

Abstract
Compared with traditional therapies, targeted therapy has merits in selectivity, efficacy, and tolerability. Small molecule inhibitors are one of the primary targeted therapies for cancer. Due to their advantages in a wide range of targets, convenient medication, and the ability to penetrate into the central nervous system, many efforts have been devoted to developing more small molecule inhibitors. To date, 88 small molecule inhibitors have been approved by the United States Food and Drug Administration to treat cancers. Despite remarkable progress, small molecule inhibitors in cancer treatment still face many obstacles, such as low response rate, short duration of response, toxicity, biomarkers, and resistance. To better promote the development of small molecule inhibitors targeting cancers, we comprehensively reviewed small molecule inhibitors involved in all the approved agents and pivotal drug candidates in clinical trials arranged by the signaling pathways and the classification of small molecule inhibitors. We discussed lessons learned from the development of these agents, the proper strategies to overcome resistance arising from different mechanisms, and combination therapies concerned with small molecule inhibitors. Through our review, we hoped to provide insights and perspectives for the research and development of small molecule inhibitors in cancer treatment.
Keywords: combination therapy, multikinase molecule inhibitors, resistance, small molecule inhibitors, small molecule kinase inhibitors
Small molecule inhibitors have been permitted to be used in post‐line therapy, first‐line treatment, and adjuvant therapy. Compared with post‐line and first‐line treatment, attempts at adjuvant therapy are just beginning. Several factors should be considered in the adjuvant setting, including other standard treatments, subsequent therapy, financial benefit ratio, and treatment‐related side effects.
1. INTRODUCTION
Tumors are complex, and there are some ways to treat them. Cancer chemotherapy and radiation therapy have some selectivity for tumor cells because of their increased proliferation rate. 1 With the advent of modern cell biology since the 1980s, lots of molecular drivers of cancer have been obtained and targeted cancer therapy has become dominant in novel drug development. 2
Antibody therapies and small molecule inhibitors are the two main methods for targeted cancer treatment. 3 The mechanism of small molecule inhibitors is to inhibit the target proteins’ function by binding to the “pocket” on their surface. Small molecule inhibitors can bind a wider range of extracellular and intracellular targets compared with antibodies due to their smaller size. Besides, most small molecule inhibitors can be taken orally, while antibodies are administered subcutaneously or intravenously. What is more, some of small molecule inhibitors can penetrate the blood–brain barrier to control intracranial lesions. 1 , 2 , 3 , 4
The targets of these drugs cover a large scope. Most small molecule inhibitors belong to protein kinase inhibitors. 5 , 6 , 7 In addition, drugs involved in deoxyribonucleic acid (DNA) repair, epigenetics, apoptosis, tumor metabolism, and beyond are also being discovered. 8 , 9 , 10 , 11 Surprisingly, targets considered untargetable or difficult to target in the past, such as RAS, have also been approved recently. 12 , 13 It is undeniable that small molecule inhibitors still encounter many challenges such as low response rate and drug resistance.
In our review, we first describe the classification of small molecule inhibitors, including selective small molecule kinase inhibitors, selective small molecule nonkinase inhibitors, and multikinase small molecule inhibitors. Then, we discuss the small molecule inhibitors according to signaling pathways and the classification of small molecule inhibitors. In each part of specific inhibitors, we first introduce this target and the pathways involved in the target, as well as its significance in tumors. Followed by discussing United States Food and Drug Administration (US FDA)‐approved small molecule inhibitors. We subsequently introduce small molecule inhibitors in clinical trials summarized from clinical trials website. At the end of our review, we also discuss the main issues and future directions concerned with small molecule inhibitors.
2. CLASSIFICATION OF SMALL MOLECULE INHIBITORS
To our knowledge, there are 88 small molecule inhibitors approved by the US FDA for oncology indications by August 2022 5 , 6 , 7 , 8 , 9 , 10 , 11 (Figure 1). According to target selectivity, small molecule inhibitors can be divided into selective small molecule inhibitors and multikinase small molecule inhibitors. According to whether the substrate is a protein kinase, selective small molecule inhibitors are further divided into selective small molecule kinase inhibitors and selective small molecule nonkinase inhibitors. 10
FIGURE 1.
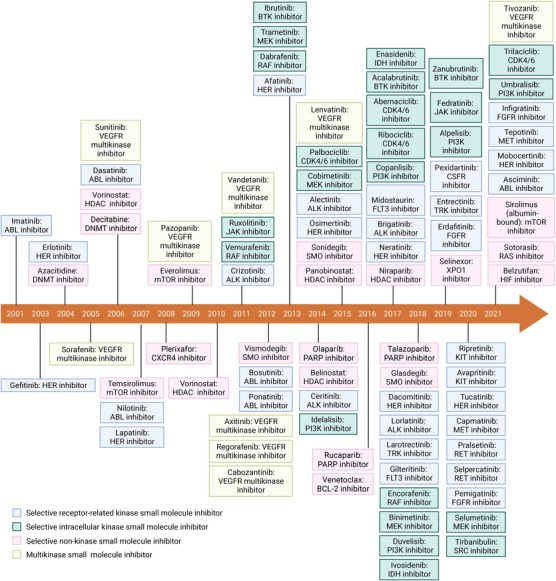
Timeline for the US FDA‐approved small molecule inhibitors targeting the cancers. Figure created with BioRender.com
Selective small molecule inhibitors usually bind to a single target and inhibit the target‐related cell signaling. A subset of cancers strongly relies on a few dysfunctions related to growth, survival, apoptosis, differentiation, cancer metabolism, and even immune modulation. 14 , 15 Selective small inhibitors can antagonize the critical target to inhibit its unusual function or reverse its regular action, correspondingly, to treat tumors. Patients commonly need strict screening for the presence or absence of specific gene alteration detected from solid tumor tissue or circulating tumor cells in the blood or other body fluid when treated by selective small molecule inhibitors. 16 Under this condition, selective small molecule inhibitors can effectively target tumors and avoid side effects brought by off‐target inhibition. 17 , 18
Selective small molecule inhibitors are further divided into selective small molecule kinase inhibitors and selective small molecule nonkinase inhibitors. 10 Protein kinase inhibitors are the main category of small molecule inhibitors and the criterion for a protein kinase inhibitors to be a multikinase or selective small molecule kinase inhibitor is the number of kinases whose values of IC50 of inhibitory activity are below 10 nM. 19 Multikinase small molecule inhibitors exert anticancer activity by repressing multiple protein kinases in the tumor. Multikinase inhibitors do not require precise detection but rely on histology. 20 , 21 Most approved drugs are multikinase inhibitors of the vascular endothelial growth factor receptors (VEGFR) owning antiangiogenic and antiproliferative effects. Selective small molecule kinase inhibitors account for most selective small molecule inhibitors. 10 The selective small molecule kinase inhibitors contains receptor‐related kinase inhibitors, kinase inhibitors targeted intracellular signaling pathways, and inhibitors targeting other cytoplasmic kinases. 22 Beyond the kinome, many molecular targets with proven roles in cancer have gradually developed into selective small molecule nonkinase inhibitors. The molecular targets involved in the nucleus, whose mechanisms are gene transcription, DNA repair, epigenetic modification, and nuclear protein exportation. Some of receptor and intracellular signaling inhibitors and agents involved in triggering apoptosis are also fall into this category. 23 The details of mechanisms will be discussed below and have been depicted in Figure 2.
FIGURE 2.
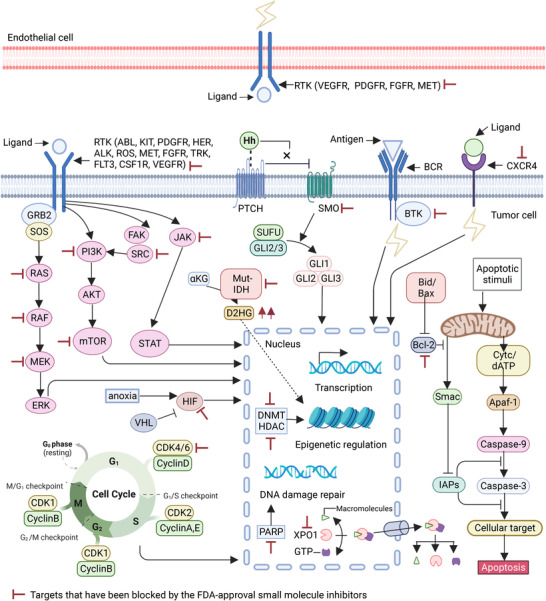
The mechanisms of the US FDA‐approved small molecule inhibitors targeting the cancers. The cell surface receptors and signaling pathways, DNA transcription, DNA damage repair, epigenetic modifications, nuclear transport, blood vessels, and apoptosis are involved in the targets of the US FDA‐approved small molecule inhibitors. Figure created with BioRender.com
Both selective small molecule kinase inhibitors and multikinase small molecule inhibitors belong to protein kinase inhibitors. 24 560 protein kinases in the human kinome are divided into 500 eukaryotic protein kinases (ePKs) and 60 atypical protein kinases (aPKs). Tyrosine kinases and threonine/serine kinases are two essential classes of eight categories of ePKs, while lipid kinases belong to aPKs. 25 Protein kinases bind adenosine triphosphate (ATP) through their ATP‐binding pocket and then transfer the γ phosphate group at the end of ATP to the substrate, thereby activating the substrate and the signal transduction pathway. 26 Reversible protein phosphorylation mediated by kinases and phosphatases has a crucial role in regulating cellular functions. 27 Deregulation of kinases’ function owing to their mutations, translocations, or overexpression in cancer offers an opportunity for small molecule kinase inhibitors, which can block the binding of ATP to protein kinases. 28
The highly dynamic nature of protein kinases allows for the design of inhibitors that recognize the active or multiple inactive conformations. Correspondingly, protein kinase inhibitors are classified into six types (type I–VI) based on their biochemical mechanisms of action. 29 Type I inhibitors interact directly with the ATP binding site and target the kinase active state, which is characterized by the DFG‐in conformation. Besides, αC is in their active “in” position and the G‐loop is not in an active conformation. 30 Type II inhibitors also bind to the ATP binding site but with DFG‐out catalytically inactive conformation. The state of αC and G‐loop of type II inhibitors maintains the same as type I inhibitors. The binding modes of type I1/2 inhibitors are between those of canonical type I and type II inhibitors. 31 Type I1/2 inhibitors definitely disrupt the kinase's R‐spine, and the DFG motif remains in a DFG‐in position. 32 Both type III and IV inhibitors belong to allosteric inhibitors, which bind to the hydrophobic pocket of kinase and are more selective than ATP‐competitive inhibitors. The difference between type III and IV inhibitors is the distance between the hydrophobic pocket and the ATP‐binding site. Type III inhibitors’ hydrophobic binding pocket is adjacent to the ATP‐binding site, while type IV inhibitors bind away from the ATP binding pocket. 33 Type V inhibitors, also known as bi‐substrate inhibitors, interact with both the allosteric and ATP binding pockets. 34 The above classes of inhibitors interact reversibly, while type VI inhibitors form an irreversible covalent bond with cysteine residues in and around the ATP‐binding site of the kinase. These covalent approaches make type VI inhibitors more potent and more specific. 35
In our review, we will introduce them in the order of selective small molecule kinase inhibitors, selective small molecule nonkinase inhibitors, and multikinase small molecule inhibitors.
3. SELECTIVE SMALL MOLECULE KINASE INHIBITORS
The number of selective small molecule kinase inhibitors is the largest in the US FDA‐approved small molecule inhibitors. This part will initially describe the first approved ABL kinase inhibitor, followed by receptor‐related kinase inhibitors like human epidermal growth factor recepter (HER), anaplastic lymphoma kinase (ALK), and fibroblast growth factor receptor (FGFR) kinase inhibitors. Then, attention will be given to kinase inhibitors targeted intracellular signaling pathways involving RAF/MEK/ERK, PI3K/AKT/mTOR, and JAK/STAT3 signaling. Finally, inhibitors targeting other cytoplasmic kinases will be introduced. The selective small molecule kinase inhibitors approved by the US FDA are summarized in Table 1 and the pivotal candidates of small molecule kinase inhibitors are listed in Table S1.
TABLE 1.
Summary of approved selective small molecule kinase inhibitors
| Class | Drug name | Company | First approval | Target | Protein substrate | Administration pathway | Indications |
|---|---|---|---|---|---|---|---|
| ABL | Imatinib (Gleevec) | Novartis | 2001 | BCR–ABL, PDGFR, SCF, KIT | Tyrosine | Oral | Ph‐positive CML, Ph‐positive ALL, PDGFR rearrangements MDS/MPD, ASM, HES, CEL, DFSP, KIT‐positive GIST |
| ABL | Dasatinib (Sprycel) | Bristol‐Myers Squibb | 2006 | BCR–ABL, SRC family (SRC, LCK, YES, FYN), and KIT, EPHA2, PDGFRβ | Tyrosine | Oral | Ph‐positive CML, Ph‐positive ALL, |
| ABL | Nilotinib (Tasigna) | Novartis | 2007 | BCR–ABL, PDGFRB, KIT | Tyrosine | Oral | Ph‐positive CML |
| ABL | Bosutinib (Bosulif) | Wyeth Inc | 2012 | BCR–ABL, SRC‐family (SRC, LYN, and HCK) | Tyrosine | Oral | Ph‐positive CML |
| ABL | Ponatinib (Iclusig) | Ariad | 2012 | BCR–ABL, BCR–ABL (T315I), VEGFR, PDGFR, FGFR, EPH receptors, SRC families of kinases, KIT, RET, TIE2, FLT3 | Tyrosine | Oral | Ph‐positive CML and Ph‐positive ALL resistant/intolerant to therapy, T315I‐positive CML, T315I‐positive/Ph‐positive ALL |
| ABL | Asciminib (Scemblix) | Novartis | 2021 | BCR–ABL, BCR–ABL (T315I) | Tyrosine | Oral | Ph‐positive CML‐CP resistant to therapy, T315I‐positive CML |
| KIT | Ripretinib (Quinlock) | Deciphera | 2020 | KIT, PDGFRA, PDGFRA mutations, PDGFRB, TIE2, VEGFR2, BRAF | Tyrosine | Oral | GIST |
| KIT | Avapritinib (Ayvakit) | Blueprint Medicines | 2020 | KIT, KIT D816V, KIT exon 11, 11/17, and 17 mutants, PDGFRA and PDGFRA D842 mutants, PDGFRB, and CSFR1 | Tyrosine | Oral | PDGFRA exon 18 mutation (including D842V) positive GIST, advanced systemic mastocytosis |
| HER | Gefitinib (Iressa) | AstraZeneca | 2003 | EGFR and HER family | Tyrosine | Oral | NSCLC |
| HER | Erlotinib (Tarceva) | OSI | 2004 | EGFR and HER family | Tyrosine | Oral | NSCLC with EGFR 19del or L858R, pancreatic cancer |
| HER | Afatinib (Gilotrif) | Boehringer Ingelheim | 2013 | EGFR and HER family | Tyrosine | Oral | NSCLC with nonresistant EGFR mutations, squamous NSCLC |
| HER | Osimertinib (Tagrisso) | AstraZeneca | 2015 | EGFR and HER family | Tyrosine | Oral | NSCLC with EGFR 19del or L858R, NSCLC with T790M positive |
| HER | Dacomitinib (Vizimpro) | Pfizer | 2018 | EGFR and HER family | Tyrosine | Oral | NSCLC with EGFR 19del or L858R |
| HER | Mobocertinib (Exkivity) | Takeda Pharmaceuticals | 2021 | EGFR and HER family | Tyrosine | Oral | NSCLC with EGFR 20 exon insertion |
| HER | Lapatinib (Tykerb) | SmithKline Beecham | 2007 | EGFR and HER family | Tyrosine | Oral | HER2‐positive breast cancer |
| HER | Neratinib (Nerlynx) | Puma Biotechnology | 2017 | EGFR and HER family | Tyrosine | Oral | HER2‐positive breast cancer |
| HER | Tucatinib (Tukysa) | Seattle Genetics | 2020 | EGFR and HER family | Tyrosine | Oral | HER2‐positive breast cancer |
| ALK | Crizotinib (Xalkori) | PF Prism CV | 2011 | ALK, HGFR, c‐Met, ROS1, RON | Tyrosine | Oral | ALK‐ or ROS1‐positive NSCLC, ALK‐positive anaplastic large cell lymphoma |
| ALK | Ceritinib (Zykadia) | Novartis | 2014 | ALK, IGF‐1R, InsR, ROS1 | Tyrosine | Oral | ALK‐positive NSCLC |
| ALK | Alectinib (Alecensa) | Roche | 2015 | ALK, RET | Tyrosine | Oral | ALK‐positive NSCLC |
| ALK | Brigatinib (Alunbrig) | ARIAD | 2017 | ALK, ROS1, IGF‐1R, FLT‐3, EGFR deletion and point mutations | Tyrosine | Oral | ALK‐positive NSCLC |
| ALK | Lorlatinib (Lorviqua) | Pfizer | 2018 | ALK, ROS1, TYK1, FER, FPS, TRKA, TRKB, TRKC, FAK, FAK2, ACK | Tyrosine | Oral | ALK‐positive NSCLC |
| MET | Capmatinib (Tabrecta) | Novartis | 2020 | MET, MET exon 14 skipping | Tyrosine | Oral | NSCLC with MET exon 14 skipping |
| MET | Tepotinib (Tepmetko) | Merck | 2021 | MET, MET exon 14 skipping | Tyrosine | Oral | NSCLC with MET exon 14 skipping |
| RET | Pralsetinib (Gavreto) | Blueprint Medicines | 2020 | wild‐type RET, oncogenic RET fusions (CCDC6‐RET), RET mutations (RET V804L, RET V804M and RET M918T) | Tyrosine | Oral | RET fusion‐positive NSCLC, RET mutant MTC, RET fusion‐positive thyroid cancer |
| RET | Selpercatinib (Retevmo) | Eli Lilly | 2020 | wild‐type RET, multiple mutated RET isoforms | Tyrosine | Oral | RET fusion‐positive NSCLC, RET mutant MTC, RET fusion‐positive thyroid cancer |
| FGFR | Erdafitinib (Balversa) | Janssen | 2019 | FGFR1, FGFR2, FGFR3, FGFR4, RET, CSF1R, PDGFRA, PDGFRB, FLT4, KIT, VEGFR2 | Tyrosine | Oral | Urothelial carcinoma with FGFR3 or FGFR2 genetic alterations |
| FGFR | Pemigatinib (Pemazyre) | Incyte | 2020 | FGFR1, FGFR2, FGFR3 | Tyrosine | Oral | Cholangiocarcinoma with FGFR2 fusion or other rearrangement |
| FGFR | Infigratinib (Truseltiq) | Helsinn Hlthcare | 2021 | FGFR1, FGFR2, FGFR3, FGFR4 | Tyrosine | Oral | Cholangiocarcinoma with FGFR2 fusion or other rearrangement |
| TRK | Larotrectinib (Vitrakvi) | Loxo Oncology | 2018 | NTRK1, NTRK2, NTRK3 | Tyrosine | Oral | NTRK fusion‐positive tumours |
| TRK | Entrectinib (Rozlytrek) | Genentech | 2019 | NTRK1, NTRK2, NTRK3, ROS1, ALK, JAK2, TNK2 | Tyrosine | Oral | NTRK fusion‐positive tumours, ROS1 positive NSCLC |
| FLT3 | Midostaurin (Rydapt) | Novartis | 2017 | FLT3 | Tyrosine | Oral | FLT mutant AML, ASM, SM‐AHN, MCL |
| FLT3 | Gilteritinib (Xospata) | Astellas | 2018 | FLT3 | Tyrosine | Oral | FLT mutant AML |
| CSF1R | Pexidartinib (Turalio) | Daiichi Sankyo | 2019 | CSF1R, KIT, FLT3 with ITD mutation | Tyrosine | Oral | Tenosynovial giant cell tumor |
| RAF | Vemurafenib (Zelboraf) | Hoffmann La Roche | 2011 | mutated forms of BRAF, wild‐type BRAFCRAF, ARAF, SRMS, ACK1, MAP4K5, FGR | Serine‐threonine | Oral | Melanoma with BRAF V600E |
| RAF | Dabrafenib (Tafinlar) | GSK | 2013 | BRAF V600E, BRAF V600K, and BRAF V600D, wild‐type BRAF, CRAF, SIK1, NEK11, LIMK1 | Serine‐threonine | Oral | Melanoma with BRAF V600E, in combination with trametinib: melanoma with BRAF V600E or V600K, NSCLC with BRAF V600E, ATC with BRAF V600E, solid tumors with BRAF V600E |
| RAF | Encorafenib (Braftovi) | Array BioPharma | 2018 | BRAF V600E, wild‐type BRAF, CRAF, JNK1, JNK2, JNK3, LIMK1, LIMK2, MEK4, STK36 | Serine‐threonine | Oral | In combination with binimetinib: melanoma with BRAF V600E or V600K, in combination with cetuximab: CRC with BRAF V600E |
| MEK | Trametinib (Mekinist) | GSK | 2013 | MEK1, MEK2 | Serine‐threonine | Oral | Melanoma with BRAF V600E or V600K, in combination with dabrafenib: melanoma with BRAF V600E or V600K, NSCLC with BRAF V600E, ATC with BRAF V600E, solid tumors with BRAF V600E |
| MEK | Cobimetinib (Cotellic) | Genentech/Exelixis | 2015 | MEK1, MEK2 | Serine‐threonine | Oral | In combination with vemurafenib: melanoma with a BRAF V600E or V600K |
| MEK | Binimetinib (Mektovi) | Array BioPharma | 2018 | MEK1, MEK2 | Serine‐threonine | Oral | In combination with encorafenib: melanoma with a BRAF V600E or V600K |
| MEK | Selumetinib (Koselugo) | Astra Zeneca | 2020 | MEK1, MEK2 | Serine‐threonine | Oral | Neurofibromatosis type 1 |
| PI3K | Idelalisib (Zydelig) | Gilead Sciences | 2014 | PI3Kδ | Phosphatidylinosi‐tol 3‐kinase | Oral | CLL |
| PI3K | Copanlisib (Aliqopa) | Bayer Healthcare | 2017 | PI3Kα, PI3Kδ | Phosphatidylinosi‐tol 3‐kinase | Intravenous | FL |
| PI3K | Duvelisib (Copiktra) | Secura | 2018 | PI3Kδ, PI3Kγ | Phosphatidylinosi‐tol 3‐kinase | Oral | CLL/SLL |
| PI3K | Alpelisib (Piqray) | Novartis | 2019 | PI3Kα | Phosphatidylinosi‐tol 3‐kinase | Oral | HR‐positive, HER2‐negative breast cancer, PIK3CA‐mutated breast cancer (in combination with fulvestrant) |
| PI3K | Umbralisib (Ukoniq) | TG Theraps | 2021 | PI3Kδ, CK1ε | Phosphatidylinosi‐tol 3‐kinase | Oral | MZL, FL |
| JAK | Ruxolitinib (Jakafi) | Incyte | 2011 | JAK1, JAK2 | Tyrosine | Oral | Myeloproliferative neoplasms |
| JAK | Fedratinib (Impact) | Impact | 2019 | JAK2 | Tyrosine | Oral | Myeloproliferative neoplasms |
| CYC | Palbociclib (Ibrance) | Pfizer | 2015 | CDK4, CDK6 | Serine‐threonine | Oral | HR‐positive, HER2‐negative breast cancer |
| CYC | Ribociclib (Kisqali) | Novartis | 2017 | CDK4, CDK6 | Serine‐threonine | Oral | HR‐positive, HER2‐negative breast cancer |
| CYC | Abemaciclib (Verzenio) | Eli Lilly | 2017 | CDK4, CDK6 | Serine‐threonine | Oral | HR‐positive, HER2‐negative breast Cancer |
| CYC | Trilaciclib (Cosela) | G1 Therap | 2021 | CDK4, CDK6 | Serine‐threonine | Intravenous | Prevent chemotherapy‐induced myelosuppression in SCLC |
| BTK | Ibrutinib (Imbruvica) | Sandoz | 2013 | BTK | Tyrosine | Oral | MCL, CLL/SLL, CLL/SLL with 17p deletion, WM, MZL |
| BTK | Acalabrutinib (Calquence) | AstraZeneca | 2017 | BTK | Tyrosine | Oral | MCL, CLL/SLL |
| BTK | Zanubrutinib (Brukinsa) | BeiGene | 2019 | BTK | Tyrosine | Oral | MCL, WM, MZL |
| IDH1 and IDH2 | Enasidenib (Idhifa) | CelGene | 2017 | IDH1 and IDH2 | Tyrosine | Oral | IDH2 mutant AML |
| IDH1 and IDH2 | Ivosidenib (Tibsovo) | Servier | 2018 | IDH1 and IDH2 | Tyrosine | Oral | IDH1 mutant AML, IDH1 mutant cholangiocarcinoma |
| SRC | Tirbanibulin (Klisyri) | Almirall | 2020 | SRC | Tyrosine | Opical | Actinic keratosis |
Abbreviations: Ph‐positive CML, Philadelphia chromosome‐positive chronic myeloid leukemia; Ph‐positive ALL, Philadelphia chromosome positive acute lymphoblastic leukemia; MDS/MPD, myelodysplastic/myeloproliferative diseases; ASM, aggressive systemic mastocytosis; HES, hypereosinophilic syndrome; CEL, chronic eosinophilic leukemia; DFSP, dermatofibrosarcoma protuberans; GIST, gastrointestinal stromal tumors; NSCLC, nonsmall cell lung cancer; MTC, medullary thyroid cancer; AML, acute myeloid leukemia; SM‐AHN, systemic mastocytosis with associated hematological neoplasm; MCL, mantle cell lymphoma; CRC, colorectal cancer; ATC, anaplastic thyroid cancer; CLL/SLL, chronic lymphocytic leukemia/small lymphocytic lymphoma; FL, follicular lymphoma; MZL, marginal zone lymphoma; MCL, mantle cell lymphoma; WM, waldenström's macroglobulinemia. Data sources: https://www.fda.gov/drugs/development‐approval‐process‐drugs/drug‐approvals‐and‐databases.
3.1. ABL1 kinase inhibitors
Philadelphia (Ph) chromosome translocation results in a fusion between the abelson murine leukemia viral oncogene homolog 1 (ABL1) on chromosome 9 and the breakpoint cluster region (BCR) gene on chromosome 22, forming an aberrant BCR–ABL1 fusion gene on chromosome 22, which encodes a 210 kDa tyrosine kinase. This BCR–ABL1 protein further triggers phosphorylation of numerous substrates to activate MAPK, PI3K/AKT/mTOR, and JAK/STAT pathways, thereby driving the uncontrolled proliferation of leukemia cells. Nearly all of patients with chronic myeloid leukemia (CML) and 20–30% of patients with acute lymphoblastic leukemia (ALL) have BCR–ABL1 gene fusions, which are the driving molecular abnormality of these diseases. Correspondingly, BCR–ABL1 fusion tyrosine kinase is a crucial target in treatment of certain leukemias.
Imatinib, initially approved for treating BCR–ABL1‐driven CML by the US FDA in 2001, opens a new era in using protein kinase inhibitors in cancer treatment. 36 Imatinib binds to a catalytically inactive conformation of the BCR–ABL1 kinase domain (type II). In the phase III IRIS study, patients with newly diagnosed CML in chronic phase (CML‐CP) were assigned to receive imatinib or interferon alfa plus low‐dose cytarabine. At 18 months, the rate of major cytogenetic response was 87.1% (95%CI: 84.1, 90.0) in the imatinib arm, as compared with 34.7% (95%CI: 29.3, 40.0) in the group that received interferon alfa plus cytarabine (p < 0.001). The rates of complete cytogenetic response (CCyR) were 76.2% (95%CI: 72.5, 79.9) and 14.5% (95%CI, 10.5, 18.5), respectively (p < 0.001). 37 Long‐term follow‐up of IRIS trial participants revealed a 5‐year survival rate of 89% and a 10‐year survival rate of 83.3% with infrequent serious side effects. The 5‐year and 10‐year CCyR rates in imatinib group were 87 and 82.8%, respectively. 38 , 39 Though imatinib significantly improves outcomes in patients with BCR–ABL1‐driven CML, resistance emerges unavoidably after long‐term treatment, such as M244, G250, Q252, Y253, and E255 located in the P loop, T315 and F317 in the ATP‐binding region, M351 and F359 in SH2 contact and C‐lobe region, and H396 in the activation loop. 40 Second‐ and third‐generation inhibitors dasatinib, nilotinib, bosutinib, and ponatinib can mitigate resistance caused by specific point mutations in BCR–ABL1 kinase domain. 41 , 42 , 43 , 44 Dasatinib and bosutinib bind at the ATP site in an active conformation of the ABL1 kinase domain (type I) and are demonstrated to bind to ABL1 kinase domain much stronger than that of imatinib. The binding mode of nilotinib is similar to imatinib, but with a 50‐fold higher BCR–ABL1 inhibitory activity in vitro than imatinib. Dasatinib, nilotinib, and bosutinib are not only efficacious after imatinib failure but have also been shown to be superior to imatinib when used in the first‐line treatment of CML‐CP, with deeper molecular responses. 45 , 46 , 47 The T315I mutant produces resistance to imatinib, dasatinib, nilotinib, and bosutinib. Ponatinib shows its advantage to overcome resistance from T315I mutation and is approved as a rescue strategy. 48 , 49 , 50 , 51 , 52 The above‐mentioned BCR–ABL1 inhibitors all belong to ATP competitive inhibitors, which are difficult to limit their targets only to BCR–ABL1. 48 , 49 , 50 , 51 , 52 In 2021, the US FDA approved an allosteric inhibitor, asciminib, to treat Ph‐positive CML‐CP with the T315I mutation and for third‐line therapy of Ph‐positive CML‐CP. As it binds to the ABL1 myristoyl pocket, its targets are limited to BCR–ABL1 and mutated BCR–ABL1, including the gatekeeper T315I mutant. The side effects associated with imprecise targets are significantly reduced. 53 , 54 , 55
Three ABL1 kinase inhibitors (imatinib, dasatinib, and ponatinib) are granted US FDA approval for Ph‐positive ALL. Though the efficacy of single‐agent tyrosine kinase inhibitor (TKI) therapy for Ph‐positive ALL is poor, the combination of ABL1 inhibitors with conventional chemotherapy largely improves the response and survival and now is standard of care for Ph‐positive ALL. 56 Adding imatinib to hyper‐CVAD led 93% (42 out of 45) of patients to achieve complete remission (CR) and 48% of patients to live beyond 3 years. 57 The combination of dasatinib with hyper‐CVAD further improved the prognosis of Ph‐positive ALL, with the CR rate and 3‐year survival rate reaching 96 and 60%, respectively. 58 The occurrence of T315I kinase domain mutations in up to 75% of patients and low complete molecular remission (CMR) rates (∼50% with high‐intensity combination) after treatment with first‐ and second‐generation ABL1 inhibitors requires the outcome of third‐generation ABL1 inhibitor ponatinib, which is more potent and might further improve outcomes by inducing higher CMR rates and suppress the emergence of T315I mutations. 52 , 59 The CMR rate was 84%, the CR rate reached 98%, and the 3‐year survival rate was 79% for patients in the ponatinib plus hyper‐CVAD group. 60 However, careful dose adjustment was recommended for ponatinib when combined with hyper‐CVAD in order to avoid serious toxic effects, such as vascular events and pancreatitis. 61 , 62 Furthermore, the high remission rate of ABL1 inhibitor plus chemotherapy decreases the proportion of patients underwent allogeneic hematopoietic stem cell transplantation (allo‐HSCT) and increases the probability of patients to proceed to allo‐HSCT. 63 There are other combination regimes for treating Ph‐positive ALL, including low‐intensity chemotherapy and chemotherapy‐free combination. ABL1 kinase inhibitor is combined with low‐intensity chemotherapy or steroids in patients 60 years and older and those unable to receive intensive chemotherapy. 64 The chemotherapy‐free regimens of ABL1 inhibitor dasatinib or ponatinib and a CD3–CD19 bispecific antibody blinatumomab represent a new trend because of their safety and excellent results in frontline setting. Notably, phase II studies showed that patients received a CMR rate of 86% and a 3‐year survival rate of 94% in the combination of ponatinib plus blinatumomab group. 52
Researches on ABL1 kinase inhibitors are ongoing, mainly focused on three directions. First, new ABL1 kinase inhibitors still arouse great interest. Both flumatinib and radotinib belong to second‐generation ABL1 kinase inhibitors and have been approved for Ph‐positive CML‐CP in China and Korea, respectively. In addition, both of them have been proved to be superior to imatinib in first‐line therapy for newly diagnosed CML‐CP. 65 , 66 Resistance to ponatinb requires the development of new T315I‐targeted inhibitors. The third‐generation ABL1 kinase inhibitor olverembatinib was licensed in China for TKI‐resistant CML‐CP or accelerated‐phase CML harboring the T315I mutation. 67 Vodobatinib is a third‐generation ABL1 kinase inhibitor and is in phase I/II stage for treatment‐refractory/intolerant CML (NCT02629692). Rebastinib is a noncompetitive conformational control inhibitor designed to overcome BCR–ABL1 gatekeeper mutations. However, phase I study (NCT00827138) failed to show sufficient clinical benefit. 68 PF‐114 is a fourth‐generation ABL1 kinase inhibitor. Phase I/II study (NCT02885766) of this agent is recruiting patients with Ph‐positive CML whose disease is resistant to the second‐generation ABL1 inhibitors or has T315I mutation in the BCR–ABL gene. Second, combination with other mechanism drugs to increase efficacy or overcome drug resistance is also popular. A phase III study (NCT04530565) is recruiting patients with Ph‐positive ALL to compare the effect of usual treatment of chemotherapy and steroids and an ABL1 inhibitor to the same treatment plus blinatumomab. Combined with BCL‐2 inhibitor venetoclax is another choice. The efficacy and safety of dasatinib plus venetoclax for Ph‐positive CML is assessed in a phase II study (NCT02689440). A potent combination regime concluding decitabine, venetoclax, and ponatinib for Ph‐positive ALL or Ph‐positive myeloid blast phase or accelerate phase CML is under estimation in a phase II study (NCT04188405). Combination with immune checkpoint inhibitors (ICIs) is also worth expectance. A phase II study (NCT03516279) is assessing pembrolizumab and ABL1 kinase inhibitor (dasatinib, imatinib, or nilotinib) for patients with CML and persistently detectable minimal residual disease. Third, dose reduction or discontinuation has also been studied. The standard dose of dasatinib for CML‐CP in adults is 100 mg once daily, which is associated with myelosuppression and pleural effusions. Dasatinib at a lower dose of 50 mg daily was demonstrated to be active and well tolerated in patients with newly diagnosed CML‐CP in phase II studies. 69 , 70 Cessation of ABL1 kinase inhibitors has been thoroughly and continuously studied. The conclusion of clinical trials supported discontinuation in patients with a confirmed deep molecular response for at least 1 year. 71 , 72
3.2. KIT receptor kinase inhibitors
Stem cell factor, also known as c‐KIT ligand, exerts its biological functions by binding to and activating the receptor tyrosine kinase (RTK) c‐KIT, also known as stem cell growth factor receptor. Dimerization of c‐KIT leads phosphorylation and activation of downstream kinases in PI3K/AKT/mTOR, MAPK, and JAK/STAT pathways and further maintains cell survival, migration, and proliferation. 73 c‐KIT is mainly expressed in stem cells, progenitor cells, and other cells with self‐renewal potency. However, dysregulation of c‐KIT can promote tumor formation and progression. Overexpression or gain of function mutations of c‐KIT has been reported in various cancers, such as gastrointestinal stromal tumors (GISTs), small‐cell lung carcinomas (SCLC), advanced systemic mastocytosis, and acute myeloid leukemia (AML). 74
About 99% of GISTs have an identifiable driver alteration. The KIT (mostly in exon 11, followed by exon 9) and PDGFRA (mainly in exon 18 and less frequently in exon 12 or 14) mutations are the two major molecular subtypes that disrupt autoinhibitory regions of the RTK and thereby result in ligand‐independent activation, occurring in around 70 and 15% of all GISTs, respectively. GISTs without KIT and PDGFRA mutations can be divided into SDH‐deficient and SDH‐competent GISTs. 75 , 76
Before 2000, no effective therapies were available for patients with advanced GISTs, while imatinib revolutionized the treatment of patients with this disease. 77 Imatinib inhibits not only BCR–ABL1 but also the kinases of the KIT and PDGFA receptors. 78 , 79 Mutations in KIT exon 11, 9, and 13 confer sensitivity to imatinib, while most KIT exon 17 mutations are considered to be resistant to imatinib. 76 In advanced GISTs with KIT mutations, the response to imatinib of advanced GISTs with KIT exon 11 mutations is higher than that of GISTs with exon 9 mutations, especially when using standard‐dose imatinib (400 mg total daily dose). Heinrich et al. reported that the partial response (PR) rate in patients with GISTs harboring exon 11 KIT mutations was 83.5%, whereas the percent decreased to 47.8% in patients with tumors containing an exon 9 KIT mutation. With a median follow‐up of 19 months, patients whose tumors contained exon 11 KIT mutations had a longer event‐free survival (EFS) (687 vs. 200 days; p < 0.0001) and overall survival (OS) (p = 0.0034) than those whose tumors expressed exon 9 KIT mutations. 80 The data from the North American phase III SWOG S0033/CALGB 150105 study further confirmed the favorable impact of KIT exon 11 mutations when compared with KIT exon 9 mutations for the objective response rate (ORR) (71.7 vs. 44.4%; p = 0.007), median time to tumor progression (24.7 vs. 16.7 months; p = 0.0013), and median OS (60.0 vs. 38.4 months; p = 0.011). 81 Imatinib is also used in the adjuvant setting for patients with GISTs harboring KIT or PDGFRA mutations that confer imatinib sensitivity. Compared with placebo, 12‐month imatinib prolonged recurrence‐free survival (RFS) compared with placebo (98 vs. 83%; HR, 0.35; 95%CI: 0.22, 0.53; p < 0.0001). 82 Patients with KIT exon 11 deletions assigned to 1 year of adjuvant imatinib had a longer RFS. 83 Three‐year imatinib further prolonged 5‐year RFS (65.6 vs. 47.9%; HR, 0.46; 95%CI: 0.32, 0.65; p < 0.001), significantly prolonged 5‐year OS (92.0 vs. 81.7%; HR, 0.45; 95%CI: 0.22, 0.89; p = 0.02), compared with 1‐year imatinib. 84 In the 3‐year imatinib group, 5‐year OS and 10‐year OS rates were 92.0 and 79.0%, respectively, and in the 1‐year imatinib group 85.5 and 65.3%, respectively. 85 Research also showed that the benefit only existed in patients with KIT exon 11 deletion or insertion‐deletion mutation whose 5‐year RFS increased from 41.3% in the 1‐year group to 71.0% in the 3‐year group (p < 0.001). 86
The main imatinib‐resistance mechanisms are activating other signaling pathways and secondary KIT mutations. Second‐line sunitinib and third‐line regorafenib are both multikinase inhibitors and will be discussed below. 87 The new type II kinase inhibitor, ripretinib, can bind to a novel region of both the KIT and PDGFRA kinases to force the activation loop into an inactive conformation and target a broad spectrum of KIT and PDGFRA mutations. 88 In 2020, ripretinib was approved by the US FDA for fourth‐line therapy for patients with advanced GISTs who have received prior treatment with three or more kinase inhibitors, including imatinib, based on the data that ripretinib resulted in an ORR of 9% versus 0% with placebo (p = 0.05), a median progression‐free survival (PFS) of 6.3 months versus 1.0 months (HR, 0.15; 95%CI: 0.09,0.5; p < 0.0001) and median OS of 15.1 months versus 6.6 months (HR, 0.36; 95%CI: 0.21–0.62) in the phase III INVICTUS trial. 89 , 90 Subsequent analysis of the phase III INVICTUS trial uncovered that ripretinib inhibits a broad range of KIT/PDGFRα mutations based on the data that ripretinib provided PFS benefit regardless of mutation status in patients with advanced GISTs. 91
Currently, there are still many researches on KIT kinase inhibitors. Like imatinib, other ABL1 kinase inhibitors also inhibit c‐KIT, such as dasatinib, nilotinib, and ponatinib. In a single‐arm phase II trial, ponatinib demonstrated activity in advanced GISTs with failure of prior TKI, particularly in subtype with KIT exon 11 mutations. 92 However, nilotinib failed to show its advantages over imatinib in treating first‐line GISTs in a phase III trial. 93 In addition to GISTs, KIT inhibitors have also been tried in other tumor types with KIT alterations, particularly in KIT‐altered melanoma. Imatinib pioneered the clinical trials in melanoma, followed by nilotinib, dasatinib, and regorafenib. Phase II trials showed that imatinib was effective in melanoma with KIT amplification and/or mutations, with ORR fluctuating from 16 to 29%. 94 , 95 , 96 The value of ORR was similar in nilotinib‐treated/KIT‐altered melanoma but was much lower in dasatinib‐treated individuals. 97 , 98 A phase II study is recruiting patients with c‐KIT‐mutated melanoma for second‐line therapy with regorafenib (NCT02501551). The combination of KIT inhibitors with PD‐1 inhibitors is a new trend for melanoma with c‐KIT gene mutations (NCT05274438). In addition, a phase I trial (NCT02571036) is designed to evaluate ripretinib in patients with advanced malignancies. Most importantly, new KIT kinase inhibitors are still being developed. Masitinib is a potent and highly selective TKI with activity against the wild‐type c‐Kit receptor and its juxtamembrane mutation. A phase II study (NCT00998751) evaluated masitinib as the first‐line treatment of advanced GIST, which showed masitinib was comparable with imatinib in terms of safety and response. 99 Masitinib also have been trialed in advanced GISTs after failure of imatinib. The results of a phase III trial (NCT01694277) ensured that the masitinib arm could satisfy a prespecified PFS threshold and received significantly longer OS with lower occurrence of severe adverse events compared with sunitinib. 100 Masitinib has also been explored in many other tumor types, such as colorectal cancer (CRC), systemic mastocytosis, prostate cancer, ovarian cancer, and pancreatic cancer (NCT03556956, NCT04333108, NCT03761225, NCT02490488, and NCT00789633). Amuvatinib is a novel TKI with in vitro pharmacological activity against mutant KIT, PDGFRA, and Rad51. Phase I trial (NCT00894894) proved its safety profile and transient response in refractory GISTs and more trials are needed to prove its efficacy. 101 AZD3229 is a pan‐KIT mutant kinase inhibitor that also targets PDGFRα. It is 15–60 times more potent than imatinib in inhibiting KIT primary mutations and has low nanomolar activity against a broad spectrum of secondary mutations. However, it is still in the preclinical stage. 102 In total, the expansion of the indications of existing drugs and the development of novel KIT inhibitors are the main research directions.
3.3. PDGFR kinase inhibitors
PDGFRs (PDGFRA and PDGFRB), activated in PDGF‐dependent or PDGF‐independent modes, phosphorylate substrates and engage in signaling cascades that drive physiological or pathological functions. 103 In cancers, the PDGF/PDGFR system influences tumor growth, metastasis, and drug response through direct impact on tumor cells or indirect impact on tumor stromal fibroblasts and perivascular cells. Point mutations, rearrangements, and amplification of genetic alterations in tumor cells are known to activate PDGFRs. 104 PDGFR‐signaling in malignant cells with PDGFRA alterations are the main targets, such as PDGFRA‐mutated GISTs. At the same time, the expression of PDGFR in the extracellular matrix is associated with angiogenesis. The activation of PDGF/PDGFR pathway is one potential resistance mechanism to VEGFR2 inhibition. High expression of PDGFR in stromal fibroblasts and perivascular cells can be found in various cancers, such as breast, gastric, colorectal, kidney, ovarian, and pancreatic cancer, and predicts poor prognosis in these tumor types. 105
As mentioned above, 15% of all GISTs occur PDGFRA mutations. Imatinib‐sensitive PDGFRA mutants include mutations in exon 12, 14, or indels in exon 18. GISTs with PDGFRA exon 18 D842V variant leads to primary resistance to imatinib and other type II PDGFRA/KIT TKIs and predicts poor outcomes for patients with GISTs. The D842V mutation, the most common PDGFRA mutation (9–10% of all primary GISTs), became a key target for rational drug design. 76 , 106 In 2020, the US FDA approved the type I PDGFRA/KIT TKI avapritinib for patients with advanced GISTs harboring a PDGFRA exon 18 mutation (including D842V). 107 , 108 In the phase I NAVIGATOR trial, the ORR reached 91 and 61% of patients responded over 6 months. 109 After a median follow‐up of 27.5 months, the ORR, median duration of response (DOR), and median PFS were 91%, 27.6 months, and 34.0 months, respectively. Median OS was not reached with a manageable safety profile. 110 The above data showed avapritinib resulted in an unprecedented and durable clinical benefit in patients with PDGFRA D842V‐mutant GISTs. However, avapritinib failed to prolong PFS in patients with molecularly unselected, late‐line GISTs compared with regorafenib. 111 The effect of avapritinib in treating malignant solid tumors with c‐KIT or PDGFRA mutations is under research (NCT04771520).
A large number of multikinase TKIs with inhibitory activity toward PDGFR are used to target stromal PDGFR expression, including sorafenib for hepatocellular carcinoma (HCC), renal cell carcinoma (RCC), and radioiodine‐refractory DTC and pazopanib for RCC and soft tissue sarcoma (STS). However, tumors often cannot be controlled through single inhibition of PDGFR expression in the stroma, which is proved by two PDGFRA antibody, olaratumab and MEDI‐575. Olaratumab, combined with doxorubicin, is approved for the treatment of adult patients with STS based on a phase II trial, in which the researchers found olaratumab plus doxorubicin improved PFS and OS compared with monotherapy with doxorubicin. 112 But the conclusion was overturned by the phase III ANNOUNCE trial. In this phase III clinical trial, the addition of olaratumab to doxorubicin resulted in no significant difference in OS. 113 Though the safety profile of PDGFRA antibodies is acceptable, the prognosis cannot be improved by olaratumab and MEDI‐575 in various tumor types, including ovarian cancer, glioma, lung cancer, and prostate cancer. 114 , 115 , 116 , 117 On the contrary, positive results can be obtained by using olaratumab to inhibit PDGFR alterations in tumor cells. Olaratumab prolonged disease control in previously treated patients with PDGFRA D842V‐mutant GISTs compared with historical data. 118
Some PDGFR kinase inhibitors are undergoing clinical trials. Crenolanib is a highly specific PDGFR kinase inhibitor and is proved to be safe in several phase I clinical trials. 119 , 120 A phase II study (NCT01243346) evaluated the antitumor efficacy and pharmacokinetics of crenolanib in patients with D842‐mutant GISTs. A phase III trial (NCT02847429) is ongoing to assess crenolanib in D842‐mutant GISTs. Similar to PDGFR antibodies, single inhibition of PDGFR in tumor stroma by crenolanib is not an ideal method to control tumors. Another phase II study (NCT01229644) was designed to evaluate the antitumor efficacy of crenolanib in patients with recurrent high‐grade glioma and in patients with low‐grade glioma, which was terminated ahead because of poor efficacy. The same situation happens to a type III PDGFRβ kinase inhibitor, tandutinib, which can only be used to inhibit the expression of PDGFR in tumor stroma. The results of phase II studies (NCT00379080 and NCT00408902) of tandutinib on glioblastomas or RCC did not meet the primary end points. 121 , 122 In the future, selective PDGFR kinase inhibitors should preferably only target tumor cells with PDGFR alterations.
3.4. HER kinase inhibitors
The EGFR or HER family members comprise four structurally related RTKs that are EGFR/HER1, HER2, HER3, and HER4. 123 PI3K/AKT/mTOR, MAPK, PLCγ, and JAK/STAT are the four major representative downstream signaling pathways activated by EGFR or HER family members, which are related with tumorigenesis, tumor growth, and progression. Mutations in EGFR/HER1 tyrosine kinases play an essential role in tumor growth and progression, especially for nonsmall cell lung cancer (NSCLC). 124 , 125 HER2 overexpression or amplification has been observed in approximately 15–30% of breast cancer and the HER2‐positive subtype predicts a worse prognosis than the HER2‐negative breast cancer. 126 , 127
From 2003, the treatment for NSCLC progressed significantly. First‐generation EGFR TKIs, gefitinib and erlotinib, were originally designed for patients with overexpression of wild‐type EGFR. 128 In the following clinical trials, researchers found that these inhibitors are more sensitive to tumors with EGFR exon 19 deletion or exon 21 (L858R) mutations. 129 , 130 , 131 The US FDA has approved both gefitinib and erlotinib as first‐line treatment for NSCLC with EGFR exon 19 deletion or exon 21 (L858R) mutations, based on the evidence that they can significantly improve median PFS and ORR, compared with chemotherapy. First‐line gefitinib prolonged median PFS from 5.4 months in the paclitaxel plus carboplatin group to 10.8 months (HR, 0.30; 95%CI: 0.22, 0.41; p < 0.001) and ORR from 30.7% to 73.7% (p < 0.001). 132 In Chinese patients with advanced EGFR mutation‐positive NSCLC, median PFS was 13.1 months with erlotinib and 4.6 months with chemotherapy (HR, 0.16; 95%CI: 0.10, 0.26; p < 0.0001), which was 9.7 months and 5.2 months (HR, 0.37; 95%CI: 0.25, 0.54; p < 0.0001), respectively, in European patients. 133 , 134 As pancreatic cancer overexpresses EGFR, erlotinib combined with gemcitabine was also approved for first‐line treatment for pancreatic cancer because OS was significantly longer with erlotinib plus gemcitabine than with gemcitabine (median OS: 6.24 vs. 5.91 months; HR, 0.82; 95%CI: 0.69, 0.99; p = 0.038). 135
Second‐generation EGFR TKIs, afatinib and dacomitinib, bind irreversibly to the ATP pocket of EGFR TK, developed to circumvent drug resistance or increase efficacy. 136 , 137 In 2018, the US FDA approved afatinib to treat NSCLC with nonresistant EGFR mutations. The LUX‐Lung 3 trial showed the median PFS increased from 6.9 months in the pemetrexed plus cisplatin arm to 11.1 months in the afatinib arm (HR, 0.58; 95%CI: 0.43, 0.78; p = 0.001), ORR from 19.1 to 50.4%. 138 The LUX‐Lung 6 trial further consolidated afatinib in first‐line treatment of NSCLC harboring EGFR mutations, which showed first‐line afatinib significantly improved PFS compared with gemcitabine plus cisplatin (median PFS: 11.0 vs. 5.6 months; HR, 0.28; 95%CI: 0.2, 0.39; p < 0.0001). 139 Post‐hoc analysis of LUX‐Lung 2, LUX‐Lung 3, and LUX‐Lung 6 indicated that afatinib was active in NSCLC harboring certain types of uncommon EGFR mutations, such as Gly719Xaa, Leu861Gln, and Ser768Ile, but was inactive in NSCLC with T790M and exon 20 insertion mutations. 140 The LUX‐Lung 7 trial demonstrated that afatinib slightly prolonged PFS, time‐to‐treatment failure, and ORR and exhibited a manageable tolerability profile when compared with gefitinib for first‐line treatment of EGFR mutation‐positive NSCLC. 141 What is more, afatinib has also been approved to treat patients with squamous NSCLC who progressed on platinum‐based doublet chemotherapy in 2016, based on the LUX‐Lung 8 trial, in which median OS (8.4 vs. 6.6 months; HR, 0.81; 95%CI: 0.62, 1.05; p = 0.12) and median PFS (3.5 vs. 2.5 months; HR, 0.69; 95%CI: 0.51, 0.92; p = 0.01) of afatinib were slightly better than those of erlotinib. 142 While another second‐generation EGFR TKI dacomitinib, a pan‐HER inhibitor, demonstrated its advantages over gefitinib in median PFS (14.7 vs. 9.2 months; HR, 0.59; 95%CI: 0.47, 0.74; p < 0.0001) and median OS (34.1 vs. 26.8 months; HR, 0.76; 95%CI: 0.58, 0.99; p = 0.04), was also approved for first‐line treatment for NSCLC with EGFR exon 19 deletion or exon 21 (L858R) mutations. 143 , 144
Osimertinib, third‐generation EGFR‐TKI, binds irreversibly to the activating and T790M mutation at approximately ninefold lower concentrations than wild‐type EGFR. Surprisingly, osimertinib can distribute to the brain at plasma AUC ratios of approximately 2 following oral dosing. 145 The phase III FLAURA trial demonstrated that osimeritinib significantly improved median PFS from 10.2 months in erlotinib or gefitinib to 18.9 months in osimeritinib (HR, 0.46; 95%CI: 0.37, 0.57; p < 0.0001) in first‐line therapy. In FLAURA, osimeritinib also showed its advantage in controlling central nervous system (CNS) lesions over erlotinib or gefitinib. 146 After a long‐term follow‐up for OS, the duration of median OS in the osimeritinib group was 38.6 months and 31.8 months in the comparator group (erlotinib or gefitinib) (HR, 0.80; 95.05%CI: 0.64 to 1.00; p = 0.046). 147 When used for previously treated EGFR T790M mutation‐positive NSCLC, osimeritinib increased median PFS from 4.4 months in chemotherapy to 10.1 months in osimeritinib (HR, 0.30; 95%CI: 0.23, 0.41; p < 0.001), ORR from 29 to 65%. Because of its merits, it has been approved for first‐line treatment for NSCLC with EGFR exon 19 deletion or exon 21 (L858R) mutations and EGFR T790M mutation‐positive NSCLC in 2018 and 2015, respectively. Osimertinib has also made great strides in adjuvant therapy and has already been allowed for adjuvant therapy for NSCLC with EGFR exon 19 deletion or exon 21 (L858R) mutations in 2020. The phase III ADAURA trial showed that median disease‐free survival (DFS) was not reached and 19.6 months in placebo (HR, 0.17; 95%CI: 0.12, 0.23; p < 0.0001). However, the OS data were immature. 146 , 147 , 148
EGFR exon 20 insertion is another observable mutation type and associated with a poor prognosis, accounting for about 1–12% of EGFR mutations. 149 The above EGFR TKIs cannot be applied for NSCLC with EGFR exon 20 insertion. A new EGFR TKI, mobocertinib, changed this situation in 2021. The ORR was 28% and the investigator‐assessed confirmed response rate was 43% with median DOR of 14 months in patients with EGFR exon 20 insertion mutations whose disease has progressed on or after platinum‐based chemotherapy. 150 However, more clinical trials are needed to verify its efficacy.
HER2‐targeted therapy is important for patients with HER2‐positive advanced breast cancer. First‐line treatment with the combination of trastuzumab and pertuzumab plus taxane and second‐line treatment with trastuzumab deruxtecan significantly improve the outcome of patients with advanced or metastatic HER2‐positive breast cancer, leading to great enthusiasm to develop novel anti‐HER2 agents. HER kinase inhibitors for breast cancer are primarily approved in patients whose disease refractory to traditional HER2‐directed therapies. The addition of lapatinib to capecitabine after trastuzumab plus chemotherapy in patients with HER2‐positive breast cancer has advantages in the time to progression (TTP), from 4.4 months in the placebo plus capecitabine group to 8.4 months in the lapatinib plus capecitabine group (HR,0.49; 95%CI: 0.34, 0.71; p < 0.001). 151 Furthermore, the phase III NALA trial demonstrated that neratinib combined with capecitabine improved PFS (HR, 0.76; 95%CI: 0.63, 0.93; p = 0.0059) and time to intervention for CNS disease compared with lapatinib plus capecitabine. 152 In the phase II HER2CLIMB trial, another HER kinase inhibitor, tucatinib, was added to trastuzumab plus capecitabine after one or more prior anti‐HER2‐based regimens not only extended PFS (HR, 0.54; 95%CI: 0.42, 0.71; p < 0.001) and OS (HR, 0.66; 95%CI: 0.50, 0.88; p = 0.005) in patients with HER2‐positive metastatic breast cancer but also resulted in better PFS (median PFS: 7.6 vs. 5.4 months; HR, 0.48; 95%CI: 0.34, 0.69; p < 0.001) in those with brain metastases. 153 Further analyses in patients with brain metastases showed that the addition of tucatinib to trastuzumab and capecitabine reduced the risk of intracranial progression or death by 68%, doubled duration of median CNC‐PFS (intracranial progression or death), and prolonged OS. 154 Based on the above clinical trials, they are allowed to use in patients with advanced or metastatic HER2‐positive breast cancer who have received prior anti‐HER2‐based regimens. In addition, lapatinib combined with letrozole is granted approval by the US FDA for first‐line therapy of HR‐positive and HER2‐positive metastatic breast cancer. 155 The US FDA also approved neratinib in using extended adjuvant treatment of HER2‐positive breast cancer following adjuvant trastuzumab‐based therapy, based on its ability to prolong invasive DFS. 156
Research on already approved HER kinase inhibitors and novel HER kinase inhibitors will give us more choices. Already approved HER kinase inhibitors are mainly for EGFR‐mutant NSCLC and HER2‐positive breast cancer. To treat NSCLC, monotherapy with gefitinib, erlotinib, afatinib, or dacomitinib, is successfully used in advanced NSCLC with EGFR exon 19 deletion or exon 21 (L858R) mutations. The third‐generation EGFR TKI osimertinib is demonstrated to be superior to the first‐ and second‐generation EGFR TKIs and used in EGFR T790M mutation‐positive NSCLC and first‐line treatment and adjuvant therapy for NSCLC with EGFR exon 19 deletion or exon 21 (L858R) mutations. 124 Combination therapy is widely studied for EGFR TKIs. First, the results of EGFR TKI combined with chemotherapy are encouraging. Compared with gefitinib alone, gefitinib combined with carboplatin plus pemetrexed improved PFS in patients with untreated advanced NSCLC with EGFR mutations. 157 However, the continuation of gefitinib after progression on first‐line gefitinib did not prolong PFS in patients who received platinum‐based doublet chemotherapy as second‐line treatment. 158 Dual inhibition of EGFR and VEGF pathways presents another trend. Bevacizumab plus erlotinib significantly improved PFS in patients with untreated metastatic EGFR‐mutated NSCLC but not OS. 159 On the contrary, adding bevacizumab to osimertinib failed to show prolongation of PFS in patients with advanced lung adenocarcinoma with EGFR T790M mutation. 160 Previous studies showed that ICIs was not active in patients with EGFR‐mutant or ALK‐rearrangement NSCLC. 161 Several combinations of ICIs and EGFR‐TKIs are being evaluated in TKI‐failed EGFR mutant patients. Preclinical research suggested that EGFR inhibitors could increase the efficacy of immunotherapy in lung adenocarcinomas. 162 However, the phase Ib TATTON study indicated that osimertinib plus durvalumab was not feasible due to increased reporting of interstitial lung disease. 163 More trials are needed to show whether they can be combined. Studies offer us several single agents or combination regimes to treat previously untreated patients with advanced EGFR‐mutated NSCLC. However, which one is the most suitable? Zhao included 18 trials involved 4628 patients and 12 treatments and used network meta‐analysis to compare the efficacy and safety of first‐line treatments for these individuals. Twelve treatments were EGFR TKIs (osimertinib, dacomitinib, afatinib, erlotinib, gefitinib, and icotinib), pemetrexed‐based chemotherapy, pemetrexed free chemotherapy, and combination treatments (afatinib plus cetuximab, erlotinib plus bevacizumab, gefitinib plus pemetrexed‐based chemotherapy, and gefitinib plus pemetrexed). The authors found that osimertinib resulted in the best PFS for patients with EGFR exon 19 deletion and gefitinib plus pemetrexed‐based chemotherapy gave superiority for patients with EGFR exon 21 (L858R) mutations. 164 In addition, many new third‐generation EGFR TKIs for EGFR T790M‐mutated NSCLC are emerging. Icotinib, furmonertinib, and almonertinib have been approved in China and olmutinib got approval in Korea. 165 , 166 Abivertinib also belongs to third‐generation EGFR TKI and is in the phase III stage for NSCLC (NCT03856697). To date, mobocertinib is the only approved EGFR TKI for second‐line therapy of NSCLC with EGFR exon 20 insertions. 150 A phase III study (NCT04129502) is underway to evaluate mobocertinib as a first‐line treatment versus platinum‐based chemotherapy for NSCLC with EGFR exon 20 insertions. A new EGFR‐TKI poziotinib is an irreversible pan‐HER TKI that targets EGFR, HER2, and HER4 and showed an ORR of 27.8% in second‐line therapy for NSCLC with EGFR exon 20 insertions in a phase II study (NCT03318939). 167 A phase III study (NCT05378763) is ongoing to further verify the efficacy of poziotinib for NSCLC with EGFR exon 20 insertions. As a pan‐HER TKI, poziotinib has also been tested in other tumor types and yielded promising antitumor efficacy with manageable toxicity in HER2‐positive tumors, such as gastric cancer, breast cancer, and head and neck squamous cell carcinoma. 168 , 169 HER kinase inhibitors are permitted for post‐line treatment and subsequent adjuvant therapy of HER2‐positive breast cancer and first‐line therapy of HER2‐positive, HR‐positive breast cancer. Particularly, the neoadjuvant phase III ALTERNATIVE trial showed the addition of lapatinib to trastuzumab plus an aromatase inhibitor in postmenopausal women with HER2‐positive, HR‐positive metastatic breast cancer doubled the duration of PFS without adding adverse events. 170 Other HER kinase inhibitors are also explored. Pyrotinib is an emerging irreversible EGFR/HER2 dual TKI and has been approved in HER2‐positive breast cancer in China. 171
3.5. ALK and ROS1 kinase inhibitors
ALK is a RTK within the insulin receptor family, comprised of an extracellular region, a single transmembrane domain, and an intracellular kinase region. ALK fusion proteins interact with a complex network of proteins and thereby drive aberrant proliferation and survival through PI3K/AKT/mTOR, MAPK, and JAK/STAT pathways. ROS proto‐oncogene 1 (ROS1) is a RTK with an unknown physiological role in humans, while ROS1 fusion proteins drive occurrence of various tumors, including glioblastomas, NSCLC, and IMTs. Though ALK and ROS1 fusion proteins existed in multiple tumor types, ALK and ROS1 kinases inhibitors are only approved for ALK or ROS1‐positive NSCLC before July 14, 2022. ALK and ROS1 rearrangements occur in 3–8% and 0.9‐2.6% of the overall NSCLC population, mostly on adenocarcinomas, with higher prevalence in nonsmokers, younger age and early‐stage brain metastasis. 124 , 172
The first ALK/ROS1 inhibitor, crizotinib, was initially approved in 2011. The PROFILE 1014 and the PROFILE 1007 trials confirmed a survival superiority of crizotinib over platinum‐based chemotherapy in first‐line and second‐line therapy (after 1 platinum‐based chemotherapy regimen) for ALK‐positive NSCLC, respectively. 173 , 174 Compared with chemotherapy, crizotinib extended the median PFS from 7.0 months to 10.9 months (HR, 0.45; 95%CI: 0.35, 0.60; p < 0.001) and upgraded ORR from 45% to 74% in first‐line treatment, median PFS from 3.0 months to 7.7 months (HR, 0.49; 95%CI: 0.37, 0.64; p < 0.001) and ORR from 20 to 65% in second‐line treatment. 173 , 174 Final OS analysis of the phase III PROFILE 1014 trial showed that there was an improvement in OS that favored crizotinib after crossover adjustment. 175 While crizotinib induced resistance in approximately a third of ALK‐rearranged NSCLC owing to on‐target mutation or progression in the CNS. 176 Unlike T790M gatekeeper mutation predominating in EGFR‐mutant NSCLC, a much broader spectrum of on‐target mutations has been detected in ALK TKI‐resistant NSCLC. The subsequent development of more selective and potent second‐generation ALK inhibitors, such as ceritinib, alectinib, and brigatinib, is efficacious after crizotinib‐driven resistance, including Leu1196Met and Gly1269Ala mutations in ALK. 177 , 178 In second‐line therapy, nearly half of the patients resistant to crizotinib can receive a complete or PR when using second‐generation ALK inhibitors. However, they did not target all the ALK mutants resistant to crizotinib. 179 , 180 , 181 , 182 The third‐generation ALK inhibitor, lorlatinib, is effective against resistance mutations (including p.G1202r) generated by first‐ and second‐generation ALK inhibitors. 183 When lorlatinib was used in patients with ALK‐positive NSCLC previously treated with one or more ALK kinase inhibitors, the ORR can still reach 48%. 184
Second‐ and third‐generation ALK inhibitors are not just for post‐line therapy. Continuous researches give more choice for patients with ALK‐positive NSCLC in first‐line treatment. The first second‐generation ALK inhibitor ceritinib showed a statistically significant and clinically meaningful improvement in PFS (median PFS: 16.6 vs. 8.1 months; HR, 0.55; 95%CI: 0.42, 0.73; p < 0.00001) versus chemotherapy. 185 Then, alectinib, brigatinib, and lorlatinib were demonstrated to be superior to crizotinib on 12‐month EFS rate, ORR, and intracranial response in untreated ALK‐positive NSCLC. 186 , 187 , 188 For example, second interim analysis of the phase III ALTA‐1L trial showed that median PFS was 24 months with brigatinib and 11 months with crizotinib (HR, 0.49; 95%CI: 0.35, 0.68; p < 0.0001). 189 In addition, alectinib, brigatinib, and lorlatinib also have potent CNS penetration to control intracranial lesions even after resistance to crizotinib. 190 Together with ceritinib, they have been approved to treat ALK‐positive NSCLC in first‐line treatment.
As the kinase domains of ALK is closely related to that of ROS1, ALK inhibitors regularly also bind to ROS1. 191 The US FDA has approved two ROS1 kinase TKIs, crizotinib and entrectinib, for ROS1‐positive NSCLC. As mentioned above, crizotinib also inhibits ROS1 activity. 191 Given a dose of 250 mg twice daily, the ORR was 72%, median PFS was 19.3 months, with median OS at 51.4 months. Progression in CNS because of its poor brain penetration makes the need for new ROS1‐targeting inhibitors with better cerebral penetration urgently. 192 Entrectinib shows similar activity against TRK, ROS1, and ALK in vitro. Entrectinib inhibits NTRK‐fusion tumors will be discussed below. In vitro, its anti‐ROS1 activity is 40 times more potent than that of crizotinib. The phase II basket STARTRK‐2 trial demonstrated the efficacy of entrectinib in ROS1‐arranged NSCLC with no prior therapy with a ROS1 inhibitor. The ORR was 67.1% and median PFS was 15.7 months in the entire population. For the 24 patients with measurable brain metastases at diagnosis, the ORR was 79.2% and median PFS was 12 months. 193 , 194
Many efforts have been made to broaden the indications and enhance the antitumor activity of already approved ALK/ROS1 inhibitors. Inflammatory myofibroblastic tumors (IMTs) are a kind of rare mesenchymal tumor consisting of a variable mixture of myofibroblasts and inflammatory infiltrates that can occur throughout the body, mainly in the mesentery, retroperitoneum, and pelvis. ALK rearrangements occur in ≥50% of IMTs. On July 14, 2022, the US FDA approved crizotinib for patients with unresectable, recurrent, or refractory ALK‐positive IMTs based on a phase Ib trial (NCT01121588). The ORR was 67% for ALK‐positive IMTs with a consistent safety profile, the efficacy of which was confirmed by a phase II study (NCT01524926). 195 , 196 In addition, a phase I–II study (NCT01970865) showed clinical activity of lorlatinib in advanced ROS1‐positive NSCLC, including those with CNS metastases and those previously treated with crizotinib. 197 Like EGFR TKIs plus ICIs for NSCLC, it is uncertain whether ALK/ROS1 TKIs could combine with ICIs. The outcome of group E in CheckMate 370 showed that 38% (five out of 13) of patients treated with nivolumab plus crizotinib developed severe hepatic toxicities, including two deaths. 198 Another phase Ib trial evaluated the possibility of combination of nivolumab and ceritinib and subsequently showed a potent efficacy (ORR reaching up to 83%) with increasing toxicity in the treatment of naïve ALK‐rearranged NSCLC. 199 Further studies are needed to determine the possibility and the pattern of combination regimes.
Researches are ongoing for developing new ALK/ROS1 inhibitors. Ensartinib has been approved in China for ALK‐positive NSLCL. A phase III trial (NCT02767804) showed that first‐line treatment with ensartinib for ALK‐positive NSCLC had superior efficacy to crizotinib in both systemic and intracranial disease. 200 A phase II study (NCT03215693) demonstrated the efficacy and safety of ensartinib in crizotinib‐resistant, ALK‐positive NSCLC. 201 Repotrectinib belongs to fourth‐generation ALK inhibitor that targets ALK, ROS1, and TRK with similar IC50 values for ALK, ALK (G1202R), and ALK (L1196M). It is in phase II clinical trials for solid tumors with ALK, ROS1, or NTRK1‐3 rearrangements (NCT03093116). A new ALK/ROS1 inhibitor XZP‐3621 is underestimation in patients with ALK or ROS1 rearrangement NSCLC (NCT05204628). Taletrectinib is a ROS1/TRK inhibitor with potent activity against ROS1 G2032R mutation. A phase I study of this agent showed preliminary efficacy in patients with crizotinib‐refractory ROS1‐positive NSCLC with manageable toxicities. 202 Those novel ALK/ROS1/TRK, ALK/ROS1, or ROS1/TRK will provide more chance for tumors with ALK or ROS1 alterations.
3.6. MET receptor kinase inhibitors
Mesenchymal–epithelial transition factor (MET), also known as the tyrosine receptor of HGF, is a single‐pass transmembrane receptor. MET homodimerization activates the MAPK and PI3K/AKT/mTOR pathways, which promote cell migration, proliferation, and survival. 203 MET dysregulation through gene amplification and/or overexpression, mutation, and rearrangement can promote cancer initiation, progression, and malignancy in epithelial cancers. 204 Of these, MET exon 14 skipping mutations promote oncogenic activity by suppressing MET receptor degradation and occur in 3–4% of patients with NSCLC. 203
Two small molecule kinase inhibitors targeting MET, capmatinib and tepotinib, got accelerated approval for NSCLC with MET exon 14 skipping in 2020 and 2021, respectively. For capmatinib, the phase II GEOMETRY mono‐1 trial demonstrated that the ORR in the treatment‐naïve population was 68% and DOR was 12.6 months, with 41% and 9.7 months, respectively, in the previously treated group. At the same time, low‐grade peripheral edema and nausea were the main toxic effects. 205 To expand the scope of capmatinib in NSCLC with MET exon 14 skipping, a phase II trial (NCT04926831) is recruiting patients to evaluate the possibility of using capmatinib in neoadjuvant and adjuvant treatment. When NSCLC with MET exon 14 skipping treated by tepotinib, the ORR was 56% and was similar between the treatment‐naïve arm and the previously treated arm. 206
Researches on MET inhibitors are focused in three directions. First, selective MET inhibitors are explored in other types of MET‐dysregulated NSCLC and different kinds of tumors. MET amplification or overexpression attributes to one of the predominant EGFR‐TKI resistance mechanisms in patients with EGFR‐mutated NSCLC. 124 Phase Ib/II studies evaluated the efficacy and safety of gefitinib plus capmatinib or tepotinib after the failure of EGFR‐TKI therapy in patients with EGFR‐mutated, MET‐amplified/overexpressing NSCLC, the results of which showed these combination regimes were a promising treatment, especially for patients with a MET gene copy number ≥6 or high (IHC3+) MET overexpression. 207 , 208 Adding MET inhibitors to the continuous medication of previous EGFR TKIs may be another method to overcome resistance to EGFR TKIs induced by MET overexpression, and many researches are ongoing (NCT04816214 and NCT03940703). In addition, MET alterations also occur in other types of tumors. A phase Ib/II trial (NCT01988493) showed that tepotinib improved TTP versus sorafenib in treatment‐naïve HCC with MET overexpression. 209 Another phase Ib/II (NCT02115373) trial indicated that tepotinib was efficacious in sorafenib pretreated HCC with MET overexpression. 210 Then, combination strategies are investigated to enhance efficacy. Combination of MET inhibitors with ICIs, other targeted therapies (including VEGFR inhibitors, MEK inhibitors, and HER inhibitors), and chemotherapy are ongoing. Advanced NSCLC patients with MET exon 14 skipping mutations are prepared to receive capmatinib plus spartalizumab (a new PD‐1 inhibitor) (NCT04139317). A phase I trial (NCT05435846) is ready to recruit NSCLC patients with MET exon 14 skipping mutations to receive capmatinib plus trametinib. A phase I/II trial (NCT05439993) of paclitaxel and tepotinib is recruiting patients with advanced gastric and gastroesophageal junction carcinomas with MET amplification or MET exon 14 alterations. There are still many new MET inhibitors under development. Savolitinib is a selective MET TKI and yielded promising activity in pulmonary sarcomatoid carcinomas and NSCLC with MET exon 14 skipping with ORR reaching 42.9% and an acceptable safety profile (NCT02897479), which has already been approved in China for this kind of NSCLC. 211 Phase III trials (NCT04923945 and NCT05261399) of savolitinib for NSCLC patients with MET exon 14 mutations or NSCLC patients whose disease progressed on osimertinib are recruiting. It is also tested for RCC, gastric cancer, and esophagogastric junction adenocarcinoma (NCT05043090 and NCT04923932). Tivantinib is the first non‐ATP competitive and selective oral MET kinase inhibitor. It has been explored in various types of tumors, such as NSLCL and HCC (NCT01395758 and NCT01755767). However, phase III study of tivantinib plus erlotinib versus placebo plus erlotinib for nonsquamous NSCLC was terminated (NCT01244191). Glumetinib is also a potent and highly selective Met inhibitor. Bozitinib is a highly selective ATP‐competitive Met inhibitor with blood‐brain barrier permeability. Both of them are on their phase II stage for NSCLC with MET exon 14 mutations (NCT04270591 and NCT04258033).
3.7. RET receptor kinase inhibitors
Rearranged during transfection (RET) is a transmembrane RTK whose homodimerization activates several downstream pathways, including MAPK, PI3K/AKT/mTOR, JAK/STAT, PKA, and PKC pathways, essential for the normal development and maturation of diverse tissues. 203 Aberrant RET signaling in cancers, due to RET mutations, gene fusions, and overexpression, results in the activation of downstream pathways promoting proliferation, differentiation, and survival. RET fusions occur in 1–2% of NSCLC and up to 20% of papillary thyroid cancer. RET mutations may cause multiple endocrine neoplasia 2, characterized by a high risk of developing medullary thyroid cancer (MTC). 212 , 213
Multikinase inhibitors with anti‐RET activity, such as cabozantinib and vandetanib, were initially tested in patients with advanced RET‐rearranged NSCLC. 214 , 215 In 2020, RET‐selective inhibitors pralsetinib and selpercatinib received clinical approval for RET fusion‐positive NSCLC, RET‐mutant MTC, and RET fusion‐positive thyroid cancer based on the results of the phase I/II ARROW study and the phase I/II LIBRETTO‐001 trial, respectively. 213 In RET fusion‐positive NSCLC, the ORR of pralsetinib was 53% in patients with previous platinum‐based chemotherapy and 70% in patients who were treatment naïve with no treatment‐related deaths. Correspondingly, the ORR of selpercatinib increased to 64 and 85%, respectively. The ORRs of pralsetinib were 71 and 60%, respectively, in patients with treatment‐naive RET‐mutant MTC and in patients who had previously received cabozantinib or vandetanib. The LIBRETTO‐001 trial showed that previous use of cabozantinib or vandetanib for RET‐mutant MTC has little impact on selpercatinib‐related ORR (69 vs. 73%). In patients with previously treated RET fusion‐positive thyroid cancer, the percentages who had a response were 89% for pralsetinib and 79% for selpercatinib, respectively. 213 , 216 , 217 , 218 In addition, selpercatinib is tested in advanced solid tumors, lymphomas, and histiocytic disorders with activating RET gene alterations (NCT04320888). Both pralsetinib and selpercatinib are under verification in the approved indications (NCT05170204, NCT04194944, and NCT04211337).
Many MET inhibitors are undergoing clinical trials. RXDX‐105 is a multikinase RET inhibitor. In phase I/Ib trial for RET inhibitor‐naïve patients with RET fusion‐positive NSCLCs, RXDX‐105 showed different ORRs between KIF5B‐RET‐containing and non‐KIF5B‐RET‐containing tumors. The ORRs were 0 and 67% for these two gene alterations, respectively (NCT01877811). The reason contributing to the differential responses is not clear. 219 TPX‐0046 is a RET/SRC inhibitor. This agent is assessed in phase I/II clinical trial (NCT04161391) for advanced solid tumors harboring RET fusions or mutations. A RET inhibitor TAS0953/HM06 is evaluated clinically to treat advanced solid tumors with RET gene abnormalities (NCT04683250). Through the above efforts, it is expected that the prognosis of RET‐altered tumors can be improved.
3.8. FGFR kinase inhibitors
Fibroblast growth factor (FGF) receptors (FGFR1, FGFR2, FGFR3, and FGFR4) are RTKs consisting of three extracellular immunoglobulin (Ig)‐like domains (I, II, III), a transmembrane domain and two intracellular tyrosine kinase domains (TK1 and TK2). 220 In the presence of FGF or other ligands, FGFRs dimerization further induces the activation of downstream signaling cascades, including PI3K/AKT/mTOR, MAPK, and JAK/STAT pathways, with a role in a variety of physiological processes, such as embryonic development, metabolic homeostasis, tissue repair, and regeneration. 19 The aberrantly activated FGF/FGFR signaling plays a pivotal role in the oncogenic process, including proliferation, survival, migration, and invasion of cancer cells, angiogenesis and immune evasion in the tumor microenvironment (TME). 220 Multikinase inhibitors with anti‐FGFR activity often simultaneously target VEGF/VEGFR signaling, thereby collaboratively interfering with tumor angiogenesis and regulating the immune microenvironment. 19 The details of multikinase inhibitors will be discussed below, and we focus on selective FGFR inhibitors in this part. An analysis of 4853 solid tumors by the next‐generation sequencing (NGS) technique demonstrated that FGFR aberrations occurred in 7.1% of cancers. Among them, gene amplification, gene mutations, and gene rearrangement accounted for 66, 26, and 8%, respectively. Selective FGFR inhibitors are a promising method to treat FGFR‐ altered tumors. 19 , 221
To date, three selective FGFR kinase inhibitors, erdafitinib, pemigatinib, and infigratinib, are approved for treating urothelial carcinoma or cholangiocarcinoma. 222 , 223 , 224 Erdafitinib is a pan‐FGFR inhibitor. A phase I study (NCT01703481) showed that erdafitinib‐related response rates in urothelial carcinoma and cholangiocarcinoma were highest among advanced solid tumors with genomic changes in the FGFR pathway. 225 In the phase II BLC2001 trial, erdafitinib brought an ORR of 40% to patients with urothelial carcinoma susceptible to FGFR3 or FGFR2 genetic alterations whose diseases progressed during or following at least one line of prior platinum‐containing chemotherapy. While treatment‐related grade 3 or higher adverse events were reported in nearly half the patients. 222 Analysis of long‐term efficacy and safety of erdafitinib showed consistent activity and a manageable safety profile. 226 Pemigatinib and infigratinib are FGFR1‐3 inhibitors approved for previously treated cholangiocarcinoma with an FGFR2 fusion or other rearrangements. The ORR was 35.5% in pemigatinib and 23% in infigratinib. Hyperphosphataemia was the most common all‐grade adverse event irrespective of the cause in both pemigatinib and infigratinib and more than half of patients had a grade 3 or worse adverse event, including hyperphosphatemia, stomatitis, and fatigue. 223 , 224 In other words, FGFR inhibitors are mildly effective but have comparatively serious side effects.
These selective FGFR inhibitors have been explored in other tumors. Preclinical trials showed that erdafitinib inhibited the growth of glioma cells with FGFR3‐TACC3 fusions in vitro and in vivo. Erdafitinib manifested clinical improvement with stable disease and minor response for glioblastoma patients with FGFR3‐TACC3 rearrangements. 227 However, infigratinib had limited efficacy in patients with recurrent gliomas and different FGFR genetic alterations. 228 A phase I trial (NCT01004224) demonstrated antitumor activity of infigratinib for FGFR1‐amplified squamous cell NSCLC and FGFR3‐mutant bladder/urothelial cancers. 229 The three approved FGFR inhibitors are also under evaluation in breast cancer, bladder cancer, prostate cancer, myeloid/lymphoid neoplasms, and NSCLC and in combination with PD‐1 monoclonal antibody for advanced solid tumors (NCT03238196, NCT04917809, NCT04754425, NCT03011372, NCT05210946, and NCT03547037).
There are many selective FGFR inhibitors at different stages of clinical trials. FGFR inhibitors with clinical activity are classified into FGFR1‐4, FGFR1‐3, and FGFR4 inhibitors. Futibatinib, rogaratinib, LY2874455, and ASP5878 belong to FGFR1‐4 inhibitors. Futibatinib is an irreversible FGFR1‐4 inhibitor. Phase I trial (NCT02052778) of futibatinib recruited patients with advanced solid tumors harboring FGF/FGFR aberrations. This trial highlighted that futibatinib was also effective in patients whose disease was already resistant to prior FGFR inhibitors. 230 Phase II trials of futibatinib for urothelial carcinoma and breast cancer are ongoing (NCT04601857 and NCT04024436). In addition, it is in phase III trial (NCT04093362) for advanced cholangiocarcinoma harboring FGFR2 gene rearrangements. Rogaratinib is a pan‐FGFR inhibitor. Phase I trial of rogaratinib (NCT01976741) showed clinically active against several types of cancer, especially for the subgroup selected by FGFR mRNA expression. 231 A phase II/III trial (NCT03410693) of rogaratinib has completed for urothelial carcinoma. Both LY2874455 and ASP5878 are on their phase I stage (NCT03125239, NCT01212107, and NCT02038673). AZD4547 and derazantinib are potent and selective FGFR1‐3 inhibitors. AZD4547 has been clinically evaluated as a second‐line treatment in FGFR1‐amplified squamous cell NSCLC, gastric adenocarcinoma with FGFR2 polysomy or gene amplification, malignant pleural mesothelioma, the results of which showed AZD4547 was well tolerated but had minimal antitumor activity. 232 , 233 , 234 Derazantinib is on phase II stage for cholangiocarcinoma (NCT03230318). Selective FGFR4 inhibitors are mainly developed to inhibit the growth of HCC. Phase I trials of H3B‐6527 and FGF401 for HCC have completed (NCT02834780 and NCT02325739). According to previous studies, we conclude that finding the specific tumor histology with FGFR alterations is essential for the efficacy of FGFR inhibitors.
3.9. TRK receptor kinase inhibitors
NTRK1, NTRK2, and NTR3 genes encode tropomyosin receptor kinase A, B, and C (TRKA, TRKB, and TRKC), which are the members of the cell surface RTK family, regulating cell proliferation, differentiation, and apoptosis through activate pathways of PLCγ, MAPK, PI3K/AKT/mTOR, and PKC. 235 NTRK gene fusions lead to TRK fusion proteins with constitutive, ligand‐independent activation of the intrinsic TK. The incidence of NTRK gene rearrangements is very low across a wide range of different tumor types. 236 , 237 , 238 TRK TKIs are developed based on the tumor's molecular characteristics rather than a tumor primary site. 239
Two agents, larotrectinib and entrectinib, have received US FDA approval for NTRK‐gene fusion locally advanced or metastatic tumors. A pooled analysis of three phase I/II clinical trials (NCT02122913, NCT02637687, and NCT02576431) showed that the ORR of larotrectinib was 79% in patients with TRK fusion‐positive solid tumors, with 16% of patients having CR. 240 Subgroup analysis of those trials yielded the rapid and durable responses and high disease control rate for patients with TRK fusion‐positive CNS tumors. 241 An integrated analysis of three entrectinib‐related phase I/II trials (ALKA‐372‐001, STARTRK‐1, and STARTRK‐2) showed that 57% of patients had an objective response, of which 7% were CR and 50% PR and median DOR was 10 months. 193 What is more, grade 3 or 4 adverse events of any kind for larotrectinib and entrectinib happened in less than 10% of patients. 193 , 240 In total, the treatment of patients with NTRK fusion‐positive cancers with larotrectinib or entrectinib is associated with high response rates and well‐tolerated.
Similarly, developing new indications for existing drugs and exploiting new drugs are two important research directions. Trials have been done or are ongoing to find new usage scenarios. A pediatric phase I trial of neoadjuvant larotrectinib presented a promising medication for children with newly diagnosed and locally advanced TRK fusion sarcomas, which might expand the use of TRK inhibitors in neoadjuvant therapy. 242 Though larotrectinib and entrectinib have been approved for TRK fusion‐positive solid tumors, studies are ongoing to assess their efficacy and safety in histologically specific tumors, such as glioma and non‐Hodgkin lymphoma (NHL) (NCT04655404 and NCT03155620). Acquired resistance to prior TRK kinase inhibitors has spawned the development of next‐generation TRK TKI. TRKA G595R mutation was found in LMNA‐NTRK1 fusion‐positive CRC treated with larotrectinib. ETV6‐NTRK3 fusion‐positive infantile fibrosarcoma progressed on larotrectinib because of TRKC G623R mutation. Selitrectinib is a second‐generation TRK TKI and was demonstrated to inhibit these two recurrent tumors. 243 A novel potent TRK inhibitor, AZD7451, completely blocked TRC activation and inhibited the growth of high TRKC‐expressed adenoid cystic carcinoma in the preclinical trial. 244 Phase I trial (NCT01468324) of this agent for patients with recurrent gliomas has been completed.
3.10. FLT3 receptor kinase inhibitors
FMS‐like tyrosine kinase 3 (FLT3), expressed in various lymphohematopoietic cells and tissues, belongs to the class III RTK family and plays an important role in regulating cell survival, cell proliferation, and differentiation of hematopoietic progenitor cells through activating PI3K/AKT/ mTOR, MAPK, and JAK/STAT downstream signalings. 245 Mutations of the FLT3 gene exist in about 30% of patients with AML, with 25% having the FLT3 internal tandem duplication (ITD) mutation and the other 5–10% presenting with the FLT3 tyrosine kinase domain (TKD) mutation, which lead ligand‐independent activating of the RTK and the downstream proliferative signaling pathways. 246 , 247
Correspondingly, FLT3 inhibitors are developed to treat FLT3‐mutated AML. 248 Midostaurin is the first FLT3 inhibitor approved for newly diagnosed FLT3 mutation‐positive AML. The phase IIB trial of midostaurin for wild‐type or mutated FLT3 AML and high‐risk myelodysplastic syndrome (MDS) showed that the rate of reduction in peripheral blood or bone marrow blasts by ≥50% was 71% in patients with FLT3‐mutant and 42% in patients with FLT3 wild‐type. The success of the following phase III trial led to the approval of midostaurin. Using midostaurin together with standard chemotherapy regimens significantly improved patients’ EFS from 3.0 months for placebo plus standard chemotherapy to 8.2 months for midostaurin plus standard chemotherapy (HR, 0.78; 95%CI: 0.66, 0.93; one‐sided p = 0.002) and OS from 25.6 months to 74.7 months (HR, 0.78; 95%CI: 0.63, 0.96; one‐sided p = 0.009). The rate of severe adverse events was similar in the two groups. 249 , 250 Given the structural similarity of FLT3 and KIT, midostaurin also inhibits D816V‐mutated KIT and has been approved for aggressive systemic mastocytosis. 251 Rapid generation of resistance mutations, particularly in codon Asp835 (D835), gave rise to second‐generation FLT3 inhibitors. 252 Gilteritinib, a second‐generation inhibitor, is the only US FDA‐approved FLT3 inhibitor to be used as a single agent for patients with relapsed or refractory AML having an FLT3 ITD, D835, or I836 mutation based on the results of the phase III ADMIRAL trial. In this trial, gilteritinib significantly prolonged survival compared with chemotherapy (median OS: 9.3 vs. 5.6 months; HR, 0.64; 95%CI: 0.49, 0.81; p < 0.001). The percentages of patients with CR (21.1 vs. 10.5%) and CR with full or partial hematologic recovery (34.0 vs. 15.3%) were higher in the gilteritinib than in the chemotherapy group. In addition, grade 3 or higher and serious adverse events occurred less frequently in the gilteritinib group than in the chemotherapy group. 253 Taken together, FLT3 inhibitors are suitable for treating FLT3‐mutated AML in first‐ and second‐line treatment because of their excellent efficacy and low toxicity.
Researches on midostaurin and gilteritinib are ongoing. Only midostaurin has been approved for first‐line treatment of FLT3 mutant AML. A phase II study (NCT03836209) is recruiting untreated patients with FLT3 mutant AML to compare the effectiveness of gilteritinib to midostaurin in first‐line therapy. Enhancing efficacy through combination therapy is another trend. A phase I/II trial for patients with AML and MDS showed that the combination of midostaurin and a DMNT inhibitor 5‐azacytidine was an effective and safe regime, especially for patients with FLT3 mutations but not previously exposed to other FLT3 inhibitors. 254 Combining gilteritinib with the anti‐CD33 antibody gemtuzumab plus cytarabine for FLT3‐ITD‐mutated relapsed/refractory AML is in the phase II stage (NCT05199051).
New FLT3 inhibitors are under different research stages. The second‐generation FLT3 inhibitor quizartinib is widely studied and is the most promising drug for approval. FLT3 inhibitors are classified as either type I or II based on their interaction with their kinase targets. Type I FLT3 inhibitors target either FLT3‐ITD or FLT3‐TKD point mutations, while type II inhibitors commonly only target FLT3‐ITD. 248 Quizartinib is a selective and highly potent type II FLT3 inhibitor. Phase III trial (NCT02039726) of monotherapy with quizartinib has been proved to be efficacious and safe in patients with FLT3‐ITD‐mutated, relapsed/refractory AML. OS was longer for quizartinib than for salvage chemotherapy (median OS: 6.2 vs. 4.7 months; HR, 0.76; 95%CI: 0.58, 0.98; p = 0.02). 255 A phase I trial (NCT01468467) yielded that following allogeneic hematopoietic‐cell transplant with quizartinib had acceptable tolerability and reduced relapse rate. 256 Quizartinib, combined with standard chemotherapy, is a promising treatment for newly diagnosed AML because of its safety and tolerability (NCT 01390337). 257 Another second‐generation FLT3 kinase inhibitor, crenolanib, belongs to type I inhibitor with activity against FLT3‐ITD mutants and FLT3‐D835 point mutants. 258 Clinical trials similar to quizartinib are also underway in crenolanib, with results not yet available or published. For example, a Phase III trial (NCT03258931) comparing crenolanib with midostaurin following induction chemotherapy and consolidation therapy is ongoing in newly diagnosed FLT3‐mutated AML. Phase II studies (NCT01657682 and NCT01522469) of crenolanib in relapsed or refractory AML with FLT3 activating mutations have been finished, and the results have not yet been published. Besides, a novel FLT3 kinase inhibitor G‐749 has antitumor activity against the FLT3 wild type and mutants, including FLT3‐ITD, FLT3‐D835Y, FLT3‐ITD‐N676D, and FLT3‐ITD/F691L and can overcome drug resistance in preclinical models. 259 As AML cells failed to develop resistant clones under incubation with quziartinib plus the CDK4 inhibitor PD0332991, a dual FLT3/CDK4 inhibitor AMG 925 came up and was demonstrated to overcome FLT3 inhibitor resistance. 260
3.11. CSF1R kinase inhibitors
The TME is rich in cytokines and their receptors, which exert complex functions in antitumor response or tumor‐promoting effect. 261 For example, cytokines such as IL‐2, IFNα, and IFNγ are involved in the antitumor response, while TGF‐β is correlated to the lymphocyte‐deficient phenotype. 262 , 263 , 264 Most antitumor treatments utilizing cytokines and their receptors show limited therapeutic efficacy, perhaps because patients with advanced‐stage disease might not be suitable for cytokine‐based therapy. 265 Colony‐stimulating factor 1 receptor (CSF1R) is a lineage marker for monocytes and macrophages and the receptor for the growth factor CSF1. CSF1/CSF1R axis promotes survival, differentiation, and proliferation of macrophages. The high levels of macrophage infiltration in human tumors and the numerous tumor‐promoting actions of these extremely plastic immune cells have provided a rationale for CSF1R inhibitors. 266
Tenosynovial giant cell tumor (TGCT) is a rare and locally aggressive neoplasm with overexpression of CSF1. Before 2019, surgery was standard with no approved systemic therapy. The CSF1R kinase inhibitor, pexidartinib, is indicated for patients with TGCT based on the data from the phase III ENLIVEN trial in 2019, which pioneers the way for harnessing cytokines and chemokines for cancer therapy. 267 Overexpression of the CSF1R ligand promotes cell proliferation and pexidartinib can inhibit the proliferation of CSF1R‐dependent cell lines in vitro and in vivo. 268 , 269 In the clinical setting, the ORR was 39% with pexidartinib versus 0% with placebo in patients with TGCT (p < 0.0001). Serious adverse events occurred in 13% of patients in the pexidartinib group, including increasing levels of aspartate aminotransferase, alanine aminotransferase, and alkaline phosphatase and hypertension. In the ENLIVEN trial, hepatotoxicity was a dose limiting toxicity. 270 To further clarify the liver toxicity, a long‐term study is ongoing to evaluate hepatotoxicity associated with pexidartinib (NCT04635111).
Pexidartinib has also been explored in other tumor types. Trials of pexidartinib in advanced castration‐resistant prostate cancer, recurrent glioblastoma, and relapsed or refractory Hodgkin lymphoma have been finished, and the results are waiting to be published (NCT01499043, NCT01349036, and NCT01217229). A similar situation is for the phase I/II trial (NCT02452424) of pexidartinib plus anti‐PD‐1 antibody pembrolizumab for advanced melanoma and some other solid tumors.
The development of novel CSF1R kinase inhibitors is relatively slow and a few monoclonal antibodies against CSF1R are also exploited to target the CSF1/CSF1R axis. Edicotinib is a selective inhibitor of the CSF1R tyrosine kinase. Phase I/II study of this agent for relapsed/refractory classical Hodgkin lymphoma demonstrated its safety profile but limited antitumor activity in treating classical Hodgkin lymphoma. 271 Phase II trial (NCT03557970) of evaluating edicotinib in patients with AML is under the writing stage. CSF1R monoclonal antibodies conclude AMG 820 and emactuzumab. AMG 820 was tolerated with manageable toxicities up to 20 mg/kg every 2 weeks but had limited antitumor activity. 272 Emactuzumab showed its preliminary activity against TGCT with acceptable toxicities in a phase I trial and also resulted in poor response in solid tumors (NCT01494688). 273 , 274
3.12. RAF/MEK/ERK kinase inhibitors
The MAPK cascade, also known as the RAS/RAF/MEK/ERK signaling pathway, regulates cell proliferation, differentiation, and survival. 275 As the first component of MAPK signaling, RAS has three gene isoforms: HRAS, KRAS, and NRAS and they are small GTPases that initiate RAF/MEK/ERK kinase cascade. The RAF family includes three RAF isoforms, ARAF, BRAF, and CRAF, and is the direct downstream of RAS. RAF activates two dual‐specific kinases MEK1/2, which contain a docking site for substrate ERK1/2. Activated ERK phosphorylates a number of substrates that regulate cell functions. 276 Alterations in key genes of this pathway can lead to a consequential constitutional activation of MAPK, inducing transformation and tumor progression. 277 Aberrant activation of this signaling pathway plays an important role in over 40% of human cancer cases, including melanoma, NSCLC, anaplastic thyroid cancer (ATC), and other types of cancer. 278 RAS small molecule inhibitors are not protein kinase inhibitors and will be discussed below, and none of the ERK inhibitors have been approved by the US FDA to date. 279 , 280 In this part, we will focus on RAF and MEK kinase inhibitors.
BRAF, MEK, and ERK are oncogenes encoding serine‐threonine protein kinases. Some mutations in the BRAF gene result in constitutively activated BRAF proteins and then triggering downstream MEK and ERK. 276 , 277 Up to 66% of melanomas have BRAF mutations, BRAF V600E occupying nearly 80% of total BRAF mutations, followed by BRAF V600K (20%). 281 , 282 The main indication for BRAF and MEK inhibitors is BRAF V600 mutant melanomas. Three BRAF inhibitors (vemurafenib, dabrafenib, and encorafenib) and three MEK inhibitors (trametinib, cobimetinib, and binimetinib) are permitted for metastatic melanoma with BRAF V600E or V600K mutations as a single agent or in combination with other agents and exhibit excellent efficacies. 283 Three drugs (vemurafenib, dabrafenib, and trametinib) are allowed as single agents for melanoma. In first‐line therapy, compared with chemotherapy (dacarbazine or paclitaxel), they increased median PFS by at least 3 months, from less than 2 months to over 5 months, and ORR from less than 20% to more than 50%. The rates of OS were also proved to be improved with BRAF or MEK inhibitors. 284 , 285 , 286
Vertical blockade of BRAF and MEK in the MAPK pathway reduces paradoxical downstream activations and efficiently targets the MAPK pathway. 282 , 283 Three combinations of BRAF and MEK inhibitors have been approved by the US FDA for metastatic melanoma with BRAF V600E or V600K mutations: cobimetinib plus vemurafenib, dabrafenib plus trametinib, and encorafenib plus binimetinib. 278 , 281 Vemurafenib was the first BRAF kinase inhibitor approved for melanoma. 287 Combination of cobimetinib with vemurafenib prolonged median PFS from 7.2 months to 12.3 months (HR, 0.56; 95%CI: 0.46, 0.72; p < 0.0001) and median OS from 17.4 months to 22.3 months (HR, 0.70; 95%CI: 0.55, 0.90; p = 0.005) and the safety profile for cobimetinib plus vemurafenib was tolerable and manageable in the phase III coBRIM trial. 288 Compared with dabrafenib monotherapy in the phase III COMBI‐d study or vemurafenib in the phase III COMBI‐v study, the addition of trametinib to dabrafenib was demonstrated to promote ORR and prolong PFS and OS. 289 , 290 The COMBI‐v study also concluded that dabrafenib plus trametinib significantly alleviated disease‐associated and adverse‐event‐associated symptoms. 290 Furthermore, trametinib to dabrafenib is effective against melanoma brain metastases. 291 Similarly, the phase III COLUMBUS trial illustrated benefits in terms of PFS and OS in the combination of encorafenib with binimetinib compared with vemurafenib when treating patients who were BRAF‐V600E mutant melanoma and were treatment naïve or progressed on or after first‐line immunotherapy. Median PFS increased from 7.3 months in monotherapy to 14.9 months in the doublets (HR, 0.54; 95%CI: 0.41, 0.71; p < 0.0001), median OS from 16.9 months to 33.6 months (HR, 0.61; 95%CI: 0.47, 0.79; p < 0.0001). 292 In short, dual inhibition of BRAF and MEK to unresectable or metastatic melanoma with BRAF V600 mutation is superior to inhibiting BRAF or MEK separately in the form of ORR, PFS, OS, and quality of life. At the same time, combination therapy could reduce toxicities by avoiding paradoxical downstream activations. 293 Besides, adjuvant treatment with BRAF inhibitor dabrafenib and MEK inhibitor trametinib is approved in resected stage III melanoma with BRAF V600E or V600K mutations. Five‐year follow‐up of phase III COMBI‐AD trial demonstrated that 12 months of adjuvant therapy with dabrafenib plus trametinib resulted in a longer duration of relapse‐free survival and survival without distant metastasis. 294
As mentioned above, other types of cancer may harbor BRAF mutations or MAPK pathway aberrations. Correspondingly, monotherapy with BRAF or MEK inhibitors and BRAF plus MEK inhibitors also have indications in other types of tumors. Vemurafenib is indicated for the treatment of patients with BRAF V600 mutant erdheim‐chester disease. 295 Selumetinib, a new MEK inhibitor, broadens indication to neurofibromatosis type 1 with inoperable plexiform neurofibromas and the ORR reached 66% in the SPRINT phase II Stratum 1 trial. 296 Dabrafenib in combination with trametinib is approved for advanced or metastatic BRAF V600E mutant NSCLC and ATC with BRAF V600E mutation. 297 , 298 The US FDA recently granted accelerated approval to dabrafenib in combination with trametinib for unresectable or metastatic solid tumors with BRAF V600E mutation after standard therapies. 299
Though having not been permitted, many clinical trials have proven efficacy of BRAF or MEK inhibitors or BRAF plus MEK inhibitors in more cancer types. Phase II clinical trials showed that vemurafenib represented a potential new treatment option for BRAF‐V600E‐positive papillary thyroid cancer and NSCLC with BRAF V600 mutations. 300 , 301 Trametinib prolonged PFS of patients with recurrent low‐grade serous ovarian cancer compared with standard of care. 302 Patients with BRAF V600E‐mutant glioma and BRAF V600E‐mutated biliary tract cancer benefited from the dabrafenib plus trametinib based on the results of the phase II ROAR trial. 303 , 304
However, BRAF or MEK inhibitors are not suitable for all BRAF‐mutant cancers. Phase II clinical trials showed that single‐agent vemurafenib did not show meaningful clinical activity in patients with BRAF V600E mutant‐CRC, which may attribute to tumor specificity and feedback mechanisms and may be conquered by combination strategies. 305 Fortunately, the phase III BEACON study showed BRAF inhibitor encorafenib plus EGFR antibody cetuximab improved OS, ORR, and PFS compared with standard chemotherapy and is a new standard of care for previously treated BRAF V600E‐mutant metastatic CRC. 306
The clinical benefit of combining PD‐1/PD‐L1 antibody with RAF and MEK inhibitors is inconsistent. Preclinical trials provide a rationale for these combining strategies. 307 The phase III IMspire150 trial showed that treating BRAF V600‐mutant melanoma with atezolizumab, vemurafenib, and cobimetinib significantly increased PFS from 10.6 months with vemurafenib and cobimetinib to 15.1 months (HR, 0.78; 95%CI: 0.63, 0.97; p = 0.025) without obviously adding adverse events. 308 However, the phase III COMBI‐i trial evaluated spartalizumab plus dabrafenib and trametinib for BRAF V600‐mutant melanoma, the results of which showed that the addition of spartalizumab to targeted therapy with dabrafenib and trametinib did not significantly prolong PFS but increased grade ≥3 adverse events by 22% compared with the dabrafenib plus dabrafenib group. 309 Further details and trials are waiting to clarify this combination's possibility.
Novel kinase inhibitors involved in RAF, MEK, and ERK are under development. New RAF inhibitors are developed to resolve more molecular alterations in RAS‐RAF‐MEK signaling. Belvarafenib is a type II RAF inhibitor and is effective in BRAF V600E‐ and NRAS‐mutant melanoma. 310 Mutations in ARAF contributed to resistance to belvarafenib, which could be addressed by combination strategies. Related experiments are executed in NRAS‐mutant melanoma and solid tumors (NCT04835805 and NCT03284502). Another RAF inhibitor, lifirafenib, is a reversible inhibitor of BRAF V600E, wild‐type ARAF, BRAF, CRAF, and EGFR and showed antitumor activity in BRAF V600‐mutated solid tumors (NCT02610361). 311 Further trials may compare lifirafenib with first‐generation BRAF inhibitors and explore the possibility of lifirafenib as second‐line therapy (NCT03905148). Research on new MEK drugs is not always successful. The study on MEK inhibitor CI‐1040 has been halted because of its insufficient antitumor activity. 312 Monotherapy with a second‐generation MEK inhibitor, mirdametinib, is not recommended. 313 Trials are ongoing as combination regimes (NCT03905148 and NCT03170206). Refametinib is an allosteric MEK inhibitor and yields preliminary efficacy in some kinds of solid tumors, such as HCC and pancreatic cancers. 314 , 315 A phase II study (NCT01204177) showed that HCC patients with RAS mutations could benefit from refametinib/sorafenib combination with a DCR of 44.8%. 314 However, follow‐up studies of this drug are lacking. Dual inhibition of RAF and MEK is a trend. RO5126766, also known as CH5126766 or VS‐6766, is a dual RAF/MEK inhibitor and has antitumor activity across various cancers with RAF‐RAS‐MEK pathway mutations. 316 A combination of RO5126766 with PD‐1/PD‐L1 antibody may increase the antitumor effect. 317 The development of selective ERK inhibitors lags far behind compared with the RAF and MEK inhibitors. The clinical trials of ERK inhibitors are in their early stages, such as ulixertinib, GDC0994, and LY3214996. Ulixertinib showed potent preclinical activity in BRAF‐ and RAS‐ mutant cell lines and had clinical activity in NRAS‐ and BRAF V600‐ and non‐V600‐mutant solid‐tumor malignancies with an acceptable safety profile in the phase I trial (NCT01781429). 318 Phase II studies of this agent involved in solid tumors, NHL, AML, and MDS are under‐evaluated (NCT03155620, NCT04488003, NCT02296242, and NCT03155620). GDC0994 is another selective ERK inhibitor and only has been tested in phase I trials. The single‐agent activity was observed in BRAF‐mutant CRC (NCT01875705). 319 The combination of cobimetinib and GDC‐0994 led to cumulative toxicity, which restricted further development (NCT02457793). 320 LY3214996 is a potent and selective ATP‐competitive inhibitor of ERK with IC50 values for ERK1and ERK2 below 0.001 mmol/L. 321 Clinical trials of this agent are ongoing (NCT02857270, NCT04616183, and NCT04956640).
3.13. PI3K/AKT/mTOR kinase inhibitors
The PI3K/AKT/mTOR signaling pathway triggered by various extracellular stimuli regulates cell proliferation, survival, and angiogenesis. PI3Ks are the family of vital lipid kinases widely distributed in mammalian cells that phosphorylate PIP2 into PIP3, which activates PDK1 and then influences AKT. Activation of PI3K and AKT leads to mTOR activation and phosphorylation of S6K1 and 4e‐BP1. 322 Among the three classes of PI3K, class I is the most important, with four isoforms (PI3Kα, PI3Kβ, PI3Kδ, and PI3Kγ), the catalytic isoforms of which are encoded by PIK3CA, PIK3CB, PIK3CG, and PIK3CD, respectively. PI3Kα and PI3Kβ are ubiquitously expressed in various tissues. PI3Kδ expression is limited mainly to the B cells and their precursors, whereas PI3Kγ is expressed in T lymphocytes. 323 , 324 Hyperactivation of the PI3K/AKT/mTOR pathway in tumors provides a rationale to target key elements involved in this cascade. 325 , 326 AKT and mTOR kinase inhibitors are in clinical trials. 327 Rapamycin and its analogs (temsirolimus and everolimus) are allosteric inhibitors of mTOR will be discussed below. 328 Here, we focus on PI3K kinase inhibitors and briefly introduce AKT and mTOR kinase inhibitors.
It is reported that PIK3CA mutations occur in 40% of HR‐positive, HER2‐negative breast cancer. 329 , 330 The selective PI3Kα inhibitor alpelisib combined with fulvestrant is permitted to treat breast HR‐positive, HER2‐negative breast cancer harboring PIK3CA mutation after an endocrine‐based regimen. The phase III SOLAR‐1 trial demonstrated that the PFS and ORR of the alpelisib plus fulvestrant arm were better than those of the placebo plus fulvestrant arm (median PFS: 11.0 vs. 5.7 months, HR, 0.65, 95%CI: 0.50, 0.85, p < 0.001; ORR: 35.7 vs. 16.2%). 331 Acquired of PTEN may attribute to resistance of alpelisib, which could be reversed by PTEN knockdown. 332 Several critical phase II trials have received positive results concerned with PI3Kα inhibitor alpelisib. Monotherapy with alpelisib in later lines of therapy for PI3K‐altered, pretreated advanced cancer received an ORR of 30%. 333 In another phase II study, alpelisib plus fulvestrant proved their antitumor activity and manageable toxicity in patients with PI3KCA‐mutated, HR‐positive, HER2‐negative advanced breast cancer after progression on a CDK4/6 inhibitor plus an aromatase inhibitor, which offered a choice for these kinds of patients (NCT03056755). 334 As PI3Kδ is widely expressed in B cells and their precursors, idelalisib, copanlisib, duvelisib, and umbralisib are PI3Kδ inhibitors approved for the post line therapy of leukemia or lymphoma derived from B cells, including chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma (SLL), follicular lymphoma (FL), and marginal zone lymphoma (MZL). 335 , 336 , 337 , 338 The standard of care for patients with relapsed or refractory CLL is bendamustine plus rituximab. Idelalisib plus CD20 antibody rituximab for patients with relapsed CLL whose disease only suitable for rituximab alone received 19.4 months of median PFS and 81% of ORR, while the values were 6.5 months and 13%, respectively, for the placebo plus rituximab group. 339 Final observation showed that OS favored the patients in the idelalisib plus rituximab group (median OS: 40.6 vs. 34.6 months; HR, 0.8; 95%CI: 0.5, 1.1; p = 0.1343). 340 Good results from clinical trials resulted in the approval of idelalisib plus rituximab for CLL. Besides, adding idelalisib to bendamustine plus rituximab prolonged median PFS from 11.1 months in the bendamustine plus rituximab group to 20.8 months (HR, 0.33; 95%CI: 0.25, 0.44; p < 0.0001) (NCT01569295). In contrast, the accompanying serious adverse events and infections may prevent its approval. Compared with the PI3Kδ specific inhibitor idelalisib, the PI3Kδ/γ dual duvelisib shows a more potent inhibitory activity of PI3K protein and the US FDA has approved duvelisib for CLL or SLL after at least two prior therapies. 341 In addition, the phase II DYNAMO study showed that duvelisib monotherapy may provide a new option for patients with heavily pretreated, double‐refractory indolent NHL. The ORR was 47.3%, which even reached 67.9% for SLL. 342 Another PI3Kα/δ dual inhibitor, copanlisib, was granted US FDA accelerated approval in 2017 for FL after at least two prior systemic therapies based on an ORR of 59% under monotherapy with copanlisib in the phase II CHRONOS‐1 trial. 343 In addition, the phase III CHRONOS‐3 trial supported the combination of copanlisib with rituximab for relapsed indolent NHL, in which the median PFS was increased by 7.7 months compared with rituximab monotherapy. 344 Umbralisib is a selective PI3Kδ inhibitor indicated for MZL and FL and reduces the incidence of autoimmune complications as it additionally inhibits CK1ε. 345 , 346 Except for copanlisib, the other four PI3K‐approved inhibitors are orally bioavailable. 335 However, the incidence of grade 3 or 4 adverse events is much higher in the PI3K inhibitor group than in the placebo group. The role of the PI3K/AKT/mTOR signaling pathway in the immune system renders inhibitors bring significant on‐target toxicities, such as pneumonitis, stomatitis, and infections, which also limit their use in the clinic. 347
Though several PI3K inhibitors have been approved, efforts never stop to exploit more suitable inhibitors. Buparlisib and pictilisib belong to pan‐PI3K kinase inhibitors, which have been tested as a single agent or combination regimes in many tumor types, such as breast cancer, squamous cell carcinoma of the head and neck, glioblastoma, and lymphoma. 348 , 349 , 350 Preclinical and early‐stage clinical trials have demonstrated the safety profile and preliminary activity of monotherapy with pan‐PI3K inhibitors in some tumor types, especially for breast cancer. 351 , 352 , 353 However, two phase III trials (BELLE‐2 and BELLE‐3) did not support buparlisib plus fulvestrant for further development in breast cancer because of toxicities. 354 , 355 Taselisib is a selective PI3Kα inhibitor and yields antitumor activity toward PIK3CA‐mutant tumors, especially for breast cancer. 356 Phase II LORELEI trial found that neoadjuvant letrozole plus taselisib for oestrogen receptor‐positive, HER2‐negative, early‐stage breast cancer increased the proportion of patients who achieved an objective response. 357 But phase III SANDPIPER trial showed the addition of taselisib to fulvestrant for oestrogen receptor‐positive, HER2‐negative, PI3KCA‐mutant, advanced breast cancer was not suggested given its toxicities and modest clinical benefit. 358 GSK2636771 is a PI3Kβ inhibitor. The safety profile was acceptable, but the efficacy in metastatic castration‐resistant prostate cancer was limited. 359 More clinical trials are ongoing (NCT04439188 and NCT02951091).
To date, no AKT inhibitors have been approved. Several AKT inhibitors are in different clinical stages for various tumor types, especially for breast cancer, prostate cancer, and RCC, such as ipatasertib, capivasertib, afuresertib, and MK‐2206. 360 , 361 Their toxicities are acceptable as a single agent or in combination with other agents. The activation of the PI3K/AKT/mTOR signaling pathway caused by gene mutation and/or deletions of PTEN is associated with drug sensitivity. 362 For example, ipatasertib is a selective ATP‐competitive inhibitor of AKT and has received positive results in treating triple‐negative breast cancer and prostate cancer. 362 , 363 Notably, the only published phase III clinical trial for AKT inhibitor is ipatasertib for previously untreated metastatic castration‐resistant prostate cancer. In this trial, ipatasertib plus abiraterone significantly improved PFS compared with placebo plus abiraterone among patients with PTEN‐loss tumors. Many trials are ongoing, and four out of five phase III trials are for breast cancer. 363 The phase III IPATunity130 is evaluating ipatasertib plus paclitaxel as first‐line therapy for PI3KCA/AKT/PTEN‐altered triple‐negative breast cancer or HR‐positive, HER2‐negative breast cancer. Another phase III trial of ipatasertib in combination with atezolizumab and paclitaxel for triple‐negative breast cancer is ready to recruit. Two phase III trials (NCT04060862 and NCT04650581) are for HR‐positive or ER‐positive, HER2‐negative breast cancer. MK‐2206 is an allosteric inhibitor of AKT and resulted in a higher pathologic complete response rate in HR‐negative and HER2‐positive breast cancer when combined with standard neoadjuvant therapy (NCT01042379). 364 However, it was not superior to standard RCC and pancreatic cancer treatment. 365 , 366
The approved mTOR inhibitors only inhibit TORC1, which could inversely activate AKT. mTOR kinase inhibitors can inhibit TORC1 and TORC2 and are supposed to avoid resistance caused by AKT phosphorylation. 328 Currently, no mTOR kinase inhibitors are approved, but some are under evaluation, such as AZD2014, sapanisertib, and vistusertib. 367 , 368 , 369 Notably, vistusertib plus anastrozole improved was superior to anastrozole alone in HR‐positive, recurrent or metastatic endometrial cancer in the form of PFS and ORR with manageable adverse events. 369 Dual inhibition of PI3K and mTOR is a direction, and many dual PI3K/mTOR inhibitors are in their early‐stage trials, such as gedatolisib, paxalisib, samotolisib, voxtalisib, and apitolisib. 370 For example, samotolisib is a promising dual PI3K/mTOR inhibitor and had a clinical benefit on PFS in metastatic castration‐resistant prostate cancer with tolerable side effects when combined with enzalutamide. 371 Trials on advanced solid tumors, NHL, and histiocytic disorders are ongoing (NCT03155620 and NCT03213678). Paxalisib is a brain‐penetrant dual PI3K/mTOR inhibitor and was proved to cross the blood‐brain barrier in phase I clinical trial (NCT01547546). 372 Further trials of paxalisib are undergoing in tumors with primary and secondary brain metastases, including glioblastoma and breast cancer with brain metastases (NCT03970447, NCT05009992, and NCT03994796). Dual inhibition is supposed to achieve better antitumor activity than inhibiting one mechanism alone but also increases toxicity. Phase II trial (NCT01442090) comparing apitolisib with everolimus in RCC failed to show better outcomes due to multiple on‐target adverse events.
3.14. JAK kinase inhibitors
The Janus kinase (JAK)/signal transducer and activators of transcription (STAT) pathway plays essential roles in tumorigenesis and immune function by mediating the cellular response to cytokines, interferons, and growth factors. 373 JAKs belong to the intracellular nonreceptor protein tyrosine kinase family and consist of four isoforms (JAK1‐3 and TYK2). 374 The STAT protein family comprises seven members, including STAT1‐6, STA5a, and STA5b. 375 Upon ligands binding to the surface receptors, dimerization of JAK‐associated receptors induces activation of JAK kinases, which in turn recruit and phosphorylate cytosolic STAT proteins and lead to nuclear translocation of STATs, which function as transcription factors. Mutation or amplification of JAK1, JAK2, STAT3, and STAT5 frequently occurs in malignant tumors, which induces dysregulated JAK/STAT signaling and therefore provides targets for tumor treatment. 376
Myelofibrosis is a kind of hematological malignancy that includes primary myelofibrosis, postpolycythaemia vera myelofibrosis, and postessential thrombocytosis myelofibrosis, and ectopically activates the JAK/STAT pathway. 377 , 378 Hyperactivation of JAK/STAT pathway provides a rationale for targeting JAK or STAT in myelofibrosis. Two JAK inhibitors are approved for myelofibrosis. Ruxolitinib, a selective JAK1/JAK2 inhibitor, significantly alleviates palpable splenomegaly, ameliorated debilitating myelofibrosis‐related symptoms, and improved OS compared with placebo. The primary endpoint was the proportion of patients with a reduction in spleen volume of 35% or more at 24 weeks. 41.9% of patients in the ruxolitinib group achieved a 35% or greater reduction in spleen volume from baseline, while less than 1% of patients had such curative effect (p < 0.001). 379 , 380 , 381 Fedratinib, a JAK2‐selective inhibitor, has a similar role in controlling palpable splenomegaly when treating myelofibrosis. 382 , 383 Though JAK inhibitors have also been trialed and received mild response in patients with chronic neutrophilic leukemia, atypical CML, and metastatic pancreatic cancer, none of them are formally permitted. 384 , 385 Except for cancer, the JAK/STAT pathway also has impact on immunity, JAK inhibitors are therefore used in treating autoimmune diseases, which are beyond our discussion and will not be described in our review. 386 Adverse events related to JAK inhibitors include anemia, thrombocytopenia, gastrointestinal symptoms, increased levels of liver transaminases, serum creatinine, and pancreatic enzymes. Among these, serious and fatal encephalopathy caused by fedratinib received a black‐box warning indicating that reliable prevention and monitoring strategies are needed during the medication process. 381 , 383
Several JAK inhibitors have been explored, but some of them discontinued further research due to serious adverse events or limited efficacy, such as JAK1/2 inhibitor AZD1480 and momelotinib. AZD1480 ceased in the phase I study because of unusual dose‐limiting toxicities and the lack of clinical activity. 387 Momelotinib failed in two published III trials for the treatment of myelofibrosis. The phase III SIMPLIFY‐1 trial of momelotinib versus ruxolitinib in JAK inhibitor‐naïve myelofibrosis demonstrated that 24 weeks of momelotinib treatment was noninferior to ruxolitinib for spleen response but not for symptom response. 388 The phase III SIMPLIFY‐2 trial of comparing momelotinib with the best available therapy in treating myelofibrosis previously treated with ruxolitinib showed that momelotinib was not superior to the best available therapy. 389 Luckily, pacritinib is a promising JAK2/JAK2 (V617F) inhibitor and has received positive outcomes in clinical trials. Two phase III trials (PERSIST‐1 and PERSIST‐2) showed that pacritinib was well tolerated and induced significant and sustained spleen volume reduction and symptom reduction, even in patients with thrombocytopenia or resistance to ruxolitinib. 390 , 391 Phase II study of this agent for prostate cancer is ongoing (NCT04635059). Itacitinib is a JAK1 inhibitor and has shown preliminary antitumor activity and safety profile in early‐stage trials for B‐cell lymphoma and pancreatic cancers. 392 Now it is under evaluation in many clinical trials for various tumor types, such as myelofibrosis, HCC, NSCLC, sarcoma, Hodgkin lymphoma, T‐cell prolymphocytic leukemia, and diffuse large B‐cell lymphoma (NCT04640025, NCT04358185, NCT02917993, NCT03670069, NCT03697408, NCT03989466, and NCT02760485).
3.15. CDK kinase inhibitors
Cell cycle is crucial for cell proliferation process and mainly mediated by cyclin‐dependent kinases (CDKs), cyclins, and CDK inhibitors. CDKs are the cell cycle activators and require cyclin proteins for phosphorylation of the key substrates. CDK inhibitors negatively regulates the activity of CDKs. In the human cell, at least 20 CDKs and 29 cyclins have been identified. 393 Complexes of CDK1‐cyclin A/B, CDK2‐cyclin E/A, and CDK4/CDK6‐cyclin D are required in phase progression, completion of which is verified by three checkpoints at G1‐S, G2‐M, and metaphase‐to‐anaphase transitions. 394 Increased expression levels of CDKs or cyclins, or decreased endogenous CDK inhibitors’ levels, have been observed in cancers. CDKs are protein‐serine/threonine kinases and have become a target for anticancer therapy based on their role in cell proliferation. 395
To date, the CDK4/6 inhibitors have been proved to interfere with the proliferation of breast cancer cells by decreasing pRb phosphorylation and arresting the cell cycle in the G1 phase. 396 Since 2015, three CDK4/6 inhibitors, palbociclib, ribociclib, and abemaciclib have been approved for HR‐positive/HER2‐negative breast cancer in various settings and combination regimens. 397 , 398 The addition of the CDK4/6 inhibitors palbociclib, ribociclib, and abemaciclib to an aromatase inhibitor were studied in postmenopausal HR‐positive/HER2‐negative advanced breast cancer in the first‐line setting in PALOMA‐2, MONALEESA‐2, and MONARCH‐3 trials, respectively. The three combination regimens nearly doubled the duration of median PFS and improved ORR compared with the single‐use of an aromatase inhibitor, with or without prolonged OS. 399 , 400 , 401 In second‐line treatment, CDK4/6 inhibitors combined with the selective estrogen receptor degrader fulvestrant for HR‐positive/HER2‐negative breast cancer were studied in three trials: PALOMA‐3, MONALEESA‐3, and MONARCH‐2. The phase III trials showed the CDK4/6 inhibitors prolonged the PFS with or without OS. 402 , 403 , 404 Besides, monotherapy with abemaciclib was also permitted for HR‐positive/HER2‐negative advanced breast cancer after prior endocrine therapy and 1–2 chemotherapy regimens according to an ORR of 19.7% in the MONARCH‐1 trial. 405 The regime of abemaciclib plus standard endocrine therapy can be used as adjuvant treatment for postmenopausal, high‐risk, and HR‐positive/HER2‐negative early breast cancer. 406 However, CDK4/6 inhibitors largely increased the rates of myelotoxic effects, especially for neutropenia and leukopenia. Fortunately, neutropenia and leukopenia are relatively easy to manage in clinic. 398 In 2021, the newest CDK4/CDK6 inhibitor, trilaciclib, was permitted for patients with SCLC indicated to prevent chemotherapy‐induced myelosuppression. 407 SCLC is an ideal disease model to assess the myeloprotective effect of trilaciclib. First, chemotherapy for SCLC is highly hematologically toxic and SCLC is a chemosensitive tumor, providing a chance to demonstrate that trilaciclib does not antagonize chemotherapy efficacy. Second, SCLC tumor cells replicate independently of CDK4/6 and trilaciclib will not directly have effects on the tumor. 408 Trilaciclib administered before chemotherapy could transiently maintain hematopoietic stem and progenitor cells in G1 arrest and protect them from damage by cytotoxic chemotherapy, leading to faster hematopoietic recovery and enhanced antitumor immunity. 407
In addition to the approved indications, these CDK4/6 inhibitors are also trying to broaden their use scopes. The phase II PALLET trial evaluated adding palbociclib to letrozole as neoadjuvant therapy in patients with ER‐positive early breast cancer. However, this regime did not increase the clinical response rate over 14 weeks. 409 Phase II trials supported the use of this agent in HPV‐unrelated HNSCC and liposarcoma (NCT02101034 and NCT01209598). 410 , 411 Phase II trial of ribociclib in combination with docetaxel plus prednisone had acceptable toxicity and encouraging efficacy in metastatic castration‐resistant prostate cancer. 412 Abemaciclib even showed promising clinical activity in patients with p16ink4A‐deficient mesothelioma in a single‐arm phase II trial (NCT03654833). 413 These inhibitors are ongoing clinical trials for ovarian cancer, NSCLC, RCC, AML, and so on (NCT03936270, NCT03170206, NCT05468697, and NCT03844997).
New CDK4/6 inhibitors are still in development. Dalpiciclib is also a CDK4/6 inhibitor and has been approved in combination with fulvestrant for HR‐positive and HER2‐negative breast cancer progressing on endocrine therapy in China. 414 Lerociclib is a novel CDK4/CDK6 inhibitor with IC50 values in the nanomolar range. 415 It showed an antiproliferation role in ER‐positive breast cancer in animal models and is under clinical evaluation in treating breast cancer and NSCLC (NCT05085002 and NCT03455829). PF‐06873600 is a CDK4/6 and CDK2 inhibitor, which may overcome resistance to CDK4/6 inhibition. 416 Phase II study (NCT03519178) of this agent for cancers in recruiting. In addition to CDK4/CDK6 inhibitors, inhibitors of other CDK isoforms are emerging. Dinaciclib is a CDK1/2/5/9 inhibitor with the values of IC50 below 4nM. It has shown preliminary efficacy and acceptable toxicity in treating CML in early‐stage trials and a phase III trial for CML has been completed (NCT01580228). 417 Fadraciclib is a second‐generation inhibitor of CDK2/9, with IC50s of 5nM and 26nM, respectively, and two phase I/II trials (NCT04983810 and NCT05168904) for solid and hematological tumors are ongoing. 418 CDKI‐73 is a CDK9 inhibitor, which is CDKI‐73 has been studied a lot in preclinical in recent years and may be a promising agent. 419
3.16. BTK tyrosine inhibitors
Bruton's agammaglobulinemia tyrosine kinase (BTK) belongs to the Tec non‐RTK family, which is one of the largest kinase families in mammals and includes BTK, ITK, TEC, BMX, and TXK. 420 BTK is also a key cytoplasmatic kinase in the B cell antigen receptor signal transduction pathway and regulates various signals, such as PI3K/AKT/mTOR, MAPK, and NF‐κB pathways. 421 As these signaling pathways regulate the proliferation, differentiation, and apoptosis of B cells, the secretion of proinflammatory cytokines, as well as degranulation and histamine release, BTK inhibition is an essential method in treating B‐cell tumors and B‐cell immune diseases with expected effect and low toxicity. 422 In our article, we focus on tumor‐related indications.
In 2013, a first‐generation BTK inhibitor, ibrutinib, was first approved by the US FDA for mantle cell lymphoma (MCL), CLL/SLL with or without 17p deletion, waldenström's macroglobulinemia (WM), and MZL. 421 Ibrutinib irreversibly and covalently binds to Cys481 within the ATP‐binding pocket of BTK. The ORR achieved at least 60% in the second‐line treatment and can be beyond 80% in the first‐line therapy using ibrutinib alone, which was increased after a longer follow‑up. 422 Combination approaches of ibrutinib with other drugs (mostly BCL‐2 inhibitor venetoclax or CD20 antibody rituximab or obinutuzumab or standard chemoimmunotherapy further explored clinical benefits. The aims of combination regimes are to induce CR for minimal residual disease and then enable treatment discontinuation. 421 Ibrutinib's administration largely improved ORR, PFS, and even OS compared with prior standardized therapies. 423 , 424 , 425 , 426 , 427 Notably, ibrutinib represents an important therapeutic advance for the treatment of CLL/SLL. 428 , 429 However, ibrutinib could also bring side effects, such as bleeding, diarrhea, rash, infection, atrial fibrillation, and even ventricular arrhythmias and sudden cardiac death caused by simultaneously inhibiting the activity of other kinases in the Tec family and/or several non‐BTK kinases, including c‐terminal Src kinase, HER2, and JAK3. 430 In addition, mutations in BTK (C481S, C481R, C481F, and C481Y) and PLCG2 (R665W, L845F, and S707Y) region confer resistance to ibrutinib. Therefore, there is a need to develop the next‐generation BTK inhibitors to overcome drug resistance, improve the selectivity of BTK, and reduce drug toxicity. 420 Under such circumstances, second‐generation BTK inhibitors with more selectivity and more potent binding ability in BTK, acalabrutinib and zanubrutinib, were launched in the market in 2017 and 2019, respectively. 430 Acalabrutinib was approved for CLL/SLL based on the phase III ELEVATE TN trial for treatment‐naïve CLL/SLL and the phase III ASCEND trial for relapsed or refractory CLL/SLL, both of which demonstrated prolonged PFS in the acalabrutinib group compared with the traditional treatment group. 431 , 432 Direct comparison of acalabrutinib and ibrutinib in patients with previously treated CLL/SLL further verified that acalabrutinib received noninferior PFS with fewer cardiovascular adverse events. 433 The other indication of acalabrutinib is MCL, according to 81% of patients achieving an ORR treated by acalabrutinib alone. The indications for zanubrutinib include MCL, WM, and MZL. 434 , 435 Though not approved for CLL/SLL, the recent results of the phase III SEQUOIA trial proved that zanubrutinib significantly improved PFS versus bendamustine–rituximab and is a potential new treatment option for untreated CLL/SLL. 436 Similar to the relationship between acalabrutinib and ibrutinib, zanubrutinib treatment was associated with a better response rate and less toxicity when compared with ibrutinib in treating patients with WM. 437
Tirabrutinib and orelabrutinib also belong to second‐generation BTK inhibitors. Tirabrutinib binds to BTK with an IC50 of 2.2nM. The data from phase II trials indicated favorable efficacy of tirabrutinib in patients with WM and primary CNS lymphoma. 438 , 439 This agent has completed evaluation for previously treated CLL/SLL or NHL in a phase II study (NCT01659255). Orelabrutinib has already received approval in China for patients with MCL or CLL/ SLL who have received at least one prior treatment. 440 The binding of first‐ and second‐generation agents to BTK rely on Cys481. Mutations at Cys481 disrupt the covalent binding between BTK and BTK inhibitors, which further induce resistance. 430
In contrast to the covalent inhibitors, the third‐generation BTK inhibitors, pirtobrutinib and nemtabrutinib, noncovalently and reversibly bind to BTK kinase, do not rely on Cys481, and may further overcome the resistance caused by previous BTK inhibitors. 420 Pirtobrutinib was demonstrated to circumvent resistance caused by C481S and C481R. Besides, this agent's phase I/II study (NCT03740529) showed it was safe and active in multiple B‐cell malignancies, even for patients previously treated with covalent BTK inhibitors. 441 , 442 Pirtobrutinib is under phase III clinical trials for multiple B‐cell malignancies (NCT05023980, NCT04965493, and NCT04662255). Compared with pirtobrutinib, nemtabrutinib is in early‐stage clinical trials for B‐cell malignancies (NCT04728893 and NCT05458297).
3.17. IDH tyrosine inhibitor
Isocitrate dehydrogenase 1 (IDH1) and isocitrate dehydrogenase 2 (IDH2) are important metabolic enzymes that catalyze isocitrate to α‐ketoglutarate (αKG). 443 Mutations in the enzymatic active site of IDH1 and IDH2 block the cycle reaction of isocitrate to αKG but trigger the conversion of αKG to 2‐hydroxyglutarate (2HG). 2HG inhibits αKG‐dependent dioxygenases involved in DNA and histone methylation and is thought to block cellular differentiation to promote leukemogenesis and tumorigenesis. 444 Mutations in IDH1 and IDH2 can be detected in various human cancers, including AML (20%), cholangiocarcinoma (20%), chondrosarcoma (80%), and glioma (80%). 445 , 446 Mutations in IDH1 and IDH2 occur at conserved arginine residues within the enzymatic active site, such as IDH1 R132 and IDH2 R140 or R172. Therefore, mutated IDH as a therapeutic target stirs great interest in cancer treatment research. IDH inhibitors may effectively prevent the 2HG oncometabolite, subsequently reversing epigenetic dysregulation and restoring normal cellular differentiation.
To date, two IDH inhibitors have been approved for AML and/or cholangiocarcinoma.
Enasidenib is an IDH2 inhibitor approved by the US FDA for relapsed or refractory AML with IDH2 mutation in 2017. At 100 mg daily dose of enasidenib, 19% of patients achieved CR and CR with partial hematological recovery (CRh) in another 4% of patients in a phase I/II study (NCT01915498). 447 , 448 A phase III study (NCT02577406) is ongoing to verify the efficacy. In the following phase II trial (NCT02677922) for newly diagnosed, IDH2‐mutant AML ineligible for intensive chemotherapy, the addition of enasidenib to azacitidine significantly improved ORR (74 vs. 36%) without adding serious treatment‐related adverse events compared with azacitidine monotherapy. 449 But enasidenib has not been granted for newly diagnosed AML. Phase I/II trials of enasidenib for other tumor types, such as IDH2‐mutated MDS, glioma, or angioimmunoblastic T‐cell lymphoma with an IDH2 mutation, and advanced hematologic malignancies with an IDH2 mutation, are under evaluation (NCT03744390, NCT02273739, and NCT01915498). The indications for the IDH1 inhibitor, ivosidenib, extend to IDH1 mutant AML and IDH1 mutant cholangiocarcinoma. Ivosidenib, given at a dose of 500 mg/daily as a single agent for newly‐diagnosed AML with an IDH1 mutation, demonstrated CR and CRh rates of 28.6 and 14.3%, respectively. The rates of CR and CRh were 24.7 and 8.0% in relapsed or refractory IDH1 mutant AML. Specially, ivosidenib in combination with azacitidine has been approved for newly diagnosed AML with an IDH1 mutation. The phase III AGILE trial for newly diagnosed IDH1‐mutated AML ineligible for intensive induction chemotherapy showed the combination of ivosidenib and azacitidine was superior to placebo and azacitidine in terms of EFS (HR, 0.33; p = 0.002) and OS (median OS: 24.0 vs. 7.9 months; HR, 0.44; 95%CI: 0.27, 0.73; p = 0.001). CR was 47% in the ivosidenib plus azacitidine arm and 15% in the placebo plus azacitidine arm. The other indication of ivosidenib is for the treatment of locally advanced or metastatic, chemotherapy refractory cholangiocarcinoma with an IDH1 mutation based on data from the phase III study AG120‐C‐005 (ClarIDHy). Ivosidenib at a dose of 500mg daily significantly improved median PFS (HR, 0.37; 95%CI: 0.25, 0.54; p < 0.0001) and resulted in a favorable OS benefit (median OS: 10.3 vs. 7.5 months; HR, 0.79; 95%CI: 0.56, 1.12; one‐sided p = 0.09) compared with placebo. 450 , 451 In addition, phase I studies of ivosidenib also showed clinical benefits in IDH1‐mutated advanced glioma and advanced mutant IDH1 chondrosarcoma. Ivosidenib is trying to expand to solid and other hematological tumors with an IDH1 mutation, such as glioma, pancreatic adenocarcinoma, and myedysplastic syndrome (NCT02073994, NCT05209074, and NCT03839771). Two phase II studies (NCT04056910 and NCT04044209) are evaluating the possibility of combining ivosidenib with PD‐1 inhibitor nivolumab for IDH1‐mutated tumors.
Research on drug sensitivity and resistance found several factors were associated with the IDH inhibitors. First, cooccurring RAS pathway mutations decreased the possibility of responding to ivosidenib monotherapy. Interestingly, patients with a JAK2 mutation achieved a higher percentage of a CR or CRh. 452 Second, approximately 30% of patients had mutations in transcription factors and/or chromatin regulators, such as RUNX1, CEBPA, GATA2, DNMT3A, and ASXL1, which were associated with acquired relapse. 453 Third, second‐site mutations in IDH1 (i.e., D279N and S280F) and IDH2 (i.e., Q316E and I318M) interfere with the binding of the IDH inhibitors to the enzymes attributed to the therapeutic resistance. 444
Additional small molecule IDH inhibitors are under various preclinical and clinical development stages. IDH‐305, BAY‐1436032, and DS‐1001b belong to IDH1 inhibitors. In the phase I trial (NCT02381886) for IDH1‐R132‐mutant AML and MDS, IDH‐305 exhibited a narrow therapeutic window, and the following trials were hindered. 454 Phase I studies (NCT02746081 and NCT03127735) of BAY‐1436032 for IDH1‐mutant solid tumors and AML demonstrated it was well tolerated and showed durable ORR in lower grade glioma. 455 But the low ORR did not support further clinical development of this agent in AML. 456 DS‐1001b showed antitumor activity toward recurrent gliomas and chondrosarcoma in xenograft models, and clinical trials are ongoing (NCT04458272 and NCT03030066). 457 , 458 IDH2 inhibitor AGI‐6780 has not yet advanced to the clinical stage and was demonstrated to induce cellular differentiation in leukemia cells. 459 Vorasidenib is a dual IDH1/IDH2 inhibitor and showed preliminary antitumor activity in recurrent or progressive nonenhancing lower grade gliomas with good tolerance (NCT02481154). 460 Phase I study (NCT05484622) of vorasidenib and pembrolizumab combination in recurrent or progressive enhancing IDH‐1 mutant astrocytomas is ready to recruit.
3.18. Src tyrosine inhibitor
Nine structurally similar nonreceptor protein TKs (Src, Fyn, Lyn, Yes, Blk, Lck, Fgr, and Yrk) constitute the Src family kinases, which interact with multiple tyrosine kinase, growth factor, integrin, and G‐protein‐coupled receptors and participate in pathways promoting cell proliferation and survival. 461 Src is not a primary driver of tumorigenesis and mutations in Src are very rare. Thus, Src inhibitors may exert an auxiliary role in cancer treatments with limiting efficacy when used alone. 462 , 463
The selective Src inhibitor was not approved until 2 years ago for patients with actinic keratosis, a precancerous lesion of invasive cutaneous squamous cell carcinoma (iCSCC). Actinic keratosis may progress to iCSCC at a rate of 0.025–16% per lesion per year. Because of its unpredictable nature of progression, treatment is recommended. Cryosurgery is a common method for individual actinic keratosis lesions. Photodynamic therapy and topical agents (fluorouracil, diclofenac, imiquimod, and ingenol mebutate) are recommended for multiple lesions and surrounding solar‐damaged skin. However, local reactions or long‐term medication may reduce adherence and undermine treatment success. Hence, there is a need to find other methods for actinic keratosis. In 2020, tirbanibulin, a Src‐specific kinase and tubulin polymerization inhibitor, was approved for the topical treatment of actinic keratosis based on the data from two phase III trials. 464 In trial 1, Src inhibition induced a significant difference in the rates of complete clearance between the tirbanibulin and vehicle arm (44 vs. 5%) after 5 consecutive days of treatment when evaluating at Day 57. The percentages were 54 and 13%, respectively, in trial 2. However, the rate of recurrence of lesions at 1 year was 47% among patients who had a complete response to tirbanibulin. The most common local reactions were erythema and flaking or scaling. But it is safe to use tirbanibulin, as severe local reactions were infrequent among tirbanibulin‐treated patients. 465 Taken together, the Src inhibitor tirbanibulin is of mild efficacy, low toxicity, and short‐term medication for treating actinic keratosis.
Combined with other agents to increase other agents’ efficacy or avoid resistance caused by the principal agents, maybe a future for selective Src inhibitors and some studies have shown the possibilities in preclinical models. Saracatinib is a selective Src inhibitor, and it was well tolerated in patients with advanced solid malignancies in the phase I trial. In the follow‐up single‐drug studies, no good results were obtained. 466 In ER‐positive ovarian cancer models, combined Src and ER blockade by saracatinib and fulvestrant inhibited ovarian cancer xenograft growth more effectively than monotherapy. 467 Dual Src and MEK inhibition in ovarian cancer models addressed bypass activation. 468 Attenuating Src kinase activity could increase the efficacy of poly‐ADP‐ribose polymerase (PARP) inhibitors in BRCA2‐altered prostate cancer cells. 469 However, it failed to improve the activity of weekly paclitaxel in platinum‐resistant ovarian cancer and increase the efficacy of a VEGF‐targeted therapy in patients with relapsed metastatic clear RCC in clinical settings. 470 , 471 More clinical trials are needed to verify the possibility of combination.
Selective small molecule kinase inhibitors are supposed to inhibit a single protein kinase, but they usually bind to a few structurally related kinases. 10 This feature brings two obvious clinical benefits, enhancing their efficacy for particular cancers and indicating for cancers harboring different molecular alterations. 6 For example, targeting both CDK4 and CDK6 by palbociclib, ribociclib, abemaciclib, and trilaciclib is believed to increase the effectiveness in treating breast cancer. 397 In addition, entrectinib, which is a TRK receptor kinase inhibitor indicated for NTRK fusion‐positive tumors, is also a potent inhibitor of ROS1, enabling it to be approved for ROS1‐positive NSCLC. 193 , 194 , 472 On the other hand, off‐target toxicity deserves attention. 1 As mentioned above, serious cardiovascular side effects induced by poor selectivity of first‐generation BTK inhibitor ibrutinib could be reduced by second‐generation BTK inhibitor acalabrutinib or zanubrutinib because of their high selectivity to BTK. 430
4. SELECTIVE SMALL MOLECULE NONKINASE INHIBITORS
Selective small molecule nonkinase inhibitors bind to targets beyond the kinome, thereby blocking subsequent functions to control tumors. 10 This part will first introduce the molecular targets involved in the nucleus, whose mechanisms are gene transcription, DNA repair, epigenetic modification, and nuclear protein exportation. Next, receptor and intracellular signaling inhibitors will be described. Finally, you can find agents involved in triggering apoptosis (Table 2 and Table S2).
TABLE 2.
Summary of approved small molecule nonkinase inhibitors
| Class | Drug name | Company | First approval | Targets | Administration pathway | Indications |
|---|---|---|---|---|---|---|
| HIF | Belzutifan (Welireg) | Merck | 2021 | HIF | Oral | VHL‐associated RCC, CNS hemangioblastomas and pNET |
| PARP | Olaparib (Lynparza) | AstraZeneca | 2014 | PARP1, PARP2, PARP3 | Oral | Ovarian, fallopian tube or primary peritoneal cancer, breast cancer, pancreatic cancer, prostate cancer |
| PARP | Rucaparib (Rubraca) | Clovis Oncology | 2016 | PARP1, PARP2, PARP3 | Oral | Ovarian cancer, prostate cancer |
| PARP | Niraparib (Zejula) | GlaxoSmithkline | 2017 | PARP1, PARP2 | Oral | Ovarian cancer |
| PARP | Talazoparib (Talzenna) | Pfizer | 2018 | PARP1, PARP2 | Oral | Breast cancer |
| DNMT | Azacitidine (Vidaza) | Celgene | 2004 | DNMT | Subcutaneous or intravenous | Myelodysplastic syndromes, AML, JMML |
| DNMT | Decitabine (Dacogen) | Otsuka | 2006 | DNMT | Intravenous | Myelodysplastic syndromes |
| HDAC | Vorinostat (Zolinza) | Merck | 2006 | HDAC1, HDAC2, HDAC3, HDAC6 | Oral | cutaneous T‐cell lymphoma |
| HDAC | Romidepsin (Istodax) | Celgene | 2010 | HDACs | Intravenous | Cutaneous T‐cell lymphoma |
| HDAC | Belinostat (Beleodaq) | Acrotech | 2014 | HDACs | Intravenous | Peripheral T‐cell lymphoma |
| HDAC | Panobinostat (Farydak) | Secura | 2015 | HDACs | Oral | Multiple myeloma |
| XPO1 | Selinexor (Xpovio) | Karyopharm Theraps | 2019 | XPO1 | Oral | Multiple myeloma, diffuse large B‐cell lymphoma |
| CXCR4 | Plerixafor (Mozobil) | Dr Reddys Labs | 2008 | CXCR4 | Subcutaneous | Autologous transplantation for NHL and MM |
| KRASG12C | Sotorasib (Lumakras) | Amgen | 2021 | KRASG12C | Oral | NSCLC with KRAS G12C‐mutation |
| mTOR | Temsirolimus (Torisel) | PF Prism CV | 2007 | mTOR | Intravenous | RCC |
| mTOR | Everolimus (Afinitor) | Novartis | 2009 | mTOR | Oral | HR‐positive, HER2‐negative breast cancer, neuroendocrine tumors, RCC, |
| mTOR | Sirolimus (albumin‐bound) (Fyarro) | DR Reddys Labs | 2021 | mTOR | Intravenous | Perivascular epithelioid cell tumor |
| SMO | Vismodegib (Erivedge) | GenenTech | 2012 | SMO | Oral | (Locally advanced)/metastatic basal cell carcinoma |
| SMO | Sonidegib (Odomzo) | Sun Pharm | 2015 | SMO | Oral | Local advanced basal cell carcinoma |
| SMO | Glasdegib (Daurismo) | Pfizer | 2018 | SMO | Oral | AML |
| BCL‐2 | Venetoclax (Venclexta) | Abbvie | 2016 | BCL‐2 | Oral | CLL/SLL, AML |
Abbreviations: RCC, renal cell carcinoma; pNET, pancreatic neuroendocrine tumor; NHL, non‐Hodgkin's lymphoma; MM, multiple myeloma; AML, acute myeloid leukemia; CLL/SLL, chronic lymphocytic leukemia/small lymphocytic lymphoma; JMML, juvenile myelomonocytic leukemia. Data sources: https://www.fda.gov/drugs/development‐approval‐process‐drugs/drug‐approvals‐and‐databases.
4.1. Hypoxia‐inducible factor 1α inhibitors
Hypoxia‐inducible factor 1α (HIF‐1α) and HIF‐2α trigger HIF‐1 transcription to promote induction of various proangiogenic genes, such as VEGF, VEGFR, EGF, ANGPT, Tie‐2, TIMP‐1, and PAI‐1, and thereby play critical roles in tumor proliferation, apoptosis, metabolism, and metastasis in response to low‐oxygen concentrations in the TME, which can be hydrolysis by VHL protein. 473 HIF‐1 translation is also modulated by PI3K/AKT/mTOR, MAPK, JAK/STAT, NF‐Kβ, and Ca2+/CaM pathways in the cytoplasm. HIF‐1 accumulation in VHL‐associated tumors is a therapeutic target in cancer therapy. 474 , 475
Four strategies can be used for the interference of HIF‐1 accumulation. Direct inhibition of HIF‐1 transcription by HIF‐1α or HIF‐2α inhibitor is an important method. Blocking HIF‐1 translation or signaling pathways associated with HIF‐1 collection and enhancing HIF‐1 degradation can also circumvent the roles of HIF‐1. 474 In 2021, the HIF‐2α inhibitor belzutifan was approved as monotherapy for patients with VHL‐associated RCC, CNS hemangioblastomas, and pancreatic neuroendocrine tumor (pNET) based on the basket phase II trial (NCT03401788) showing an ORR of 49, 63, and 83%, respectively. The most common adverse events were anemia and fatigue. Meanwhile, embryo‐fetal toxicity deserves attention. 476
Clinical studies of belzutifan (as monotherapy or combination therapy) for particular tumor types are ongoing, especially for RCC and pNET. Among them, 12 out of all 18 trials are for RCC. A phase III study (NCT04195750) is ready to recruit 736 participants with RCC to compare belzutifan with everolimus. Combination of belzutifan with PD‐1 inhibitor and/or multikinase inhibitor (lenvatinib or cabozantinib) is a trend to increase efficacy and avoid resistance and is under estimation in several phase II or III trials (NCT05239728, NCT04976634, and NCT03634540).
The factors affecting the above four aspects of HIF‐1 may impact HIF‐1 accumulation. There are also HIF‐1α inhibitors under study, such as IDF‐11774 and PX‐478. IDF‐11774 is a HIF‐1α inhibitor that suppresses HSP70 chaperone activity to disrupt HIF‐1α refolding and stimulate HIF‐1α degradation. It showed antitumor activity on melanoma, prostate cancer, and thyroid cancer in vitro and/or in vivo. 477 , 478 PX‐478 is also an inhibitor of constitutive and HIF‐1α levels and thus HIF‐1 activity. It showed antitumor activity in various cancer types and could synergize with anti‐PD‐1 to impair tumor growth in preclinical trials. 479 , 480 A phase I trial (NCT00522652) of this agent for solid tumors has been completed, but the results have not been published.
4.2. PARP inhibitors
PARP contributes to DNA damage repair (DDR) and the maintenance of genomic stability, which allow cancer cells to develop resistance to radiation and DNA damaging chemotherapeutics. 481 BRCA1/2 and ATM are tumor suppressors that are commonly mutated or dysregulated in breast or ovarian cancers. Tumors with BRCA, ATM, or other mutations have a deficiency in homologous recombination, which repairs the DNA double‐strand breaks, DNA and protein‐DNA cross‐links, and collapses replication forks, and thereby induces genomic instability. PARP protein is overexpressed in various types of cancers, such as breast, ovarian, and oral cancers compared with their normal surrounding healthy tissues. PARP inhibitors are employed to disturb the DDR and stall DNA replication to trigger apoptosis in cancers (also called synthetic lethality), specifically for cancers with homologous recombination deficiency (HRD), which refers to tumor BRCA, ATM, or other mutant, or a genomic instability score ≥42. 482 , 483 Therefore, inhibition of PARP activity will become a promising strategy for cancer therapy. Four PARP inhibitors (olaparib, rucaparib, niraparib, and talazoparib) have been licensed to date for use in the treatment of ovarian, breast, pancreatic cancer, and prostate cancer. PARP inhibitors for ovarian and breast cancer are studied more than other tumors. 482
Three PARP inhibitors (olaparib, rucaparib, and niraparib) for ovarian cancer can be used as a single agent during maintenance therapy after response to first‐line platinum‐based chemotherapy or as monotherapy in the post‐line treatment. 484 , 485 , 486 , 487 In the maintenance setting, three pivotal phase III trials (SOLO‐2, NOVA, and ARIEL) demonstrated PFS was significantly prolonged in the PARP inhibitor group than in the placebo group (median PFS: 16.6–21.0 months vs. 5.4–5.5 months). Although BRCA mutations or HRD are not required for rucaparib and niraparib, subgroup analyses showed longer PFS in patients with these changes. 486 , 487 Monotherapy with rucaparib or niraparib is suitable for BRCA‐mutated or HRD‐positive ovarian cancer in post line therapy. The ORR ranged from 24% after 3 or more chemotherapies with niraparib to 54% after 2 or more chemotherapies with rucaparib. The main common adverse events of these drugs in ovarian cancer were nausea, fatigue, vomiting, and anemia. 488 , 489 , 490
In breast cancer, olaparib and talazoparib demonstrated significant clinical benefits over chemotherapy in PFS and ORR for germline BRCA‐mutated, HER2‐negative metastatic or locally advanced breast cancer based on the phase III OlympiAD and EMBRACA trials, respectively. Notably, grade ≥3 adverse events in the olaparib arm were fewer than in the chemotherapy arm (36.6 vs. 50.5%). Compared with chemotherapy, talazoparib brought a 17% increase in grade 3–4 hematologic adverse events and a 6% decrease in the rate of nonhematologic grade 3 events. 491 , 492 In addition, olaparib was also approved for the adjuvant treatment of patients with germline BRCA‐mutated, HER2‐negative metastatic breast cancer after chemotherapy on the basis of better iDFS and OS compared with placebo. 493
In addition, two PARP inhibitors, olaparib and rucaparib, received US FDA designation for metastatic castration‐resistant prostate cancer with homologous recombination repair gene mutation or BRCA mutation or ATM mutation. 494 , 495 The phase III PROfound trial recruited men with metastatic castration‐resistant prostate cancer who progressed on a hormonal agent, such as enzalutamide or abiraterone. Cohort A included the patients with at least one alteration in BRCA1, BRCA2, or ATM. In these individuals, olaparib was associated with longer PFS (median PFS: 7.4 vs. 3.6 months; HR, 0.34; 95%CI: 0.25, 0.7; p < 0.001) and OS (median OS: 19.1 vs. 14.7 months; HR, 0.69; 95%CI: 0.50, 0.97; p = 0.02) compared with enzalutamide or abiraterone. Anemia and nausea were the main toxic effects in patients who received olaparib. 496 The phase II single‐arm trial TRITON2 showed an ORR of 44%, with 59% of patients occurring grade 3–4 adverse events. 497 The phase III trial TRITON3 is ongoing to verify the clinical benefit of this agent (NCT02975934). In addition, olaparib was also approved for BRCA‐mutated pancreatic cancer in the maintenance setting after response to platinum‐based chemotherapy based on improved PFS and ORR compared with placebo. 498
The approved PARP inhibitors are developing in two main directions. First, the therapeutic scope of PARP inhibitors is expanding to other tumor types, most with BRCA or ATM mutations, such as endometrial, urothelial, SCLC, STS, mesothelioma, and gastric cancer. 499 , 500 Take mesothelioma as an example. A phase II study (NCT03654833) yielded that the rate of patients with BAP1‐negative or BRCA1‐negative heavily treated mesothelioma who received disease control from rucaparib treatment at 12 weeks was 58%, and the ratio was 23% at 24 weeks. 501 Second, combination with other agents is a promising method to increase efficacy. Chemotherapy, other targeted agents, and ICIs are the three major categories of combination partners. Phase II study (NCT01063517) of olaparib plus paclitaxel could improve OS compared with placebo plus paclitaxel in recurrent or metastatic gastric cancer, with a greater OS benefit in ATM low patients. 502 Combined with VEGFR inhibitors is a deserving attempt. Phase III trial (NCT02477644) of adding olaparib to bevacizumab as first‐line maintenance for HRD‐positive ovarian cancer provided a significant PFS survival compared with bevacizumab alone. 503 ICIs are another worthy companion. In the phase I/II MEDIOLA trial, the combination of olaparib with PD‐L1 inhibitor durvalumab showed promising antitumor activity and safety profile in patients with germline BRCA‐mutated metastatic breast cancer. 504 Besides, a phase Ib/II trial (NCT03404960) demonstrated that 6‐month PFS of niraparib plus CTLA‐4 antibody ipilimumab was superior to that of niraparib plus PD‐1 antibody nivolumab for platinum‐sensitive advanced pancreatic cancer. 505
New PARP inhibitors are ongoing in different clinical stages, such as, pamiparib, fluzoparib, iniparib, veliparib, and stenoparib. 482 Among them, pamiparib and fluzoparib are the most representative, and they are all licensed in China. Pamiparib displays excellent PARP‐1 and PARP‐2 inhibition with IC50 of 1.3 and 0.9 nM, respectively. It is approved for the treatment of germline BRCA‐mutant recurrent advanced ovarian cancer based on a phase I/II trial (NCT03333915). This agent showed an ORR of 68.3% in the platinum‐sensitive cohort and 31.6% in the platinum‐resistant cohort. 506 Phase III maintenance treatment trial (NCT03519230) with pamiparib versus placebo for platinum‐sensitive recurrent ovarian cancer is ongoing. Fluzoparib is a PARP‐1 inhibitor with an IC50 of 1.46 nM. The indication of fluzoparib is the same as pamiparib. The ORR reached 69.9%, with 4.4% of patients achieving a CR and 65.5% with a PR. 507 Besides, the recently published phase III FZOCUS‐2 trial (NCT03863860) supported fluzoparib as maintenance therapy in patients with platinum‐sensitive, recurrent ovarian carcinoma, regardless of germline BRCA 1/2 mutation. 508
Two other questions about PARP inhibitors are also important. First, the factors associated with the sensitivity of PARP inhibitors are studied extensively. As mentioned above, patients whose tumors harbor BRCA or ATM or other mutations or HRD are likely to respond to PARP inhibition. An HRD phenotype is associated with genomic scars and mutational signatures, which can be used for defining broader populations that may benefit from DDR‐targeted drugs. Moreover, there is a strong association between HRD and ovarian cancer platinum sensitivity. Therefore, platinum sensitivity functions as a surrogate marker for HRD. 483 Second, ways to address resistance are under research. After the initial response to PARP inhibitors, tumors often develop drug resistance by developing compensatory mechanisms or restoring HR function that allows the cancer cell to repair the damage and proliferate. Combination therapy may address the bypassing activated signaling. Approximately 450 proteins that sense, signal, and/or repair DNA damage are involved in the DDR, such as MLH1, CHK1/2, WEE1, and RAD51. Therefore, inhibition of other proteins in the DDR may overcome resistance caused by PARP inhibitors. 509
4.3. Epigenetic inhibitors
Tumor cells are not only activated by genetic alterations but also often use epigenetic processes to ensure their survival despite of various interference. Since epigenetic pathways exhibit greater flexibility of several orders of magnitude relative to genetic alterations, epigenetic alterations are receiving increasing attention. 510 Epigenetics defines the regulation of chromatin structure and gene expression through modifying histone proteins and nucleic acids that regulate chromatin structure without affecting the underlying DNA sequence. 511 Dysregulation of the epigenome drives aberrant transcriptional programs that promote cancer onset and progression, allowing for suppression of tumor suppressor genes and the increased expression of oncogenes, which leads to the development of targeted epigenetic drugs for the treatment of cancer. 512 , 513 Although epigenetic therapy has taken a long time to be accepted and proved effective in treating blood and solid tumors, recent drug discovery efforts have increasingly focused on DNA and histone modification. The DNA methyltransferase (DNMT) and histone deacetylase (HDAC) inhibitors are the most clinically advanced epigenetic therapies. 514 , 515
4.3.1. DNMT inhibitors
DNA methylation is one of the best‐described epigenetic events that controls gene expression and is dysregulated in diseases like cancer. 516 Regulation of DNA methylation depends on the family of DNMTs, including DNMT1, DNMT2, and DNMT3. DNMTs are a class of enzymes that transfer methyl groups from S‐adenosylmethionine to cytosine bases of CpG dinucleotides in gene promoters and regulatory regions. 517 CpG promoter islands can be methylated during development, leading to long‐term gene silencing. DNA hypermethylation is also associated with silencing tumor suppressor genes and differentiation genes in various cancers. 518 DNMT inhibitors, commonly known as hypomethylating agents, are the most widely used epigenetic therapies for cancer treatment. The DNMT inhibitors are mainly divided into two categories: (i) cytosine analog inhibitors and (ii) non‐nucleotide analog inhibitors. 519 As a classical anticancer drug, DNMT inhibitors have been in clinical trials for more than 40 years with little effect. However, in the past 20 years, they have been revitalized due to the discovery of their mechanism of action. 520 DNMT inhibitors mainly exert anticancer activity through the following mechanisms: (I) hypomethylation, (II) induction of double stranded DNA breaks, (III) cell cycle or G2 phase arrest, and (IV) stimulation of immune signals. 521 To date, the US FDA approved two DNMT inhibitors, namely azacytidine and decitabine. 522
Azacytidine is the first approved DNMT inhibitor for MDS, AML, and juvenile myelomonocytic leukemia (JMML). The phase III cancer and leukemia group B trial for higher‐risk MDS showed azacytidine treatment enhanced treatment response (60 vs. 5%, p < 0.001), delayed time to transformation to AML or death (21 vs. 13 months, p = 0.007), and improved quality of life compared with supportive care. 523 , 524 Moreover, OS was increased with azacitidine treatment relative to conventional care in another phase III trial (NCT00071799). 525 In the following studies, it was found that treatment with azacitidine could substantially delay hematological relapse in measurable residual disease‐positive patients with MDS or AML who were at high risk of relapse. 526 Patients with lower‐risk MDS could also benefit from azacitidine. The results of a phase III trial (NCT01566695) showed azacitidine significantly improved RBC transfusion independence and induced durable bilineage improvements. In addition to MDS, many efforts have been devoted to treating AML. Azacitidine was granted approval for maintenance therapy of AML ineligible for complete intensive curative therapy in 2020 based on the phase III QUAZAR AML‐001 trial. Azacitidine treatment favored in OS (median OS: 24.7 vs. 14.8 months; p < 0.001) and RFS (10.2 vs. 4.8 months; p < 0.001) compared with placebo. 527 Azacitidine for elderly patients’ AML significantly prolongs OS (median OS: 24.5 vs. 16.0 months; HR, 0.47; 95%CI: 0.28, 0.79; p = 0.005) compared with conventional care regimes. 528 Besides, azacitidine in combination with BCL‐2 inhibitor venetoclax or IDH inhibitor ivosidenib has been approved for AML, which has been discussed in other parts of this review. 529 Failure also exists in the development process. TP53 inhibitor eprenetapopt combined with azacytidine for TP53‐mutated MDS and AML was reported to be safe and superior to azacytidine alone in the form of ORR, CR rate, and OS in a phase II trial (NCT03588078). However, it failed in the phase III trial. 530 The newest indication of azacitidine is monotherapy before allo‐HSCT in pediatric patients with newly diagnosed JMML. The data from the phase II AZA‐JMML‐001 trial showed azacitidine provided clinical benefit to JMML patients prior to HSCT in terms of clinical CR or PR. 531
Similarly, decitabine was licensed for MDS based on several studies demonstrating an ORR of 49% in elderly patients with high‐risk MDS, reduction of AML transformation, and improvements in patient‐reported quality of life. 532 Besides, as the medication of decitabine via intravenous is inconvenient, the phase II ADOPT trial provided an alternative dosing schedule that can be administered in an outpatient setting with comparable efficacy and safety. 533 Decitabine for the treatment of AML has been researched extensively. A phase II study of decitabine investigated the efficacy and toxicity of decitabine as initial therapy in older patients with AML showing an ORR of 25%, median OS of 7.7 months, and 7% of 30‐day mortality. 534 A phase III trial compared decitabine with treatment choice in older patients with newly diagnosed AML and showed an improvement in response rates with decitabine compared with standard therapies (17.8 vs. 7.8%; HR, 2.5; 95%CI: 1.4, 4.8; p = 0.001). Moreover, a phase II study (ChiCTR‐IIR‐16008182) found that rhG‐CSF combined with minimal‐dose decitabine maintenance after allo‐HSCT can reduce the incidence of relapse in patients with high‐risk AML. 535 Decitabine has also been studied in solid tumors, but only refined in phase I stage. 536
Several aspects concerned with DNMT inhibitors deserve attention, including toxicity, efficacy, and combination therapy. First, the most common grade 3–4 adverse event was cytopenia, including neutropenia and thrombocytopenia. Clinically, there are corresponding drugs for myelosuppression, so it is not difficult to address. Second, several factors may affect the efficacy of DNMT inhibitors. Unfavorable‐risk cytogenetic abnormalities and TP53 mutations were correlated with favorable clinical response and robust (but incomplete) mutation clearance in AML and MDS treated by decitabine. 537 We also observed that poor efficacy was seen in cases of solid tumors, primarily as single agents, including the following reasons: (i) the function of DNMT inhibitors depends upon DNA incorporation, while solid tumors divide relatively slowly; (ii) lower stability of DNMT inhibitors. Third, research on the combination of DNMT inhibitor with PD‐1 inhibitor provides a novel strategy for cancer treatment. DNMT inhibitors could upregulate PD‐1 and IFNγ signaling. Phase II trial showed azacitidine plus nivolumab brought an ORR of 33% for relapsed or refractory AML, which was 58 and 22% for hypomethylating agent‐naïve and hypomethylating‐pretreated patients, respectively. 538 The addition of decitabine to PD‐1 antibody camrelizumab in patients with relapsed or refractory classical Hodgkin lymphoma who were clinically naïve to PD‐1 antibody received a higher CR rate than camrelizumab alone (71 vs. 32%; p = 0.003). 539
Many other DNMT inhibitors are under preclinical or different clinical stages, such as guadecitabine, zebularine, and CP‐4200. The latter two drugs are in the preclinical phase. 514 Specifically, guadecitabine is a decitabine analog and showed antitumor activity toward MDS, AML, peripheral T‐cell lymphoma, urothelial bladder carcinoma, ovarian cancer, and melanoma in clinical settings. 511 The safety profile and efficacy of guadecitabine were initially demonstrated in MDS and AML (NCT01261312). 540 , 541 Even phase III trial (NCT02348489) for AML has been completed. Combination strategies broaden the use scope. Guadecitabine, in combination with ICI pembrolizumab or ipilimumab, resulted in a durable clinical benefit and reversed previous resistance to ICIs in solid tumors (NCT02998567). 542 , 543 Guadecitabine combined with standard chemotherapy enhanced antitumor immunity in platinum‐resistant ovarian cancer, urothelial carcinoma, and germ cell cancer. 544 , 545
4.3.2. HDAC inhibitors
Proteins of the histone family are conserved alkaline proteins in eukaryotes and play pivotal roles in DNA packaging and gene regulation. Histone acetylation is associated with active transcription and is mainly localized at enhancer regions, promoters, and the gene body. 546 The positively charged histone is neutralized after adding a negatively charged acetyl group to a histone tail, which further weakens the electrostatic interaction between histones and the negatively charged DNA, resulting in an open conformation of chromatin that is more accommodating to transcription factors. HDAC is an enzyme that inhibits the process of histone acetylation. 547 At least 18 human HDACs have been identified, with varying function, localization, and substrates, and further classified into four classes of HDACs, namely class I, II, III, and IV. Classes I, II, and IV contain a zinc molecule in their active site and are inhibited by HDAC inhibitors. In the contrary, class III HDACs do not have a zinc molecule and are not hindered by any current HDAC inhibitors. 514 Overexpression of HDAC is found in various cancers associated with the invasiveness and migration of cancer and is an indicator of poor prognosis. 548 Inhibition of HDAC can reverse the process of histone acetylation and induce open chromatin conformation at tumor suppressor gene loci and thereby inducing tumor suppression through cell‐cycle arrest, apoptosis, and chemo/radio‐sensitization. 549
To date, four HDAC inhibitors (vorinostat, romidepsin, belinostat, and panobinostat) have been approved to treat hematological malignancies. 550 Vorinostat and romidepsin received US FDA approval for patients with cutaneous T‐cell lymphoma after two or one systemic therapy, respectively. The phase IIb trial (NCT00091559) showed the vorinostat‐related ORR was 29.5%, median TTP was 4.9 months, and 32% of patients had pruritus relief. The most common adverse events were diarrhea, fatigue, nausea, and anorexia. 551 Romidepsin was approved based on a phase II trial, which demonstrated that the ORR of romidepsin was 34% and the median DOR was 13.7 months with pruritus improvement in 43% of patients. Drug‐related adverse events were generally mild, including nausea, vomiting, fatigue, transient thrombocytopenia, and granulocytopenia. 552 However, adding romidepsin to CHOP (the standard first‐line treatment of peripheral T‐cell lymphoma) as first‐line treatment for cutaneous T‐cell lymphoma did not improve PFS, response rates, and OS but increased adverse events. 553 Belinostat brought an ORR of 25.8% to patients with relapsed or refractory peripheral T‐cell lymphoma. The median DOR, PFS, and OS were 13.6, 1.6, and 7.9 months, respectively. The most common grade 3–4 adverse events were anemia, thrombocytopenia, dyspnea, and neutropenia. 554 Based on the above data from the phase II study, the US FDA approved belinostat for relapsed or refractory peripheral T‐cell lymphoma. Finally, panobinostat is a potent oral pan‐deacetylase inhibitor and was licensed for multiple myeloma in combination with bortezomib and dexamethasone. The phase III PANORAMA1 trial showed that the addition of panobinostat to bortezomib and dexamethasone prolonged the median PFS from 8.08 months in the placebo, bortezomib, and dexamethasone arm to 11.99 months in the panobinostat, bortezomib, and dexamethasone arm (HR, 0.63; 95%CI 0.52, 0.76; p < 0.0001) when treating patients with relapsed or refractory multiple myeloma. 555
HDAC inhibitors as a single agent or a part of combination regimes have been explored in other solid and hematological tumors, such as NSCLC, gastrointestinal carcinoma, malignant pleural mesothelioma, neuroblastoma, AML, MDS, and so on. 514 High‐quality published research mainly revolves around the first approved selective HDAC inhibitor, vorinostat. The efficacy of monotherapy with HDAC inhibitor vorinostat was assessed in relapsed or refractory indolent NHL and MCL in a phase II study (NCT00253630). Only patients with FL and MZL responded to vorinostat, with no formal responders among patients with MCL. 556 Monotherapy with vorinostat given as a second‐line or third‐line treatment also failed in the phase III VANTAGE‐014 trial for advanced malignant pleural mesothelioma. Vorinostat treatment did not prolong OS compared with placebo (median OS: 30.7 vs. 27.1 weeks; HR, 0.98; 95%CI: 0.83, 1.17; p = 0.86). 557 Considering the limitation of vorinostat monotherapy, vorinostat combined with other agents is widely studied. First, adding vorinostat to standard chemotherapy has received positive outcomes in treating multiple myeloma, AML, MDS, and NSLCL. The phase III VANTAGE 088 trial showed that the combination of vorinostat and bortezomib for relapsed or refractory multiple myeloma prolonged PFS relative to bortezomib plus placebo (median PFS: 7.63 vs. 6.83 months; HR, 0.77; 95%CI: 0.64, 0.94; p = 0.01). 558 The ORR in the phase II trial of vorinostat with idarubicin and cytarabine for newly diagnosed AML or MDS reached 85%. 559 Vorinostat enhanced the efficacy of carboplatin plus paclitaxel in first‐line therapy of advanced NSLCL in confirmed response rate (34 vs. 12.5%). 560 Second, vorinostat improves sensitivity to radiotherapy. In a phase I trial (NCT00455351) for gastrointestinal carcinoma, vorinostat combined with pelvic palliative radiotherapy was safe. 561 As shown in a phase II trial (NCT02035137), the response rate of vorinostat plus 131I‐metaiodobenzylguanidine for patients with relapsed or refractory neuroblastoma was 32% and the value was 14% in the 131I‐metaiodobenzylguanidine arm. 562 Third, HDAC inhibitor combined with ICI is a trend. The studies of pembrolizumab with vorinostat in phase I/Ib study for advanced renal or urothelial cell carcinoma and in a phase II study for progressive advanced mucosal cancer are ongoing (NCT02619253 and NCT04357873). In addition, the combination of HDAC inhibitor and targeted agent of other mechanisms is investigated. Phase I/II trial (NCT01266031) of bevacizumab versus bevacizumab plus vorinostat in adults with recurrent glioblastoma has been completed. The efficacy of DNMT inhibitor plus HDAC inhibitor is not certain. A phase I/II trial (NCT00387465) of combining epigenetic therapy with azacitidine and entinostat resulted in objective and durable responses in patients with heavily pretreated NSCLC. 563 However, two phase II trials (NCT00313586 and NCT01522976) showed that adding entinostat to azacitidine did not increase clinical response for patients with MDS or AML with myelodysplasia‐related changes. Further studies are needed to determine the feasibility of combination therapy. 564 , 565
Many HDAC inhibitors are underdeveloped, including, but not limited to, pracinostat, quisinostat, dacinostat, AR‐42, mocetinostat, and citarinostat. Pracinostat is a pan‐HDAC inhibitor and has been evaluated in the phase II clinical stage for various of solid and hematological tumors, including MDS, AML, prostate cancer, and sarcoma (NCT01993641, NCT01912274, NCT01075308, and NCT01112384). 566 , 567 , 568 Dacinostat and quisinostat belong to the second‐generation and pan‐HDAC inhibitors for class I, II, and IV HDACs. 568 , 569 Preclinical evaluation of dacinostat verified an improved efficacy compared with other HDAC inhibitors. Another pan‐HDAC inhibitor, quisinostat, proved its safety and tolerability in phase I trial (NCT00677105) for solid tumors and showed efficacy and safety in patients with previously treated cutaneous T‐cell lymphoma in a phase II trial (NCT01486277). 569 , 570 , 571 AR‐42 is also a pan‐HDAC inhibitor and has been investigated in phase I stage for multiple myeloma, T‐ and B‐cell lymphomas, AML, and solid tumors (NCT01129193 and NCT01798901). 572 , 573 Mocetinostat is a class I/IV HDAC inhibitor in the clinical phase II stage. 574 Citarinostat is a selective HDAC6 inhibitor, and a phase I trial for smoldering multiple myeloma is ongoing (NCT02886065). 575
4.4. XPO1 inhibitors
The nuclear export receptor XPO1, also known as CRM1, with a pleiotropic role in transporting a plethora of proteins and RNA species, including rRNAs, snRNAs, mRNA, microRNAs, and tRNAs, is in charge of the nuclear export of tumor suppressor protein and growth regulators from the nucleus to the cytoplasm. 576 XPO1 functions together with RAN GTPase, which provides the energy for transport and ensures the directionality of nuclear export. XPO1 is involved in the export of nearly 220 proteins. Among these proteins, several tumor suppressors, including p53, BRCA1/2, and p27, have been extensively studied. 577 It is overexpressed in multiple cancers, including pancreatic, gastric, prostate, and CRC, and such overexpression is associated with disease progression, treatment resistance, and inferior PFS and/or OS. 578 Overexpression of XPO1 in various cancers can lead to decreased tumor suppressors and aberrant cytoplasmic localization. Nuclear export blockade of tumor suppressor proteins has been postulated as the primary mechanism of action for XPO1 inhibitors. 579
The XPO1 inhibitor selinexor covalently and reversibly interacts with Cys528 in the nuclear export signal‐binding pocket of XPO1. Therefore, it restores tumor suppressor function and eventually causes tumor cell apoptosis. 580 Selinexor was initially proved to be of safety and preliminary antitumor activity in three critical phase I trials for advanced solid tumors, bone or STS, and relapsed or refractory acute leukemia (NCT01607905, NCT01896505, and NCT02212561). The most common adverse events were nausea, vomiting, anorexia, and fatigue. Grade 3–4 toxicities were fatigue, thrombocytopenia, anemia, lymphopenia, and leukopenia. 580 , 581 , 582 Subsequently, many large clinical trials have been launched, and some of their results have been published. In 2019, the phase II STORM trial of selinexor in combination with dexamethasone for patients with triple‐class refractory multiple myeloma showed that a PR or better was observed in 26% of patients with a median DOR of 4.4 months. 583 Another phase III trial for multiple myeloma was the BOSTON trial. This trial recruited patients with previously treated multiple myeloma to receive once‐per‐week selinexor, bortezomib, and dexamethasone or twice‐per‐week bortezomib and dexamethasone. Median PFS was 13.93 months with selinexor, bortezomib, and dexamethasone and 9.46 months with bortezomib and dexamethasone (HR, 0.70; 95%CI: 0.53, 0.93, p = 0.0075). Grade 3–4 adverse events, including thrombocytopenia, fatigue, and anemia, were higher in the three‐drug combination. 584 Based on the outcomes of STORM and BOSTON trials, the US FDA granted selinexor for the treatment of multiple myeloma. Besides, monotherapy with selinexor is licensed in patients with relapsed or refractory diffuse large B‐cell lymphoma based on the phase II SADAL trial. In this study, single‐agent selinexor improved OS compared with historical data, with an ORR of 28%. 585 , 586 Phase III study (NCT04442022) for relapsed or refractory diffuse large B‐cell lymphoma is ongoing to evaluate the value of adding selinexor to R‐GDP.
Clinical trials of selinexor for dedifferentiated liposarcoma, recurrent glioblastoma, NHL, and AML also received positive results. Among them, the studies of dedifferentiated liposarcoma and recurrent glioblastoma have advanced to clinical phase II and/or phase III. 587 , 588 , 589 Notably, in the recent published phase II/III trial SEAL, patients with heavily treated dedifferentiated liposarcoma were assigned to receive selinexor or placebo. PFS with selinexor was longer than that with placebo (median PFS: 2.8 vs. 2.1 months; HR, 0.70; 95%CI: 0.52, 0.95; one‐sided p = 0.011). Besides, the absence of CALB1 predicted a longer PFS with selinexor compared with placebo (median PFS: 6.9 vs. 2.2 months; HR, 0.19; p = 0.001). Therefore, the expression of CALB1 may be used as a biomarker to choose suitable patients with dedifferentiated liposarcoma for the treatment of selinexor. 588 Besides, other predictive biomarkers have been investigated in different situations. Cytogenetically normal AML is characterized by mutated NPM1, the protein product of which results in abnormal cytoplasmic localization and hinders differentiation of AML cells. XPO1 inhibition relocalizes it to the nucleus and promotes the differentiation of AML cells. Therefore, XPO1 inhibitors may be effective against cytogenetically normal AML with NPM1 mutations. 590 In addition, overexpression of XPO1 or mutant (E571K or E571G) XPO1 can drive leukemogenesis and can be obstructed by the XPO1 inhibitor selinexor. 591 On the contrary, expression of mutant‐p53 in diffuse large B‐cell lymphoma confers resistance to selinexor treatment. 592
Eltanexor and verdinexor also belong to XPO1 inhibitors, the binding mode of which is similar to that of selinexor. 579 Etanexor is an investigational XPO1 inhibitor with low CNS penetrance and an acceptable tolerability profile, which yields antitumor activity in preclinical models of human multiple myeloma, ALL, AML, and gastric cancer. 593 , 594 , 595 , 596 A phase I/II study (NCT02649790) of the safety, tolerability, and efficacy of eltanexor in relapsed or refractory cancer is ongoing. MDS is one of the tumor types in the ongoing trial. The preliminary results were published in August 2022, showing that single‐agent etanexor met the presetting goals. 594 Verdinexor is another XPO1 inhibitor and shows antitumor efficacy against esophageal carcinoma and neuroblastoma in xenograft models. 597 , 598 Phase I trial (NCT02431364) is designed to evaluate the safety and tolerability of etanexor in healthy adults, but it has been terminated.
4.5. CXCR4 inhibitors
C‐X‐C‐motif chemokine receptor 4 (CXCR4) is a G‐protein‐coupled receptor involved in a number of physiological processes in the hematopoietic and immune systems and also has important roles in promoting tumor cell proliferation, metastasis, and angiogenesis. 599 , 600 CXCR4 is found to be a prognostic marker in many different cancers, including leukemia, breast, lung, prostate, ovarian and CRC, where the stromal cell‐derived factor‐1 (SDF‐1)/CXCR4 axis (also called CXCL12/CXCR4 axis) initiates divergent signaling pathways, including PI3K/AKT, MAPK, and JAK/STAT signals, which can result in a variety of responses such as chemotaxis, cell survival and/or proliferation, increase in intracellular calcium, and gene transcription. 601 , 602 In addition, the organs and tissues that possess high levels of SDF‐1, such as liver, lung, bone marrow, and lymph nodes, attract the migration of CXCR4‐expressing cancer cells. 603 CXCR4 is a promising target for imaging and therapy of both hematologic and solid tumors. 604
To date, none of the CXCR4 inhibitors are approved directly to target tumors. The CXCR4 inhibitor plerixafor is approved for patients with NHL or multiple myeloma to mobilize hematopoietic stem cells into the peripheral blood for autologous transplantation. 605 , 606 However, it has also been investigated in several tumor types. The CXCL12/CXCR4 axis can transactivate HER2 and promote intraosseous tumor growth in prostate cancer. Inhibition of the CXCL12/CXCR4 axis through the CXCR4 inhibitor plerixafor could abrogate the initial establishment of tumor growth in vivo. In other words, plerixafor can prevent bone metastasis in prostate cancer. 607 Besides, adding plerixafor to radio‐chemotherapy for cervical cancer improved primary tumor response and reduced lymph node metastases with no increase in toxicity in xenograft models. 608 In addition, the SDF‐1/CXCR4 axis may mediate resistance to VEGFR inhibition in recurrent high‐grade glioma. Preclinical data showed plerixafor could inhibit glioma progression after anti‐VEGF pathway inhibition. Other studies demonstrated that SDF‐1/CXCR4 was responsible for postirradiation tumor revascularization in glioblastoma. 609 , 610 To date, the phase I trial of plerixafor and bevacizumab showed the combination regime was well tolerated in high‐grade glioma. 610 Phase I/II of infusing plerixafor to standard chemoradiation improved local control of tumor recurrences in patients with newly diagnosed glioblastoma. 609 A clinical study of plerixafor is ongoing for pancreatic cancer (NCT04177810).
There are many other small molecule CXCR4 inhibitors for cancers in development, namely small peptide molecule and small nonpeptide molecule inhibitors. 611 LY2510924 is one of the peptide molecule inhibitors. It showed preclinical antitumor activity in multiple cancer types and proved safe in phase I trials (NCT02652871,). 612 However, two phase II studies (NCT01439568 and NCT01391130) for SCLC and RCC failed to deliver the additional role with LY2510924. 613 , 614 Mavorixafor belongs to a nonpeptide molecule inhibitor and is a selective, allosteric CXCR4 inhibitor with promising anticancer properties. Phase Ib trial (NCT02923531) of the combination of mavorixafor with nivolumab demonstrated potential antitumor activity and a manageable safety profile in patients with advanced RCC. 615 Phase I study (NCT04274738) mavorixafor in combination with ibrutinib in patients with WM harboring mutations in MYD88 and CXCR4 is ongoing. Some CXCR4 inhibitors are in preclinical stages, even in the fundamental experiments, such as WZ811 and AMD3465. 616 , 617
4.6. RAS inhibitors
The principal upstream factor of the MAPK pathway is RAS protein, which is a small, membrane‐bound guanine nucleotide‐binding GTPase and concludes four members, including HRAS, NRAS, KRAS4A, and KRAS4B. The KRAS4A and KRAS4B are two isoforms of KRAS. 279 About 19% of cancer patients have a RAS mutation, KRAS mutations accounting for the majority of RAS gene mutations (75%), followed by NRAS mutations (17%) and HRAS mutations (7%). 70% of RAS mutations are G12D, G12V, G12C, G13D, and Q61R. 618 KRAS G12C mutation happened in 13.8% of NSCLC. 619
Once, RAS protein was thought to be untargetable because of lacking binding pockets on its protein surface. 280 , 620 Luckily, the KRAS G12C mutant provides a cysteine residue for designing covalent inhibitors. Sotorasib is a selective and irreversible inhibitor of the KRAS G12C and showed antitumor activity against advanced solid tumors harboring the KRAS G12C mutation. 13 This agent obtained approval by the US FDA in 2021 for patients with KRAS G12C‐mutated NSCLC who have already received an ICI and/or platinum‐based chemotherapy. The phase II CodeBreak 100 trial showed that the ORR was 37.1% and the median DOR was 11.1 months without new safety signals. 621 , 622 Clinical trials involving neoadjuvant therapy and first‐line therapy with sotorasib, and combination therapy with sotorasib and other agents are underway (NCT05400577, NCT04933695, and NCT05074810).
There are other RAS inhibitors or agents of other mechanisms under development for targeting RAS mutant tumors. Adagrasib is the most promising agent. It is also a selective and irreversible KRAS G12C inhibitor. In the phase II study (NCT03785249) of this agent for patients with KRAS G12C mutant NSCLC previously treated with PD‐1/PD‐L1 antibody and platinum‐based chemotherapy, the ORR reached 42.9% and the median DOR was 8.5 months. Besides, the intracranial confirmed ORR was 33.3% and the safety profile was acceptable. 623 Phase III study (NCT04685135) of adagrasib for patients with KRAS G12C mutant NSCLC is ongoing. JDQ443 is a selective covalent inhibitor of KRAS G12C and forms novel interactions with the switch II pocket. 624 It showed potent antiproliferative activity in KRAS G12C‐mutated and KRAS G12C/H95 double mutated cell lines and is in clinical trials for KRAS G12C‐mutated tumors (NCT04699188 and NCT05132075). Prenylation of mutant RAS is the primary activator of downstream signaling. The predominant form of prenylation is farnesylation, which is unique in HRAS mutations. Tipifarnib is a highly potent and selective farnesyltransferase inhibitor that disrupts HRAS function. Phase II trial of this agent for head and neck squamous cell carcinoma showed an ORR of 55% in patients with high mutant HRAS variant allele frequency. Median PFS was 5.6 months and median OS was 15.4 months. The most frequent adverse events included anemia and lymphopenia (NCT02383927). 625 In addition, tipifarnib also resulted in modest clinical activity in HRAS‐mutant salivary gland cancer and urothelial carcinoma harboring HRAS mutations. 626 , 627
4.7. mTOR inhibitors
mTOR, a classical downstream effector of the PI3K pathway, is usually assembled in two complexes, mTOR complex 1 (mTORC1) and mTORC2, with different regulatory mechanisms. 322 It is hyperactivated in various solid tumors. Therefore, targeting mTOR in cancer treatment has become a promising strategy. 328 , 370
Rapamycin, also known as sirolimus, and its analogs (temsirolimus and everolimus) constitute the first generation of mTOR inhibitors, which bind to FKBP12 and preferentially inhibit mTORC1. 328 Both temsirolimus and everolimus have been approved to treat RCC. Temsirolimus improved OS (median OS: 10.9 vs. 7.3 months; HR, 0.73; 95%CI: 0.58, 0.92; p = 0.0078) and PFS (median PFS: 5.5 vs. 3.1 months; HR, 0.66; 95%CI: 0.53, 0.81; p = 0.0001) compared with interferon in previously untreated RCC. Everolimus was superior to placebo for PFS in metastatic RCC after treatment with sunitinib, sorafenib, or both. 628 , 629 Besides, the addition of everolimus to exemestane can overcome resistance due to constitutive activation of the PI3K/AKT/mTOR pathway after prior therapy with letrozole or anastrozole in HR‐positive, HER2‐negative breast cancer. 630 Due to low bioavailability and patent expiration, sirolimus in cancer treatment was hardly seen. 328 While in 2021, the sirolimus protein‐bound particle was approved for malignant perivascular epithelioid cell tumor based on the phase II AMPECT trial showing an ORR of 39%. 631 But both the on‐target immunosuppressive roles and resistance caused by subsequent phosphorylation of AKT ser473 by mTORC2 restrict the use of rapamycin and its analogs. mTOR kinase inhibitors are under development, which can simultaneously suppress the activity of both mTORC1 and mTORC2 and have been discussed above. 328 , 370
4.8. SMO inhibitors
Hedgehog (Hh) signaling plays a pivotal role in embryonic development, tissue homeostasis, regeneration, and stem maintenance in adults. In the absence of Hh ligands, the transmembrane Patched receptor (PTCH) antagonize the activation of smoothened (SMO). Upon Hh ligand binding, the PTCH receptor is exported from the cilium allowing SMO to enter and to activate downstream elements of the pathway. SMO activation promotes the dissociation of glioma‐associated oncogene (GLI) transcriptional factors GLI1, GLI2, and GLI3, from their negative regulator SUFU, and subsequently induce nuclear translocation. Its deregulation in its regulators such as PTCH, SMO, GLI1, and GLI2 causes many different tumors, including basal cell carcinoma (BCC), AML, and several solid cancers. 632 , 633 The SMO protein represents a bottleneck in the canonical Hh signal transduction system, and its genetic deletion or pharmacological blockade is associated with a complete abrogation of ligand‐induced target gene expression. A great deal of work has been devoted to the development of SMO inhibitors, which is hoped to be able to block the Hh signaling at upstream level.
The US FDA accepted three SMO antagonists (vismodegib, sonidegib, and glasdegib), to treat BCC or AML. 634 , 635 , 636 , 637 , 638 Specifically, Hp signaling is aberrantly activated in around 95% of BCC. Data showed an ORR of 43% in locally advanced BCC and 30% in metastatic BCC in phase II study (NCT00833417) of vismodegib. However, adverse effects often lead to drug discontinuation. Serious adverse events were reported in 25% of patients, including seven deaths. 634 , 635 The phase II BOLT study showed that 200 mg sonidegib can also be used in locally advanced or metastatic BCC with an ORR of 43%. 635 In combination with low‐dose cytarabine, glasdegib was capable of bringing an OS of 8.3 months and an ORR of 18.2% to patients with newly diagnosed AML in adults ≥75 years or with comorbidities that preclude use of intensive induction chemotherapy. Therefore, the US FDA approved glasdegib in combination with low‐dose cytarabine for the treatment of AML. 636 , 637 , 638
While serious adverse events and drug resistance hinder their use in the clinic. Many efforts have been paid to solve these problems. 639 , 640 With the high frequency of serious adverse events in SMO inhibitors, researchers tried to find ways to address this problem. First, discontinuation of SMO inhibitors after CR of locally advanced BCC and rechallenging when relapse after discontinuation may be a good way. The data showed the median RFS of discontinuation of vismodegib was 18.4 months, and 85% of relapsed patients could still respond to vismodegib rechallenge. Second, intermittent use of medication ensures efficacy and reduces side effects. 641 The phase II MIKIE trial showed that two long‐term intermittent vismodegib dosing regimens for BCC showed good activity and largely reduced the percentages of side effects. 642 The SMO inhibitors’ resistance is mainly from three aspects: drug‐resistance mutations, bypassing signaling activation, and SMO‐independent Hh signaling activation. 640 The secondary or acquired resistance of the SMO inhibitors in BCC is relatively rare than EGFR inhibitors in NSCLC. SMO‐D473H and SMO‐A459V mutations showed a reduction in sensitivity to vismodegib. 643 Moreover, the preclinical study found a slow‐cycling tumor population characterized by LGR5 expression and active Wnt signaling was responsible for resistance to vismodegib in treating BCC. Therefore, the synergy between Wnt and smoothened inhibitors is a strategy for overcoming tumor relapse. 644 Another preclinical trial showed sonic Hh and PI3K pathways were activated in PTEN‐deficient glioblastoma, simultaneously targeting both pathways resulted in markedly growth reduction of PTEN‐deficient glioblastomas. 645 In addition to the canonical signaling cascade, SMO‐independent activation of GLI cross‐talks with pathways such as MAPK, PI3K/AKT, and NF‐κB and mediates the resistance to the SMO inhibitors. Under this condition, GLI inhibitors can be used for treating cancers. 633
The approved SMO inhibitors are being tested in other tumor types, and novel SMO inhibitors are developing. Phase I study (NCT01125800) of sonidegib in pediatric brain and solid tumors demonstrated that sonidegib was well tolerated. 646 Phase II study in children and adults with relapsed medulloblastoma showed that the treatment of sonidegib was effective, with five of the 10 medulloblastomas with activated Hh pathway demonstrating a CR or PR. 646 However, the phase II studies (NCT00939484 and NCT01239316) of vismodegib for medulloblastoma showed that not all patients responded to vismodegib, indicating the necessity to identify target populations that will genuinely benefit. 647 Besides, the phase I/II trial (NCT01064622) for pancreatic cancers failed to show any clinical benefit if adding vismodegib to gemcitabine in an unselected cohort. 648 A phase III study (NCT03416179) evaluating intensive chemotherapy with or without glasdegib or azacitidine with or without glasdegib for patients with untreated AML has been finished. However, the results have not been published. Two phase III studies (NCT04168502 and NCT04842604) of glasdegib for hematological tumors are ongoing. Several new SMO inhibitors exist, such as taladegib, PF‐5274857, Sant‐1, and MRT‐83. Among them, taladegib is in the clinical stage. Phase I trial of this agent for locally advanced or metastatic BCC showed that responses were observed in patients previously treated with Hh therapy (11 out of 31) and in Hh treatment‐naïve (11 out of 16) patients. 649 Phase II studies of this agent on various types of tumors, are ongoing (NCT05199584). At the same time, the other SMO inhibitors are still in the preclinical stage.
4.9. BCL‐2 inhibitors
Anticancer therapies mainly involve viral and cellular oncogenes, cellular proliferation, and transformation. However, programmed cell death was deemed a logical and realistic therapeutic strategy until the discovery of apoptosis after tumor cells exposure to glucocorticoids, cytotoxic agents, or radiation. 650 The BCL‐2 family contains two subgroups with opposite functions: proapoptotic and antiapoptotic members. BAX, BAK1, BIM, BID, and BBC3 belong to proapoptotic molecules. On the contrary, antiapoptotic molecules conclude BCL‐2, BCL‐XL, BCL‐W, BCL‐2‐A1, and MCL1. 651 Prosurvival and proapoptotic proteins synergistically balance cellular apoptosis and determine whether apoptosis occurs. Once apoptosis happens, cytochrome C releases from the mitochondria and thereby triggers the activation of proteins of the caspase family, which proteins from the IAP family can negatively regulate. 652 However, evasion of apoptosis is a hallmark of cancer that arises when this balance is tipped in favor of survival. Many antiapoptotic proteins associated with tumor cell survival and resistance to chemoradiotherapy, such as BCL‐2, BCL‐ XL, MCL1, or the IAP proteins, are overexpressed in human tumors. Therefore, many therapeutic agents have been designed to block these proteins. 653
To date, only the BCL‐2 inhibitor venetoclax has been approved for treating hematological tumors. Venetoclax belongs to BH3 mimetics, which are small molecules that mimic the binding of BH3‐only proteins to antiapoptotic BCL‐2 proteins and therefore block antiapoptotic function. BH3‐only proteins are a subclass of proapoptotic BCL‐2 proteins that contain only one BCL‐2 homology domain, which can bind to the groove formed by four BCL‐2 homology domains of antiapoptotic members of the BCL‐2 family. The BCL‐2 inhibitors are designed to imitate the structure of BH3‐only proteins. 650 Venetoclax monotherapy was permitted for the treatment of adult patients with CLL/SLL based on one phase I trial (NCT01328626) and two phase II trials (NCT02141282 and NCT01889186), which demonstrated the efficacy and safety profile of venetoclax in patients with previously untreated CLL harboring 17p deletion and relapsed or refractory CLL/SLL, including those with poor prognostic features or progressing on BTK inhibitors. The ORR ranged from 65 to 79%. The pooled analysis of four early‐phase trials showed that the estimated median PFS, DOR, and TTP were 30.2, 38.4, and 36.9 months, respectively. Serious adverse events occurred in half of the patients, including neutropenia, infection, anemia, and thrombocytopenia. Another adverse event, tumor lysis syndrome, should be avoided by controlling the drug dose. 652 , 654 , 655 , 656 Subsequent trials of venetoclax combinations further expanded the benefits of venetoclax in the setting of both refractory and untreated CLL/SLL. The phase III CLL14 trial showed that patients with previously untreated CLL and coexisting conditions received longer PFS with venetoclax and obinutuzumab than with chlorambucil and obinutuzumab. 657 In another phase III MURANO trial, patients with relapsed or refractory CLL were randomly assigned to receive venetoclax plus rituximab or bendamustine plus rituximab. The results showed that venetoclax plus rituximab resulted in significantly higher rates of PFS than bendamustine plus rituximab. 658 The other indication of venetoclax is for patients with newly diagnosed AML. Venetoclax combined with hypomethylating drugs or chemotherapy (azacytidine, decitabine, or low‐dose cytarabine) was demonstrated to be superior to the chemical drug alone when treating patients with newly diagnosed AML whose disease was ineligible for intensive chemotherapy. The ORR of combining venetoclax with hypomethylating agent therapy (decitabine or azacitidine) was 60% in a phase Ib trial (NCT02203773). 659 The phase III VIALE‐A trial for previously untreated AML showed OS was significantly longer in patients treated with azacitidine plus venetoclax than those with azacitidine plus placebo (median OS: 14.7 vs. 9.6 months; HR, 0.66; 95%CI: 0.52, 0.85; p < 0.001). 529 Similarly, another phase III trial (NCT03069352) verified the clinical benefits in venetoclax plus low‐dose cytarabine in terms of OS, CR rate, and reduction in risk of death compared with placebo plus low‐dose cytarabine. 660
Expanding the indications of venetoclax and achieving durable and deep response through combination regimes are two developmental directions for venetoclax. Many tumor types, especially for other hematological tumors are under investigation, such as WM, ALL, and multiple myeloma. Venetoclax was tested in patients with previously treated WM in a phase II study (NCT02677324), which found venetoclax was safe and high active, even in patients progressing on BTK inhibitors. 661 Single‐agent venetoclax was active in relapsed or refractory multiple myeloma with t (11;14) translocation. Phase I study (NCT03314181) combining venetoclax with CD38 antibody daratumumab and dexamethasone demonstrated a high rate of deep and durable responses in patients with relapsed or refractory multiple myeloma, especially for patients with relapsed or refractory multiple myeloma those with t (11;14). 662 Investigating novel combined therapy for the approved indications is another measure. For example, combination of venetoclax with BTK inhibitor ibrutinib or acalabrutinib with or without chemoimmunotherapy with obinutuzumab as frontline treatment for CLL has received positive outcomes in several phase II trials (NCT02910583, NCT03580928, and NCT02756897). 429 , 663 , 664 This combination regime is under investigation in the phase III trial (NCT03836261).
Several resistant mechanisms to venetoclax have been described, including alterations of other BCL‐2 proteins’ expression, mutations in the BCL‐2 protein, and modifications in mitochondrial function. For example, increased expression of BCL‐XL is associated with resistance to venetoclax in MCL. Mutations in the BCL‐2 protein, such as G101V and F104L, could largely reduce the BCL‐2 binding affinity for venetoclax compared with wild‐type BCL‐2. Combinatorial strategies may conquer the resistance. 650 As mentioned above, agents targeting BCL‐XL, MCL1, or the IAP proteins are also under investigation, such as BCL‐XL inhibitors ABBV‐155, WEHI‐539, and A‐1155463, MCL1 inhibitors AMG176, MIK665, and AZD5991, and IAP inhibitors and SMAC mimetics LCL161 and birinapant. Moreover, there are also dual BCL‐2/BCL‐XL inhibitors, including navitoclax, APG‐1252, and AZD4320. 529 Take navitoclax as an example. It was initially proved to be safe and effective as a single agent in several phase I studies (NCT00406809 and NCT00481091) for hematological or solid tumors. 665 , 666 Another phase I study (NCT03181126) utilized venetoclax with low‐dose navitoclax and chemotherapy to treat patients with relapsed/refractory ALL or lymphoblastic lymphoma. The results showed the combination regimes were well tolerated and had promising efficacy. 667 Phase II study (NCT03222609) evaluated the addition of navitoclax to ongoing ruxolitinib for patients with myelofibrosis with progression or suboptimal response. 668 Two phase III studies (NCT04472598 and NCT04468984) are assessing first‐line or post‐line therapy of navitoclax plus ruxolitinib for patients with myelofibrosis.
Selective small molecule inhibitors are often used in patients with the presence or absence of specific predictive molecular alteration detected from tumor tissue or body fluid sampling such as blood. 669 , 670 With the development of NGS, it is much easier and more convenient to unlock alterations that exist in genes. 671 , 672 However, not all selective small molecule inhibitors require individualized patient selection due to alteration of a particular gene or hyperactivation of a kind of signaling pathway that existed in nearly all cases with a specific tumor. 10 For example, almost all patients with CLL and AML are characterized by overexpression of BCL‐2, so additional tests are not required when patients use BCL‐2 inhibitor venetoclax. 529 , 673
5. MULTIKINASE SMALL MOLECULE INHIBITORS
As mentioned above, multikinase inhibitors have a wide range of targets. They block the VEGF/VEGFR signaling pathway while also inhibiting other pathways to exert antiangiogenic and antiproliferative effects. 20 , 21 Multikinase inhibitors have various indications, and their development is mainly empirical. None of these drugs make use of a biomarker to identify patients that respond. 10 To date, there are nine multikinase inhibitors approved by the US FDA, namely sorafenib, sunitinib, pazopanib, vandetanib, axitinib, regorafenib, cabozantinib, lenvatinib, and tivozanib. Clear cell RCC, HCC, and thyroid cancer are the most studied in the clinic 21 , 674 , 675 (Table 3).
TABLE 3.
Summary of approved multikinase small molecule inhibitors
| Class | Drug name | Company | First approval | Targets | Protein substrate | Administration pathway | Indications |
|---|---|---|---|---|---|---|---|
| VEGFR multikinase | Sorafenib (Nexavar) | Bayer Healthcare | 2005 | c‐CRAF, BRAF, mutant BRAF, KIT, FLT‐ 3, RET, RET/PTC, VEGFR1, VEGFR2, VEGFR3, PDGFRβ | Tyrosine | Oral | HCC, RCC, radioiodine‐ refractory DTC |
| VEGFR multikinase | Sunitinib (Sutent) | CPPI CV (Pfizer) | 2006 | PDGFRα, PDGFRβ, VEGFR1, VEGFR2, VEGFR3, KIT, FLT3, CSF‐1R, RET | Tyrosine | Oral | GIST, RCC, pNET |
| VEGFR multikinase | Pazopanib (Votrient) | Novartis | 2009 | VEGFR1, VEGFR2, VEGFR3, PDGFRα, PDGFRβ, FGFR1, FGFR3, Kit, Itk, Lck, c‐Fms | Tyrosine | Oral | RCC, soft tissue sarcoma |
| VEGFR multikinase | Vandetanib (Caprelsa) | Genzyme Corp | 2011 | EGFR, VEGFR, RET, BRK, TIE2, EPH receptor, Src, | Tyrosine | Oral | MTC |
| VEGFR multikinase | Axitinib (Inlyta) | PF Prism CV | 2012 | VEGFR1, VEGFR2, VEGFR3 | Tyrosine | Oral | RCC |
| VEGFR multikinase | Regorafenib (Stivarga) | Bayer Healthcare | 2012 | RET, VEGFR1, VEGFR2, VEGFR3, KIT, PDGFR‐alpha, PDGFR‐beta, FGFR1, FGFR2, TIE2, DDR2, TrkA, Eph2A, RAF‐1, BRAF, BRAF V600E, SAPK2, PTK5, Abl, CSF1R | Tyrosine | Oral | CRC, GIST, HCC |
| VEGFR multikinase | Cabozantinib (Cabometyx, Comertiq) | Exelixis | 2012 | MET, VEGFR1, VEGFR2, VEGFR3, AXL, RET, ROS1, TYRO3, MER, KIT, TRKB, FLT‐3, TIE‐2 | Tyrosine | Oral | RCC, HCC, radioiodine‐ refractory DTC |
| VEGFR multikinase | Lenvatinib (Lenvima) | Eisai | 2015 | VEGFR1, VEGFR2, VEGFR3, FGFR1, FGFR2, FGFR3, FGFR4, PDGFRα, KIT, RET | Tyrosine | Oral | Radioiodine‐ refractory DTC, RCC, HCC, endometrial carcinoma |
| VEGFR multikinase | Tivozanib (Fotvida) | Aveo Pharms | 2021 | VEGFR1, VEGFR2, VEGFR3, c‐kit, PDGFRβ | Tyrosine | Oral | RCC |
Abbreviations: HCC, hepatocellular carcinoma; RCC, renal cell carcinoma; DTC, differentiated thyroid carcinoma; GIST, gastrointestinal stromal tumor; pNET, pancreatic neuroendocrine tumor; MTC, medullary thyroid cancer; CRC, colorectal cancer. Data sources: https://www.fda.gov/drugs/development‐approval‐process‐drugs/drug‐approvals‐and‐databases.
Clear cell RCC is the most common type of RCC and is rich in vascular networks induced by dysregulation of the VHL/HIF pathway, which provides a scientific rationale for developing VEGFR‐focused multikinase inhibitors (sorafenib, sunitinib, pazopanib, axitinib, cabozantinib, lenvatinib, and tivozanib) in this tumor type. 475 , 674 , 676 , 677 Sunitinib is the only agent used in the adjuvant treatment setting based on a phase III S‐TRAC trial. Compared with placebo, sunitinib given on a 4‐weeks‐on, 2‐weeks‐off schedule for 1‐year prolonged duration of DFS (median DFS: 6.8 vs. 5.6 years; HR, 0.76; 95%CI: 0.59, 0.98; p = 0.03). 678 The US FDA approved sunitinib and four other combination regimes for patients with treatment‐naïve RCC in the first‐line therapy. 677 Monotherapy with sunitinib had a statistically significant advantage over interferon alfa in the major endpoint of PFS (median PFS: 11 vs. 5 months; HR, 0.42; 95%CI: 0.32, 0.54; p < 0.001). 679 , 680 Compared with sunitinib, the four combination regimes (axitinib plus avelumab, axitinib plus pembrolizumab, cabozantinib plus nivolumab, and lenvatinib plus pembrolizumab) not only have advantages in PFS but also in OS. 681 , 682 , 683 , 684 Adding a PD‐1/PD‐L1 inhibitor to a multikinase inhibitor can broadly promote the prognosis of the patient with RCC. 685 The median PFS of lenvatinib plus pembrolizumab was the longest, reaching 23.9 months in the phase III CLEAR trial. 684 In addition to tivozanib, the other six multikinase inhibitors approved for RCC can be used in second‐line therapy. Most of them were used in monotherapy, while lenvatinib plus everolimus is the only approved combination form and also brings the best median PFS duration time reaching 14.6 months. 686 Monotherapy with tivozanib is indicated for RCC after two or more prior systemic therapies. In phase III TIVO‐3 trial, median PFS was significantly longer with tivozanib than with sorafenib (5.6 vs. 3.9 months; HR, 0.73; 95%CI: 0.56, 0.94; p = 0.016). Moreover, the proportion of hypertension with tivozanib (the most common grade 3 or 4 treatment‐related adverse event) was lower than with sorafenib (14 vs. 20%). 687
Sorafenib was the first agent approved for patients with advanced HCC based on the phase III SHARP trial, which revealed survival benefits with sorafenib versus placebo (median OS: 10.7 vs. 7.9 months; HR, 0.69; 95%CI: 0.55, 0.87; p < 0.001). 688 , 689 In 2018, lenvatinib was allowed to treat advanced HCC in the frontline. In the phase III REFLECT trial, lenvatinib was noninferior to sorafenib in terms of OS (13.6 vs. 12.3 months; HR, 0.92; 95%CI: 0.79, 1.06). The median PFS and ORR on the lenvatinib arm were also superior to the sorafenib arm. 690 Besides, regorafenib and cabozantinib were available in patients with HCC after treatment with sorafenib. The efficacy and tolerability of regorafenib were demonstrated by the phase III RESORCE trial. Regorafenib improved OS (median OS: 10.6 vs. 7.8 months) with a HR of 0.63 (95%CI: 0.50, 0.79; one‐sided p < 0.0001) compared with placebo. 691 In the phase III CELESTIAL trial, duration of median OS (10.2 vs. 8.0 months; HR, 0.76; 95%CI: 0.63, 0.92; p = 0.005) and median PFS (5.2 vs. 1.9 months; HR, 0.44; 95%CI: 0.36, 0.52; p < 0.001) was longer with cabozantinib than with placebo. The ORRs were 4% and less than 1%, respectively (p = 0.009). 692 Median OS durations with these agents were significantly prolonged than the placebo, reaching around 1 year. 691 , 692 , 693 , 694
In thyroid cancer, sorafenib, lenvatinib, and cabozantinib have received US FDA approval to treat radioiodine‐refractory differential thyroid cancer based on phase III trials (NCT00984282, NCT01321554, and NCT03690388). Median PFS in the sorafenib arm was 10.8 months and the value was 5.8 months in the placebo arm (HR, 0.59; 95%CI: 0.45, 0.76; p < 0.0001). 695 Lenvatinib, as compared with placebo, improved PFS (median PFS: 18.6 vs. 3.6 months; HR, 0.21; 95%CI: 0.14, 0.31; p < 0.001) and increased response rate (64.8 vs. 1.5%; p < 0.001). 696 Similarly, cabozantinib significantly prolonged PFS compared with placebo (HR, 0.22; 95%CI: 0.13, 0.36; p < 0.0001). 697 Two multikinase inhibitors, vandetanib and cabozantinib, were on the market for radiographic progression of metastatic MTC. 21 , 698 Vandetanib was superior to placebo in terms of PFS (HR, 0.46; 95%CI: 0.31, 0.69; p < 0.001) and ORR (p < 0.001). 699 The phase III trial (NCT00704730) showed that the median PFS and response rate were 11.2 months and 28% for cabozantinib versus 4.0 months and 0% for placebo. 700 In total, all the drugs showed significant improvement in PFS with or without an increase in ORR compared with the placebo. However, none of them demonstrated a significant difference in OS. 696 , 697 , 699 , 700
Advanced imatinib‐resistant GISTs respond to second‐line sunitinib and third‐line regorafenib, both of which not only inhibit KIT but also PDGFR and/or VEGFR family members. 76 , 701 , 702 Compared with placebo, sunitinib prolonged median time to tumor progression from 6.4 weeks to 27.3 weeks (HR, 0.33; 95%CI: 0.23, 0.47; p < 0.0001) and median PFS from 6.0 weeks to 24.1 weeks (HR, 0.33; 95%CI: 0.24, 0.47; p < 0.0001). 703 In the following studies, researchers found that sunitinib preferentially suppresses KIT with secondary resistance mutations affecting the ATP‐binding pocket (exons 13 and 14) and exon 9 mutations. Unlike imatinib, PFS and OS of patients with KIT exon 9 mutations or with a wild‐type genotype were longer than that of those with KIT exon 11 mutations. Median PFS and median OS for patients with primary KIT exon 9 mutations reached 19.4 months and 26.9 months, 19.0 months and 30.5 months for patients with a wild‐type genotype, and 5.1 months and 12.3 months for patients with KIT exon 11 mutations. In addition, PFS and OS of patients with secondary KIT exon 13 or 14 mutations were longer than for those with exon 17 or 18 mutations. 704 For regorafenib, the phase III GRID trial showed that third‐line therapy with regorafenib largely prolonged median PFS compared with placebo (4.8 vs. 0.9 months; HR,0.27; 95%CI: 0.19, 0.39; p < 0.0001) and DCR was 52.6 and 9.1%, respectively. No significant difference in OS between study arms was found, because 84.8% of patients in the control arm crossed over to regorafenib. 705 A clinical study involving 33 patients showed patients whose tumors harbored a KIT exon 11 mutation had longer median PFS than patients with KIT/PDGFRA wild‐type and non‐SDH‐deficient tumors (13.4 vs. 1.6 months, p < 0.0001). 706 While the definite correlation between kinase genotypes and the biological and clinical activity of regorafenib needs more researches.
Several other factors concerned with multikinase inhibitors deserve attention. First, in addition to the above diseases for multikinase inhibitors, they also have other indications: sunitinib for pNET, pazopanib for STS, regorafenib for metastatic CRC, and lenvatinib for endometrial carcinoma. 707 , 708 , 709 , 710 Second, the most common toxicities include hypertension, hand‐foot skin reaction, diarrhea, decreased weight and appetite, nausea, and fatigue, mainly attributed to on‐targeted mechanisms. 697 Corresponding medications can treat these side effects to relieve symptoms, such as antihypertensive drugs for high blood pressure. Side effects can also be alleviated by reducing or discontinuing multikinase inhibitors. 711 Third, as mentioned above, no markers are established to indicate the efficacy of multikinase inhibitors, especially for the only vascular‐targeted multikinase inhibitors. The resistance mechanisms of multikinase inhibitors are different from that of other small molecular inhibitors. Resistance to VEGFR inhibitors usually does not involve secondary mutation of the target but instead activates bypassing pathways that stimulate angiogenesis. Therefore, combining other mechanisms to synergistically inhibit tumor blood vessels may overcome the resistance of multikinase inhibitors. 220
Many small molecule inhibitors targeting VEGFR are still in clinical trials and can be divided into selective VEGFR kinase inhibitors, dual mechanism inhibitors, and multikinase inhibitors (Table S3). Among them, multikinase inhibitors are the best developed. Selective VEGFR kinase and dual mechanism inhibitors relatively lag behind.
The development of selective VEGFR kinase inhibitors is limited. Vatalanib and semaxanib belong to selective VEGFR2 kinase inhibitors. Vatalanib mainly targets VEGFR tyrosine kinases in submicromolar range but it also inhibits PDGFR, KIT, and c‐Fms at higher concentrations. The antiangiogenic drug vatalanib added to chemotherapy with FOLFOX4 failed to show advantages in OS and PFS compared with FOLFOX4 in two phase III trials for first‐ and second‐line treatment of CRC (CONFIRM 1 and 2). 712 , 713 Similarly, trials of semaxanib for patients with advanced STS or head and neck cancers showed that semaxanib was well tolerated but with limited antitumor activity. 714 , 715 Therefore, the development of vatalanib and semaxanib was suspended and predicted limited roles of small molecule inhibitors that only target VEGFR2. Luckily, fruquintinib, a VEGFR1‐3 inhibitor, has been approved in China for NSCLC and CRC. Phase II study (NCT02590965) of third‐ and fourth‐line fruquintinib in patients with advanced nonsquamous NSCLC was superior to placebo in the form of PFS, survival rate, and DCR. 716 The phase III FRESCO trial demonstrated that third‐line or later therapy with fruquintinib significantly prolonged PFS (median PFS: 3.7 vs. 1.8 months; HR, 0.26; 95%CI: 0.21, 0.34; p < 0.001) and OS (median OS: 9.3 vs. 6.6 months; HR, 0.65; 95%CI: 0.51, 0.83; p < 0.001) compared with placebo. 717 Fruquintinib was licensed based on these two clinical trials.
As mentioned above, multikinase inhibitors have made great success in treating cancers. In addition to the US FDA‐approved multikinase inhibitors, anlotinib, apatinib, donafenib, and surufatinib have received approval in China. Anlotinib was licensed for NSCLC and STS. The phase III ALTER 0303 trial showed that anlotinib improved PFS (median PFS: 5.4 vs. 1.4 months; HR, 0.25; 95%CI: 0.19, 0.31, p < 0.001) and OS (median OS: 9.6 vs. 6.3 months; HR, 0.68; 95%CI: 0.54, 0.87; p = 0.002) in patients with advanced NSCLC progressing after second‐line or further treatment compared with a matched placebo. 718 A phase II trial (NCT01878448) of this agent showed progression‐free rate at 12 weeks for refractory STS was 68% and ORR was 13%. 719 Apatinib approved for gastric adenocarcinoma and gastroesophageal junction adenocarcinoma was based on a phase III trial, which demonstrated a longer duration of PFS (median PFS: 2.6 vs. 1.8 months; HR, 0.44; 95%CI: 0.331, 0.595; p < 0.001) and OS (median OS: 6.5 vs. 4.7 months; HR, 0.709; 95%CI: 0.537, 0.937; p = 0.0156) with apatinib than with a placebo. 720 Donafenib is a deuterated sorafenib derivative and was allowed for first‐line treatment of HCC, as it exhibited superior OS outcomes (median OS: 12.1 vs. 10.3 months; HR, 0.831; 95%CI: 0.699, 0.988; p = 0.245) and fewer adverse events (38 vs. 50%; p = 0.0018) versus sorafenib. 721 The last approved agent, surufatinib, is for neuroendocrine tumor, including extrapancreatic and pNETs based on two phase III trials (SANET‐ep and SANET‐p). 722 , 723 In addition, many multikinase inhibitors are in the developmental stage, such as altiratinib, cediranib, dovitinib, lucitanib, ningetinib, nintedanib, and telatinib. Cediranib is under phase III stage for ovarian, fallopian tube, and primary peritoneal cancer (NCT03278717, NCT02502266, and NCT02446600). Phase III studies of this agent for CRC, glioblastoma, biliary tract neoplasm, and NSCLC have been completed (NCT00399035, NCT00384176, NCT00777153, NCT00939848, NCT00795340, and NCT00245154). The primary endpoints were not met in glioblastoma and biliary tract neoplasm. 724 , 725 Dovitinib has been tried in many tumor types and has even been submitted an application to the US FDA for third‐line treatment of RCC based on positive results from a phase III study (NCT01223027). 726 Lucitanib is an inhibitor of VEGFR1‐3, FGFR1‐3, and PDGFRα/β. Previous studies (NCT02053636 and NCT01283945) of lucitanib for breast cancer and solid tumors indicated that FGFR alteration was correlated with the sensitivity of lucitanib. 727 Phase II/III trial (NCT04254471) of lucitanib in patients with SCLC is recruiting. Ningetinib is also a multikinase inhibitor with potent activity against MET, VEGFR2, and Axl. It has just completed a phase I trial for solid tumors (NCT04577703) and is ready to recruit patients with NSCLC harboring MET exon 14 skipping mutations for phase II study (NCT04992858). Similarly, a phase I trial (NCT03175497) of telatinib for solid tumors has been completed, and patients with gastric cancer and HCC are under phase II stage (NCT04798781) with telatinib. Nintedanib is successfully used in treating idiopathic pulmonary fibrosis and interstitial lung disease, but the indications for cancers are far from reaching. 728 , 729 Altiratinib is in preclinical stage. The development of these drugs will give more chances to patients.
The limited efficacy of selective VEGFR inhibitors and side effects companied with multikinase inhibitors urge the research on dual mechanism inhibitors, such as dual VEGFR/MET inhibitors foretinib and golvatinib, dual VEGFR/PDGFR inhibitors linifanib and vorolanib, and dual VEGFR/FGFR inhibitors brivanib and ODM‐203. The trials of the dual VEGFR/MET inhibitors stalled in phase II stage. Foretinib demonstrated activity in patients with advanced papillary RCC, especially in patients with germline MET mutations, and a manageable toxicity profile (NCT00726323). 730 However, negative results were obtained from trials of triple‐negative breast cancer and gastric cancer (NCT01147484 and NCT00725712). 731 , 732 Although golvatinib has completed two phase II clinical studies (NCT01332266 and NCT01271504) for HCC and squamous cell carcinoma of the head and neck, the results have not yet been published. Clinical trials of dual VEGFR/PDGFR inhibitor linifanib are basically not continued because of limited efficacy and increasing toxicity. The only phase III trial (NCT01009593) of linifanib compared efficacy and tolerability of linifanib versus sorafenib in HCC without prior systemic therapy. However, linifanib did not meet predefined superiority and noninferiority OS boundaries. 733 Vorolanib is a promising dual VEGFR/PDGFR inhibitor and has received preliminary efficacy in solid tumors with acceptable safety profiles (NCT03511222). 734 Phase II study (NCT04373369) of vorolanib plus atezolizumab for extensive‐stage SCLC is ongoing. Dual inhibition of VEGFR and FGFR is another trend and may be beneficial for FGF/FGFR‐altered tumors. Brivanib received positive results in both phase I and II clinical trials but failed in all phase III clinical trials for HCC, all of which set OS as the primary endpoint, including frontline and second‐line therapies for advanced HCC and adjuvant therapy for unresectable HCC after transcatheter arterial chemoembolization (NCT00355238, NCT00858871, NCT00908752, and NCT00825955). 735 , 736 , 737 , 738 It is now under stage II trial for cervical cancer (NCT04395612). ODM‐203 is a pan‐VEGFR/FGFR inhibitor and has shown preliminary signs of therapeutic activity in solid tumors with acceptable tolerability (NCT02264418). 739 More researches are needed to verify the safety and effectiveness of ODM‐203. In general, dual inhibitors can theoretically increase efficacy and reduce side effects, but the practical application remains to be verified.
6. CONCLUSIONS AND PERSPECTIVES
The development and approval of small molecule inhibitors have essentially transformed multiple cancers’ clinical care and improved prognosis in many tumors. 124 Take NSCLC as an example. Chemotherapy was used to be the most popular treatment for locally advanced or metastatic NSCLC. While specific small molecule inhibitors now are widely used for NSCLC with EGFR mutation, ALK or ROS1 rearrangement, MET mutation, which can improve ORRs and PFS and reduce adverse side effects compared with chemotherapy. 740 , 741 Despite substantial progress in discovering small molecule inhibitors, many challenges and opportunities remain in the field.
6.1. Targets
It is reported that at least one alteration in genes can be detected in approximately 40% of cancers. 742 Although we have made significant progress in developing small molecule inhibitors targeting cancers, minimal gene alterations have successfully translated into clinical use. 743 Take protein kinase inhibitors as an example, approved drugs have targeted only 5% of protein kinases. 5 At the same time, a network of proteins contributes to repair DNA damage, only PARP inhibitors of this category have been approved. 9 New therapeutic targets are to be investigated. With the development of computer‐aided drug design, medicinal chemistry, biochemical assays, animal experiments, and clinical trials, we believe more and more small molecular inhibitors with new targets will be developed to treat cancers. 1 Actually, small molecule inhibitors targeting the aurora kinases, transcriptional kinases, immuno‐regulatory kinases, and EZH2 are actively pursued by pharmaceutical and academic groups 669 , 744 (Figure 3A).
FIGURE 3.
Some of the challenges and future directions of small molecule inhibitors. (A) Approaches to develop small molecule inhibitors with new targets. (B) Factors for adjuvant therapy with small molecule inhibitors. (C) Precautions in the clinical use of small molecule inhibitors. Figure created with BioRender.com
6.2. Adjuvant and neoadjuvant therapy
The development of small molecule inhibitors is often initially used for post‐line therapy, gradually transitioning to first‐line treatment, and finally trying to be applied in adjuvant and neoadjuvant settings. 745 , 746
To date, several agents have received the permits for patients with early malignancies after surgery, such as imatinib for KIT or PDGFRA mutant GISTS, osimertinib for NSCLC with EGFR exon 19 deletion or exon 21 (L858R) mutations, neratinib for HER2‐positive breast cancer, dabrafenib plus trametinib for melanoma with BRAF V600E or V600K mutations, and sunitinib for RCC. 82 , 156 , 294 , 678 , 747 Some of them can bring obvious clinical benefits to patients, like imatinib for patients with KIT or PDGFRA mutations. 82 But more factors should be considered for patients who have traditional treatment in the adjuvant setting or whose advantages are insignificant. 747 In NSCLC, adjuvant chemotherapy is still the standard for NSCLC and osimertinib is the most potent EGFR‐TKI for locally advanced or metastatic NSCLC. Subsequent therapies will be complex once patients progress to the early stage after treatment with osimertinib. 748 Another example is sunitinib for RCC. Patients with RCC were treated for nine cycles (approximately 1 year) after surgery and 5‐year DFS rate only increased from 51.3% with placebo to 59.3% with sunitinib. 678 In addition, more drugs did not meet the primary endpoints in clinical trials and were not approved for adjuvant therapy. For example, the phase III BRIM8 trial showed that the benefit of DFS in adjuvant vemurafenib monotherapy for 52 weeks was not consistency compared with placebo in patients with resected, BRAFV600 mutation‐positive melanoma. 749 Taken together, the economic benefit ratio and treatment‐related side effects deserve more consideration in this situation 750 (Figure 3B).
Though many efforts have been made for the neoadjuvant treatment of cancers with promising outcomes, no targeted drug has yet been approved. Although not yet approved, scientists have not stopped exploring. As mentioned above, numerous small molecule inhibitors have been explored in the field of neoadjuvant therapy, such as MET inhibitor capmatinib for NSCLC, TRK inhibitor larotrectinib for sarcomas, PI3K inhibitor taselisib, CDK4/6 inhibitor palbociclib, and AKT inhibitor MK‐2206 for breast cancer. 242 , 357 , 409 Another example is the use of neoadjuvant dabrafenib plus trametinib therapy in resectable stage III BRAF V600 mutant melanoma, which led to a high proportion of patients achieving a complete response (46%) and a complete pathological response (49%) without progression in a single‐arm phase II study. 751 These trials are only in early stage, and these is still a long way from the indications for neoadjuvant therapy.
6.3. Efficacy and toxicity
It is crucial to balance the drugs’ efficacy and toxicity. Nearly all the small molecule inhibitors have a chance to be halted in more or less percent of patients because of severe side effects. 6 In the clinic, detailed advice, careful observation, and regular testing of relevant indicators should be given to patients on medication. Dosage reduction or complete drug discontinuation should be adopted according to adverse reaction grade 5 (Figure 3C).
6.4. Treatment interruption
We should avoid abrupt discontinuation of small molecule inhibitors without any other systematic treatment when the patients encounter the onset of progression. Because of differences in growth kinetics of responsive and resistant tumor subclones, abrupt discontinuation may lead to a disease flare. 1 , 10 Topical therapies such as radiotherapy are recommended for oligoprogression without cessation of small molecule inhibitors 752 , 753 (Figure 3C). However, small molecule inhibitors are not impossible to interrupt, they just need to be stopped at the right time. Treatment discontinuation of imatinib can be safe in patients with CML who have been in deep molecular remission for at least 2 years. 754 A series of trials evaluated the safety of treatment discontinuation in nearly 4000 patients, which showed that approximately 50% of individuals experienced long‐term treatment‐free remission (TFR). 755 What is more, combination ABL1 kinase inhibitors with agents of other mechanisms, such as JAK/STAT inhibitors, BCL‐2 inhibitors, and IFN‐α is demonstrated to achieve better elimination of CML stem cells and deeper molecular response, which in turn offer more possibilities for patients to discontinue therapy. 756 In addition, advances in technology also help patients with CML achieve TFR. Disease undetectable verified by both conventional and droplet digital PCR (ddPCR) showed only a 10% risk of disease relapse. In comparison, the risk of relapse was 50–65% through ddPCR or conventional RT‐PCR. 757 Whereas, imatinib discontinuation is not suitable for patients with GISTSs free of progression after 3 years of imatinib. Two‐year PFS rate was higher in the continuation group than in the interruption group (80 vs. 16%; p < 0.0001). 758
6.5. Drug resistance and countermeasures
Resistance to small molecule inhibitors can be divided into primary and secondary resistance. 759 Combination therapies are often needed to tackle primary resistance because of the molecular heterogeneity of tumors. 760 The small molecular inhibitors are not curative, and the emergence of secondary resistance is a significant barrier to achieve long‐term remission in cancer. The reasons for secondary resistance are relatively complex. 6 In this part, we focus on secondary resistance and corresponding solutions (Figure 4).
FIGURE 4.
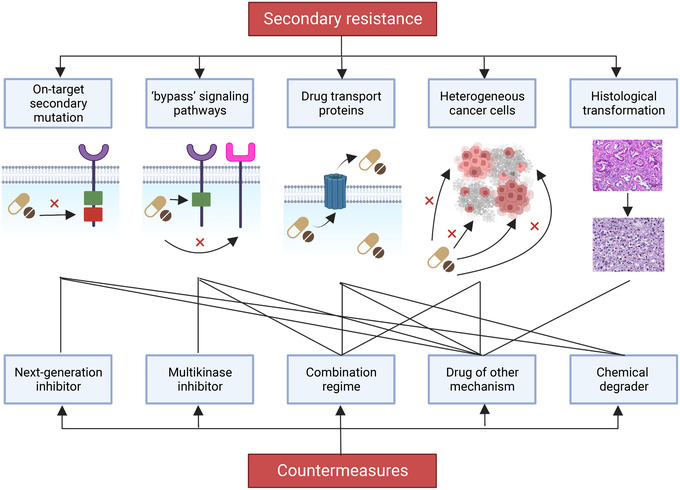
Mechanisms of secondary resistance of small molecule inhibitors and countermeasures. Figure created with BioRender.com
First, on‐target secondary mutations have been studied a lot, which spurs the enthusiasm for finding the next‐generation inhibitors with fewer resistance liabilities, such as ABL TKIs in CML, EGFR and ALK TKIs in lung cancer, and KIT TKIs in GISTs. 75 , 756 , 761 Second, overexpression or amplification of other kinases with similar functions as the drugged kinase can cause the acquisition of “bypass” signaling pathways. 762 , 763 Multikinase inhibitors having the ability to target multiple targets can be applied for targeting “bypass” signaling pathways. For example, second‐line sunitinib and third‐line regorafenib are used for imatinib‐resistance GISTs. 76 The use of a combination regimen is another approach. MET amplification is the second main reason for resistance to first‐generation EGFR TKIs, which may be controlled by dual EGFR/MET inhibition. 764 One general mechanism is the production of drug transport proteins. 765 In clinic, we can observe the phenomenon that different sites of tumor cells show different responses to small molecule inhibitors. 766 , 767 Next‐generation agents with markedly improved CNS penetration are capable of controlling central lesions. In rare cases, histological transformation also accounts for secondary resistance. 768 However, some researchers found that other tissue types existed before treatment but became the primary type after medication. 769 In short, next‐generation inhibitors, combination regimes, or drugs of other mechanisms can deal with secondary resistance under their individual conditions.
In addition to traditional ways to address resistance, new methods which are capable of inducing loss of the total proteins, rather than just loss of their functions, are ideal for overcoming resistance caused by mutation or overexpression. 770 , 771 Proteolysis‐targeting chimeras (PROTACs) fall into the class of chemical degraders and are heterobifunctional small molecules containing two ligands linked by a linker. The two ligands recruit and bind an E3 ligase complex and a specific protein of interest, respectively, promoting ubiquitination and subsequent degradation of the target proteins. 772 , 773 It is reported that E3 ligase–PROTAC–target ternary complexes induce approximately 200 ePKs degradations. Although none of these kinds of inhibitors are on the market and a small number of them are in the clinical development, it is believed that more PROTACs will enter clinical development in the near future. 774
6.6. Combination therapy
Combination therapy aims to increase and prolong the therapeutic benefits or reversal of primary and acquired resistance through the utility of agents with different mechanisms. 775 Several strategies can be used to achieve this goal.
First, vertical blockade of one signaling pathway is a strategy to increase the curative effect of small molecule inhibitors. 287 Both BRAF and MEK are the key molecules in the MAPK pathway. Three combinations of BRAF and MEK inhibitors, cobimetinib plus vemurafenib, dabrafenib plus trametinib, and encorafenib plus binimetinib, have shown their superior to BRAF or MEK inhibitor monotherapy and have been approved by the US FDA for metastatic melanoma with BRAF V600E or V600K mutations. 275 , 282
Second, small molecule inhibitors’ horizontal inhibition of two or more pathways is another schedule. 75 Multikinase inhibitors are a good example, which have been approved for various indications. 76 Although the combination of different selective small molecule inhibitors that inhibit different signaling pathways have not been approved, many similar efforts have been made. 764 For example, the combination of ABL1 inhibitor imatinib and MEK inhibitor binimetinib for treatment‐naive adult patients with confirmed advanced GISTs received 69.0% of ORR, which was 20% improvement in the ORR over imatinib alone. 776 Addition of BCL‐2 inhibitor navitoclax to ongoing ruxolitinib therapy could reverse the resistance for patients with myelofibrosis, whose disease had been progressed or suboptimal response to ruxolitinib monotherapy. 777 In addition, the combination of antibody drugs and small molecule inhibitors is also an effective way to jointly inhibit multiple signaling pathways. Dual blockade of the EGFR and VEGF pathways in EGFR‐mutated metastatic NSCLC is an example. The trials enrolled patients with metastatic EGFR‐mutant NSCLC to receive bevacizumab (or ramucirumab) plus erlotinib or erlotinib alone and the results showed that the addition of bevacizumab (or ramucirumab) to erlotinib significantly prolonged PFS but failed to prolong survival (median OS: 50.7 vs. 46.2 months; HR, 1.007; 95%CI: 0.681, 1.490; p = 0.97). 159 , 778 , 779 Prolonged PFS was also found in the PI3Kα/δ copanlisib plus CD20 antibody rituximab group for indolent NHL, as compared with rituximab alone. 780 However, not all targets can be used in combination. The combination of DNMT inhibitor and HDAC inhibitor failed to show clinical benefits for patients with MDS and AML compared with monotherapy with HDAC inhibitor. 564 In other words, simultaneous horizontal inhibition of multiple targets or pathways is not always successful in all endpoints. Whether dual blockade suitable for clinical treatment deserves more consideration, including efficacy, cost‐effectiveness, compliance, and safety.
Combined small molecule inhibitors with ICIs is a prevailing direction. 781 The PD‐1/PD‐L1 inhibitors are the most commonly used ICIs, restoring the ability of immune cells to destroy tumors. But the ORR of monotherapy with PD‐1/PD‐L1 inhibitors is less than 20%. 782 On the other hand, small molecule inhibitors have pleiotropic effects on the immune system. VEGFR inhibitors modulate the TME by normalizing blood vessels. PI3K/mTOR inhibitors themselves are crucial for immune activation. 783 Besides, most small molecule agents have the potential to block oncogenic signaling pathways and modulate PD‐L1 expression. For example, PARP inhibitors and MET inhibitors can upregulate PD‐L1 expression in breast cancer and HCC, respectively. 784 Therefore, combining ICIs with appropriate small molecule inhibitors may confer a synergistic response in oncogenic addiction cancers. In RCC, four combination regimes (axitinib plus avelumab, axitinib plus pembrolizumab, cabozantinib plus nivolumab, and lenvatinib plus pembrolizumab) represent the best therapeutic effect for local advanced or metastatic RCC. 681 , 684 , 685 Another example is unresectable advanced BRAFV600 mutation‐positive melanoma, the addition of atezolizumab to targeted therapy with vemurafenib and cobimetinib significantly increased PFS (median PFS: 15.1 vs. 10.6 months; HR, 0.78; 95%CI: 0.63, 0.97; p = 0.025) and was proved to be safe in phase III IMspire 150 trial. 308 However, monotherapy with PD‐1/PD‐L1 inhibitors has poor efficacy in NSCLC with EGFR mutations because of a noninflamed TME formed in EGFR‐mutated cancer. 785 The phase I TATTON trial also showed that osimertinib plus durvalumab was not suggested due to increased interstitial lung disease in patients with EGFR‐mutant NSCLC progression on a prior EGFR‐TKI. Downregulation of PD‐L1 due to EGFR inhibitors may make it better not to use it in combination. 163 In addition, more experiments and clinical trials are required to determine whether a specific small molecule inhibitor is appropriately combined with a PD‐1/PD‐L1 inhibitor. 775
Combination with other traditional therapies is also prevalent, such as a CDK4/6 inhibitor plus an aromatase inhibitor for HR‐positive, HER2‐negative breast cancer, BCL‐2 inhibitor plus chemotherapy for AML, HDAC inhibitor plus radiotherapy for neuroblastoma. 397 , 562 , 786 Like combination of small molecule inhibitors with PD‐1/PD‐L1 inhibitors, small molecule inhibitors are not always suitable to combine with chemotherapy. For example, sunitinib plus FOLFIRI was not superior to FOLFIRI alone in median PFS (8.4 vs. 7.8 months; HR, 1.095; 95%CI: 0.892, 1.344; p = 0.807) and had a poor safety profile in previously untreated metastatic CRC. 787 Though gefitinib plus pemetrexed or pemetrexed and carboplatin improved PFS compared with gefitinib alone in patients with untreated advanced NSCLC with EGFR mutations, the phase III IMPRESS trial indicated that the continuation of gefitinib plus chemotherapy was detrimental to OS when compared with placebo plus chemotherapy in patients with EGFR mutation‐positive NSCLC resistant to first‐line gefitinib (median OS: 13.4 vs. 19.5 months; HR, 1.44; 95%CI: 1.07, 1.94; p = 0.016), warning that first‐generation EGFR TKIs cannot be continued when chemotherapy is initiated after disease progression. 158 , 788 What is more, some inhibitors are mechanistically not expected to cooperate with specific mechanisms of chemotherapy. For example, CDK4/6 inhibitors are inappropriate in combination with DNA‐damaging or antimitotic chemotherapies as the former prevent cell‐cycle entry, thereby disturbing S‐phase‐ or mitosis‐targeting agents. 789 Taken together, combination small molecule inhibitors with chemotherapies needs rational mechanisms and should be verified by clinical trials.
There are multiple ways to achieve combination therapy, but whether combination therapy is effective still needs to be verified by trials, especially large clinical trials, rather than a blind combination. Although clinical trials of combination therapy are at risk of failure, they are still a potential direction. We should continue researching combination therapies to improve patients’ prognoses.
In summary, small molecule inhibitors bind to a wide range of targets to control the proliferation, survival, apoptosis, differentiation, metabolism, and TME of malignancies, which offer convenience and good curative effect to patients with tumors harboring specific gene alterations or patients with specific histological tumors. 670 , 760 Although some problems and obstacles exist in the field, small molecule inhibitors will undoubtedly continue to play an essential role in treating cancers in the following decades. 10
AUTHOR CONTRIBUTION
H. S. S. offered direction and guidance of the manuscript. G. H. L. and T. C. drafted the initial manuscript. X. L. M. revised the manuscript. G. H. L. and X. Z. illustrated the figures and tables for the manuscript. All authors approved the final manuscript.
CONFLICT OF INTEREST
The authors have no conflict of interest to declare.
ETHICS STATEMENT
No ethical approval was required for this study.
Supporting information
Supplementary Table S1 Summary of the pivotal candidates of small molecule kinase inhibitors. Data sources: https://clinicaltrials.gov/
Supplementary Table S2 Summary of the pivotal candidates of small molecule non‐kinase inhibitors. Data sources: https://clinicaltrials.gov/
Supplementary Table S3 Summary of the pivotal candidates of small molecule inhibitors targeting VEGFR.Data sources: https://clinicaltrials.gov/
ACKNOWLEDGMENT
This work was financially supported by National Natural Science Foundation of China (81201788).
Liu G‐H, Chen T, Zhang X, Ma X‐L, Shi H‐S. Small molecule inhibitors targeting the cancers. MedComm. 2022;3:e181. 10.1002/mco2.181
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Shi DD, Guo JA, Hoffman HI, et al. Therapeutic avenues for cancer neuroscience: translational frontiers and clinical opportunities. Lancet Oncol. 2022;23(2):e62‐e74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Martínez‐Reyes I, Chandel NS. Cancer metabolism: looking forward. Nat Rev Cancer. 2021;21(10):669‐680. [DOI] [PubMed] [Google Scholar]
- 3. Berger MF, Mardis ER. The emerging clinical relevance of genomics in cancer medicine. Nat Rev Clin Oncol. 2018;15(6):353‐365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gharwan H, Groninger H. Kinase inhibitors and monoclonal antibodies in oncology: clinical implications. Nat Rev Clin Oncol. 2016;13(4):209‐227. [DOI] [PubMed] [Google Scholar]
- 5. Cohen P, Cross D, Jänne PA. Kinase drug discovery 20 years after imatinib: progress and future directions. Nat Rev Drug Discov. 2021;20(7):551‐569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Attwood MM, Fabbro D, Sokolov AV, Knapp S, Schiöth HB. Trends in kinase drug discovery: targets, indications and inhibitor design. Nat Rev Drug Discov. 2021;20(11):839‐861. [DOI] [PubMed] [Google Scholar]
- 7. Ferguson FM, Gray NS. Kinase inhibitors: the road ahead. Nat Rev Drug Discov. 2018;17(5):353‐377. [DOI] [PubMed] [Google Scholar]
- 8. Hogg SJ, Beavis PA, Dawson MA, Johnstone RW. Targeting the epigenetic regulation of antitumour immunity. Nat Rev Drug Discov. 2020;19(11):776‐800. [DOI] [PubMed] [Google Scholar]
- 9. Greene BL, Kang G, Cui C, et al. Ribonucleotide reductases: structure, chemistry, and metabolism suggest new therapeutic targets. Annu Rev Biochem. 2020;89:45‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bedard PL, Hyman DM, Davids MS, Siu LL. Small molecules, big impact: 20 years of targeted therapy in oncology. Lancet. 2020;395(10229):1078‐1088. [DOI] [PubMed] [Google Scholar]
- 11. Merino D, Kelly GL, Lessene G, Wei AH, Roberts AW, Strasser A. BH3‐mimetic drugs: blazing the trail for new cancer medicines. Cancer Cell. 2018;34(6):879‐891. [DOI] [PubMed] [Google Scholar]
- 12. Nakayama A, Nagashima T, Nishizono Y, et al. Characterisation of a novel KRAS G12C inhibitor ASP2453 that shows potent anti‐tumour activity in KRAS G12C‐mutated preclinical models. Br J Cancer. 2022;126(5):744‐753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hong DS, Fakih MG, Strickler JH, et al. KRAS(G12C) inhibition with sotorasib in advanced solid tumors. N Engl J Med. 2020;383(13):1207‐1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Case LB. Membranes regulate biomolecular condensates. Nat Cell Biol. 2022;24(4):404‐405. [DOI] [PubMed] [Google Scholar]
- 15. Kurreck A, Stintzing S, Modest DP. Efficacy, molecular biology, quality of life, or economic aspects: what do we really focus on? J Clin Oncol. 2022;40(11):1260‐1262. [DOI] [PubMed] [Google Scholar]
- 16. Aggarwal C, Albelda SM. Molecular characterization of malignant mesothelioma: time for new targets? Cancer Discov. 2018;8(12):1508‐1510. [DOI] [PubMed] [Google Scholar]
- 17. Dietlein F, Wang AB, Fagre C, et al. Genome‐wide analysis of somatic noncoding mutation patterns in cancer. Science. 2022;376(6589):eabg5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Veerman GDM, Hussaarts K, Jansman FGA, Koolen SWL, van Leeuwen RWF, Mathijssen RHJ. Clinical implications of food‐drug interactions with small‐molecule kinase inhibitors. Lancet Oncol. 2020;21(5):e265‐e279. [DOI] [PubMed] [Google Scholar]
- 19. Katoh M. Fibroblast growth factor receptors as treatment targets in clinical oncology. Nat Rev Clin Oncol. 2019;16(2):105‐122. [DOI] [PubMed] [Google Scholar]
- 20. Cully M. Trial watch: Multikinase‐targeting therapy finds potential niche in thyroid cancer. Nat Rev Drug Discov. 2015;14(4):229. [DOI] [PubMed] [Google Scholar]
- 21. Gild ML, Tsang VHM, Clifton‐Bligh RJ, Robinson BG. Multikinase inhibitors in thyroid cancer: timing of targeted therapy. Nat Rev Endocrinol. 2021;17(4):225‐234. [DOI] [PubMed] [Google Scholar]
- 22. Klaeger S, Heinzlmeir S, Wilhelm M, et al. The target landscape of clinical kinase drugs. Science. 2017;358(6367):eaan4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Laetsch TW, DuBois SG, Bender JG, Macy ME, Moreno L. Opportunities and challenges in drug development for pediatric cancers. Cancer Discov. 2021;11(3):545‐559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Battisti NML, Decoster L, Williams GR, Kanesvaran R, Wildiers H, Ring A. Targeted therapies in older adults with solid tumors. J Clin Oncol. 2021;39(19):2128‐2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McGee SL, Hargreaves M. Exercise adaptations: molecular mechanisms and potential targets for therapeutic benefit. Nat Rev Endocrinol. 2020;16(9):495‐505. [DOI] [PubMed] [Google Scholar]
- 26. Lee JH, Paull TT. Cellular functions of the protein kinase ATM and their relevance to human disease. Nat Rev Mol Cell Biol. 2021;22(12):796‐814. [DOI] [PubMed] [Google Scholar]
- 27. Vervoort SJ, Devlin JR, Kwiatkowski N, Teng M, Gray NS, Johnstone RW. Targeting transcription cycles in cancer. Nat Rev Cancer. 2022;22(1):5‐24. [DOI] [PubMed] [Google Scholar]
- 28. Suski JM, Braun M, Strmiska V, Sicinski P. Targeting cell‐cycle machinery in cancer. Cancer Cell. 2021;39(6):759‐778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Knighton DR, Zheng JH, Ten Eyck LF, et al. Crystal structure of the catalytic subunit of cyclic adenosine monophosphate‐dependent protein kinase. Science. 1991;253(5018):407‐414. [DOI] [PubMed] [Google Scholar]
- 30. Knighton DR, Zheng JH, Ten Eyck LF, Xuong NH, Taylor SS, Sowadski JM. Structure of a peptide inhibitor bound to the catalytic subunit of cyclic adenosine monophosphate‐dependent protein kinase. Science. 1991;253(5018):414‐420. [DOI] [PubMed] [Google Scholar]
- 31. Blencke S, Zech B, Engkvist O, et al. Characterization of a conserved structural determinant controlling protein kinase sensitivity to selective inhibitors. Chem Biol. 2004;11(5):691‐701. [DOI] [PubMed] [Google Scholar]
- 32. Nolen B, Taylor S, Ghosh G. Regulation of protein kinases; controlling activity through activation segment conformation. Mol Cell. 2004;15(5):661‐675. [DOI] [PubMed] [Google Scholar]
- 33. Kornev AP, Taylor SS, Ten Eyck LF. A helix scaffold for the assembly of active protein kinases. Proc Natl Acad Sci USA. 2008;105(38):14377‐14382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cully M. Rational drug design: Tuning kinase inhibitor residence time. Nat Rev Drug Discov. 2015;14(7):457. [DOI] [PubMed] [Google Scholar]
- 35. Abdeldayem A, Raouf YS, Constantinescu SN, Moriggl R, Gunning PT. Advances in covalent kinase inhibitors. Chem Soc Rev. 2020;49(9):2617‐2687. [DOI] [PubMed] [Google Scholar]
- 36. Druker BJ, Talpaz M, Resta DJ, et al. Efficacy and safety of a specific inhibitor of the BCR‐ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344(14):1031‐1037. [DOI] [PubMed] [Google Scholar]
- 37. O'Brien SG, Guilhot F, Larson RA, et al. Imatinib compared with interferon and low‐dose cytarabine for newly diagnosed chronic‐phase chronic myeloid leukemia. N Engl J Med. 2003;348(11):994‐1004. [DOI] [PubMed] [Google Scholar]
- 38. Hochhaus A, Larson RA, Guilhot F, et al. Long‐term outcomes of imatinib treatment for chronic myeloid leukemia. N Engl J Med. 2017;376(10):917‐927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Druker BJ, Guilhot F, O'Brien SG, et al. Five‐year follow‐up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355(23):2408‐2417. [DOI] [PubMed] [Google Scholar]
- 40. Druker BJ, Sawyers CL, Kantarjian H, et al. Activity of a specific inhibitor of the BCR‐ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med. 2001;344(14):1038‐1042. [DOI] [PubMed] [Google Scholar]
- 41. Shah NP, Kantarjian HM, Kim DW, et al. Intermittent target inhibition with dasatinib 100 mg once daily preserves efficacy and improves tolerability in imatinib‐resistant and ‐intolerant chronic‐phase chronic myeloid leukemia. J Clin Oncol. 2008;26(19):3204‐3212. [DOI] [PubMed] [Google Scholar]
- 42. Kantarjian H, Giles F, Wunderle L, et al. Nilotinib in imatinib‐resistant CML and Philadelphia chromosome‐positive ALL. N Engl J Med. 2006;354(24):2542‐2551. [DOI] [PubMed] [Google Scholar]
- 43. Cortes JE, Kantarjian HM, Brümmendorf TH, et al. Safety and efficacy of bosutinib (SKI‐606) in chronic phase Philadelphia chromosome‐positive chronic myeloid leukemia patients with resistance or intolerance to imatinib. Blood. 2011;118(17):4567‐4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cortes J, Apperley J, Lomaia E, et al. Ponatinib dose‐ranging study in chronic‐phase chronic myeloid leukemia: a randomized, open‐label phase 2 clinical trial. Blood. 2021;138(21):2042‐2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kantarjian H, Shah NP, Hochhaus A, et al. Dasatinib versus imatinib in newly diagnosed chronic‐phase chronic myeloid leukemia. N Engl J Med. 2010;362(24):2260‐2270. [DOI] [PubMed] [Google Scholar]
- 46. Saglio G, Kim DW, Issaragrisil S, et al. Nilotinib versus imatinib for newly diagnosed chronic myeloid leukemia. N Engl J Med. 2010;362(24):2251‐2259. [DOI] [PubMed] [Google Scholar]
- 47. Cortes JE, Gambacorti‐Passerini C, Deininger MW, et al. Bosutinib versus imatinib for newly diagnosed chronic myeloid leukemia: results from the randomized before trial. J Clin Oncol. 2018;36(3):231‐237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lombardo LJ, Lee FY, Chen P, et al. Discovery of N‐(2‐chloro‐6‐methyl‐ phenyl)‐2‐(6‐(4‐(2‐hydroxyethyl)‐ piperazin‐1‐yl)‐2‐methylpyrimidin‐4‐ ylamino)thiazole‐5‐carboxamide (BMS‐354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J Med Chem. 2004;47(27):6658‐6661. [DOI] [PubMed] [Google Scholar]
- 49. Golas JM, Arndt K, Etienne C, et al. SKI‐606, a 4‐anilino‐3‐quinolinecarbonitrile dual inhibitor of Src and Abl kinases, is a potent antiproliferative agent against chronic myelogenous leukemia cells in culture and causes regression of K562 xenografts in nude mice. Cancer Res. 2003;63(2):375‐381. [PubMed] [Google Scholar]
- 50. O'Hare T, Walters DK, Stoffregen EP, et al. In vitro activity of Bcr‐Abl inhibitors AMN107 and BMS‐354825 against clinically relevant imatinib‐resistant Abl kinase domain mutants. Cancer Res. 2005;65(11):4500‐4505. [DOI] [PubMed] [Google Scholar]
- 51. Golemovic M, Verstovsek S, Giles F, et al. AMN107, a novel aminopyrimidine inhibitor of Bcr‐Abl, has in vitro activity against imatinib‐resistant chronic myeloid leukemia. Clin Cancer Res. 2005;11(13):4941‐4947. [DOI] [PubMed] [Google Scholar]
- 52. O'Hare T, Shakespeare WC, Zhu X, et al. AP24534, a pan‐BCR‐ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation‐based resistance. Cancer Cell. 2009;16(5):401‐412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Réa D, Mauro MJ, Boquimpani C, et al. A phase 3, open‐label, randomized study of asciminib, a STAMP inhibitor, vs bosutinib in CML after 2 or more prior TKIs. Blood. 2021;138(21):2031‐2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wylie AA, Schoepfer J, Jahnke W, et al. The allosteric inhibitor ABL001 enables dual targeting of BCR‐ABL1. Nature. 2017;543(7647):733‐737. [DOI] [PubMed] [Google Scholar]
- 55. Hughes TP, Mauro MJ, Cortes JE, et al. Asciminib in chronic myeloid leukemia after ABL kinase inhibitor failure. N Engl J Med. 2019;381(24):2315‐2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Jabbour E, Pui CH, Kantarjian H. Progress and innovations in the management of adult acute lymphoblastic leukemia. JAMA Oncol. 2018;4(10):1413‐1420. [DOI] [PubMed] [Google Scholar]
- 57. Daver N, Thomas D, Ravandi F, et al. Final report of a phase II study of imatinib mesylate with hyper‐CVAD for the front‐line treatment of adult patients with Philadelphia chromosome‐positive acute lymphoblastic leukemia. Haematologica. 2015;100(5):653‐661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ravandi F, O'Brien SM, Cortes JE, et al. Long‐term follow‐up of a phase 2 study of chemotherapy plus dasatinib for the initial treatment of patients with Philadelphia chromosome‐positive acute lymphoblastic leukemia. Cancer. 2015;121(23):4158‐4164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Rousselot P, Coudé MM, Gokbuget N, et al. Dasatinib and low‐intensity chemotherapy in elderly patients with Philadelphia chromosome‐positive ALL. Blood. 2016;128(6):774‐782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Jabbour E, Short NJ, Ravandi F, et al. Combination of hyper‐CVAD with ponatinib as first‐line therapy for patients with Philadelphia chromosome‐positive acute lymphoblastic leukaemia: long‐term follow‐up of a single‐centre, phase 2 study. Lancet Haematol. 2018;5(12):e618‐e627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Jabbour E, Kantarjian H, Ravandi F, et al. Combination of hyper‐CVAD with ponatinib as first‐line therapy for patients with Philadelphia chromosome‐positive acute lymphoblastic leukaemia: a single‐centre, phase 2 study. Lancet Oncol. 2015;16(15):1547‐1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Cortes JE, Kantarjian H, Shah NP, et al. Ponatinib in refractory Philadelphia chromosome‐positive leukemias. N Engl J Med. 2012;367(22):2075‐2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Fielding AK, Rowe JM, Buck G, et al. UKALLXII/ECOG2993: addition of imatinib to a standard treatment regimen enhances long‐term outcomes in Philadelphia positive acute lymphoblastic leukemia. Blood. 2014;123(6):843‐850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Martinelli G, Papayannidis C, Piciocchi A, et al. INCB84344‐201: ponatinib and steroids in frontline therapy for unfit patients with Ph+ acute lymphoblastic leukemia. Blood Adv. 2022;6(6):1742‐1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zhang L, Meng L, Liu B, et al. Flumatinib versus imatinib for newly diagnosed chronic phase chronic myeloid leukemia: a phase III, randomized, open‐label, multi‐center FESTnd study. Clin Cancer Res. 2021;27(1):70‐77. [DOI] [PubMed] [Google Scholar]
- 66. Kwak JY, Kim SH, Oh SJ, et al. Phase III clinical trial (RERISE study) results of efficacy and safety of radotinib compared with imatinib in newly diagnosed chronic phase chronic myeloid leukemia. Clin Cancer Res. 2017;23(23):7180‐7188. [DOI] [PubMed] [Google Scholar]
- 67. Dhillon S. Olverembatinib: first approval. Drugs. 2022;82(4):469‐475. [DOI] [PubMed] [Google Scholar]
- 68. Cortes J, Talpaz M, Smith HP, et al. Phase 1 dose‐finding study of rebastinib (DCC‐2036) in patients with relapsed chronic myeloid leukemia and acute myeloid leukemia. Haematologica. 2017;102(3):519‐528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Naqvi K, Jabbour E, Skinner J, et al. Long‐term follow‐up of lower dose dasatinib (50 mg daily) as frontline therapy in newly diagnosed chronic‐phase chronic myeloid leukemia. Cancer. 2020;126(1):67‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Naqvi K, Jabbour E, Skinner J, et al. Early results of lower dose dasatinib (50 mg daily) as frontline therapy for newly diagnosed chronic‐phase chronic myeloid leukemia. Cancer. 2018;124(13):2740‐2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Saussele S, Richter J, Guilhot J, et al. Discontinuation of tyrosine kinase inhibitor therapy in chronic myeloid leukaemia (EURO‐SKI): a prespecified interim analysis of a prospective, multicentre, non‐randomised, trial. Lancet Oncol. 2018;19(6):747‐757. [DOI] [PubMed] [Google Scholar]
- 72. Imagawa J, Tanaka H, Okada M, et al. Discontinuation of dasatinib in patients with chronic myeloid leukaemia who have maintained deep molecular response for longer than 1 year (DADI trial): a multicentre phase 2 trial. Lancet Haematol. 2015;2(12):e528‐e535. [DOI] [PubMed] [Google Scholar]
- 73. Lennartsson J, Ronnstrand L. Stem cell factor receptor/c‐Kit: from basic science to clinical implications. Physiol Rev. 2012;92(4):1619‐1649. [DOI] [PubMed] [Google Scholar]
- 74. Pathania S, Pentikainen OT, Singh PK. A holistic view on c‐Kit in cancer: structure, signaling, pathophysiology and its inhibitors. Biochim Biophys Acta Rev Cancer. 2021;1876(2):188631. [DOI] [PubMed] [Google Scholar]
- 75. Klug LR, Khosroyani HM, Kent JD, Heinrich MC. New treatment strategies for advanced‐stage gastrointestinal stromal tumours. Nat Rev Clin Oncol. 2022;19(5):328‐341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Blay JY, Kang YK, Nishida T, von Mehren M. Gastrointestinal stromal tumours. Nat Rev Dis Primers. 2021;7(1):22. [DOI] [PubMed] [Google Scholar]
- 77. Dagher R, Cohen M, Williams G, et al. Approval summary: imatinib mesylate in the treatment of metastatic and/or unresectable malignant gastrointestinal stromal tumors. Clin Cancer Res. 2002;8(10):3034‐3038. [PubMed] [Google Scholar]
- 78. Druker BJ, Tamura S, Buchdunger E, et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr‐Abl positive cells. Nat Med. 1996;2(5):561‐566. [DOI] [PubMed] [Google Scholar]
- 79. Tuveson DA, Willis NA, Jacks T, et al. STI571 inactivation of the gastrointestinal stromal tumor c‐KIT oncoprotein: biological and clinical implications. Oncogene. 2001;20(36):5054‐5058. [DOI] [PubMed] [Google Scholar]
- 80. Heinrich MC, Corless CL, Demetri GD, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21(23):4342‐4349. [DOI] [PubMed] [Google Scholar]
- 81. Heinrich MC, Owzar K, Corless CL, et al. Correlation of kinase genotype and clinical outcome in the North American Intergroup Phase III Trial of imatinib mesylate for treatment of advanced gastrointestinal stromal tumor: CALGB 150105 Study by Cancer and Leukemia Group B and Southwest Oncology Group. J Clin Oncol. 2008;26(33):5360‐5367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Dematteo RP, Ballman KV, Antonescu CR, et al. Adjuvant imatinib mesylate after resection of localised, primary gastrointestinal stromal tumour: a randomised, double‐blind, placebo‐controlled trial. Lancet. 2009;373(9669):1097‐1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Corless CL, Ballman KV, Antonescu CR, et al. Pathologic and molecular features correlate with long‐term outcome after adjuvant therapy of resected primary GI stromal tumor: the ACOSOG Z9001 trial. J Clin Oncol. 2014;32(15):1563‐1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Joensuu H, Eriksson M, Sundby Hall K, et al. One vs three years of adjuvant imatinib for operable gastrointestinal stromal tumor: a randomized trial. Jama. 2012;307(12):1265‐1272. [DOI] [PubMed] [Google Scholar]
- 85. Joensuu H, Eriksson M, Sundby Hall K, et al. Survival outcomes associated with 3 years vs 1 year of adjuvant imatinib for patients with high‐risk gastrointestinal stromal tumors: an analysis of a randomized clinical trial after 10‐year follow‐up. JAMA Oncol. 2020;6(8):1241‐1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Joensuu H, Wardelmann E, Sihto H, et al. Effect of KIT and PDGFRA mutations on survival in patients with gastrointestinal stromal tumors treated with adjuvant imatinib: an exploratory analysis of a randomized clinical trial. JAMA Oncol. 2017;3(5):602‐609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. von Mehren M, Joensuu H. Gastrointestinal stromal tumors. J Clin Oncol. 2018;36(2):136‐143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Smith BD, Kaufman MD, Lu WP, et al. Ripretinib (DCC‐2618) is a switch control kinase inhibitor of a broad spectrum of oncogenic and drug‐resistant KIT and PDGFRA variants. Cancer Cell. 2019;35(5):738‐751. e739. [DOI] [PubMed] [Google Scholar]
- 89. Janku F, Abdul Razak AR, Chi P, et al. Switch control inhibition of KIT and PDGFRA in patients with advanced gastrointestinal stromal tumor: a phase I study of ripretinib. J Clin Oncol. 2020;38(28):3294‐3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Blay JY, Serrano C, Heinrich MC, et al. Ripretinib in patients with advanced gastrointestinal stromal tumours (INVICTUS): a double‐blind, randomised, placebo‐controlled, phase 3 trial. Lancet Oncol. 2020;21(7):923‐934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Bauer S, Heinrich MC, George S, et al. Clinical activity of ripretinib in patients with advanced gastrointestinal stromal tumor harboring heterogeneous KIT/PDGFRA Mutations in the phase III INVICTUS study. Clin Cancer Res. 2021;27(23):6333‐6342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. George S, von Mehren M, Fletcher JA, et al. Phase II study of ponatinib in advanced gastrointestinal stromal tumors: efficacy, safety, and impact of liquid biopsy and other biomarkers. Clin Cancer Res. 2022;28(7):1268‐1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Blay JY, Shen L, Kang YK, et al. Nilotinib versus imatinib as first‐line therapy for patients with unresectable or metastatic gastrointestinal stromal tumours (ENESTg1): a randomised phase 3 trial. Lancet Oncol. 2015;16(5):550‐560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Hodi FS, Corless CL, Giobbie‐Hurder A, et al. Imatinib for melanomas harboring mutationally activated or amplified KIT arising on mucosal, acral, and chronically sun‐damaged skin. J Clin Oncol. 2013;31(26):3182‐3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Guo J, Si L, Kong Y, et al. Phase II, open‐label, single‐arm trial of imatinib mesylate in patients with metastatic melanoma harboring c‐Kit mutation or amplification. J Clin Oncol. 2011;29(21):2904‐2909. [DOI] [PubMed] [Google Scholar]
- 96. Carvajal RD, Antonescu CR, Wolchok JD, et al. KIT as a therapeutic target in metastatic melanoma. Jama. 2011;305(22):2327‐2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Guo J, Carvajal RD, Dummer R, et al. Efficacy and safety of nilotinib in patients with KIT‐mutated metastatic or inoperable melanoma: final results from the global, single‐arm, phase II TEAM trial. Ann Oncol. 2017;28(6):1380‐1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Kalinsky K, Lee S, Rubin KM, et al. A phase 2 trial of dasatinib in patients with locally advanced or stage IV mucosal, acral, or vulvovaginal melanoma: a trial of the ECOG‐ACRIN Cancer Research Group (E2607). Cancer. 2017;123(14):2688‐2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Le Cesne A, Blay JY, Bui BN, et al. Phase II study of oral masitinib mesilate in imatinib‐naïve patients with locally advanced or metastatic gastro‐intestinal stromal tumour (GIST). Eur J Cancer. 2010;46(8):1344‐1351. [DOI] [PubMed] [Google Scholar]
- 100. Adenis A, Blay JY, Bui‐Nguyen B, et al. Masitinib in advanced gastrointestinal stromal tumor (GIST) after failure of imatinib: a randomized controlled open‐label trial. Ann Oncol. 2014;25(9):1762‐1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Tibes R, Fine G, Choy G, et al. A phase I, first‐in‐human dose‐escalation study of amuvatinib, a multi‐targeted tyrosine kinase inhibitor, in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2013;71(2):463‐471. [DOI] [PubMed] [Google Scholar]
- 102. Kettle JG, Anjum R, Barry E, et al. Discovery of N‐(4‐{[5‐Fluoro‐7‐(2‐methoxyethoxy)quinazolin‐4‐yl]amino}phenyl)‐2‐[4‐(propan‐2‐yl)‐1 H‐1,2,3‐triazol‐1‐yl]acetamide (AZD3229), a potent pan‐KIT mutant inhibitor for the treatment of gastrointestinal stromal tumors. J Med Chem. 2018;61(19):8797‐8810. [DOI] [PubMed] [Google Scholar]
- 103. Kazlauskas A. PDGFs and their receptors. Gene. 2017;614:1‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Cao Y. Multifarious functions of PDGFs and PDGFRs in tumor growth and metastasis. Trends Mol Med. 2013;19(8):460‐473. [DOI] [PubMed] [Google Scholar]
- 105. Ostman A. PDGF receptors in tumor stroma: biological effects and associations with prognosis and response to treatment. Adv Drug Deliv Rev. 2017;121:117‐123. [DOI] [PubMed] [Google Scholar]
- 106. Corless CL, Fletcher JA, Heinrich MC. Biology of gastrointestinal stromal tumors. J Clin Oncol. 2004;22(18):3813‐3825. [DOI] [PubMed] [Google Scholar]
- 107. Wu CP, Lusvarghi S, Wang JC, et al. Avapritinib: a selective inhibitor of KIT and PDGFRα that reverses ABCB1 and ABCG2‐mediated multidrug resistance in cancer cell lines. Mol Pharm. 2019;16(7):3040‐3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Gebreyohannes YK, Wozniak A, Zhai ME, et al. Robust activity of avapritinib, potent and highly selective inhibitor of mutated KIT, in patient‐derived xenograft models of gastrointestinal stromal tumors. Clin Cancer Res. 2019;25(2):609‐618. [DOI] [PubMed] [Google Scholar]
- 109. Heinrich MC, Jones RL, von Mehren M, et al. Avapritinib in advanced PDGFRA D842V‐mutant gastrointestinal stromal tumour (NAVIGATOR): a multicentre, open‐label, phase 1 trial. Lancet Oncol. 2020;21(7):935‐946. [DOI] [PubMed] [Google Scholar]
- 110. Jones RL, Serrano C, von Mehren M, et al. Avapritinib in unresectable or metastatic PDGFRA D842V‐mutant gastrointestinal stromal tumours: Long‐term efficacy and safety data from the NAVIGATOR phase I trial. Eur J Cancer. 2021;145:132‐142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Kang YK, George S, Jones RL, et al. Avapritinib versus regorafenib in locally advanced unresectable or metastatic GI stromal tumor: a randomized, open‐label phase III study. J Clin Oncol. 2021;39(28):3128‐3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Tap WD, Jones RL, Van Tine BA, et al. Olaratumab and doxorubicin versus doxorubicin alone for treatment of soft‐tissue sarcoma: an open‐label phase 1b and randomised phase 2 trial. Lancet. 2016;388(10043):488‐497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Tap WD, Wagner AJ, Schöffski P, et al. Effect of doxorubicin plus olaratumab vs doxorubicin plus placebo on survival in patients with advanced soft tissue sarcomas: the ANNOUNCE randomized clinical trial. Jama. 2020;323(13):1266‐1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. McGuire WP, Penson RT, Gore M, et al. Randomized phase II study of the PDGFRα antibody olaratumab plus liposomal doxorubicin versus liposomal doxorubicin alone in patients with platinum‐refractory or platinum‐resistant advanced ovarian cancer. BMC Cancer. 2018;18(1):1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Gerber DE, Swanson P, Lopez‐Chavez A, et al. Phase II study of olaratumab with paclitaxel/carboplatin (P/C) or P/C alone in previously untreated advanced NSCLC. Lung Cancer. 2017;111:108‐115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Hakenberg OW, Perez‐Gracia JL, Castellano D, et al. Randomised phase II study of second‐line olaratumab with mitoxantrone/prednisone versus mitoxantrone/prednisone alone in metastatic castration‐resistant prostate cancer. Eur J Cancer. 2019;107:186‐195. [DOI] [PubMed] [Google Scholar]
- 117. Phuphanich S, Raizer J, Chamberlain M, et al. Phase II study of MEDI‐575, an anti‐platelet‐derived growth factor‐α antibody, in patients with recurrent glioblastoma. J Neurooncol. 2017;131(1):185‐191. [DOI] [PubMed] [Google Scholar]
- 118. Wagner AJ, Kindler H, Gelderblom H, et al. A phase II study of a human anti‐PDGFRα monoclonal antibody (olaratumab, IMC‐3G3) in previously treated patients with metastatic gastrointestinal stromal tumors. Ann Oncol. 2017;28(3):541‐546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Lewis NL, Lewis LD, Eder JP, et al. Phase I study of the safety, tolerability, and pharmacokinetics of oral CP‐868,596, a highly specific platelet‐derived growth factor receptor tyrosine kinase inhibitor in patients with advanced cancers. J Clin Oncol. 2009;27(31):5262‐5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Moy RH, Greally M, Chou JF, et al. Phase I/Ib study of crenolanib with ramucirumab and paclitaxel as second‐line therapy for advanced esophagogastric adenocarcinoma. Cancer Chemother Pharmacol. 2022;89(2):255‐265. [DOI] [PubMed] [Google Scholar]
- 121. Batchelor TT, Gerstner ER, Ye X, et al. Feasibility, phase I, and phase II studies of tandutinib, an oral platelet‐derived growth factor receptor‐β tyrosine kinase inhibitor, in patients with recurrent glioblastoma. Neuro Oncol. 2017;19(4):567‐575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Shepard DR, Cooney MM, Elson P, et al. A phase II study of tandutinib (MLN518), a selective inhibitor of type III tyrosine receptor kinases, in patients with metastatic renal cell carcinoma. Invest New Drugs. 2012;30(1):364‐367. [DOI] [PubMed] [Google Scholar]
- 123. Yarden Y, Pines G. The ERBB network: at last, cancer therapy meets systems biology. Nat Rev Cancer. 2012;12(8):553‐563. [DOI] [PubMed] [Google Scholar]
- 124. Tan AC, Tan DSW. Targeted therapies for lung cancer patients with oncogenic driver molecular alterations. J Clin Oncol. 2022;40(6):611‐625. [DOI] [PubMed] [Google Scholar]
- 125. Oh DY, Bang YJ. HER2‐targeted therapies: a role beyond breast cancer. Nat Rev Clin Oncol. 2020;17(1):33‐48. [DOI] [PubMed] [Google Scholar]
- 126. Gingras I, Gebhart G, de Azambuja E, Piccart‐Gebhart M. HER2‐positive breast cancer is lost in translation: time for patient‐centered research. Nat Rev Clin Oncol. 2017;14(11):669‐681. [DOI] [PubMed] [Google Scholar]
- 127. Goutsouliak K, Veeraraghavan J, Sethunath V, et al. Towards personalized treatment for early stage HER2‐positive breast cancer. Nat Rev Clin Oncol. 2020;17(4):233‐250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Cappuzzo F, Gregorc V, Rossi E, et al. Gefitinib in pretreated non‐small‐cell lung cancer (NSCLC): analysis of efficacy and correlation with HER2 and epidermal growth factor receptor expression in locally advanced or metastatic NSCLC. J Clin Oncol. 2003;21(14):2658‐2663. [DOI] [PubMed] [Google Scholar]
- 129. Mitsudomi T, Kosaka T, Endoh H, et al. Mutations of the epidermal growth factor receptor gene predict prolonged survival after gefitinib treatment in patients with non‐small‐cell lung cancer with postoperative recurrence. J Clin Oncol. 2005;23(11):2513‐2520. [DOI] [PubMed] [Google Scholar]
- 130. Tsao MS, Sakurada A, Cutz JC, et al. Erlotinib in lung cancer ‐ molecular and clinical predictors of outcome. N Engl J Med. 2005;353(2):133‐144. [DOI] [PubMed] [Google Scholar]
- 131. Eberhard DA, Johnson BE, Amler LC, et al. Mutations in the epidermal growth factor receptor and in KRAS are predictive and prognostic indicators in patients with non‐small‐cell lung cancer treated with chemotherapy alone and in combination with erlotinib. J Clin Oncol. 2005;23(25):5900‐5909. [DOI] [PubMed] [Google Scholar]
- 132. Sequist LV, Martins RG, Spigel D, et al. First‐line gefitinib in patients with advanced non‐small‐cell lung cancer harboring somatic EGFR mutations. J Clin Oncol. 2008;26(15):2442‐2449. [DOI] [PubMed] [Google Scholar]
- 133. Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as first‐line treatment for European patients with advanced EGFR mutation‐positive non‐small‐cell lung cancer (EURTAC): a multicentre, open‐label, randomised phase 3 trial. Lancet Oncol. 2012;13(3):239‐246. [DOI] [PubMed] [Google Scholar]
- 134. Zhou C, Wu YL, Chen G, et al. Erlotinib versus chemotherapy as first‐line treatment for patients with advanced EGFR mutation‐positive non‐small‐cell lung cancer (OPTIMAL, CTONG‐0802): a multicentre, open‐label, randomised, phase 3 study. Lancet Oncol. 2011;12(8):735‐742. [DOI] [PubMed] [Google Scholar]
- 135. Moore MJ, Goldstein D, Hamm J, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007;25(15):1960‐1966. [DOI] [PubMed] [Google Scholar]
- 136. Li D, Ambrogio L, Shimamura T, et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene. 2008;27(34):4702‐4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Engelman JA, Zejnullahu K, Gale CM, et al. PF00299804, an irreversible pan‐ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib. Cancer Res. 2007;67(24):11924‐11932. [DOI] [PubMed] [Google Scholar]
- 138. Yang JC, Hirsh V, Schuler M, et al. Symptom control and quality of life in LUX‐Lung 3: a phase III study of afatinib or cisplatin/pemetrexed in patients with advanced lung adenocarcinoma with EGFR mutations. J Clin Oncol. 2013;31(27):3342‐3350. [DOI] [PubMed] [Google Scholar]
- 139. Wu YL, Zhou C, Hu CP, et al. Afatinib versus cisplatin plus gemcitabine for first‐line treatment of Asian patients with advanced non‐small‐cell lung cancer harbouring EGFR mutations (LUX‐Lung 6): an open‐label, randomised phase 3 trial. Lancet Oncol. 2014;15(2):213‐222. [DOI] [PubMed] [Google Scholar]
- 140. Yang JC, Sequist LV, Geater SL, et al. Clinical activity of afatinib in patients with advanced non‐small‐cell lung cancer harbouring uncommon EGFR mutations: a combined post‐hoc analysis of LUX‐Lung 2, LUX‐Lung 3, and LUX‐Lung 6. Lancet Oncol. 2015;16(7):830‐838. [DOI] [PubMed] [Google Scholar]
- 141. Park K, Tan EH, O'Byrne K, et al. Afatinib versus gefitinib as first‐line treatment of patients with EGFR mutation‐positive non‐small‐cell lung cancer (LUX‐Lung 7): a phase 2B, open‐label, randomised controlled trial. Lancet Oncol. 2016;17(5):577‐589. [DOI] [PubMed] [Google Scholar]
- 142. Soria JC, Felip E, Cobo M, et al. Afatinib versus erlotinib as second‐line treatment of patients with advanced squamous cell carcinoma of the lung (LUX‐Lung 8): an open‐label randomised controlled phase 3 trial. Lancet Oncol. 2015;16(8):897‐907. [DOI] [PubMed] [Google Scholar]
- 143. Wu YL, Cheng Y, Zhou X, et al. Dacomitinib versus gefitinib as first‐line treatment for patients with EGFR‐mutation‐positive non‐small‐cell lung cancer (ARCHER 1050): a randomised, open‐label, phase 3 trial. Lancet Oncol. 2017;18(11):1454‐1466. [DOI] [PubMed] [Google Scholar]
- 144. Mok TS, Cheng Y, Zhou X, et al. Improvement in overall survival in a randomized study that compared dacomitinib with gefitinib in patients with advanced non‐small‐cell lung cancer and EGFR‐activating mutations. J Clin Oncol. 2018;36(22):2244‐2250. [DOI] [PubMed] [Google Scholar]
- 145. Cross DA, Ashton SE, Ghiorghiu S, et al. AZD9291, an irreversible EGFR TKI, overcomes T790M‐mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014;4(9):1046‐1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Soria JC, Ohe Y, Vansteenkiste J, et al. Osimertinib in untreated EGFR‐mutated advanced non‐small‐cell lung cancer. N Engl J Med. 2018;378(2):113‐125. [DOI] [PubMed] [Google Scholar]
- 147. Ramalingam SS, Vansteenkiste J, Planchard D, et al. Overall survival with osimertinib in untreated, EGFR‐mutated advanced NSCLC. N Engl J Med. 2020;382(1):41‐50. [DOI] [PubMed] [Google Scholar]
- 148. Reungwetwattana T, Nakagawa K, Cho BC, et al. CNS response to osimertinib versus standard epidermal growth factor receptor tyrosine kinase inhibitors in patients with untreated EGFR‐mutated advanced non‐small‐cell lung cancer. J Clin Oncol. 2018:Jco2018783118. [DOI] [PubMed] [Google Scholar]
- 149. Friedlaender A, Subbiah V, Russo A, et al. EGFR and HER2 exon 20 insertions in solid tumours: from biology to treatment. Nat Rev Clin Oncol. 2022;19(1):51‐69. [DOI] [PubMed] [Google Scholar]
- 150. Riely GJ, Neal JW, Camidge DR, et al. Activity and safety of mobocertinib (TAK‐788) in previously treated non‐small cell lung cancer with EGFR exon 20 insertion mutations from a phase I/II trial. Cancer Discov. 2021;11(7):1688‐1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Geyer CE, Forster J, Lindquist D, et al. Lapatinib plus capecitabine for HER2‐positive advanced breast cancer. N Engl J Med. 2006;355(26):2733‐2743. [DOI] [PubMed] [Google Scholar]
- 152. Saura C, Oliveira M, Feng YH, et al. Neratinib plus capecitabine versus lapatinib plus capecitabine in HER2‐positive metastatic breast cancer previously treated with ≥ 2 HER2‐directed regimens: phase III NALA trial. J Clin Oncol. 2020;38(27):3138‐3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Murthy RK, Loi S, Okines A, et al. Tucatinib, trastuzumab, and capecitabine for HER2‐positive metastatic breast cancer. N Engl J Med. 2020;382(7):597‐609. [DOI] [PubMed] [Google Scholar]
- 154. Lin NU, Borges V, Anders C, et al. Intracranial efficacy and survival with tucatinib plus trastuzumab and capecitabine for previously treated HER2‐positive breast cancer with brain metastases in the HER2CLIMB trial. J Clin Oncol. 2020;38(23):2610‐2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Johnston S, Pippen J, Jr. , Pivot X, et al. Lapatinib combined with letrozole versus letrozole and placebo as first‐line therapy for postmenopausal hormone receptor‐positive metastatic breast cancer. J Clin Oncol. 2009;27(33):5538‐5546. [DOI] [PubMed] [Google Scholar]
- 156. Chan A, Delaloge S, Holmes FA, et al. Neratinib after trastuzumab‐based adjuvant therapy in patients with HER2‐positive breast cancer (ExteNET): a multicentre, randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet Oncol. 2016;17(3):367‐377. [DOI] [PubMed] [Google Scholar]
- 157. Hosomi Y, Morita S, Sugawara S, et al. Gefitinib Alone Versus Gefitinib Plus Chemotherapy for Non‐Small‐Cell Lung Cancer With Mutated Epidermal Growth Factor Receptor: NEJ009 Study. J Clin Oncol. 2020;38(2):115‐123. [DOI] [PubMed] [Google Scholar]
- 158. Noronha V, Patil VM, Joshi A, et al. Gefitinib versus gefitinib plus pemetrexed and carboplatin chemotherapy in EGFR‐mutated lung cancer. J Clin Oncol. 2020;38(2):124‐136. [DOI] [PubMed] [Google Scholar]
- 159. Zhou Q, Xu CR, Cheng Y, et al. Bevacizumab plus erlotinib in Chinese patients with untreated, EGFR‐mutated, advanced NSCLC (ARTEMIS‐CTONG1509): A multicenter phase 3 study. Cancer Cell. 2021;39(9):1279‐1291. e1273. [DOI] [PubMed] [Google Scholar]
- 160. Akamatsu H, Toi Y, Hayashi H, et al. Efficacy of osimertinib plus bevacizumab vs osimertinib in patients with EGFR T790M‐mutated non‐small cell lung cancer previously treated with epidermal growth factor receptor‐tyrosine kinase inhibitor: West Japan Oncology Group 8715L Phase 2 Randomized Clinical Trial. JAMA Oncol. 2021;7(3):386‐394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Garassino MC, Cho BC, Kim JH, et al. Durvalumab as third‐line or later treatment for advanced non‐small‐cell lung cancer (ATLANTIC): an open‐label, single‐arm, phase 2 study. Lancet Oncol. 2018;19(4):521‐536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Sugiyama E, Togashi Y, Takeuchi Y, et al. Blockade of EGFR improves responsiveness to PD‐1 blockade in EGFR‐mutated non‐small cell lung cancer. Sci Immunol. 2020;5(43):eaav3937. [DOI] [PubMed] [Google Scholar]
- 163. Oxnard GR, Yang JC, Yu H, et al. TATTON: a multi‐arm, phase Ib trial of osimertinib combined with selumetinib, savolitinib, or durvalumab in EGFR‐mutant lung cancer. Ann Oncol. 2020;31(4):507‐516. [DOI] [PubMed] [Google Scholar]
- 164. Zhao Y, Liu J, Cai X, et al. Efficacy and safety of first line treatments for patients with advanced epidermal growth factor receptor mutated, non‐small cell lung cancer: systematic review and network meta‐analysis. Bmj. 2019;367:l5460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Kim ES. Olmutinib: first global approval. Drugs. 2016;76(11):1153‐1157. [DOI] [PubMed] [Google Scholar]
- 166. Deeks ED. Furmonertinib: first approval. Drugs. 2021;81(15):1775‐1780. [DOI] [PubMed] [Google Scholar]
- 167. Le X, Cornelissen R, Garassino M, et al. Poziotinib in non‐small‐cell lung cancer harboring HER2 exon 20 insertion mutations after prior therapies: ZENITH20‐2 trial. J Clin Oncol. 2022;40(7):710‐718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Kim TY, Han HS, Lee KW, et al. A phase I/II study of poziotinib combined with paclitaxel and trastuzumab in patients with HER2‐positive advanced gastric cancer. Gastric Cancer. 2019;22(6):1206‐1214. [DOI] [PubMed] [Google Scholar]
- 169. Park YH, Lee KH, Sohn JH, et al. A phase II trial of the pan‐HER inhibitor poziotinib, in patients with HER2‐positive metastatic breast cancer who had received at least two prior HER2‐directed regimens: results of the NOV120101‐203 trial. Int J Cancer. 2018;143(12):3240‐3247. [DOI] [PubMed] [Google Scholar]
- 170. Fernandez‐Martinez A, Krop IE, Hillman DW, et al. Survival, pathologic response, and genomics in CALGB 40601 (Alliance), a neoadjuvant phase III trial of paclitaxel‐trastuzumab with or without lapatinib in HER2‐positive breast cancer. J Clin Oncol. 2020;38(35):4184‐4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Xu B, Yan M, Ma F, et al. Pyrotinib plus capecitabine versus lapatinib plus capecitabine for the treatment of HER2‐positive metastatic breast cancer (PHOEBE): a multicentre, open‐label, randomised, controlled, phase 3 trial. Lancet Oncol. 2021;22(3):351‐360. [DOI] [PubMed] [Google Scholar]
- 172. Martínez P, Mak RH, Oxnard GR. Targeted therapy as an alternative to whole‐brain radiotherapy in EGFR‐mutant or ALK‐positive non‐small‐cell lung cancer with brain metastases. JAMA Oncol. 2017;3(9):1274‐1275. [DOI] [PubMed] [Google Scholar]
- 173. Costa DB, Shaw AT, Ou SH, et al. Clinical experience with crizotinib in patients with advanced ALK‐rearranged non‐small‐cell lung cancer and brain metastases. J Clin Oncol. 2015;33(17):1881‐1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Solomon BJ, Mok T, Kim DW, et al. First‐line crizotinib versus chemotherapy in ALK‐positive lung cancer. N Engl J Med. 2014;371(23):2167‐2177. [DOI] [PubMed] [Google Scholar]
- 175. Solomon BJ, Kim DW, Wu YL, et al. Final overall survival analysis from a study comparing first‐line crizotinib versus chemotherapy in ALK‐mutation‐positive non‐small‐cell lung cancer. J Clin Oncol. 2018;36(22):2251‐2258. [DOI] [PubMed] [Google Scholar]
- 176. Frampton JE. Crizotinib: a review of its use in the treatment of anaplastic lymphoma kinase‐positive, advanced non‐small cell lung cancer. Drugs. 2013;73(18):2031‐2051. [DOI] [PubMed] [Google Scholar]
- 177. Friboulet L, Li N, Katayama R, et al. The ALK inhibitor ceritinib overcomes crizotinib resistance in non‐small cell lung cancer. Cancer Discov. 2014;4(6):662‐673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Sakamoto H, Tsukaguchi T, Hiroshima S, et al. CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell. 2011;19(5):679‐690. [DOI] [PubMed] [Google Scholar]
- 179. Kim DW, Mehra R, Tan DSW, et al. Activity and safety of ceritinib in patients with ALK‐rearranged non‐small‐cell lung cancer (ASCEND‐1): updated results from the multicentre, open‐label, phase 1 trial. Lancet Oncol. 2016;17(4):452‐463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Gadgeel SM, Shaw AT, Govindan R, et al. Pooled analysis of CNS response to alectinib in two studies of pretreated patients with ALK‐positive non‐small‐cell lung cancer. J Clin Oncol. 2016;34(34):4079‐4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Gadgeel SM, Gandhi L, Riely GJ, et al. Safety and activity of alectinib against systemic disease and brain metastases in patients with crizotinib‐resistant ALK‐rearranged non‐small‐cell lung cancer (AF‐002JG): results from the dose‐finding portion of a phase 1/2 study. Lancet Oncol. 2014;15(10):1119‐1128. [DOI] [PubMed] [Google Scholar]
- 182. Camidge DR, Kim DW, Tiseo M, et al. Exploratory analysis of brigatinib activity in patients with anaplastic lymphoma kinase‐positive non‐small‐cell lung cancer and brain metastases in two clinical trials. J Clin Oncol. 2018;36(26):2693‐2701. [DOI] [PubMed] [Google Scholar]
- 183. Johnson TW, Richardson PF, Bailey S, et al. Discovery of (10R)‐7‐amino‐12‐fluoro‐2,10,16‐trimethyl‐15‐oxo‐10,15,16,17‐tetrahydro‐2H‐8,4‐(metheno)pyrazolo[4,3‐h][2,5,11]‐benzoxadiazacyclotetradecine‐3‐carbonitrile (PF‐06463922), a macrocyclic inhibitor of anaplastic lymphoma kinase (ALK) and c‐ros oncogene 1 (ROS1) with preclinical brain exposure and broad‐spectrum potency against ALK‐resistant mutations. J Med Chem. 2014;57(11):4720‐4744. [DOI] [PubMed] [Google Scholar]
- 184. Shaw AT, Felip E, Bauer TM, et al. Lorlatinib in non‐small‐cell lung cancer with ALK or ROS1 rearrangement: an international, multicentre, open‐label, single‐arm first‐in‐man phase 1 trial. Lancet Oncol. 2017;18(12):1590‐1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Soria JC, Tan DSW, Chiari R, et al. First‐line ceritinib versus platinum‐based chemotherapy in advanced ALK‐rearranged non‐small‐cell lung cancer (ASCEND‐4): a randomised, open‐label, phase 3 study. Lancet. 2017;389(10072):917‐929. [DOI] [PubMed] [Google Scholar]
- 186. Shaw AT, Bauer TM, de Marinis F, et al. First‐line lorlatinib or crizotinib in advanced ALK‐positive lung cancer. N Engl J Med. 2020;383(21):2018‐2029. [DOI] [PubMed] [Google Scholar]
- 187. Camidge DR, Kim HR, Ahn MJ, et al. Brigatinib versus crizotinib in ALK‐positive non‐small‐cell lung cancer. N Engl J Med. 2018;379(21):2027‐2039. [DOI] [PubMed] [Google Scholar]
- 188. Peters S, Camidge DR, Shaw AT, et al. Alectinib versus crizotinib in untreated ALK‐positive non‐small‐cell lung cancer. N Engl J Med. 2017;377(9):829‐838. [DOI] [PubMed] [Google Scholar]
- 189. Camidge DR, Kim HR, Ahn MJ, et al. Brigatinib versus crizotinib in advanced ALK inhibitor‐naive ALK‐positive non‐small cell lung cancer: second interim analysis of the phase III ALTA‐1L trial. J Clin Oncol. 2020;38(31):3592‐3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Golding B, Luu A, Jones R, Viloria‐Petit AM. The function and therapeutic targeting of anaplastic lymphoma kinase (ALK) in non‐small cell lung cancer (NSCLC). Mol Cancer. 2018;17(1):52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Chin LP, Soo RA, Soong R, Ou SH. Targeting ROS1 with anaplastic lymphoma kinase inhibitors: a promising therapeutic strategy for a newly defined molecular subset of non‐small‐cell lung cancer. J Thorac Oncol. 2012;7(11):1625‐1630. [DOI] [PubMed] [Google Scholar]
- 192. Shaw AT, Riely GJ, Bang YJ, et al. Crizotinib in ROS1‐rearranged advanced non‐small‐cell lung cancer (NSCLC): updated results, including overall survival, from PROFILE 1001. Ann Oncol. 2019;30(7):1121‐1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Doebele RC, Drilon A, Paz‐Ares L, et al. Entrectinib in patients with advanced or metastatic NTRK fusion‐positive solid tumours: integrated analysis of three phase 1–2 trials. Lancet Oncol. 2020;21(2):271‐282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Drilon A, Siena S, Dziadziuszko R, et al. Entrectinib in ROS1 fusion‐positive non‐small‐cell lung cancer: integrated analysis of three phase 1–2 trials. Lancet Oncol. 2020;21(2):261‐270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Schöffski P, Sufliarsky J, Gelderblom H, et al. Crizotinib in patients with advanced, inoperable inflammatory myofibroblastic tumours with and without anaplastic lymphoma kinase gene alterations (European Organisation for Research and Treatment of Cancer 90101 CREATE): a multicentre, single‐drug, prospective, non‐randomised phase 2 trial. Lancet Respir Med. 2018;6(6):431‐441. [DOI] [PubMed] [Google Scholar]
- 196. Gambacorti‐Passerini C, Orlov S, Zhang L, et al. Long‐term effects of crizotinib in ALK‐positive tumors (excluding NSCLC): a phase 1b open‐label study. Am J Hematol. 2018;93(5):607‐614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Shaw AT, Solomon BJ, Chiari R, et al. Lorlatinib in advanced ROS1‐positive non‐small‐cell lung cancer: a multicentre, open‐label, single‐arm, phase 1–2 trial. Lancet Oncol. 2019;20(12):1691‐1701. [DOI] [PubMed] [Google Scholar]
- 198. Spigel DR, Reynolds C, Waterhouse D, et al. Phase 1/2 study of the safety and tolerability of nivolumab plus crizotinib for the first‐line treatment of anaplastic lymphoma kinase translocation ‐ positive advanced non‐small cell lung cancer (CheckMate 370). J Thorac Oncol. 2018;13(5):682‐688. [DOI] [PubMed] [Google Scholar]
- 199. Felip E, de Braud FG, Maur M, et al. Ceritinib plus nivolumab in patients with advanced ALK‐rearranged non‐small cell lung cancer: results of an open‐label, multicenter, phase 1B study. J Thorac Oncol. 2020;15(3):392‐403. [DOI] [PubMed] [Google Scholar]
- 200. Horn L, Wang Z, Wu G, et al. Ensartinib vs crizotinib for patients with anaplastic lymphoma kinase‐positive non‐small cell lung cancer: a randomized clinical trial. JAMA Oncol. 2021;7(11):1617‐1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Yang Y, Zhou J, Zhou J, et al. Efficacy, safety, and biomarker analysis of ensartinib in crizotinib‐resistant, ALK‐positive non‐small‐cell lung cancer: a multicentre, phase 2 trial. Lancet Respir Med. 2020;8(1):45‐53. [DOI] [PubMed] [Google Scholar]
- 202. Papadopoulos KP, Borazanci E, Shaw AT, et al. U.S. Phase I First‐in‐human Study of Taletrectinib (DS‐6051b/AB‐106), a ROS1/TRK Inhibitor, in Patients with Advanced Solid Tumors. Clin Cancer Res. 2020;26(18):4785‐4794. [DOI] [PubMed] [Google Scholar]
- 203. Recondo G, Che J, Janne PA, Awad MM. Targeting MET dysregulation in cancer. Cancer Discov. 2020;10(7):922‐934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Comoglio PM, Trusolino L, Boccaccio C. Known and novel roles of the MET oncogene in cancer: a coherent approach to targeted therapy. Nat Rev Cancer. 2018;18(6):341‐358. [DOI] [PubMed] [Google Scholar]
- 205. Wolf J, Seto T, Han JY, et al. Capmatinib in MET Exon 14‐mutated or MET‐amplified non‐small‐cell lung cancer. N Engl J Med. 2020;383(10):944‐957. [DOI] [PubMed] [Google Scholar]
- 206. Paik PK, Felip E, Veillon R, et al. Tepotinib in non‐small‐cell lung cancer with MET Exon 14 skipping mutations. N Engl J Med. 2020;383(10):931‐943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Wu YL, Zhang L, Kim DW, et al. Phase Ib/II study of capmatinib (INC280) plus gefitinib after failure of epidermal growth factor receptor (EGFR) inhibitor therapy in patients with EGFR‐mutated, MET factor‐dysregulated non‐small‐cell lung cancer. J Clin Oncol. 2018;36(31):3101‐3109. [DOI] [PubMed] [Google Scholar]
- 208. Wu YL, Cheng Y, Zhou J, et al. Tepotinib plus gefitinib in patients with EGFR‐mutant non‐small‐cell lung cancer with MET overexpression or MET amplification and acquired resistance to previous EGFR inhibitor (INSIGHT study): an open‐label, phase 1b/2, multicentre, randomised trial. Lancet Respir Med. 2020;8(11):1132‐1143. [DOI] [PubMed] [Google Scholar]
- 209. Ryoo BY, Cheng AL, Ren Z, et al. Randomised Phase 1b/2 trial of tepotinib vs sorafenib in Asian patients with advanced hepatocellular carcinoma with MET overexpression. Br J Cancer. 2021;125(2):200‐208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Decaens T, Barone C, Assenat E, et al. Phase 1b/2 trial of tepotinib in sorafenib pretreated advanced hepatocellular carcinoma with MET overexpression. Br J Cancer. 2021;125(2):190‐199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Lu S, Fang J, Li X, et al. Once‐daily savolitinib in Chinese patients with pulmonary sarcomatoid carcinomas and other non‐small‐cell lung cancers harbouring MET exon 14 skipping alterations: a multicentre, single‐arm, open‐label, phase 2 study. Lancet Respir Med. 2021;9(10):1154‐1164. [DOI] [PubMed] [Google Scholar]
- 212. Drilon A, Hu ZI, Lai GGY, Tan DSW. Targeting RET‐driven cancers: lessons from evolving preclinical and clinical landscapes. Nat Rev Clin Oncol. 2018;15(3):151‐167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Subbiah V, Cote GJ. Advances in targeting RET‐dependent cancers. Cancer Discov. 2020;10(4):498‐505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Yoh K, Seto T, Satouchi M, et al. Vandetanib in patients with previously treated RET‐rearranged advanced non‐small‐cell lung cancer (LURET): an open‐label, multicentre phase 2 trial. Lancet Respir Med. 2017;5(1):42‐50. [DOI] [PubMed] [Google Scholar]
- 215. Drilon A, Rekhtman N, Arcila M, et al. Cabozantinib in patients with advanced RET‐rearranged non‐small‐cell lung cancer: an open‐label, single‐centre, phase 2, single‐arm trial. Lancet Oncol. 2016;17(12):1653‐1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Subbiah V, Hu MI, Wirth LJ, et al. Pralsetinib for patients with advanced or metastatic RET‐altered thyroid cancer (ARROW): a multi‐cohort, open‐label, registrational, phase 1/2 study. Lancet Diabetes Endocrinol. 2021;9(8):491‐501. [DOI] [PubMed] [Google Scholar]
- 217. Subbiah V, Velcheti V, Tuch BB, et al. Selective RET kinase inhibition for patients with RET‐altered cancers. Ann Oncol. 2018;29(8):1869‐1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Wirth LJ, Sherman E, Robinson B, et al. Efficacy of selpercatinib in RET‐altered thyroid cancers. N Engl J Med. 2020;383(9):825‐835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Drilon A, Fu S, Patel MR, et al. A Phase I/Ib trial of the VEGFR‐sparing multikinase RET inhibitor RXDX‐105. Cancer Discov. 2019;9(3):384‐395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Liu G, Chen T, Ding Z, Wang Y, Wei Y, Wei X. Inhibition of FGF‐FGFR and VEGF‐VEGFR signalling in cancer treatment. Cell Prolif. 2021;54(4):e13009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Geng L, Lam KSL, Xu A. The therapeutic potential of FGF21 in metabolic diseases: from bench to clinic. Nat Rev Endocrinol. 2020;16(11):654‐667. [DOI] [PubMed] [Google Scholar]
- 222. Loriot Y, Necchi A, Park SH, et al. Erdafitinib in locally advanced or metastatic urothelial carcinoma. N Engl J Med. 2019;381(4):338‐348. [DOI] [PubMed] [Google Scholar]
- 223. Abou‐Alfa GK, Sahai V, Hollebecque A, et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: a multicentre, open‐label, phase 2 study. Lancet Oncol. 2020;21(5):671‐684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Javle M, Lowery M, Shroff RT, et al. Phase II study of BGJ398 in patients with FGFR‐altered advanced cholangiocarcinoma. J Clin Oncol. 2018;36(3):276‐282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Bahleda R, Italiano A, Hierro C, et al. Multicenter phase I study of erdafitinib (JNJ‐42756493), oral pan‐fibroblast growth factor receptor inhibitor, in patients with advanced or refractory solid tumors. Clin Cancer Res. 2019;25(16):4888‐4897. [DOI] [PubMed] [Google Scholar]
- 226. Siefker‐Radtke AO, Necchi A, Park SH, et al. Efficacy and safety of erdafitinib in patients with locally advanced or metastatic urothelial carcinoma: long‐term follow‐up of a phase 2 study. Lancet Oncol. 2022;23(2):248‐258. [DOI] [PubMed] [Google Scholar]
- 227. Di Stefano AL, Fucci A, Frattini V, et al. Detection, characterization, and inhibition of FGFR‐TACC fusions in IDH wild‐type glioma. Clin Cancer Res. 2015;21(14):3307‐3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Lassman AB, Sepúlveda‐Sánchez JM, Cloughesy TF, et al. Infigratinib in patients with recurrent gliomas and FGFR alterations: a multicenter phase II study. Clin Cancer Res. 2022;28(11):2270‐2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Nogova L, Sequist LV, Perez Garcia JM, et al. Evaluation of BGJ398, a fibroblast growth factor receptor 1–3 kinase inhibitor, in patients with advanced solid tumors harboring genetic alterations in fibroblast growth factor receptors: results of a global phase i, dose‐escalation and dose‐expansion study. J Clin Oncol. 2017;35(2):157‐165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Meric‐Bernstam F, Bahleda R, Hierro C, et al. Futibatinib, an irreversible FGFR1‐4 inhibitor, in patients with advanced solid tumors harboring FGF/FGFR aberrations: a phase I dose‐expansion study. Cancer Discov. 2022;12(2):402‐415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Schuler M, Cho BC, Sayehli CM, et al. Rogaratinib in patients with advanced cancers selected by FGFR mRNA expression: a phase 1 dose‐escalation and dose‐expansion study. Lancet Oncol. 2019;20(10):1454‐1466. [DOI] [PubMed] [Google Scholar]
- 232. Lam WS, Creaney J, Chen FK, et al. A phase II trial of single oral FGF inhibitor, AZD4547, as second or third line therapy in malignant pleural mesothelioma. Lung Cancer. 2020;140:87‐92. [DOI] [PubMed] [Google Scholar]
- 233. Aggarwal C, Redman MW, Lara PN, Jr. , et al. SWOG S1400D (NCT02965378), a phase II study of the fibroblast growth factor receptor inhibitor AZD4547 in previously treated patients with fibroblast growth factor pathway‐activated stage IV squamous cell lung cancer (Lung‐MAP Substudy). J Thorac Oncol. 2019;14(10):1847‐1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Van Cutsem E, Bang YJ, Mansoor W, et al. A randomized, open‐label study of the efficacy and safety of AZD4547 monotherapy versus paclitaxel for the treatment of advanced gastric adenocarcinoma with FGFR2 polysomy or gene amplification. Ann Oncol. 2017;28(6):1316‐1324. [DOI] [PubMed] [Google Scholar]
- 235. Amatu A, Sartore‐Bianchi A, Bencardino K, Pizzutilo EG, Tosi F, Siena S. Tropomyosin receptor kinase (TRK) biology and the role of NTRK gene fusions in cancer. Ann Oncol. 2019;30(Suppl 8):viii5‐viii15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Cocco E, Scaltriti M, Drilon A. NTRK fusion‐positive cancers and TRK inhibitor therapy. Nat Rev Clin Oncol. 2018;15(12):731‐747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Drilon A. TRK inhibitors in TRK fusion‐positive cancers. Ann Oncol. 2019;30(Suppl 8):viii23‐viii30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Solomon JP, Benayed R, Hechtman JF, Ladanyi M. Identifying patients with NTRK fusion cancer. Ann Oncol. 2019;30(Suppl 8):viii16‐viii22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Huang FW, Feng FY. A tumor‐agnostic NTRK (TRK) inhibitor. Cell. 2019;177(1):8. [DOI] [PubMed] [Google Scholar]
- 240. Hong DS, DuBois SG, Kummar S, et al. Larotrectinib in patients with TRK fusion‐positive solid tumours: a pooled analysis of three phase 1/2 clinical trials. Lancet Oncol. 2020;21(4):531‐540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Doz F, van Tilburg CM, Geoerger B, et al. Efficacy and safety of larotrectinib in TRK fusion‐positive primary central nervous system tumors. Neuro Oncol. 2022;24(6):997‐1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. DuBois SG, Laetsch TW, Federman N, et al. The use of neoadjuvant larotrectinib in the management of children with locally advanced TRK fusion sarcomas. Cancer. 2018;124(21):4241‐4247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Drilon A, Nagasubramanian R, Blake JF, et al. A next‐generation TRK kinase inhibitor overcomes acquired resistance to prior TRK kinase inhibition in patients with TRK fusion‐positive solid tumors. Cancer Discov. 2017;7(9):963‐972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Ivanov SV, Panaccione A, Brown B, et al. TrkC signaling is activated in adenoid cystic carcinoma and requires NT‐3 to stimulate invasive behavior. Oncogene. 2013;32(32):3698‐3710. [DOI] [PubMed] [Google Scholar]
- 245. Bullinger L, Döhner K, Döhner H. Genomics of acute myeloid leukemia diagnosis and pathways. J Clin Oncol. 2017;35(9):934‐946. [DOI] [PubMed] [Google Scholar]
- 246. Döhner H, Wei AH, Löwenberg B. Towards precision medicine for AML. Nat Rev Clin Oncol. 2021;18(9):577‐590. [DOI] [PubMed] [Google Scholar]
- 247. Kazi JU, Rönnstrand L. FMS‐like tyrosine kinase 3/FLT3: from basic science to clinical implications. Physiol Rev. 2019;99(3):1433‐1466. [DOI] [PubMed] [Google Scholar]
- 248. Wu M, Li C, Zhu X. FLT3 inhibitors in acute myeloid leukemia. J Hematol Oncol. 2018;11(1):133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. Schlenk RF, Weber D, Fiedler W, et al. Midostaurin added to chemotherapy and continued single‐agent maintenance therapy in acute myeloid leukemia with FLT3‐ITD. Blood. 2019;133(8):840‐851. [DOI] [PubMed] [Google Scholar]
- 250. Fischer T, Stone RM, Deangelo DJ, et al. Phase IIB trial of oral Midostaurin (PKC412), the FMS‐like tyrosine kinase 3 receptor (FLT3) and multi‐targeted kinase inhibitor, in patients with acute myeloid leukemia and high‐risk myelodysplastic syndrome with either wild‐type or mutated FLT3. J Clin Oncol. 2010;28(28):4339‐4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Gotlib J, Kluin‐Nelemans HC, George TI, et al. Efficacy and safety of midostaurin in advanced systemic mastocytosis. N Engl J Med. 2016;374(26):2530‐2541. [DOI] [PubMed] [Google Scholar]
- 252. Perl AE. Availability of FLT3 inhibitors: how do we use them? Blood. 2019;134(9):741‐745. [DOI] [PubMed] [Google Scholar]
- 253. Perl AE, Martinelli G, Cortes JE, et al. Gilteritinib or chemotherapy for relapsed or refractory FLT3‐mutated AML. N Engl J Med. 2019;381(18):1728‐1740. [DOI] [PubMed] [Google Scholar]
- 254. Strati P, Kantarjian H, Ravandi F, et al. Phase I/II trial of the combination of midostaurin (PKC412) and 5‐azacytidine for patients with acute myeloid leukemia and myelodysplastic syndrome. Am J Hematol. 2015;90(4):276‐281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Cortes JE, Khaled S, Martinelli G, et al. Quizartinib versus salvage chemotherapy in relapsed or refractory FLT3‐ITD acute myeloid leukaemia (QuANTUM‐R): a multicentre, randomised, controlled, open‐label, phase 3 trial. Lancet Oncol. 2019;20(7):984‐997. [DOI] [PubMed] [Google Scholar]
- 256. Sandmaier BM, Khaled S, Oran B, Gammon G, Trone D, Frankfurt O. Results of a phase 1 study of quizartinib as maintenance therapy in subjects with acute myeloid leukemia in remission following allogeneic hematopoietic stem cell transplant. Am J Hematol. 2018;93(2):222‐231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257. Altman JK, Foran JM, Pratz KW, Trone D, Cortes JE, Tallman MS. Phase 1 study of quizartinib in combination with induction and consolidation chemotherapy in patients with newly diagnosed acute myeloid leukemia. Am J Hematol. 2018;93(2):213‐221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258. Galanis A, Ma H, Rajkhowa T, et al. Crenolanib is a potent inhibitor of FLT3 with activity against resistance‐conferring point mutants. Blood. 2014;123(1):94‐100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Lee HK, Kim HW, Lee IY, et al. G‐749, a novel FLT3 kinase inhibitor, can overcome drug resistance for the treatment of acute myeloid leukemia. Blood. 2014;123(14):2209‐2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Li C, Liu L, Liang L, et al. AMG 925 is a dual FLT3/CDK4 inhibitor with the potential to overcome FLT3 inhibitor resistance in acute myeloid leukemia. Mol Cancer Ther. 2015;14(2):375‐383. [DOI] [PubMed] [Google Scholar]
- 261. Hanahan D. Hallmarks of cancer: new dimensions. Cancer Discov. 2022;12(1):31‐46. [DOI] [PubMed] [Google Scholar]
- 262. Hernandez R, Põder J, LaPorte KM, Malek TR. Engineering IL‐2 for immunotherapy of autoimmunity and cancer. Nat Rev Immunol. 2022. 10.1038/s41577-022-00680-w [DOI] [PubMed] [Google Scholar]
- 263. Garlanda C, Mantovani A. Interleukin‐1 in tumor progression, therapy, and prevention. Cancer Cell. 2021;39(8):1023‐1027. [DOI] [PubMed] [Google Scholar]
- 264. Batlle E, Massagué J. Transforming growth factor‐β signaling in immunity and cancer. Immunity. 2019;50(4):924‐940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265. Propper DJ, Balkwill FR. Harnessing cytokines and chemokines for cancer therapy. Nat Rev Clin Oncol. 2022;19(4):237‐253. [DOI] [PubMed] [Google Scholar]
- 266. Buechler MB, Fu W, Turley SJ. Fibroblast‐macrophage reciprocal interactions in health, fibrosis, and cancer. Immunity. 2021;54(5):903‐915. [DOI] [PubMed] [Google Scholar]
- 267. Lamb YN. Pexidartinib: First Approval. Drugs. 2019;79(16):1805‐1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Parker BS, Rautela J, Hertzog PJ. Antitumour actions of interferons: implications for cancer therapy. Nat Rev Cancer. 2016;16(3):131‐144. [DOI] [PubMed] [Google Scholar]
- 269. Smith CC, Zhang C, Lin KC, et al. Characterizing and overriding the structural mechanism of the quizartinib‐resistant FLT3 “gatekeeper” F691L mutation with PLX3397. Cancer Discov. 2015;5(6):668‐679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270. Tap WD, Gelderblom H, Palmerini E, et al. Pexidartinib versus placebo for advanced tenosynovial giant cell tumour (ENLIVEN): a randomised phase 3 trial. Lancet. 2019;394(10197):478‐487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271. von Tresckow B, Morschhauser F, Ribrag V, et al. An open‐label, multicenter, phase I/II study of JNJ‐40346527, a CSF‐1R inhibitor, in patients with relapsed or refractory hodgkin lymphoma. Clin Cancer Res. 2015;21(8):1843‐1850. [DOI] [PubMed] [Google Scholar]
- 272. Papadopoulos KP, Gluck L, Martin LP, et al. First‐in‐human study of AMG 820, a monoclonal anti‐colony‐stimulating factor 1 receptor antibody, in patients with advanced solid tumors. Clin Cancer Res. 2017;23(19):5703‐5710. [DOI] [PubMed] [Google Scholar]
- 273. Gomez‐Roca CA, Italiano A, Le Tourneau C, et al. Phase I study of emactuzumab single agent or in combination with paclitaxel in patients with advanced/metastatic solid tumors reveals depletion of immunosuppressive M2‐like macrophages. Ann Oncol. 2019;30(8):1381‐1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274. Cassier PA, Italiano A, Gomez‐Roca CA, et al. CSF1R inhibition with emactuzumab in locally advanced diffuse‐type tenosynovial giant cell tumours of the soft tissue: a dose‐escalation and dose‐expansion phase 1 study. Lancet Oncol. 2015;16(8):949‐956. [DOI] [PubMed] [Google Scholar]
- 275. Ronkina N, Gaestel M. MAPK‐activated protein kinases: servant or partner? Annu Rev Biochem. 2022;91:505‐540. [DOI] [PubMed] [Google Scholar]
- 276. Yuan J, Dong X, Yap J, Hu J. The MAPK and AMPK signalings: interplay and implication in targeted cancer therapy. J Hematol Oncol. 2020;13(1):113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277. Dillon M, Lopez A, Lin E, Sales D, Perets R, Jain P. Progress on Ras/MAPK signaling research and targeting in blood and solid cancers. Cancers (Basel). 2021;13(20):5059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278. Yaeger R, Corcoran RB. Targeting alterations in the RAF‐MEK Pathway. Cancer Discov. 2019;9(3):329‐341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279. Lavoie H, Gagnon J, Therrien M. ERK signalling: a master regulator of cell behaviour, life and fate. Nat Rev Mol Cell Biol. 2020;21(10):607‐632. [DOI] [PubMed] [Google Scholar]
- 280. Moore AR, Rosenberg SC, McCormick F, Malek S. RAS‐targeted therapies: is the undruggable drugged? Nat Rev Drug Discov. 2020;19(8):533‐552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281. Schadendorf D, van Akkooi ACJ, Berking C, et al. Melanoma. Lancet. 2018;392(10151):971‐984. [DOI] [PubMed] [Google Scholar]
- 282. Patton EE, Mueller KL, Adams DJ, et al. Melanoma models for the next generation of therapies. Cancer Cell. 2021;39(5):610‐631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283. Luke JJ, Flaherty KT, Ribas A, Long GV. Targeted agents and immunotherapies: optimizing outcomes in melanoma. Nat Rev Clin Oncol. 2017;14(8):463‐482. [DOI] [PubMed] [Google Scholar]
- 284. Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507‐2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285. Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF‐mutated metastatic melanoma: a multicentre, open‐label, phase 3 randomised controlled trial. Lancet. 2012;380(9839):358‐365. [DOI] [PubMed] [Google Scholar]
- 286. Flaherty KT, Robert C, Hersey P, et al. Improved survival with MEK inhibition in BRAF‐mutated melanoma. N Engl J Med. 2012;367(2):107‐114. [DOI] [PubMed] [Google Scholar]
- 287. Giugliano F, Crimini E, Tarantino P, et al. First line treatment of BRAF mutated advanced melanoma: does one size fit all? Cancer Treat Rev. 2021;99:102253. [DOI] [PubMed] [Google Scholar]
- 288. Larkin J, Ascierto PA, Dréno B, et al. Combined vemurafenib and cobimetinib in BRAF‐mutated melanoma. N Engl J Med. 2014;371(20):1867‐1876. [DOI] [PubMed] [Google Scholar]
- 289. Long GV, Flaherty KT, Stroyakovskiy D, et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K‐mutant melanoma: long‐term survival and safety analysis of a phase 3 study. Ann Oncol. 2017;28(7):1631‐1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290. Grob JJ, Amonkar MM, Karaszewska B, et al. Comparison of dabrafenib and trametinib combination therapy with vemurafenib monotherapy on health‐related quality of life in patients with unresectable or metastatic cutaneous BRAF Val600‐mutation‐positive melanoma (COMBI‐v): results of a phase 3, open‐label, randomised trial. Lancet Oncol. 2015;16(13):1389‐1398. [DOI] [PubMed] [Google Scholar]
- 291. Davies MA, Saiag P, Robert C, et al. Dabrafenib plus trametinib in patients with BRAF(V600)‐mutant melanoma brain metastases (COMBI‐MB): a multicentre, multicohort, open‐label, phase 2 trial. Lancet Oncol. 2017;18(7):863‐873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292. Dummer R, Ascierto PA, Gogas HJ, et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF‐mutant melanoma (COLUMBUS): a multicentre, open‐label, randomised phase 3 trial. Lancet Oncol. 2018;19(5):603‐615. [DOI] [PubMed] [Google Scholar]
- 293. Ribas A, Lawrence D, Atkinson V, et al. Combined BRAF and MEK inhibition with PD‐1 blockade immunotherapy in BRAF‐mutant melanoma. Nat Med. 2019;25(6):936‐940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294. Dummer R, Hauschild A, Santinami M, et al. Five‐year analysis of adjuvant dabrafenib plus trametinib in stage III melanoma. N Engl J Med. 2020;383(12):1139‐1148. [DOI] [PubMed] [Google Scholar]
- 295. Diamond EL, Subbiah V, Lockhart AC, et al. Vemurafenib for BRAF V600‐mutant erdheim‐chester disease and langerhans cell histiocytosis: analysis of data from the histology‐independent, phase 2, open‐label VE‐BASKET study. JAMA Oncol. 2018;4(3):384‐388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296. Fangusaro J, Onar‐Thomas A, Young Poussaint T, et al. Selumetinib in paediatric patients with BRAF‐aberrant or neurofibromatosis type 1‐associated recurrent, refractory, or progressive low‐grade glioma: a multicentre, phase 2 trial. Lancet Oncol. 2019;20(7):1011‐1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297. Planchard D, Besse B, Groen HJM, et al. Dabrafenib plus trametinib in patients with previously treated BRAF(V600E)‐mutant metastatic non‐small cell lung cancer: an open‐label, multicentre phase 2 trial. Lancet Oncol. 2016;17(7):984‐993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298. Subbiah V, Kreitman RJ, Wainberg ZA, et al. Dabrafenib and trametinib treatment in patients with locally advanced or metastatic BRAF V600‐mutant anaplastic thyroid cancer. J Clin Oncol. 2018;36(1):7‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299. Salama AKS, Li S, Macrae ER, et al. Dabrafenib and trametinib in patients with tumors with BRAF(V600E) mutations: results of the NCI‐MATCH trial subprotocol H. J Clin Oncol. 2020;38(33):3895‐3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300. Brose MS, Cabanillas ME, Cohen EE, et al. Vemurafenib in patients with BRAF(V600E)‐positive metastatic or unresectable papillary thyroid cancer refractory to radioactive iodine: a non‐randomised, multicentre, open‐label, phase 2 trial. Lancet Oncol. 2016;17(9):1272‐1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301. Hyman DM, Puzanov I, Subbiah V, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med. 2015;373(8):726‐736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302. Gershenson DM, Miller A, Brady WE, et al. Trametinib versus standard of care in patients with recurrent low‐grade serous ovarian cancer (GOG 281/LOGS): an international, randomised, open‐label, multicentre, phase 2/3 trial. Lancet. 2022;399(10324):541‐553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303. Wen PY, Stein A, van den Bent M, et al. Dabrafenib plus trametinib in patients with BRAF(V600E)‐mutant low‐grade and high‐grade glioma (ROAR): a multicentre, open‐label, single‐arm, phase 2, basket trial. Lancet Oncol. 2022;23(1):53‐64. [DOI] [PubMed] [Google Scholar]
- 304. Subbiah V, Lassen U, Élez E, et al. Dabrafenib plus trametinib in patients with BRAF(V600E)‐mutated biliary tract cancer (ROAR): a phase 2, open‐label, single‐arm, multicentre basket trial. Lancet Oncol. 2020;21(9):1234‐1243. [DOI] [PubMed] [Google Scholar]
- 305. Kopetz S, Desai J, Chan E, et al. Phase II pilot study of vemurafenib in patients with metastatic BRAF‐mutated colorectal cancer. J Clin Oncol. 2015;33(34):4032‐4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306. Tabernero J, Grothey A, Van Cutsem E, et al. Encorafenib plus cetuximab as a new standard of care for previously treated BRAF V600E‐mutant metastatic colorectal cancer: updated survival results and subgroup analyses from the BEACON study. J Clin Oncol. 2021;39(4):273‐284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307. Callahan MK, Chapman PB. PD‐1 or PD‐L1 blockade adds little to combination of BRAF and MEK inhibition in the treatment of BRAF V600‐mutated melanoma. J Clin Oncol. 2022;40(13):1393‐1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308. Gutzmer R, Stroyakovskiy D, Gogas H, et al. Atezolizumab, vemurafenib, and cobimetinib as first‐line treatment for unresectable advanced BRAF(V600) mutation‐positive melanoma (IMspire150): primary analysis of the randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet. 2020;395(10240):1835‐1844. [DOI] [PubMed] [Google Scholar]
- 309. Dummer R, Long GV, Robert C, et al. Randomized phase III trial evaluating spartalizumab plus dabrafenib and trametinib for BRAF V600‐mutant unresectable or metastatic melanoma. J Clin Oncol. 2022;40(13):1428‐1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310. Yen I, Shanahan F, Lee J, et al. ARAF mutations confer resistance to the RAF inhibitor belvarafenib in melanoma. Nature. 2021;594(7863):418‐423. [DOI] [PubMed] [Google Scholar]
- 311. Desai J, Gan H, Barrow C, et al. Phase I, open‐label, dose‐escalation/dose‐expansion study of lifirafenib (BGB‐283), an RAF family kinase inhibitor, in patients with solid tumors. J Clin Oncol. 2020;38(19):2140‐2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312. Lorusso PM, Adjei AA, Varterasian M, et al. Phase I and pharmacodynamic study of the oral MEK inhibitor CI‐1040 in patients with advanced malignancies. J Clin Oncol. 2005;23(23):5281‐5293. [DOI] [PubMed] [Google Scholar]
- 313. van Geel R, van Brummelen EMJ, Eskens F, et al. Phase 1 study of the pan‐HER inhibitor dacomitinib plus the MEK1/2 inhibitor PD‐0325901 in patients with KRAS‐mutation‐positive colorectal, non‐small‐cell lung and pancreatic cancer. Br J Cancer. 2020;122(8):1166‐1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314. Lim HY, Merle P, Weiss KH, et al. Phase II studies with refametinib or refametinib plus sorafenib in patients with RAS‐mutated hepatocellular carcinoma. Clin Cancer Res. 2018;24(19):4650‐4661. [DOI] [PubMed] [Google Scholar]
- 315. Van Laethem JL, Riess H, Jassem J, et al. Phase I/II study of refametinib (BAY 86–9766) in combination with gemcitabine in advanced pancreatic cancer. Target Oncol. 2017;12(1):97‐109. [DOI] [PubMed] [Google Scholar]
- 316. Guo C, Chénard‐Poirier M, Roda D, et al. Intermittent schedules of the oral RAF‐MEK inhibitor CH5126766/VS‐6766 in patients with RAS/RAF‐mutant solid tumours and multiple myeloma: a single‐centre, open‐label, phase 1 dose‐escalation and basket dose‐expansion study. Lancet Oncol. 2020;21(11):1478‐1488. [DOI] [PubMed] [Google Scholar]
- 317. Ono H, Horinaka M, Sukeno M, et al. Novel RAF/MEK inhibitor CH5126766/VS‐6766 has efficacy in combination with eribulin for the treatment of triple‐negative breast cancer. Cancer Sci. 2021;112(10):4166‐4175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318. Sullivan RJ, Infante JR, Janku F, et al. First‐in‐class ERK1/2 inhibitor ulixertinib (BVD‐523) in patients with MAPK mutant advanced solid tumors: results of a phase I dose‐escalation and expansion study. Cancer Discov. 2018;8(2):184‐195. [DOI] [PubMed] [Google Scholar]
- 319. Varga A, Soria JC, Hollebecque A, et al. A first‐in‐human phase I study to evaluate the ERK1/2 inhibitor GDC‐0994 in patients with advanced solid tumors. Clin Cancer Res. 2020;26(6):1229‐1236. [DOI] [PubMed] [Google Scholar]
- 320. Weekes C, Lockhart A, LoRusso P, et al. A phase Ib study to evaluate the MEK inhibitor cobimetinib in combination with the ERK1/2 inhibitor GDC‐0994 in patients with advanced solid tumors. Oncologist. 2020;25(10):833‐e1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321. Bhagwat SV, McMillen WT, Cai S, et al. ERK inhibitor LY3214996 targets ERK pathway‐driven cancers: a therapeutic approach toward precision medicine. Mol Cancer Ther. 2020;19(2):325‐336. [DOI] [PubMed] [Google Scholar]
- 322. Dancey J. mTOR signaling and drug development in cancer. Nat Rev Clin Oncol. 2010;7(4):209‐219. [DOI] [PubMed] [Google Scholar]
- 323. Bilanges B, Posor Y, Vanhaesebroeck B. PI3K isoforms in cell signalling and vesicle trafficking. Nat Rev Mol Cell Biol. 2019;20(9):515‐534. [DOI] [PubMed] [Google Scholar]
- 324. Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov. 2014;13(2):140‐156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325. Hoxhaj G, Manning BD. The PI3K‐AKT network at the interface of oncogenic signalling and cancer metabolism. Nat Rev Cancer. 2020;20(2):74‐88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326. Alzahrani AS. PI3K/Akt/mTOR inhibitors in cancer: At the bench and bedside. Semin Cancer Biol. 2019;59:125‐132. [DOI] [PubMed] [Google Scholar]
- 327. Hillmann P, Fabbro D. PI3K/mTOR pathway inhibition: opportunities in oncology and rare genetic diseases. Int J Mol Sci. 2019;20(22):5792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328. Shams R, Ito Y, Miyatake H. Mapping of mTOR drug targets: featured platforms for anti‐cancer drug discovery. Pharmacol Ther. 2022;232:108012. [DOI] [PubMed] [Google Scholar]
- 329. Turner NC, Neven P, Loibl S, Andre F. Advances in the treatment of advanced oestrogen‐receptor‐positive breast cancer. Lancet. 2017;389(10087):2403‐2414. [DOI] [PubMed] [Google Scholar]
- 330. Kono M, Fujii T, Lim B, Karuturi MS, Tripathy D, Ueno NT. Androgen receptor function and androgen receptor‐targeted therapies in breast cancer: a review. JAMA Oncol. 2017;3(9):1266‐1273. [DOI] [PubMed] [Google Scholar]
- 331. Narayan P, Prowell TM, Gao JJ, et al. FDA approval summary: alpelisib plus fulvestrant for patients with HR‐positive, HER2‐negative, PIK3CA‐mutated, advanced or metastatic breast cancer. Clin Cancer Res. 2021;27(7):1842‐1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332. Juric D, Castel P, Griffith M, et al. Convergent loss of PTEN leads to clinical resistance to a PI(3)Kα inhibitor. Nature. 2015;518(7538):240‐244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333. Savas P, Lo LL, Luen SJ, et al. Alpelisib monotherapy for PI3K‐altered, pre‐treated advanced breast cancer: a phase 2 study. Cancer Discov. 2022;12(9):2058‐2073. [DOI] [PubMed] [Google Scholar]
- 334. Rugo HS, Lerebours F, Ciruelos E, et al. Alpelisib plus fulvestrant in PIK3CA‐mutated, hormone receptor‐positive advanced breast cancer after a CDK4/6 inhibitor (BYLieve): one cohort of a phase 2, multicentre, open‐label, non‐comparative study. Lancet Oncol. 2021;22(4):489‐498. [DOI] [PubMed] [Google Scholar]
- 335. Hus I, Pula B, Robak T. PI3K inhibitors for the treatment of chronic lymphocytic leukemia: current status and future perspectives. Cancers (Basel). 2022;14(6):1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336. Janku F, Yap TA, Meric‐Bernstam F. Targeting the PI3K pathway in cancer: are we making headway? Nat Rev Clin Oncol. 2018;15(5):273‐291. [DOI] [PubMed] [Google Scholar]
- 337. Meng D, He W, Zhang Y, et al. Development of PI3K inhibitors: advances in clinical trials and new strategies (Review). Pharmacol Res. 2021;173:105900. [DOI] [PubMed] [Google Scholar]
- 338. Roskoski R, Jr. Properties of FDA‐approved small molecule phosphatidylinositol 3‐kinase inhibitors prescribed for the treatment of malignancies. Pharmacol Res. 2021;168:105579. [DOI] [PubMed] [Google Scholar]
- 339. Furman RR, Sharman JP, Coutre SE, et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med. 2014;370(11):997‐1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340. Sharman JP, Coutre SE, Furman RR, et al. Final results of a randomized, phase III study of rituximab with or without idelalisib followed by open‐label idelalisib in patients with relapsed chronic lymphocytic leukemia. J Clin Oncol. 2019;37(16):1391‐1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341. Flinn IW, Hillmen P, Montillo M, et al. The phase 3 DUO trial: duvelisib vs ofatumumab in relapsed and refractory CLL/SLL. Blood. 2018;132(23):2446‐2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342. Flinn IW, Miller CB, Ardeshna KM, et al. DYNAMO: a phase II study of duvelisib (IPI‐145) in patients with refractory indolent non‐Hodgkin lymphoma. J Clin Oncol. 2019;37(11):912‐922. [DOI] [PubMed] [Google Scholar]
- 343. Patnaik A, Appleman LJ, Tolcher AW, et al. First‐in‐human phase I study of copanlisib (BAY 80–6946), an intravenous pan‐class I phosphatidylinositol 3‐kinase inhibitor, in patients with advanced solid tumors and non‐Hodgkin's lymphomas. Ann Oncol. 2016;27(10):1928‐1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344. Matasar MJ, Capra M, Özcan M, et al. Copanlisib plus rituximab versus placebo plus rituximab in patients with relapsed indolent non‐Hodgkin lymphoma (CHRONOS‐3): a double‐blind, randomised, placebo‐controlled, phase 3 trial. Lancet Oncol. 2021;22(5):678‐689. [DOI] [PubMed] [Google Scholar]
- 345. Burris HA, 3rd , Flinn IW, Patel MR, et al. Umbralisib, a novel PI3Kδ and casein kinase‐1ε inhibitor, in relapsed or refractory chronic lymphocytic leukaemia and lymphoma: an open‐label, phase 1, dose‐escalation, first‐in‐human study. Lancet Oncol. 2018;19(4):486‐496. [DOI] [PubMed] [Google Scholar]
- 346. Fowler NH, Samaniego F, Jurczak W, et al. Umbralisib, a dual PI3Kδ/CK1ε inhibitor in patients with relapsed or refractory indolent lymphoma. J Clin Oncol. 2021;39(15):1609‐1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347. Busaidy NL, Farooki A, Dowlati A, et al. Management of metabolic effects associated with anticancer agents targeting the PI3K‐Akt‐mTOR pathway. J Clin Oncol. 2012;30(23):2919‐2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348. Bendell JC, Rodon J, Burris HA, et al. Phase I, dose‐escalation study of BKM120, an oral pan‐Class I PI3K inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2012;30(3):282‐290. [DOI] [PubMed] [Google Scholar]
- 349. Sarker D, Ang JE, Baird R, et al. First‐in‐human phase I study of pictilisib (GDC‐0941), a potent pan‐class I phosphatidylinositol‐3‐kinase (PI3K) inhibitor, in patients with advanced solid tumors. Clin Cancer Res. 2015;21(1):77‐86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350. Shapiro GI, Rodon J, Bedell C, et al. Phase I safety, pharmacokinetic, and pharmacodynamic study of SAR245408 (XL147), an oral pan‐class I PI3K inhibitor, in patients with advanced solid tumors. Clin Cancer Res. 2014;20(1):233‐245. [DOI] [PubMed] [Google Scholar]
- 351. Wen PY, Touat M, Alexander BM, et al. Buparlisib in patients with recurrent glioblastoma harboring phosphatidylinositol 3‐kinase pathway activation: an open‐label, multicenter, multi‐arm, phase II trial. J Clin Oncol. 2019;37(9):741‐750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352. Soulières D, Faivre S, Mesía R, et al. Buparlisib and paclitaxel in patients with platinum‐pretreated recurrent or metastatic squamous cell carcinoma of the head and neck (BERIL‐1): a randomised, double‐blind, placebo‐controlled phase 2 trial. Lancet Oncol. 2017;18(3):323‐335. [DOI] [PubMed] [Google Scholar]
- 353. Mayer IA, Abramson VG, Isakoff SJ, et al. Stand up to cancer phase Ib study of pan‐phosphoinositide‐3‐kinase inhibitor buparlisib with letrozole in estrogen receptor‐positive/human epidermal growth factor receptor 2‐negative metastatic breast cancer. J Clin Oncol. 2014;32(12):1202‐1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354. Di Leo A, Johnston S, Lee KS, et al. Buparlisib plus fulvestrant in postmenopausal women with hormone‐receptor‐positive, HER2‐negative, advanced breast cancer progressing on or after mTOR inhibition (BELLE‐3): a randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet Oncol. 2018;19(1):87‐100. [DOI] [PubMed] [Google Scholar]
- 355. Bissig‐Choisat B, Kettlun‐Leyton C, Legras XD, et al. Novel patient‐derived xenograft and cell line models for therapeutic testing of pediatric liver cancer. J Hepatol. 2016;65(2):325‐333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356. Juric D, Krop I, Ramanathan RK, et al. Phase I dose‐escalation study of taselisib, an oral PI3K inhibitor, in patients with advanced solid tumors. Cancer Discov. 2017;7(7):704‐715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357. Saura C, Hlauschek D, Oliveira M, et al. Neoadjuvant letrozole plus taselisib versus letrozole plus placebo in postmenopausal women with oestrogen receptor‐positive, HER2‐negative, early‐stage breast cancer (LORELEI): a multicentre, randomised, double‐blind, placebo‐controlled, phase 2 trial. Lancet Oncol. 2019;20(9):1226‐1238. [DOI] [PubMed] [Google Scholar]
- 358. Dent S, Cortés J, Im YH, et al. Phase III randomized study of taselisib or placebo with fulvestrant in estrogen receptor‐positive, PIK3CA‐mutant, HER2‐negative, advanced breast cancer: the SANDPIPER trial. Ann Oncol. 2021;32(2):197‐207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359. Sarker D, Dawson NA, Aparicio AM, et al. A Phase I, open‐label, dose‐finding study of GSK2636771, a PI3Kβ inhibitor, administered with enzalutamide in patients with metastatic castration‐resistant prostate cancer. Clin Cancer Res. 2021;27(19):5248‐5257. [DOI] [PubMed] [Google Scholar]
- 360. Howell SJ, Casbard A, Carucci M, et al. Fulvestrant plus capivasertib versus placebo after relapse or progression on an aromatase inhibitor in metastatic, oestrogen receptor‐positive, HER2‐negative breast cancer (FAKTION): overall survival, updated progression‐free survival, and expanded biomarker analysis from a randomised, phase 2 trial. Lancet Oncol. 2022;23(7):851‐864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361. Blagden SP, Hamilton AL, Mileshkin L, et al. Phase IB Dose Escalation and Expansion Study of AKT Inhibitor Afuresertib with Carboplatin and Paclitaxel in Recurrent Platinum‐resistant Ovarian Cancer. Clin Cancer Res. 2019;25(5):1472‐1478. [DOI] [PubMed] [Google Scholar]
- 362. Shi Z, Wulfkuhle J, Nowicka M, et al. Functional mapping of AKT signaling and biomarkers of response from the FAIRLANE Trial of neoadjuvant ipatasertib plus paclitaxel for triple‐negative breast cancer. Clin Cancer Res. 2022;28(5):993‐1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363. Sweeney C, Bracarda S, Sternberg CN, et al. Ipatasertib plus abiraterone and prednisolone in metastatic castration‐resistant prostate cancer (IPATential150): a multicentre, randomised, double‐blind, phase 3 trial. Lancet. 2021;398(10295):131‐142. [DOI] [PubMed] [Google Scholar]
- 364. Chien AJ, Tripathy D, Albain KS, et al. MK‐2206 and standard neoadjuvant chemotherapy improves response in patients with human epidermal growth factor receptor 2‐positive and/or hormone receptor‐negative breast cancers in the I‐SPY 2 trial. J Clin Oncol. 2020;38(10):1059‐1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365. Jonasch E, Hasanov E, Corn PG, et al. A randomized phase 2 study of MK‐2206 versus everolimus in refractory renal cell carcinoma. Ann Oncol. 2017;28(4):804‐808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366. Chung V, McDonough S, Philip PA, et al. Effect of selumetinib and MK‐2206 vs oxaliplatin and fluorouracil in patients with metastatic pancreatic cancer after prior therapy: SWOG S1115 study randomized clinical trial. JAMA Oncol. 2017;3(4):516‐522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367. Powles T, Wheater M, Din O, et al. A randomised phase 2 study of AZD2014 versus everolimus in patients with VEGF‐refractory metastatic clear cell renal cancer. Eur Urol. 2016;69(3):450‐456. [DOI] [PubMed] [Google Scholar]
- 368. Davis SL, Ionkina AA, Bagby SM, et al. Preclinical and dose‐finding phase I trial results of combined treatment with a TORC1/2 inhibitor (TAK‐228) and aurora A kinase inhibitor (alisertib) in solid tumors. Clin Cancer Res. 2020;26(17):4633‐4642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369. Heudel P, Frenel JS, Dalban C, et al. Safety and efficacy of the mTOR inhibitor, vistusertib, combined with anastrozole in patients with hormone receptor‐positive recurrent or metastatic endometrial cancer: the VICTORIA multicenter, open‐label, phase 1/2 randomized clinical trial. JAMA Oncol. 2022;8(7):1001‐1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370. Ediriweera MK, Tennekoon KH, Samarakoon SR. Role of the PI3K/AKT/mTOR signaling pathway in ovarian cancer: Biological and therapeutic significance. Semin Cancer Biol. 2019;59:147‐160. [DOI] [PubMed] [Google Scholar]
- 371. Sweeney CJ, Percent IJ, Babu S, et al. Phase Ib/II study of enzalutamide with samotolisib (LY3023414) or placebo in patients with metastatic castration‐resistant prostate cancer. Clin Cancer Res. 2022;28(11):2237‐2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372. Wen PY, Cloughesy TF, Olivero AG, et al. First‐in‐human phase I study to evaluate the brain‐penetrant PI3K/mTOR inhibitor GDC‐0084 in patients with progressive or recurrent high‐grade glioma. Clin Cancer Res. 2020;26(8):1820‐1828. [DOI] [PubMed] [Google Scholar]
- 373. Villarino AV, Gadina M, O'Shea JJ, Kanno Y. SnapShot: Jak‐STAT signaling II. Cell. 2020;181(7):1696‐1696. e1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374. McLornan DP, Pope JE, Gotlib J, Harrison CN. Current and future status of JAK inhibitors. Lancet. 2021;398(10302):803‐816. [DOI] [PubMed] [Google Scholar]
- 375. Levy DE, Darnell JE, Jr. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3(9):651‐662. [DOI] [PubMed] [Google Scholar]
- 376. Tzeng HT, Chyuan IT, Lai JH. Targeting the JAK‐STAT pathway in autoimmune diseases and cancers: a focus on molecular mechanisms and therapeutic potential. Biochem Pharmacol. 2021;193:114760. [DOI] [PubMed] [Google Scholar]
- 377. Spivak JL. Myeloproliferative neoplasms. N Engl J Med. 2017;376(22):2168‐2181. [DOI] [PubMed] [Google Scholar]
- 378. Greenfield G, McMullin MF, Mills K. Molecular pathogenesis of the myeloproliferative neoplasms. J Hematol Oncol. 2021;14(1):103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379. Harrison C, Kiladjian JJ, Al‐Ali HK, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. 2012;366(9):787‐798. [DOI] [PubMed] [Google Scholar]
- 380. Verstovsek S, Kantarjian H, Mesa RA, et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med. 2010;363(12):1117‐1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381. Verstovsek S, Mesa RA, Gotlib J, et al. A double‐blind, placebo‐controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012;366(9):799‐807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382. Pardanani A, Gotlib JR, Jamieson C, et al. Safety and efficacy of TG101348, a selective JAK2 inhibitor, in myelofibrosis. J Clin Oncol. 2011;29(7):789‐796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383. Pardanani A, Harrison C, Cortes JE, et al. Safety and efficacy of fedratinib in patients with primary or secondary myelofibrosis: a randomized clinical trial. JAMA Oncol. 2015;1(5):643‐651. [DOI] [PubMed] [Google Scholar]
- 384. Dao KT, Gotlib J, Deininger MMN, et al. Efficacy of ruxolitinib in patients with chronic neutrophilic leukemia and atypical chronic myeloid leukemia. J Clin Oncol. 2020;38(10):1006‐1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385. Hurwitz HI, Uppal N, Wagner SA, et al. Randomized, double‐blind, phase II study of ruxolitinib or placebo in combination with capecitabine in patients with metastatic pancreatic cancer for whom therapy with gemcitabine has failed. J Clin Oncol. 2015;33(34):4039‐4047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386. Zeiser R, von Bubnoff N, Butler J, et al. Ruxolitinib for glucocorticoid‐refractory acute graft‐versus‐host disease. N Engl J Med. 2020;382(19):1800‐1810. [DOI] [PubMed] [Google Scholar]
- 387. Plimack ER, Lorusso PM, McCoon P, et al. AZD1480: a phase I study of a novel JAK2 inhibitor in solid tumors. Oncologist. 2013;18(7):819‐820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388. Mesa RA, Kiladjian JJ, Catalano JV, et al. SIMPLIFY‐1: A phase III randomized trial of momelotinib versus ruxolitinib in janus kinase inhibitor‐naïve patients with myelofibrosis. J Clin Oncol. 2017;35(34):3844‐3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389. Harrison CN, Vannucchi AM, Platzbecker U, et al. Momelotinib versus best available therapy in patients with myelofibrosis previously treated with ruxolitinib (SIMPLIFY 2): a randomised, open‐label, phase 3 trial. Lancet Haematol. 2018;5(2):e73‐e81. [DOI] [PubMed] [Google Scholar]
- 390. Mascarenhas J, Hoffman R, Talpaz M, et al. Pacritinib vs best available therapy, including ruxolitinib, in patients with myelofibrosis: a randomized clinical trial. JAMA Oncol. 2018;4(5):652‐659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391. Mesa RA, Vannucchi AM, Mead A, et al. Pacritinib versus best available therapy for the treatment of myelofibrosis irrespective of baseline cytopenias (PERSIST‐1): an international, randomised, phase 3 trial. Lancet Haematol. 2017;4(5):e225‐e236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392. Phillips TJ, Forero‐Torres A, Sher T, et al. Phase 1 study of the PI3Kδ inhibitor INCB040093 ± JAK1 inhibitor itacitinib in relapsed/refractory B‐cell lymphoma. Blood. 2018;132(3):293‐306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393. Lukasik P, Baranowska‐Bosiacka I, Kulczycka K, Gutowska I. Inhibitors of cyclin‐dependent kinases: types and their mechanism of action. Int J Mol Sci. 2021;22(6):2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394. Barnum KJ, O'Connell MJ. Cell cycle regulation by checkpoints. Methods Mol Biol. 2014;1170:29‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395. Roskoski R, Jr. Cyclin‐dependent protein serine/threonine kinase inhibitors as anticancer drugs. Pharmacol Res. 2019;139:471‐488. [DOI] [PubMed] [Google Scholar]
- 396. Susanti NMP, Tjahjono DH. Cyclin‐dependent kinase 4 and 6 inhibitors in cell cycle dysregulation for breast cancer treatment. Molecules. 2021;26(15):4462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397. Braal CL, Jongbloed EM, Wilting SM, Mathijssen RHJ, Koolen SLW, Jager A. Inhibiting CDK4/6 in breast cancer with palbociclib, ribociclib, and abemaciclib: similarities and differences. Drugs. 2021;81(3):317‐331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398. Elfgen C, Bjelic‐Radisic V. Targeted therapy in HR+ HER2‐metastatic breast cancer: current clinical trials and their implications for CDK4/6 inhibitor therapy and beyond treatment options. Cancers (Basel). 2021;13(23):5994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399. Finn RS, Martin M, Rugo HS, et al. Palbociclib and letrozole in advanced breast cancer. N Engl J Med. 2016;375(20):1925‐1936. [DOI] [PubMed] [Google Scholar]
- 400. Hortobagyi GN, Stemmer SM, Burris HA, et al. Updated results from MONALEESA‐2, a phase III trial of first‐line ribociclib plus letrozole versus placebo plus letrozole in hormone receptor‐positive, HER2‐negative advanced breast cancer. Ann Oncol. 2018;29(7):1541‐1547. [DOI] [PubMed] [Google Scholar]
- 401. Goetz MP, Toi M, Campone M, et al. MONARCH 3: abemaciclib as initial therapy for advanced breast cancer. J Clin Oncol. 2017;35(32):3638‐3646. [DOI] [PubMed] [Google Scholar]
- 402. Cristofanilli M, Turner NC, Bondarenko I, et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone‐receptor‐positive, HER2‐negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA‐3): final analysis of the multicentre, double‐blind, phase 3 randomised controlled trial. Lancet Oncol. 2016;17(4):425‐439. [DOI] [PubMed] [Google Scholar]
- 403. Slamon DJ, Neven P, Chia S, et al. Phase III randomized study of ribociclib and fulvestrant in hormone receptor‐positive, human epidermal growth factor receptor 2‐negative advanced breast cancer: MONALEESA‐3. J Clin Oncol. 2018;36(24):2465‐2472. [DOI] [PubMed] [Google Scholar]
- 404. Sledge GW, Jr. , Toi M, Neven P, et al. MONARCH 2: abemaciclib in combination with fulvestrant in women with HR+/HER2‐advanced breast cancer who had progressed while receiving endocrine therapy. J Clin Oncol. 2017;35(25):2875‐2884. [DOI] [PubMed] [Google Scholar]
- 405. Dickler MN, Tolaney SM, Rugo HS, et al. MONARCH 1, a phase II study of abemaciclib, a CDK4 and CDK6 inhibitor, as a single agent, in patients with refractory HR(+)/HER2(‐) metastatic breast cancer. Clin Cancer Res. 2017;23(17):5218‐5224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406. Dhakal A, Falkson C, O'Regan RM. Adjuvant cyclin‐dependent kinase 4/6 inhibition in hormone receptor‐positive breast cancer: one monarch to rule them all? Cancer. 2021;127(18):3302‐3309. [DOI] [PubMed] [Google Scholar]
- 407. Weiss JM, Csoszi T, Maglakelidze M, et al. Myelopreservation with the CDK4/6 inhibitor trilaciclib in patients with small‐cell lung cancer receiving first‐line chemotherapy: a phase Ib/randomized phase II trial. Ann Oncol. 2019;30(10):1613‐1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408. Rudin CM, Brambilla E, Faivre‐Finn C, Sage J. Small‐cell lung cancer. Nat Rev Dis Primers. 2021;7(1):3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409. Johnston S, Puhalla S, Wheatley D, et al. Randomized phase II study evaluating palbociclib in addition to letrozole as neoadjuvant therapy in estrogen receptor‐positive early breast cancer: PALLET Trial. J Clin Oncol. 2019;37(3):178‐189. [DOI] [PubMed] [Google Scholar]
- 410. Adkins D, Ley J, Neupane P, et al. Palbociclib and cetuximab in platinum‐resistant and in cetuximab‐resistant human papillomavirus‐unrelated head and neck cancer: a multicentre, multigroup, phase 2 trial. Lancet Oncol. 2019;20(9):1295‐1305. [DOI] [PubMed] [Google Scholar]
- 411. Dickson MA, Schwartz GK, Keohan ML, et al. Progression‐free survival among patients with well‐differentiated or dedifferentiated liposarcoma treated with CDK4 inhibitor palbociclib: a phase 2 clinical trial. JAMA Oncol. 2016;2(7):937‐940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412. de Kouchkovsky I, Rao A, Carneiro BA, et al. A phase Ib/II study of the CDK4/6 inhibitor ribociclib in combination with docetaxel plus prednisone in metastatic castration‐resistant prostate cancer. Clin Cancer Res. 2022;28(8):1531‐1539. [DOI] [PubMed] [Google Scholar]
- 413. Fennell DA, King A, Mohammed S, et al. Abemaciclib in patients with p16ink4A‐deficient mesothelioma (MiST2): a single‐arm, open‐label, phase 2 trial. Lancet Oncol. 2022;23(3):374‐381. [DOI] [PubMed] [Google Scholar]
- 414. Xu B, Zhang Q, Zhang P, et al. Dalpiciclib or placebo plus fulvestrant in hormone receptor‐positive and HER2‐negative advanced breast cancer: a randomized, phase 3 trial. Nat Med. 2021;27(11):1904‐1909. [DOI] [PubMed] [Google Scholar]
- 415. Andreano KJ, Wardell SE, Baker JG, et al. G1T48, an oral selective estrogen receptor degrader, and the CDK4/6 inhibitor lerociclib inhibit tumor growth in animal models of endocrine‐resistant breast cancer. Breast Cancer Res Treat. 2020;180(3):635‐646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416. Freeman‐Cook K, Hoffman RL, Miller N, et al. Expanding control of the tumor cell cycle with a CDK2/4/6 inhibitor. Cancer Cell. 2021;39(10):1404‐1421. e1411. [DOI] [PubMed] [Google Scholar]
- 417. Ghia P, Scarfò L, Perez S, et al. Efficacy and safety of dinaciclib vs ofatumumab in patients with relapsed/refractory chronic lymphocytic leukemia. Blood. 2017;129(13):1876‐1878. [DOI] [PubMed] [Google Scholar]
- 418. Chen R, Chen Y, Xiong P, et al. Cyclin‐dependent kinase inhibitor fadraciclib (CYC065) depletes anti‐apoptotic protein and synergizes with venetoclax in primary chronic lymphocytic leukemia cells. Leukemia. 2022;36(6):1596‐1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419. Rahaman MH, Yu Y, Zhong L, et al. CDKI‐73: an orally bioavailable and highly efficacious CDK9 inhibitor against acute myeloid leukemia. Invest New Drugs. 2019;37(4):625‐635. [DOI] [PubMed] [Google Scholar]
- 420. Liu XJ, Xu L, Pang XJ, et al. Progress in the development of small molecular inhibitors of the Bruton's tyrosine kinase (BTK) as a promising cancer therapy. Bioorg Med Chem. 2021;47:116358. [DOI] [PubMed] [Google Scholar]
- 421. Burger JA, Wiestner A. Targeting B cell receptor signalling in cancer: preclinical and clinical advances. Nat Rev Cancer. 2018;18(3):148‐167. [DOI] [PubMed] [Google Scholar]
- 422. Hendriks RW, Yuvaraj S, Kil LP. Targeting Bruton's tyrosine kinase in B cell malignancies. Nat Rev Cancer. 2014;14(4):219‐232. [DOI] [PubMed] [Google Scholar]
- 423. Advani RH, Buggy JJ, Sharman JP, et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI‐32765) has significant activity in patients with relapsed/refractory B‐cell malignancies. J Clin Oncol. 2013;31(1):88‐94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424. Noy A, de Vos S, Thieblemont C, et al. Targeting Bruton tyrosine kinase with ibrutinib in relapsed/refractory marginal zone lymphoma. Blood. 2017;129(16):2224‐2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425. Treon SP, Gustine J, Meid K, et al. Ibrutinib monotherapy in symptomatic, treatment‐naïve patients with waldenström macroglobulinemia. J Clin Oncol. 2018;36(27):2755‐2761. [DOI] [PubMed] [Google Scholar]
- 426. Treon SP, Tripsas CK, Meid K, et al. Ibrutinib in previously treated Waldenström's macroglobulinemia. N Engl J Med. 2015;372(15):1430‐1440. [DOI] [PubMed] [Google Scholar]
- 427. Wang ML, Rule S, Martin P, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle‐cell lymphoma. N Engl J Med. 2013;369(6):507‐516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428. Hillmen P, Rawstron AC, Brock K, et al. Ibrutinib plus venetoclax in relapsed/refractory chronic lymphocytic leukemia: the CLARITY study. J Clin Oncol. 2019;37(30):2722‐2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429. Jain N, Keating M, Thompson P, et al. Ibrutinib and venetoclax for first‐line treatment of CLL. N Engl J Med. 2019;380(22):2095‐2103. [DOI] [PubMed] [Google Scholar]
- 430. Tasso B, Spallarossa A, Russo E, Brullo C. The development of BTK inhibitors: a five‐year update. Molecules. 2021;26(23):7411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431. Ghia P, Pluta A, Wach M, et al. ASCEND: phase III, randomized trial of acalabrutinib versus idelalisib plus rituximab or bendamustine plus rituximab in relapsed or refractory chronic lymphocytic leukemia. J Clin Oncol. 2020;38(25):2849‐2861. [DOI] [PubMed] [Google Scholar]
- 432. Sharman JP, Egyed M, Jurczak W, et al. Acalabrutinib with or without obinutuzumab versus chlorambucil and obinutuzmab for treatment‐naive chronic lymphocytic leukaemia (ELEVATE TN): a randomised, controlled, phase 3 trial. Lancet. 2020;395(10232):1278‐1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433. Byrd JC, Hillmen P, Ghia P, et al. Acalabrutinib versus ibrutinib in previously treated chronic lymphocytic leukemia: results of the first randomized phase III trial. J Clin Oncol. 2021;39(31):3441‐3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 434. Song Y, Zhou K, Zou D, et al. Treatment of patients with relapsed or refractory mantle‐cell lymphoma with zanubrutinib, a selective inhibitor of bruton's tyrosine kinase. Clin Cancer Res. 2020;26(16):4216‐4224. [DOI] [PubMed] [Google Scholar]
- 435. Opat S, Tedeschi A, Linton K, et al. The MAGNOLIA trial: zanubrutinib, a next‐generation bruton tyrosine kinase inhibitor, demonstrates safety and efficacy in relapsed/refractory marginal zone lymphoma. Clin Cancer Res. 2021;27(23):6323‐6332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 436. Tam CS, Brown JR, Kahl BS, et al. Zanubrutinib versus bendamustine and rituximab in untreated chronic lymphocytic leukaemia and small lymphocytic lymphoma (SEQUOIA): a randomised, controlled, phase 3 trial. Lancet Oncol. 2022;23(8):1031‐1043. [DOI] [PubMed] [Google Scholar]
- 437. Tam CS, Opat S, D'Sa S, et al. A randomized phase 3 trial of zanubrutinib vs ibrutinib in symptomatic Waldenström macroglobulinemia: the ASPEN study. Blood. 2020;136(18):2038‐2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438. Narita Y, Nagane M, Mishima K, et al. Phase I/II study of tirabrutinib, a second‐generation Bruton's tyrosine kinase inhibitor, in relapsed/refractory primary central nervous system lymphoma. Neuro Oncol. 2021;23(1):122‐133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 439. Sekiguchi N, Rai S, Munakata W, et al. A multicenter, open‐label, phase II study of tirabrutinib (ONO/GS‐4059) in patients with Waldenström's macroglobulinemia. Cancer Sci. 2020;111(9):3327‐3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 440. Dhillon S. Orelabrutinib: first approval. Drugs. 2021;81(4):503‐507. [DOI] [PubMed] [Google Scholar]
- 441. Aslan B, Kismali G, Iles LR, et al. Pirtobrutinib inhibits wild‐type and mutant Bruton's tyrosine kinase‐mediated signaling in chronic lymphocytic leukemia. Blood Cancer J. 2022;12(5):80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442. Mato AR, Shah NN, Jurczak W, et al. Pirtobrutinib in relapsed or refractory B‐cell malignancies (BRUIN): a phase 1/2 study. Lancet. 2021;397(10277):892‐901. [DOI] [PubMed] [Google Scholar]
- 443. Waitkus MS, Diplas BH, Yan H. Biological role and therapeutic potential of IDH mutations in cancer. Cancer Cell. 2018;34(2):186‐195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444. Issa GC, DiNardo CD. Acute myeloid leukemia with IDH1 and IDH2 mutations: 2021 treatment algorithm. Blood Cancer J. 2021;11(6):107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445. Cerchione C, Romano A, Daver N, et al. IDH1/IDH2 Inhibition in Acute Myeloid Leukemia. Front Oncol. 2021;11:639387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 446. Dang L, Su SM. Isocitrate dehydrogenase mutation and (R)‐2‐hydroxyglutarate: from basic discovery to therapeutics development. Annu Rev Biochem. 2017;86:305‐331. [DOI] [PubMed] [Google Scholar]
- 447. Fathi AT, DiNardo CD, Kline I, et al. Differentiation syndrome associated with enasidenib, a selective inhibitor of mutant isocitrate dehydrogenase 2: analysis of a phase 1/2 Study. JAMA Oncol. 2018;4(8):1106‐1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 448. Stein EM, DiNardo CD, Pollyea DA, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130(6):722‐731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 449. DiNardo CD, Schuh AC, Stein EM, et al. Enasidenib plus azacitidine versus azacitidine alone in patients with newly diagnosed, mutant‐IDH2 acute myeloid leukaemia (AG221‐AML‐005): a single‐arm, phase 1b and randomised, phase 2 trial. Lancet Oncol. 2021;22(11):1597‐1608. [DOI] [PubMed] [Google Scholar]
- 450. Abou‐Alfa GK, Macarulla T, Javle MM, et al. Ivosidenib in IDH1‐mutant, chemotherapy‐refractory cholangiocarcinoma (ClarIDHy): a multicentre, randomised, double‐blind, placebo‐controlled, phase 3 study. Lancet Oncol. 2020;21(6):796‐807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 451. DiNardo CD, Stein EM, de Botton S, et al. Durable remissions with ivosidenib in IDH1‐mutated relapsed or refractory AML. N Engl J Med. 2018;378(25):2386‐2398. [DOI] [PubMed] [Google Scholar]
- 452. Choe S, Wang H, DiNardo CD, et al. Molecular mechanisms mediating relapse following ivosidenib monotherapy in IDH1‐mutant relapsed or refractory AML. Blood Adv. 2020;4(9):1894‐1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 453. Intlekofer AM, Shih AH, Wang B, et al. Acquired resistance to IDH inhibition through trans or cis dimer‐interface mutations. Nature. 2018;559(7712):125‐129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454. DiNardo CD, Hochhaus A, Frattini MG, et al. A phase 1 study of IDH305 in patients with IDH1(R132)‐mutant acute myeloid leukemia or myelodysplastic syndrome. J Cancer Res Clin Oncol. 2022. 10.1007/s00432-022-03983-6 [DOI] [PubMed] [Google Scholar]
- 455. Wick A, Bähr O, Schuler M, et al. Phase I assessment of safety and therapeutic activity of BAY1436032 in patients with IDH1‐mutant solid tumors. Clin Cancer Res. 2021;27(10):2723‐2733. [DOI] [PubMed] [Google Scholar]
- 456. Heuser M, Palmisiano N, Mantzaris I, et al. Safety and efficacy of BAY1436032 in IDH1‐mutant AML: phase I study results. Leukemia. 2020;34(11):2903‐2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 457. Machida Y, Nakagawa M, Matsunaga H, et al. A potent blood‐brain barrier‐permeable mutant IDH1 inhibitor suppresses the growth of glioblastoma with IDH1 mutation in a patient‐derived orthotopic xenograft model. Mol Cancer Ther. 2020;19(2):375‐383. [DOI] [PubMed] [Google Scholar]
- 458. Nakagawa M, Nakatani F, Matsunaga H, et al. Selective inhibition of mutant IDH1 by DS‐1001b ameliorates aberrant histone modifications and impairs tumor activity in chondrosarcoma. Oncogene. 2019;38(42):6835‐6849. [DOI] [PubMed] [Google Scholar]
- 459. Wang F, Travins J, DeLaBarre B, et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science. 2013;340(6132):622‐626. [DOI] [PubMed] [Google Scholar]
- 460. Mellinghoff IK, Penas‐Prado M, Peters KB, et al. Vorasidenib, a dual inhibitor of mutant IDH1/2, in recurrent or progressive glioma; results of a first‐in‐human phase I trial. Clin Cancer Res. 2021;27(16):4491‐4499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 461. Roskoski R, Jr. Src protein‐tyrosine kinase structure, mechanism, and small molecule inhibitors. Pharmacol Res. 2015;94:9‐25. [DOI] [PubMed] [Google Scholar]
- 462. Song Y, Zhao M, Zhang H, Yu B. Double‐edged roles of protein tyrosine phosphatase SHP2 in cancer and its inhibitors in clinical trials. Pharmacol Ther. 2022;230:107966. [DOI] [PubMed] [Google Scholar]
- 463. Song Z, Wang M, Ge Y, et al. Tyrosine phosphatase SHP2 inhibitors in tumor‐targeted therapies. Acta Pharm Sin B. 2021;11(1):13‐29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 464. Markham A, Duggan S. Tirbanibulin: first approval. Drugs. 2021;81(4):509‐513. [DOI] [PubMed] [Google Scholar]
- 465. Blauvelt A, Kempers S, Lain E, et al. Phase 3 trials of tirbanibulin ointment for actinic keratosis. N Engl J Med. 2021;384(6):512‐520. [DOI] [PubMed] [Google Scholar]
- 466. Baselga J, Cervantes A, Martinelli E, et al. Phase I safety, pharmacokinetics, and inhibition of SRC activity study of saracatinib in patients with solid tumors. Clin Cancer Res. 2010;16(19):4876‐4883. [DOI] [PubMed] [Google Scholar]
- 467. Simpkins F, Hevia‐Paez P, Sun J, et al. Src Inhibition with saracatinib reverses fulvestrant resistance in ER‐positive ovarian cancer models in vitro and in vivo. Clin Cancer Res. 2012;18(21):5911‐5923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468. Simpkins F, Jang K, Yoon H, et al. Dual Src and MEK inhibition decreases ovarian cancer growth and targets tumor initiating stem‐like cells. Clin Cancer Res. 2018;24(19):4874‐4886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469. Chakraborty G, Patail NK, Hirani R, et al. Attenuation of SRC kinase activity augments PARP inhibitor‐mediated synthetic lethality in BRCA2‐altered prostate tumors. Clin Cancer Res. 2021;27(6):1792‐1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 470. Powles T, Brown J, Larkin J, et al. A randomized, double‐blind phase II study evaluating cediranib versus cediranib and saracatinib in patients with relapsed metastatic clear‐cell renal cancer (COSAK). Ann Oncol. 2016;27(5):880‐886. [DOI] [PubMed] [Google Scholar]
- 471. McNeish IA, Ledermann JA, Webber L, et al. A randomised, placebo‐controlled trial of weekly paclitaxel and saracatinib (AZD0530) in platinum‐resistant ovarian, fallopian tube or primary peritoneal cancer†. Ann Oncol. 2014;25(10):1988‐1995. [DOI] [PubMed] [Google Scholar]
- 472. Drilon A, Siena S, Ou SI, et al. Safety and antitumor activity of the multitargeted pan‐TRK, ROS1, and ALK inhibitor entrectinib: combined results from two phase I trials (ALKA‐372‐001 and STARTRK‐1). Cancer Discov. 2017;7(4):400‐409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 473. McGettrick AF, O'Neill LAJ. The role of HIF in immunity and inflammation. Cell Metab. 2020;32(4):524‐536. [DOI] [PubMed] [Google Scholar]
- 474. Ma Z, Xiang X, Li S, et al. Targeting hypoxia‐inducible factor‐1, for cancer treatment: recent advances in developing small‐molecule inhibitors from natural compounds. Semin Cancer Biol. 2022;80:379‐390. [DOI] [PubMed] [Google Scholar]
- 475. Choueiri TK, Kaelin WG, Jr. Targeting the HIF2‐VEGF axis in renal cell carcinoma. Nat Med. 2020;26(10):1519‐1530. [DOI] [PubMed] [Google Scholar]
- 476. Jonasch E, Donskov F, Iliopoulos O, et al. Belzutifan for renal cell carcinoma in von Hippel‐Lindau disease. N Engl J Med. 2021;385(22):2036‐2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 477. Shen Z, Pei Q, Zhang H, et al. Hypoxia‐inducible factor‐1α inhibition augments efficacy of programmed cell death 1 antibody in murine prostatic cancer models. Anticancer Drugs. 2022;33(6):587‐594. [DOI] [PubMed] [Google Scholar]
- 478. Kim MH, Lee TH, Lee JS, Lim DJ, Lee PC. Hif‐1α inhibitors could successfully inhibit the progression of differentiated thyroid cancer in vitro. Pharmaceuticals (Basel). 2020;13(9):208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 479. Luo F, Lu FT, Cao JX, et al. HIF‐1α inhibition promotes the efficacy of immune checkpoint blockade in the treatment of non‐small cell lung cancer. Cancer Lett. 2022;531:39‐56. [DOI] [PubMed] [Google Scholar]
- 480. Jacoby JJ, Erez B, Korshunova MV, et al. Treatment with HIF‐1alpha antagonist PX‐478 inhibits progression and spread of orthotopic human small cell lung cancer and lung adenocarcinoma in mice. J Thorac Oncol. 2010;5(7):940‐949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 481. Gourley C, Balmana J, Ledermann JA, et al. Moving from poly (ADP‐Ribose) polymerase inhibition to targeting DNA repair and DNA damage response in cancer therapy. J Clin Oncol. 2019;37(25):2257‐2269. [DOI] [PubMed] [Google Scholar]
- 482. Curtin NJ, Szabo C. Poly(ADP‐ribose) polymerase inhibition: past, present and future. Nat Rev Drug Discov. 2020;19(10):711‐736. [DOI] [PubMed] [Google Scholar]
- 483. Hengel SR, Spies MA, Spies M. Small‐molecule inhibitors targeting DNA repair and DNA repair deficiency in research and cancer therapy. Cell Chem Biol. 2017;24(9):1101‐1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 484. Moore K, Colombo N, Scambia G, et al. Maintenance olaparib in patients with newly diagnosed advanced ovarian cancer. N Engl J Med. 2018;379(26):2495‐2505. [DOI] [PubMed] [Google Scholar]
- 485. Fong PC, Boss DS, Yap TA, et al. Inhibition of poly(ADP‐ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361(2):123‐134. [DOI] [PubMed] [Google Scholar]
- 486. Coleman RL, Oza AM, Lorusso D, et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet. 2017;390(10106):1949‐1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 487. González‐Martín A, Pothuri B, Vergote I, et al. Niraparib in patients with newly diagnosed advanced ovarian cancer. N Engl J Med. 2019;381(25):2391‐2402. [DOI] [PubMed] [Google Scholar]
- 488. Swisher EM, Kwan TT, Oza AM, et al. Molecular and clinical determinants of response and resistance to rucaparib for recurrent ovarian cancer treatment in ARIEL2 (Parts 1 and 2). Nat Commun. 2021;12(1):2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 489. Swisher EM, Lin KK, Oza AM, et al. Rucaparib in relapsed, platinum‐sensitive high‐grade ovarian carcinoma (ARIEL2 Part 1): an international, multicentre, open‐label, phase 2 trial. Lancet Oncol. 2017;18(1):75‐87. [DOI] [PubMed] [Google Scholar]
- 490. Moore KN, Secord AA, Geller MA, et al. Niraparib monotherapy for late‐line treatment of ovarian cancer (QUADRA): a multicentre, open‐label, single‐arm, phase 2 trial. Lancet Oncol. 2019;20(5):636‐648. [DOI] [PubMed] [Google Scholar]
- 491. Robson M, Im SA, Senkus E, et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N Engl J Med. 2017;377(6):523‐533. [DOI] [PubMed] [Google Scholar]
- 492. Litton JK, Rugo HS, Ettl J, et al. Talazoparib in patients with advanced breast cancer and a germline BRCA mutation. N Engl J Med. 2018;379(8):753‐763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 493. Tutt ANJ, Garber JE, Kaufman B, et al. Adjuvant olaparib for patients with BRCA1‐ or BRCA2‐mutated breast cancer. N Engl J Med. 2021;384(25):2394‐2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 494. de Bono J, Mateo J, Fizazi K, et al. Olaparib for metastatic castration‐resistant prostate cancer. N Engl J Med. 2020;382(22):2091‐2102. [DOI] [PubMed] [Google Scholar]
- 495. Teyssonneau D, Margot H, Cabart M, et al. Prostate cancer and PARP inhibitors: progress and challenges. J Hematol Oncol. 2021;14(1):51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 496. Hussain M, Mateo J, Fizazi K, et al. Survival with olaparib in metastatic castration‐resistant prostate cancer. N Engl J Med. 2020;383(24):2345‐2357. [DOI] [PubMed] [Google Scholar]
- 497. Anscher MS, Chang E, Gao X, et al. FDA approval summary: rucaparib for the treatment of patients with deleterious BRCA‐mutated metastatic castrate‐resistant prostate cancer. Oncologist. 2021;26(2):139‐146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 498. Golan T, Hammel P, Reni M, et al. Maintenance olaparib for germline BRCA‐mutated metastatic pancreatic cancer. N Engl J Med. 2019;381(4):317‐327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 499. Farago AF, Yeap BY, Stanzione M, et al. Combination olaparib and temozolomide in relapsed small‐cell lung cancer. Cancer Discov. 2019;9(10):1372‐1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 500. Bang YJ, Xu RH, Chin K, et al. Olaparib in combination with paclitaxel in patients with advanced gastric cancer who have progressed following first‐line therapy (GOLD): a double‐blind, randomised, placebo‐controlled, phase 3 trial. Lancet Oncol. 2017;18(12):1637‐1651. [DOI] [PubMed] [Google Scholar]
- 501. Fennell DA, King A, Mohammed S, et al. Rucaparib in patients with BAP1‐deficient or BRCA1‐deficient mesothelioma (MiST1): an open‐label, single‐arm, phase 2a clinical trial. Lancet Respir Med. 2021;9(6):593‐600. [DOI] [PubMed] [Google Scholar]
- 502. Bang YJ, Im SA, Lee KW, et al. Randomized, double‐blind phase II trial with prospective classification by ATM protein level to evaluate the efficacy and tolerability of olaparib plus paclitaxel in patients with recurrent or metastatic gastric cancer. J Clin Oncol. 2015;33(33):3858‐3865. [DOI] [PubMed] [Google Scholar]
- 503. Ray‐Coquard I, Pautier P, Pignata S, et al. Olaparib plus bevacizumab as first‐line maintenance in ovarian cancer. N Engl J Med. 2019;381(25):2416‐2428. [DOI] [PubMed] [Google Scholar]
- 504. Domchek SM, Postel‐Vinay S, Im SA, et al. Olaparib and durvalumab in patients with germline BRCA‐mutated metastatic breast cancer (MEDIOLA): an open‐label, multicentre, phase 1/2, basket study. Lancet Oncol. 2020;21(9):1155‐1164. [DOI] [PubMed] [Google Scholar]
- 505. Reiss KA, Mick R, Teitelbaum U, et al. Niraparib plus nivolumab or niraparib plus ipilimumab in patients with platinum‐sensitive advanced pancreatic cancer: a randomised, phase 1b/2 trial. Lancet Oncol. 2022;23(8):1009‐1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 506. Markham A. Pamiparib: first approval. Drugs. 2021;81(11):1343‐1348. [DOI] [PubMed] [Google Scholar]
- 507. Lee A. Fuzuloparib: first approval. Drugs. 2021;81(10):1221‐1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 508. Li N, Zhang Y, Wang J, et al. Fuzuloparib maintenance therapy in patients with platinum‐sensitive, recurrent ovarian carcinoma (FZOCUS‐2): a multicenter, randomized, double‐blind, placebo‐controlled, phase III trial. J Clin Oncol. 2022;40(22):2436‐2446. [DOI] [PubMed] [Google Scholar]
- 509. Dias MP, Moser SC, Ganesan S, Jonkers J. Understanding and overcoming resistance to PARP inhibitors in cancer therapy. Nat Rev Clin Oncol. 2021;18(12):773‐791. [DOI] [PubMed] [Google Scholar]
- 510. Polo SE, Kaidi A, Baskcomb L, Galanty Y, Jackson SP. Regulation of DNA‐damage responses and cell‐cycle progression by the chromatin remodelling factor CHD4. EMBO J. 2010;29(18):3130‐3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 511. Dai Z, Ramesh V, Locasale JW. The evolving metabolic landscape of chromatin biology and epigenetics. Nat Rev Genet. 2020;21(12):737‐753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 512. Hogg SJ, Beavis PA, Dawson MA, Johnstone RW. Targeting the epigenetic regulation of antitumour immunity. Nature Reviews Drug Discovery. 2020;19(11):776‐800. [DOI] [PubMed] [Google Scholar]
- 513. Michalak EM, Burr ML, Bannister AJ, Dawson MA. The roles of DNA, RNA and histone methylation in ageing and cancer. Nat Rev Mol Cell Biol. 2019;20(10):573‐589. [DOI] [PubMed] [Google Scholar]
- 514. Kaur J, Daoud A, Eblen ST. Targeting chromatin remodeling for cancer therapy. Curr Mol Pharmacol. 2019;12(3):215‐229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 515. Dawson MA. The cancer epigenome: concepts, challenges, and therapeutic opportunities. Science. 2017;355(6330):1147‐1152. [DOI] [PubMed] [Google Scholar]
- 516. Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128(4):683‐692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 517. Issa JP, Kantarjian HM. Targeting DNA methylation. Clin Cancer Res. 2009;15(12):3938‐3946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 518. Zemach A, McDaniel IE, Silva P, Zilberman D. Genome‐wide evolutionary analysis of eukaryotic DNA methylation. Science. 2010;328(5980):916‐919. [DOI] [PubMed] [Google Scholar]
- 519. Ahuja N, Easwaran H, Baylin SB. Harnessing the potential of epigenetic therapy to target solid tumors. J Clin Invest. 2014;124(1):56‐63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 520. Allis CD, Jenuwein T. The molecular hallmarks of epigenetic control. Nat Rev Genet. 2016;17(8):487‐500. [DOI] [PubMed] [Google Scholar]
- 521. Jones PA, Issa JP, Baylin S. Targeting the cancer epigenome for therapy. Nat Rev Genet. 2016;17(10):630‐641. [DOI] [PubMed] [Google Scholar]
- 522. Berdasco M, Esteller M. Clinical epigenetics: seizing opportunities for translation. Nat Rev Genet. 2019;20(2):109‐127. [DOI] [PubMed] [Google Scholar]
- 523. Silverman LR, Demakos EP, Peterson BL, et al. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group B. J Clin Oncol. 2002;20(10):2429‐2440. [DOI] [PubMed] [Google Scholar]
- 524. Kornblith AB, Herndon JE, 2nd , Silverman LR, et al. Impact of azacytidine on the quality of life of patients with myelodysplastic syndrome treated in a randomized phase III trial: a Cancer and Leukemia Group B study. J Clin Oncol. 2002;20(10):2441‐2452. [DOI] [PubMed] [Google Scholar]
- 525. Fenaux P, Mufti GJ, Hellstrom‐Lindberg E, et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher‐risk myelodysplastic syndromes: a randomised, open‐label, phase III study. Lancet Oncol. 2009;10(3):223‐232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 526. Platzbecker U, Middeke JM, Sockel K, et al. Measurable residual disease‐guided treatment with azacitidine to prevent haematological relapse in patients with myelodysplastic syndrome and acute myeloid leukaemia (RELAZA2): an open‐label, multicentre, phase 2 trial. Lancet Oncol. 2018;19(12):1668‐1679. [DOI] [PubMed] [Google Scholar]
- 527. Wei AH, Döhner H, Pocock C, et al. Oral azacitidine maintenance therapy for acute myeloid leukemia in first remission. N Engl J Med. 2020;383(26):2526‐2537. [DOI] [PubMed] [Google Scholar]
- 528. Fenaux P, Mufti GJ, Hellström‐Lindberg E, et al. Azacitidine prolongs overall survival compared with conventional care regimens in elderly patients with low bone marrow blast count acute myeloid leukemia. J Clin Oncol. 2010;28(4):562‐569. [DOI] [PubMed] [Google Scholar]
- 529. DiNardo CD, Jonas BA, Pullarkat V, et al. Azacitidine and venetoclax in previously untreated acute myeloid leukemia. N Engl J Med. 2020;383(7):617‐629. [DOI] [PubMed] [Google Scholar]
- 530. Cluzeau T, Sebert M, Rahmé R, et al. Eprenetapopt plus azacitidine in TP53‐mutated myelodysplastic syndromes and acute myeloid leukemia: a phase II Study by the Groupe Francophone des Myélodysplasies (GFM). J Clin Oncol. 2021;39(14):1575‐1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 531. Niemeyer CM, Flotho C, Lipka DB, et al. Response to upfront azacitidine in juvenile myelomonocytic leukemia in the AZA‐JMML‐001 trial. Blood Adv. 2021;5(14):2901‐2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 532. Wijermans P, Lübbert M, Verhoef G, et al. Low‐dose 5‐aza‐2'‐deoxycytidine, a DNA hypomethylating agent, for the treatment of high‐risk myelodysplastic syndrome: a multicenter phase II study in elderly patients. J Clin Oncol. 2000;18(5):956‐962. [DOI] [PubMed] [Google Scholar]
- 533. Steensma DP, Baer MR, Slack JL, et al. Multicenter study of decitabine administered daily for 5 days every 4 weeks to adults with myelodysplastic syndromes: the alternative dosing for outpatient treatment (ADOPT) trial. J Clin Oncol. 2009;27(23):3842‐3848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 534. Cashen AF, Schiller GJ, O'Donnell MR, DiPersio JF. Multicenter, phase II study of decitabine for the first‐line treatment of older patients with acute myeloid leukemia. J Clin Oncol. 2010;28(4):556‐561. [DOI] [PubMed] [Google Scholar]
- 535. Gao L, Zhang Y, Wang S, et al. Effect of rhG‐CSF combined with decitabine prophylaxis on relapse of patients with high‐risk MRD‐negative AML after HSCT: an open‐label, multicenter, randomized controlled trial. J Clin Oncol. 2020;38(36):4249‐4259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 536. Appleton K, Mackay HJ, Judson I, et al. Phase I and pharmacodynamic trial of the DNA methyltransferase inhibitor decitabine and carboplatin in solid tumors. J Clin Oncol. 2007;25(29):4603‐4609. [DOI] [PubMed] [Google Scholar]
- 537. Welch JS, Petti AA, Miller CA, et al. TP53 and decitabine in acute myeloid leukemia and myelodysplastic syndromes. N Engl J Med. 2016;375(21):2023‐2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 538. Daver N, Garcia‐Manero G, Basu S, et al. Efficacy, safety, and biomarkers of response to azacitidine and nivolumab in relapsed/refractory acute myeloid leukemia: a nonrandomized, open‐label, phase II study. Cancer Discov. 2019;9(3):370‐383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 539. Nie J, Wang C, Liu Y, et al. Addition of low‐dose decitabine to anti‐PD‐1 antibody camrelizumab in relapsed/refractory classical Hodgkin lymphoma. J Clin Oncol. 2019;37(17):1479‐1489. [DOI] [PubMed] [Google Scholar]
- 540. Kantarjian HM, Roboz GJ, Kropf PL, et al. Guadecitabine (SGI‐110) in treatment‐naive patients with acute myeloid leukaemia: phase 2 results from a multicentre, randomised, phase 1/2 trial. Lancet Oncol. 2017;18(10):1317‐1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 541. Issa JJ, Roboz G, Rizzieri D, et al. Safety and tolerability of guadecitabine (SGI‐110) in patients with myelodysplastic syndrome and acute myeloid leukaemia: a multicentre, randomised, dose‐escalation phase 1 study. Lancet Oncol. 2015;16(9):1099‐1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 542. Papadatos‐Pastos D, Yuan W, Pal A, et al. Phase 1, dose‐escalation study of guadecitabine (SGI‐110) in combination with pembrolizumab in patients with solid tumors. J Immunother Cancer. 2022;10(6):e004495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 543. Di Giacomo AM, Covre A, Finotello F, et al. Guadecitabine plus ipilimumab in unresectable melanoma: the NIBIT‐M4 clinical trial. Clin Cancer Res. 2019;25(24):7351‐7362. [DOI] [PubMed] [Google Scholar]
- 544. Crabb SJ, Danson S, Catto JWF, et al. Phase I trial of DNA methyltransferase inhibitor guadecitabine combined with cisplatin and gemcitabine for solid malignancies including urothelial carcinoma (SPIRE). Clin Cancer Res. 2021;27(7):1882‐1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 545. Oza AM, Matulonis UA, Alvarez Secord A, et al. A randomized phase II trial of epigenetic priming with guadecitabine and carboplatin in platinum‐resistant, recurrent ovarian cancer. Clin Cancer Res. 2020;26(5):1009‐1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 546. Gallinari P, Di Marco S, Jones P, Pallaoro M, Steinkuhler C. HDACs, histone deacetylation and gene transcription: from molecular biology to cancer therapeutics. Cell Res. 2007;17(3):195‐211. [DOI] [PubMed] [Google Scholar]
- 547. Nebbioso A, Clarke N, Voltz E, et al. Tumor‐selective action of HDAC inhibitors involves TRAIL induction in acute myeloid leukemia cells. Nat Med. 2005;11(1):77‐84. [DOI] [PubMed] [Google Scholar]
- 548. El‐Khoury V, Pierson S, Szwarcbart E, et al. Disruption of autophagy by the histone deacetylase inhibitor MGCD0103 and its therapeutic implication in B‐cell chronic lymphocytic leukemia. Leukemia. 2014;28(8):1636‐1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 549. West AC, Johnstone RW. New and emerging HDAC inhibitors for cancer treatment. J Clin Invest. 2014;124(1):30‐39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 550. Falkenberg KJ, Johnstone RW. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov. 2014;13(9):673‐691. [DOI] [PubMed] [Google Scholar]
- 551. Olsen EA, Kim YH, Kuzel TM, et al. Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T‐cell lymphoma. J Clin Oncol. 2007;25(21):3109‐3115. [DOI] [PubMed] [Google Scholar]
- 552. Piekarz RL, Frye R, Turner M, et al. Phase II multi‐institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T‐cell lymphoma. J Clin Oncol. 2009;27(32):5410‐5417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 553. Bachy E, Camus V, Thieblemont C, et al. Romidepsin plus CHOP versus CHOP in Patients with previously untreated peripheral T‐cell lymphoma: results of the Ro‐CHOP phase III study (conducted by LYSA). J Clin Oncol. 2022;40(3):242‐251. [DOI] [PubMed] [Google Scholar]
- 554. O'Connor OA, Horwitz S, Masszi T, et al. Belinostat in patients with relapsed or refractory peripheral T‐cell lymphoma: results of the pivotal phase II BELIEF (CLN‐19) Study. J Clin Oncol. 2015;33(23):2492‐2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 555. Tan D, Phipps C, Hwang WY, et al. Panobinostat in combination with bortezomib in patients with relapsed or refractory peripheral T‐cell lymphoma: an open‐label, multicentre phase 2 trial. Lancet Haematol. 2015;2(8):e326‐e333. [DOI] [PubMed] [Google Scholar]
- 556. Kirschbaum M, Frankel P, Popplewell L, et al. Phase II study of vorinostat for treatment of relapsed or refractory indolent non‐Hodgkin's lymphoma and mantle cell lymphoma. J Clin Oncol. 2011;29(9):1198‐1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 557. Krug LM, Kindler HL, Calvert H, et al. Vorinostat in patients with advanced malignant pleural mesothelioma who have progressed on previous chemotherapy (VANTAGE‐014): a phase 3, double‐blind, randomised, placebo‐controlled trial. Lancet Oncol. 2015;16(4):447‐456. [DOI] [PubMed] [Google Scholar]
- 558. Dimopoulos M, Siegel DS, Lonial S, et al. Vorinostat or placebo in combination with bortezomib in patients with multiple myeloma (VANTAGE 088): a multicentre, randomised, double‐blind study. Lancet Oncol. 2013;14(11):1129‐1140. [DOI] [PubMed] [Google Scholar]
- 559. Garcia‐Manero G, Tambaro FP, Bekele NB, et al. Phase II trial of vorinostat with idarubicin and cytarabine for patients with newly diagnosed acute myelogenous leukemia or myelodysplastic syndrome. J Clin Oncol. 2012;30(18):2204‐2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 560. Ramalingam SS, Maitland ML, Frankel P, et al. Carboplatin and Paclitaxel in combination with either vorinostat or placebo for first‐line therapy of advanced non‐small‐cell lung cancer. J Clin Oncol. 2010;28(1):56‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 561. Ree AH, Dueland S, Folkvord S, et al. Vorinostat, a histone deacetylase inhibitor, combined with pelvic palliative radiotherapy for gastrointestinal carcinoma: the Pelvic Radiation and Vorinostat (PRAVO) phase 1 study. Lancet Oncol. 2010;11(5):459‐464. [DOI] [PubMed] [Google Scholar]
- 562. DuBois SG, Granger MM, Groshen S, et al. Randomized phase II trial of MIBG versus MIBG, vincristine, and irinotecan versus MIBG and vorinostat for patients with relapsed or refractory neuroblastoma: a report from NANT Consortium. J Clin Oncol. 2021;39(31):3506‐3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 563. Juergens RA, Wrangle J, Vendetti FP, et al. Combination epigenetic therapy has efficacy in patients with refractory advanced non‐small cell lung cancer. Cancer Discov. 2011;1(7):598‐607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 564. Sekeres MA, Othus M, List AF, et al. Randomized phase II study of azacitidine alone or in combination with lenalidomide or with vorinostat in higher‐risk myelodysplastic syndromes and chronic myelomonocytic leukemia: North American Intergroup Study SWOG S1117. J Clin Oncol. 2017;35(24):2745‐2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 565. Prebet T, Sun Z, Figueroa ME, et al. Prolonged administration of azacitidine with or without entinostat for myelodysplastic syndrome and acute myeloid leukemia with myelodysplasia‐related changes: results of the US Leukemia Intergroup trial E1905. J Clin Oncol. 2014;32(12):1242‐1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 566. Yalniz FF, Berdeja JG, Maris MB, et al. A phase II study of addition of pracinostat to a hypomethylating agent in patients with myelodysplastic syndromes who have not responded to previous hypomethylating agent therapy. Br J Haematol. 2020;188(3):404‐412. [DOI] [PubMed] [Google Scholar]
- 567. Garcia‐Manero G, Abaza Y, Takahashi K, et al. Pracinostat plus azacitidine in older patients with newly diagnosed acute myeloid leukemia: results of a phase 2 study. Blood Adv. 2019;3(4):508‐518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 568. Chu QS, Nielsen TO, Alcindor T, et al. A phase II study of SB939, a novel pan‐histone deacetylase inhibitor, in patients with translocation‐associated recurrent/metastatic sarcomas‐NCIC‐CTG IND 200†. Ann Oncol. 2015;26(5):973‐981. [DOI] [PubMed] [Google Scholar]
- 569. Venugopal B, Baird R, Kristeleit RS, et al. A phase I study of quisinostat (JNJ‐26481585), an oral hydroxamate histone deacetylase inhibitor with evidence of target modulation and antitumor activity, in patients with advanced solid tumors. Clin Cancer Res. 2013;19(15):4262‐4272. [DOI] [PubMed] [Google Scholar]
- 570. Moreau P, Facon T, Touzeau C, et al. Quisinostat, bortezomib, and dexamethasone combination therapy for relapsed multiple myeloma. Leuk Lymphoma. 2016;57(7):1546‐1559. [DOI] [PubMed] [Google Scholar]
- 571. Child F, Ortiz‐Romero PL, Alvarez R, et al. Phase II multicentre trial of oral quisinostat, a histone deacetylase inhibitor, in patients with previously treated stage IB‐IVA mycosis fungoides/Sézary syndrome. Br J Dermatol. 2016;175(1):80‐88. [DOI] [PubMed] [Google Scholar]
- 572. Collier KA, Valencia H, Newton H, et al. A phase 1 trial of the histone deacetylase inhibitor AR‐42 in patients with neurofibromatosis type 2‐associated tumors and advanced solid malignancies. Cancer Chemother Pharmacol. 2021;87(5):599‐611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 573. Liva SG, Coss CC, Wang J, et al. Phase I study of AR‐42 and decitabine in acute myeloid leukemia. Leuk Lymphoma. 2020;61(6):1484‐1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 574. Younes A, Oki Y, Bociek RG, et al. Mocetinostat for relapsed classical Hodgkin's lymphoma: an open‐label, single‐arm, phase 2 trial. Lancet Oncol. 2011;12(13):1222‐1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 575. Gordon MS, Shapiro GI, Sarantopoulos J, et al. Phase Ib study of the histone deacetylase 6 inhibitor citarinostat in combination with paclitaxel in patients with advanced solid tumors. Front Oncol. 2021;11:786120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 576. Klemm SL, Shipony Z, Greenleaf WJ. Chromatin accessibility and the regulatory epigenome. Nat Rev Genet. 2019;20(4):207‐220. [DOI] [PubMed] [Google Scholar]
- 577. Becskei A, Mattaj IW. The strategy for coupling the RanGTP gradient to nuclear protein export. Proc Natl Acad Sci USA. 2003;100(4):1717‐1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 578. Saulino DM, Younes PS, Bailey JM, Younes M. CRM1/XPO1 expression in pancreatic adenocarcinoma correlates with survivin expression and the proliferative activity. Oncotarget. 2018;9(30):21289‐21295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 579. Azmi AS, Uddin MH, Mohammad RM. The nuclear export protein XPO1 ‐ from biology to targeted therapy. Nat Rev Clin Oncol. 2021;18(3):152‐169. [DOI] [PubMed] [Google Scholar]
- 580. Abdul Razak AR, Mau‐Soerensen M, Gabrail NY, et al. First‐in‐class, first‐in‐human phase i study of selinexor, a selective inhibitor of nuclear export, in patients with advanced solid tumors. J Clin Oncol. 2016;34(34):4142‐4150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 581. Alexander TB, Lacayo NJ, Choi JK, Ribeiro RC, Pui CH, Rubnitz JE. Phase I study of selinexor, a selective inhibitor of nuclear export, in combination with fludarabine and cytarabine, in pediatric relapsed or refractory acute leukemia. J Clin Oncol. 2016;34(34):4094‐4101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 582. Gounder MM, Zer A, Tap WD, et al. Phase IB study of selinexor, a first‐in‐class inhibitor of nuclear export, in patients with advanced refractory bone or soft tissue sarcoma. J Clin Oncol. 2016;34(26):3166‐3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 583. Chari A, Vogl DT, Gavriatopoulou M, et al. Oral selinexor‐dexamethasone for triple‐class refractory multiple myeloma. N Engl J Med. 2019;381(8):727‐738. [DOI] [PubMed] [Google Scholar]
- 584. Grosicki S, Simonova M, Spicka I, et al. Once‐per‐week selinexor, bortezomib, and dexamethasone versus twice‐per‐week bortezomib and dexamethasone in patients with multiple myeloma (BOSTON): a randomised, open‐label, phase 3 trial. Lancet. 2020;396(10262):1563‐1573. [DOI] [PubMed] [Google Scholar]
- 585. Maerevoet M, Zijlstra JM, Follows G, et al. Survival among patients with relapsed/refractory diffuse large B cell lymphoma treated with single‐agent selinexor in the SADAL study. J Hematol Oncol. 2021;14(1):111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 586. Kalakonda N, Maerevoet M, Cavallo F, et al. Selinexor in patients with relapsed or refractory diffuse large B‐cell lymphoma (SADAL): a single‐arm, multinational, multicentre, open‐label, phase 2 trial. Lancet Haematol. 2020;7(7):e511‐e522. [DOI] [PubMed] [Google Scholar]
- 587. Seymour EK, Khan HY, Li Y, et al. Selinexor in combination with R‐CHOP for frontline treatment of non‐Hodgkin lymphoma: results of a phase I study. Clin Cancer Res. 2021;27(12):3307‐3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 588. Gounder MM, Razak AA, Somaiah N, et al. Selinexor in advanced, metastatic dedifferentiated liposarcoma: a multinational, randomized, double‐blind, placebo‐controlled trial. J Clin Oncol. 2022;40(22):2479‐2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 589. Lassman AB, Wen PY, van den Bent MJ, et al. A Phase II study of the efficacy and safety of oral selinexor in recurrent glioblastoma. Clin Cancer Res. 2022;28(3):452‐460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 590. Brunetti L, Gundry MC, Sorcini D, et al. Mutant NPM1 maintains the leukemic state through HOX expression. Cancer Cell. 2018;34(3):499‐512. e499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 591. Walker JS, Hing ZA, Harrington B, et al. Recurrent XPO1 mutations alter pathogenesis of chronic lymphocytic leukemia. J Hematol Oncol. 2021;14(1):17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 592. Deng M, Zhang M, Xu‐Monette ZY, et al. XPO1 expression worsens the prognosis of unfavorable DLBCL that can be effectively targeted by selinexor in the absence of mutant p53. J Hematol Oncol. 2020;13(1):148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 593. Cornell RF, Baz R, Richter JR, et al. A phase 1 clinical trial of oral eltanexor in patients with relapsed or refractory multiple myeloma. Am J Hematol. 2022;97(2):E54‐E58. [DOI] [PubMed] [Google Scholar]
- 594. Lee S, Mohan S, Knupp J, et al. Oral eltanexor treatment of patients with higher‐risk myelodysplastic syndrome refractory to hypomethylating agents. J Hematol Oncol. 2022;15(1):103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 595. Turner JG, Cui Y, Bauer AA, et al. Melphalan and exportin 1 inhibitors exert synergistic antitumor effects in preclinical models of human multiple myeloma. Cancer Res. 2020;80(23):5344‐5354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 596. Verbeke D, Demeyer S, Prieto C, et al. The XPO1 inhibitor KPT‐8602 synergizes with dexamethasone in acute lymphoblastic leukemia. Clin Cancer Res. 2020;26(21):5747‐5758. [DOI] [PubMed] [Google Scholar]
- 597. Ou L, Wang X, Cheng S, et al. Verdinexor, a selective inhibitor of nuclear exportin 1, inhibits the proliferation and migration of esophageal cancer via XPO1/c‐Myc/FOSL1 axis. Int J Biol Sci. 2022;18(1):276‐291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 598. Pan L, Cheng C, Duan P, Chen K, Wu Y, Wu Z. XPO1/CRM1 is a promising prognostic indicator for neuroblastoma and represented a therapeutic target by selective inhibitor verdinexor. J Exp Clin Cancer Res. 2021;40(1):255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 599. Alsayed R, Khan AQ, Ahmad F, et al. Epigenetic regulation of CXCR4 signaling in cancer pathogenesis and progression. Semin Cancer Biol. 2022:S1044‐579X(22)00075‐X. [DOI] [PubMed] [Google Scholar]
- 600. Daniel SK, Seo YD, Pillarisetty VG. The CXCL12‐CXCR4/CXCR7 axis as a mechanism of immune resistance in gastrointestinal malignancies. Semin Cancer Biol. 2020;65:176‐188. [DOI] [PubMed] [Google Scholar]
- 601. Barbolina MV, Kim M, Liu Y, et al. Microenvironmental regulation of chemokine (C‐X‐C‐motif) receptor 4 in ovarian carcinoma. Mol Cancer Res. 2010;8(5):653‐664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 602. Li YM, Pan Y, Wei Y, et al. Upregulation of CXCR4 is essential for HER2‐mediated tumor metastasis. Cancer Cell. 2004;6(5):459‐469. [DOI] [PubMed] [Google Scholar]
- 603. Burger M, Glodek A, Hartmann T, et al. Functional expression of CXCR4 (CD184) on small‐cell lung cancer cells mediates migration, integrin activation, and adhesion to stromal cells. Oncogene. 2003;22(50):8093‐8101. [DOI] [PubMed] [Google Scholar]
- 604. Wang J‐F, Park I‐W, Groopman JE. Stromal cell‐derived factor‐1α stimulates tyrosine phosphorylation of multiple focal adhesion proteins and induces migration of hematopoietic progenitor cells: roles of phosphoinositide‐3 kinase and protein kinase C. Blood. 2000;95(8):2505‐2513. [PubMed] [Google Scholar]
- 605. DiPersio JF, Micallef IN, Stiff PJ, et al. Phase III prospective randomized double‐blind placebo‐controlled trial of plerixafor plus granulocyte colony‐stimulating factor compared with placebo plus granulocyte colony‐stimulating factor for autologous stem‐cell mobilization and transplantation for patients with non‐Hodgkin's lymphoma. J Clin Oncol. 2009;27(28):4767‐4773. [DOI] [PubMed] [Google Scholar]
- 606. Devine SM, Flomenberg N, Vesole DH, et al. Rapid mobilization of CD34+ cells following administration of the CXCR4 antagonist AMD3100 to patients with multiple myeloma and non‐Hodgkin's lymphoma. J Clin Oncol. 2004;22(6):1095‐1102. [DOI] [PubMed] [Google Scholar]
- 607. Conley‐LaComb MK, Semaan L, Singareddy R, et al. Pharmacological targeting of CXCL12/CXCR4 signaling in prostate cancer bone metastasis. Mol Cancer. 2016;15(1):68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 608. Chaudary N, Pintilie M, Jelveh S, Lindsay P, Hill RP, Milosevic M. Plerixafor improves primary tumor response and reduces metastases in cervical cancer treated with radio‐chemotherapy. Clin Cancer Res. 2017;23(5):1242‐1249. [DOI] [PubMed] [Google Scholar]
- 609. Thomas RP, Nagpal S, Iv M, et al. Macrophage exclusion after radiation therapy (MERT): a first in human phase I/II trial using a CXCR4 inhibitor in glioblastoma. Clin Cancer Res. 2019;25(23):6948‐6957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 610. Lee EQ, Duda DG, Muzikansky A, et al. Phase I and biomarker study of plerixafor and bevacizumab in recurrent high‐grade glioma. Clin Cancer Res. 2018;24(19):4643‐4649. [DOI] [PubMed] [Google Scholar]
- 611. Ghasemi K, Ghasemi K. MSX‐122: Is an effective small molecule CXCR4 antagonist in cancer therapy? Int Immunopharmacol. 2022;108:108863. [DOI] [PubMed] [Google Scholar]
- 612. Boddu P, Borthakur G, Koneru M, et al. Initial report of a phase I study of LY2510924, idarubicin, and cytarabine in relapsed/refractory acute myeloid leukemia. Front Oncol. 2018;8:369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 613. Salgia R, Stille JR, Weaver RW, et al. A randomized phase II study of LY2510924 and carboplatin/etoposide versus carboplatin/etoposide in extensive‐disease small cell lung cancer. Lung Cancer. 2017;105:7‐13. [DOI] [PubMed] [Google Scholar]
- 614. Hainsworth JD, Reeves JA, Mace JR, et al. A randomized, open‐label phase 2 study of the CXCR4 inhibitor LY2510924 in combination with sunitinib versus sunitinib alone in patients with metastatic renal cell carcinoma (RCC). Target Oncol. 2016;11(5):643‐653. [DOI] [PubMed] [Google Scholar]
- 615. Choueiri TK, Atkins MB, Rose TL, et al. A phase 1b trial of the CXCR4 inhibitor mavorixafor and nivolumab in advanced renal cell carcinoma patients with no prior response to nivolumab monotherapy. Invest New Drugs. 2021;39(4):1019‐1027. [DOI] [PubMed] [Google Scholar]
- 616. Gaines T, Garcia F, Virani S, et al. Synthesis and evaluation of 2,5‐furan, 2,5‐thiophene and 3,4‐thiophene‐based derivatives as CXCR4 inhibitors. Eur J Med Chem. 2019;181:111562. [DOI] [PubMed] [Google Scholar]
- 617. Zhang Z, Wu H, Huang S, et al. AMD3465 (hexahydrobromide) rescues the MG63 osteoblasts against the apoptosis induced by high glucose. Biomed Pharmacother. 2021;138:111476. [DOI] [PubMed] [Google Scholar]
- 618. Nissley DV, McCormick F. RAS at 40: Update from the RAS Initiative. Cancer Discov. 2022;12(4):895‐898. [DOI] [PubMed] [Google Scholar]
- 619. Reck M, Carbone DP, Garassino M, Barlesi F. Targeting KRAS in non‐small‐cell lung cancer: recent progress and new approaches. Ann Oncol. 2021;32(9):1101‐1110. [DOI] [PubMed] [Google Scholar]
- 620. Palma G, Khurshid F, Lu K, Woodward B, Husain H. Selective KRAS G12C inhibitors in non‐small cell lung cancer: chemistry, concurrent pathway alterations, and clinical outcomes. NPJ Precis Oncol. 2021;5(1):98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 621. Akhave NS, Biter AB, Hong DS. Mechanisms of resistance to KRAS(G12C)‐targeted therapy. Cancer Discov. 2021;11(6):1345‐1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 622. Skoulidis F, Li BT, Dy GK, et al. Sotorasib for lung cancers with KRAS p.G12C mutation. N Engl J Med. 2021;384(25):2371‐2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 623. Jänne PA, Riely GJ, Gadgeel SM, et al. Adagrasib in non‐small‐cell lung cancer harboring a KRAS(G12C) mutation. N Engl J Med. 2022;387(2):120‐131. [DOI] [PubMed] [Google Scholar]
- 624. Weiss A, Lorthiois E, Barys L, et al. Discovery, preclinical characterization, and early clinical activity of JDQ443, a structurally novel, potent, and selective covalent oral inhibitor of KRASG12C. Cancer Discov. 2022;12(6):1500‐1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 625. Ho AL, Brana I, Haddad R, et al. Tipifarnib in head and neck squamous cell carcinoma with HRAS mutations. J Clin Oncol. 2021;39(17):1856‐1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 626. Lee HW, Sa JK, Gualberto A, et al. A phase II trial of tipifarnib for patients with previously treated, metastatic urothelial carcinoma harboring HRAS mutations. Clin Cancer Res. 2020;26(19):5113‐5119. [DOI] [PubMed] [Google Scholar]
- 627. Hanna GJ, Guenette JP, Chau NG, et al. Tipifarnib in recurrent, metastatic HRAS‐mutant salivary gland cancer. Cancer. 2020;126(17):3972‐3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 628. Hudes G, Carducci M, Tomczak P, et al. Temsirolimus, interferon alfa, or both for advanced renal‐cell carcinoma. N Engl J Med. 2007;356(22):2271‐2281. [DOI] [PubMed] [Google Scholar]
- 629. Motzer RJ, Escudier B, Oudard S, et al. Efficacy of everolimus in advanced renal cell carcinoma: a double‐blind, randomised, placebo‐controlled phase III trial. Lancet. 2008;372(9637):449‐456. [DOI] [PubMed] [Google Scholar]
- 630. Baselga J, Campone M, Piccart M, et al. Everolimus in postmenopausal hormone‐receptor‐positive advanced breast cancer. N Engl J Med. 2012;366(6):520‐529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 631. Wagner AJ, Ravi V, Riedel RF, et al. nab‐Sirolimus for patients with malignant perivascular epithelioid cell tumors. J Clin Oncol. 2021;39(33):3660‐3670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 632. Quaglio D, Infante P, Di Marcotullio L, Botta B, Mori M. Hedgehog signaling pathway inhibitors: an updated patent review (2015‐present). Expert Opin Ther Pat. 2020;30(4):235‐250. [DOI] [PubMed] [Google Scholar]
- 633. Galperin I, Dempwolff L, Diederich WE, Lauth M. Inhibiting hedgehog: an update on pharmacological compounds and targeting strategies. J Med Chem. 2019;62(18):8392‐8411. [DOI] [PubMed] [Google Scholar]
- 634. Sekulic A, Migden MR, Oro AE, et al. Efficacy and safety of vismodegib in advanced basal‐cell carcinoma. N Engl J Med. 2012;366(23):2171‐2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 635. Migden MR, Guminski A, Gutzmer R, et al. Treatment with two different doses of sonidegib in patients with locally advanced or metastatic basal cell carcinoma (BOLT): a multicentre, randomised, double‐blind phase 2 trial. Lancet Oncol. 2015;16(6):716‐728. [DOI] [PubMed] [Google Scholar]
- 636. Cortes JE, Heidel FH, Hellmann A, et al. Randomized comparison of low dose cytarabine with or without glasdegib in patients with newly diagnosed acute myeloid leukemia or high‐risk myelodysplastic syndrome. Leukemia. 2019;33(2):379‐389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 637. Savona MR, Pollyea DA, Stock W, et al. Phase Ib study of glasdegib, a hedgehog pathway inhibitor, in combination with standard chemotherapy in patients with AML or high‐risk MDS. Clin Cancer Res. 2018;24(10):2294‐2303. [DOI] [PubMed] [Google Scholar]
- 638. Cortes JE, Douglas Smith B, Wang ES, et al. Glasdegib in combination with cytarabine and daunorubicin in patients with AML or high‐risk MDS: Phase 2 study results. Am J Hematol. 2018;93(11):1301‐1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 639. Hall ET, Cleverdon ER, Ogden SK. Dispatching sonic hedgehog: molecular mechanisms controlling deployment. Trends Cell Biol. 2019;29(5):385‐395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 640. Qi X, Li X. Mechanistic insights into the generation and transduction of hedgehog signaling. Trends Biochem Sci. 2020;45(5):397‐410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 641. Herms F, Lambert J, Grob JJ, et al. Follow‐up of patients with complete remission of locally advanced basal cell carcinoma after vismodegib discontinuation: a multicenter french study of 116 patients. J Clin Oncol. 2019;37(34):3275‐3282. [DOI] [PubMed] [Google Scholar]
- 642. Dréno B, Kunstfeld R, Hauschild A, et al. Two intermittent vismodegib dosing regimens in patients with multiple basal‐cell carcinomas (MIKIE): a randomised, regimen‐controlled, double‐blind, phase 2 trial. Lancet Oncol. 2017;18(3):404‐412. [DOI] [PubMed] [Google Scholar]
- 643. Sharpe HJ, Pau G, Dijkgraaf GJ, et al. Genomic analysis of smoothened inhibitor resistance in basal cell carcinoma. Cancer Cell. 2015;27(3):327‐341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 644. Sánchez‐Danés A, Larsimont JC, Liagre M, et al. A slow‐cycling LGR5 tumour population mediates basal cell carcinoma relapse after therapy. Nature. 2018;562(7727):434‐438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 645. Filbin MG, Dabral SK, Pazyra‐Murphy MF, et al. Coordinate activation of Shh and PI3K signaling in PTEN‐deficient glioblastoma: new therapeutic opportunities. Nat Med. 2013;19(11):1518‐1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 646. Kieran MW, Chisholm J, Casanova M, et al. Phase I study of oral sonidegib (LDE225) in pediatric brain and solid tumors and a phase II study in children and adults with relapsed medulloblastoma. Neuro Oncol. 2017;19(11):1542‐1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 647. Robinson GW, Orr BA, Wu G, et al. Vismodegib exerts targeted efficacy against recurrent sonic hedgehog‐subgroup medulloblastoma: results from phase II Pediatric Brain Tumor Consortium Studies PBTC‐025B and PBTC‐032. J Clin Oncol. 2015;33(24):2646‐2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 648. Catenacci DV, Junttila MR, Karrison T, et al. Randomized phase Ib/II study of gemcitabine plus placebo or vismodegib, a hedgehog pathway inhibitor, in patients with metastatic pancreatic cancer. J Clin Oncol. 2015;33(36):4284‐4292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 649. Bendell J, Andre V, Ho A, et al. Phase I Study of LY2940680, a Smo antagonist, in patients with advanced cancer including treatment‐naïve and previously treated basal cell carcinoma. Clin Cancer Res. 2018;24(9):2082‐2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 650. Carneiro BA, El‐Deiry WS. Targeting apoptosis in cancer therapy. Nat Rev Clin Oncol. 2020;17(7):395‐417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 651. Diepstraten ST, Anderson MA, Czabotar PE, Lessene G, Strasser A, Kelly GL. The manipulation of apoptosis for cancer therapy using BH3‐mimetic drugs. Nat Rev Cancer. 2022;22(1):45‐64. [DOI] [PubMed] [Google Scholar]
- 652. Roberts AW, Davids MS, Pagel JM, et al. Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia. N Engl J Med. 2016;374(4):311‐322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 653. Delbridge AR, Grabow S, Strasser A, Vaux DL. Thirty years of BCL‐2: translating cell death discoveries into novel cancer therapies. Nat Rev Cancer. 2016;16(2):99‐109. [DOI] [PubMed] [Google Scholar]
- 654. Roberts AW, Ma S, Kipps TJ, et al. Efficacy of venetoclax in relapsed chronic lymphocytic leukemia is influenced by disease and response variables. Blood. 2019;134(2):111‐122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 655. Jones JA, Mato AR, Wierda WG, et al. Venetoclax for chronic lymphocytic leukaemia progressing after ibrutinib: an interim analysis of a multicentre, open‐label, phase 2 trial. Lancet Oncol. 2018;19(1):65‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 656. Stilgenbauer S, Eichhorst B, Schetelig J, et al. Venetoclax in relapsed or refractory chronic lymphocytic leukaemia with 17p deletion: a multicentre, open‐label, phase 2 study. Lancet Oncol. 2016;17(6):768‐778. [DOI] [PubMed] [Google Scholar]
- 657. Al‐Sawaf O, Zhang C, Tandon M, et al. Venetoclax plus obinutuzumab versus chlorambucil plus obinutuzumab for previously untreated chronic lymphocytic leukaemia (CLL14): follow‐up results from a multicentre, open‐label, randomised, phase 3 trial. Lancet Oncol. 2020;21(9):1188‐1200. [DOI] [PubMed] [Google Scholar]
- 658. Kater AP, Seymour JF, Hillmen P, et al. Fixed duration of venetoclax‐rituximab in relapsed/refractory chronic lymphocytic leukemia eradicates minimal residual disease and prolongs survival: post‐treatment follow‐up of the MURANO phase III study. J Clin Oncol. 2019;37(4):269‐277. [DOI] [PubMed] [Google Scholar]
- 659. DiNardo CD, Pratz KW, Letai A, et al. Safety and preliminary efficacy of venetoclax with decitabine or azacitidine in elderly patients with previously untreated acute myeloid leukaemia: a non‐randomised, open‐label, phase 1b study. Lancet Oncol. 2018;19(2):216‐228. [DOI] [PubMed] [Google Scholar]
- 660. Wei AH, Panayiotidis P, Montesinos P, et al. 6‐month follow‐up of VIALE‐C demonstrates improved and durable efficacy in patients with untreated AML ineligible for intensive chemotherapy (141/150). Blood Cancer J. 2021;11(10):163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 661. Castillo JJ, Allan JN, Siddiqi T, et al. Venetoclax in previously treated Waldenström Macroglobulinemia. J Clin Oncol. 2022;40(1):63‐71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 662. Bahlis NJ, Baz R, Harrison SJ, et al. Phase I study of venetoclax plus daratumumab and dexamethasone, with or without bortezomib, in patients with relapsed or refractory multiple myeloma with and without t(11;14). J Clin Oncol. 2021;39(32):3602‐3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 663. Wierda WG, Allan JN, Siddiqi T, et al. Ibrutinib plus venetoclax for first‐line treatment of chronic lymphocytic leukemia: primary analysis results from the minimal residual disease cohort of the randomized phase II CAPTIVATE study. J Clin Oncol. 2021;39(34):3853‐3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 664. Davids MS, Lampson BL, Tyekucheva S, et al. Acalabrutinib, venetoclax, and obinutuzumab as frontline treatment for chronic lymphocytic leukaemia: a single‐arm, open‐label, phase 2 study. Lancet Oncol. 2021;22(10):1391‐1402. [DOI] [PubMed] [Google Scholar]
- 665. Wilson WH, O'Connor OA, Czuczman MS, et al. Navitoclax, a targeted high‐affinity inhibitor of BCL‐2, in lymphoid malignancies: a phase 1 dose‐escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol. 2010;11(12):1149‐1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 666. Gandhi L, Camidge DR, Ribeiro de Oliveira M, et al. Phase I study of Navitoclax (ABT‐263), a novel Bcl‐2 family inhibitor, in patients with small‐cell lung cancer and other solid tumors. J Clin Oncol. 2011;29(7):909‐916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 667. Roberts AW, Seymour JF, Brown JR, et al. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: results of a phase I study of navitoclax in patients with relapsed or refractory disease. J Clin Oncol. 2012;30(5):488‐496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 668. Pullarkat VA, Lacayo NJ, Jabbour E, et al. Venetoclax and navitoclax in combination with chemotherapy in patients with relapsed or refractory acute lymphoblastic leukemia and lymphoblastic lymphoma. Cancer Discov. 2021;11(6):1440‐1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 669. Rosenbaum MI, Clemmensen LS, Bredt DS, Bettler B, Strømgaard K. Targeting receptor complexes: a new dimension in drug discovery. Nat Rev Drug Discov. 2020;19(12):884‐901. [DOI] [PubMed] [Google Scholar]
- 670. Bianco G, Goodsell DS, Forli S. Selective and effective: current progress in computational structure‐based drug discovery of targeted covalent inhibitors. Trends Pharmacol Sci. 2020;41(12):1038‐1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 671. Logsdon GA, Vollger MR, Eichler EE. Long‐read human genome sequencing and its applications. Nat Rev Genet. 2020;21(10):597‐614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 672. Finotello F, Rieder D, Hackl H, Trajanoski Z. Next‐generation computational tools for interrogating cancer immunity. Nat Rev Genet. 2019;20(12):724‐746. [DOI] [PubMed] [Google Scholar]
- 673. Fischer K, Al‐Sawaf O, Bahlo J, et al. Venetoclax and obinutuzumab in patients with CLL and coexisting conditions. N Engl J Med. 2019;380(23):2225‐2236. [DOI] [PubMed] [Google Scholar]
- 674. Posadas EM, Limvorasak S, Figlin RA. Targeted therapies for renal cell carcinoma. Nat Rev Nephrol. 2017;13(8):496‐511. [DOI] [PubMed] [Google Scholar]
- 675. Llovet JM, Montal R, Sia D, Finn RS. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat Rev Clin Oncol. 2018;15(10):599‐616. [DOI] [PubMed] [Google Scholar]
- 676. Signoretti S, Flaifel A, Chen YB, Reuter VE. Renal cell carcinoma in the era of precision medicine: from molecular pathology to tissue‐based biomarkers. J Clin Oncol. 2018:JCO2018792259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 677. Kotecha RR, Motzer RJ, Voss MH. Towards individualized therapy for metastatic renal cell carcinoma. Nat Rev Clin Oncol. 2019;16(10):621‐633. [DOI] [PubMed] [Google Scholar]
- 678. Ravaud A, Motzer RJ, Pandha HS, et al. Adjuvant sunitinib in high‐risk renal‐cell carcinoma after nephrectomy. N Engl J Med. 2016;375(23):2246‐2254. [DOI] [PubMed] [Google Scholar]
- 679. Motzer RJ, Michaelson MD, Redman BG, et al. Activity of SU11248, a multitargeted inhibitor of vascular endothelial growth factor receptor and platelet‐derived growth factor receptor, in patients with metastatic renal cell carcinoma. J Clin Oncol. 2006;24(1):16‐24. [DOI] [PubMed] [Google Scholar]
- 680. Motzer RJ, Hutson TE, Tomczak P, et al. Sunitinib versus interferon alfa in metastatic renal‐cell carcinoma. N Engl J Med. 2007;356(2):115‐124. [DOI] [PubMed] [Google Scholar]
- 681. Motzer RJ, Penkov K, Haanen J, et al. Avelumab plus axitinib versus sunitinib for advanced renal‐cell carcinoma. N Engl J Med. 2019;380(12):1103‐1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 682. Rini BI, Plimack ER, Stus V, et al. Pembrolizumab plus axitinib versus sunitinib for advanced renal‐cell carcinoma. N Engl J Med. 2019;380(12):1116‐1127. [DOI] [PubMed] [Google Scholar]
- 683. Choueiri TK, Powles T, Burotto M, et al. Nivolumab plus cabozantinib versus sunitinib for advanced renal‐cell carcinoma. N Engl J Med. 2021;384(9):829‐841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 684. Motzer R, Alekseev B, Rha SY, et al. Lenvatinib plus pembrolizumab or everolimus for advanced renal cell carcinoma. N Engl J Med. 2021;384(14):1289‐1300. [DOI] [PubMed] [Google Scholar]
- 685. Cheng AL, Hsu C, Chan SL, Choo SP, Kudo M. Challenges of combination therapy with immune checkpoint inhibitors for hepatocellular carcinoma. J Hepatol. 2020;72(2):307‐319. [DOI] [PubMed] [Google Scholar]
- 686. Escudier B, Eisen T, Stadler WM, et al. Sorafenib in advanced clear‐cell renal‐cell carcinoma. N Engl J Med. 2007;356(2):125‐134. [DOI] [PubMed] [Google Scholar]
- 687. Rini BI, Pal SK, Escudier BJ, et al. Tivozanib versus sorafenib in patients with advanced renal cell carcinoma (TIVO‐3): a phase 3, multicentre, randomised, controlled, open‐label study. Lancet Oncol. 2020;21(1):95‐104. [DOI] [PubMed] [Google Scholar]
- 688. Cheng AL, Kang YK, Chen Z, et al. Efficacy and safety of sorafenib in patients in the Asia‐Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double‐blind, placebo‐controlled trial. Lancet Oncol. 2009;10(1):25‐34. [DOI] [PubMed] [Google Scholar]
- 689. Llovet JM, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378‐390. [DOI] [PubMed] [Google Scholar]
- 690. Kudo M, Finn RS, Qin S, et al. Lenvatinib versus sorafenib in first‐line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non‐inferiority trial. Lancet. 2018;391(10126):1163‐1173. [DOI] [PubMed] [Google Scholar]
- 691. Bruix J, Qin S, Merle P, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet. 2017;389(10064):56‐66. [DOI] [PubMed] [Google Scholar]
- 692. Abou‐Alfa GK, Meyer T, Cheng AL, et al. Cabozantinib in patients with advanced and progressing hepatocellular carcinoma. N Engl J Med. 2018;379(1):54‐63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 693. Duffaud F, Mir O, Boudou‐Rouquette P, et al. Efficacy and safety of regorafenib in adult patients with metastatic osteosarcoma: a non‐comparative, randomised, double‐blind, placebo‐controlled, phase 2 study. Lancet Oncol. 2019;20(1):120‐133. [DOI] [PubMed] [Google Scholar]
- 694. Finn RS, Merle P, Granito A, et al. Outcomes of sequential treatment with sorafenib followed by regorafenib for HCC: Additional analyses from the phase III RESORCE trial. J Hepatol. 2018;69(2):353‐358. [DOI] [PubMed] [Google Scholar]
- 695. Brose MS, Nutting CM, Jarzab B, et al. Sorafenib in radioactive iodine‐refractory, locally advanced or metastatic differentiated thyroid cancer: a randomised, double‐blind, phase 3 trial. Lancet. 2014;384(9940):319‐328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 696. Schlumberger M, Tahara M, Wirth LJ, et al. Lenvatinib versus placebo in radioiodine‐refractory thyroid cancer. N Engl J Med. 2015;372(7):621‐630. [DOI] [PubMed] [Google Scholar]
- 697. Brose MS, Robinson B, Sherman SI, et al. Cabozantinib for radioiodine‐refractory differentiated thyroid cancer (COSMIC‐311): a randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet Oncol. 2021;22(8):1126‐1138. [DOI] [PubMed] [Google Scholar]
- 698. Lamartina L, Grani G, Durante C, Filetti S, Cooper DS. Screening for differentiated thyroid cancer in selected populations. The Lancet Diabetes & Endocrinology. 2020;8(1):81‐88. [DOI] [PubMed] [Google Scholar]
- 699. Wells SA, Jr. , Robinson BG, Gagel RF, et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: a randomized, double‐blind phase III trial. J Clin Oncol. 2012;30(2):134‐141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 700. Elisei R, Schlumberger MJ, Müller SP, et al. Cabozantinib in progressive medullary thyroid cancer. J Clin Oncol. 2013;31(29):3639‐3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 701. Sun L, Liang C, Shirazian S, et al. Discovery of 5‐[5‐fluoro‐2‐oxo‐1,2‐ dihydroindol‐(3Z)‐ylidenemethyl]‐2,4‐ dimethyl‐1H‐pyrrole‐3‐carboxylic acid (2‐diethylaminoethyl)amide, a novel tyrosine kinase inhibitor targeting vascular endothelial and platelet‐derived growth factor receptor tyrosine kinase. J Med Chem. 2003;46(7):1116‐1119. [DOI] [PubMed] [Google Scholar]
- 702. Wilhelm SM, Dumas J, Adnane L, et al. Regorafenib (BAY 73–4506): a new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int J Cancer. 2011;129(1):245‐255. [DOI] [PubMed] [Google Scholar]
- 703. Demetri GD, van Oosterom AT, Garrett CR, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368(9544):1329‐1338. [DOI] [PubMed] [Google Scholar]
- 704. Heinrich MC, Maki RG, Corless CL, et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib‐resistant gastrointestinal stromal tumor. J Clin Oncol. 2008;26(33):5352‐5359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 705. Demetri GD, Reichardt P, Kang YK, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo‐controlled, phase 3 trial. Lancet. 2013;381(9863):295‐302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 706. Ben‐Ami E, Barysauskas CM, von Mehren M, et al. Long‐term follow‐up results of the multicenter phase II trial of regorafenib in patients with metastatic and/or unresectable GI stromal tumor after failure of standard tyrosine kinase inhibitor therapy. Ann Oncol. 2016;27(9):1794‐1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 707. Raymond E, Dahan L, Raoul JL, et al. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N Engl J Med. 2011;364(6):501‐513. [DOI] [PubMed] [Google Scholar]
- 708. van der Graaf WT, Blay JY, Chawla SP, et al. Pazopanib for metastatic soft‐tissue sarcoma (PALETTE): a randomised, double‐blind, placebo‐controlled phase 3 trial. Lancet. 2012;379(9829):1879‐1886. [DOI] [PubMed] [Google Scholar]
- 709. Grothey A, Van Cutsem E, Sobrero A, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo‐controlled, phase 3 trial. Lancet. 2013;381(9863):303‐312. [DOI] [PubMed] [Google Scholar]
- 710. Makker V, Colombo N, Casado Herráez A, et al. Lenvatinib plus pembrolizumab for advanced endometrial cancer. N Engl J Med. 2022;386(5):437‐448. [DOI] [PubMed] [Google Scholar]
- 711. Brose MS, Bible KC, Chow LQM, et al. Management of treatment‐related toxicities in advanced medullary thyroid cancer. Cancer Treat Rev. 2018;66:64‐73. [DOI] [PubMed] [Google Scholar]
- 712. Van Cutsem E, Bajetta E, Valle J, et al. Randomized, placebo‐controlled, phase III study of oxaliplatin, fluorouracil, and leucovorin with or without PTK787/ZK 222584 in patients with previously treated metastatic colorectal adenocarcinoma. J Clin Oncol. 2011;29(15):2004‐2010. [DOI] [PubMed] [Google Scholar]
- 713. Hecht JR, Trarbach T, Hainsworth JD, et al. Randomized, placebo‐controlled, phase III study of first‐line oxaliplatin‐based chemotherapy plus PTK787/ZK 222584, an oral vascular endothelial growth factor receptor inhibitor, in patients with metastatic colorectal adenocarcinoma. J Clin Oncol. 2011;29(15):1997‐2003. [DOI] [PubMed] [Google Scholar]
- 714. Heymach JV, Desai J, Manola J, et al. Phase II study of the antiangiogenic agent SU5416 in patients with advanced soft tissue sarcomas. Clin Cancer Res. 2004;10(17):5732‐5740. [DOI] [PubMed] [Google Scholar]
- 715. Fury MG, Zahalsky A, Wong R, et al. A Phase II study of SU5416 in patients with advanced or recurrent head and neck cancers. Invest New Drugs. 2007;25(2):165‐172. [DOI] [PubMed] [Google Scholar]
- 716. Lu S, Chang J, Liu X, et al. Randomized, double‐blind, placebo‐controlled, multicenter phase II study of fruquintinib after two prior chemotherapy regimens in Chinese patients with advanced nonsquamous non‒small‐cell lung cancer. J Clin Oncol. 2018;36(12):1207‐1217. [DOI] [PubMed] [Google Scholar]
- 717. Li J, Qin S, Xu RH, et al. Effect of fruquintinib vs placebo on overall survival in patients with previously treated metastatic colorectal cancer: the FRESCO randomized clinical trial. Jama. 2018;319(24):2486‐2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 718. Han B, Li K, Wang Q, et al. Effect of anlotinib as a third‐line or further treatment on overall survival of patients with advanced non‐small cell lung cancer: the ALTER 0303 Phase 3 randomized clinical trial. JAMA Oncol. 2018;4(11):1569‐1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 719. Chi Y, Fang Z, Hong X, et al. Safety and efficacy of anlotinib, a multikinase angiogenesis inhibitor, in patients with refractory metastatic soft‐tissue sarcoma. Clin Cancer Res. 2018;24(21):5233‐5238. [DOI] [PubMed] [Google Scholar]
- 720. Li J, Qin S, Xu J, et al. Randomized, double‐blind, placebo‐controlled phase III trial of apatinib in patients with chemotherapy‐refractory advanced or metastatic adenocarcinoma of the stomach or gastroesophageal junction. J Clin Oncol. 2016;34(13):1448‐1454. [DOI] [PubMed] [Google Scholar]
- 721. Qin S, Bi F, Gu S, et al. Donafenib versus sorafenib in first‐line treatment of unresectable or metastatic hepatocellular carcinoma: a randomized, open‐label, parallel‐controlled phase II‐III trial. J Clin Oncol. 2021;39(27):3002‐3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 722. Xu J, Shen L, Zhou Z, et al. Surufatinib in advanced extrapancreatic neuroendocrine tumours (SANET‐ep): a randomised, double‐blind, placebo‐controlled, phase 3 study. Lancet Oncol. 2020;21(11):1500‐1512. [DOI] [PubMed] [Google Scholar]
- 723. Xu J, Shen L, Bai C, et al. Surufatinib in advanced pancreatic neuroendocrine tumours (SANET‐p): a randomised, double‐blind, placebo‐controlled, phase 3 study. Lancet Oncol. 2020;21(11):1489‐1499. [DOI] [PubMed] [Google Scholar]
- 724. Valle JW, Wasan H, Lopes A, et al. Cediranib or placebo in combination with cisplatin and gemcitabine chemotherapy for patients with advanced biliary tract cancer (ABC‐03): a randomised phase 2 trial. Lancet Oncol. 2015;16(8):967‐978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 725. Batchelor TT, Mulholland P, Neyns B, et al. Phase III randomized trial comparing the efficacy of cediranib as monotherapy, and in combination with lomustine, versus lomustine alone in patients with recurrent glioblastoma. J Clin Oncol. 2013;31(26):3212‐3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 726. Motzer RJ, Porta C, Vogelzang NJ, et al. Dovitinib versus sorafenib for third‐line targeted treatment of patients with metastatic renal cell carcinoma: an open‐label, randomised phase 3 trial. Lancet Oncol. 2014;15(3):286‐296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 727. Soria JC, DeBraud F, Bahleda R, et al. Phase I/IIa study evaluating the safety, efficacy, pharmacokinetics, and pharmacodynamics of lucitanib in advanced solid tumors. Ann Oncol. 2014;25(11):2244‐2251. [DOI] [PubMed] [Google Scholar]
- 728. Flaherty KR, Wells AU, Cottin V, et al. Nintedanib in progressive fibrosing interstitial lung diseases. N Engl J Med. 2019;381(18):1718‐1727. [DOI] [PubMed] [Google Scholar]
- 729. Richeldi L, du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2071‐2082. [DOI] [PubMed] [Google Scholar]
- 730. Choueiri TK, Vaishampayan U, Rosenberg JE, et al. Phase II and biomarker study of the dual MET/VEGFR2 inhibitor foretinib in patients with papillary renal cell carcinoma. J Clin Oncol. 2013;31(2):181‐186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 731. Rayson D, Lupichuk S, Potvin K, et al. Canadian Cancer Trials Group IND197: a phase II study of foretinib in patients with estrogen receptor, progesterone receptor, and human epidermal growth factor receptor 2‐negative recurrent or metastatic breast cancer. Breast Cancer Res Treat. 2016;157(1):109‐116. [DOI] [PubMed] [Google Scholar]
- 732. Shah MA, Wainberg ZA, Catenacci DV, et al. Phase II study evaluating 2 dosing schedules of oral foretinib (GSK1363089), cMET/VEGFR2 inhibitor, in patients with metastatic gastric cancer. PLoS One. 2013;8(3):e54014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 733. Cainap C, Qin S, Huang WT, et al. Linifanib versus Sorafenib in patients with advanced hepatocellular carcinoma: results of a randomized phase III trial. J Clin Oncol. 2015;33(2):172‐179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 734. Bagegni NA, Park H, Kraft K, et al. Phase 1b trial of anti‐VEGF/PDGFR vorolanib combined with immune checkpoint inhibitors in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2022;89(4):487‐497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 735. Kudo M, Han G, Finn RS, et al. Brivanib as adjuvant therapy to transarterial chemoembolization in patients with hepatocellular carcinoma: A randomized phase III trial. Hepatology. 2014;60(5):1697‐1707. [DOI] [PubMed] [Google Scholar]
- 736. Johnson PJ, Qin S, Park JW, et al. Brivanib versus sorafenib as first‐line therapy in patients with unresectable, advanced hepatocellular carcinoma: results from the randomized phase III BRISK‐FL study. J Clin Oncol. 2013;31(28):3517‐3524. [DOI] [PubMed] [Google Scholar]
- 737. Finn RS, Kang YK, Mulcahy M, et al. Phase II, open‐label study of brivanib as second‐line therapy in patients with advanced hepatocellular carcinoma. Clin Cancer Res. 2012;18(7):2090‐2098. [DOI] [PubMed] [Google Scholar]
- 738. Park JW, Finn RS, Kim JS, et al. Phase II, open‐label study of brivanib as first‐line therapy in patients with advanced hepatocellular carcinoma. Clin Cancer Res. 2011;17(7):1973‐1983. [DOI] [PubMed] [Google Scholar]
- 739. Bono P, Massard C, Peltola KJ, et al. Phase I/IIa, open‐label, multicentre study to evaluate the optimal dosing and safety of ODM‐203 in patients with advanced or metastatic solid tumours. ESMO Open. 2020;5(6):e001081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 740. Arbour KC, Riely GJ. Systemic therapy for locally advanced and metastatic non‐small cell lung cancer: a review. Jama. 2019;322(8):764‐774. [DOI] [PubMed] [Google Scholar]
- 741. Wang M, Herbst RS, Boshoff C. Toward personalized treatment approaches for non‐small‐cell lung cancer. Nat Med. 2021;27(8):1345‐1356. [DOI] [PubMed] [Google Scholar]
- 742. Zhao S, Allis CD, Wang GG. The language of chromatin modification in human cancers. Nat Rev Cancer. 2021;21(7):413‐430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 743. Hahn WC, Bader JS, Braun TP, et al. An expanded universe of cancer targets. Cell. 2021;184(5):1142‐1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 744. Tanaka K, Yu HA, Yang S, et al. Targeting Aurora B kinase prevents and overcomes resistance to EGFR inhibitors in lung cancer by enhancing BIM‐ and PUMA‐mediated apoptosis. Cancer Cell. 2021;39(9):1245‐1261. e1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 745. Franzoi MA, Agostinetto E, Perachino M, et al. Evidence‐based approaches for the management of side‐effects of adjuvant endocrine therapy in patients with breast cancer. Lancet Oncol. 2021;22(7):e303‐e313. [DOI] [PubMed] [Google Scholar]
- 746. Saw SPL, Ong BH, Chua KLM, Takano A, Tan DSW. Revisiting neoadjuvant therapy in non‐small‐cell lung cancer. Lancet Oncol. 2021;22(11):e501‐e516. [DOI] [PubMed] [Google Scholar]
- 747. Wu YL, Tsuboi M, He J, et al. Osimertinib in resected EGFR‐mutated non‐small‐cell lung cancer. N Engl J Med. 2020;383(18):1711‐1723. [DOI] [PubMed] [Google Scholar]
- 748. Chaft JE, Shyr Y, Sepesi B, Forde PM. Preoperative and postoperative systemic therapy for operable non‐small‐cell lung cancer. J Clin Oncol. 2022;40(6):546‐555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 749. Maio M, Lewis K, Demidov L, et al. Adjuvant vemurafenib in resected, BRAF(V600) mutation‐positive melanoma (BRIM8): a randomised, double‐blind, placebo‐controlled, multicentre, phase 3 trial. Lancet Oncol. 2018;19(4):510‐520. [DOI] [PubMed] [Google Scholar]
- 750. Pun SC, Neilan TG. Cardiovascular side effects of small molecule therapies for cancer. Eur Heart J. 2016;37(36):2742‐2745. [DOI] [PubMed] [Google Scholar]
- 751. Long GV, Saw RPM, Lo S, et al. Neoadjuvant dabrafenib combined with trametinib for resectable, stage IIIB‐C, BRAF(V600) mutation‐positive melanoma (NeoCombi): a single‐arm, open‐label, single‐centre, phase 2 trial. Lancet Oncol. 2019;20(7):961‐971. [DOI] [PubMed] [Google Scholar]
- 752. Bogart JA, Waqar SN, Mix MD. Radiation and Systemic Therapy for Limited‐Stage Small‐Cell Lung Cancer. J Clin Oncol. 2022;40(6):661‐670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 753. Zhu X, Cao Y, Liu W, et al. Stereotactic body radiotherapy plus pembrolizumab and trametinib versus stereotactic body radiotherapy plus gemcitabine for locally recurrent pancreatic cancer after surgical resection: an open‐label, randomised, controlled, phase 2 trial. Lancet Oncol. 2022;23(3):e105‐e115. [DOI] [PubMed] [Google Scholar]
- 754. Braun TP, Druker BJ. Tyrosine kinase inhibitor discontinuation in patients with chronic myeloid leukemia: updates from the LAST study on patient‐reported outcomes and biomarkers for relapse. JAMA Oncol. 2021;7(1):50‐51. [DOI] [PubMed] [Google Scholar]
- 755. Saussele S, Richter J, Guilhot J, et al. Discontinuation of tyrosine kinase inhibitor therapy in chronic myeloid leukaemia (EURO‐SKI): a prespecified interim analysis of a prospective, multicentre, non‐randomised, trial. The Lancet Oncology. 2018;19(6):747‐757. [DOI] [PubMed] [Google Scholar]
- 756. Braun TP, Eide CA, Druker BJ. Response and resistance to BCR‐ABL1‐targeted therapies. Cancer Cell. 2020;37(4):530‐542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 757. Etienne G, Guilhot J, Rea D, et al. Long‐term follow‐up of the French stop imatinib (STIM1) study in patients with chronic myeloid leukemia. J Clin Oncol. 2017;35(3):298‐305. [DOI] [PubMed] [Google Scholar]
- 758. Le Cesne A, Ray‐Coquard I, Bui BN, et al. Discontinuation of imatinib in patients with advanced gastrointestinal stromal tumours after 3 years of treatment: an open‐label multicentre randomised phase 3 trial. Lancet Oncol. 2010;11(10):942‐949. [DOI] [PubMed] [Google Scholar]
- 759. Heersche N, Veerman GDM, de With M, et al. Clinical implications of germline variations for treatment outcome and drug resistance for small molecule kinase inhibitors in patients with non‐small cell lung cancer. Drug Resist Updat. 2022;62:100832. [DOI] [PubMed] [Google Scholar]
- 760. Weiss F, Lauffenburger D, Friedl P. Towards targeting of shared mechanisms of cancer metastasis and therapy resistance. Nat Rev Cancer. 2022;22(3):157‐173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 761. Herbst RS, Morgensztern D, Boshoff C. The biology and management of non‐small cell lung cancer. Nature. 2018;553(7689):446‐454. [DOI] [PubMed] [Google Scholar]
- 762. Gu X, Qiu Y, Lin M, et al. CuS nanoparticles as a photodynamic nanoswitch for abrogating bypass signaling to overcome gefitinib resistance. Nano Lett. 2019;19(5):3344‐3352. [DOI] [PubMed] [Google Scholar]
- 763. Kuwano M, Sonoda K, Murakami Y, Watari K, Ono M. Overcoming drug resistance to receptor tyrosine kinase inhibitors: learning from lung cancer. Pharmacol Ther. 2016;161:97‐110. [DOI] [PubMed] [Google Scholar]
- 764. Rosell R, Chaib I, Santarpia M. Targeting MET amplification in EGFR‐mutant non‐small‐cell lung cancer. Lancet Respir Med. 2020;8(11):1068‐1070. [DOI] [PubMed] [Google Scholar]
- 765. Weiss F, Lauffenburger D, Friedl P. Towards targeting of shared mechanisms of cancer metastasis and therapy resistance. Nat Rev Cancer. 2022;22(3):157‐173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 766. Yalcin‐Ozkat G. Molecular modeling strategies of cancer multidrug resistance. Drug Resist Updat. 2021;59:100789. [DOI] [PubMed] [Google Scholar]
- 767. Hanssen KM, Haber M, Fletcher JI. Targeting multidrug resistance‐associated protein 1 (MRP1)‐expressing cancers: beyond pharmacological inhibition. Drug Resist Updat. 2021;59:100795. [DOI] [PubMed] [Google Scholar]
- 768. Oser MG, Niederst MJ, Sequist LV, Engelman JA. Transformation from non‐small‐cell lung cancer to small‐cell lung cancer: molecular drivers and cells of origin. Lancet Oncol. 2015;16(4):e165‐172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 769. Li R, Jiang L, Zhou X, Lu Y, Zhang Y. Pseudo‐small cell transformation in EGFR‐mutant adenocarcinoma. Lung Cancer. 2021;153:120‐125. [DOI] [PubMed] [Google Scholar]
- 770. Sosič I, Bricelj A, Steinebach C. E3 ligase ligand chemistries: from building blocks to protein degraders. Chem Soc Rev. 2022;51(9):3487‐3534. [DOI] [PubMed] [Google Scholar]
- 771. Cowan AD, Ciulli A. Driving E3 ligase substrate specificity for targeted protein degradation: lessons from nature and the laboratory. Annu Rev Biochem. 2022;91:295‐319. [DOI] [PubMed] [Google Scholar]
- 772. Bekes M, Langley DR, Crews CM. PROTAC targeted protein degraders: the past is prologue. Nat Rev Drug Discov. 2022;21(3):181‐200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 773. Schneider M, Radoux CJ, Hercules A, et al. The PROTACtable genome. Nat Rev Drug Discov. 2021;20(10):789‐797. [DOI] [PubMed] [Google Scholar]
- 774. Dale B, Cheng M, Park KS, Kaniskan H, Xiong Y, Jin J. Advancing targeted protein degradation for cancer therapy. Nat Rev Cancer. 2021;21(10):638‐654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 775. Shah MP, Neal JW. Targeting acquired and intrinsic resistance mechanisms in epidermal growth factor receptor mutant non‐small‐cell lung cancer. Drugs. 2022;82(6):649‐662. [DOI] [PubMed] [Google Scholar]
- 776. Chi P, Qin LX, Nguyen B, et al. Phase II trial of imatinib plus binimetinib in patients with treatment‐naive advanced gastrointestinal stromal tumor. J Clin Oncol. 2022;40(9):997‐1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 777. Harrison CN, Garcia JS, Somervaille TCP, et al. Addition of navitoclax to ongoing ruxolitinib therapy for patients with myelofibrosis with progression or suboptimal response: phase II safety and efficacy. J Clin Oncol. 2022;40(15):1671‐1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 778. Kawashima Y, Fukuhara T, Saito H, et al. Bevacizumab plus erlotinib versus erlotinib alone in Japanese patients with advanced, metastatic, EGFR‐mutant non‐small‐cell lung cancer (NEJ026): overall survival analysis of an open‐label, randomised, multicentre, phase 3 trial. Lancet Respir Med. 2022;10(1):72‐82. [DOI] [PubMed] [Google Scholar]
- 779. Nakagawa K, Garon EB, Seto T, et al. Ramucirumab plus erlotinib in patients with untreated, EGFR‐mutated, advanced non‐small‐cell lung cancer (RELAY): a randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet Oncol. 2019;20(12):1655‐1669. [DOI] [PubMed] [Google Scholar]
- 780. Flinn IW, O'Brien S, Kahl B, et al. Duvelisib, a novel oral dual inhibitor of PI3K‐δ,γ, is clinically active in advanced hematologic malignancies. Blood. 2018;131(8):877‐887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 781. Fang H, Guo Z, Chen J, et al. Combination of epigenetic regulation with gene therapy‐mediated immune checkpoint blockade induces anti‐tumour effects and immune response in vivo. Nat Commun. 2021;12(1):6742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 782. Dall'Olio FG, Marabelle A, Caramella C, et al. Tumour burden and efficacy of immune‐checkpoint inhibitors. Nat Rev Clin Oncol. 2022;19(2):75‐90. [DOI] [PubMed] [Google Scholar]
- 783. Zarrin AA, Bao K, Lupardus P, Vucic D. Kinase inhibition in autoimmunity and inflammation. Nat Rev Drug Discov. 2021;20(1):39‐63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 784. Yamaguchi H, Hsu JM, Yang WH, Hung MC. Mechanisms regulating PD‐L1 expression in cancers and associated opportunities for novel small‐molecule therapeutics. Nat Rev Clin Oncol. 2022;19(5):287‐305. [DOI] [PubMed] [Google Scholar]
- 785. Lisberg A, Cummings A, Goldman JW, et al. A Phase II Study of Pembrolizumab in EGFR‐Mutant, PD‐L1+, Tyrosine Kinase Inhibitor Naive Patients With Advanced NSCLC. J Thorac Oncol. 2018;13(8):1138‐1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 786. Wei AH, Montesinos P, Ivanov V, et al. Venetoclax plus LDAC for newly diagnosed AML ineligible for intensive chemotherapy: a phase 3 randomized placebo‐controlled trial. Blood. 2020;135(24):2137‐2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 787. Carrato A, Swieboda‐Sadlej A, Staszewska‐Skurczynska M, et al. Fluorouracil, leucovorin, and irinotecan plus either sunitinib or placebo in metastatic colorectal cancer: a randomized, phase III trial. J Clin Oncol. 2013;31(10):1341‐1347. [DOI] [PubMed] [Google Scholar]
- 788. Mok TSK, Kim SW, Wu YL, et al. Gefitinib plus chemotherapy versus chemotherapy in epidermal growth factor receptor mutation‐positive non‐small‐cell lung cancer resistant to first‐line gefitinib (IMPRESS): overall survival and biomarker analyses. J Clin Oncol. 2017;35(36):4027‐4034. [DOI] [PubMed] [Google Scholar]
- 789. Salvador‐Barbero B, Álvarez‐Fernández M, Zapatero‐Solana E, et al. CDK4/6 inhibitors impair recovery from cytotoxic chemotherapy in pancreatic adenocarcinoma. Cancer Cell. 2020;37(3):340‐353. e346. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table S1 Summary of the pivotal candidates of small molecule kinase inhibitors. Data sources: https://clinicaltrials.gov/
Supplementary Table S2 Summary of the pivotal candidates of small molecule non‐kinase inhibitors. Data sources: https://clinicaltrials.gov/
Supplementary Table S3 Summary of the pivotal candidates of small molecule inhibitors targeting VEGFR.Data sources: https://clinicaltrials.gov/
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.