

Abstract
Background
Globally, liver cancer is one of the most common malignant tumors and is the third leading cause of cancer deaths. RNA-binding protein (RBP) is a general term for a class of proteins that bind to RNA to regulate metabolic processes. The expression of RNA-binding proteins is related to the prognosis of liver cancer patients.
Methods
The RBP gene expression data of liver cancer were extracted from the TCGA database. First, the differentially expressed RBPs (DE RBPs) were selected through enrichment analysis and volcano mapping. Then, the prognosis-related RBP genes were selected through single-factor Cox regression analysis. The key prognosis-related RBPs were further screened by multifactor Cox regression analysis, and a formula for the patient’s risk coefficient was obtained. Finally, based on the patient’s risk score, a nomogram was established and verified.
Results
We extracted 374 cancer tissue samples and 50 normal tissue samples with the clinical information from each sample. Through enrichment analysis, we screened 208 upregulated RBPs and 122 downregulated RBPs. Prognosis-related high-risk genes were EEF1E1, NOP56, UPF3B, SF3B4, SMG5, CD3EAP, BRCA1, BARD1, XPO5, CSTF2, EZH2, EXO1, RRP12, PRIM1, LIN28B, NROB1 and TCOF1, and the low-risk genes were MRPL46, RCL1, MRPL54, CPEB3, IFIT5, PPARGC1A, EIF2AK4, SEPSECS, ACO1, SECISBP2 L and ZCCHC24. Further multivariate Cox regression analysis was performed on the prognosis-related RBPs, and the three key prognosis-related RBPs were screened out, which were BARD1, NR0B1 and EIF2AK4. A patient risk coefficient calculation formula was obtained: risk score = (1.207×BARD1 Exp) + (0.483×NR0B1 Exp) + (-0.720×EIF2AK4 Exp). Finally, a nomogram was established based on the risk score to predict the survival time of patients from 1 to 5 years.
Conclusions
The nomogram has good predictive value for the survival time of liver cancer patients.
Keywords: TCGA, RNA-binding protein (RBP), prognostic model, nomogram, liver cancer
Introduction
Globally, liver cancer is one of the most common malignant tumors and the third leading cause of cancer deaths. According to the global malignant tumor statistics of the International Agency for Research on Cancer (IARC), 905,677 cases of liver cancers were newly diagnosed, and 830,180 patients died in 2020 worldwide (1). Although treatments such as surgical resection and liver transplantation are intended to cure liver cancer (2), the postoperative recurrence rate is still high, and the long-term effect is unsatisfactory for many reasons and the limited beneficiary groups (3).
With the continuous development of molecular biology, tumor research has gradually moved from macroscopic research at the overall organ and tissue levels to microscopic research at the subcellular structure and molecular levels, thereby fundamentally seeking a cure for the disease. Therefore, the study of genes and their related proteins has become a hot spot in today’s research (4-6). Although some scholars have studied the relationship between RNA-binding protein (RBP) and liver cancer, they have focused on the relationship between genes and immune cell infiltration (7,8), and there is no reliable prognostic model to predict patient outcome.
RBP is a general term for a class of proteins that bind to RNA to regulate metabolic processes. RBPs main roles are to mediate RNA maturation, transport, positioning and translation (9). In addition, RBPs can interact with proteins and various types of RNA (mRNAs, ncRNAs, tRNAs, snRNAs, snoRNAs, etc.) to form ribonucleoprotein (RNP) complexes. Therefore, changes in RBP expression or RBP mutations may lead to cancers or other diseases (10,11). Studies have shown that RBP dysregulation is related to the poor prognosis of liver cancer (12-14). Therefore, a prognostic model can be established through RBP to predict the outcome of liver cancer patients.
With the rapid development of sequencing technology and the establishment of TCGA (https://cancergenome.nih.gov), the production of large-scale tumor genomic datasets and comprehensive biological information analysis has become possible (15). This study extracted liver cancer RBP-encoding gene expression profile data from the TCGA database to establish a prognostic model of liver cancer, predict the survival time of liver cancer patients, and provide a theoretical reference for clinicians. We present the following article in accordance with the TRIPOD reporting checklist (available at https://tcr.amegroups.com/article/view/10.21037/tcr-21-2820/rc).
Methods
Data download and analysis
The TCGA database (https://portal.gdc.cancer.gov/) (15) was jointly established by the US National Cancer Institute and the US National Human Genome Research Institute. It is the world’s largest cancer gene information database, incorporating information obtained from various genomic analysis techniques. TCGA has not only developed a large-scale genome sequence dataset but also contains rich samples and information from more than 30 kinds of cancers. Most importantly, for our purposes, TCGA includes very detailed prognostic information. In this study, the gene expression profiles and associated clinical data for liver cancer were collected from the TCGA-GDC (Genomic Data Commons) database (up to May 21, 2020), and the PERL language script was used to handle the raw data. Finally, the clinical information [including ID (identity document) number, survival time, survival status, age, sex, clinical installment, T (tumor) staging, lymph node transfer state, and distant transfer state] of 374 cancer tissue samples and 50 adjacent tissue samples was obtained. The sample mutation data were acquired, analyzed, and visualized using the “maftools” tool (version 2.8.05, https://github.com/PoisonAlien/maftools) in the R package [R version 4.0.0 (2020-04-24), https://mirrors.tuna.tsinghua.edu.cn/CRAN/] (16). Then, the R package “limma” and the Wilcoxon test were used for differential analysis to identify the differential expression of RBP between liver cancer and normal liver tissues. The threshold was |log2 FC|≥0.5, and the adjusted P value was <0.05.
GO and KEGG enrichment analysis of differentially expressed RBPs
GO analysis mainly includes three parts: biological process (BP), cell compositions (CCS) and molecular functions (MFS) (17). KEGG enrichment analysis provides biological interpretation of genome sequences and other high-throughput data (18). We used the R package “clusterProfiler” [R version 4.0.0 (2020-04-24), https://mirrors.tuna.tsinghua.edu.cn/CRAN/] to perform GO and KEGG enrichment analyses on the differentially expressed RBPs, and the significance threshold and false discovery rate (FDR) of P were set to less than 0.05.
Protein-protein interaction (PPI) network construction and subnetwork enrichment analysis and visualization
After we obtained the differentially expressed genes through enrichment analysis, we studied the interactions among these differentially expressed RBPs in the STRING database (19). Then, based on the data in the STRING database, we used Cytoscape (version 3.6.1, https://cytoscape.org/) to construct the PPI network (20) and subnetworks.
Screening and visualization of prognosis-related RBPs
First, RBP gene expression in the prognosis-related PPI network obtained from the TCGA-GDO database was combined with survival time, and “survival” was used in the R software package to perform single-factor Cox regression analysis to determine the differentially expressed RBPs and prognosis-related RBPs. Then, LASSO regression analysis was performed, and the P value was set to <0.01. In addition, we used the R package “glmnet” to screen RBP genes related to prognosis. Then, the forest map was visualized through the HR value, and the prognosis-related RBP genes were obtained.
Model construction and survival analysis of prognosis-related RBPs
The prognosis-related RBPs obtained from the TCGA database were divided into a training group and a test group. The training group was used to construct the model, and the test group was used to verify the accuracy of the model. First, the obtained prognosis-related RBPs were subjected to multivariate Cox regression analysis, the core prognosis-related RBPs were screened out, and their standardized regression coefficients were obtained. In addition, the risk score was calculated by the following formula: Risk Score=Expression of Gene 1×Coefficient of Gene 1+Expression of Gene 2×Coefficient of Gene 2+…Expression of Gene N×Coefficient of Gene N (21). The risk value of each patient was calculated according to the formula of the expression level of each patient’s gene and the risk value, and then the patients were divided into high- and low-risk groups according to the median risk value of the training group. For patients in the test group, the expression levels of these genes are also known, and the risk value of each patient can also be calculated according to the risk value formula. Then, according to the median risk of the training group, patients in the test group were divided into high-risk groups and low-risk groups. The R package “survival” was used to perform Kaplan-Meier curve analysis to analyze the difference in survival between the two groups, and P<0.05 was set as a difference. In addition, the ROC curve was drawn, and the area under the curve (AUC) value was calculated using the R software package “survival ROC” to evaluate the predictive ability (22). If AUC >0.65, it indicates that the curve has a certain accuracy, and if AUC >0.7, the curve has a higher predictive ability. A risk curve was drawn based on the risk values of the high- and low-risk groups to further verify the accuracy of the model.
To verify whether the model can be used as an independent prognostic factor without taking into account other clinical traits, we performed an independent prognostic analysis. Single-factor and multifactor prognostic analyses were performed for the training group and the test group, respectively.
Nomogram establishment of key prognosis-related RBPs
To predict the score of each patient based on the expression of the model gene, we then predicted the survival period of each patient. According to the results of multivariate Cox analysis, we used the R package “rms” to predict the overall survival (OS) at 1, 2, 3, 4, and 5 years in the TCGA cohort of liver cancer patients, and on this basis, a prognostic nomogram of key prognosis-related RBPs was generated to predict the patient’s survival time.
Verification of expression level and prognostic significance
We used the online data of the Human Protein Atlas (HPA) (23) to detect the expression of the central RBP at the translation level and verify whether the normal tissues and tumor tissues were different from each other through immunohistochemical images.
Statistical analysis
The PERL language script was used to handle the gene expression profiles and associated clinical data for liver cancer. And the R package “limma” and the Wilcoxon test were used for differential analysis. The threshold was |log2 FC|≥0.5, and the adjusted P value was <0.05. Also, the R package was used to perform GO and KEGG enrichment analyses on the differentially expressed RBPs, and the significance threshold and FDR of P were set to less than 0.05. The “survival” was used in the R software package to perform single-factor Cox regression analysis to determine the differentially expressed RBPs and prognosis-related RBPs. The multivariate Cox regression analysis was used to screen out the core prognosis-related RBPs.
Ethical statement
The study was conducted in accordance with the Declaration of Helsinki (as revised in 2013).
Results
Screening of differentially expressed RBPs
Using the R package “LIMMA” and Wilcoxon tests, we screened 330 differentially expressed RBP genes between liver cancer tissues and normal liver tissues, including 208 upregulated RBP genes and 122 downregulated RBP genes (as shown online at: https://cdn.amegroups.cn/static/public/tcr-21-2820-01.pdf), and there were significant differences (P<0.05). The heatmap and volcano map are shown in Figure 1.
Figure 1.

Identification of DE RBPs in the TCGA database and enrichment analysis. (A) Heatmap of the DE RBPs based on P values and fold change. Red represents high expression, and green represents low expression. (B) Volcano plot of all DE RBPs between liver cancer and normal samples. Red: upregulated RBPs; black: unchanged RBPs; green: downregulated RBPs. DE RBPs, differentially expressed RNA binding proteins.
GO and KEGG functional enrichment analyses
To understand which functions the differentially expressed RBP genes shown in Figure 1 are involved in, we used the R package to draw a histogram of the differentially expressed RBP upregulated and downregulated genes to perform GO enrichment analysis. The results of GO enrichment analysis of the differentially expressed RBP upregulated genes are shown (Figure 2A): they are significantly enriched in RNA splicing, ncRNA processing, catalytic activity, acting on RNA, RNA splicing, via transesterification reactions with bulged adenosine as nucleophile, mRNA splicing, via spliceosome, RNA splicing, via transesterification reactions, RNA catabolic process, mRNA catabolic process and spliceosomal complex. The results of GO enrichment analysis of the differentially expressed RBP downregulated genes are shown (Figure 2B). The downregulated genes are significantly enriched in regulation of translation, regulation of cellular amide metabolic process, catalytic activity, acting on RNA, RNA catabolic process, regulation of mRNA metabolic process, nucleic acid phosphodiester bond hydrolysis, response to virus, negative regulation of translation, negative regulation of cellular amide metabolic process and defense response to virus.
Figure 2.

Enrichment analysis of GO and KEGG pathways that differentially express RBPs. (A) Histogram of GO enrichment analysis of upregulated genes. (B) Histogram of GO enrichment analysis of downregulated genes. (C) Histogram of KEGG enrichment analysis of upregulated genes. (D) Histogram of KEGG enrichment analysis of downregulated genes. GO, gene ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; RBPs, RNA-binding proteins.
In addition, we also drew a histogram of the differentially expressed RBP genes for the upregulated and downregulated genes to perform KEGG enrichment analysis. The results showed that the differentially expressed RBP upregulated genes are significantly enriched (Figure 2C) in spliceosome, RNA transport, mRNA surveillance pathway, ribosome, ribosome biogenesis in eukaryotes and RNA degradation, while rarely enriched in homologous recombination; the downregulated genes of differentially expressed RBP are significantly enriched in influenza A, RNA degradation, ribosome biogenesis in eukaryotes and hepatitis C (as shown in Figure 2D) but rarely enriched in RNA transport.
PPI network and subnetwork
After we obtained the differentially expressed RBP genes, to understand whether there was a protein interaction relationship among these differentially expressed genes, we used Cytoscape software to build a PPI network based on the data in the STRING database and visualized it to understand the upregulation and downregulation of the RBP genes (shown in Figure 3). In total, the PPI network included 311 nodes and 2942 edges (shown in Figure 3A). We used the MCODE tool to construct a subnetwork and screened out three hub modules. The results showed that subnetwork 1 (shown in Figure 3B) contains 53 genes and 709 nodes, including 39 upregulated genes and 12 downregulated genes. Subnet 2 (Figure 3C) contains 19 genes and 97 nodes, including 4 upregulated genes and 13 downregulated genes. Subnet 3 (Figure 3D) contains 27 genes and 134 nodes, including 16 upregulated genes and 10 downregulated genes.
Figure 3.

PPI network of differentially expressed genes and subnetworks. (A) PPI network of differentially expressed genes. (B) PPI network of subnet 1. (C) PPI network of subnet 2. (D) PPI network of subnet 3. Red: upregulated RBPs; Yellow: unchanged RBPs; Green: downregulated RBPs. PPI, protein-protein interaction; RBPs, RNA-binding proteins.
Prognosis-related RBP screening
To obtain the correlation between RBP genes and survival, we obtained prognosis-related RBP genes according to single factor Cox regression analysis and then visualized the forest map through HR values (Figure 4). The results showed that the prognosis-related high-risk genes were EEF1E1, NOP56, UPF3B, SF3B4, SMG5, CD3EAP, BRCA1, BARD1, XPO5, CSTF2, EZH2, EXO1, RRP12, PRIM1, LIN28B, NR0B1 and TCOF1; the prognosis-related low-risk genes were MRPL46, RCL1, MRPL54, CPEB3, IFIT5, PPARGC1A, EIF2AK4, SEPSECS, ACO1, SECISBP2 L and ZCCHC24.
Figure 4.

Forest plot of prognosis-related RBP genes. RBP, RNA-binding protein.
Model construction and survival analysis of prognosis-related RBPs
We performed multivariate Cox regression analysis on the prognosis-related RBP genes and identified three key prognosis-related RBP genes, BARD1, NR0B1 and EIF2AK4, and the results are shown in Figure 5. Meanwhile, the formula for calculating the patient risk coefficient was obtained: risk score= (1.207×BARD1 Exp) +(0.483×NR0B1 Exp) +(-0.720×EIF2AK4 Exp), where BARD1 and NR0B1 are prognostic-related high-risk factors (HR >1), and EIF2AK4 is a protective factor (HR <1).
Figure 5.
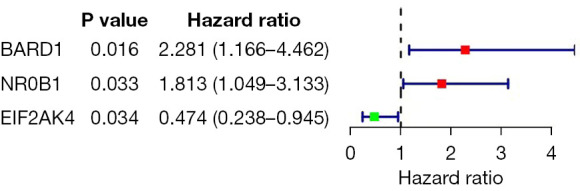
Forest plot of prognosis-related key RBP genes. RBP, RNA-binding protein.
We drew a Kaplan-Meier curve to analyze the difference in survival between the test group and the training group. The results of the test group (as shown in Figure 6A), P=3.67e−03<0.05, were statistically significant; the results of the training group (Figure 6B) showed that P=9.671e−07<0.05, which were also statistically significant. Moreover, both groups showed that the OS of low-risk patients was significantly longer than that of high-risk patients (P=3.67e−03; P=9.671e−07). In addition, we drew an ROC curve to evaluate the predictive ability. The results of the test group (Figure 6C) showed that AUC =0.740>0.7, which is statistically significant, indicating that the model has good predictive ability; the results of the training group are shown in Figure 6D, AUC =0.717>0.7, which is statistically significant, indicating that the model has good predictive ability.
Figure 6.

Survival difference between the test group and the training group. (A) Kaplan-Meier survival curve of the test group. (B) Kaplan-Meier survival curve of the training group. (C) ROC curve of the test group. (D) ROC curve of the training group. ROC, receiver operating characteristic; AUC, area under the curve.
In addition, we drew a risk curve to show the risk scores and survival status of the high-risk and low-risk groups in the TCGA database. The results of the training group are shown in Figure 7A,7B, indicating that as the risk score increases, the number of deaths from hepatocellular carcinoma (HCC) also increases. Then, a heatmap was drawn to show the expression levels of the three key genes in the two groups, as shown in Figure 7C.
Figure 7.

Risk curve analysis of the two groups. (A) Risk score distribution of the training group. (B) Survival time of the training group. (C) Three-gene expression heatmap of the training group. (D) Risk score distribution of the test group. (E) Survival time of the test group. (F) Three-gene expression heatmap of the test group.
To verify the predictive ability of the model, we drew the risk curve of the test group. The results are shown in Figure 7D,7E. The OS of the high-risk group was significantly lower than that of the low-risk group. A heatmap was drawn to show the expression levels of the three core genes of the two groups, as shown in Figure 7F. The results of the training and test groups were comparable, indicating that our prognostic markers have considerable stability in predicting the OS of liver cancer patients.
We performed an independent prognostic analysis to verify whether the model can be used as an independent prognostic factor independent of other clinical traits. The results of the independent prognostic analysis of the training group are shown (Figure 8A,8B). The P values of tumor stage and risk value were all <0.001, which was statistically significant, while the P values of patient age, sex and degree of differentiation were all >0.05, not statistically significant. The independent prognostic analysis of the test group also obtained the same result (Figure 8C,8D).
Figure 8.

Independent prognostic analysis of the two groups. (A) Univariate analysis of the training group. (B) Multivariate analysis of the training group. (C) Univariate analysis of the test group. (D) Multivariate analysis of the test group.
Nomogram establishment for the three key prognosis-related RBP genes
We drew a prognostic nomogram of the prognosis-related RBP genes to predict the survival time of each patient based on each patient’s risk score, which was calculated from the formula we obtained before. The score corresponding to each gene and the survival time corresponding to the total score of all genes of the patients are shown in Figure 9.
Figure 9.

A nomogram of liver cancer in the TCGA database.
Verification of the expression level in the HPA database
To verify whether there were differences between normal tissues and tumor tissues, we collected BARD1 and EIF2AK4 immunohistochemical specimen images for comparison by using the online data of the HPA (21), but unfortunately, images of NR0B1 immunohistochemistry specimens have not been included so far. The collected pathological results show that the tissue structures of the normal tissue and tumor tissue are different, as shown in Figure 10.
Figure 10.
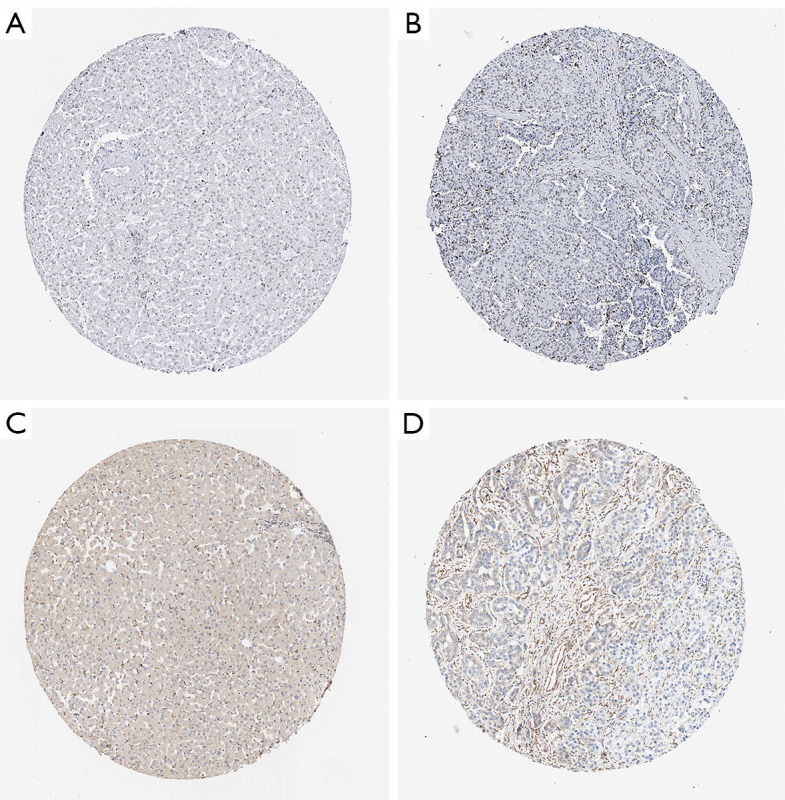
Immunohistochemistry of two RBPs using the HPA database. (A) Normal tissue expression of BRAD1. (B) Tumor tissue expression of BRAD1. (C) Normal tissue expression of EIF2AK4. (D) Tumor tissue expression of EIF2AK4. RBP, RNA-binding protein.
Discussion
With the rapid development of high-throughput sequencing technology, bioinformatics technology has become a powerful tool for screening biomarkers. Caruso et al. (24) used this technology to find the response markers of liver cancer and determined the genetic changes and gene expression patterns related to the drug response. Many studies have found a relationship between specific molecular markers of liver cancer and its treatment and prognosis (25-27). However, there is no relevant previous research on an RBP prognostic model of liver cancer. Therefore, the purpose of this study was to establish an RBP prognostic model for liver cancer to predict the survival of patients.
First, we used the genes and clinical information in the TCGA database to obtain 208 upregulated RBP genes and 122 downregulated RBP genes. Then, the related biological pathways were further analyzed, and a PPI network of RBPs was constructed. Next, through univariate Cox regression analysis, we found that a total of 28 prognosis-related RBPs were significantly related to liver cancer. Afterward, through LOSSO analysis and multivariate Cox regression analysis, three key RBP genes (BARD1, EIF2AK4 and NR0B1) were identified. Subsequently, we conducted survival and ROC curve analysis to further explore their clinical significance. Finally, we constructed a risk model based on 3 prognosis-related key RBP genes to construct a nomogram to predict the survival of liver cancer patients. After verification, we found that the model is useful for predicting the survival of liver cancer patients.
We obtained through GO enrichment analysis that the upregulated genes are significantly enriched in RNA splicing, ncRNA processing, catalytic activity, acting on RNA, RNA splicing, via transesterification reactions with bulged adenosine as nucleophile, mRNA splicing, via spliceosome, RNA splicing, via transesterification reactions, RNA catabolic process, mRNA catabolic process and spliceosomal complex, while the downregulated genes are significantly enriched in regulation of translation, regulation of cellular amide metabolic process, catalytic activity, acting on RNA, RNA catabolic process, regulation of mRNA metabolic process, nucleic acid phosphodiester bond hydrolysis, response to virus, negative regulation of translation, negative regulation of cellular amide metabolic process and defense response to virus.
In recent years, many studies have confirmed the role of RBPs in various diseases (28-30). An RBP is generally considered to be a protein that binds to RNA through one or more spherical RNA binding domains and changes the fate or function of the bound RNA. These effects on the RNAs include changing their subcellular localization, causing alternative splicing, affecting their translation efficiency, and altering their metabolism, which all play key roles in RNA dynamics (31).
Fragile×mental retardation protein (FMRP) mainly plays a role in the nervous system; it plays the role of an RBP and controls the translation of its target mRNAs (32). Therefore, circZKSCAN1 inhibits the cell stemness of liver cancer by regulating the function of RBP FMRP, inhibiting the progression of liver cancer (33).
The analysis of the enriched KEGG pathways showed that the upregulated genes are significantly enriched in spliceosome, RNA transport, mRNA surveillance pathway, ribosome, ribosome biogenesis in eukaryotes and RNA degradation, while the downregulated genes are significantly enriched in influenza A, RNA degradation, ribosome biogenesis in eukaryotes and hepatitis C. On this issue, there are similar conclusions in the research on colon cancer (34).
Through univariate Cox regression analysis, we found 28 prognosis-related RBPs, and through multivariate Cox regression analysis, we identified three key prognosis-related RBPs, including BARD1, EIF2AK4 and NR0B1.
In 1996, Wu et al. (35) discovered the BARD1 gene while investigating the biological function of the BRCA1 protein, and they also found that BARD1 directly interacts with BRCA1 through its N-terminal loop domain and that the BRCA1/BARD1 complex is involved in DNA repair and centrosome regulation. The centrosome is the main microtubule organization center in animal cells and is essential for the formation of the bipolar mitotic spindle. BRCA1 and BARD1 are located in the centrosome during the cell cycle, and BRCA1/BARD1 dimers ubiquitinate centrosome proteins to regulate centrosome function and then participate in the process of tumorigenesis (36). BARD1 is considered to be a potential pathogenic gene in colorectal cancer (37,38), endometrial cancer (39) and pancreatic cancer (40). However, there is no related research on BARD1 and liver cancer, so multicenter experiments should be conducted to further confirm its role in liver cancer.
Mutations in the NR0B1 gene were first mentioned in adrenal hypoplasia congenita (AHC) (41). NR0B1 has an unusual structure; the carboxy terminal region of the protein contains 12 helices typical of other nuclear receptors, while the amino terminal region contains 3.5 repeats, approximately 66–67 amino acids, and contains the LXXLL motif (42). NR0B1 is expressed in progenitor stem cells, where it can inhibit differentiation, thereby allowing the stem cell population to expand (43). Related to this process, some mechanism is involved in tumorigenesis. Zhang et al. (44) found that high expression of NR0B1 is associated with a better prognosis in operable node-negative breast cancer. However, its role in liver cancer needs further research.
EIF2AK4 gene mutation has been confirmed in pulmonary vein occlusive disease (PVOD) and pulmonary capillary hemangioma (PCH) (45,46), but there are no relevant studies confirming its pathogenesis and prognostic roles in liver cancer. Huang et al. (47) also confirmed through similar studies that CNOT6, UPF3B, MRPL54, IFIT5 and PPARGC1A are the key RBP coding genes for primary HCC and can also predict the survival of patients. Their results are different from ours, which may be caused by using different reference databases. Research on genes related to the prognosis of liver cancer is in the initial stage. There is no relevant literature to explain the mechanisms of these genes. Therefore, to verify the pathogenesis of these genes, more multicenter basic experiments are needed on prognosis-related genes of liver cancer.
In summary, we obtained prognosis-related RBP genes of liver cancer through screening. Further screening identified three key prognosis-related genes of liver cancer, BARD1, EIF2AK4 and NR0B1. Among them, high expression of BARD1 and NR0B1 is associated with a poor prognosis, and high expression of EIF2AK4 is associated with a better prognosis. In addition, a nomogram was constructed based on the risk coefficient calculation formula to predict the survival of patients from 1 to 5 years. Even though this research provided reference information for the prognosis of liver cancer, more basic research is necessary to verify these outcomes.
Supplementary
The article’s supplementary files as
Acknowledgments
Funding: This work was supported by the Xinjiang Uygur Autonomous Region Health Commission Youth Science and Technology Innovation Project (No. WJWY-202014).
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. The study was conducted in accordance with the Declaration of Helsinki (as revised in 2013).
Footnotes
Reporting Checklist: The authors have completed the TRIPOD reporting checklist. Available at https://tcr.amegroups.com/article/view/10.21037/tcr-21-2820/rc
Conflicts of Interest: All authors have completed the ICMJE uniform disclosure form (available at https://tcr.amegroups.com/article/view/10.21037/tcr-21-2820/coif). The authors have no conflicts of interest to declare.
References
- 1.Sung H, Ferlay J, Siegel RL, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin 2021;71:209-49. 10.3322/caac.21660 [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin 2019;69:7-34. 10.3322/caac.21551 [DOI] [PubMed] [Google Scholar]
- 3.Zhang T, Liu Z, Zhao X, et al. A novel prognostic score model based on combining systemic and hepatic inflammation markers in the prognosis of HBV-associated hepatocellular carcinoma patients. Artif Cells Nanomed Biotechnol 2019;47:2246-55. 10.1080/21691401.2019.1573174 [DOI] [PubMed] [Google Scholar]
- 4.Fu J, Wang H. Precision diagnosis and treatment of liver cancer in China. Cancer Lett 2018;412:283-8. 10.1016/j.canlet.2017.10.008 [DOI] [PubMed] [Google Scholar]
- 5.Pipas JM. DNA Tumor Viruses and Their Contributions to Molecular Biology. J Virol 2019;93:e01524-18. 10.1128/JVI.01524-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang W, Wang C, Xu H, et al. Aldehyde Dehydrogenase, Liver Disease and Cancer. Int J Biol Sci 2020;16:921-34. 10.7150/ijbs.42300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu Y, Yang Y, Luo Y, et al. Prognostic potential of PRPF3 in hepatocellular carcinoma. Aging (Albany NY) 2020;12:912-30. 10.18632/aging.102665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jiang Y, Chen S, Li Q, et al. TANK-Binding Kinase 1 (TBK1) Serves as a Potential Target for Hepatocellular Carcinoma by Enhancing Tumor Immune Infiltration. Front Immunol 2021;12:612139. 10.3389/fimmu.2021.612139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mohibi S, Chen X, Zhang J. Cancer the'RBP'eutics-RNA-binding proteins as therapeutic targets for cancer. Pharmacol Ther 2019;203:107390. 10.1016/j.pharmthera.2019.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gerstberger S, Hafner M, Tuschl T. A census of human RNA-binding proteins. Nat Rev Genet 2014;15:829-45. 10.1038/nrg3813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Neelamraju Y, Gonzalez-Perez A, Bhat-Nakshatri P, et al. Mutational landscape of RNA-binding proteins in human cancers. RNA Biol 2018;15:115-29. 10.1080/15476286.2017.1391436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dang H, Takai A, Forgues M, et al. Oncogenic Activation of the RNA Binding Protein NELFE and MYC Signaling in Hepatocellular Carcinoma. Cancer Cell 2017;32:101-114.e8. 10.1016/j.ccell.2017.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kang C, Jia X, Liu H. Development and validation of a RNA binding protein gene pair-associated prognostic signature for prediction of overall survival in hepatocellular carcinoma. Biomed Eng Online 2020;19:68. 10.1186/s12938-020-00812-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang X, Zhang J, Gao F, et al. KPNA2-Associated Immune Analyses Highlight the Dysregulation and Prognostic Effects of GRB2, NRAS, and Their RNA-Binding Proteins in Hepatocellular Carcinoma. Front Genet 2020;11:593273. 10.3389/fgene.2020.593273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tomczak K, Czerwińska P, Wiznerowicz M. The Cancer Genome Atlas (TCGA): an immeasurable source of knowledge. Contemp Oncol (Pozn) 2015;19:A68-77. 10.5114/wo.2014.47136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mayakonda A, Koeffler HP. Maftools: efficient analysis, visualization and summarization of MAF files from large-scale cohort based cancer studies. BioRxiv 052662 . Available online: https://www.biorxiv.org/content/10.1101/052662v1 10.1101/052662 [DOI]
- 17.Ashburner M, Ball CA, Blake JA, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 2000;25:25-9. 10.1038/75556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kanehisa M, Sato Y, Kawashima M, et al. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res 2016;44:D457-62. 10.1093/nar/gkv1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Szklarczyk D, Gable AL, Nastou KC, et al. The STRING database in 2021: customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res 2021;49:D605-12. 10.1093/nar/gkaa1074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 2003;13:2498-504. 10.1101/gr.1239303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu M, Li X, Zhang T, et al. Identification of a Nine-Gene Signature and Establishment of a Prognostic Nomogram Predicting Overall Survival of Pancreatic Cancer. Front Oncol 2019;9:996. 10.3389/fonc.2019.00996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hanley JA, McNeil BJ. The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology 1982;143:29-36. 10.1148/radiology.143.1.7063747 [DOI] [PubMed] [Google Scholar]
- 23.Thul PJ, Åkesson L, Wiking M, et al. A subcellular map of the human proteome. Science 2017;356:eaal3321. 10.1126/science.aal3321 [DOI] [PubMed] [Google Scholar]
- 24.Caruso S, Calatayud AL, Pilet J, et al. Analysis of Liver Cancer Cell Lines Identifies Agents With Likely Efficacy Against Hepatocellular Carcinoma and Markers of Response. Gastroenterology 2019;157:760-76. 10.1053/j.gastro.2019.05.001 [DOI] [PubMed] [Google Scholar]
- 25.Yao M, Yang JL, Wang L, et al. Carcinoembryonic type specific markers and liver cancer immunotherapy. Zhonghua Gan Zang Bing Za Zhi 2020;28:466-70. [DOI] [PubMed] [Google Scholar]
- 26.Xu RH, Wei W, Krawczyk M, et al. Circulating tumour DNA methylation markers for diagnosis and prognosis of hepatocellular carcinoma. Nat Mater 2017;16:1155-61. 10.1038/nmat4997 [DOI] [PubMed] [Google Scholar]
- 27.Gu Y, Zheng F, Zhang Y, et al. Novel Nomogram Based on Inflammatory Markers for the Preoperative Prediction of Microvascular Invasion in Solitary Primary Hepatocellular Carcinoma. Cancer Manag Res 2022;14:895-907. 10.2147/CMAR.S346976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Z, Lei X. Matrix factorization with neural network for predicting circRNA-RBP interactions. BMC Bioinformatics 2020;21:229. 10.1186/s12859-020-3514-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zang J, Lu D, Xu A. The interaction of circRNAs and RNA binding proteins: An important part of circRNA maintenance and function. J Neurosci Res 2020;98:87-97. 10.1002/jnr.24356 [DOI] [PubMed] [Google Scholar]
- 30.Zhang M, Wang T, Xiao G, et al. Large-Scale Profiling of RBP-circRNA Interactions from Public CLIP-Seq Datasets. Genes (Basel) 2020;11:54. 10.3390/genes11010054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hentze MW, Castello A, Schwarzl T, et al. A brave new world of RNA-binding proteins. Nat Rev Mol Cell Biol 2018;19:327-41. 10.1038/nrm.2017.130 [DOI] [PubMed] [Google Scholar]
- 32.Alpatov R, Lesch BJ, Nakamoto-Kinoshita M, et al. A chromatin-dependent role of the fragile X mental retardation protein FMRP in the DNA damage response. Cell 2014;157:869-81. 10.1016/j.cell.2014.03.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhu YJ, Zheng B, Luo GJ, et al. Circular RNAs negatively regulate cancer stem cells by physically binding FMRP against CCAR1 complex in hepatocellular carcinoma. Theranostics 2019;9:3526-40. 10.7150/thno.32796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu D, Chen J, Hou T. Development and Validation of a Prognostic Model of RNA-Binding Proteins in Colon Adenocarcinoma: A Study Based on TCGA and GEO Databases. Cancer Manag Res 2021;13:7709-22. 10.2147/CMAR.S330434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu LC, Wang ZW, Tsan JT, et al. Identification of a RING protein that can interact in vivo with the BRCA1 gene product. Nat Genet 1996;14:430-40. 10.1038/ng1296-430 [DOI] [PubMed] [Google Scholar]
- 36.Otsuka K, Yoshino Y, Qi H, et al. The Function of BARD1 in Centrosome Regulation in Cooperation with BRCA1/OLA1/RACK1. Genes (Basel) 2020;11:842. 10.3390/genes11080842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Son HJ, Choi EJ, Yoo NJ, et al. Somatic frameshift mutations of cancer-related genes KIF3C and BARD1 in colorectal cancers. Pathol Res Pract 2019;215:152579. 10.1016/j.prp.2019.152579 [DOI] [PubMed] [Google Scholar]
- 38.Zhang YQ, Pilyugin M, Kuester D, et al. Expression of oncogenic BARD1 isoforms affects colon cancer progression and correlates with clinical outcome. Br J Cancer 2012;107:675-83. 10.1038/bjc.2012.297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ring KL, Bruegl AS, Allen BA, et al. Germline multi-gene hereditary cancer panel testing in an unselected endometrial cancer cohort. Mod Pathol 2016;29:1381-9. 10.1038/modpathol.2016.135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hu C, Hart SN, Bamlet WR, et al. Prevalence of Pathogenic Mutations in Cancer Predisposition Genes among Pancreatic Cancer Patients. Cancer Epidemiol Biomarkers Prev 2016;25:207-11. 10.1158/1055-9965.EPI-15-0455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Muscatelli F, Strom TM, Walker AP, et al. Mutations in the DAX-1 gene give rise to both X-linked adrenal hypoplasia congenita and hypogonadotropic hypogonadism. Nature 1994;372:672-6. 10.1038/372672a0 [DOI] [PubMed] [Google Scholar]
- 42.Zanaria E, Muscatelli F, Bardoni B, et al. An unusual member of the nuclear hormone receptor superfamily responsible for X-linked adrenal hypoplasia congenita. Nature 1994;372:635-41. 10.1038/372635a0 [DOI] [PubMed] [Google Scholar]
- 43.Suntharalingham JP, Buonocore F, Duncan AJ, et al. DAX-1 (NR0B1) and steroidogenic factor-1 (SF-1, NR5A1) in human disease. Best Pract Res Clin Endocrinol Metab 2015;29:607-19. 10.1016/j.beem.2015.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang H, Slewa A, Janssen E, et al. The prognostic value of the orphan nuclear receptor DAX-1 (NROB1) in node-negative breast cancer. Anticancer Res 2011;31:443-9. [PubMed] [Google Scholar]
- 45.Ma L, Bao R. Pulmonary capillary hemangiomatosis: a focus on the EIF2AK4 mutation in onset and pathogenesis. Appl Clin Genet 2015;8:181-8. 10.2147/TACG.S68635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Best DH, Sumner KL, Smith BP, et al. EIF2AK4 Mutations in Patients Diagnosed With Pulmonary Arterial Hypertension. Chest 2017;151:821-8. 10.1016/j.chest.2016.11.014 [DOI] [PubMed] [Google Scholar]
- 47.Huang Y, Chen S, Qin W, et al. A Novel RNA Binding Protein-Related Prognostic Signature for Hepatocellular Carcinoma. Front Oncol 2020;10:580513. 10.3389/fonc.2020.580513 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The article’s supplementary files as