

Abstract
Background and Objectives
This study aimed to examine whether baseline CSF measures of Alzheimer disease (AD)–related pathology are associated with the time to onset of mild cognitive impairment (MCI) and whether these associations differ by age, sex, Apolipoprotein E (ApoE4) status, and proximal (≤7 years) vs distal (>7 years) time to symptom onset.
Methods
Measures of amyloid (Aβ1-42 and Aβ1-40), phospho-tau (ptau181), and total tau (t-tau) were determined from CSF samples obtained at baseline from participants in an ongoing longitudinal project, known as the Biomarkers for Older Controls at Risk for Alzheimer Disease study (BIOCARD) study. The fully automated, Lumipulse G immunoassay was used to analyze the specimens. Cox regression models were used to examine the relationship of baseline biomarker levels with time to symptom onset of MCI and interactions with age, sex, and ApoE allelic status in subjects who progressed from normal cognition to MCI.
Results
Analyses included 273 participants from the BIOCARD cohort, who were cognitively normal and predominantly middle-aged at baseline, and have been followed for an average of 16 years (max = 23.6). During follow-up, 94 progressed to MCI (median time to symptom onset = 6.9 years). In Cox regression models, elevated ptau181 and t-tau levels were associated with time to MCI symptom onset if it occurred within 7 years of baseline (HR 1.386 and 1.329; p = 0.009 and 0.017, respectively), while a lower Aβ42/Aβ40 ratio was associated with symptom onset if it occurred >7 years from baseline (HR 0.596, p = 0.003). There were also significant 3-way CSF × age × sex interactions for ptau181 and Aβ42/Aβ40, with follow-up analyses indicating that associations between these biomarkers and progression to MCI were stronger among men than among women, but this difference between sexes diminished with increasing age.
Discussion
The lengthy follow-up of BIOCARD participants permitted an examination of time-varying associations between CSF AD biomarkers with MCI symptom onset and the influence of sex, baseline age, and ApoE4 genotype on these associations. These factors may inform clinical trial enrollment strategies, or trial duration and outcomes, which may use these measures as surrogate markers of treatment response.
AD-related pathologic processes begin in middle age, decades before the onset of cognitive symptoms.1-3 CSF measures of amyloid beta protein (Aβ) and phosphorylated tau (ptau) reflect the primary pathologic hallmarks of AD (amyloid plaques and neurofibrillary tangles). These measures are altered many years before the onset of clinical symptoms of mild cognitive impairment (MCI).4
Most previous studies have used CSF assays based on ELISA or immunosorbent assays. Although requiring extensive user interaction, intralaboratory values are generally consistent. However, several international studies have demonstrated considerable interlaboratory variability.5-7 In attempt to mitigate these issues, fully automated assays have been developed, which use standardized reference samples that reduce variability within and between laboratories.8-10 For example, the Lumipulse G CSF assays have demonstrated good analytical performance,11 which is comparable with manual and other automated immunoassays that have been used in the AD research field.9,12,13 In addition, Lumipulse and Elecsys methods are highly concordant with clinical diagnoses, with recent work suggesting that the Lumipulse Aβ42 and ptau measures provide the highest discriminating power.10 Diagnostic accuracy of these CSF biomarkers have also been demonstrated for AD vs other neurodegenerative dementias,14 and validated cut points for the ratio of total tau (t-tau)/Aβ42 have been shown to correspond to amyloid-PET status in clinical and observational cohorts.9,15
Previous studies of CSF biomarkers, measured using the fully automated assays, have not examined whether these markers are associated with the onset of MCI symptoms among cognitively normal individuals over both short term and long term. This study addresses this gap by leveraging a unique data set derived from the Biomarkers for Older Controls at Risk for Alzheimer Disease study (BIOCARD) study, which began collecting CSF specimens in cognitively normal individuals in 1995. As most participants have been followed to the present time, this affords a particularly long duration of follow-up (median = 15.9 years, max = 23.6 years). It is important that 94 participants have progressed from normal cognition to MCI or dementia, thus allowing an examination of potential interactions of these CSF measures with other variables in relationship to the risk of progression to MCI, including baseline age, sex, and Apolipoprotein E (ApoE) genotype. This has been difficult to achieve in previous studies because of more limited numbers of progressors, which reduces the power to provide estimates of interaction effects. The long duration of follow-up also allows an examination of whether these measures are differentially associated with risk of progression proximally (within 7 years) vs more distally (>7 years) from baseline. Such analyses may contribute useful information for clinical trial strategies of cognitively normal individuals, ensuring enrollment of a sufficient number of subjects who are likely to progress during the time frame of this study.
Methods
Standard Protocol Approvals, Registrations, and Patient Consents
The BIOCARD study was approved by the Johns Hopkins Medical Institutional Review Board #3. Written informed consent was obtained from all participants in this study.
Study Design and Participant Selection
Data for this study were derived from the BIOCARD study, which was designed to recruit and follow a cohort of cognitively normal individuals to identify variables that could predict the subsequent development of mild to moderate symptoms of AD. By design, approximately 75% of the participants had a first-degree relative with dementia of the AD type. The BIOCARD study was initiated at the NIH in 1995. Recruitment procedures, baseline evaluations, and annual clinical and cognitive assessments were previously presented in detail.16 In brief, recruitment was conducted from 1995 to 2005 by staff at the Geriatric Psychiatry Branch of the National Institutes of Mental Health. Individuals were excluded from participation if they were cognitively impaired or had significant medical problems such as severe cardiovascular disease (e.g., atrial fibrillation), chronic neurologic disorders (e.g., epilepsy and multiple sclerosis), or severe cerebrovascular disease (based on MRI scan). After providing written informed consent, 349 individuals were enrolled in this study (eTable 1, links.lww.com/WNL/C197). At baseline, participants completed a comprehensive evaluation consisting of a physical, neurologic, and psychiatric examination; neuropsychological testing; an electrocardiogram; and standard laboratory studies. CSF specimens and MRI scans were obtained approximately every 2 years. This study was stopped in 2005 for administrative reasons. In 2009, a research team from the Johns Hopkins School of Medicine was funded to re-establish the cohort, continue annual clinical and cognitive assessments and blood draws, and evaluate the previously acquired data. Biannual collection of CSF specimens and MRI scans were reinitiated in 2015. Amyloid PET imaging was instituted in 2015, and tau PET imaging was instituted in 2021. eFigure 1, links.lww.com/WNL/C197, presents a schematic representation of the study design. Data collection is ongoing.
Clinical and Cognitive Assessments
Annual clinical examinations included medical history, record of medication use, physical and neurologic examination, behavioral and mood assessments, and the Clinical Dementia Rating (CDR) scale (based on a semistructured interview with the participant and collateral source).17,18 The neuropsychological battery covered all major cognitive domains.16
Participants received a consensus diagnosis at each study visit by the staff of the BIOCARD Clinical Core at Johns Hopkins (prospectively for subjects evaluated starting in 2009 and retrospectively for subjects evaluated at the NIH), as previously described.16 A syndromic diagnosis (e.g., normal, MCI, and dementia) was established by review of (1) clinical data pertaining to the medical, neurologic, and psychiatric status; (2) reports of changes in cognition by the subject and by a collateral source; and (3) decline in cognitive performance, based on longitudinal cognitive testing from multiple domains (and by comparison with published norms). A diagnosis of “impaired not MCI” was made for individuals with contrasting information between the CDR interview and the cognitive test scores. These participants were included in the group of cognitively normal individuals because they do not meet MCI criteria, but all primary analyses were rerun excluding these participants.
If a subject was deemed to be impaired, the decision about the likely etiology of the syndrome was based on the medical, neurologic, and psychiatric information collected at each visit, as well as medical records obtained from the subject, where necessary. More than one etiology could be endorsed for each subject (e.g., AD and vascular disease). Consensus diagnosis procedures followed the diagnostic recommendations incorporated in the National Institute on Aging–Alzheimer Association workgroup reports for the diagnosis of MCI19 and dementia due to AD.20 Consensus diagnoses were blinded to participants' biomarker measures and ApoE status.
The primary outcome variable was time from baseline to onset of MCI clinical symptoms. This determination was established separately, based primarily on information from the CDR interview. In these analyses, “baseline” was defined as time of first CSF measurement.
CSF Biomarker Measures
CSF samples used in the present analyses were collected between 1995 and 2005, while this study was at the NIH. CSF was collected through lumbar puncture and aliquoted into polypropylene cryotubes, which were kept on dry ice, and transferred to a −80°C freezer for long-term storage immediately after collection. For this study, samples were thawed for the first time since collection to run the fully automated chemiluminescence enzyme immunoassays (Lumipulse G1200, Fujirebio Diagnostics, Inc.) for Aβ40, Aβ42, ptau181, and t-tau. The coefficients of variation of an internal CSF standard run every day over the 10 days the assays were performed were Aβ1-42, 3.4%; Aβ1-40, 2.7%; ptau181, 1.8%; and t-tau, 8.0%.
Previous reports indicate that the Aβ42/Aβ40 ratio is more informative than either Aβ measure alone because it accounts for interindividual differences in total CSF Aβ levels and may mitigate against preanalytic variability.9,21-23 Thus, in addition to examining the individual Aβ and tau measures, we examined the following ratios: Aβ42/Aβ40, ptau181/Aβ42, and t-tau/Aβ42. The Supplementary Materials include analyses of additional ratios: ptau181/Aβ40, ptau181/Aβ42:Aβ40, and t-tau/Aβ42:Aβ40.
ApoE Genotype
ApoE genotypes were determined by restriction endonuclease digestion of PCR-amplified genomic DNA (performed by Athena Diagnostics). ApoE4 carrier status was coded by an indicator variable, with E4 carriers coded as 1 and noncarriers coded as 0.
Statistical Analyses
Group differences in baseline characteristics were assessed using t tests or Wilcoxon rank-sum tests for continuous variables and χ2 tests for dichotomous variables. Cox regression models (i.e., proportional hazard models) were used to examine the probability of MCI symptom onset in the observed period and which variables were associated with the time (in years) to this outcome. All CSF variables were standardized into z-scores by subtracting their baseline means and then dividing by the SD, based on their baseline distributions before model fitting, to facilitate comparison of the results across CSF variables. Hazard ratios (HRs; i.e., the relative hazard) and 95% CIs were calculated for variables in the Cox models. Separate models were run for each CSF variable.
First, Cox models (adjusted for baseline age, years of education, ApoE4 status, and sex) were used to determine whether each baseline CSF measures were associated with time to onset of MCI clinical symptoms. Next, the models examined whether the association between baseline CSF measures and risk of clinical symptom onset differed by baseline age and sex, by including a 3-way CSF × age × sex interaction term (as well as all relevant lower-order interaction terms); when significant 3-way interactions were found, follow-up analyses, stratified by sex and age (younger than 60 years vs 60 years or older), were run. If the 3-way interaction was not significant, the model was rerun excluding this term, as well as the age × sex interaction term, to test 2-way interactions (i.e., CSF × age and CSF × sex) in reduced models. A separate set of models tested interactions involving ApoE4 status by including a 3-way CSF × ApoE4 × sex interaction term (as well as all relevant lower-order interaction terms). Nonsignificant 3-way interaction terms were removed, and reduced models tested 2-way interactions (i.e., ApoE4 × sex and ApoE × CSF). Multiple comparison corrections for p values used the Simes method that controls the false discovery rate24 (see the eMethods, links.lww.com/WNL/C197, for details regarding the interpretation of HRs.
In a second set of analyses, Cox models examined whether the CSF measures were associated with clinical symptom onset for transitions occurring closer vs later in time, relative to baseline, by including time-varying coefficients for the CSF biomarkers in the Cox models. These models included an indicator variable for time, as part of the coefficient for the CSF measure, reflecting progression proximal to baseline (within 7 years) vs more distal from baseline (>7 years). Seven years was selected as the cut-off point because it reflected the median time to MCI clinical symptom onset and resulted in a nearly equivalent number of subjects falling within the 2 subgroups (see Results). This analytic approach is equivalent to treating CSF biomarker levels with time-dependent HRs, allowing an examination of whether baseline CSF measures were differentially associated with risk of progression within 7 years vs after 7 years from baseline, as in previous reports.25,26
All analyses were performed in Stata (version 16.1). Significance was set at p < 0.05.
Data Availability
Data used in these analyses are available through standard application procedures described on the BIOCARD website (biocard-se.org).
Results
Descriptive and Baseline Statistics
The analyses presented here include data from 273 participants who were cognitively normal at their baseline CSF measure. Subjects were excluded for the following reasons: Those who withdrew or did not re-enroll (n = 26), whose symptom onset was determined to be before the baseline visit (n = 12) or first CSF measure (n = 4), whose last diagnosis was before their first CSF measure (n = 1), and who did not have CSF collected while this study was at NIH (n = 33).
Table 1 presents the descriptive and baseline characteristics for all participants included in these analyses and for participants stratified by follow-up diagnosis. On average, participants were followed for 15.6 years (maximum = 23.6 years); 179 remained cognitively normal and 94 progressed to MCI or dementia (mean [SD] time from baseline to clinical symptom onset = 7.6 [4.5] years). At baseline, there were no significant differences between participants who remained normal and those who progressed to MCI or dementia in mini-mental state examination (MMSE) scores, sex, ApoE4 status, education, or race (all p > 0.1). However, participants who progressed were older than those who remained cognitively unimpaired (p < 0.001) and had significantly higher levels of CSF ptau181 and t-tau and lower Aβ42/Aβ40 ratios at baseline (all p < 0.001).
Table 1.
Descriptive and Baseline Characteristics for All Participants Included in These Analyses

Associations Between Baseline CSF Biomarker Measures and Progression to MCI Symptom Onset
The results of the Cox regression models examining each of the CSF measures in relation to time to MCI symptom onset are presented in Table 2. With the exception of Aβ40 and Aβ42 alone, all measures were significantly associated with time to MCI clinical symptom onset (all p < 0.02). Specifically, lower Aβ42/Aβ40, higher ptau181 and t-tau, and higher ratios of ptau181 and t-tau to Aβ42 were associated with a faster time to symptom onset. Kaplan-Meier plots reflecting these associations are shown in Figures 1 and 2.
Table 2.
Hazard Ratios for Baseline CSF Measures in Relation to Time to MCI Symptom Onset

Figure 1. Kaplan-Meier Plots Showing the Relationship Between Baseline Levels of CSF t-tau, ptau181, and the Ratio Aβ42/Aβ40 and Time to Onset of Clinical Symptoms of MCI.
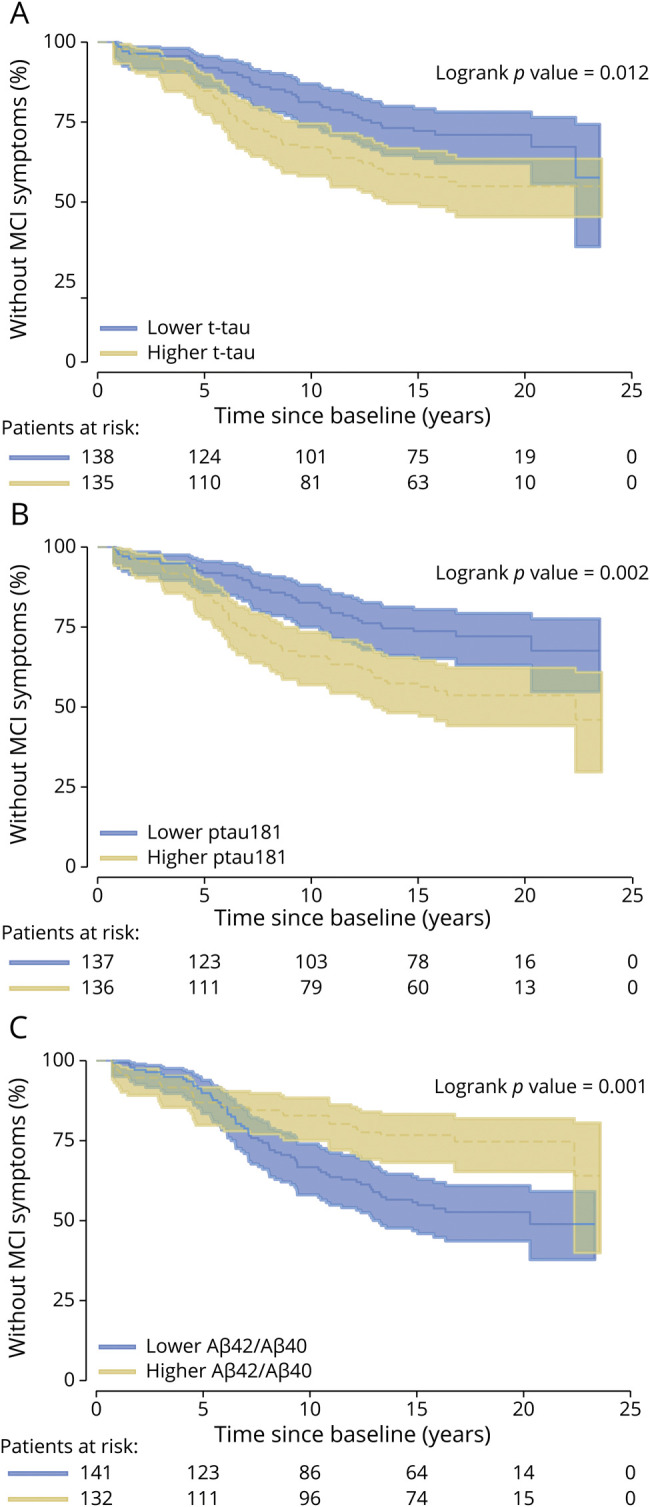
Higher levels of CSF t-tau (A) and ptau181 (B) and lower levels of Aβ42/Aβ40 (C) are significantly associated with a shorter time to onset of clinical symptoms of MCI among individuals who were cognitively normal at baseline. MCI = mild cognitive impairment; t-tau = total tau.
Figure 2. Kaplan-Meier Plots Showing the Relationship Between Baseline CSF Ratios of t-tau/Aβ42 and ptau181/Aβ42 and Time to Onset of Clinical Symptoms of MCI.
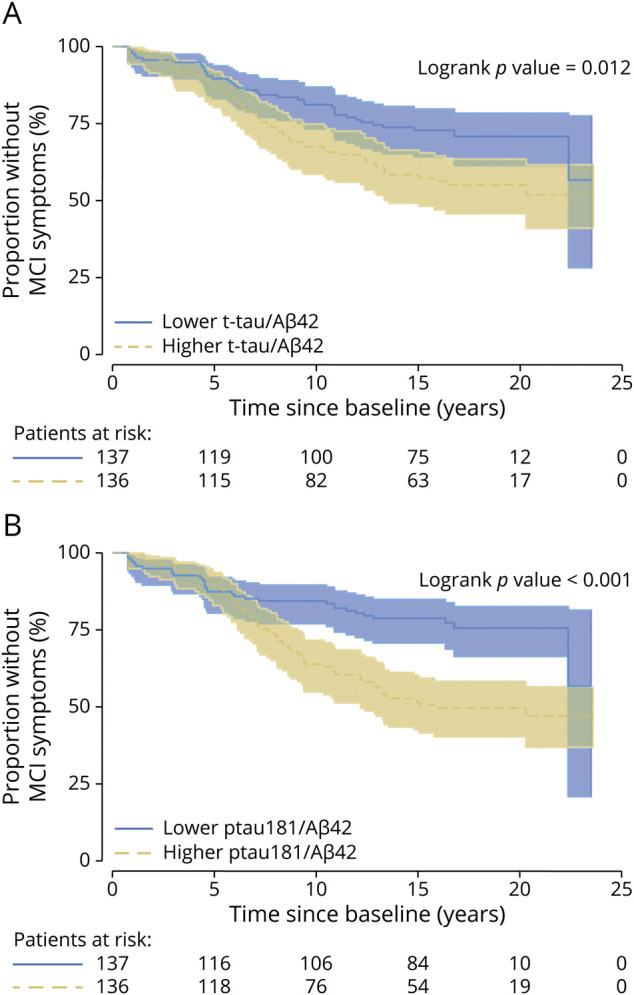
Higher levels of ratios of t-tau/Aβ42 (A) and ptau181/Aβ42 (B) are significantly associated with a shorter time to onset of clinical symptoms of MCI among individuals who were cognitively normal at baseline. MCI = mild cognitive impairment; t-tau = total tau.
The pattern of results was the same when participants with a diagnosis of impaired not MCI (n = 23) were excluded from the analysis, except that the association for Aβ42 reached significance (HR = 0.79, p = 0.04) (eTable 2A, links.lww.com/WNL/C197, for analyses of the ptau181/Aβ40, t-tau/Aβ42:Aβ40, and ptau181/Aβ42:Aβ40 ratios, which were also associated with time to symptom onset. Corresponding Kaplan-Meier plots reshown in eFigure 2, links.lww.com/WNL/C197).
For covariate effects, in all models, older baseline age was associated with reduced time to symptom onset, while more years of education were associated with increased time to onset of MCI symptoms (data not shown). There were also trends for main effects of sex, indicating a tendency for women to show a weaker association between the CSF measures and time to symptom onset than men (eTables 5–6, links.lww.com/WNL/C197, for baseline CSF biomarker values by sex). The main effect of ApoE4 status was not significant in any model (all p > 0.1).
Interactions Between CSF Biomarkers, Baseline Age, Sex, and ApoE4 With Time to MCI Symptom Onset
3-Way CSF × Age × Sex Interaction Terms
Examining whether associations between CSF biomarkers and MCI symptom onset differed by baseline age and sex revealed significant 3-way interactions only for ptau181 (HR for the interaction term = 1.28, p = 0.014) and Aβ42/Aβ40 (HR for the interaction term = 0.74, p = 0.017), as presented in Figure 3. For male participants, there was a significant main effect of ptau181 (HR = 1.89, p < 0.001) and an interaction between ptau181 and age (HR = 0.84, p = 0.008), indicating that higher ptau181 was associated with greater risk of progression but that the strength of this association decreased among men with an older baseline age. For female participants, the main effect of ptau181 and its interaction with age were not significant (both p > 0.30). Holding age constant, the difference in the risk of progression between male participants and female participants, based on ptau181 levels, was significant (HR = 0.53, p = 0.016 for sex × ptau181 interaction), but this difference decreased with increasing age (Figure 3A).
Figure 3. Hazard Ratios for the Baseline CSF Measures at Specific Baseline Ages, Stratified by Sex.

In male participants (blue line), compared with female participants (red line), higher CSF ptau (ptau181) levels (A) and lower ratios of CSF Aβ42/Aβ40 (B) are associated with a shorter time to symptom onset, with the difference between the sexes diminishing as baseline age increases. For illustration purposes, hazard ratios were calculated from the Cox regression models at specific age points (age 50, 60, and 70 years) stratified by sex, using the Cox regression models with the 3-way interactions (CSF measures × age × sex, years of education, and ApoE4 carrier status). ApoE = Apolipoprotein E.
A similar pattern of results was found for Aβ42/Aβ40. Analyses stratified by sex showed that for male participants, lower Aβ42/40 was associated with increased risk of progression (HR = 0.40, p < 0.001). This association was weaker among older men, as indicated by a significant interaction between Aβ42/40 and age (HR = 1.32, p = 0.006). For female participants, neither the main effect of Aβ42/40 nor its interaction with age reached significance (both p > 0.1). Holding age constant, the difference in the risk of progression between male participants and female participants associated with low Aβ42/Aβ40 was significant (HR = 1.90, p = 0.017 for sex × Aβ42/Aβ40 interaction). The difference between sexes diminished with increasing baseline age to a point where they were roughly equivalent by age 65–70 years (Figure 3B).
Finally, the results of reduced models (for the CSF measures for which there were no 3-way interactions) are presented in eTable 3, links.lww.com/WNL/C197. For t-tau, there was a stronger association for male participants than for female participants, as indicated by sex × t-tau interaction (HR = 0.68, p = 0.044), but the interaction between t-tau and age was not significant (p = 0.221). The interaction results for the secondary models are presented in eTable 2B, links.lww.com/WNL/C197.
3-Way CSF × ApoE4 × Sex Interaction Terms
There were no significant 3-way interactions between ApoE4 status, sex, and any of the primary CSF measures (all p > 0.12; data not shown). In the reduced models, none of the ApoE4 × CSF (all p > 0.11) or ApoE4 × sex interactions were significant (all p > 0.2; data not shown) (eTable 2C, links.lww.com/WNL/C197, for the interaction results of the ratios of t-tau/Aβ42:Aβ40 and ptau181/Aβ42 and the risk of progression to MCI, which differed among ApoE4 carriers vs noncarriers).
CSF Biomarkers in Relation to Risk of Progression Within vs After 7 Years
Among the 94 participants who developed symptoms of MCI or dementia over time, 48 became symptomatic within 7 years of baseline and 46 after 7 years from baseline. Descriptive and baseline statistics for these 2 subgroups are presented in eTables 5 and 6, links.lww.com/WNL/C197. There were no differences in baseline age, sex, education, race, ApoE4 status, baseline MMSE score, or any CSF measure, except that the group who progressed within 7 years had higher ptau181 levels (p = 0.045) and marginally higher t-tau levels (p = 0.096). The mean age of MCI symptom onset was significantly younger for the proximal vs the distal progressors.
The results from the Cox regression models with time-varying coefficients for CSF biomarkers are presented in Table 3. The ratio of Aβ42/Aβ40 was strongly associated with MCI symptom onset in those who progressed >7 years from baseline (HR = 0.60, p = 0.003), but not in those who progressed more proximally to baseline (HR = 0.74, p = 0.051). By contrast, elevated ptau181 and t-tau, as well as the ratios of t-tau and ptau181 to Aβ42, were associated with MCI symptom onset in those who progressed proximally (i.e., within 7 years) to baseline (all HR > 1.32, all p ≤ 0.02). However, the only tau ratio that was associated with MCI symptom onset occurring more distally from baseline was the ratio t-tau/Aβ42 (HR = 1.42, p = 0.032; all others p > 0.05). The results were similar when participants with a diagnosis of impaired not MCI were excluded, except that the associations of Aβ42 with risk of progression reached significance in the >7 year group (HR = 0.68, p = 0.026). The results for other secondary ratios are presented in eTable 2D, links.lww.com/WNL/C197.
Table 3.
Hazard Ratios for Baseline CSF Measures in Relation to Onset of MCI Symptoms Proximally (Within 7 Years) vs Distally (>7 Years)

Discussion
In this study, the fully automated Lumipulse G assay for Aβ40, Aβ42, ptau181, and t-tau was used to investigate relationships between baseline CSF measures and time to MCI symptom onset in a cohort of 273 well-characterized, longitudinally followed individuals who were cognitively normal and largely middle-aged at baseline, 94 of whom subsequently progressed to MCI or dementia. There were several notable findings. First, extending previous studies27 baseline levels of CSF ptau181, t-tau, and the ratios of Aβ42/Aβ40 and tau/Aβ42 were significantly associated with time to MCI clinical symptom onset. Second, the strength of the relationships of some of these CSF measures to time to MCI symptom onset varied with baseline age, sex, and ApoE4 genetic status. Finally, measures of CSF tau and Aβ42/Aβ40 had different time-varying relationships with MCI symptom onset.
Notably, CSF tau-based measures (i.e., ptau181 and t-tau) were associated with MCI symptom onset within 7 years of baseline, but not with MCI symptom onset >7 years from baseline. Conversely, CSF Aβ-based measures showed the opposite temporal association, with Aβ42/Aβ40 being strongly associated with progression >7 years from baseline, but not within 7 years of baseline. These time-dependent associations suggest that the Aβ42/Aβ40 ratio provides information regarding more distal progression, whereas tau measures, as well as the tau/Aβ ratios, are associated with more proximal progression. This is consistent with hypothetical models suggesting that the changes in Aβ levels occur earlier in the disease course than tau (e.g., Ref. 28) and with findings indicating that elevations in tau, or the presence of both amyloid and tau pathology, have a closer temporal relationship to cognitive decline among cognitively normal individuals, relative to amyloid alone.25,29-32 For example, a recent large multicohort cross-sectional study of 3,284 cognitively normal individuals that included CSF biomarker measurements estimated that changes in Aβ markers occur further in advance of the onset of cognitive decline than t-tau and ptau181 during preclinical stages of AD.33 In addition, of note, the only baseline difference between individuals who progressed within 7 years vs after 7 years was higher ptau181 levels (and marginally higher t-tau) among proximal progressors. This suggests that ptau181 levels may be informative in estimating the time to symptom onset (after accounting for other risk factors, including age, education, and Aβ levels).
Given the number of individuals who progressed from normal cognition to MCI or dementia over the long duration of BIOCARD study follow-up, these analyses offered the unique opportunity to examine whether associations between baseline CSF measures and risk of progression to MCI symptom onset were modified by other factors, including ApoE genotype, sex, and baseline age. The results indicated that the relationship between ptau181 and the ratio of Aβ42/Aβ40, with the risk of progression to MCI symptom onset, differed by both baseline age and sex. For both biomarkers, the strength of the association was stronger in male participants than in female participants at younger baseline ages, but the differences between sexes diminished with increasing baseline age to a point where they were roughly equivalent by age 65–70 years (Figure 3). Although the strengths of these associations decreased with older baseline ages in male participants, in female participants, by comparison, the relationship between ptau181 and Aβ42/Aβ40 with the risk of progression MCI symptom onset did not significantly differ by baseline age. These interactions are unlikely to be due to sex differences in absolute CSF biomarker levels, given that baseline levels of t-tau, ptau181, and Aβ42/Aβ40 did not differ between men and women, either in the full cohort (eTable 4, links.lww.com/WNL/C197) or among the 94 subjects who progressed to MCI or dementia (eTable 5, links.lww.com/WNL/C197).
A possible explanation for these observed 3-way interactions may pertain to age-related differences in cerebrovascular disease among men vs women. Cardiovascular risk factors and cerebrovascular changes are more prevalent in men than in women younger than 60 years, whereas at older ages, sex differences in cerebrovascular changes and cardiovascular risks decrease.34,35 Evidence suggests that the presence of both AD and vascular pathology has additive effects on the threshold for cognitive impairment.36,37 Thus, at younger ages, the additive effect of vascular disease and AD pathology may lower the threshold for cognitive impairment in men, resulting in a stronger relationship between AD biomarkers and risk of progression from normal cognition to MCI symptom onset in men vs women. By contrast, at older ages, the additive effect of vascular disease and AD pathology may be similar in men and women, resulting in AD biomarkers showing equally strong associations with the risk of progression to MCI symptom onset. Future studies are needed to replicate these findings and further explore this possibility.
This study found no sex difference in the risk of progression associated with ApoE4 genotype, although sex differences in AD risk factors have been described for ApoE genotype (e.g., Ref. 34, 38, 39). For example, previous studies have suggested that ApoE4 genotype is a stronger risk factor for AD in women, especially among women of younger ages (e.g., Refs. 40-42). Several recent studies have also suggested a stronger relationship between ApoE4 genotype and CSF tau levels among women, especially in the presence of greater amyloid burden,43-46 but the results have been mixed,40,47 and overall, the literature is sparse. Despite the sizeable number of individuals who progressed from normal cognition to MCI symptom onset, this study may have been underpowered to detect significant 2-way and 3-way interactions. Future studies with larger sample sizes are needed to address the possible interactions between CSF biomarkers, ApoE genotype, age, and sex regarding the risk of progressing to MCI or dementia.
As presented in eTable 2C, links.lww.com/WNL/C197, there was a stronger relationship between the ratios of ptau181/Aβ42:Aβ40 and t-tau/Aβ42:Aβ40 and the risk of MCI symptom onset among ApoE4 carriers than noncarriers. In addition, although the other CSF measures did not show significant interactions with ApoE4 genotype, models stratified by ApoE4 demonstrated trends for stronger relationships between these CSF measures and risk of symptom onset among ApoE4 carriers (data not shown), suggesting these effects may be evident in larger sample sizes. A stronger association between CSF biomarkers and risk of progression among ApoE4 carriers than noncarriers is consistent with ApoE4 being a risk factor for AD,41 while among noncarriers, other risk factors (not examined in this study) may have alternative or additive impacts on predicting progression to MCI.
Elucidating the natural history of AD in its presymptomatic phases is essential for the design of clinical trials in which cognitively normal individuals would be enrolled, because such data provide information about those most likely to progress within specific periods. Of note, an analysis of placebo data from a large number of MCI clinical trials demonstrated that many subjects had limited progression over the trial time frame, making it difficult to determine whether the treatments were efficacious.48 Thus, it will be critical to enroll subjects likely to progress during the time frame of a clinical trial among cognitively normal individuals, selected on the basis of their biomarker profiles, to identify differences in outcomes between those on placebo vs those on an interventional therapy. Establishing temporal relationships between amyloid, tau, and other biomarkers of disease stage and progression and clarifying interactions with other risk factors such as sex, age, and ApoE4 genotype will be important in this regard, particularly in cognitively normal individuals, where heterogeneity is likely to be a particularly critical factor (e.g., Ref. 49). A recent example of baseline measures guiding trial design was implemented in the phase 2 study of the antiamyloid immunotherapy donanemab, in which enrollment was based on specific levels of both amyloid and tau, as measured by PET scans.50
This study has several strengths, including the long duration of follow-up, and the relatively large number of well-characterized, cognitively normal individuals who were primarily in middle age at baseline. These findings should also be interpreted within the context of their limitations. Participants were well-educated and primarily White, which limits the generalizability of these results. Although these analyses included a total of 273 participants, the number of individuals who progressed to MCI (n = 94) was modest for examining complex interaction effects, such as age, sex, or ApoE genetic status. Therefore, these interaction results should be considered preliminary because they require replication in future studies with larger sample sizes and more outcomes. In addition, CSF biomarkers are reflective of global changes. Brain region-specific effects were not examined. Finally, this study focused on baseline CSF levels, rather than longitudinal changes in CSF biomarkers. Future studies evaluating longitudinal measures regarding the risk of progressing to MCI are needed.
In summary, these findings confirm and extend prior results in several important ways. Across the 94 subjects who progressed from normal cognition to MCI or dementia, elevated tau levels were associated with MCI symptom onset if it occurred within 7 years of baseline measure, but less so if onset occurred after 7 years. Conversely, Aβ measures were associated with symptom onset if it occurred after, rather than before 7 years. Moreover, the associations of CSF AD biomarkers to time to MCI symptom onset are influenced by sex, baseline age, and ApoE4 genotype factors potentially affecting clinical trial enrollment and anticipated outcomes, either for amyloid-based and tau-based therapies or alternative therapeutic approaches that may use these measures as surrogate markers of treatment response.
Acknowledgment
The BIOCARD Study consists of 7 Cores with the following members: (1) The Administrative Core (Marilyn Albert, Barbara Rodzon), (2) the Clinical Core (Marilyn Albert, Anja Soldan, Corinne Pettigrew, Leonie Farrington, Maura Grega, Gay Rudow, Rostislav Brichko, Scott Rudow, and Nicole Johnson), (3) the Imaging Core (Michael Miller, Susumu Mori, Tilak Ratnanather, Anthony Kolasny, Hanzhang Lu, Peter vanZijl, and Laurent Younes), (4) the Biospecimen Core (Abhay Moghekar, Jacqueline Darrow, and Alexandria Lewis), (5) the Informatics Core (Ann Ervin, David Shade, Jennifer Jones, Hamadou Coulibaly, and Kathy Moser), the (6) Biostatistics Core (Mei-Cheng Wang, Yuxin Zhu, and Jiangxia Wang), and (7) the Neuropathology Core (Juan Troncoso, David Nauen, and Karen Fisher).
Glossary
- Aβ
β-amyloid
- AD
Alzheimer disease
- ApoE
Apolipoprotein E
- BIOCARD
Biomarkers for Older Controls at Risk for Alzheimer Disease study
- CDR
Clinical Dementia Rating scale
- HR
hazard ratio
- MCI
mild cognitive impairment
- MMSE
mini-mental state examination
- ptau
phospho-tau
- t-tau
total tau
Appendix. Authors
Study Funding
This study was funded by the National Institute on Aging (Grant Number: U19–AG033655).
Disclosure
The authors report no relevant disclosures. Go to Neurology.org/N for full disclosures.
References
- 1.Barthélemy NR, Li Y, Joseph-Mathurin N, et al. , Dominantly Inherited Alzheimer Network. A soluble phosphorylated tau signature links tau, amyloid and the evolution of stages of dominantly inherited Alzheimer's disease. Nat Med. 2020;26(3):398-407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gordon BA, Blazey TM, Su Y, et al. Spatial patterns of neuroimaging biomarker change in individuals from families with autosomal dominant Alzheimer's disease: a longitudinal study. Lancet Neurol. 2018;17(3):241-250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Quiroz YT, Zetterberg H, Reiman EM, et al. Plasma neurofilament light chain in the presenilin 1 E280Aautosomal dominant Alzheimer's disease kindred: a cross-sectional and longitudinal cohort study. Lancet Neurol. 2020;19(6):513-521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alzheimer's Association. Alzheimer's disease facts and figures. Alzheimers Dement. 2021;17:327-406. [DOI] [PubMed] [Google Scholar]
- 5.Mattsson N, Andreasson U, Persson S, et al. The Alzheimer's Association external quality control program for cerebrospinal fluid biomarkers. Alzheimers Dement. 2011;7(4):386-395.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.del Campo M, Mollenhauer B, Engelborghs S, et al. Recommendations to standardize preanalytical confounding factors in Alzheimer's and Parkinson's disease cerebrospinal fluid biomarkers: an update. Biomark Med. 2012;6(4):419-430. [DOI] [PubMed] [Google Scholar]
- 7.Janelidze S, Stomrud E, Brix B, Hansson O. Towards a unified protocol for handling of CSF before β-amyloid measurements. Alzheimers Res Ther. 2019;11(1):63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bittner T, Zetterberg H, Teunissen CE, et al. Technical performance of a novel, fully automated eletrochemiluminescence immunoassay for the quantitation of -amyloid (1-42) in human cerebrospinal fluid. Alzheimers Dement. 2016;12:517-526. [DOI] [PubMed] [Google Scholar]
- 9.Willemse EAJ, Tijms BM, van Berckel BNM, et al. Comparing CSF amyloid-beta biomarker ratios for two automated immunoassays, Elecsys and Lumipulse, with amyloid PET status. Alzheimers Dement. 2021;13(1):e12182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dakterzada F, López-Ortega R, Arias A, et al. Assessment of the concordance and diagnostic accuracy between Elecsys and Lumipulse fully automated platforms and Innotest. Front Aging Neurosci. 2021;13:604119. 10.3389/fnagi.2021.604119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tão MJ, Silva-Spinola A, Santana I, et al. Clinical validation of the Lumipulse G cerebrospinal fluid assays for routine diagnosis of Alzheimer's disease. Alzheimers Res Ther. 2019;11:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Paciotti S, Sepe FN, Eusebi P, et al. Diagnostic performance of a fully automated chemiluminescent enzyme immunoassay for Alzheimer's disease diagnosis. Clin Chim Acta. 2019;494:74-78. [DOI] [PubMed] [Google Scholar]
- 13.Keshavan A, Wellington H, Chen Z, et al. Concordance of CSF measures of Alzheimer's pathology with amyloid PET status in a preclinical cohort: a comparison of Lumipulse and established immunoassays. Alzheimers Dement. 2021;13(1):e12131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Agnello L, Piccoli T, Vidali M, et al. Diagnostic accuracy of cerebrospinal fluid biomarkers measured by chemiluminescent enzyme immunoassay for Alzheimer disease diagnosis. Scand J Clin Lab Invest. 2020;80(4):313-317. [DOI] [PubMed] [Google Scholar]
- 15.Kaplow J, Vandijck M, Gray J, et al. Concordance of Lumipulse cerebrospinal fluid t-tau/Aβ42 ratio with amyloid PET status. Alzheimers Dement. 2019;16:144-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Albert M, Soldan A, Gottesman R, et al. Cognitive changes preceding clinical symptom onset of mild cognitive impairment and relationship to ApoE genotype. Curr Alzheimer Res. 2014;11(8):773-784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hughes CP, Berg L, Danziger WL, Coben LA, Martin RL. A new clinical scale for the staging of dementia. Br J Psychiatry J Ment Sci. 1982;140:566-572. [DOI] [PubMed] [Google Scholar]
- 18.Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology. 1993;43(11):2412-2414. [DOI] [PubMed] [Google Scholar]
- 19.Albert MS, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7(3):270-279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7(3):263-269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dumurgier J, Schraen S, Gabelle A, et al. Cerebrospinal fluid amyloid-β 42/40 ratio in clinical setting of memory centers: a multicentric study. Alzheimers Res Ther. 2014;7:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lewczuk P, Matzen A, Blennow K, et al. Cerebrospinal fluid Aβ42/40 corresponds better than Aβ42 to amyloid PET in Alzheimer's disease. J Alzheimers Dis. 2017;55(2):813-822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gervaise-Henry C, Watfa G, Albuisson E, et al. Cerebrospinal fluid Aβ42/Aβ40 as a means to limiting tube- and storage-dependent pre-analytical variability in clinical setting. J Alzheimers Dis. 2017;57(2):437-445. [DOI] [PubMed] [Google Scholar]
- 24.Newson RB. Frequentist Q-values for multiple-test procedures. Stata J. 2010;10:568-584. [Google Scholar]
- 25.Pettigrew C, Soldan A, Zhu Y, et al. , BIOCARD Research Team. Cortical thickness in relation to clinical symptom onset in preclinical AD. Neuroimage. 2016;12:116-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chan CK, Soldan A, Pettigrew C, et al. Depressive symptoms in relation to clinical symptom onset of mild cognitive impairment. Int Psychogeriatr. 2019;31(4):561-569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moghekar A, Li S, Lu Y, et al. , BIOCARD Research Team. CSF biomarker changes precede symptom onset of mild cognitive impairment. Neurology. 2013;81(20):1753-1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jack CR, Knopman DS, Jagust WJ, et al. Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12(2):207-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Soldan A, Pettigrew C, Cai Q, et al. , BIOCARD Research Team. Hypothetical preclinical Alzheimer disease groups and longitudinal cognitive change. JAMA Neurol. 2016;73(6):698-705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Soldan A, Pettigrew C, Fagan AM, et al. ATN profiles among cognitively normal individuals and longitudinal cognitive outcomes. Neurology. 2019;92(14):e1567-e1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sperling RA, Mormino EC, Schultz AP, et al. The impact of amyloid-beta and tau on prospective cognitive decline in older individuals. Ann Neurol. 2019;85(2):181-193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vos SJB, Xiong C, Visser PJ, et al. Preclinical Alzheimer's disease and its outcome: a longitudinal cohort study. Lancet Neurol. 2013;12(10):957-965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luo J, Agboola F, Grant E, et al. Sequence of Alzheimer disease biomarker changes in cognitively normal adults. A cross-sectional study. Neurology. 2020;95(23):e3104-e3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ferretti MT, Iulita MF, Cavedo E, et al. , Women's Brain Project and the Alzheimer Precision Medicine Initiative. Sex differences in Alzheimer' disease–the gateway to precision medicine. Nat Rev Neurol. 2018;14(8):457-469. [DOI] [PubMed] [Google Scholar]
- 35.Oveisgharan S, Arvanitakis Z, Yu L, Farfel J, Schneider JA, Bennett DA. Sex differences in Alzheimer's disease and common neuropathologies of aging. Acta Neuropathol. 2018;136(6):887-900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kapasi A, DeCarli C, Schneider JA. Impact of multiple pathologies on the threshold for clinically overt dementia. Acta Neuropathol. 2017;134(2):171-186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Troncoso JC, Zonderman AB, Resnick SM, Crain B, Pletnikova O, O'Brien RJ. Effect of infarcts on dementia in the Baltimore longitudinal study of aging. Ann Neurol. 2008;64(2):168-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Riedel BC, Thompson PM, Brinton RD. Age, APOE and sex: triad of risk of Alzheimer's disease. J Steroid Biochem Mol Biol. 2016;160:134-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Duarte-Guterman P, Albert AY, Barha CK, Galea LAM, On Behalf of the Alzheimer's Disease Neuroimaging Initiative. Sex influences the effects of APOE genotype and Alzheimer's diagnosis on neuropathology and memory. Psychoneuroendocrinology. 2021;129:105248. [DOI] [PubMed] [Google Scholar]
- 40.Altmann A, Tian L, Henderson VW, Greicius MD, Alzheimer's Disease Neuroimaging Initiative Investigators. Sex modifies the APOE-related risk of developing Alzheimer disease. Ann Neurol. 2014;75(4):563-573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Farrer LA, Cupples LA, Haines JL, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA. 1997;278(16):1349-1356. [PubMed] [Google Scholar]
- 42.Neu SC, Pa J, Kukull W, et al. Apolipoprotein E genotype and sex risk factors for Alzheimer disease. A meta-analysis. JAMA Neurol. 2017;74(10):1178-1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Buckley RF, Mormino EC, Chhatwal J, et al. , Alzheimer's Disease Neuroimaging Initiative. Associations between baseline amyloid, sex, and APOE on subsequent tau accumulation in cerebrospinal fluid. Neurobiol Aging. 2019;78:178-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hohman TJ, Dumitrescu L, Barnes LL, et al. , Alzheimer's Disease Genetics Consortium and the Alzheimer's Disease Neuroimaging Initiative. Sex-specific association of apolipoprotein E with cerebrospinal fluid levels of tau. JAMA Neurol. 2018;75(8):989-998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mofrad RB, Tijms BM, Scheltens P, et al. Sex differences in CSF biomarkers vary by Alzheimer disease stage and APOE ԑ4 genotype. Neurology. 2020;95:e2378–e2388. [DOI] [PubMed] [Google Scholar]
- 46.Sundermann EE, Panizzon MS, Chen X, Andrews M, Galasko D, Banks SJ, Alzheimer's Disease Neuroimaging Initiative. Sex differences in Alzheimer's-related Tau biomarkers and a mediating effect of testosterone. Biol Sex Differ. 2020;11(1):33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li G, Shofer JB, Petrie EC, et al. Cerebrospinal fluid biomarkers for Alzheimer's and vascular disease vary by age, gender, and APOE genotype in cognitively normal adults. Alzheimers Res Ther. 2017;9(1):48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Petersen RC, Thomas RG, Aisen PS, Mohs RC, Carrillo MC, Albert MS, Alzheimer's Disease Neuroimaging Initiative ADNI and Foundation for NIH FNIH Biomarkers Consortium AD MCI Placebo Data Analysis Project Team. Randomized controlled trials in mild cognitive impairment: sources of variability. Neurology. 2017;88(18):1751-1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ryan L, Hay M, Huentelman MJ, et al. Precision aging: applying precision medicine to the field of cognitive aging. Front Aging Neurosci. 2019;11:128. doi: 10.3389/fnagi.2019.00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mintun MA, Lo AC, Duggan Evans C, et al. Donanemab in early Alzheimer's disease. New Engl J Med. 2021;384(18):1691-1704. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data used in these analyses are available through standard application procedures described on the BIOCARD website (biocard-se.org).