, we noted a complete reversal in the prognostic effect direction for MGA alterations between Europeans and Asians, which was recently identified as a putative biomarker in Europeans(Sun et al., 2021) and further validated herein in the DFCI and MSKCC cohorts. The lack of trans-ancestry portability of biomarkers demonstrated herein calls for further efforts to include non-Europeans in clinical trials and publicly available databases. This ancestry-specific association between TMB or MGA genomic alterations on one hand, and clinical outcomes in patients treated with ICIs on the other hand, is likely due to 1) lack of statistical power (much less non-European patients were treated with ICI at DFCI and MSKCC compared to the European patients) 2) remnant technical concerns in TMB calculations (e.g. Need for better reference genome characterization in non-Europeans) 3) differential treatment patterns and healthcare disparities (quality of care and access to care is inferior among non-Europeans) 4) and biological underpinnings that are yet to be unraveled.
Our work has several limitations. First, non-White populations were markedly under-represented in our cohort and, despite the advantages of genetic ancestry estimates over binary race categories, our ICI cohorts were still underpowered for definitive genomic and survival associations in non-Europeans. Second, tumors sequenced at DFCI were formalin-fixed paraffin embedded (FFPE) while TCGA samples used for calibration were based on fresh frozen samples. As such, estimation of TMB even after calibration may be confounded by artifactual variant calls due to poorer quality and formalin modification of DNA. Future efforts should focus on calibrating using paired tumor/normal FFPE tissue as a reference especially that the current clinical workflow relies on FFPE samples for next-generation sequencing and clinical testing. Third, in addition to technical differences, differences in biomarker performance by ancestry can be caused by a range of social and environmental confounding factors for which ancestry is merely a proxy(Bach et al., 2001; Berry et al., 2009; Borrell et al., 2021) and require further study. Fourth, our approach to TMB recalibration relied on heuristic ancestry groupings to translate between cohorts and will likely benefit from richer admixture data and more granular recalibration incorporating local ancestry. However, our findings validated in an independent cohort from MSKCC with paired tumor/normal sequencing, and sensitivity analyses using fine-scale ancestry showed that most of the TMB bias was explained by broad continental ancestry.
Our work builds on prior studies demonstrating that tumor-only sequencing approaches overestimate TMB(Parikh et al., 2020) and expands on recent cross-racial comparisons in multiple myeloma(Asmann et al., 2021) to a wide spectrum of solid tumors. Differential estimation between ancestries is driven by under-representation of non-European populations in germline databases(Lek et al., 2016) (Sherry et al., 2001) and is directly mediated by patient ancestry rather than race/ethnicity. We thus suggest revising TMB calculations for tumor-only samples using the tumor-based ancestry inference employed herein, when paired germline-tumor samples or large reference panels are unavailable. More broadly, biomarker studies of under-represented populations are critical to ensure that disparities are minimized in the era of precision medicine.
STAR METHODS
Resource Availability
Lead Contact
Further information and requests for resources should be directed to and will be fulfilled by the corresponding author Alexander Gusev (alexander_gusev@dfci.harvard.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
This paper analyzes existing, publicly available data from the 1000 Genomes project(Genomes Project et al., 2015), ExAC(Lek et al., 2016), gnomAD v2.1.1(Karczewski et al., 2021), Trans-Omics for Precision Medicine (TOPMed) program(Taliun et al., 2021), Partners HealthCare Biobank (RRID:SCR_001316), TCGA whole-exome sequencing (RRID: SCR_014555), and GENIE v11.1(Consortium, 2017). These accession numbers for the datasets are listed in the key resources table. The raw ancestry assignments generated for the TCGA cohorts can be found at https://portal.gdc.cancer.gov. The published article includes also includes datasets generated or analyzed from Dana-Farber Cancer Institute and Memorial Sloan Kettering Cancer Center, currently not publicly available. While targeted sequencing raw data were not deposited in a public repository since full sequencing data are not consented to be shared, the data that support the findings of this study are available from the corresponding author upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| 1000 Genomes project data | The International Genome Sample Resource | RRID: SCR_006828 |
| ExAC | Lek et al. 2016 | https://gnomad.broadinstitute.org/ |
| gnomAD v2.1.1 | Karczewski et al. 2020 | https://gnomad.broadinstitute.org/ |
| Partners HealthCare Biobank | Mass General Brigham | RRID:SCR_00131 6 |
| Trans-Omics for Precision Medicine (TOPMed) program | NHLBI Trans-Omics for Precision Medicine | RRID:SCR_01567 7 |
| TCGA whole-exome sequencing | Genomic Data Commons | RRID: SCR_014555 |
| TCGA SNP 6.0 array | TCGA legacy archive | https://portal.gdc.cancer.gov/legacy-archive/search/f |
| TCGA clinical and subtype data | Genomic Data Commons | https://gdc.cancer.gov/about-data/publications/pancanatlas |
| Additional somatic validation datasets | GENIE v11.1 | https://www.aacr.org/professionals/research/aacr-project-genie/ |
| Software and algorithms | ||
| PLINK v2.0 | Chang et al., 2015 | https://www.cog-genomics.org/plink/2.0/ |
| MuTect | Cibulskis et al. 2013 | https://software.broadinstitute.org/cancer/cga/mutect_download |
| Picard | Broad Institute | http://broadinstitute.github.io/picard/ |
| GATK | Poplin et al., 2017 | https://gatk.broadinstitute.org/hc |
| Burrows Wheeler Aligner | Li and Durbin, 2009 | http://bio-bwa.sourceforge.net/ |
| Bcftools v1.10.2 | Danecek et al., 2021 | https://samtools.github.io/bcftools/bcftools.html |
| VCFtools v0.1.15 | Danecek et al., 2011 | https://vcftools.github.io/ |
METHOD DETAILS
Study Design and Patient Cohorts
A summary of the clinical cohorts is presented below and in Figure S1B.
1. DFCI ICI cohort
We identified patients with one of seven solid tumor types treated with ICIs at Dana-Farber Cancer Institute (DFCI, Boston, MA), who also had Oncopanel somatic mutation analysis. Solid tumor types included colorectal cancer (CRC), esophagogastric cancer (EGC), head and neck squamous cell carcinoma (HNSCC), melanoma, non-small cell lung cancer (NSCLC), urothelial carcinoma (UC), and renal cell carcinoma (RCC). All DFCI patients received an anti–PD-1/PD-L1 or anti–CTLA-4 agent in the metastatic setting, either alone or in combination with chemotherapy or targeted therapy. Overall, 1840 tumor samples met the inclusion criteria and had next generation sequencing with a CLIA-certified panel, Oncopanel, across three incremental versions of the panel targeting 275–447 genes. The DFCI institutional review board granted a waiver of informed consent for this study.
2. DFCI/PROFILE Entire Cohort
To study the impact of TMB calibration more broadly and by continuous ancestry, we expanded the DFCI cohort to all patients sequenced (n=32,366 tumors). Among the entire cohort, 8193 patients were diagnosed with one of the seven cancer types above, and sequenced by Oncopanel, regardless of stage or treatment regimens.
3. The Cancer Genome Atlas (TCGA) Cohorts
To study TMB calibration with and without matched normal data, we utilized the following cohorts from the TCGA: bladder UC(Robertson et al., 2018), colorectal adenocarcinoma(Cancer Genome Atlas, 2012), esophageal carcinoma(Cancer Genome Atlas Research et al., 2017), gastric adenocarcinoma(Cancer Genome Atlas Research, 2014), HNSCC(Cancer Genome Atlas, 2015a), kidney chromophobe(Davis et al., 2014), clear cell(Cancer Genome Atlas Research, 2013), and papillary carcinoma(Cancer Genome Atlas Research et al., 2016), lung adenocarcinoma, lung squamous cell carcinoma(Campbell et al., 2016), and melanoma(Cancer Genome Atlas, 2015b). As MSI tumors were included as part of the pembrolizumab approval for TMB-high tumors (≥10)(Subbiah et al., 2020), we opted to include them in the TCGA cohort. The MSI tumors constitute 3.8% of the TCGA cohort (139/3618), with no significant difference between Europeans and non-Europeans (Non-Europeans which include East Asians and Africans, 17/582 (2.9%) versus Europeans: 122/3036 (4.0%), p=0.24). Overall, 3618 patients were evaluated for TMB calibration. Genetic ancestry indices for these TCGA subjects were computed as described previously(Yuan et al., 2018).
4. MSKCC Cohort
For validation, a cohort of 1898 patients with NSCLC treated with ICIs underwent targeted tumor/normal paired sequencing using one of three versions of MSK-IMPACT.
A separate cohort of 466 patients (n=327 NSCLC and n=139 other cancer types) had data on paired tumor/normal TMB and tumor-only TMB. Among these, 234 patients with NSCLC were treated with ICI.
Estimates of Continental and Sub-Continental Genetic Ancestry
a. Constructing a rich genetic ancestry reference panel
A genetic ancestry reference panel was constructed using germline genotype data from: 2,504 international samples from the 1000 Genomes Project (whole-genome sequencing); 10,585 normal samples from TCGA (Affymetrix SNP 6.0 array); and 58,178 patients in the Partners Biobank (Illumina Multi-Ethnic Genotyping Array followed by imputation to the 1000 Genomes). These populations were selected to maximize global diversity (1000 Genomes) as well as reflect patients from the target population (Partners Biobank). All genotyped individuals were merged (total n=71,267) and restricted to variants directly genotyped by the TCGA, followed by stringent LD pruning (removing variants with an r^2 of >0.2 within 500kb).
Genetic ancestry was inferred from the combined genotype dataset using Principal Component Analysis (PCA) to infer the 30 lead PCs. Inference was carried out using the fast randomized approximation of PCA(Galinsky et al., 2016) implemented in the PLINK software, which is accurate for top PCs in large datasets. As observed in prior multi-ancestry studies, PC1 and PC2 captured continental ancestry across European, African, and Asian populations (Figure S6A). Notably, the additional samples beyond the 1000 Genomes data substantially increased the number of samples with moderate African ancestry as well as samples with multi-continental ancestry, which had not been broadly sampled by the 1000 Genomes (Figure S6A, black points). For presentation purposes, PC1 and PC2 were linearly rotated and rescaled so that EUR samples have mean 0.0 for both PCs and AFR/EAS individuals have mean 1.0 for PC1/PC2 respectively (Figure S6B-C). These rescaled PCs thus correspond to percentage ancestry, as previously shown(Chen et al., 2013), though it is important to note that the rescaling does not influence the statistical significance of any downstream analysis.
b. Estimating the number of meaningful ancestry PCs and classifying populations
We sought to determine the minimum number of PCs that corresponded to fine-scale ancestry (i.e. when to stop considering lower PCs) through classification of the labeled 1000 Genomes populations. A non-linear classifier was built between an increasing number of PCs and the population/sub-population labels as the response. Random Forests were used to train the classifier as they can directly accommodate categorical response variables. Accuracy was quantified using the “out-of-bag” error statistic, which evaluates performance on held-out samples while training the classifier. For the five major 1000 Genomes populations (AFR=African, AMR=Admixed American, EAS=East Asian, EUR=European, SAS=South Asian) classification accuracy was optimized using the first 4 PCs and did not improve subsequently (Figure S6D). The overall error rate was nearly zero, consistent with broad continental ancestry being well estimated with the leading PCs. For the 26 subpopulations studied in 1000 Genomes, classification accuracy hit diminishing returns after 11 PCs (Figure S6E). The remaining classification error was attributable to recently diverged populations with shared genetic ancestry: within northern Europe (CEU/GBR), within southern Europe (ITU/STU), within China (CHB/CHS), within West Africa (MSL/ESN/YRI), and within African Americans (ACB/ASW), and within India (GIH/PJL). We thus focused on PC1-PC4 and PC1-PC11 for all subsequent analyses of “broad” (continental) and “fine-scale” (subcontinental) ancestry, respectively. Finally, the random forest classifier of broad continental ancestry was applied to the remaining non-1000 Genomes samples without population labels to define general ancestry groups (Figure S6F). We note that due to the absence of individuals with moderate African ancestry in the 1000 Genomes reference, individuals were arbitrarily classified as African versus European at approximately 50% African ancestry, even though a gradient of ancestry was clearly observed in the target data (Figure S6F). This further underscored our use of quantitative ancestry components for recalibration and led us to consider multiple continental ancestry thresholds when defining population groups.
c. Projecting and verifying ancestry for the DFCI PROFILE data
For PROFILE, quantitative genetic ancestry for African, Admixed American, East Asian, European, and South Asian populations was inferred using common germline polymorphisms called from off-target and on-target sequencing reads from the tumor-only sequencing. To guard against potential artifacts in the tumor-imputed DFCI PROFILE data, these samples were not used to compute ancestry components but were instead projected into the ancestry components inferred in the reference data (see above). For each reference component (x, ranging from PC1 to PC11) and SNP (i), factor loadings (wxi) were estimated in the reference data as part of the PC analysis. Each (DFCI PROFILE) target individual was then projected into this ancestry space as a simple linear combination of the loadings: projected PCx = Σigiwxi, where g is the genotype at SNP i. This procedure was carried out for PC1 to PC11 for each target individual. Benchmarking was conducted using a subset of 872 individuals that had germline genotyping as part of the Partners Healthcare Biobank and also tumor sequencing as part of the DFCI PROFILE cohort. Each sample was processed through the reference/target workflows respectively as described above. Accuracy was then assessed by Pearson correlation between the germline ground truth and tumor imputed data for each PC (Figure S6G). For each of the informative PCs (PC1-PC11), the correlation between the germline ground truth and the tumor imputed PC was >0.90, and for the subset of continental PCs (PC1-PC4) the correlation was >0.98. The tumor-inferred PCs were thus highly accurate for all PCs used in our analyses. This approach for genetic ancestry inference was previously shown to be highly reliable (Gusev et al., 2021). Ancestry was significantly correlated with self-reported race (Pearson correlation=0.90; p<1x10−10) but provided additional admixture variation and was not susceptible to missingness (Figure S7A). Continental ancestry groups were defined with European ancestry treated as the reference group and African/Asian groups defined based on inflection points in the ancestry distribution relative to self-reported race (Figure S7B,C).
For the MSKCC cohort, genetic ancestry was estimated by a previously described method(Srinivasan et al., 2021). Briefly, continental and sub-continental ancestry was determined by ADMIXTURE, using SNP markers within captured regions of the targeted MSK-IMPACT panel, with 1000 Genome samples as reference. Quantitative genetic ancestry for African, Admixed American, East Asian, European, and South Asian populations was inferred.
TMB Calculation
For the DFCI/PROFILE and TCGA cohorts, TMB was defined as the total number of exonic non-synonymous variants divided by the total number megabases sequenced. We used TCGA whole-exome sequencing (WES) data to quantify the bias in Oncopanel tumor-only TMB and developed linearly recalibrated TMB (TMB-c) that accounted for both tumor-only and ancestry-specific error. We used the TCGA cohorts for CRC, EGC, HNSCC, melanoma, NSCLC, UC, and RCC as our benchmark. Using TCGA genomic variant calls, we computed TMB estimates for each sample using two methods: 1) A gold standard paired tumor-normal approach, hereby referred to as TMB-paired approach; 2) a tumor-only approach blinded to germline variant filtering, hereby referred to as TMB tumor-only approach. For the TMB tumor-only approach, we applied filters for variant quality and putative germline variant removal mirroring the in-house pipeline used for reporting TMB to physicians. VCF files for TCGA samples with available ancestry calls from Yuan et. al (Yuan et al., 2018) were obtained from the GDC data portal (https://portal.gdc.cancer.gov/). Somatic variants were called using Mutect2(Benjamin et al., 2019). Filtering of variants followed the below algorithm:
I. Variants not passing the following quality measures were filtered out:
bPcr: "Variant allele shows a bias towards one PCR template strand."
bSeq: "Variant allele shows a bias towards one sequencing strand."
t_lod_star: "Tumor does not meet likelihood threshold."
germline_risk: "Evidence indicates this site is germline, not somatic."
Variants tagged with other quality issues (listed next) were retained as those filters were not adopted in the PROFILE pipeline:
clustered_events: "Clustered events observed in the tumor."
triallelic_site: “Site filtered because more than two alt alleles pass tumor LOD."
str_event: "Site filtered due to contraction of short tandem repeat region"
Evidently, variants tagged with “alt_allele_in_normal” (Evidence seen in the normal sample) were retained to blind the calling to matched normal germline variants.
II. Next, the following filters were applied:
Variants overlapping with PROFILE designed panel-of-normals (PON) loci were filtered out.
Variants in tier5 list were filtered out.
- Remaining variants were checked for their functional consequence and only those with the following consequences (from VEP) are retained:
- missense_variant
- stop_gained
- stop_lost
- start_lost
- frameshift_variant
- inframe_insertion
- inframe_deletion
- coding_sequence_variant
- protein_altering_variant
- incomplete_terminal_codon_variant
III. Variants were subject to population frequency checks.
-
If max population frequency across the European Standard Population (ESP) and gnomAD (maxPOPAF) is <= 0.1%, variant allele frequency (VAF) was checked.
If VAF is >= 3%, variant was retained.
If VAF < 3% and variant is present in COSMIC at least twice, the variant was retained.
If max population frequency across ESP and gnomAD (maxPOPAF) is > 0.1% but <= 10% and variant is present in COSMIC at least twice, the variant was retained.
Variants occurring in >0.1% of the TOPMED database were excluded (https://topmed.nhlbi.nih.gov/topmed-whole-genome-sequencing-methods-freeze-8)
IV. Subsequently, the variants were restricted to those occurring within the target intervals for each panel version of Oncopanel. The number of variants is divided by the size of the exonic bait set (0.76 Mb or v1, 0.82 Mb for v2, 1.31 Mb for v3/3.1) to calculate the final TMB estimate. TMB was calculated separately for each panel version for every TCGA sample (see Table S1). For the MSKCC cohort, tumor/normal and tumor-only TMB were computed for patients with sequenced using MSK-IMPACT. For tumor-only TMB, somatic mutations were called by the tumor-only mode of MuTect v.1.1.5 and annotated by ANNOVAR. This was followed by filtering out variants seen in gnomAD or with frequency greater than 0.1% in TOPMed Freeze 8. Only nonsynonymous SNVs were counted in the TMB calculation. Tumor/normal TMB was calculated for the same samples by incorporating the matched normal sample in the mutation calling step by MuTect to filter out variants detected in normal tissue.
TMB calibration by continuous genetic ancestry
First, we constructed a large reference population to enrich for admixed individuals (see Constructing a rich genetic ancestry reference panel), by combining the Partners Healthcare Biobank(RRID:SCR_001316), 1000 Genomes Project(Genomes Project et al., 2015), and TCGA cohorts (total n=71,267). This reference data is sufficiently large and reflective of the DFCI population to capture realistic admixture. We then used PC analysis to identify 11 PCs associated with fine-scale ancestry, of which 4 were associated with continental ancestry. We then projected the entire DFCI cohort regardless of cancer type (n=32366) into this 11-dimensional space. Consistent with our previous analyses, PC1 and PC2 (corresponding to African and Asian ancestry) contributed the majority of the continental classification accuracy.
Second, we modified our TMB recalibration algorithm to model continuous ancestry components rather than dichotomous ancestry populations using regression. This makes no assumptions on hard population definitions beyond which ancestry components are included in the model, allowing admixed individuals to be modeled as mixtures of components. We evaluated models with an increasing number of PCs in TCGA data (TMBd ~ PC1+PC2+PC3…PC11) and found that PC1–3 were significantly associated with TMB differences (Table S4); again consistent with our assumption that the TMB miscalibration is primarily driven by continental ancestry.
Third, we evaluated TMB recalibration in the DFCI data for the seven cancer types of interest (n=8193 patients) using the continuous components with (i) a model using African + Asian ancestry; (ii) a model additionally including PC3. The inclusion of PC3 led to reclassification of 1.2% individuals compared to the model with African + Asian ancestry alone (Table S4).
Given the minimal contribution of PC3 to TMB recalibration and the fact that PC3 captures admixed subcontinental ancestry that is difficult to define and may be reference dependent, we elected to use the African + Asian ancestry model for our primary analyses.
TMB Recalibration
Ancestry-specific TMB calibration coefficients were estimated as follows in TCGA. For each panel version (Oncopanel v1, v2, v3/3.1), the linear relationship between TCGA paired TMB (Thorsson et al., 2018) and simulated panel (tumor-only) TMB was estimated separately for the individuals defined as European and non-European by linear regression:
Where m corresponds to the coefficient on the TMB tumor-only estimate and b is the overall constant bias in the TMB estimate. The estimated calibration coefficients for each panel version were:
Europeans: Oncopanel v1: m=0.989, b=−2.18; Oncopanel v2: m= 0.994, b= −2.12; Oncopanel v3 m= 1.094, b= −1.94
Non-Europeans: Oncopanel v1: m= 0.821, b= −1.71; Oncopanel v2: m=0.84, b= −1.71; Oncopanel v3: m=0.895, b=−1.29.
In the DFCI/PROFILE cohort, TMB-c was computed for each sample using the coefficients corresponding to their called ancestry and version of Oncopanel. In the MSKCC cohort, TMB-c was computed for each patient using calibration coefficients from Oncopanel v2 given the similarity in bait-set size (Oncopanel v2 bait-set=0.83; MSK-IMPACT average bait-set=0.89).
Data collection
Clinical outcomes included overall survival (OS) and time to ICI failure (TTF) for the DFCI ICI cohort. OS was linked to the National Death Index (NDI) and calculated from the date of ICI initiation to the date of death. TTF was calculated from the data of ICI initiation to the date of next line treatment or death. Alive patients were censored at the date of last follow-up.
Genomic analysis
Details of the tissue collection, DNA extraction and tumor targeted sequencing by Oncopanel were previously described (Sholl et al., 2016). Specifics about genomic profiling are derived verbatim from the AACR Project GENIE Data Guide(2020; Consortium, 2017), “DFCI uses a custom, hybridization-based capture panel (Oncopanel) to detect single nucleotide variants, small indels, copy number alterations, and structural variants from tumor-only sequencing data. Three (3) versions of the panel have been submitted to GENIE: version 1 containing 275 genes, version 2 containing 300 genes, version 3 containing 447 genes. Specimens are reviewed by a pathologist to ensure tumor cellularity of at least 20%. Tumors are sequenced to an average unique depth of coverage of approximately 200x for version 1 and 350x for version 2. Reads are aligned using BWA, flagged for duplicate read pairs using Picard Tools, and locally realigned using GATK. Sequence mutations are called using MuTect for SNVs and GATK SomaticIndelDetector for small indels. Putative germline variants are filtered out using a panel of historical normals or if present in ESP at a frequency ≥ 0.1%, unless the variant is also present in COSMIC. Copy number alterations are called using a custom pipeline and reported for fold-change >1. Structural rearrangements are called using BreaKmer. Testing is performed for all patients across all solid tumor types. Version 3 includes the exonic regions of 447 genes and 191 intronic regions across 60 genes targeted for rearrangement detection. 52 genes present in previous versions were retired in the v3 test.”
Separate from the TMB calculation, individual gene carriers were defined as follows: For tumor suppressor genes, pathogenic variants included nonsense mutations, frameshift insertions or deletions, splice-site variants affecting consensus nucleotides, or homozygous deletions. For proto-oncogenes, the functional impact of missense variants was determined using SIFT(Kumar et al., 2009) and Polyphen-2(Adzhubei et al., 2010). Missense mutations classified as “damaging” in SIFT and/or “probably damaging” in Polyphen-2 were deemed pathogenic. All genes covered by Oncopanel v3 were included in the analysis. For genomic alteration frequency calculation and survival analyses, all patients that were not assayed for alterations in a gene of interest were excluded from the frequency calculations and comparisons pertaining to that gene.
Interaction analysis
Cox proportional hazards models accounting for covariates (prior lines of therapy, ICI type, treatment prior to sequencing (yes/no), TMB-c, and histologic subtype) were employed to detect any interaction between ancestry scores and genomic alterations (GAs) and its subsequent effect on survival outcomes. ANOVA was used to estimate interaction p-values across the following models:
Downsampling power analysis
To assess whether some biomarker associations were not detected in specific patient subgroups (ex. African patients with NSCLC, Ngroup=64) due to lack of power, we ran a permutation analysis (1000 permutations) using random subsets of a larger cohort where an effect was detected (ex. European with NSCLC); nsubset = Ngroup. For each random subset, we ran a cox regression model using the specific biomarker as a predictor variable and generated the corresponding p-values for the association between this biomarker and survival outcomes. We then reported the fraction of sampling where a significant effect was detected.
In the case of African patients with NSCLC (nsubset = Ngroup = ), 1000 random subsets of 64 European patients with NSCLC were analyzed. Out of 1000 cox regression tests, 101 showed a statistically significant association between TMB and overall survival.
Quantification and Statistical Analysis
Logistic regression models were used to study the association between continental ancestry and somatic alterations with histologic subtype used as a covariate. Multivariable Cox regression analysis was used to examine associations between ancestry indices, somatic alterations, and clinical outcomes, with prior lines of therapy, ICI type, treatment prior to sequencing (yes/no) and TMB-c used as covariates. Interaction term analysis was performed for gene associations with ancestry and clinical outcomes in the NSCLC cohort. The models were used to calculate the odds ratios, 95% confidence intervals (CI), and p-values. False discovery rate (FDR) correction was applied using Benjamini Hochberg for the number of independent tests conducted (Significant q-value<0.1). Statistical analyses were performed using R version 4.0.1.
Supplementary Material
Table S1: Related to Figure 1. TCGA TMB Estimates.
Table S2: Related to Figure 1. Cohort Breakdown and TMB Calculation in the MSKCC Cohort (n=672.
Table S3: Related to Table 1 and Figure 2. Clinical, demographic, and TMB estimates of patients treated from the DFCI cohort.
Table S4: Related to Figure 2 and STAR Methods. TMB recalibration in the DFCI data for the seven cancer types of interest (n=8193 patients) using continuous components.
Table S5: Related to Figure 4. Associations between covariates analyzed and clinical outcomes in the DFCI-ICI treated cohort of patients (n=1840).
Table S6: Related to Figure 5. Impact of genetic ancestry on associations between TMB and clinical outcomes in the DFCI-ICI treated cohort of patients with NSCLC (n=879).
Table S7: Related to Figure 6. Genomic correlates of OS and TTF by ancestry in the DFCI-ICI treated cohort of patients with NSCLC (n=879).
Table S8: Related to Figure 6. Raw SNV and CNV calls reported by Oncopanel for ICI-treated patients at DFCI (n=1840).
Highlights.
Across tumor-only panels, TMB inflation is more pronounced in non-Europeans
There are ancestry-specific differences in clinical outcomes by TMB in lung cancer
Calibration of tumor-only TMB off paired tumor/normal TMB improves ancestral biases
MGA alterations do not generalize across ancestries as immunotherapy biomarkers
ACKNOWLEDGMENTS
We are grateful for all our patients treated at Dana-Farber Cancer Institute and Memorial Sloan Kettering Cancer Center. A.G. was supported by R01CA227237, R01CA244569, R01HG006399, R01HG012133, and awards from The Louis B. Mayer Foundation, Doris Duke Charitable Foundation, the Phi Beta Psi Sorority, and the Emerson Collective. S.G. was supported by the Dana-Farber Trustee Science Committee Fellowship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
Guru Sonpavde reports the following disclosures : Advisory Board: BMS, Genentech, EMD Serono, Merck, Sanofi, Seattle Genetics/Astellas, Astrazeneca, Exelixis, Janssen, Bicycle Therapeutics, Pfizer, Immunomedics/Gilead, Scholar Rock, G1 Therapeutics; Research Support to Institution: Sanofi, Astrazeneca, Immunomedics/Gilead, QED, Predicine, BMS; Steering committee of studies: BMS, Bavarian Nordic, Seattle Genetics, QED, G1 Therapeutics (all unpaid), and Astrazeneca, EMD Serono, Debiopharm (paid); Data safety monitoring committee: Mereo; Travel costs: BMS (2019), Astrazeneca (2018); Writing/Editor fees: Uptodate, Editor of Elsevier Practice Update Bladder Cancer Center of Excellence; Speaking fees: Physicians Education Resource (PER), Onclive, Research to Practice, Medscape (all educational)
Marios Giannakis receives research funding from Bristol-Myers Squibb, Merck, Servier and Janssen.
Frank Stephen Hodi reports grants and other from Bristol-Myers Squibb, personal fees from Merck, personal fees from EMD Serono, grants, personal fees and other from Novartis, personal fees from Surface, personal fees from Compass Therapeutics, personal fees from Apricity, personal fees from Aduro, personal fees from Sanofi, personal fees from Pionyr, personal fees from Torque, personal fees from Bicara, other from Pieris Pharmacutical, personal fees from Eisai, personal fees from Checkpoint Therapeutics, personal fees from Idera, personal fees from Genentech/Roche, personal fees from Bioentre, personal fees from Gossamer, personal fees from Phio, personal fees from Iovance, personal fees from Trillium, from Abcuro, personal fees from Catalym, from Immunocore, outside the submitted work; In addition, Dr. Hodi has a patent Methods for Treating MICA-Related Disorders (#20100111973) with royalties paid, a patent Tumor antigens and uses thereof (#7250291) issued, a patent Angiopoiten-2 Biomarkers Predictive of Anti-immune checkpoint response (#20170248603) pending, a patent Compositions and Methods for Identification, Assessment, Prevention, and Treatment of Melanoma using PD-L1 Isoforms (#20160340407) pending, a patent Therapeutic peptides (#20160046716) pending, a patent Therapeutic Peptides (#20140004112) pending, a patent Therapeutic Peptides (#20170022275) pending, a patent Therapeutic Peptides (#20170008962) pending, a patent THERAPEUTIC PEPTIDES
Therapeutic Peptides
Patent number: 9402905 issued, a patent METHODS OF USING PEMBROLIZUMAB AND TREBANANIB pending, a patent Vaccine compositions and methods for restoring NKG2D pathway function against cancers
Patent number: 10279021 issued, a patent
Antibodies that bind to MHC class I polypeptide-related sequence A
Patent number: 10106611 issued, and a patent
Anti-Galectin antibody biomarkers predictive of anti-immune checkpoint and anti-angiogenesis responses
Toni K. Choueiri: Research/advisory boards/consultancy/Honorarium (Institutional and personal, paid and unpaid): AstraZeneca, Aveo, Bayer, Bristol Myers-Squibb, Eisai, EMD Serono, Exelixis, GlaxoSmithKline, IQVA, Ipsen, Kanaph, Lilly, Merck, Nikang, Novartis, Pfizer, Roche, Sanofi/Aventis, Takeda, Tempest. Travel, accommodations, expenses, medical writing in relation to consulting, advisory roles, or honoraria. Stock options: Pionyr, Tempest. Other: Up-to-Date royalties, CME-related events (e.g.: OncLIve, PVI, MJH Life Sciences) honorarium. NCI GU Steering Committee. Patents filed, royalties or other intellectual properties (No income as of current date): related to biomarkers of immune checkpoint blockers and ctDNA. No speaker’s bureau. T. K. Choueiri is supported in part by the Dana-Farber/Harvard Cancer Center Kidney SPORE and Program, the Kohlberg Chair at Harvard Medical School and the Trust Family, Michael Brigham, and Loker Pinard Funds for Kidney Cancer Research at DFCI.
INCLUSION AND DIVERSITY
We worked to ensure gender balance in the recruitment of human subjects. We worked to ensure ethnic or other types of diversity in the recruitment of human subjects. One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community. While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list.
References
- (2020). AACR GENIE 8.0-public Data Guide In.
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, and Sunyaev SR (2010). A method and server for predicting damaging missense mutations. Nat Methods 7, 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asmann YW, Parikh K, Bergsagel PL, Dong H, Adjei AA, Borad MJ, and Mansfield AS (2021). Inflation of tumor mutation burden by tumor-only sequencing in under-represented groups. NPJ Precis Oncol 5, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach PB, Cramer LD, Schrag D, Downey RJ, Gelfand SE, and Begg CB (2001). The influence of hospital volume on survival after resection for lung cancer. N Engl J Med 345, 181–188. [DOI] [PubMed] [Google Scholar]
- Benjamin D, Sato T, Cibulskis K, Getz G, Stewart C, and Lichtenstein L (2019). Calling Somatic SNVs and Indels with Mutect2. bioRxiv, 861054
- Berry J, Bumpers K, Ogunlade V, Glover R, Davis S, Counts-Spriggs M, Kauh J, and Flowers C (2009). Examining racial disparities in colorectal cancer care. J Psychosoc Oncol 27, 59–83. [DOI] [PubMed] [Google Scholar]
- Borrell LN, Elhawary JR, Fuentes-Afflick E, Witonsky J, Bhakta N, Wu AHB, Bibbins-Domingo K, Rodriguez-Santana JR, Lenoir MA, Gavin JR 3rd, et al. (2021). Race and Genetic Ancestry in Medicine - A Time for Reckoning with Racism. N Engl J Med 384, 474–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell JD, Alexandrov A, Kim J, Wala J, Berger AH, Pedamallu CS, Shukla SA, Guo G, Brooks AN, Murray BA, et al. (2016). Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat Genet 48, 607–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas N (2012). Comprehensive molecular characterization of human colon and rectal cancer. Nature 487, 330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas N (2015a). Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 517, 576–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas N (2015b). Genomic Classification of Cutaneous Melanoma. Cell 161, 1681–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research, N. (2013). Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 499, 43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research, N. (2014). Comprehensive molecular characterization of gastric adenocarcinoma. Nature 513, 202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research, N., Analysis Working Group: Asan, U., Agency, B. C. C., Brigham, Women's, H., Broad, I., Brown, U., Case Western Reserve, U., Dana-Farber Cancer, I., Duke, U., et al. (2017). Integrated genomic characterization of oesophageal carcinoma. Nature 541, 169–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research, Linehan N, Spellman WM, Ricketts PT, Creighton CJ, Fei CJ, Davis SS, Wheeler C, Murray DA, Schmidt BA, L., et al. (2016). Comprehensive Molecular Characterization of Papillary Renal-Cell Carcinoma. N Engl J Med 374, 135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrot-Zhang J, Chambwe N, Damrauer JS, Knijnenburg TA, Robertson AG, Yau C, Zhou W, Berger AC, Huang KL, Newberg JY, et al. (2020). Comprehensive Analysis of Genetic Ancestry and Its Molecular Correlates in Cancer. Cancer Cell 37, 639–654 e636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrot-Zhang J, Soca-Chafre G, Patterson N, Thorner AR, Nag A, Watson J, Genovese G, Rodriguez J, Gelbard MK, Corrales-Rodriguez L, et al. (2021). Genetic Ancestry Contributes to Somatic Mutations in Lung Cancers from Admixed Latin American Populations. Cancer Discov 11, 591–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, Schrock A, Campbell B, Shlien A, Chmielecki J, et al. (2017). Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med 9, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CY, Pollack S, Hunter DJ, Hirschhorn JN, Kraft P, and Price AL (2013). Improved ancestry inference using weights from external reference panels. Bioinformatics 29, 1399–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium APG (2017). AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov 7, 818–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristescu R, Aurora-Garg D, Albright A, Xu L, Liu XQ, Loboda A, Lang L, Jin F, Rubin EH, Snyder A, and Lunceford J (2022). Tumor mutational burden predicts the efficacy of pembrolizumab monotherapy: a pan-tumor retrospective analysis of participants with advanced solid tumors. J Immunother Cancer 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis CF, Ricketts CJ, Wang M, Yang L, Cherniack AD, Shen H, Buhay C, Kang H, Kim SC, Fahey CC, et al. (2014). The somatic genomic landscape of chromophobe renal cell carcinoma. Cancer Cell 26, 319–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fancello L, Gandini S, Pelicci PG, and Mazzarella L (2019). Tumor mutational burden quantification from targeted gene panels: major advancements and challenges. J Immunother Cancer 7, 183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galinsky KJ, Bhatia G, Loh PR, Georgiev S, Mukherjee S, Patterson NJ, and Price AL (2016). Fast Principal-Component Analysis Reveals Convergent Evolution of ADH1B in Europe and East Asia. Am J Hum Genet 98, 456–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi L, Rodriguez-Abreu D, Gadgeel S, Esteban E, Felip E, De Angelis F, Domine M, Clingan P, Hochmair MJ, Powell SF, et al. (2018). Pembrolizumab plus Chemotherapy in Metastatic Non-Small-Cell Lung Cancer. N Engl J Med 378, 2078–2092. [DOI] [PubMed] [Google Scholar]
- Garofalo A, Sholl L, Reardon B, Taylor-Weiner A, Amin-Mansour A, Miao D, Liu D, Oliver N, MacConaill L, Ducar M, et al. (2016). The impact of tumor profiling approaches and genomic data strategies for cancer precision medicine. Genome Med 8, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genomes Project C, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, and Abecasis GR (2015). A global reference for human genetic variation. Nature 526, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusev A, Groha S, Taraszka K, Semenov YR, and Zaitlen N (2021). Constructing germline research cohorts from the discarded reads of clinical tumor sequences. medRxiv, 2021.2004.2009.21255197 [DOI] [PMC free article] [PubMed]
- Hellmann MD, Paz-Ares L, Bernabe Caro R, Zurawski B, Kim SW, Carcereny Costa E, Park K, Alexandru A, Lupinacci L, de la Mora Jimenez, E., et al. (2019). Nivolumab plus Ipilimumab in Advanced Non-Small-Cell Lung Cancer. N Engl J Med 381, 2020–2031. [DOI] [PubMed] [Google Scholar]
- Jorde LB, and Bamshad MJ (2020). Genetic Ancestry Testing: What Is It and Why Is It Important? JAMA 323, 1089–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, et al. (2021). Author Correction: The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 590, E53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, and Ng PC (2009). Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4, 1073–1081. [DOI] [PubMed] [Google Scholar]
- Kumar R, Seibold MA, Aldrich MC, Williams LK, Reiner AP, Colangelo L, Galanter J, Gignoux C, Hu D, Sen S, et al. (2010). Genetic ancestry in lung-function predictions. N Engl J Med 363, 321–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O'Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marabelle A, Fakih M, Lopez J, Shah M, Shapira-Frommer R, Nakagawa K, Chung HC, Kindler HL, Lopez-Martin JA, Miller WH Jr., et al. (2020a). Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol 21, 1353–1365. [DOI] [PubMed] [Google Scholar]
- Marabelle A, Le DT, Ascierto PA, Di Giacomo AM, De Jesus-Acosta A, Delord JP, Geva R, Gottfried M, Penel N, Hansen AR, et al. (2020b). Efficacy of Pembrolizumab in Patients With Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. J Clin Oncol 38, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus L, Fashoyin-Aje LA, Donoghue M, Yuan M, Rodriguez L, Gallagher PS, Philip R, Ghosh S, Theoret MR, Beaver JA, et al. (2021). FDA Approval Summary: Pembrolizumab for the Treatment of Tumor Mutational Burden-High Solid Tumors. Clin Cancer Res 27, 4685–4689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin AR, Kanai M, Kamatani Y, Okada Y, Neale BM, and Daly MJ (2019). Clinical use of current polygenic risk scores may exacerbate health disparities. Nature genetics 51, 584–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazha B, Mishra M, Pentz R, and Owonikoko TK (2019). Enrollment of Racial Minorities in Clinical Trials: Old Problem Assumes New Urgency in the Age of Immunotherapy. Am Soc Clin Oncol Educ Book 39, 3–10. [DOI] [PubMed] [Google Scholar]
- Parikh K, Huether R, White K, Hoskinson D, Beaubier N, Dong H, Adjei AA, and Mansfield AS (2020). Tumor Mutational Burden From Tumor-Only Sequencing Compared With Germline Subtraction From Paired Tumor and Normal Specimens. JAMA Netw Open 3, e200202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz-Ares L, Luft A, Vicente D, Tafreshi A, Gumus M, Mazieres J, Hermes B, Cay Senler F, Csoszi T, Fulop A, et al. (2018). Pembrolizumab plus Chemotherapy for Squamous Non-Small-Cell Lung Cancer. N Engl J Med 379, 2040–2051. [DOI] [PubMed] [Google Scholar]
- Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, and Reich D (2006). Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 38, 904–909. [DOI] [PubMed] [Google Scholar]
- Robert C (2020). A decade of immune-checkpoint inhibitors in cancer therapy. Nat Commun 11, 3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson AG, Kim J, Al-Ahmadie H, Bellmunt J, Guo G, Cherniack AD, Hinoue T, Laird PW, Hoadley KA, Akbani R, et al. (2018). Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell 174, 1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaue S, Kanai M, Karjalainen J, Akiyama M, Kurki M, Matoba N, Takahashi A, Hirata M, Kubo M, Matsuda K, et al. (2020). Trans-biobank analysis with 676,000 individuals elucidates the association of polygenic risk scores of complex traits with human lifespan. Nat Med 26, 542–548. [DOI] [PubMed] [Google Scholar]
- Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, and Sirotkin K (2001). dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 29, 308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sholl LM, Do K, Shivdasani P, Cerami E, Dubuc AM, Kuo FC, Garcia EP, Jia Y, Davineni P, Abo RP, et al. (2016). Institutional implementation of clinical tumor profiling on an unselected cancer population. JCI Insight 1, e87062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan P, Bandlamudi C, Jonsson P, Kemel Y, Chavan SS, Richards AL, Penson AV, Bielski CM, Fong C, Syed A, et al. (2021). The context-specific role of germline pathogenicity in tumorigenesis. Nat Genet 53, 1577–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbiah V, Solit DB, Chan TA, and Kurzrock R (2020). The FDA approval of pembrolizumab for adult and pediatric patients with tumor mutational burden (TMB) >/=10: a decision centered on empowering patients and their physicians. Ann Oncol 31, 1115–1118. [DOI] [PubMed] [Google Scholar]
- Sun L, Li M, Deng L, Niu Y, Tang Y, Wang Y, and Guo L (2021). MGA Mutation as a Novel Biomarker for Immune Checkpoint Therapies in Non-Squamous Non-Small Cell Lung Cancer. Front Pharmacol 12, 625593–625593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taliun D, Harris DN, Kessler MD, Carlson J, Szpiech ZA, Torres R, Taliun SAG, Corvelo A, Gogarten SM, Kang HM, et al. (2021). Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature 590, 290–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang TH, Porta-Pardo E, Gao GF, Plaisier CL, Eddy JA, et al. (2018). The Immune Landscape of Cancer. Immunity 48, 812–830 e814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Hu Z, Mahal BA, Zhao SD, Kensler KH, Pi J, Hu X, Zhang Y, Wang Y, Jiang J, et al. (2018). Integrated Analysis of Genetic Ancestry and Genomic Alterations across Cancers. Cancer Cell 34, 549–560 e549. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Related to Figure 1. TCGA TMB Estimates.
Table S2: Related to Figure 1. Cohort Breakdown and TMB Calculation in the MSKCC Cohort (n=672.
Table S3: Related to Table 1 and Figure 2. Clinical, demographic, and TMB estimates of patients treated from the DFCI cohort.
Table S4: Related to Figure 2 and STAR Methods. TMB recalibration in the DFCI data for the seven cancer types of interest (n=8193 patients) using continuous components.
Table S5: Related to Figure 4. Associations between covariates analyzed and clinical outcomes in the DFCI-ICI treated cohort of patients (n=1840).
Table S6: Related to Figure 5. Impact of genetic ancestry on associations between TMB and clinical outcomes in the DFCI-ICI treated cohort of patients with NSCLC (n=879).
Table S7: Related to Figure 6. Genomic correlates of OS and TTF by ancestry in the DFCI-ICI treated cohort of patients with NSCLC (n=879).
Table S8: Related to Figure 6. Raw SNV and CNV calls reported by Oncopanel for ICI-treated patients at DFCI (n=1840).
Data Availability Statement
This paper analyzes existing, publicly available data from the 1000 Genomes project(Genomes Project et al., 2015), ExAC(Lek et al., 2016), gnomAD v2.1.1(Karczewski et al., 2021), Trans-Omics for Precision Medicine (TOPMed) program(Taliun et al., 2021), Partners HealthCare Biobank (RRID:SCR_001316), TCGA whole-exome sequencing (RRID: SCR_014555), and GENIE v11.1(Consortium, 2017). These accession numbers for the datasets are listed in the key resources table. The raw ancestry assignments generated for the TCGA cohorts can be found at https://portal.gdc.cancer.gov. The published article includes also includes datasets generated or analyzed from Dana-Farber Cancer Institute and Memorial Sloan Kettering Cancer Center, currently not publicly available. While targeted sequencing raw data were not deposited in a public repository since full sequencing data are not consented to be shared, the data that support the findings of this study are available from the corresponding author upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| 1000 Genomes project data | The International Genome Sample Resource | RRID: SCR_006828 |
| ExAC | Lek et al. 2016 | https://gnomad.broadinstitute.org/ |
| gnomAD v2.1.1 | Karczewski et al. 2020 | https://gnomad.broadinstitute.org/ |
| Partners HealthCare Biobank | Mass General Brigham | RRID:SCR_00131 6 |
| Trans-Omics for Precision Medicine (TOPMed) program | NHLBI Trans-Omics for Precision Medicine | RRID:SCR_01567 7 |
| TCGA whole-exome sequencing | Genomic Data Commons | RRID: SCR_014555 |
| TCGA SNP 6.0 array | TCGA legacy archive | https://portal.gdc.cancer.gov/legacy-archive/search/f |
| TCGA clinical and subtype data | Genomic Data Commons | https://gdc.cancer.gov/about-data/publications/pancanatlas |
| Additional somatic validation datasets | GENIE v11.1 | https://www.aacr.org/professionals/research/aacr-project-genie/ |
| Software and algorithms | ||
| PLINK v2.0 | Chang et al., 2015 | https://www.cog-genomics.org/plink/2.0/ |
| MuTect | Cibulskis et al. 2013 | https://software.broadinstitute.org/cancer/cga/mutect_download |
| Picard | Broad Institute | http://broadinstitute.github.io/picard/ |
| GATK | Poplin et al., 2017 | https://gatk.broadinstitute.org/hc |
| Burrows Wheeler Aligner | Li and Durbin, 2009 | http://bio-bwa.sourceforge.net/ |
| Bcftools v1.10.2 | Danecek et al., 2021 | https://samtools.github.io/bcftools/bcftools.html |
| VCFtools v0.1.15 | Danecek et al., 2011 | https://vcftools.github.io/ |